

Abstract
The NusG protein family is structurally and functionally conserved in all domains of life. Its members directly bind RNA polymerases and regulate transcription processivity and termination. RfaH, a divergent sub‐family in its evolutionary history, is known for displaying distinct features than those in NusG proteins, which allows them to regulate the expression of virulence factors in enterobacteria in a DNA sequence‐dependent manner. A striking feature is its structural interconversion between an active fold, which is the canonical NusG three‐dimensional structure, and an autoinhibited fold, which is distinctively novel. How this novel fold is encoded within RfaH sequence to encode a metamorphic protein remains elusive. In this work, we used publicly available genomic RfaH protein sequences to construct a complete multiple sequence alignment, which was further augmented with metagenomic sequences and curated by predicting their secondary structure propensities using JPred. Coevolving pairs of residues were calculated from these sequences using plmDCA and GREMLIN, which allowed us to detect the enrichment of key metamorphic contacts after sequence filtering. Finally, we combined our coevolutionary predictions with molecular dynamics to demonstrate that these interactions are sufficient to predict the structures of both native folds, where coevolutionary‐derived non‐native contacts may play a key role in achieving the compact RfaH novel fold. All in all, emergent coevolutionary signals found within RfaH sequences encode the autoinhibited and active folds of this protein, shedding light on the key interactions responsible for the action of this metamorphic protein.
Keywords: direct coupling analysis, evolution, fold‐switch, metamorphic proteins, protein folding, transcription factor
1. INTRODUCTION
Protein evolution is at the cornerstone of organism adaptation and gain of function. It diversifies proteins into entire families, whose members can branch out into proteins carrying distinct functions than their predecessors. This is the case of RfaH, a transcription and virulence factor in enterobacteria that evolved from a highly conserved family of transcription regulators called NusG. 1 This protein family is universally conserved in all domains of life, regulating transcription by directly binding to RNA polymerase (RNAP), 2 and an ancestor of this protein family is thought to have been present in the last universal common ancestor (LUCA). 3
In Escherichia coli, NusG is an essential protein that regulates virtually all transcription processes. 3 Meanwhile, its RfaH paralog is quite unique as it regulates transcription in a sequence‐dependent manner. 4 RfaH, unlike NusG, is not directly recruited to RNAP, but to the entire ops‐paused transcription elongation complex (TEC), 5 with the ops (operon polarity suppressor) corresponding to a DNA sequence commonly found in pathogenicity islands and xenogenes incorporated by enterobacteria. 1
This striking feature of RfaH is achieved by its three‐dimensional structure, which differs from that of the canonical NusG fold. As in NusG, it consists of an N‐terminal domain (NTD) comprising an hydrophobic depression that binds RNAP, but that in RfaH is blocked by its own C‐terminal domain (CTD) folded as an α‐helical hairpin, constituting an autoinhibited state (αRfaH). 6 Notably, when RfaH encounters the ops‐paused TEC, it binds to it and relieves itself from autoinhibition, upon which the released CTD refolds into a β‐barrel (βRfaH) in a process that is known as fold‐switching 7 (Figure 1).
FIGURE 1.
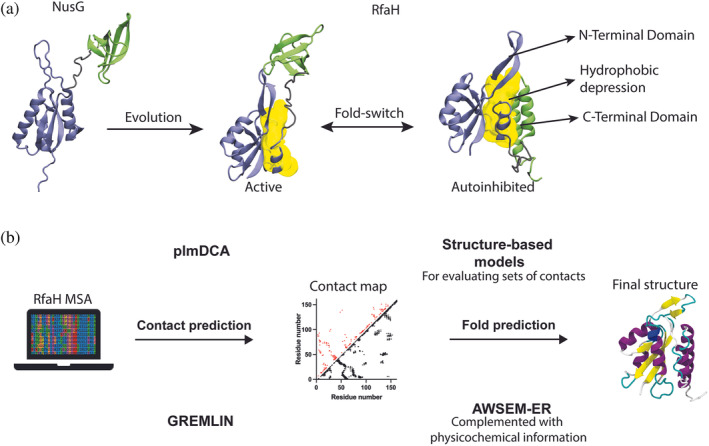
Summary of the research and methods used in this work. (a) NusG, a non‐metamorphic protein, evolved into its paralog RfaH, whose fold‐switch is characterized to take place between an autoinhibited fold (αRfaH) that has interdomain contacts at the hydrophobic patch (yellow) and an active fold that does not establish interdomain contacts (βRfaH). We hypothesized that the emergence of these intradomain and interdomain contacts can be inferred via coevolutionary analysis. (b) By constructing a metagenomic‐enriched multiple sequence alignment (MSA) of RfaH and filtering out non‐metamorphic sequences based on secondary structure predictions, we inferred a contact map of coevolving residue pairs that we used to predict the structures of the autoinhibited and active states of RfaH through molecular dynamics using two different pipelines, namely DCA/SBM and GREMLIN/AWSEM‐ER, which capture the distinctive features of RfaH folding.
It is estimated that between 0.5% and 4% of proteins whose structures are deposited in the PDB are likely to exhibit fold‐switching, or metamorphic, behavior. 8 Among them, RfaH is one of the most studied cases due to its dramatic all α‐helix (αCTD) to all β‐strand (βCTD) conversion of a whole domain. Computational approaches have sought to determine the fold‐switching mechanism of this protein, 9 , 10 , 11 , 12 , 13 , 14 , 15 and experimental structural work has been performed to characterize its binding to the TEC. 6 , 16 Recent reports suggest that during evolution, protein metamorphosis emerges as the connecting path between two distinct folds. 17 Nevertheless, the fold‐switching process of RfaH is key for its function, as it allows RfaH to become active upon specific recognition of a DNA sequence while avoiding its spurious binding to RNAP. 16
Experimental and computational approaches have shown that interdomain contacts formed between the CTD and the RNAP‐binding interface of the NTD are essential for the formation of the autoinhibited state of RfaH. Particularly, the electrostatic interaction E48‐R138 is responsible for stabilizing the α‐folded state, as its disruption leads to roughly equally populated βRfaH and αRfaH in solution. 7 Furthermore, removal of interdomain contacts in coarse‐grained simulations of the full‐length protein is enough to give rise to βRfaH. 11 , 18 Consequently, the autoinhibiting interdomain interactions, absent in the RfaH paralog NusG, are essential to stabilize the novel αRfaH fold, giving rise to its structural duality.
