

Abstract
Background/Objective
AKT2 is a serine/threonine kinase that plays a key role in regulating insulin signaling. The phenotype related to the gain-of-function alteration in the AKT2 gene (c.49G>A, p.Glu17Lys) has been described in 5 patients with clinical findings that mimic hyperinsulinemic hypoglycemia but with undetectable levels of insulin and C-peptide. One of the reports highlights the facial dysmorphic features. We report the case of a new patient with the same activating AKT2 alteration leading to autonomous activation of the insulin signaling pathway and dysmorphic features. Moreover, to our knowledge, this is the first report using waxy maize heat-modified starch (WMHMS) in this condition.
Case Report
A previously healthy child was evaluated at 6 months of age for episodes of hypoglycemia. The laboratory test results for the critical samples showed hypoketotic hypoglycemia (glucose level, 2.16 mmol/L [38 mg/dL]) with undetectable levels of insulin (<0.2 mU/L) and C-peptide (<0.033 nmol/L [reference range, 0.37-1.47 nmol/L]). Physical examination revealed hypertelorism, prominent proptosis of the eyes, a flat nasal bridge, delayed psychomotor development, and postnatal symmetrical overgrowth. The genetic study of AKT2 showed a pathogenic variant (c.49G>A, p.Glu17Lys). To achieve euglycemia, a diet of regular uncooked cornstarch (UCCS) carbohydrate was started. Subsequently, waxy maize heat-modified starch (WMHMS; Glycosade Vitaflo) was used to increase the fasting period to 4 hours. However, we did not find any advantages in comparison with UCCS.
Discussion
The range of phenotypes of this gain-of-function alteration in AKT2 may be broad, including dysmorphic features, although the patients harbor the same pathogenic variant.
Conclusion
Regarding the treatment, we observed a similar response with WMHMS compared with UCCS, with no adverse effects.
Key words: hyperinsulinism, hypoglycemia, overgrowth
Abbreviations: UCCS, uncooked cornstarch; WMHMS, waxy maize heat-modified starch
Introduction
Hyperinsulinemic hypoglycemia represents an important cause of persistent and severe hypoglycemia in newborns and is characterized by unregulated insulin secretion, leading to persistently low blood glucose concentrations, and low serum levels of free fatty acids and β-hydroxybutyrate.1 However, in some cases, the levels of serum insulin may be undetectable due to its short half-life and because of alterations in the PI3K-AKT-mTOR pathway (phosphatidylinositol 3-kinase/protein kinase B/rapamycin target in mammals), which is a key mediator of the action of insulin on target tissues.2,3 In such cases, we observe hypoglycemia with undetectable levels of serum insulin and C-peptide because of the autonomous activity of this pathway in the absence of the physiologic ligand insulin, as the result of increased glucose uptake through redistribution of glucose transporter 4 (GLUT4) from intracellular sites to the plasma membrane, and reduced hepatic gluconeogenesis.4
AKT2 is a serine/threonine kinase that plays a key role in regulating cell survival, insulin signaling, angiogenesis, and tumor formation and a downstream mediator of PI3K phosphorylation, which regulates glucose transport. Most pathogenic variants of the gene (AKT2) encoding this enzyme have been reported in the context of cancer; however, only 1 alteration (p.Glu17Lys) has been reported in relation to nonketotic hypoglycemia.5
In this report, we describe the clinical features and management of a patient with hypoinsulinemic hypoglycemia due to the same pathogenic variant of AKT2gene.
Case Report
The patient was a full-term boy, with a birth weight of 3800 g (75th percentile) and height of 51 cm (59th percentile), born to nonconsanguineous parents. No family medical records were noted.
At the age of 6 months, he presented with generalized tonic-clonic seizures, evaluated by a telehealth pediatric neurologist who began treatment with valproic acid. Electroencephalography and metabolic screening were performed, both of which yielded normal results. At the age of 7 months, he had another seizure. Interictal electroencephalography was performed, and it was normal; however, laboratory tests showed hypoglycemia with a glucose level of 1.67 mmol/L (30 mg/dL). A glucose infusion of 5 mg/kg/min was needed to achieve normoglycemia. Nonetheless, frequent hypoglycemic episodes occurred after stopping the glucose infusion in-between feedings every 3 hours. Critical blood samples were obtained, showing hypoketotic hypoglycemia with undetectable levels of insulin and C-peptide (Table, episode 1). A glucagon test showed a glycemic increment from 1.67 mmol/L (30 mg/dL) to 4.62 mmol/L (83 mg/dL) following glucagon administration, which confirmed glycogen storage in the liver. Regarding other findings of the critical sample, a growth hormone concentration of 15 mIU/L (reference value, >21 mIU/L) and insulin-like growth factor 1 level of 28 ng/mL (reference range, 15-129 ng/mL) were found (Table). Other parameters of the critical sample were normal.
