

Summary
Different effector arms of the immune system are optimized to protect from different classes of pathogens. In some cases, pathogens manipulate the host immune system to promote the wrong type of effector response – a phenomenon known as immune deviation. Typically, immune deviation helps pathogens to avoid destructive immune responses. Here we report on a type of immune deviation whereby an opportunistic pathogen, Pseudomonas aeruginosa, induces the type-2 immune response resulting in mucin production that is used as an energy source by the pathogen. Specifically, P. aeruginosa secreted toxin, LasB, processed and activated epithelial amphiregulin to induce type-2 inflammation and mucin production. This ‘niche remodeling’ by P. aeruginosa promoted colonization, and as a by-product, allergic sensitization. Our study thus reveals a type of bacterial immune deviation by increasing nutrient supply. It also uncovers a mechanism of allergic sensitization by a bacterial virulence factor.
Keywords: opportunistic pathogen, allergic inflammation, P. aeruginosa, LasB, amphiregulin, mucin, type-2 immunity
Introduction
Microbial pathogens rely on host-derived nutrients for successful colonization. In acute infections, this typically results in overt tissue destruction. Chronic infections and colonization by commensal microbiota require more nuanced long-term adaptations with pathogens actively modifying their niches within the host. Well-studied examples include chronic infections with Mycobacterium and Staphylococcus, which impact the structure and composition of colonized host tissues (Geoghegan et al., 2018; Ravimohan et al., 2018). These bacteria exploit host metabolic nutrients including monosaccharides, amino and fatty acids to persist in a tissue (Ehrt et al., 2018; Tan et al., 2020). Commensal microbes are also known to actively modify the gut epithelium (Martens et al., 2018; Tropini et al., 2017). Thus, colonization of germ-free mice with Bacteroides thetaiotaomicron results in induction of fucosyltransferase-2 (Fut2) and fucose utilization by the bacteria (Bry et al., 1996). In the context of infection, activation of toll-like receptors (TLRs) on intestinal dendritic cells leads to production of interleukin-23 (IL-23) and subsequent IL-22 mediated induction of Fut2, resulting in decreased expression of virulence genes in intestinal pathogens (Pickard et al., 2014).
Optimal defense from infections requires activation of an appropriate class of effector response. Thus, defense from acute bacterial infections relies on the myeloid cell derived cytokines IL-1α, IL-6, TNF, and IL-12 or IL-23, and lymphocyte-derived cytokines IFN-γ, IL-17 and IL-22 (Annunziato et al., 2015). Defense from parasitic worm infections, on the other hand, relies on epithelial-derived cytokines IL-25, IL-33 and TSLP, and lymphoid cytokines IL-4, IL-5, and IL-13 (Van Dyken et al., 2016; von Moltke and Locksley, 2014). As a convenient short-hand, these two kinds of immune response are referred to as type-1 and type-2 immunity, respectively. The type-2 immune response is engaged against both parasitic worms and environmental toxins (the latter referred to as allergic immune response) (Palm et al., 2012; Profet, 1991). Meanwhile, the type-2 immune response orchestrates not only worm and allergen expulsion, but also metabolic adaptations and tissue reparative response (Allen and Wynn, 2011; Cheng and Locksley, 2014; Palm et al., 2012). Chronic Mycobacterium and Staphylococcus infections can result in a biased type 2 immune response (Benmerzoug et al., 2019; Cronan et al., 2021; Lan et al., 2018).
Pathogens use a variety of strategies to evade recognition and destruction by the host immune system. One such strategy, the immune deviation, is based on activation of the wrong type of effector response that is ineffective in defense against a given pathogen class. How bacterial pathogens manipulate the host response to induce type-2 immunity is poorly understood. Bacteria are detected by the host pattern-recognition receptors, whereas recognition of allergens (and likely helminths) is often based on sensing of enzymatic activities as well as other features of allergens, rather than on direct molecular recognition (Florsheim et al., 2021; Palm et al., 2012). However, many bacterial and fungal pathogens secrete virulence factors and toxins that have various biochemical activities and may conceivably be detected by the sensors of the type-2 immune system.
Here we explored the hypothesis that bacterial toxins might trigger type-2 immune response to benefit the bacterial pathogens. To investigate that, we used a major opportunistic human pathogen, Pseudomonas aeruginosa, that can cause chronic lung infections in immunocompromised cohorts, such as cystic fibrosis patients (Folkesson et al., 2012; Jensen et al., 2010). These patients have been reported to exhibit pulmonary T helper-2 (Th2) cell responses (Hartl et al., 2006; Moser et al., 2000). P. aeruginosa produces a group of virulence toxins regulated by transcription factor LasR (Gambello and Iglewski, 1991; Rutherford and Bassler, 2012). The major virulence toxin necessary for successful colonization is metalloprotease LasB or elastase (Bever and Iglewski, 1988). We found that LasB promoted type-2 immune response by generating the active form of amphiregulin. Amphiregulin induced airway mucin production, that can serve as an energy source for Pseudomonas. Subsequently, P. aeruginosa induced type 2 immune response that was dependent on LasR-LasB virulence system.
Results
P. aeruginosa toxin induces tissue repair gene program in epithelial cells
The major virulence factor of P. aeruginosa, LasB, belongs to M4 family of bacterial metalloproteases with a known specific chemical inhibitor, phosphoramidon (Morihara and Tsuzuki, 1978; Oldak and Trafny, 2005). To determine the effect of LasB on airway epithelial cells, we treated human airway epithelial cell line H292 or ex vivo mouse lung organ cultures with P. aeruginosa LasB and measured changes in gene expression (Fig. 1A). LasB induced expression of TSLP and Afgan which are involved in allergic inflammation and tissue repair (Fig. 1B, S1G). Phosphoramidon efficiently inhibited proteolytic activity of LasB based on elastin cleavage assay (Fig. S1A) and blocked the induction of TSLP and Areg (Fig. 1C, S1H) suggesting that LasB enzymatic activity is crucial for promoting innate type-2 responses. To test whether LasB-mediated induction of Areg and TSLP is dependent on toll-like receptors, we performed siRNA silencing of MyD88, major signaling component of toll-like receptor pathway (Medzhitov, 2001). Silencing of MyD88 reduced expression of TSLP and Areg stimulated by a toll-like receptor ligand, bacterial flagellin, but not by LasB (Fig. S1K). Trypsin protease receptor PAR-2 had a modest effect on LasB-stimulated gene expression and signaling (Fig. S1L).
Figure 1. P. aeruginosa toxin induces tissue repair gene program in epithelial cells that is dependent on EGFR pathway.
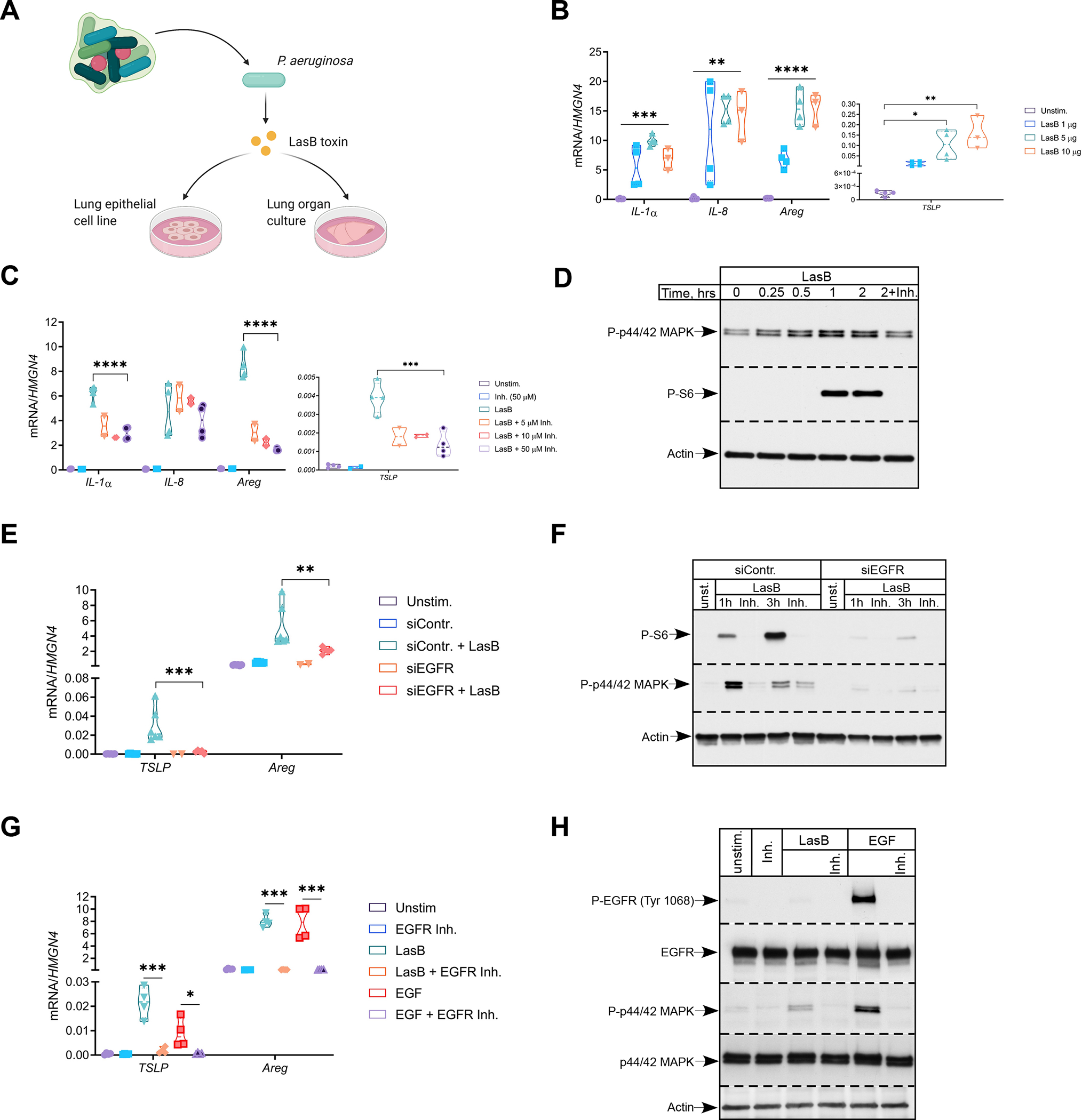
(A) Schematics of in vitro experimental system.
(B and C) Relative mRNA expression of pro-inflammatory (IL-1a, IL-8) and tissue repair (TSLP, Areg) genes in H292 cells treated with indicated amounts of LasB (B) or LasB (2 μg/ml) +/− indicated concentrations of inhibitor (C) for 3 hr (n=3–4).
(D) Immunoblot analysis of P-p44/42 MAPK and P-S6 in H292 cells treated with LasB (2 μg/ml) +/− inhibitor (50 μM) at different time points (n=4).
(E and F) Relative mRNA expression of TSLP and Areg (E) and immunoblot analysis of P-p44/42 MAPK and P-S6 (F) in H292 cells treated with control or EGFR siRNA and stimulated with LasB (2 μg/ml) +/−inhibitor (50 μM) for 1 or 3 hr (n=6).
(G and H) Relative mRNA expression of TSLP and Areg (G) and immunoblot analysis of P-EGFR Tyr1068 and P-p44/42 MAPK (H) in H292 cells pre-treated with EGFR kinase inhibitor (20 μM) for 20 min and stimulated with LasB (5 μg/ml) or EGF (10 ng/ml) for 3hr (n=4).
mRNA expression was measured relative to high mobility group nucleosome-binding domain-containing protein 4 (HMGN4). ß-actin, total EGFR and p44/42 MAPK were used as loading controls. n = independent experiments. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
See also Figure S1.
LasB induced activation of both mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) signaling pathways (Fig. 1D). Activation of these pathways was dependent on LasB proteolytic activity (Fig. 1D) and suggested involvement of receptor tyrosine kinase in LasB induced signaling. Initial siRNA silencing screening for potential receptors unexpectedly revealed that ‘control siRNA’ had a major effect (Fig. S1I, left). The manufacturer of the ‘control siRNA’ (Dharmacon, USA) informed us that this siRNA can reduce expression of mRNA encoding epidermal growth factor receptor (EGFR). To confirm this serendipitous finding, we performed experiments with newly designed EGFR specific siRNA. siRNA silencing of EGFR abrogated expression of TSLP and Areg in response to LasB (Fig. 1E) and nearly eliminated activation of MAPK and mTOR pathways (Fig. 1F). Selective EGFR kinase inhibitor diminished expression of TSLP and Areg and activation of MAPK cascade in response to LasB (Fig. 1G, H), suggesting that EGFR kinase activity is required for LasB signaling. LasB stimulation did not result in detectable phosphorylation of EGFR in contrast to a specific ligand, EGF (Fig. 1H, S1F). EGFR-mediated gene expression and signaling were not affected by LasB inhibitor (Fig. S1E). siRNA silencing of components responsible for clathrin- (AP2M) and caveolin- (PACSIN3) mediated endocytosis did not alter activation of mTOR pathway or reduce expression of TSLP and Areg (Fig. S1I, right; S1J) suggesting that EGFR endocytosis is not involved in LasB response. Taken together, these data suggest that LasB activates EGFR pathway to induce expression of tissue repair genes (TSLP and Areg) associated with innate type-2 responses in epithelial cells.
P. aeruginosa toxin employs endogenous amphiregulin to stimulate epithelial cells
Requirement of EGFR kinase activity suggested that LasB might mimic the effect of mammalian metalloproteases, which cleave and release EGF family precursors from their membrane-bound form (Khokha et al., 2013). EGF family ligands are classified as high affinity (EGF and transforming growth factor-α) and low affinity (Areg and Epiregulin) based on their dissociation constant (Krall et al., 2011). Since LasB stimulation did not induce visible EGFR phosphorylation but activated downstream pathways (Fig. 1H, S1F), we reasoned that the effect of LasB was mediated by a low affinity ligand. Using published RNA-seq data for H292 cells (Klijn et al., 2015), we found that Areg is the only low affinity ligand expressed by these cells at baseline (Fig. S2A). EGF but not Areg induced strong phosphorylation of EGFR confirming low affinity nature of latter (Fig. S2G). Both EGF and Areg similarly activated MAPK pathway (Fig. S2G). Using flow cytometric (FACS) analysis, we found that LasB stimulation elevated Areg protein amounts relative to unstimulated cells not affecting cell viability (Fig. 2A, B).
Figure 2. P. aeruginosa toxin utilizes amphiregulin to activate epithelial cells.