We sought to determine if intradomain and interdomain interactions stabilizing both RfaH folds can be inferred from the coevolutionary analysis of their amino acid sequences, and further evaluate their sufficiency to encode both RfaH folds via molecular dynamics (MD) that explicitly incorporate this information. Coevolutionary inference methods, such as direct coupling analysis (DCA) 19 and generative regularized models of proteins (GREMLIN), 20 have been developed for the statistical analysis of large multiple protein sequence alignments in two essential terms: sequence conservation and correlated mutations. Given that spatially proximate residues in the native state of a given protein family tend to coevolve, 21 these methods have been widely successful in inferring the structural proximity of coevolving residue pairs that are fundamental for folding, function, and dynamics from sequence information alone. 22 , 23
In this work, RfaH sequences deposited in the Interpro database 24 were used to predict coevolutionary contacts with pyDCA 25 and GREMLIN 20 (Figure 1). The number of RfaH sequences was further increased by constructing a hidden Markov model (HMM) profile to use as input for a subsequent search of RfaH sequences in the metagenomic database metaclust. 26 Finally, in line with recent works, 27 all sequences were filtered using secondary structure prediction in JPred 28 to select only metamorphic candidates. This metamorphic enrichment protocol yielded an alignment of 3,570 nonredundant sequences that display four coevolving pairs of residues involved in interdomain interactions in the experimentally solved structure of αRfaH.
The inferred contacts for RfaH were used as restraints for protein structure prediction in simulations based on coevolutionary structure‐based models (SBMs) 29 and coarse‐grained force fields, 30 whose final configurations largely reproduced the experimentally solved structures of both RfaH folds and the NTD‐CTD binding of αRfaH. Furthermore, choosing subsets of coevolutionary interactions to guide these simulations led to the observation that contacts between residue pairs not observed in the crystallographic structure of RfaH, that is, non‐native contacts, are important to reach a compact native state having the correct topology and that CTD compactness is essential for forming the autoinhibiting interface of RfaH.
In summary, our results effectively demonstrate that coevolutionary signals encode the metamorphic behavior of RfaH, replicating the distinct features of the active and autoinhibited folds that are essential for the biological function of this transcription factor.
2. RESULTS
2.1. Sequence retrieval and coevolutionary analysis
Retrieving enough sequences to predict robust evolutionary signals is not trivial. Research using the GREMLIN algorithm suggests that a number of nonredundant sequences at least 20 times the length of the protein (L) is needed to achieve a true positive (TP) rate (i.e., coevolving pairs forming a native contact in an experimentally solved structure) of ~0.7 for the top L/2 contact predictions. 31
This is an issue for the RfaH subfamily, considering that its most studied representative from E. coli is 162 residues long. The predicted members deposited in the Interpro database 24 make up nearly 3,000 sequences that, when clustered at 90% identity to reduce redundancy, decreases to ~1,000 sequences. This is less than one third of what is needed according to the criteria above.
Therefore, HMMER 32 was used to build an HMM profile with the nonredundant Interpro sequences, allowing to search for additional RfaH protein sequences in metaclust, a large metagenomic database of 1.59 billion sequences. 26 Using several e‐value cutoffs, 3 sets were retrieved, containing 3,865 (e‐value 10−30), 5,378 (e‐value 10−25), and 8,516 (e‐value 10−20) sequences clustered at 90% identity.
Using pyDCA 25 as a python script in Jupyter Notebooks for execution in Google Colaboratory, 33 coevolutionary interactions were calculated using pseudo‐likelihood maximization direct coupling analysis (plmDCA) on MSAs generated using hmmalign from HMMER. 32 Two MSAs were analyzed: one with only nonredundant RfaH sequences from Interpro database (Interpro, 1,005 sequences), and another one complemented with the sequences found in metaclust (Interpro + MG, 5,379 sequences). Given that GREMLIN has been shown to be more accurate than other coevolution‐based residue‐residue contact prediction methods, 31 we also performed our coevolutionary analyses with this algorithm.
Using the Interpro database alone, plmDCA correctly predicts 17 CTD contacts below 8 Å that are formed either in αCTD or βCTD, 1 interdomain (ID) contact below 10 Å and 34 NTD contacts below 8 Å, reaching a fraction of TP (with L = 162) of 0.32, whereas GREMLIN correctly predicts 4 additional contacts for RfaH CTD and 10 additional contacts for the NTD, reaching a TP of 0.40 (Figure 2, Tables S1 and S2). The choice of 10 Å for ID contacts was due to the observation that the average NTD‐CTD distance in randomized residue‐residue pairs is 32 Å, in contrast with CTD and NTD contacts that take place at 10–12 Å (Figures S1 and S2).
FIGURE 2.