Table.
Critical Sample During Hypoglycemia
| Hormonal and biochemical parameters | Episodes |
|
|---|---|---|
| 1 | 2 | |
| Glucose, mmol/L (3.5-5.5) [mg/dL] | 2.16 [38] | 2.10 [37] |
| Insulin, mU/L | <0.2 | <1 |
| C-peptide, nmol/L (0.37-1.47) | <0.033 | <0.006 |
| BOHB, mmol/L (0.01-0.26) | 0.14 | 0.16 |
| Growth hormone, mIU/L (>21) | 15 | 8.1 |
| IGF-1, ng/mL (15-129) | 28 | 31 |
| Cortisol, nmol/L (>497) | 518 | 513 |
| Fasting time, h | 2 | 3 |
Abbreviations: BOHB = β-Hydroxybutyrate; IGF-1 = insulin-like growth factor 1.
Reference values are provided in parenthesis.
During this time, the child failed to respond to diazoxide (starting dosage of 8 mg/kg/d that was increased to 18 mg/kg/d) and hydrochlorothiazide (3 mg/kg/d), which was stopped because of pericardial effusion. Subsequently, effusion subcutaneous octreotide (33 μg/kg/d) was started; however, overnight euglycemia could only be maintained with intravenous infusions of 5% dextrose at a rate of 3 to 4 mg/kg/min. Due to treatment failure, he was transferred to a tertiary center.
Physical examination revealed hypertelorism, prominent proptosis of his eyes, flat nasal bridge, and delayed psychomotor development (Fig. 1). The rest of the clinical physical examination was unremarkable; there was no macroglossia or other stigmas of Beckwith-Wiedemann phenotype and no asymmetrical extremities. According to the World Health Organization growth charts, at the age of 8 months, he reached a +3.9 SD weight-for-length z-score, a +1.7 SD length-for-age z-score, and a +3.8 head circumference-for-age z-score. Due to the macrocephaly, magnetic resonance imaging of the brain was performed, which showed normal findings; an abdominal ultrasound showed no visceromegaly. Repeated measurements of hormones and intermediary metabolites after 3 days of octreotide suspension confirmed spontaneous and fasting hypoketotic, hypoinsulinemic hypoglycemia (Table, episode 2). Regarding the clinical and biochemical findings, sequencing of the AKT2 gene was performed at the University of Exeter in the United Kingdom, showing a de novo pathogenic variant (c.49G>A, p.Glu17Lys NM_001626.5) in the patient, which was not detected in his parents.5
Fig. 1.
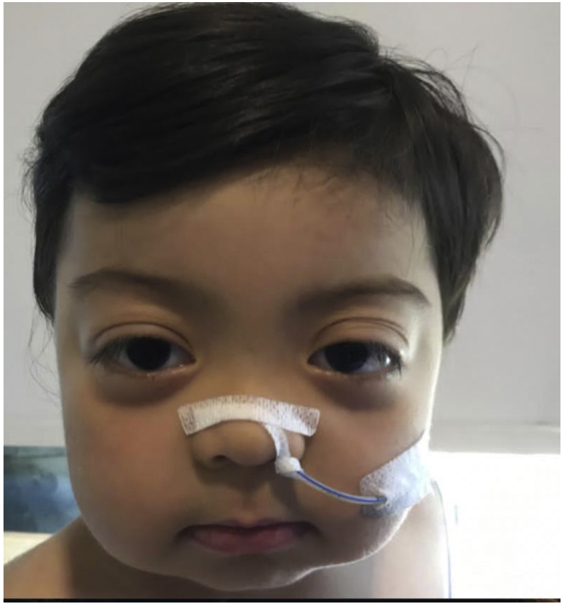
Dysmorphic findings in our patient.