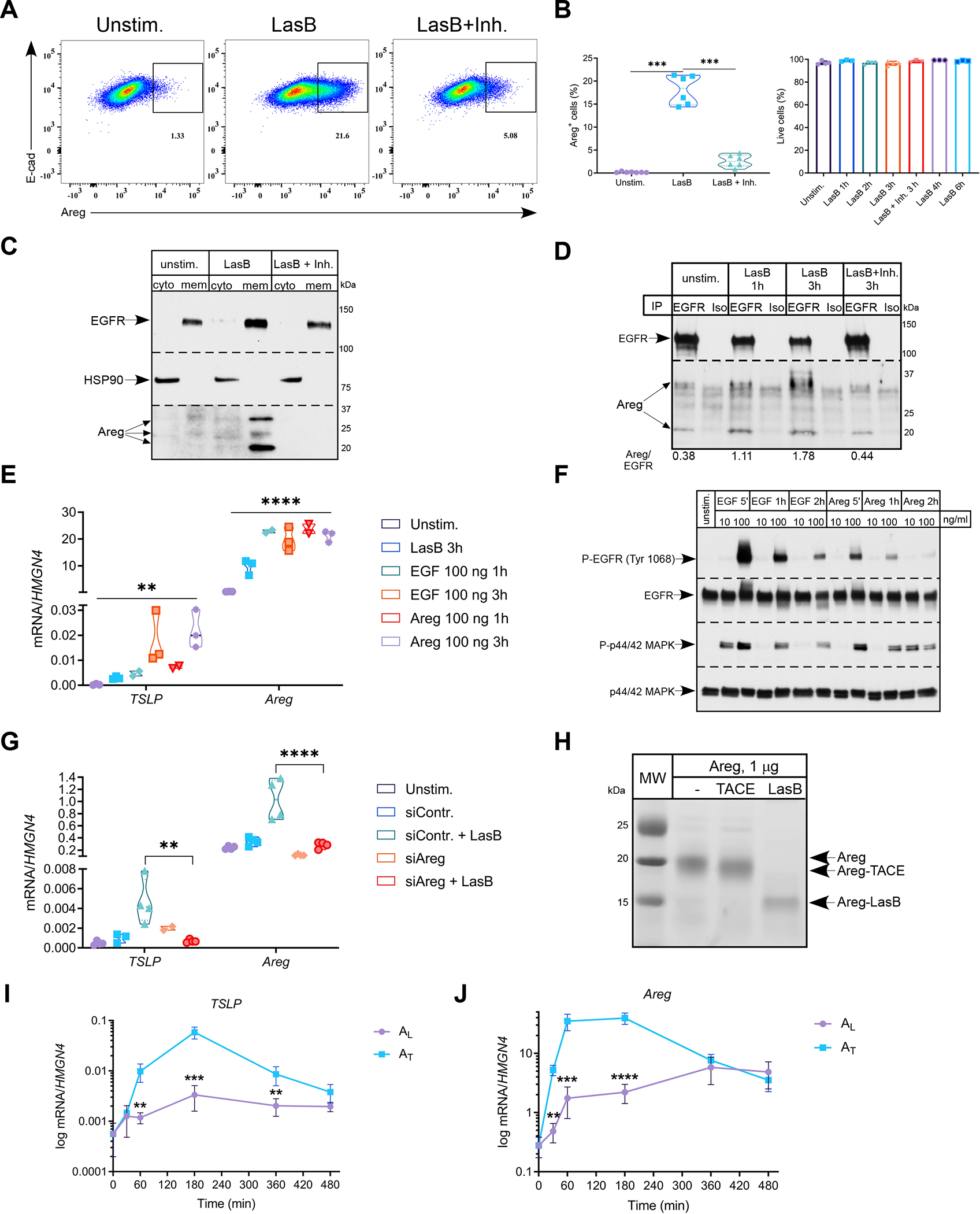
(A and B) Representative FACS plots (n=6) of frequency of Areg+ H292 cells (A) stimulated with LasB (2 μg/ml) +/− inhibitor (50 μM) for 3hr and quantification (B) of Areg+ (left) and total live cells (right) (n=3–6).
(C and D) Immunoblot analysis of Areg in cytosolic and membrane fractions (C) and after immunoprecipitation with anti-EGFR antibodies (D) in H292 cells stimulated with LasB (3 μg/ml) +/− inhibitor (50 μM) for 3hr (n=3).
(E) Relative mRNA expression of TSLP and Areg in H292 cells stimulated with LasB (2 μg/ml) for 3hr, EGF and Areg (100 ng/ml) for 1 or 3 hr (n=3).
(F) Immunoblot analysis of P-EGFR Tyr1068 and P-p44/42 MAPK in H292 cells stimulated with EGF and Areg (10 or 100 ng/ml) at indicated time points (n=3).
(G) Relative mRNA expression of TSLP and Areg in H292 cells treated with control or Areg siRNA and stimulated with LasB (5 μg/ml) for 3hr (n=4).
(H) In vitro cleavage of recombinant human Areg (1 μg) by LasB and ADAM17 or TACE (0.5 μg) for 30 min at RT (n=4).
(I and J) Kinetics of relative mRNA expression of TSLP (I) and Areg (J) in H292 cells treated with AL or AT (50 ng/ml) (n=3).
mRNA expression was measured relative to HMGN4. ß-actin, total EGFR and p44/42 MAPK and HSP90 were used as loading controls. n = independent experiments. Data are mean ± SD and were analyzed by one-way ANOVA with Tukey’s multiple comparisons test (B, E, G) or two-tailed unpaired Student’s t test (I, J). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
See also Figures S1 and S2.
To determine the localization of induced Areg in cells, we performed cytosolic and membrane fractionation of H292 cells. We found that Areg species, likely originated due to post-translational modifications, were exclusively present in the membrane fraction after LasB stimulation (Fig. 2C). Using immunoprecipitation with antibodies to extracellular portion of EGFR, we found that membrane-retained Areg species were bound by EGFR in LasB-activated cells (Fig. 2D, S2C). Accordingly, since Areg was retained in the membrane fraction and bound by EGFR upon LasB stimulation, we did not detect Areg in supernatants of LasB-activated H292 cells by ELISA (Fig. S2H). Together, these data suggest that proteolytic processing of Areg by LasB results in Areg activation and subsequent binding to EGFR. Both high affinity (EGF) and low affinity (Areg) EGFR ligands induced TSLP and Areg gene expression, similar to LasB (Fig. 2E). However, kinetic analysis showed that, in contrast to EGF, Areg induced much lower phosphorylation of EGFR despite similar activation of MAPK cascade (Fig. 2F). Finally, siRNA silencing of Areg in H292 reduced expression of TSLP and Areg in response to LasB (Fig. 2G). Using immunoblotting, we confirmed successful siRNA silencing of Areg protein amounts in response to LasB and other proteases (papain and subtilisin) (Fig. S2B).
Areg production is known to be regulated by the metalloprotease ADAM17 or TACE (Blobel, 2005). To test whether TACE is involved in LasB-mediated Areg induction, we performed siRNA silencing of ADAM17 or TACE and found it to be dispensable for the induction of TSLP and Areg (Fig. S2I). Thus, we concluded that LasB-mediated signaling is independent of ADAM17 or TACE. To test whether LasB can cleave Areg and, thus, mediate its release, we performed in vitro cleavage assay. LasB specifically cleaved recombinant human and mouse Areg in vitro resulting in generation of ~17–18 and 10–12 kDa fragments, respectively (Fig. S2D, E). Other proteases, papain and subtilisin, non-specifically degraded recombinant human Areg (Fig. S2F). When we compared LasB and TACE cleavage products, we found that these two proteases generated Areg fragments of distinct sizes suggesting different cleavage sites (Fig. 2H). TACE cleavage sites of Areg have been mapped to Lys residues K184 and K187 at C-terminal end of Areg (Fig. S2L) (Berasain and Avila, 2014). Using mass-spectrometry we found approximate cleavage sites of LasB at one of the Gly residues (G178 and G182) at C-terminal end of Areg, in accordance with LasB cleavage site sequence (Fig. S2K, L) (Rawlings et al., 2018). Alignment of Areg ligands from different species demonstrated high conservation of these Gly residues (Fig. S2K). LasB-cleaved Areg (AL) stimulated sustained kinetics of gene expression and signaling of MAPK cascade compared to TACE-cleaved Areg (AT) (Fig. 2I–J, S2J). Collectively, these data demonstrate that LasB generates distinct form of Areg to provide sustained signaling and expression of TSLP and Areg.
Amphiregulin is required for mucin expression and recruitment of eosinophils
Various polysaccharides and host mucins can serve as a nutrient source for colonizing bacteria (Desai et al., 2016; Sonnenburg and Sonnenburg, 2014). Chronic Pseudomonas infections during cystic fibrosis are characterized by increased mucus production (Folkesson et al., 2012). Muc5AC and Muc5B are the primary airway mucins secreted by goblet cells or submucosal glands, respectively. Muc5B is expressed constitutively whereas Muc5AC expression is inducible and is upregulated during different pathologies such as asthma and cystic fibrosis (Jaramillo et al., 2018). Thus, we hypothesized that P. aeruginosa virulence protease LasB and LasB-activated Areg can stimulate Muc5AC production in lung epithelial cells. Both AL and AT stimulated Muc5AC expression in H292 cells. AL induced prolonged high expression of Muc5AC (Fig. 3A). LasB stimulated Muc5AC expression in H292 cells, similar to AL and AT (Fig. 3B). Using ex vivo mouse lung organ cultures (Fig. 1A), we confirmed that LasB stimulated expression of tissue repair genes (TSLP, Areg) and Muc5AC (Fig. 3C). Moreover, TSLP and Muc5AC expression was dependent on Areg as expression of these genes was diminished in Areg-deficient (Areg−/−) lung organ cultures (Fig. 3D). Addition of exogenous recombinant mouse Areg partially restored expression of TSLP and Muc5AC in Areg−/−lung organ cultures (Fig. S4A).
Figure 3. Amphiregulin is required for mucin expression and recruitment of eosinophils.
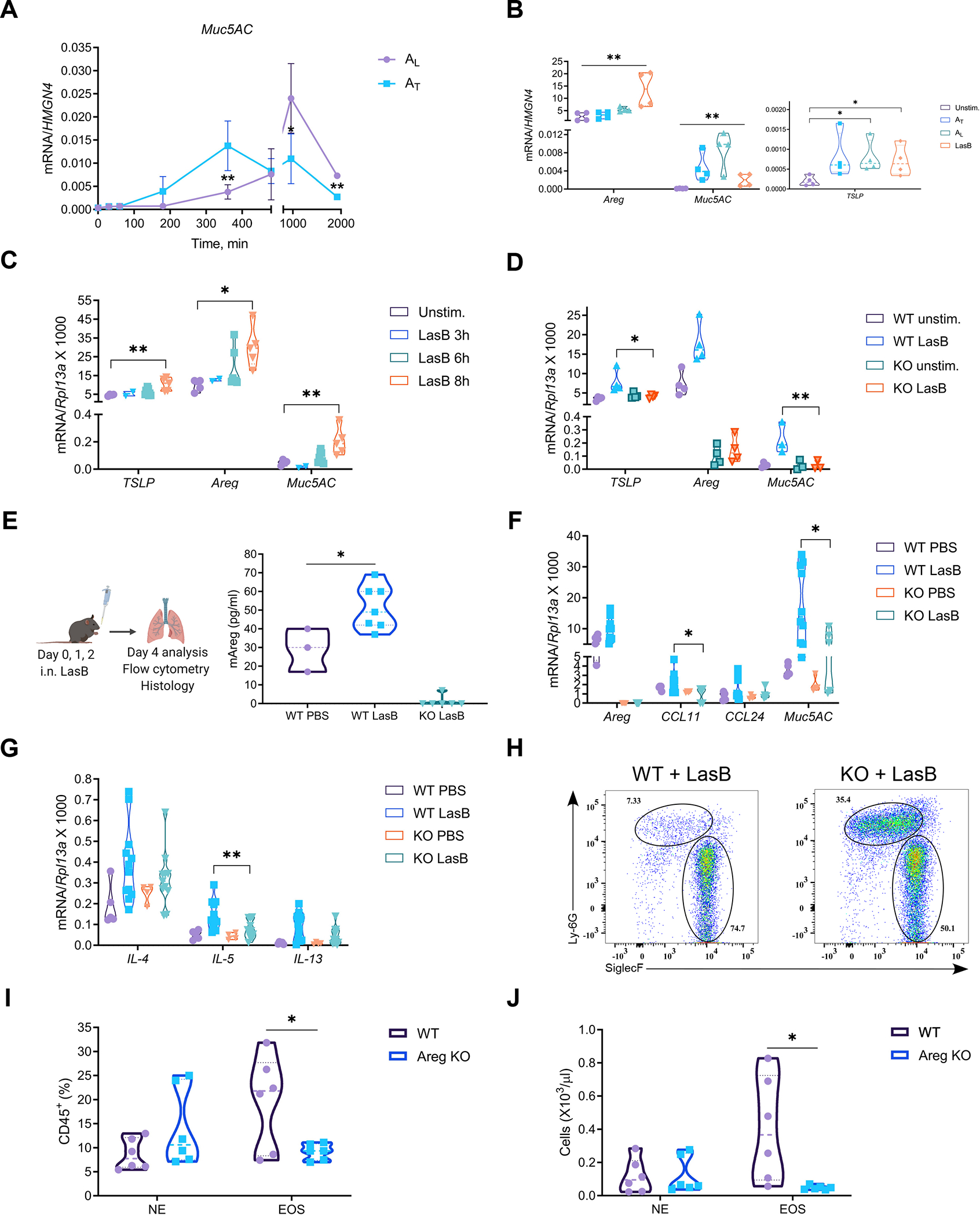
(A) Kinetics of relative mRNA expression of Muc5AC in H292 cells treated with 50 ng/ml of AL or AT (n=3).
(B) Relative mRNA expression of TSLP, Areg and Muc5AC in H292 cells stimulated with LasB (0.5 μg/ml), AL or AT (50 ng/ml) for 12hr (n=4).
(C) Relative mRNA expression of TSLP, Areg and Muc5AC in ex vivo mouse lung organ cultures stimulated with LasB (5 μg/ml) for indicated times (n=5).
(D) Relative mRNA expression of TSLP, Areg and Muc5AC in WT and Areg−/− (KO) mouse lung organ cultures stimulated with LasB (5 μg/ml) for 8 hr (n=3–4).
(E) (Left) Experimental protocol for the development of innate airway inflammation and (Right) ELISA of Areg concentrations in the BAL of WT and Areg−/− (KO) mice (combined of 3 experiments yielding 3–7 mice per group).
(F and G) Relative mRNA expression of Areg, Ccl11, Ccl24 and Muc5AC (F) and type 2 cytokines (IL-4, 5, 13) (G) in the lungs of LasB-treated WT and Areg−/− (KO) mice (combined of 3 experiments yielding 4–12 mice per group).
(H-J) Representative FACS plots (n=5) (H) and graphs showing cell frequency (I) and numbers (J) of eosinophils (CD45+MHCII–?CD11b+Siglec-F+) and neutrophils (CD45+MHCII–CD11b+Ly6G+) out of CD45+ cells in the BAL of LasB-treated WT and Areg−/− (KO) mice (combined of 2 experiments yielding 6 mice per group).
mRNA expression was measured relative to HMGN4 in H292 or Rpl13a in mouse lung organ cultures/lungs. n = independent experiments. Data are mean ± SD and were analyzed by two-tailed unpaired Student’s t test (A, I, J) or one-way ANOVA with Tukey’s multiple comparisons test (B-G). *p < 0.05; **p < 0.01; ***p < 0.001.