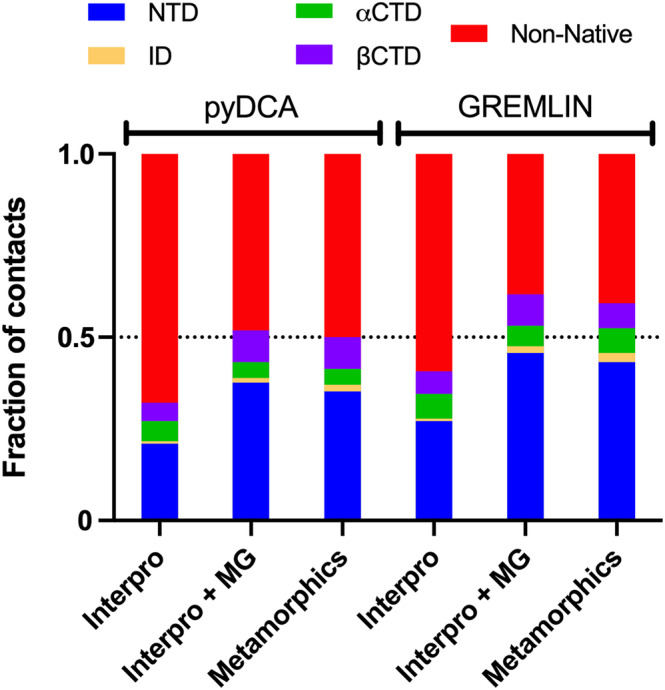
Summary of the coevolutionary analysis results for the RfaH subfamily. The stacked bar graphs show the fraction of coevolving residue pairs (L = 162) that are found forming a native contact (Cα distance <8 Å) in the NTD, αCTD, or βCTD; an ID contact (Cα distance <10 Å) or a non‐native contact. The contact distances are calculated based on the crystal structure of full‐length αRfaH (PDB 5OND) and the cryo‐EM structure of βRfaH (PDB 6C6S).
The addition of metagenomic RfaH sequences from metaclust led to an increase in TP up to 0.52 for plmDCA and 0.62 for GREMLIN (Figure 2), including one additional ID contact for plmDCA (Table S1) and two additional ID contacts for GREMLIN (Table S2). Special attention was paid to the increase in ID contacts due to their relevance in stabilizing the autoinhibited state of RfaH. 11 , 18 , 34
In this regard, it is worth noting that although increasing the number of metagenomic sequences beyond those retrieved by HMM search with an e‐value of 10−30 increases the TP rate for both RfaH folds, that is, from 0.63 to 0.71 for αRfaH and from 0.70 to 0.82 for βRfaH, there is no increase in the number of predicted ID contacts (Figure S3). Therefore, we opted to use this MSA to avoid leakage of NusG sequences into our alignment at lower e‐values, which could potentially disrupt the coevolutionary signals between the NTD and CTD of RfaH, 22 and to reduce the computing time of coevolutionary contacts through the plmDCA algorithm.
In fact, addition of the metagenomic RfaH sequences led to a 70% increase in the number of correctly predicted NTD contacts whereas the number of CTD contacts remained roughly the same, which could be an indication that non‐metamorphic protein sequences have leaked into the alignment, as the NTD fold is highly conserved throughout the NusG family.
A recent work employed a secondary structure prediction approach to filter out potential non‐metamorphic RfaH homologs. 27 Based on this work, we used JPred 28 to identify which protein sequences from the Interpro + MG MSA exhibit both β‐strand and α‐helical propensity in a short section of the CTD, comprising residues 126–162 in the representative sequence of E. coli RfaH, that reports its metamorphic duality. This filtering process led to a third MSA containing 3,570 sequences clustered at 90% identity (Metamorphics), a reduction of ~1,000 sequences from the starting MSA.
Despite this important reduction in the number of sequences, the resulting MSA almost completely replicates the coevolutionary information and TP obtained using either plmDCA or GREMLIN on the Interpro + MG MSA while correctly predicting an additional ID contact (Figure 2, Tables S1 and S2). Altogether, our best TP for L contacts (L = 162) with the highest number of ID contacts was achieved with the Metamorphics MSA and GREMLIN, obtaining 70 contacts for the NTD, 22 for the CTD in either fold, and 4 ID contacts (Figure 2 and Table S2). In comparison, a recent work on coevolutionary analysis on RfaH using EVcouplings 35 on sequences collected using iterative BLAST 36 and filtered by secondary structure propensity with JPred 28 led to the prediction of CTD and NTD contacts but did not report any correctly predicted ID contacts. 27
To rationalize the successful prediction of ID contacts in our coevolutionary analysis, we took into consideration the differences between pyDCA and GREMLIN and the increase and identity of the ID contacts predicted upon addition of metagenomic RfaH sequences and secondary structure filtering (Table 1).
TABLE 1.
TP ID contacts found for each dataset.
| Dataset | pyDCA j > i + 2 | pyDCA j > i + 3 | GREMLIN |
|---|---|---|---|
| Interpro | V92—I146 | V92—I146 | P52—S139 |
| Interpro + MG | P52—S139, E48—G135 | P52—S139, E48—G135 | P52—S139, P52—A137, E48—G135 |
| Metamorphics | P52—S139, E48—G135 | P52—S139, P52—A137, E48—G135 | P52—S139, P52—A137, N53—S139, E48—G135 |
One relevant difference between both coevolution‐based methods is that pyDCA recommends using a sequence separation for residue pairs of j > i + 3, while the sequence separation used in GREMLIN is j > i + 2. Setting the residue separation for plmDCA at j > i + 2 shows that most predicted contacts are false positives or short‐ranged (Figure S4). Despite this observation, one correctly predicted ID contact (residue pair 92–146) was retrieved from Interpro MSA and two from Interpro + MG and Metamorphics MSAs, corresponding to residue pairs 48–135 and 52–139 that are also obtained using GREMLIN. Thus, filtering out short‐range contacts at low sequence separation is required for predicting long‐range ID contacts in pyDCA.
Analysis of the Interpro MSA using plmDCA led to the correct prediction of ID residue pair 92–146. The addition of metagenomic sequences led to the disappearance of the previous residue pair and the correct prediction of ID residue pairs 48–135 and 52–139. Lastly, upon filtering the sequences via Jpred, one additional ID contact is correctly predicted for residue pair 52–137 (Table 1). This analysis suggests that some of the ID contacts that are likely important for αRfaH may have low DCA scores because they are buried in the dominant coevolutionary signals of the canonical βCTD found in both metamorphic and non‐metamorphic NusG family members.