Nutritional Management
Initially, the patient needed regular feedings every 2 hours during the day and night, with continuous overnight nasogastric feeding. However, blood glucose levels fluctuated, requiring the addition of uncooked cornstarch (UCCS) to his treatment. The fasting period tolerated during the day and at night was not >3 hours, and a dosage of 0.5 g/kg/d of UCCS was needed. Fingerstick blood glucose testing revealed no events of hypoglycemia.
After discharge, we maintained the fasting period at 3 hours, achieving blood glucose targets without any change until he was 1 year old. We subsequently tried to increase these fasting periods to 4 hours using waxy maize heat-modified starch (WMHMS; Glycosade, Vitaflo) to provide a source of slow-release carbohydrates. The dose of WMHMS was calculated by replacing the traditional UCCS as dosing standards for WMHMS have not been established.
For a week, we only used WMHMS at night, starting at 11:15 PM with a loading dose of 0.5 g/kg, and subsequently continued with a dose of 0.3 g/kg every 4 hours. In the second week, we changed to UCCS with the same dose; both were administered via a nasogastric tube with cow’s milk formula. To evaluate the response to a 4-hour fasting period with both products, capillary blood glucose was measured after 3 hours of fasting and subsequently every 15 to 20 minutes until completing 4 hours. We did not observe any differences between UCCS and WMHMS. Also, no gastrointestinal symptoms were observed, except for mild constipation with WMHMS. As we did not observe any advantage with the use of WMHMS, we decided to use UCCS (Fig. 2).
Fig. 2.

Metabolic control after using uncooked cornstarch and waxy maize heat-modified starch. UCCS = uncooked cornstarch; WMHMS = waxy maize heat-modified starch.
Discussion
The presentation of hypoketotic hypoglycemia, undetectable levels of serum insulin and C-peptide, and postnatal overgrowth suggests the autonomous activity of downstream insulin signaling, which may be related to the alteration in the PI3K/AKT/mTOR pathway. Typical findings related to PI3K alterations include megalencephaly-capillary malformation and megalencephaly-polydactyly polymicrogyria-hydrocephalus, which are absent when the AKT2 gene is affected. Here, we describe the case of a patient showing the same pathogenic variant (c.49G>A, p.Glu17Lys) in AKT2 in the context of hypoglycemic hypoinsulinemia; the patient also presented with facial anomalies, including hypertelorism, prominent proptotic eyes, and a flat nasal bridge.6
The phenotype of this gain-of-function alteration in AKT2 has been previously described in 5 patients with clinical findings that mimic hyperinsulinemic hypoglycemia, such as postnatal overgrowth, hemihypertrophy, and hypoglycemia related to fasting, but without detectable levels of serum insulin and C-peptide. One of the reports highlights the facial dysmorphic features that are similar to those of our patient but are not found in other affected patients, although they harbor the same pathogenic variant.3,5,6
Regarding the laboratory results of the critical samples of the 5 patients previously described, no alterations in other counter-regulatory hormones were present. Nonetheless, in our patient, we found an attenuated growth hormone response during hypoglycemia, which may reflect tonically active AKT2 or PI3K in the hypothalamus and/or pituitary, mimicking insulin-like growth factor 1 action that activates negative feedback inhibition of growth hormone release.7
Another consequence of excess downstream insulin signaling is the proband postnatal overgrowth with obesity and tall stature, with almost no changes at 13 months of age. Unlike other affected patients with this variant, this child does not have an asymmetrical overgrowth; nonetheless, different phenotypes found in patients with germline or mosaic AKT2 p.Glu17Lys could be related to the different functions of AKT2 through the different tissues in which it is expressed.8
Given the mechanism of hypoglycemia, these patients do not respond to diazoxide or octreotide; euglycemia can only be achieved with a regular carbohydrate diet.9 In this patient, we used WMHMS (Glycosade), a drug approved since 2007 to treat hypoglycemia in children aged >2 years with glycogen storage disease.10 This is a modified cornstarch that provides a slow-release carbohydrate source over approximately 8 hours, in contrast to other cornstarches with a duration of approximately 4 hours. This treatment allows better glycemic control with fewer adverse effects, such as abdominal bloating or flatulence related to cornstarch, thus improving tolerance and treatment adherence.11,12 Unfortunately, we did not find improvement in his glycemia with WMHMS, which could be due to the different metabolism of modified starch by the gastric enzyme according to his age.