Since P. aeruginosa toxin induced TSLP and Areg in epithelial cells and lung organ cultures, we next tested whether LasB can promote type-2 immune responses in vivo and whether it is dependent on Areg. We used innate airway inflammation model (Fig. 3E, left). Animals were intranasally instilled with LasB for three consecutive days and their response was analyzed 48hr after last challenge. Areg production was increased in the BAL of LasB-treated animals (Fig. 3B, right). We found induction of type 2 cytokines (IL-4, 5, 13), eotaxins (Ccl11, 24) and Muc5AC in the lungs of LasB-treated animals (Fig. 3F, G). Areg-deficient animals had reduced expression of Muc5AC, eosinophil-attracting chemokines (Ccl11) as well as IL-5 (Fig. 3F, G). In accordance with gene expression, eosinophil recruitment in the BAL was impaired in Areg-deficient animals (Fig. 3H–J), while baseline numbers of circulating eosinophils were comparable in WT and Areg−/−mice (Fig. S3A). Lung H&E and PAS staining demonstrated reduced cell infiltration and mucin production, respectively, in Areg-deficient animals (Fig. S3B, C). TSLP receptor-deficient animals had reduced eosinophil infiltration and expression of Ccl24 and Muc5AC in response to LasB instillation as well (Fig. S4J). Taken together, these data demonstrate that LasB induces mucin production and innate type-2 response in vivo that are dependent on Areg.
LasB is an adjuvant for allergic response
Based on our findings, P. aeruginosa LasB can induce innate type-2 immune response resulting in eosinophil infiltration and mucin production. Therefore, we reasoned that LasB might serve as an adjuvant in airway allergic response. To test that, we used a standard airway allergy model (Fig. 4A). Animals were immunized subcutaneously with ovalbumin (OVA) +/− LasB on days 0 and 7 to sensitize them to OVA. On day 13 animals were subjected to anaphylaxis by intravenous injection of OVA. Animals were intranasally challenged with OVA on days 21 and 28 and their response was assessed 24 hr after last challenge. Administration of intravenous OVA resulted in decreased body temperature, indicating a systemic anaphylaxis response (Fig. S3D). In accordance, LasB with OVA immunized animals had increased titers of total IgE, OVA-specific IgE and IgG1 antibodies in their serum (Fig. 4B–D). Eosinophil infiltration was also increased in the BAL of LasB with OVA-immunized animals (Fig. S3E–F). The concentrations of IL-4 and IL-5 but not IL-13 cytokines were increased in the BAL of immunized animals (Fig. 4E–G, S3G). The concentrations of Th1 cell-specific cytokines (IFN-γ) and antibodies (OVA-IgG2b) were not affected or mildly induced, respectively (Fig. S3G). Expression of type 2 cytokines (IL-4, 5, 13), eotaxins (Ccl11, 24) and Muc5AC was upregulated in the lungs of LasB with OVA-immunized animals (Fig. S3J). Lung H&E and PAS staining also demonstrated increased cell infiltration and mucin production (Fig. S3H, I).
Figure 4. LasB is an adjuvant for allergic response.
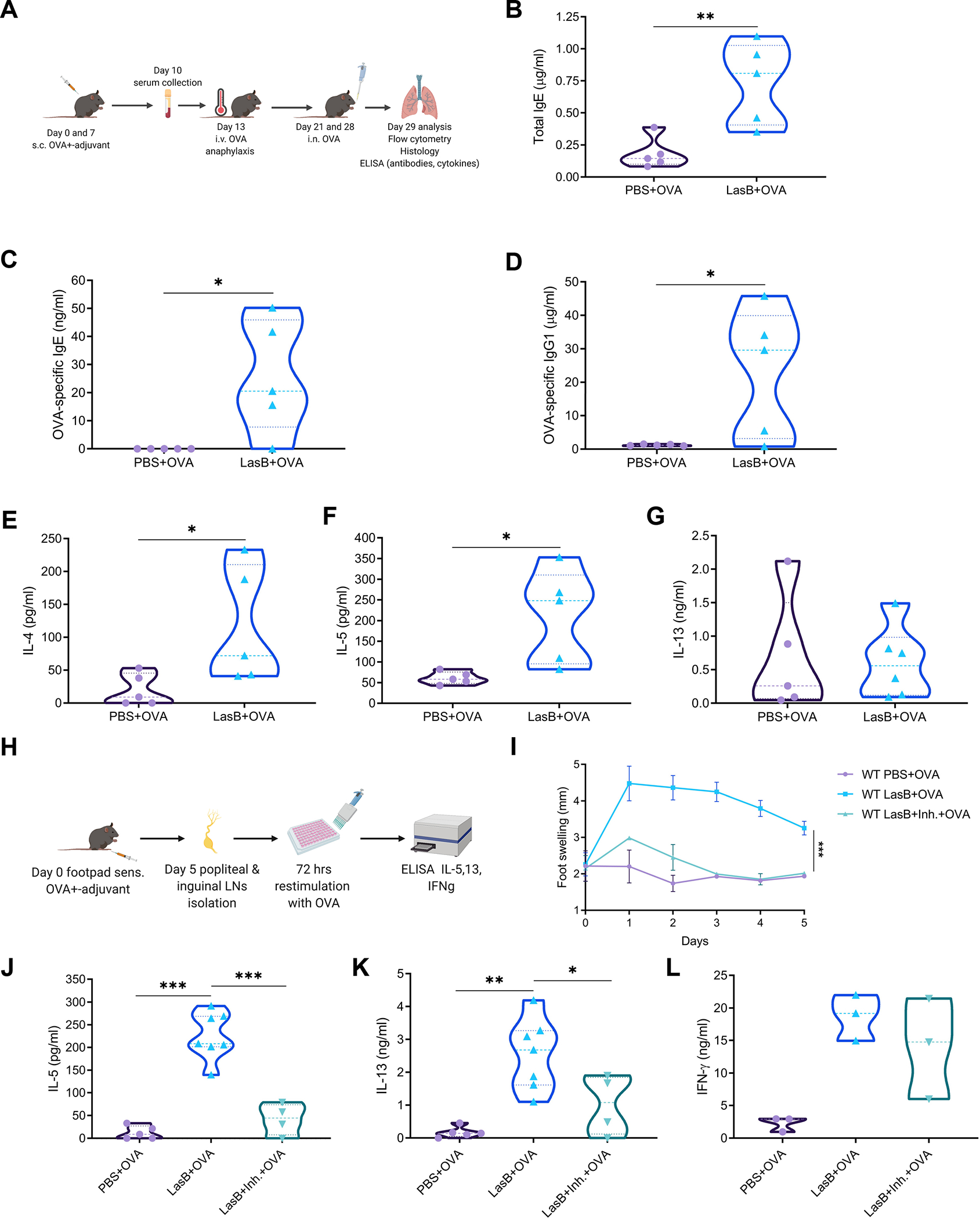
(A) Experimental protocol of LasB allergic airway inflammation.
(B-D) ELISA of total IgE (B), OVA-specific IgE (C) and OVA-specific IgG1 (D) amounts in the serum of mice immunized with OVA +/− LasB.
(E-G) ELISA of IL-4 (E), IL-5 (F) and IL-13 (G) amounts in the BAL of mice immunized with OVA +/− LasB.
(H) Experimental protocol of skin allergic inflammation.
(I) Kinetics of swelling in the left footpad of mice immunized with LasB/OVA +/− LasB inhibitor (combined of 2 experiments yielding 3–6 mice per group).
(J-L) IL-5 (J), IL-13 (K) and IFN-γ (L) production after ex vivo stimulation of total lymph node cells from mice immunized as in I (J and K - combined of 2 experiments with 4–7 mice per group, L - representative of 3 experiments, 3 mice per group).
For (B-G) data combined of 2 experiments yielding 5 mice per group. Data are mean ± SD and were analyzed by two-tailed unpaired Student t-test (B-G) or one-way ANOVA with Tukey’s multiple comparisons test (I-L). *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figures S3 and S5.
To further test type 2 adjuvant effect of LasB, we used footpad immunization model (Fig. 4H). Animals were immunized into the left hind footpads with OVA with or without LasB at day 0 and popliteal and inguinal lymph nodes were isolated on day 5, total cells were counted and re-stimulated with OVA for 72 hr to detect T cell cytokine production. First, we observed that LasB induced significant footpad swelling that was abolished by LasB inhibitor, phosphoramidon (Fig. 4I, S5C). No detectable swelling was observed in non-injected right footpad (Fig. S5A). Combination of LasB with OVA resulted in activation of a Th2 cell response, as indicated by secretion of IL-5 and IL-13 but not IFN-γ that was induced by LPS/IFA/OVA injection (Fig. 4J, K, S5B). The induction of a Th2 cell response was dependent on the LasB enzymatic activity as the response was abrogated using the LasB inhibitor (Fig. 4J, K), whereas modest induction of the Th1 cell response was not affected by inhibitor (Fig. 4L). Thus, these data demonstrate that LasB serves as an adjuvant to induce type-2 responses in the lung and skin and that the adjuvant effect is dependent on protease activity.
Areg can play a critical role in promoting adaptive type-2 responses (Zaiss et al., 2006). To determine whether the induction of adaptive type-2 responses by LasB is dependent on Areg, we immunized WT and Areg-deficient animals as above (Fig. 4H). Foot swelling was significantly reduced in Areg-deficient animals (Fig. 6A, S5D). Secretion of IL-5 but not IL-13 or IFN-γ was reduced upon LasB with OVA immunization (Fig. 6B–D). Selective reduction of IL-5 in Areg-deficient animals suggested that eosinophil recruitment is impaired which is consistent with our previous data. In conclusion, these data suggest that LasB-mediated induction of adaptive type 2 responses is partially dependent on Areg.
Figure 6. Amphiregulin is required for successful P. aeruginosa infection.
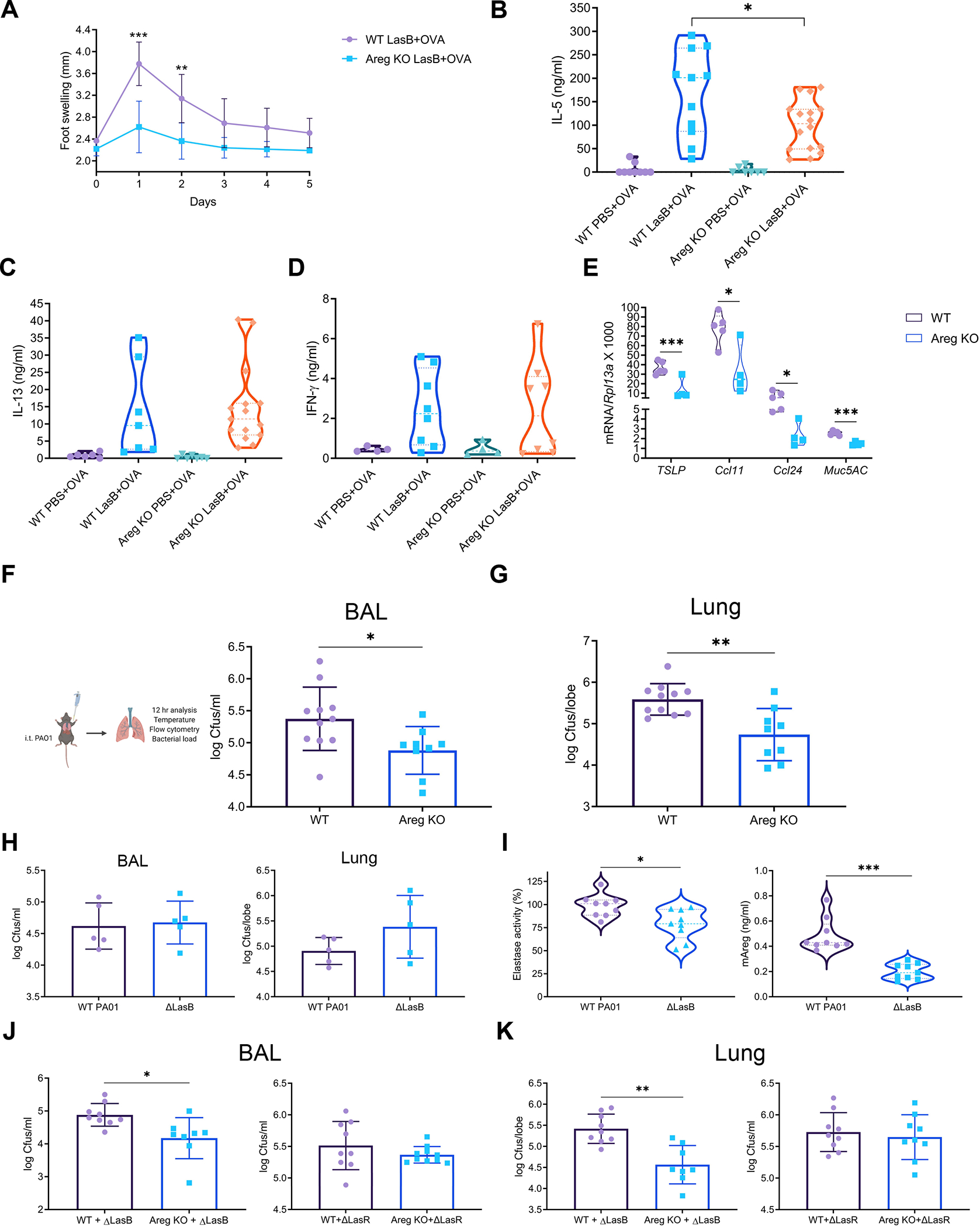
(A) Kinetics of left footpad swelling of WT and Areg−/− (KO) mice immunized with LasB/OVA (combined of 2 experiments yielding 4–7 mice per group).
(B-D) IL-5 (B), IL-13 (C) and IFN-γ (D) production after ex vivo stimulation of total lymph node cells from WT and Areg−/− (KO) mice immunized as in A (B and C - combined of 3 experiments yielding 6–16 mice per group, D - combined of 2 experiments yielding 4–8 mice per group).
(E) Relative mRNA expression of TSLP, Ccl11, Ccl24 and Muc5AC in WT and Areg−/− (KO) lungs infected with WT PA01 for 12 hr (representative of 5 experiments, 4–5 mice per group).
(F and G) Experimental protocol of WT PA01 infection (F, left), BAL (F, right) and lung (G) bacterial loads of WT PA01 in WT and Areg−/− (KO) mice 12 hr after infection (combined of 2 experiments yielding 9–11 mice per group).
(H and I) BAL (H, left) and lung (H, right) bacterial loads and BAL elastase activity (I, left) and Areg amounts (I, right) of WT and ΔLasB PA01 in WT mice 12 hr after infection (H - representative of 3 experiments, 5 mice per group; I - combined of 2 experiments yielding 9 mice per group).
(J and K) BAL (G) and lung (H) bacterial loads of ΔLasB and ΔLasR PA01 in WT and Areg−/− (KO) mice 12 hr after infection (combined of 2 experiments yielding 8–10 mice per group).
mRNA expression was measured relative to Rpl13a. Data are mean ± SD and were analyzed by two-tailed unpaired Student’s t test (A, E-K) or one-way ANOVA with Tukey’s multiple comparisons test (B-D). *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S5.