Besides the increase in ID contacts upon enriching the number of RfaH sequences and their subsequent filtering based on secondary structure predictions, we were also interested on the intradomain contacts predicted by these methods, as they may be key in stabilizing each fold. For αRfaH, it is consistently observed that both plmDCA and GREMLIN only yield helical contacts, that is, with sequence separations of 3 or 4 residues, and no interhelical contacts. Meanwhile, coevolutionary contacts in the β‐folded CTD are formed between β2‐β3, β3‐β4, and β4‐β5 (Figure S5). It is also worth noting that most of the helical contacts inferred for αCTD are exclusive to this fold, that is, the interacting residue pairs are significantly more separated in distance in the βCTD. Therefore, these findings suggest that the helical propensity of RfaH CTD is encoded within its sequence coevolution, unlike the hairpin formation, which likely results from compaction of this domain against the hydrophobic ID surface in the NTD.
For both plmDCA and GREMLIN, there are about 40%–50% coevolutionary signals that do not correspond to any known contact in αRfaH or βRfaH when using the ~3,500 sequences of the Metamorphics MSA. These apparent non‐native interactions are all significant and contribute to a large fraction of the total predicted interactions. To assess if the same rate of false positive contacts is observed in the non‐metamorphic NusG protein family members, the NusG sequences deposited in Pfam 37 were clustered at 90% identity (10,593 sequences), aligned and used as input for plmDCA (Figure S6). The results show that out of the top L = 181 residues, 157 contacts are correctly predicted, representing a TP of 0.86, which is higher than the TP rate observed for αRfaH. However, this TP rate is similar to the one obtained for βRfaH (0.82) using the Interpro and metagenomic sequences retrieved at e‐value 10−20 (8,516 sequences, Figure S3). These results suggest that it would be required to further increase the number of true metamorphic RfaH sequences to overwhelm the coevolutionary signals coming from βRfaH or non‐metamorphic homologs.
2.2. Structure prediction through coevolution‐based MD
To determine if the coevolutionary signals identified for RfaH are enough to correctly predict the αRfaH fold, which is the novel topology in the NusG family, two MD pipelines were used: Cα coarse‐grained simulations using SBMs guided by DCA‐predicted RfaH contacts, 22 and Cβ coarse‐grained semi‐empirical simulations using AWSEM‐ER guided by GREMLIN‐predicted RfaH contacts. 30 Briefly, each coevolutionary contact is used as a bias to guide the MD simulation to form such contact in a simulated annealing, that is, a descending temperature gradient that allows the formation of the selected contacts during protein folding. These coevolution‐guided MD simulations also rely on secondary structure biases for higher accuracy. Thus, we employed the secondary structure observed in PDB 5OND and 6C6S to model αRfaH and βRfaH, respectively.
A total of 10 simulations for αRfaH and βRfaH were produced for each pipeline. Regardless of the MD pipeline and the low number of ID contacts obtained with either coevolutionary analysis, most of the final configurations of αRfaH after simulated annealing exhibit the formation of an incipient NTD‐CTD interaction in the same location of the hydrophobic depression of the NTD (Figures 3 and S7). For the best predicted structure, obtained using the GREMLIN‐AWSEM‐ER pipeline, the RMSD of the NTD and CTD against the experimental structure of αRfaH reached 4.2 and 4.1 Å, respectively (Tables S3 and S4).
FIGURE 3.
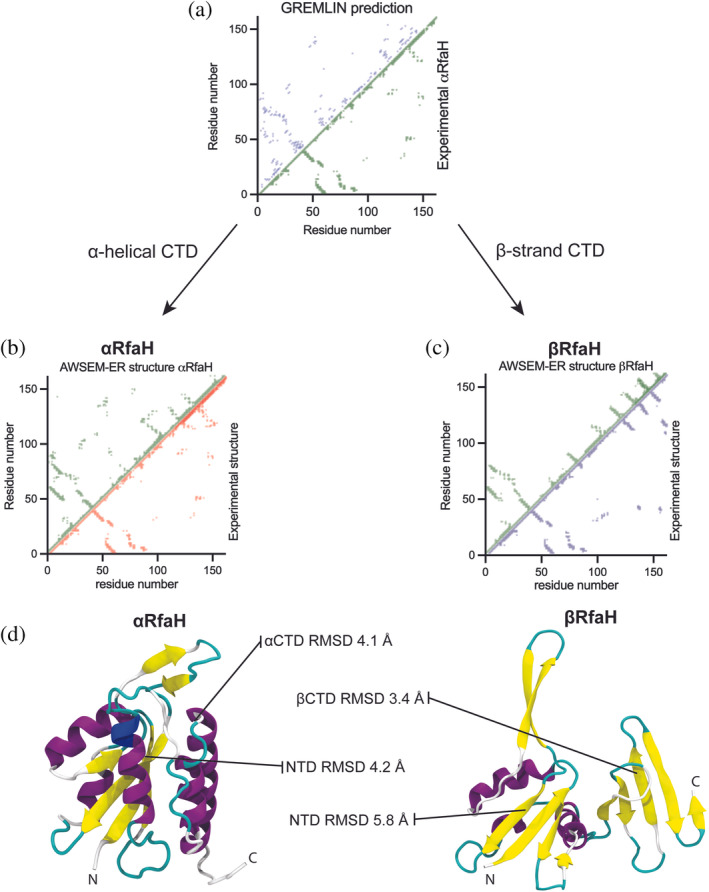
Best predicted structures for αRfaH and βRfaH based on the GREMLIN‐AWSEM‐ER pipeline. (a) Comparison of the coevolution‐based and the experimental contact maps of αRfaH (PDB 5OND). (b,c) Comparison of the contact maps generated using the AWSEM‐ER‐predicted structures and experimental structures for αRfaH (PDB 5OND, (b) and βRfaH (PDB 6C6S, c). (d) Cartoon representation of the best predicted structures for both RfaH folds, and their respective RMSD to the experimental structures.
Compaction of the CTD allows for the formation of an incipient NTD‐CTD interaction in 80% of the simulations with the GREMLIN‐AWSEM‐ER pipeline and 70% for the DCA‐SBM pipeline. However, the hairpin αCTD structure is only achieved in a few cases, supporting the idea that NTD‐CTD binding can occur even in the absence of an αCTD with all native intrahelical contacts formed, as observed in previous simulations using dual‐basin SBM 11 and experiments using hydrogen‐deuterium exchange mass spectrometry, 18 which would likely give rise to RfaH autoinhibition.