Conclusion
This is the report of a new patient, who presented with dysmorphic features similar to those of another studied patient, with the same activating AKT2 alteration leading to autonomous activation of the downstream insulin signaling pathway. This case report highlights that in the presence of the same pathogenic variant, the phenotype could be variable, including dysmorphic facial features, symmetrical overgrowth, and an attenuated growth hormone response to hypoglycemia. The use of WMHMS, not previously described in any patient aged <2 years with an AKT2 variant, did not show an increase in the fasting period; however, its long duration of action could be an advantage later in life, as in the cases in which the benefits have been reported. We observed a glycemic response similar to that with the use of UCCS with no adverse effects.
Author Contributions
M.F.O.M. designed, conducted, reported, and drafted the work; H.P., V.D.T., and C.M. drafted and reviewed the work; K.H. supervised and reviewed the final approval of the manuscript version to be published.
Disclosure
The authors have no multiplicity of interest to disclose.
Statement of Ethics
Written informed consent was obtained from the patient's parent or guardian for publication of the images.
Supplementary Material
References
- 1.Gϋemes M., Rahman S.A., Kapoor R.R., et al. Hyperinsulinemic hypoglycemia in children and adolescents: recent advances in understanding of pathophysiology and management. Rev Endocr Metab Disord. 2020;21(4):577–597. doi: 10.1007/s11154-020-09548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Otaibi H., Senniappan S., Alam S., Hussain K. Biochemical studies in patients with hyperinsulinaemic hypoglycaemia. Eur J Pediatr. 2013;172(11):1435–1440. doi: 10.1007/s00431-013-2053-0. [DOI] [PubMed] [Google Scholar]
- 3.Arya V.B., Flanagan S.E., Schober E., Rami-Merhar B., Ellard S., Hussain K. Activating AKT2 mutation: hypoinsulinemic hypoketotic hypoglycemia. J Clin Endocrinol Metab. 2014;99(2):391–394. doi: 10.1210/jc.2013-3228. [DOI] [PubMed] [Google Scholar]
- 4.Huang S., Czech M.P. The GLUT4 glucose transporter. Cell Metab. 2007;5(4):237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Hussain K., Challis B., Rocha N., et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334(6055):474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garg N., Bademci G., Foster J., Sıklar Z., Berberoglu M., Tekin M. MORFAN syndrome: an infantile hypoinsulinemic hypoketotic hypoglycemia due to an AKT2 mutation. J Pediatr. 2015;167(2):489–491. doi: 10.1016/j.jpeds.2015.04.069. [DOI] [PubMed] [Google Scholar]
- 7.Leiter S.M., Parker V.E.R., Welters A., et al. Hypoinsulinaemic, hypoketotic hypoglycaemia due to mosaic genetic activation of PI3-kinase. Eur J Endocrinol. 2017;177(2):175–186. doi: 10.1530/EJE-17-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharya K., Orton R.C., Qi X., et al. A novel starch for the treatment of glycogen storage diseases. J Inherit Metab Dis. 2007;30(3):350–357. doi: 10.1007/s10545-007-0479-0. [DOI] [PubMed] [Google Scholar]
- 9.Minic M., Rocha N., Harris J., et al. Constitutive activation of AKT2 in humans leads to hypoglycemia without fatty liver or metabolic dyslipidemia. J Clin Endocrinol Metab. 2017;102(8):2914–2921. doi: 10.1210/jc.2017-00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng J.Q., Godwin A.K., Bellacosa A., et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci U S A. 1992;89(19):9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derks T.G.J., Smit G.P.A. Dietary management in glycogen storage disease type III: what is the evidence? J Inherit Metab Dis. 2015;38(3):545–550. doi: 10.1007/s10545-014-9756-x. [DOI] [PubMed] [Google Scholar]
- 12.Hijazi G., Pai N., Nagy L.L., et al. Use of waxy maize heat modified starch in the treatment of children between 2 and 5 years with glycogen storage disease type I: a retrospective study. Mol Genet Metab Rep. 2019;21:100536. doi: 10.1016/j.ymgmr.2019.100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.