P. aeruginosa can serve as an adjuvant for an allergic response to an innocuous protein
Because P. aeruginosa toxin, LasB, can promote type-2 responses, we asked whether non-infectious amounts of P. aeruginosa (PA01 strain) can promote allergic response. To test that, we used airway allergy inflammation model and compared LasB toxin and WT PA01 as adjuvants (Fig. 5A). On days 0 and 7, animals were immunized intratracheally with OVA +/− PA01 or LasB. Animals were intratracheally challenged with OVA on days 14 and 21 and their response was analyzed 24 hr after the last challenge. We found increased eosinophil infiltration in the BAL of LasB and PA01-immunized animals (Fig. 5B). The response of PA01-immunized animals was lower that of LasB-immunized mice but significantly higher than OVA immunization alone. Expression of type 2 cytokines, eotaxins and Muc5AC were significantly upregulated in the lungs of both LasB and PA01-immunized animals (Fig. S4B). The amounts of IL-4 were increased in the BAL of LasB-immunized animals (Fig. 5C). The amounts of IL-5 were increased in the BAL of both LasB- and PA01-immunized animals (Fig. 5D). The amounts of IFN-γ were not changed (Fig. 5E). Antibody titers of total IgE, OVA-specific IgE and IgG1 were increased in the serum of immunized animals (Fig. 5F–H), whereas the titers of OVA-specific IgG2b were induced (Fig. 5I). Thus, PA01 can serve as a type-2 adjuvant and promote an allergic response. Lower allergic inflammation in PA01-immunzed animals might be due to non-infectious numbers of PA01 and sub-optimal production of virulence factors in contrast to relatively high amounts of recombinant LasB.
Figure 5. P. aeruginosa can serve as an adjuvant for an allergic response.
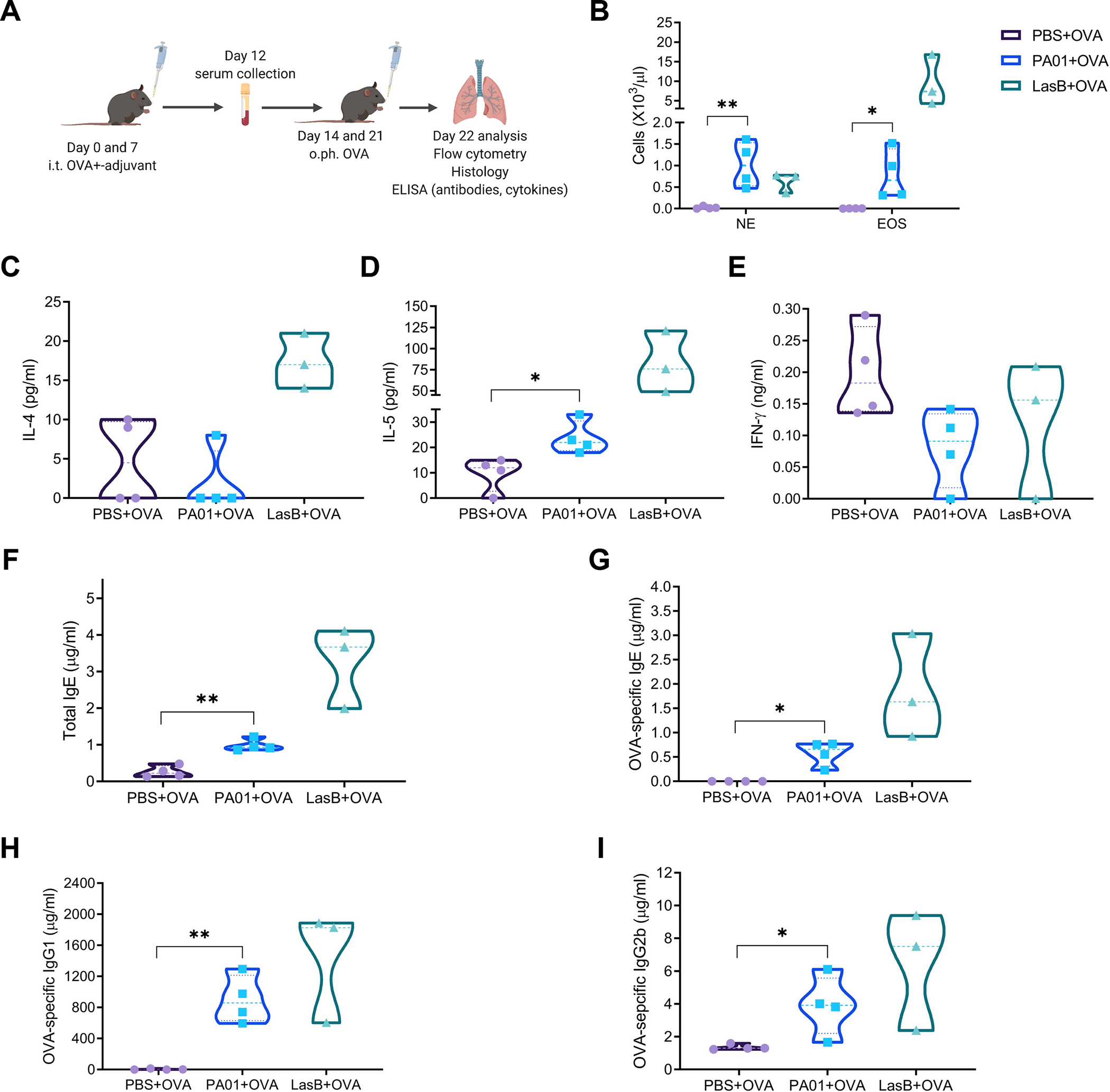
(A) Experimental protocol of PA01 allergic airway inflammation.
(B) Cell frequency of eosinophils (CD45+MHCII–CD11b+CD64-Siglec-F+) and neutrophils (CD45+MHCII–CD11b+CD64-Ly6G+) out of CD45+ cells in the BAL of mice immunized with OVA +/− PA01 or LasB.
(C-E) ELISA of IL-4 (C), IL-5 (D) and IFN-γ amounts in the BAL of mice immunized as in (A).
(F-I) ELISA of total IgE (F), OVA-specific IgE (G), OVA-specific IgG1 (H) and IgG2b (I) amounts in the serum of mice immunized as in (A).
For (B-I) representative of 3 independent experiments, 3–4 mice per group. Data were analyzed by two-tailed unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S4.
To determine whether PA01 adjuvant activity is dependent on Areg for type-2 responses, we compared allergic inflammation (Fig. 5A) in WT and Areg-deficient animals. Eosinophil infiltration in the BAL was similar in WT and Areg−/− mice, while neutrophil numbers were increased in Areg-deficient animals (Fig. S4C, D). Antibody titers of total and OVA-specific IgE were also similar in the serum of WT and Areg−/−mice (Fig. S4E, F). However, the titers of OVA-specific IgG2b, that is tailored for type-1 immunity, were increased in the serum of Areg-deficient animals (Fig. S4G, H). Finally, lung gene expression of IL-4 and Muc5AC were reduced in Areg-deficient animals (Fig. S4I). Together, these data indicate that P. aeruginosa activates type-2 immune response that is partially dependent on Areg.
P. aeruginosa uses amphiregulin to promote colonization
To test the role of Areg during acute PA01 infection, we infected WT and Areg-deficient mice intratracheally with WT PA01 (Fig. 6F, left panel). Infected animals were analyzed 12 hr after infection. We found reduced expression of TSLP, eotaxins and Muc5AC in the lungs of Areg-deficient animals (Fig. 6E). Areg-deficient animals better maintained their body temperature (Fig. S5E) and had lower bacterial loads in the BAL and lung (Fig. 6F right panel, 6G). These data suggest that PA01 takes advantage of the host Areg to promote bacterial infection. To determine whether LasB is required for PA01 infection, we infected WT and Areg−/−mice with ΔLasB PA01 that is deficient in the elastase production measured by in vitro elastin cleavage activity of bacterial supernatants (Fig. S5F). WT mice infected with WT and ΔLasB PA01 had similar BAL and lung bacterial loads (Fig. 6H). As expected ΔLasB PA01 had reduced elastase activity resulting in lower Areg amounts in BAL (Fig. 6I). Areg−/−mice infected with ΔLasB PA01 better maintained their body temperature and had lower BAL and lung bacterial loads compared to WT animals (Fig. S5G, 6J, K). Moreover, lung gene expression of TSLP, eotaxins and Muc5AC of WT and Areg-deficient animals infected with ΔLasB PA01 was not significantly different (Fig. S5H). These data suggest that other virulence proteases (LasA, ArpA or PIV) controlled by transcription regulator LasR (Oldak and Trafny, 2005) might compensate for LasB function in PA01 infection. Thus, we infected animals with ΔLasR PA01 that is deficient in transcription factor LasR and production of any secreted proteases (Fig. S5F). WT and Areg−/−mice infected with ΔLasR PA01 had similar BAL and lung bacterial loads suggesting that LasR is required for Areg-mediated infection of PA01 (Fig 6J, K). Similarly, ΔLasB and ΔLasR PA01 capacity to promote type-2 immunity is diminished based on reduced gene expression of eotaxins and Muc5AC in the lungs relative to WT PA01 (Fig. S5H).
STAT6 transcription factor is a general regulator of type-2 responses and is responsible for IL-13-mediated mucin expression (Kuperman et al., 2002). To test whether IL-13 and general impairment of type-2 responses are critical for PA01 infection, we infected WT and STAT6-deficient (STAT6−/− animals with WT PA01. WT and STAT6-deficient animals maintained their body temperature similarly (Fig. S5I) and had similar bacterial loads in the BAL and lung (Fig. S5K, L). Lung gene expression of TSLP, eotaxins and Muc5AC was not changed suggesting that IL-13 mediated mucin expression and general impairment of type-2 responses are not critical for PA01 infection. Taken together, these data demonstrate that Areg but not STAT6 and IL-13 is crucial for PA01 infection.
LasB-deficient P. aeruginosa have reduced adjuvant activity to stimulate eosinophil recruitment and mucin production
To determine whether PA01 LasB is required to promote allergic responses, we compared WT and ΔLasB PA01 as adjuvants in airway allergic inflammation (Fig. 6A). Antibody titers of total and OVA-specific IgE were comparably increased in the serum of WT and ΔLasB PA01-immunized animals (Fig. S6A, left panels). OVA-specific IgG1 titers were reduced in the serum of ΔLasB PA01-immunized animals (Fig. S6A, middle panel). OVA-specific IgG2b titters were not significantly increased in immunized animals (Fig. S6A, right panel). Eosinophil but not neutrophil infiltration was impaired in the BAL of ΔLasB PA01-immunized animals (Fig. 7A, B). H&E and PAS staining of lungs demonstrated decreased cell infiltration and mucin production in ΔLasB PA01-immunized animals (Fig. 7C, D). Lung gene expression of type 2 cytokines, eotaxins and Muc5AC were reduced in animals immunized with ΔLasB PA01 (Fig. 7E). IFN-γ amounts were reduced in the BAL of WT but not ΔLasB PA01-immunized mice (Fig. 7F). These data suggest that LasB is required for P. aeruginosa adjuvant activity to induce eosinophil infiltration and mucin production.
Figure 7. LasB-deficient P. aeruginosa have reduced adjuvant activity to stimulate eosinophil recruitment and mucin production.
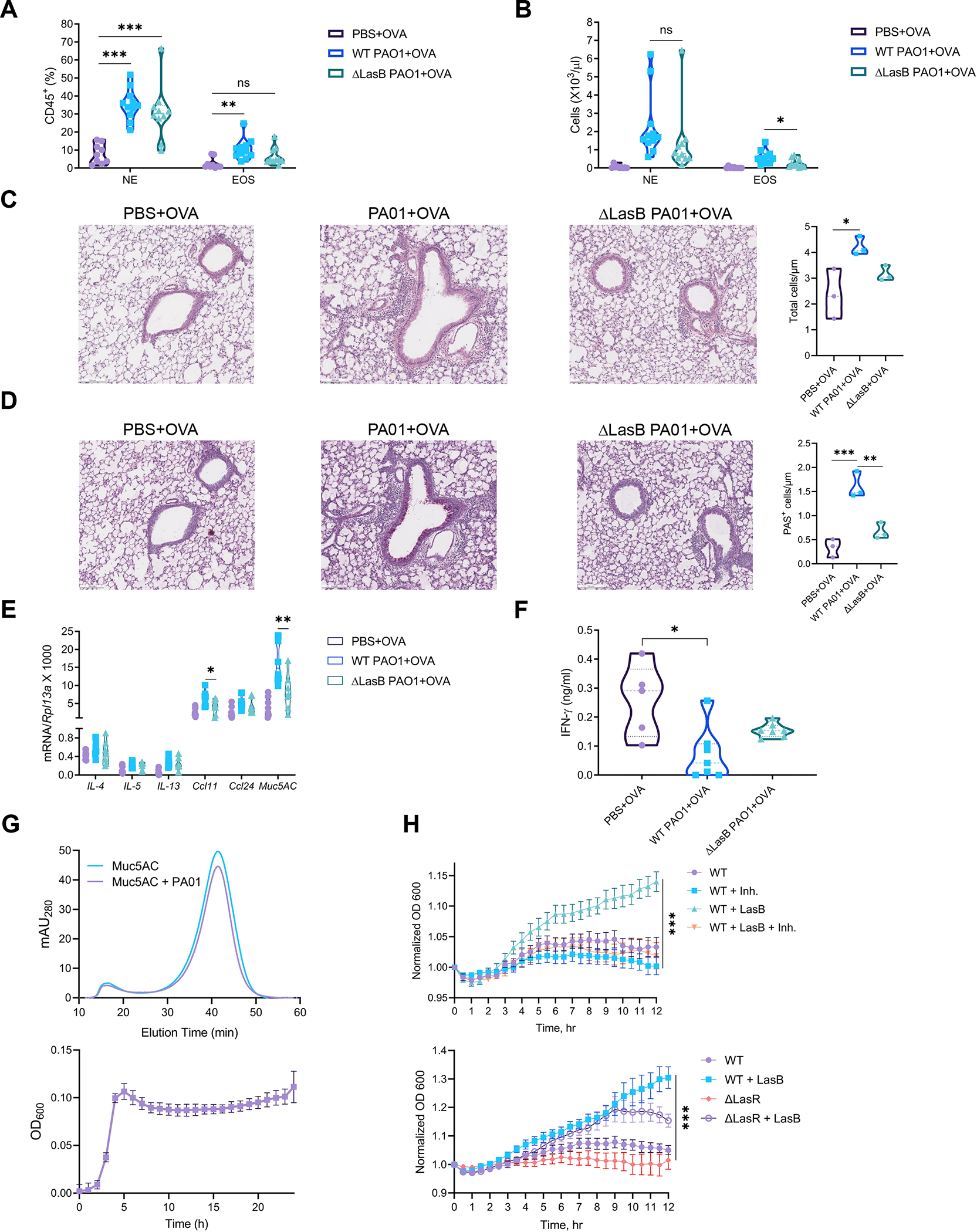
(A and B) Cell frequency (A) and numbers (B) of eosinophils (CD45+MHCII–CD11b+CD64-Siglec-F+) and neutrophils (CD45+MHCII–CD11b+CD64-Ly6G+) out of CD45+ cells in the BAL of mice immunized with OVA +/− WT or ΔLasB PA01 (combined of 3 experiments yielding 9–11 mice per group).