In the case of βRfaH, it was observed that the structure of the βCTD is too distorted to be properly folded with either MD pipeline (Tables S5 and S6), reaching instead the folding state of a three‐stranded intermediate chaperoned by the NTD in which only strands β2, β3, and β4 are formed and that has been described through multiple computational approaches in previous works. 9 , 38 , 39 Also, the RMSD of the βCTD is higher for the DCA‐SBM pipeline. As a control, we compared our DCA‐SBM simulations for full‐length βRfaH (Figure S7) with simulations of the isolated βCTD using coevolutionary contacts predicted only for the 55 C‐terminal residues of RfaH (Table S7). While in all simulations of the isolated CTD its RMSD against the experimental β‐folded state stayed above 8 Å, its RMSD against the reported β‐intermediate reaches down to 4.6 Å. This evidence suggests that βCTD folding is impeded by the idealized fully extended β‐strands that are being used as secondary structure bias in these SBM‐models, but that its main features are correctly modeled.
For the NTD, we observed that the β2‐β3 hairpin, largely responsible for its binding to the RNAP, is highly flexible in the final configurations obtained using the GREMLIN‐AWSEM‐ER pipeline, particularly in the βRfaH configuration, giving rise to higher RMSD values than in αRfaH. It should be noted, however, that the RMSD between the NTD for both experimental structures of RfaH (PDB 5OND and 6C6S) is 3.0 Å, largely due to structural differences in this hairpin. Regardless, the physicochemical potential of the AWSEM forcefield allows proper compaction of the NTD, thus exhibiting lower RMSD values for the best‐predicted structure than the DCA‐SBM pipeline.
To determine the effect of coevolution‐based native and non‐native contacts on the accuracy of RfaH structure prediction, we further employed the DCA‐SBM pipeline in which only bonded interactions and nonbonded coevolution‐based contacts are involved, in contrast to GREMLIN‐AWSEM‐ER that also incorporates physicochemical and knowledge‐based potentials. We first determined the fraction of native and non‐native contacts as a function of time for the DCA‐SBM simulation that reflects the lower RMSD to the αRfaH fold (Figure S8). We observed that the fraction of native contacts reaches 0.7 in the final native ensemble, as expected for a simulated annealing of a globular protein, whereas the fraction of non‐native contacts only reaches 0.3, indicating that the non‐native interactions inferred by coevolution are not compatible with the native contacts in this structural form.
Next, we chose different subsets of coevolution‐based predicted contacts for subsequent DCA‐SBM simulations. First, we performed simulations in which we deleted DCA‐predicted contacts that exhibit shorter distances in βCTD than in αCTD. Second, we performed additional simulations only considering TP native contacts present in the experimental αRfaH structure, effectively disregarding non‐native interactions. In the first case, we observed that the RMSD to the NTD was similar as the one achieved with the whole set of DCA‐predicted contacts (Table S8), whereas an overall increase in RMSD for the NTD and a less compact global architecture was observed for the second scenario (Table S9). Regardless, in both cases the CTD was no longer binding to the hydrophobic depression at the NTD and was less compact when compared with the initial simulations.
Finally, given that most coevolutionary‐predicted native and non‐native contacts exhibit shorter interaction distances than randomly generated contacts (Figures S1 and S2), we tested if replacing these non‐native contacts by an equal number of randomly selected non‐native interactions would replicate the compaction caused by the coevolved pairs obtained through DCA. As summarized in Table S10, we observe a similar behavior for the NTD as in the simulations in which only TP contacts for RfaH were considered, except that the topology of this domain was, in most cases, incorrect. For the CTD, we observed compaction of this domain, which is likely caused by the native interactions of the βCTD, but ID interactions occur away from the hydrophobic patch of the NTD probably due to the random nature of the non‐native contacts.
Altogether, these results suggest that non‐native coevolutionary contacts may be important to reach a compact architecture, and that compactness at the CTD may be essential for having clustered interactions that simultaneously bind the NTD at the hydrophobic depression, enabling RfaH to reach its autoinhibited state.
3. DISCUSSION
RfaH is one of the most prominent examples of metamorphic proteins, exhibiting fold‐switch of an entire domain from an α‐helical hairpin to a small β‐barrel. Since this protein subfamily is thought to have originated from non‐metamorphic NusG transcription factors, its new metamorphic fold should be stabilized by ID interactions emerging during its evolution. To assess such scenario, we sought to find RfaH sequences to infer coevolution‐based residue‐residue interactions essential for the novel RfaH autoinhibited fold.
It is worth noting that current state‐of‐the‐art structural predictors, such as AlphaFold2, 40 can predict the metamorphic behavior of RfaH. For example, using the sequence of full‐length E. coli RfaH as input into ColabFold (colabfold.com), 41 a Google Colaboratory implementation of AlphaFold2, yields the αRfaH fold as a result, whereas using only the CTD of the same protein as input yields the canonical NusG‐like β‐barrel. However, this approach is not as straightforward as the coevolutionary analysis of thousands of protein sequences in defining the key interactions stabilizing each fold and how these interactions emerged during the evolution of the NusG protein family.
Using the metagenomic database metaclust to increase the available number of RfaH sequences over those deposited in the Interpro database, and then filtering these sequences by using a secondary structure predictor to ascertain the duality of their structure propensity, it was possible to increase the number of ID contacts by enriching our coevolutionary analysis with true metamorphic sequences.
This coevolutionary information, in combination with different secondary structure biases, was sufficient to predict both the predominant autoinhibited structure of RfaH in solution and to retrieve key contacts involved in the stabilization of the βCTD in the active state and the formation of a recently described β‐intermediate. In fact, the β‐intermediate that precedes βCTD folding 38 is formed even in the presence of ID contacts. These findings suggest that the duality in secondary structure propensity of RfaH is essential for the stabilization of both folds.
We have also shown that it is not necessary to infer all interhelical CTD contacts to predict a compact αCTD that inhibits the NTD. In fact, recent experimental work has demonstrated that the ends of the αCTD hairpin are largely unstructured in solution. 7 , 18 Furthermore, it has been shown that an exactly solvable model of helical‐coil‐sheet transitions displays cooperativity in its temperature‐induced folding from helical to extended configurations, prior to reaching the coil state. 42 Altogether, these precedents suggest that nucleation of the tip of the αCTD hairpin at the NTD hydrophobic patch by coevolutionary ID contacts could trigger the formation of the autoinhibited fold of RfaH. Although the stability of the autoinhibited state over the active state of RfaH in solution cannot be derived from these coevolution‐guided simulations, its thermodynamic favorability has been thoroughly analyzed in simulations and experiments that explore its protein folding landscape in detail. 11 , 38 , 43
Our results also showed that non‐native interactions of either RfaH state were relevant to produce compactness during protein folding. In particular, non‐native contacts from the β‐barrel CTD were essential to ensure compactness of the αCTD in the autoinhibited state. Even though they account for nearly half of all coevolution‐based interactions, only 30% of these non‐native contacts are present in the final annealed configurations of αRfaH, while 70% of the correctly predicted native contacts are formed.
Although being marginally formed, some of the non‐native contacts that guide αRfaH folding are in close spatial proximity in the native state, and hence help compacting the protein structure. Thus, the local frustration brought by non‐native contacts, that is, the roughness of the potential energy landscape for protein folding arising from conflicting interactions, 44 is expected to play a fundamental role in enhancing the folding process of RfaH, as has been seen in globular proteins. 45 , 46 These pairs of significantly coevolving residues not involved in direct physical contacts in RfaH may correspond to interactions necessary to some functional aspects, presumably even fold‐switching.
4. METHODS
4.1. Sequence search
All initial RfaH sequences were retrieved from the Interpro database of protein families. 24 The choice of this database for sequence retrieval is due to a recent study that employed this database to characterize the sequence conservation in both RfaH and NusG and further experimentally tested substitutions of these residues in vitro. 47 The retrieved sequences were also used to construct an HMM 32 profile that was employed to retrieve more RfaH sequences from the metaclust database, 26 using cutoff e‐values of 10−30, 10−25, and 10−20. Lastly, the sequences from Interpro and metaclust were combined and filtered based on the duality of their secondary structure propensity, similarly to previous works. 27 To do this, a region of the CTD sequence of each protein (starting from the residue pattern FQAIF, corresponding to residue number 126 in E. coli RfaH) was used as input for secondary structure prediction on the JPred4 webserver 28 (https://www.compbio.dundee.ac.uk/jpred/) using default settings. Each unaligned sequence that had at least four consecutive helical residues in this trimmed version of the CTD was included in the Metamorphics alignment.
4.2. Coevolutionary analysis
The three datasets obtained before, Interpro with 1,005 sequences, Interpro + MG with 5,379 sequences and Metamorphics with 3,570 sequences, were used as input for the pyDCA algorithm implemented in Google Colaboratory or submitted at the GREMLIN webserver (http://gremlin.bakerlab.org). The retrieved residue‐pair list was analyzed using a homemade script to calculate the Cα distance at the target PDB file of either αRfaH or βRfaH from PDB 5OND and 6C6S, respectively (Tables S1 and S2).
4.3. Simulated annealing using structure‐based MD (DCA‐SBM)
SBMs were generated based on the secondary structure of either RfaH fold and the coevolutionary information obtained by plmDCA, 21 following a protocol already reported. 22 Besides the SBMs for αRfaH with all DCA contacts (Table S3) and βRfaH with all DCA contacts (Table S5), additional models for βCTD with predicted DCA contacts of L = 55 (Table S7), αRfaH will all DCA contacts except those of the βCTD (Table S8), αRfaH with only native DCA contacts (Table S9), and αRfaH with native contacts plus randomly generated non‐native contacts equal to the number of non‐native coevolving pairs (Table S10), were produced. All these models were run for 2 × 107 steps with a timestep 0.0005 τ in reduced units, over which a temperature gradient lowered the temperature from 200 to 0 reduced temperature units. All the simulations were performed with a modified version of GROMACS. 48
4.4. Simulated annealing using GREMLIN‐AWSEM‐ER
Following the AWSEM‐ER protocol for the GREMLIN‐derived RfaH contacts, 30 a simulated annealing was produced for αRfaH (Table S4) and βRfaH (Table S6) by decreasing the temperature from 450 to 350 temperature units over 4 × 106 steps using a timestep of 5 fs. The default AWSEM forcefield was used, except for the fragment memory potential which was turned off and the evolutionary restraints derived from GREMLIN that were added.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Pablo Galaz‐Davison: Conceptualization (supporting); data curation (lead); formal analysis (lead); funding acquisition (supporting); investigation (lead); methodology (lead); software (lead); visualization (lead); writing – original draft (lead); writing – review and editing (equal). Diego Ferreiro: Conceptualization (supporting); formal analysis (supporting); funding acquisition (supporting); methodology (supporting); writing – original draft (supporting). César A. Ramírez‐Sarmiento: Conceptualization (lead); formal analysis (supporting); funding acquisition (lead); investigation (supporting); methodology (supporting); resources (lead); software (supporting); visualization (supporting); writing – original draft (supporting); writing – review and editing (equal).
Supporting information
Data S1. Supporting Information
ACKNOWLEDGMENTS
This research was funded by the National Agency for Research and Development (ANID) through FONDECYT 1201684 (to César A. Ramírez‐Sarmiento) and Millennium Science Initiative Program ICN17_022. This work was also in part supported by the Consejo de Investigaciones Científicas y Técnicas (CONICET); the Agencia Nacional de Promoción Científica y Tecnológica (PICT2016‐1467 to Diego U. Ferreiro) and Universidad de Buenos Aires (UBACYT 2018—20020170100540BA). Pablo Galaz‐Davison was supported by an ANID doctoral scholarship (PFCHA 21181705). We would like to thank Dr. Sergey Ovchinnikov for his illuminating ideas on how to improve RfaH coevolutionary analysis.