(C and D) H&E (C) and PAS (D) staining in the lungs of mice immunized as in (A and B) (representative of 3 experiments, 4 mice per group, scale bar = 500 μm).
(E) Relative mRNA expression of type 2 cytokines (IL-4, 5, 13), eotaxins (Ccl11, 24) and Muc5AC in the lungs of mice immunized as in (A and B) (combined of 2 experiments yielding 7–9 mice per group).
(F) ELISA of IFN-γ amounts in the BAL of mice immunized with OVA +/− WT or ΔLasB PA01 (combined of 2 experiments yielding 5–7 mice per group).
(G) (Top) Representative fast protein liquid chromatography (FPLC) profile (n=3) of Muc5AC before and after incubation with PA01 and (bottom) growth curves of PA01 on Muc5AC (growth curve data are mean ± SEM, n=3; FPLC data are the mean of n=3 traces).
(H) (Top) PA01 growth +/− LasB (3 μg/ml) and/or phosphoramidon (50 mM) and (bottom) WT and ΔLasR PA01 growth +/− LasB (3 μg/ml) on human sputum samples for 12 hr at 37 °C (data are mean ± SEM, n=4).
mRNA expression was measured relative to Rpl13a. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figures S6 and S7.
The fact that P. aeruginosa secretes virulence factor that promotes type-2 immune response suggests that the pathogen can promote immune deviation. We hypothesized that P. aeruginosa might induce type-2 inflammation and mucus production to use mucin glycans as a food source. To test this, we measured P. aeruginosa growth on mucin and its degradation. WT PA01 grew on Muc5AC and efficiently degraded it relative to untreaded control (Fig. 7G). Next, we measured bacterial growth on human sputum samples containing mucus. Addition of exogenous LasB improved growth of WT and ΔLasR PA01 (deficient in any secreted protease) on human sputum samples and was dependent on enzymatic activity (Fig. 7G, H) suggesting that LasB promotes mucin utilization. To further test this, we used publicly available human microbiome datasets to investigate whether conditions mimicking P. aeruginosa chronic infection or type-2 immunity result in utilization of metabolic pathways required for mucin acquisition in human microbiota. First, we analyzed a subset of lung whole genome sequencing data of cystic fibrosis patients (Dmitrijeva et al., 2021). Taxonomic classification of bacterial species demonstrated that all patients except one (n=4) have enriched colonization of P. aeruginosa (Fig. S6B). The microbiome of cystic fibrosis patient 6 (CF6), not enriched with P. aeruginosa, served as a control to analyze abundant metabolic pathways in the lung microbiomes of other patients. Using LefSe method for analysis of human microbiome metabolic pathways (Segata et al., 2011), we found that the microbiomes of patients enriched with P. aeruginosa utilized various amino acid pathways (Fig. S6C) that were previously shown to be important for mucin utilization (Flynn et al., 2017) such as branched chain amino acids, serine and glycine and phenylalanine biosynthesis (Fig. S6D). Next, we analyzed the whole-genome sequencing of fecal microbiomes of worm-infected patients treated (n=3) or untreated (n=3) with anthelminthic (Rosa et al., 2018). Taxonomic classification showed that untreated patients are colonized by Prevotella, Streptococcus and Pseudomonas genera (Fig. S6E). Prevotella and Streptococcus possess an array of polysaccharide-degrading pathways and, thus, may help in mucin utilization by Pseudomonas (Makki et al., 2018; Shelburne et al., 2008). Metabolic pathway analysis of the microbiomes of untreated vs treated patients highlighted abundance of amino acid biosynthesis pathways in accordance with a previous dataset (Fig. S6F, J). In addition, polysaccharide degradation pathways were also enriched in untreated patients’ microbiomes (Fig. S6F, H). Specifically, superpathway of degradation of mucin glycans, such as N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminic acid, was increased in the microbiomes of untreated patients (Fig. S6H). Thus, analysis of human lung and gut microbiomes suggests that mucin utilization pathways are enriched under conditions mimicking type-2 immunity. Finally, we analyzed duodenal 16S rRNA sequencing samples of untreated control celiac disease patients (n=6) and gluten-challenged celiac patients before (n=6) and 24 hr after (n=6) hookworm infection (Giacomin et al., 2016) which is a classic inducer of type 2 immunity (Quinnell et al., 2004). We tested whether hookworm infection was associated with P. aeruginosa colonization. β-diversity analysis demonstrated separate clustering of different groups of patients (Fig. S7A). We found increased abundance of Prevotella, Streptococcus and Pseudomonas genera in duodenum of gluten-challenged celiac patients infected with worms (Fig. S7B, C) in agreement with fecal microbiome analysis. Lastly, using correlation analysis we showed that Prevotella and Pseudomonas genera are well associated with hookworm infection (Fig. S7D–F). Taken together, these data suggest that microbial mucin utilization pathways are abundant in human microbiomes under conditions promoting colonization by P. aeruginosa.
Discussion
Chronic infections require necessary nutrients to efficiently colonize a host niche. The nutrients can be acquired from exogenous sources such as diet or host tissue, manipulating metabolic pathways and resulting in long-term colonization (Ehrt et al., 2018; Geoghegan et al., 2018; Tan et al., 2020). These chronic infections are ineffectively cleared by type-1 immunity (Monack et al., 2004). Whether utilization of host nutrients or metabolites by chronic infections can result in manipulated bias towards type-2 immunity was largely unstudied. Here, using P. aeruginosa as a model system, we showed that P. aeruginosa and its toxin, LasB, induce type-2 immunity. P. aeruginosa LasB utilized host amphiregulin to promote infection and increase mucin production that can be used as an energy source for P. aeruginosa (Aristoteli and Willcox, 2003; Flynn et al., 2017; Hoffman et al., 2020). LasB-deficient bacteria induced diminished type-2 immunity and reduced mucin production. Thus, this study demonstrates that pathogenic bacteria can stimulate production of host polysaccharides that can be used as a food source by inducing type-2 immunity.
Several chronic infections such as Staphylococcus aureus and Mycobacterium tuberculosis use specific invasion site-dependent nutrient exploitation strategies to regulate virulence. Staphylococcus aureus causing atopic dermatitis (Cogen et al., 2008; Geoghegan et al., 2018) might take advantage of skin sebaceous glands producing complex lipid mixtures. Multiple reports suggest that host fatty acids and lipoproteins regulate Staphylococcus aureus pathogenesis and persistence (Delekta et al., 2018; Grayczyk et al., 2017; Kenanian et al., 2019; Lopez et al., 2017; Parsons et al., 2014). On the other hand, Mycobacterium tuberculosis utilizes host carbohydrate and amino acid pathways to acquire necessary resources to persist in lung macrophages (Borah et al., 2019; Ehrt et al., 2018; Howard and Khader, 2020; Marrero et al., 2013; Shi et al., 2015). These infections have been shown to exhibit a bias towards type-2 immunity (Abebe, 2019; Cronan et al., 2021; Davis et al., 2015; Lan et al., 2018). Whether these chronic infections can use abovementioned host metabolic pathways and metabolites to induce type-2 immunity would be an interesting question for future studies.
One of the critical pathways targeted by pathogens is polysaccharide consumption and utilization. Polysaccharides can be provided by diet or endogenous sources, in the form of mucus. Commensal microorganisms colonize mucous layer and use it as a food source under dietary limitations such as lack of fiber in a diet (Desai et al., 2016; Makki et al., 2018; Sonnenburg and Sonnenburg, 2014). Thus, specific alteration of dietary nutrients can change primary nutrient source and result in pathogenicity profile within microbiome beneficial for opportunistic pathogens. Specifically, microbiota-liberated mucin sugars generate advantageous nutrient conditions to stimulate pathogenic expansion (Ng et al., 2013; Pacheco et al., 2012). Enteric pathogens such as Salmonella and Helicobacter can specifically remodel mucus and exploit its nutritious value (Arabyan et al., 2016; Ge et al., 2018; Newton et al., 1998). We showed that Pseudomonas and its toxin enhance Muc5AC expression, thus, enabling feed forward loop to use host mucin as a nutrient. Consistent with our findings, host microbiome has been shown to assist Pseudomonas to utilize mucin (Flynn et al., 2016; Flynn et al., 2017; Hoffman et al., 2020). Importantly, it has been demonstrated that lung mucus accumulation precedes infectious pathologies in cystic fibrosis patients (Esther et al., 2019). Moreover, mucous cell metaplasia is a pre-requisite of type-2 immune responses (Sharpe et al., 2018; von Moltke et al., 2016). Thus, the availability and manipulation of host nutrient sources by infections can determine the outcome of pathogenicity and immune response.
Type 2 immune responses can afford protection against noxious environmental toxins and allergens (Marichal et al., 2013; Palm et al., 2012; Palm et al., 2013; Profet, 1991). While defense against acute bacterial infections relies on type 1-immunity, recent studies have demonstrated the role of type-2 immunity in host defense against chronic bacterial infections (Starkl et al., 2020; Wu et al., 2020). We demonstrated that Pseudomonas toxin, LasB, and bacteria can elicit type-2 immunity or allergic response. Specific mechanisms of bacterial resistance provided by type-2 immunity await future studies.
To initiate type-2 response, LasB mimics function of endogenous metalloprotease, ADAM17 or TACE, by cleaving host Areg at a unique site and activating EGFR (Blobel, 2005; Murphy, 2008). Activation of EGFR has been demonstrated to promote type-2 responses and allergic inflammation (Hur et al., 2007; Jia et al., 2021; Minutti et al., 2017). It is thus plausible that various toxins and allergens may operate through endogenous ligands or receptors or pathways to initiate type-2 responses. Possibly, these enzymes can be classified by the ligand or pathway they manipulate in the host. We showed that LasB generated different cleavage product of Areg, AL, that induced prolonged signaling and gene expression. The potential role of AL in induction of type-2 immunity and other physiological responses will be important to pursue in future studies. AL can be of special interest since it may induce differential transcriptional and tissue response relative to conventional Areg cleaved by ADAM17 or TACE. Conventional Areg is known for its tissue protective function (Arpaia et al., 2015; Monticelli et al., 2015; Zaiss et al., 2015). AL can have other, possibly, opposing functions not classically attributed to Areg. It is conceivable to speculate that endogenous proteases, other than ADAM17 or TACE, may generate AL endogenously. We found that AL induced expression of Muc5AC, a predominant component of airway epithelial mucus (Broaddus et al.) that can serve as an energy source for P. aeruginosa (Aristoteli and Willcox, 2003; Flynn et al., 2017). Thus, our findings agree with previous reports showing dependence of mucin production on Areg (Enomoto et al., 2009; Manzo et al., 2012; Monticelli et al., 2015). STAT6 and IL-13-dependent Muc5AC expression was not affected upon PA01 infection suggesting that IL-13 and Areg can operate independently to induce mucin production.
Bacterial toxins can compensate for each other’s function during infections (Fischer et al., 2020). We found that despite inability of LasB-deficient Pseudomonas to induce Muc5AC, this pathogen can still use host Areg to promote infection and trigger reduced type-2 immunity. This suggested that other toxins can compensate for LasB function (Oldak and Trafny, 2005). This group of toxins is usually regulated by bacterial quorum sensing that is also responsible for formation of biofilms (Mukherjee and Bossier, 2019; Rutherford and Bassler, 2012). Specific quorum-sensing transcription factors regulate expression of these toxins (Papenfort and Bassler, 2016). It is thus likely that certain bacterial life cycle stages (biofilm) and signaling pathways (quorum sensing) or virulence factors can contribute to induction of type-2 immunity.
In summary, this study identifies bacterial manipulation of host pathway to promote nutrient availability as a driver of immune deviation and energy source that subsequently leads to type-2 immunity. Together with recent studies (Cronan et al., 2021; Starkl et al., 2020; Wu et al., 2020) this suggests that nutrient sources, types of virulence and chronicity of infection can bias host response towards type-2 immunity. This perspective might open new avenues for type-2 immunity in developing of therapeutics against chronic bacterial infections.
Limitations, caveats, and open questions
The experiments were performed on mice in a single facility and on one genetic background (C57BL/6J). The impacts of the microbiota, genetic background, and facility-specific factors are unknown. Unnatural settings in animal facilities may affect the results and interpretation of immune response studies. It is likely that chronic infections can manipulate several metabolic pathways to induce type 2 immunity. Potential resistance mechanisms for chronic infections provided by type 2 immunity need to be explored. Open questions include the following: How different classes and life stages of bacteria elicit type-2 immunity? What is the role of distinct Areg form in other physiological processes? Can the induced immune response be classified based on the activity of the insult and not class of a pathogen? Finally, the therapeutic scope of type 2 immunity against chronic infections needs to be determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact Ruslan Medzhitov (ruslan.medzhitov@yale.edu).
Materials Availability
This study did not generate new unique reagents. All reagents generated or used in this study are available on request from the Lead Contact with a completed Materials Transfer Agreement. Information on reagents used in this study is available in the Key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal and mouse monoclonal (E10) anti-phospho-p44/42 MAPK (Thr202/Tyr204) | Cell Signaling Technology | Cat# 9101, RRID: AB_331646; Cat# 9106, RRID: AB_331768 |
|
| ||
| Rabbit polyclonal anti-p44/42 MAPK | Cell Signaling Technology | Cat# 9102, RRID: AB_330744 |
|
| ||
| Rabbit monoclonal anti-phospho-EGFR (Tyr1068) (D7A5) | Cell Signaling Technology | Cat# 3777, RRID: AB_2096270 |
|
| ||
| Rabbit monoclonal anti-EGFR (D38B1) | Cell Signaling Technology | Cat# 4267, RRID: AB_2246311 |
|
| ||
| Rabbit polyclonal anti-phospho-Akt (Ser473) | Cell Signaling Technology | Cat# 9271, RRID: AB_329825 |
|
| ||
| Rabbit polyclonal anti-phospho-S6 ribosomal protein (Ser235/236) | Cell Signaling Technology | Cat# 2211, RRID: AB_331679 |
|
| ||
| Mouse monoclonal anti-EGFR (IP specific) | Cell Signaling Technology | Cat# 2256, RRID: AB_561017 |
|
| ||
| Mouse monoclonal IgG1 isotype control (G3A1) | Cell Signaling Technology | Cat# 5415, RRID: AB_10829607 |
|
| ||
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat# A2228, RRID: AB_476697 |
|
| ||
| Mouse monoclonal anti-β-actin (8H10D10) | Cell Signaling Technology | Cat# 3700, RRID: AB_2242334 |
|
| ||
| Goat polyclonal anti-human Areg | R&D systems | Cat# AF262, RRID: AB_2243124 |
|
| ||
| Mouse monoclonal anti-human Areg (Clone 31221) | R&D systems | Cat# MAB262, RRID: AB_2060676 |
|
| ||
| Goat polyclonal anti-mouse Areg | R&D systems | Cat# AF989, RRID: AB_2060663 |
|
| ||
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 111-035-144, RRID: AB_2307391 |
|
| ||
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115-035-003, RRID: AB_10015289 |
|
| ||
| Peroxidase AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705-035-003, RRID: AB_2340390 |
|
| ||
| Goat polyclonal anti-human Areg biotin conjugated | R&D systems | Cat# BAF262, RRID: AB_2060677 |
|
| ||
| Goat polyclonal anti-mouse Areg biotinylated | R&D systems | Cat# BAF989, RRID: AB_2060662 |
|
| ||
| Mouse monoclonal anti-human Areg (AREG559), PE | Thermo Fisher Scientific | Cat# 12-5370-42, RRID: AB_2716926 |
|
| ||
| Rat monoclonal anti-mouse CD16/CD32 (93) | Thermo Fisher Scientific | Cat# 14-0161-86, RRID: AB_467135 |
|
| ||
| Rat monoclonal anti-mouse MHC Class II (I-A/I-E) (M5/114.15.2), FITC | Thermo Fisher Scientific | Cat# 11-5321-85, RRID: AB_465233 |
|
| ||
| Rat monoclonal anti-mouse Ly-6C (HK1.4), APC | Thermo Fisher Scientific | Cat# 17-5932-82, RRID: AB_1724153 |
|
| ||
| Armenian hamster anti-mouse FceR1 alpha (MAR-1), eFluor 450 | Thermo Fisher Scientific | Cat# 48-5898-82, RRID: AB_2574086 |
|
| ||
| Rat monoclonal anti-mouse F4/80 (BM8), APC-eFluor 780 | Thermo Fisher Scientific | Cat# 47-4801-82, RRID: AB_2735036 |
|
| ||
| Rat monoclonal anti-mouse IL-4 (11B11) | Thermo Fisher Scientific | Cat# 14-7041-81, RRID: AB_468410 |
|
| ||
| Rat monoclonal anti-mouse IL-13 (eBio13A) | Thermo Fisher Scientific | Cat# 14-7133-81, RRID: AB_763553 |
|
| ||
| Rat monoclonal anti-mouse IL-4 (BVD6-24G2), Biotin | Thermo Fisher Scientific | Cat# 13-7042-85, RRID: AB_466903 |
|
| ||
| Rat monoclonal anti-mouse IL-5 (TRFK4), Biotin | Thermo Fisher Scientific | Cat# 13-7051-85, RRID: AB_466907 |
|
| ||
| Rat monoclonal anti-mouse IL-13 (eBio1316H), Biotin | Thermo Fisher Scientific | Cat# 13-7135-85, RRID: AB_763556 |
|
| ||
| Rat monoclonal anti-mouse IFN gamma (R4-6A2), Biotin | Thermo Fisher Scientific | Cat# 13-7312-85, RRID: AB_466939 |
|
| ||
| Rat monoclonal IFN gamma (XMG1.2) | Thermo Fisher Scientific | Cat# 14-7311-85, RRID: AB_468468 |
|
| ||
| Mouse monoclonal anti-human EGFR, APC | BioLegend | Cat# 352906, RRID: AB_11150410 |
|
| ||
| Mouse monoclonal anti-mouse CD64 (FcgammaRI), Brilliant Violet 421 | BioLegend | Cat# 139309, RRID: AB 2562694 |
|
| ||
| Mouse monoclonal anti-cadherin E, FITC, Clone 36 | BD Biosciences | Cat# 612130, RRID: AB_399501 |
|
| ||
| Rat monoclonal anti-mouse Siglec F, PE, Clone E50-2440 | BD Biosciences | Cat# 552126, RRID: AB_394341 |
|
| ||
| Syrian hamster monoclonal anti-mouse CD3e, V500, Clone 500A2 | BD Biosciences | Cat# 560771, RRID: AB_1937314 |
|
| ||
| Rat monoclonal anti-CD11b (Mac-1), BUV 737 | BD Biosciences | Cat# 564443, RRID: AB_2738811 |
|
| ||
| Rat monoclonal anti-mouse CD45 Clone 30-F11 (RUO), BUV395 | BD Biosciences | Cat# 564279, RRID: AB_2651134 |
|
| ||
| Rat monoclonal anti-mouse Ly-6G, Clone 1A8, PerCP-Cy5.5 | BD Biosciences | Cat# 560602, RRID: AB_1727563 |
|
| ||
| Rat monoclonal anti-IL-5 | BD Biosciences | Cat# 554393, RRID: AB_398547 |
|
| ||
| Mouse IgG1, kappa Isotype Control, Clone MOPC-31C | BD Biosciences | Cat# 557273, RRID: AB_396613 |
|
| ||
| Rat monoclonal anti-mouse IgG1, Biotin, Clone A85-1 | BD Biosciences | Cat# 553441, RRID: AB_394861 |
|
| ||
| Mouse monoclonal IgE, k antibody | BD Biosciences | Cat# 557079, RRID: AB_479637 |
|
| ||
| Rat monoclonal anti-mouse IgE, Clone R35-72 | BD Biosciences | Cat# 553413, RRID: AB_394846 |
|
| ||
| Rat monoclonal anti-mouse IgE, Biotin, Clone R35-118 | BD Biosciences | Cat# 553419, RRID: AB_394850 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| NEB® 10-beta Competent E. coli (High Efficiency) | New England Biolabs | Cat# C3019I and Cat# C3019H |
|
| ||
| T7 Express lysY Competent E. coli (High Efficiency) | New England Biolabs | Cat# C3010I |
|
| ||
| PA01 | This study | N/A |
|
| ||
| MPA01 | Manoil Lab, Held et al, 2012 | https://www.gs.washington.edu/labs/manoil/libraryindex.htm |
|
| ||
| ΔLasB PA01 | Manoil Lab, Held et al, 2012 | https://www.gs.washington.edu/labs/manoil/libraryindex.htm |
|
| ||
| ΔLasR PA01 | Manoil Lab, Held et al, 2012 | https://www.gs.washington.edu/labs/manoil/libraryindex.htm |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| 123count eBeads Counting Beads | Thermo Fisher Scientific | Cat# 01-1234-42 |
|
| ||
| 16% paraformaldehyde aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
|
| ||
| IPTG | Sigma-Aldrich | Cat# I6758 |
| Cayman Chemical | Cat# 15300 | |
|
| ||
| Phosphoramidon (sodium salt) | Cayman Chemical | Cat# 15113 |
|
| ||
| Batimastat | Cayman Chemical | Cat# 14742 |
|
| ||
| Benzamidine (hydrochloride) | Cayman Chemical | Cat# 20651 |
|
| ||
| PMSF Protease Inhibitor | Thermo Fisher Scientific | Cat# 36978 |
|
| ||
| Coomassie Brilliant Blue R-250 | Bio-Rad | Cat# 1610400 |
|
| ||
| Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail (100X) | Thermo Fisher Scientific | Cat# 78442 |
|
| ||
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32106 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Cat# 34580 | |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Cat# 34094 | |
|
| ||
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
|
| ||
| Dynabeads™ Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10001D |
|
| ||
| Ni-NTA Agarose | Qiagen | Cat# 30210 |
|
| ||
| Pierce™ High Capacity Endotoxin Removal Resin | Thermo Fisher Scientific | Cat# 88270 |
|
| ||
| RPMI-1640 Medium | Sigma-Aldrich | Cat# R8758 |
|
| ||
| FBS | Thermo Fisher Scientific | Cat# 16140071 |
| R&D systems | Cat# S11550H | |
|
| ||
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Fisher Scientific | Cat# 11140050 |
|
| ||
| L-Glutamine (200 mM) | Thermo Fisher Scientific | Cat# 25030081 |
|
| ||
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat# 11360070 |
|
| ||
| HEPES | Thermo Fisher Scientific | Cat# 15630080 |
| AmericanBio | Cat# AB06021-0010 | |
|
| ||
| Dulbecco’s Phosphate Buffered Saline | Sigma-Aldrich | Cat# D8537 |
| Thermo Fisher Scientific | Cat# 14190144 | |
|
| ||
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | Cat# 25200056 |
|
| ||
| Opti-MEM™ I Reduced Serum Medium | Thermo Fisher Scientific | Cat# 31985070 |
|
| ||
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat# 13778030; Cat# 13778075 |
|
| ||
| 5X siRNA Buffer | Horizon Discovery | Cat# B-002000-UB-100 |
|
| ||
| Zombie Red™ Fixable Viability Dye | BioLegend | Cat# 423109 |
|
| ||
| RNA-Bee | Tel-Test | Cat# CS-501B |
|
| ||
| PerfeCTa SYBR Green SuperMix | QuantaBio | Cat# 95054-02K |
|
| ||
| Recombinant human amphiregulin | GeneScript | Cat# Z03103 |
|
| ||
| Recombinant mouse amphiregulin | BioLegend | Cat# 554102 |
|
| ||
| Recombinant human EGF | Thermo Fisher Scientific | Cat# PHG0311 |
|
| ||
| Recombinant mouse IL-4 | R&D systems | Cat# 404-ML |
|
| ||
| Recombinant mouse IL-5 | R&D systems | Cat# 405-ML |
|
| ||
| Recombinant mouse IL-13 | R&D systems | Cat# 413-ML |
|
| ||
| Recombinant mouse IFN-γ | R&D systems | Cat# 485-MI |
|
| ||
| Recombinant human ADAM17/TACE | BioVision | Cat# 7607-20 |
|
| ||
| Papain | Sigma-Aldrich | Cat# 10108014001 |
|
| ||
| Subtilisin | Sigma-Aldrich | Cat# P5380 |
|
| ||
| Trypsin Gold, Mass Spectrometry Grade | Promega | Cat# V5280 |
|
| ||
| E. coli flagellin | This study | N/A |
|
| ||
| Pseudomonas LasB | Elastin Products Company | Cat# PE961 |
|
| ||
| Recombinant Pseudomonas LasB | This study | N/A |
|
| ||
| Grade III and V Ovalbumin | Sigma-Aldrich | Cat# A5378 Cat# A5503 |
|
| ||
| Streptavidin-HRP | BD Biosciences | Cat# 554066 |
|
| ||
| TMB Substrate Reagent Set | BD Biosciences | Cat# 555214 |
|
| ||
| NcoI-HF® | New England Biolabs | Cat# R3193S |
|
| ||
| HindIII-HF® | New England Biolabs | Cat# R3104S |
|
| ||
| Phusion® High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0530S |
|
| ||
| SMART® MMLV Reverse Transcriptase | TakaraBio | Cat# 639524 |
|
| ||
| Endo Grade® Ovalbumin | Biovendor | Cat# LET0027 |
|
| ||
| BSA (Bovine Serum Albumin) | Thermo Fisher Scientific | Cat# BP1600-1 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| SensoLyte ® Green Elastase Assay Kit *Fluorimetric* | AnaSpec | Cat# AS-72178 |
|
| ||
| Pierce™ LAL Chromogenic Endotoxin Quantitation Kit | Thermo Fisher Scientific | Cat# 88282 |
|
| ||
| Mem-PER™ Plus Membrane Protein Extraction Kit | Thermo Fisher Scientific | Cat# 89842 |
|
| ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
|
| ||
| Deposited Data | ||
|
| ||
| LC-MS/MS dataset | This study | Table S1 |
|
| ||
| Strain-resolved Dynamics of the Lung Microbiome in Patients with Cystic Fibrosis | https://www.ebi.ac.uk/ena/browser/view/PRJEB32062 | ENA: PRJEB32062 |
|
| ||
| The bacterial community of worm infected human stool via MetaGenomic Shotgun data | Rosa et al, 2018; https://www.ebi.ac.uk/ena/browser/view/PRJNA407815 | ENA: PRJNA407815 |
|
| ||
| Changes in duodenal tissue-associated microbiota following hookworm infection and consecutive gluten challenges in humans with coeliac disease | Giacomin et al, 2016; https://www.ebi.ac.uk/ena/browser/view/PRJNA316208 | ENA: PRJNA316208 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human: NCI-H292 | ATCC | Cat# CRL-1848 |
|
| ||
| Mouse: lung organ culture from C57BL/6J mice | This study | N/A |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: C57BL/6J In-house bred | The Jackson Laboratory | Cat# 000664 |
|
| ||
| Mouse: Areg−/− C57BL/6J | Dr. Dietmar Zaiss Luetteke et al, 1999 | N/A |
|
| ||
| Mouse: STAT6−/− C57BL/6J | The Jackson Laboratory | Cat# 005977 |
|
| ||
| Mouse: Crlf24get BALB/c | Dr. Richard Locksley (Mohrs et al., 2001) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| siGENOME Non-Targeting siRNA #2 | Horizon Discovery | Cat# D-001210-02-05 |
|
| ||
| siADAM17 Silencer Select id # s13718-20 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| siAreg Silencer Select id # 57878 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| siAP2M Silencer Select id # 3114 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| siEGFR Silencer Select id # s563 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| siMyD88 Silencer Select id # s9137 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| siPACSIN3 Silencer Select id # 26562 | Thermo Fisher Scientific | Cat# 4427038 |
|
| ||
| For qPCR primers see Table S2 | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| Pseudomonas aeruginosa PAO1 genomic DNA | Dr. Barbara Kazmierczak | N/A |
|
| ||
| Plasmid: pET-21d (+) DNA | Millipore Sigma | Cat# 69743 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Prism 8.3.0-8.4.3 | GraphPad Software, Inc. | https://www.graphpad.com |
|
| ||
| FlowJo X 10.0.7-11.1.1 | FlowJo | https://www.flowjo.com/ |
|
| ||
| R | https://cran.r-project.org | v3.6.3 |
|
| ||
| Scaffold4 proteome software | Proteome Software Inc. | http://www.proteomesoftware.com/products/scaffold/ |
|
| ||
| Skyline | MacCoss Lab; https://skyline.ms/project/home/begin.view | v20.1 |
|
| ||
| Galaxy server | Afgan et al, 2016; https://usegalaxy.eu/ | UseGalaxy Europe |
|
| ||
| SortMeRNA | Kopylova et al, 2012; https://bioinfo.lifl.fr/RNA/sortmerna/ | v2.1b.6 |
|
| ||
| MetaPhlAn2 | Tin Truong et al, 2015; https://huttenhower.sph.harvard.edu/metaphlan/ | v2.6.0 |
|
| ||
| HUMAnN2 | Franzosa et al, 2018; https://huttenhower.sph.harvard.edu/humann/ | 0.11.1 |
|
| ||
| LEfSe | Segata et al, 2011; https://huttenhower.sph.harvard.edu/lefse/ | http://huttenhower.sph.harvard.edu/galaxy |
|
| ||
| Mothur | Schloss et al, 2009 | https://mothur.org/ |
|
| ||
| MicrobiomeAnalyst | Chong et al, 2020 | https://www.microbiomeanalyst.ca/ |
|
| ||
| Other | ||
|
| ||
| 3M™ Vetbond™ Tissue Adhesive | 3M | Cat# 70200742529 |
|
| ||
| Mono Q 5/50 GL | Cytiva (GE Healthcare) | Cat# 17516601 |
|
| ||
| 4-15% Mini-PROTEAN® TGX™ Precast Protein Gels 10, 12, 15-well | Bio-Rad | Cat# 4561083; Cat# 4561085; Cat# 4561086 |
|
| ||
| Trans-Blot Turbo Transfer System | Bio-Rad | Cat# 1704150 |
|
| ||
| CFX96 and CFX384 Touch Real-Time PCR Detection System | Bio-Rad | Cat# 1855195; Cat# 1855485 |
|
| ||
| NuPAGE™ 10-12%, Bis-Tris, Mini Protein Gel, 10-well | Thermo Fisher Scientific | Cat# NP0301BOX; Cat# NP0341BOX |
|
| ||
| Nunc MaxiSorp flat-bottom 96-well plate | Thermo Fisher Scientific | Cat# 44-2404-21 |
|
| ||
| Immobilon-P PVDF Membrane | Millipore Sigma | Cat# IPVH00010 |
|
| ||
| Instant Read-out Precision Digital Caliper | Electron Microscopy Sciences | Cat# 62065-40 |
|
| ||
| HemaVet 950FS Auto Blood Analyzer | Drew Scientific | N/A |
Data and Code Availability
All the data supporting the findings of the article are available within the main text or supplementary materials. The published article includes datasets generated or used/analyzed during this study. Original data for the LC/MS/MS dataset are included in this manuscript in Table S1. Original shotgun metagenomics and 16S rRNA datasets used/analyzed in this study are available on ENA indicated in Key resources table.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
Mice were bred at the Yale Animal Resources Center at Yale University in specific pathogen-free conditions, and all experiments were done in accordance with approved guidelines, regulations, and protocols as determined by the Institutional Animal Care and Use Committee at Yale. C57BL/6 mice were purchased from Jackson Laboratories and bred in-house. Areg−/− C57BL/6 mice were kindly provided by Dr. Dietmar M. Zaiss (University of Edinburgh). Crlf24get (TSLPR-deficient) mice were kindly provided by Dr. Richard Locksley (UCSF). Stat6−/− C57BL/6 mice were purchased from Jackson Laboratories. WT animals were either in-house bred C57BL/6 or BALB/c mice and/or littermate Areg wild type/heterozygotes. Mice 7–10 weeks of age, males and females were used for experiments, females were primarily used for allergic models; all animals were age-and sex-matched, and then randomized into the different groups.