Galaz‐Davison P, Ferreiro DU, Ramírez‐Sarmiento CA. Coevolution‐derived native and non‐native contacts determine the emergence of a novel fold in a universally conserved family of transcription factors. Protein Science. 2022;31(6):e4337. 10.1002/pro.4337
Review Editor: Nir Ben‐Tal
Funding information Agencia Nacional de Investigación y Desarrollo, Grant/Award Number: PFCHA 21181705; Consejo Nacional de Investigaciones Científicas y Técnicas; Fondo Nacional de Desarrollo Científico y Tecnológico, Grant/Award Number: FONDECYT 1201684; Fondo para la Investigación Científica y Tecnológica, Grant/Award Number: PICT2016‐1467; Universidad de Buenos Aires, Grant/Award Number: UBACYT 2018—20020170100540BA; Millennium Science Initiative Program, Grant/Award Number: ICN17_022
DATA AVAILABILITY STATEMENT
The retrieved RfaH sequences for coevolutionary analysis, the Jupyter Notebook for running pyDCA on Google Colaboratory, the coevolutionary analysis results, and all structure prediction simulations and final configurations are available for download at the laboratory's simulation archive in the Open Science Framework (OSF, https://osf.io/bn6u3/).
REFERENCES
- 1. Artsimovitch I, Knauer SH. Ancient transcription factors in the news. MBio. 2019;10:1–16. 10.1128/mbio.01547-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Washburn RS, Zuber PK, Sun M, et al. Escherichia coli NusG links the Lead ribosome with the transcription elongation complex. iScience. 2020;23:101352. 10.1016/j.isci.2020.101352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang B, Artsimovitch I. NusG, an ancient yet rapidly evolving transcription factor. Front Microbiol. 2021;11:1–17. 10.3389/fmicb.2020.619618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bailey MJ, Hughes C, Koronakis V. RfaH and the ops element, components of a novel system controlling bacterial transcription elongation. Mol Microbiol. 1997;26:845–851. 10.1046/j.1365-2958.1997.6432014.x [DOI] [PubMed] [Google Scholar]
- 5. Kang JY, Mooney RA, Nedialkov Y, et al. Structural basis for transcript elongation control by NusG family universal regulators. Cell. 2018;173:1650–1662. https://linkinghub.elsevier.com/retrieve/pii/S0092867418305944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belogurov GA, Vassylyeva MN, Svetlov V, et al. Structural basis for converting a general transcription factor into an operon‐specific virulence regulator. Mol Cell. 2007;26:117–129. 10.1016/j.molcel.2007.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burmann BM, Knauer SH, Sevostyanova A, et al. An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell. 2012;150:291–303. 10.1016/j.cell.2012.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter LL, Looger LL. Extant fold‐switching proteins are widespread. Proc Natl Acad Sci. 2018;115:5968–5973. 10.1073/pnas.1800168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li S, Xiong B, Xu Y, et al. Mechanism of the all‐α to all‐β conformational transition of RfaH‐CTD: Molecular dynamics simulation and markov state model. J Chem Theory Comput. 2014;10:2255–2264. 10.1021/ct5002279 [DOI] [PubMed] [Google Scholar]
- 10. Balasco N, Barone D, Vitagliano L. Structural conversion of the transformer protein RfaH: New insights derived from protein structure prediction and molecular dynamics simulations. J Biomol Struct Dyn. 2015;33:2173–2179. 10.1080/07391102.2014.994188. [DOI] [PubMed] [Google Scholar]
- 11. Ramírez‐Sarmiento CA, Noel JK, Valenzuela SL, Artsimovitch I. Interdomain contacts control native state switching of RfaH on a dual‐funneled landscape. PLoS Comput Biol. 2015;11:e1004379. 10.1371/journal.pcbi.1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seifi B, Aina A, Wallin S. Structural fluctuations and mechanical stabilities of the metamorphic protein RfaH. Proteins Struct Funct Bioinforma [Internet]. 2021;89:289–300. 10.1002/prot.26014. [DOI] [PubMed] [Google Scholar]
- 13. Xun S, Jiang F, Wu YD. Intrinsically disordered regions stabilize the helical form of the C‐terminal domain of RfaH: A molecular dynamics study. Bioorganic Med Chem. 2016;24:4970–4977. 10.1016/j.bmc.2016.08.012 [DOI] [PubMed] [Google Scholar]
- 14. Bernhardt NA, Hansmann UHE. Multifunnel landscape of the fold‐switching protein RfaH‐CTD. J Phys Chem B. 2018;122:1600–1607. 10.1021/acs.jpcb.7b11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joseph JA, Chakraborty D, Wales DJ. Energy landscape for fold‐switching in regulatory protein RfaH. J Chem Theory Comput. 2019;15:731–742. 10.1021/acs.jctc.8b00912. [DOI] [PubMed] [Google Scholar]
- 16. Zuber PK, Schweimer K, Rösch P, Artsimovitch I, Knauer SH. Reversible fold‐switching controls the functional cycle of the antitermination factor RfaH. Nat Commun. 2019;10:702. 10.1038/s41467-019-08567-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tian P, Best RB. Exploring the sequence fitness landscape of a bridge between protein folds. PLoS Comput Biol. 2020;16:e1008285. 10.1371/journal.pcbi.1008285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galaz‐Davison P, Molina JA, Silletti S, et al. Differential local stability governs the metamorphic fold switch of bacterial virulence factor RfaH. Biophys J. 2020;118:96–104. 10.1016/j.bpj.2019.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morcos F, Pagnani A, Lunt B, et al. Direct‐coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci. 2011;108:E1293–E1301. 10.1073/pnas.1111471108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue‐residue interactions across protein interfaces using evolutionary information. Elife. 2014;3:e02030. 10.7554/eLife.02030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ekeberg M, Lövkvist C, Lan Y, Weigt M, Aurell E. Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models. Phys Rev E. 2013;87:012707. 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- 22. dos Santos RN, Jiang X, Martínez L, Morcos F. Coevolutionary signals and structure‐based models for the prediction of protein native conformations. Methods Mol Biol. 2019;1851:83–103. 10.1007/978-1-4939-8736-8_5 [DOI] [PubMed] [Google Scholar]
- 23. Morcos F, Jana B, Hwa T, Onuchic JN. Coevolutionary signals across protein lineages help capture multiple protein conformations. Proc Natl Acad Sci U S A. 2013;110:20533–20538. 10.1073/pnas.1315625110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blum M, Chang H‐Y, Chuguransky S, et al. The InterPro protein families and domains database: 20years on. Nucleic Acids Res. 2021;49:D344–D354. 10.1093/nar/gkaa977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zerihun MB, Pucci F, Peter EK, Schug A. Pydca v1.0: A comprehensive software for direct coupling analysis of RNA and protein sequences. Bioinformatics. 2020;36:2264–2265. 10.1093/bioinformatics/btz892. [DOI] [PubMed] [Google Scholar]
- 26. Steinegger M, Söding J. Clustering huge protein sequence sets in linear time. Nat Commun. 2018;91(9):1–8. 10.1038/s41467-018-04964-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Porter LL, Kim AK, Looger LL, Majumdar A & Starich M. Pervasive fold switching in a ubiquitous protein superfamily. bioRxiv. 2021.06.10.447921. 10.1101/2021.06.10.447921 [DOI]
- 28. Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jana B, Morcos F, Onuchic JN. From structure to function: The convergence of structure based models and co‐evolutionary information. Phys Chem Chem Phys. 2014;16:6496–6507. 10.1039/c3cp55275f. [DOI] [PubMed] [Google Scholar]
- 30. Sirovetz BJ, Schafer NP, Wolynes PG. Protein structure prediction: Making AWSEM AWSEM‐ER by adding evolutionary restraints. Proteins Struct Funct Bioinforma. 2017;85:2127–2142. 10.1002/prot.25367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution‐based residue‐residue contact predictions in a sequence‐ and structure‐rich era. Proc Natl Acad Sci. 2013;110:15674–15679. 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engelberger F, Galaz‐Davison P, Bravo G, Rivera M, Ramírez‐Sarmiento CA. Developing and implementing cloud‐based tutorials that combine bioinformatics software, interactive coding, and visualization exercises for distance learning on structural bioinformatics. J Chem Educ. 2021;98:1801–1807. 10.1021/acs.jchemed.1c00022. [DOI] [Google Scholar]
- 34. Tomar SK, Knauer SH, NandyMazumdar M, Rösch P, Artsimovitch I. Interdomain contacts control folding of transcription factor RfaH. Nucleic Acids Res. [Internet]. 2013;41:10077–10085. 10.1093/nar/gkt779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hopf TA, Green AG, Schubert B, et al. The EVcouplings python framework for coevolutionary sequence analysis. Bioinformatics. 2019;35:1582–1584. 10.1093/bioinformatics/bty862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mistry J, Chuguransky S, Williams L, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galaz‐Davison P, Román EA, Ramírez‐Sarmiento CA. The N‐terminal domain of RfaH plays an active role in protein fold‐switching. PLoS Comput Biol. 2021;17:e1008882. 10.1371/journal.pcbi.1008882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Appadurai R, Nagesh J, Srivastava A. High resolution ensemble description of metamorphic and intrinsically disordered proteins using an efficient hybrid parallel tempering scheme. Nat Commun. 2021;12:958. 10.1038/s41467-021-21105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;5967873(596):583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S & Steinegger M ColabFold ‐ making protein folding accessible to all. bioRxiv. 2021.08.15.456425. 10.1101/2021.08.15.456425 [DOI] [PMC free article] [PubMed]
- 42. Schreck JS, Yuan JM. Exactly solvable model for helix‐coil‐sheet transitions in protein systems. Phys Rev E. 2010;81:061919. 10.1103/physreve.81.061919 [DOI] [PubMed] [Google Scholar]
- 43. Seifi B, Wallin S. The C‐terminal domain of transcription factor RfaH: Folding, fold switching and energy landscape. Biopolymers. 2021;112:e23420. 10.1002/bip.23420. [DOI] [PubMed] [Google Scholar]
- 44. Mouro PR, de Godoi CV, Chahine J, Junio de Oliveira R, Pereira Leite VB. Quantifying nonnative interactions in the protein‐folding free‐energy landscape. Biophys J. 2016;111:287–293. 10.1016/j.bpj.2016.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clementi C, Plotkin SS. The effects of nonnative interactions on protein folding rates: Theory and simulation. Protein Sci. 2004;13:1750–1766. 10.1110/ps.03580104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Contessoto VG, Lima DT, Oliveira RJ, Bruni AT, Chahine J, Leite VBP. Analyzing the effect of homogeneous frustration in protein folding. Proteins Struct Funct Bioinforma. 2013;81:1727–1737. 10.1002/prot.24309 [DOI] [PubMed] [Google Scholar]
- 47. Shi D, Svetlov D, Abagyan R, Artsimovitch I. Flipping states: A few key residues decide the winning conformation of the only universally conserved transcription factor. Nucleic Acids Res. 2017;45:8835–8843. 10.1093/nar/gkx523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Noel JK, Levi M, Raghunathan M, et al. SMOG 2: A versatile software package for generating structure‐based models. PLoS Comput Biol. 2016;12:e1004794. 10.1371/journal.pcbi.1004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Data Availability Statement
The retrieved RfaH sequences for coevolutionary analysis, the Jupyter Notebook for running pyDCA on Google Colaboratory, the coevolutionary analysis results, and all structure prediction simulations and final configurations are available for download at the laboratory's simulation archive in the Open Science Framework (OSF, https://osf.io/bn6u3/).