Innate airway inflammation model.
WT and Areg−/− C57BL/6 mice were anesthetized by isoflurane inhalation or ketamine (100 mg/kg) and were treated intranasally with PBS or recombinant LasB (1 μg) in 40 μl of PBS for 3 consecutive days. Lungs and BAL were collected 24 hr after the final intranasal challenge for flow cytometry, histological and mRNA expression analyses.
Airway allergy model.
C57BL/6 mice were subcutaneously immunized with 5 μg endograde OVA (Biovendor) alone or in combination with 10 μg of recombinant LasB on days 0 and 7. Alternatively, WT and Areg−/− C57BL/6 mice were anesthetized by isoflurane inhalation and immunized intratracheally with 5 μg endograde OVA alone or in combination with 1 μg of recombinant LasB, 2.5×104 CFUs of WT or ΔLasB PAO1 on days 0 and 7. To induce airway inflammation, ketamine or isoflurane anesthetized mice were administered intranasally or intratracheally 10 μg of endograde OVA (Biovendor) in 40 μl of PBS on days 14 and 21. On day 22, mice were euthanized and serum, lungs and BAL fluid were collected for flow cytometry, histology, mRNA expression analysis, antibody and cytokine ELISA.
Anaphylaxis measurements.
C57BL/6 mice were subcutaneously immunized with 5 μg endograde OVA (Biovendor) alone or in combination with 10 μg of recombinant LasB on days 0 and 7. To induce anaphylaxis, on day 13 mice were challenged intravenously with 100 μg of endograde OVA. Rectal temperatures were measured with a rectal thermometer (Physitemp) and followed for 5–6 hr after challenge. At rectal temperatures below 26 °C, mice were euthanized and considered to have had a lethal response.
Skin allergic inflammation model.
WT and Areg−/− C57BL/6 mice were immunized subcutaneously in left hind footpad with 50 μl PBS containing 10 μg endograde OVA (Biovendor) alone or in combination with 20–40 μg recombinant LasB +/−phosphoramidon (0.5 mM, Cayman Chemical). Footpad swelling was monitored for 5 days. Inguinal and popliteal lymph nodes were isolated 5 days after immunization.
Footpad swelling measurements.
Footpad swelling was measured before and after subcutaneous skin immunization as described above in left (injected) footpad for 5 days. Footpad swelling measurements were done in two locations around injection site using instant read-out precision digital caliper (Electron Microscopy Sciences) and the results were averaged. Right non-injected footpad swelling was measured as internal control. PA01 infections. Mice were infected with 2.5×106 CFUs of bacteria per mouse in 50 ul of PBS by intratracheal route. Briefly, mice were anesthetized using ketamine and xylazine (100 and 10 mg/kg, respectively). A vertical cut on the neck was made after ensuring proper anesthesia by toe pinch to exposure the trachea. Fifty microliter of PBS containing bacteria were instilled directly in the trachea using a Hamilton syringe. The cut was sealed suing 3M Vetbond glue (3M). Mice were euthanized at 12 hours post infection to harvest BAL and lung samples to enumerate number of bacteria and inflammation. Part of the lung was obtained in RNAlater (Thermo Fisher Scientific) to isolate mRNA.
Mouse lung organ culture.
WT and Areg−/− mice were sacrificed, cut open and the lungs were perfused with 10 ml of ice-cold sterile PBS (Sigma-Aldrich or Thermo Fisher Scientific). The lungs from each mouse were chopped to 6 pieces, transferred to 12-well plate in RPMI (Sigma-Aldrich) with 1% v/v FBS, 10 mM HEPES and sodium pyruvate (Thermo Fisher Scientific), nonessential amino acids (Thermo Fisher Scientific), additional 20 mM L-glutamine (Thermo Fisher Scientific) and 1% Pen/Strep (Thermo Fisher Scientific) and cultured for 3–4 days. All stimulations are described in the main text and figure legends. 16–20 hr prior stimulation medium was replaced to serum-free RPMI.
Cell culture and treatments.
Lung epithelial cell line H292 (ATCC) was cultured in RPMI (Thermo Fisher Scientific) with 10% v/v FBS, 10 mM HEPES and sodium pyruvate (Thermo Fisher Scientific), nonessential amino acids (Thermo Fisher Scientific) and additional 20 mM L-glutamine (Thermo Fisher Scientific) without antibiotics. For seeding, medium was aspirated and replaced with 1 ml ice cold PBS (Sigma-Aldrich or Thermo Fisher Scientific) twice. Cells were lifted using 0.25% Trypsin-EDTA (Thermo Fisher Scientific) solution that was neutralized with complete media. Cells were centrifuged (200 g, 5 min), resuspended in the complete media and counted. All stimulations, pre-treatments are described in the main text and figure legends. 16–20 hr prior stimulation medium was replaced to serum-free RPMI.
siRNA silencing.
Lung epithelial H292 cells (ATCC) were reverse transfected with Silencer Select siRNA (Thermo Fisher Scientific) (10 pmol per 1.5 ×105 cells per well or 20 pmol per 3 × 105 cell per well) in 12 or 24-well plates using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, given siRNA was resuspended in 5X siRNA buffer (Horizon Discovery) and Lipofectamine RNAiMAX was resuspended in Opti-MEM (Thermo Fisher Scientific). Then diluted siRNA was mixed with Lipofectamine RNAiMAX for 5 min at RT. After 5 min the mix was added into each well. After 20 min, cells were added on top of the mix. After 48 hours, medium was replaced to serum-free RPMI for subsequent experiments and incubated for additional 16–20 hrs. RNAi efficiency was assessed either by qPCR or immunoblotting at 72 hr post-transfection.
PA01 culture.
WT, ΔLasB and ΔLasR bacteria were cultured by plating the glycerol stock on LB plates. A single colony from the plate was picked and grown overnight in LB broth. Second day, the bacteria were subcultured for 1 hour to bring them to the linear growth phase. The number of bacteria was estimated by measuring the optical density at 600 nm. The numbers were confirmed using standard colony forming unit by plating the inoculum on agar plates.
METHOD DETAILS
Protein expression and purification.
The coding sequence of P. aeruginosa LasB was amplified by PCR using forward (CTGCTAGCAAGAAGGTTTCTACGCTTGACCTG) and reverse (GAAAGCTTCAACGCGCTCGGGCAGG) primers containing NcoI and HindIII restriction digestion sites, respectively, from P. aeruginosa PA01 genomic DNA and cloned into the expression vector pET21d (Novagen) to express C-terminally His6-tagged version of a protein. LasB was expressed using T7 Express LysY cells (New England Biolabs). The overnight cell culture (2–5 ml) was used to inoculate 0.2–0.8L of LB and the cell culture was incubated for 3–4 hr at 37 °C until OD at 600 nm reached 0.6–0.8 units. The protein expression was then induced by addition of 0.2 mM IPTG (Sigma-Aldrich or Cayman Chemical). The cells were harvested after 4 hr of incubation at 37 °C and either kept at −20 °C or disrupted by sonication. LasB was first purified by affinity chromatography on Ni-agarose beads (Qiagen) followed by anion exchange chromatography in 50–500 mM gradient of NaCl on Mono Q column (GE Healthcare). The eluted fractions were concentrated to 1–3 μg/μl concentration and endotoxin was removed using Pierce High-Capacity Endotoxin Removal Resin (Thermo Fisher Scientific). After endotoxin removal, LasB was aliquoted and stored at −80 °C until use. Endotoxin amountswere measured using Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher Scientific). Recombinantly expressed and purified P. aeruginosa LasB (Fig. S1B–D) was fully active and responsive to inhibition by phosphoramidon (Fig. S1A). Gene expression profile of commercially available and in-house purified LasB was almost identical in induction of epithelial cytokine genes (Fig. S1G). Inhibition of recombinant LasB specifically downregulated expression of tissue repair genes (TSLP and Areg) (Fig. S1H).
Enzyme activity measurements.
LasB catalytic activity was measured using SensoLyte Green Elastase Assay Kit (AnaSpec). Briefly, bronchoalveolar lavage (BAL), PA01 culture supernatants; purified LasB +/− phosphoramidon (50–100 μM final) were incubated with natural substrate elastin labeled with the 5-FAM fluorophore and the QXL™ 520 quencher for 30–60 min at RT. Proteolytic cleavage of labeled elastin yielded green fluorescence that was monitored at excitation/emission = 488 nm/520 nm. Increase in fluorescence intensity was directly proportional to LasB activity. Control samples containing only labeled elastin served as blanks.
In vitro cleavage assay.
Recombinant human (GeneScript) or mouse (BioLegend) amphiregulin (1–2 μg) was incubated with LasB (0.5–1 μg) or ADAM17/TACE (BioVision) (0.5 μg) +/− phosphoramidon (50 μM, Cayman Chemical) or batimastat (10 μM, Cayman Chemical), respectively, for 30 min at 37 °C. Reactions were stopped by addition of 4X Laemmli Sample Buffer (200 mM Tris-HCl pH 6.8, 4% SDS, 40% glycerol, 0.4% bromophenol blue, 20 % β-mercaptoethanol). Reaction products were resolved using SDS-PAGE using 4–15% TGX protein gels (Bio-Rad) or NUPAGE Novex 10–12 % gels (Thermo Fisher Scientific) and stained by Coomassie Brilliant Blue R-250 (Bio-Rad).
Mass-spectrometric analysis.
LasB-cleaved and non-cleaved amphiregulin reaction products were resolved by SDS-PAGE on 4–15% TGX protein gels (Bio-Rad). The separated bands were cut out of the gel and subjected to trypsin digestion using a ProGest robot (DigiLab). The samples were washed with 25mM ammonium bicarbonate followed by acetonitrile and reduced with 10mM dithiothreitol at 60°C followed by alkylation with 50mM iodoacetamide at RT. Digestion was performed with sequencing grade trypsin (Promega) at 37°C for 4h, the reaction was quenched with formic acid and the supernatant was analyzed directly without further processing. Half of each gel digest was analyzed by nano LC-MS/MS with a Waters NanoAcquity HPLC system interfaced to a Thermo Fisher Q Exactive mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75μm analytical column at 350nL/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 70,000 FWHM and 17,500 FWHM for MS and MS/MS respectively. The fifteen most abundant ions were selected for MS/MS. Data were searched using a local copy of Mascot (Matrix Science). Mascot DAT files were parsed into Scaffold (Proteome Software) for validation, filtering and to create a non-redundant list per sample. Data were filtered using at 1% protein and peptide FDR and requiring at least two unique peptides per protein. Chromatographic spectra were analyzed using open-source Skyline (MacCoss Lab) software. Mass-spectrometry dataset is provided in Table S1.
Immunoblotting.
Cells were plated and stimulated as indicated in respective figures in 12 and/or 24-well plates at 1.5 or 3×105 cells per well. After treatments and stimulations cells were washed twice with ice-cold PBS and lysed in cold buffer consisting of 20 mM Tris (pH 7.5), 150 mM NaCl, 1% SDS, 10 % glycerol, 0.1 % bromophenol blue, 2.5 % β-mercaptoethanol, 1 × Halt protease and phosphatase inhibitors mix (Thermo Fisher Scientific). Lysates were boiled for 10–15 minutes at 95 °C and stored at −20 °C until further use. Lysates were resolved by SDS-PAGE on 4–15% TGX protein gels (Bio-Rad) and transferred to PVDF Immobilon P membranes (Millipore Sigma) using Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in TBS-T (tris buffered saline with tween) containing 5% BSA and probed overnight at 4°C with primary antibodies in TBS-T containing 5% BSA. HRP-conjugated secondary antibodies were from Jackson ImmunoResearch. Immunoblots were visualized using the SuperSignal West ECL systems (Thermo Fisher Scientific) followed by film exposure.
Membrane protein fractionation.
Membrane protein extraction was performed using Mem-PERTM Plus Membrane Protein Extraction Kit (Thermo Fisher Scientific). Briefly, 5 × 106 cells were resuspended in the serum-free media by scraping the cells off the surface of the plate. Harvested cell suspension was centrifuged at 300 × g for 5 minutes. The cell pellet was washed and then resuspended in Cell Wash Solution. Cells were centrifuged at 300 × g for 5 minutes. The cell pellet was resuspended in 0.3–0.4 ml of Permeabilization Buffer and incubated 10 minutes at 4°C with constant mixing. Permeabilized cells were centrifuged for 15 minutes at 16,000 × g (cytosolic fraction in supernatant). The cell pellet from this step was resuspended in 0.2mL of Solubilization Buffer, incubated at 4°C for 30 minutes with constant mixing and centrifuged at 16,000 × g for 15 minutes at 4°C (solubilized membrane in supernatant). Protein concentration in cytosolic and membrane fractions was measured by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific).
Immunoprecipitation.
5×106 cells were rinsed with ice-cold PBS (Sigma or Thermo Fisher Scientific) and each plate (6 cm) was lysed with 0.4 ml of ice-cold cell lysis buffer (40 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, Halt Protease and Phosphatase Inhibitor (100X) (Thermo Fisher Scientific)) and incubated on ice for 5 min. Cells were scraped off the plate, transferred to Eppendorf tubes. Samples were sonicated on ice 3x for 5 sec each, spun down 10 min 14000g at 4C and supernatants were transferred to a new tube. Before immunoprecipitation protein A beads (Thermo Fisher Scientific - 8 μg of IgG per mg of beads) were washed twice with 500 ul of cell lysis buffer using magnetic separation rack. anti-EGFR (1:100, Cell Signaling Technologies) or isotype control antibodies (Cell Signaling Technologies) were added to cell lysate (100–250 μg of total protein). The samples were incubated with rotation O/N at 4 °C. Antibody complex was transferred to the lysate pre-washed protein A beads and incubated with rotation for 20 min at RT. Beads were pelleted using magnetic separation rack and pellet was washed 5X with 500 ul of cell lysis buffer. Pellet was resuspended in 25–30 ul of 4XSDS sample buffer. The samples were heated to 95–100 °C for 5 min. The beads were pelleted using magnetic separation rack and the supernatant was loaded on SDS-PAGE gel and subjected to immunoblotting.
RNA isolation and qPCR.
H292 cells or mouse lung organ cultures were plated and stimulated as indicated in respective figures and figure legends in 12 and/or 24-well plates at 1.5 or 3×105 cells per well. After treatments and stimulations cells were washed twice with ice-cold PBS (Sigma or Thermo Fisher Scientific) and lysed with RNA-Bee (Tel-Test) for RNA extraction. Alternatively, isolated mouse lungs from in vivo experiments were transferred to RNAlater (Thermo Fisher Scientific) and/or immediately to RNA-bee (Tel-Test) and homogenized. RNA extraction was performed according to manufacturer’s instructions. Briefly, 0.2 ml chloroform was added to 1 ml of RNA-Bee, samples were mixed for 30 seconds, kept on ice for 5 min and centrifuged 12,000g for 15 minutes at 4 °C. 0.5 ml of isopropanol was added to aqueous phase (RNA), samples were kept for 5 min at 4 °C and centrifuged as above. RNA pellet was washed twice with 75% ethanol. The final RNA pellet was air-dried for 10–15 min and resuspended in molecular biology grade water (Sigma). Total RNA was reverse transcribed with an oligo (dT) primer and a Moloney murine leukemia virus reverse transcriptase (MMLV RT, TakaraBio). cDNA was analyzed by quantitative PCR amplification using SYBR Green qPCR Master Mix (QuantaBio) on a Bio-Rad CFX96 or 384 Real-Time PCR Detection System. Primers were designed to amplify mRNA-specific sequences, and analysis of the melt-curve confirmed the amplification of single products. Unstimulated samples were used as controls. Relative expression was normalized to High mobility group nucleosome-binding domain-containing protein 4 gene (HMGN4) for H292 or ribosomal protein L13a (Rpl13a) for mouse lung organ cultures/lungs. Primer sequences used are provided in Table S2.
Human and mouse Areg ELISA.
Human lung H292 cells were plated and stimulated as indicated in respective figures in 24-well plates at 1.5 × 105 cells per well. 300–500 μl of cell supernatant was collected and frozen at −20 °C until further use. Mouse BAL samples were used for mouse Areg ELISA. For Sandwich ELISA Nunc Maxisorp 96-well plates (Thermo Fisher Scientific) were coated with capture human Areg antibody (R&D systems) at 5 μg/ml or mouse Areg antibody (R&D systems) at 0.5 μg/ml and incubated O/N at 4 °C. Plates were washed with PBS-T and blocked with 1 % BSA for 1hr at RT. Human Areg was detected with biotinylated anti-Areg antibody (R&D systems). Mouse Areg was detected with biotinylated anti-Areg antibody (R&D systems). Antibody binding was detected with streptavidin-HRP (BD Biosciences) and developed with TMB substrate set (BD Biosciences). Recombinant human (GeneScript) and mouse Areg (BioLegend) were used as standards.
Flow cytometry.
H292 cells were plated and stimulated as indicated in respective figures in 24-well plates at 1.5 × 105 cells per well. After treatments and stimulations cells were washed twice with ice-cold PBS (Sigma or Thermo Fisher Scientific), lifted with 0.25% Trypsin-EDTA, neutralized with complete media and spun down at 200 g for 5 min. For BAL samples were spun down and pelleted cells were manipulated as described below.
Spun down cell were washed with PBS and stained with Zombie Red Cell Viability Dye (BioLegend) for 10 min at RT. Cell were washed with FACS buffer (PBS supplemented with 2.5 % FBS and 0.5 mM EDTA) and stained with fluorochrome-conjugated antibodies +/− anti-CD16/CD32 (Thermo Fisher Scientific) diluted in FACS buffer with addition of 123count eBeads (Thermo Fisher Scientific) for 30 min at 4 °C. For intracellular staining, cells were permeabilized and fixed using FoxP3 fixation/permeabilization buffer (Themo Fisher Scientific)) following the manufacturer– recommended protocol and incubated with specific fluorochrome-conjugated antibodies for the specified proteins in permeabilization buffer for 30 minutes at 4 °C. Cell acquisition was performed on LSRII instrument (BD Biosciences), and data were analyzed with FlowJo software (Tree Star). Neutrophils were defined as Zombie Red– CD45+MHCII–CD11b+Ly6G + or Zombie Red–CD45+MHCII–CD11b+CD64-Ly6G+ and eosinophils as Zombie Red–CD45+MHCII–CD11b+Siglec-F+ or Zombie Red– CD45+MHCII–CD11b+CD64-Siglec-F+.
T-cell activation assays.
For in vitro restimulation of T cells, mice were immunized subcutaneously as described above. Inguinal and popliteal lymph nodes were isolated 5 days after immunization. Total lymph node cells were counted and transferred (1×106 cells per well) to round bottom 96-well plates and re-stimulated with 900 μg/ml endograde OVA (Biovendor) for 72 hr before analysis of secreted cytokines. Cells were spun down after 72 hr and supernatants were used for ELISA.
Antibody and cytokine ELISA.
Supernatants collected from re-stimulated lymph node cells or BAL samples were added to Nunc Maxisorp 96-well plates (Thermo Fisher Scientific), which had been coated with purified anti-mouse IL-4 antibody (Thermo Fisher Scientific), IL-5 (BD Biosciences), IL-13 (Thermo Fisher Scientific) and IFN-γ (Thermo Fisher Scientific) all at 2 μg/ml O/N at 4 °C. Plates were subsequently blocked with 1% BSA. Cytokines were detected with biotinylated anti-mouse IL-4 (Thermo Fisher Scientific), IL-5 (Thermo Fisher Scientific), IL-13 (Thermo Fisher Scientific) and IFN-γ (Thermo Fisher Scientific) all at 0.5 μg/ml. Recombinant IL-4, IL-5, IL-13 and IFN-γ (R&D systems) were used as a standard (starting concentration 10 ng/ml). Antibody binding was detected with streptavidin-HRP (BD Biosciences) and developed with TMB substrate set (BD Biosciences).
Serum samples.
For serum OVA-specific IgG1 antibody detection, Nunc Maxisorp 96-well plates (Thermo Fisher Scientific) were coated with 10 μg/ml OVA (Grade V, Sigma-Aldrich) O/N at 4°C, subsequently blocked with 1 % BSA and serial dilutions of sera were added (1:1000, 1:2500, 1:5000, 1:10000). Purified mouse IgG1 (BD Biosciences) was used as standard (starting concentration 500 ng/ml). Detection was achieved with biotinylated anti-IgG1 (BD Biosciences) at 0.1 μg/ml. For total IgE detection, the plates were coated with anti-mouse IgE (BD Biosciences) at 2 μg/ml and antibody was detected with biotinylated anti-IgE (BD biosciences) at 0.1 μg/ml. Purified mouse IgE (BD Biosciences) was used as standard (starting concentration 500 ng/ml). For OVA-specific IgE detection, the plates were coated with anti-mouse IgE (BD Biosciences) as above and the antibody was detected by biotin-OVA (Nanocs) at 10 μg/ml. Mouse anti-OVA IgE (Bio-Rad was used as a standard (starting concentration 250 ng/ml). Antibody binding was detected with streptavidin-HRP (BD Biosciences) and developed with TMB substrate set (BD Biosciences).
Lung histology.
Lungs were perfused through the right ventricle with 10 ml PBS, removed, and immersed in 4% PFA for 24–96 hr, followed by 70% ethanol until embedding in paraffin. Tissues were sliced, and 5-mm sections were stained with H&E or periodic acid-Schiff/Alcian blue for analysis of cellular inflammation and mucin production, respectively.
Quantification of immunochemistry images.
Quantification of cell numbers was performed using FIJI (Image J). Images were opened in FIJI and processed using “Color Deconvolution” plugin with selection of specific type of staining: H&E and PAS. Images were deconvoluted in their green, red, and blue components. The red component was further used for measurements of cell numbers. The area of the red image was measured using the “Threshold” tool. The threshold value was manually adjusted to modify the selected area. The same value of the threshold was applied to all processed images. Binary image generated after the application of the threshold value was processed using the “Watershed” function to separate individual cells. Next, the image was analyzed using “Analyze Particles” tool. The minimum particle size was set to 0.01 incĥ2 and the “Outline” was selected in the show section. The counted cells with their total area were generated as “Summary” and “Results” measurements.
PBMCs count.
Blood (50 μl) from WT and Areg−/− C57BL/6 mice was collected into Eppendorf tubes containing 50 mM EDTA to prevent coagulation. Collected blood samples were analyzed on HemaVet 950FS Auto Blood Analyzer (Drew Scientific).
Bacterial growth on Muc5AC and human sputum samples.
Human sputum was obtained from tracheal aspirates of patients who were on ventilatory support for non-pulmonary reasons. Four such patients were recruited with the exclusion criteria included current or recent use (<48 hours) of antibiotics and diagnosis of infection at lung or any other sites. The basic demographics of these patients is given in Supplementary Table S3. To loosen the sputum plugs present in the tracheal aspirate, it was passed through an 18 G needle for 10 times. The homogenous solution was centrifuged and the supernatant containing mucin was frozen in aliquots at −80 degree C until further experimentation. These processed sputum aliquots were then diluted 10 times and used in bacterial growth assays. 0.5×106 WT and ΔlasR PA01 strains were grown on diluted sputum samples +/− LasB (3 μg/ml) in 96-well plate format at 37 °C. OD600 was measured every 30 min for 12 hr. WT and ΔlasR PA01 strains grown on PBS were used as negative controls. OD600 measurements were normalized to t=0 hr.
For bacterial growth on Muc5AC, WT PA01 was grown overnight in LB at 37 degrees C, washed three times in PBS, and diluted to an OD600 of ~0.005 in a defined minimal mucin medium (MMM) in which purified Muc5AC was the sole carbon source (Flynn et al., 2016). Cultures (n=3 biological replicates, 4 technical replicates) were grown in 200μL aliquots using a 96-well plate format at 37C, and OD600 was measured every hour for 24h. Spent culture supernatant was retained for downstream mucin analysis.
Mucin analysis by size exclusion chromatography.
Replicate 200μL culture aliquots were pooled into 800μL volumes and centrifuged at 4000 ×g for 2 min to remove bacterial cells. Supernatants were spun for an additional 2 min at 14,000 × g to remove debris and filtered through a 0.45μm centrifugal filter. Eluent (500μL) was injected into an ÄKTA Pure FPLC instrument [Cytiva, Marlborough, MA] using a 10/200 Tricorn column packed with Sepharose CL-2B. Samples were subjected to an isocratic run at a flow rate of 0.4mL/min with 50mM phostphate buffer (pH 7.2) and 150mM NaCl. Absorbance was measured at 280nm. Sterile MMM was used as a control.
Taxonomic and functional profiling of metagenome sequencing data.
Raw metagenome sequencing data were downloaded from ENA browser using Project accession number to Galaxy server (Afgan et al., 2018). Quality control of sequencing data was performed using FastQC v 0.72 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Short reads (< 80 nt) and low-quality read bases were removed using Cutadapt v 1.16.5 (Martin, 2011). For downstream functional profiling, ribosomal RNA fragments were filtered using SortMeRNA v 2.1b.6 (Kopylova et al., 2012).
Quality-filtered metagenomes were taxonomically profiled using MetaPhlAn2 v 2.6.0 (Truong et al., 2015) with default parameters, which relies on approximately 1 million clade-specific marker genes derived from 17,000 microbial genomes (corresponding to >7,500 bacterial, viral, archaeal and eukaryotic species) to unambiguously classify metagenomic reads to taxonomies and yield relative abundances of taxa identified in the sample. Species and genus relative abundance data were considered in this study. Species and genera that failed to exceed 0.1% relative abundance in analyzed samples were excluded. Functional profiling of metagenomes was performed using HUMAnN2 v 0.11.1 (Franzosa et al., 2018). Briefly, the MetaPhlAn2 taxonomic profile generated from a metagenome is used to identify the set of organisms present in a sample. Metagenomic reads are then mapped using Bowtie2 (Langmead and Salzberg, 2012) to sample-specific pangenomes, including all gene families in any microorganism present. A translated search using DIAMOND (Buchfink et al., 2015) then maps unmapped reads against UniRef50 (Suzek et al., 2015). UniRef50 represents a clustering of all nonredundant protein sequences in UniProt (UniProt, 2015), such that each sequence in a cluster aligns with 50% identity and 80% coverage of the longest sequence in the cluster (the cluster seed). Hits are then counted per gene family and normalized for length and alignment quality. Gene family abundances from both the nucleotide and the translated searches are then combined into structured pathways from MetaCyc (Caspi et al., 2016) and sum-normalized to relative abundances.
HUMAnN2-generated relative abundances of metabolic pathways were next analyzed by LefSe (Segata et al., 2011). LefSe allows identification of differentially abundant metabolic pathways (subclasses) that are statistically and biologically significant between several experimental conditions (or classes). LefSe first identifies features that are statistically different among biological classes and then performs tests to assess whether these differences are consistent with respect to experimental condition. Specifically, it first uses the non-parametric factorial Kruskal-Wallis (KW) sum-rank test to detect features with significant differential abundance with respect to the biological class; biological significance is subsequently investigated using a set of pairwise tests among subclasses using the (unpaired) Wilcoxon rank-sum test. As a last step, LefSe uses linear discriminant analysis (LDA) to estimate the effect size of each differentially abundant metabolic pathway.
Analysis of 16S rRNA sequencing data.
Raw 16S rRNA sequencing data were downloaded from ENA browser using Project accession number to Galaxy server (Afgan et al., 2018). The 16S rRNA sequences were analyzed using the mothur software package v1.39.5.0 (Schloss et al., 2009). Briefly, paired-end reads were merged into contigs, screened for quality, aligned to the SILVA 16S rRNA sequence database (Quast et al., 2013), and screened for chimeras using UCHIME (Edgar et al., 2011). Sequences were classified using a naive Bayesian classifier trained against a 16S rRNA gene training set provided by the Ribosomal Database Project (Cole et al., 2014). Curated sequences were clustered into operational taxonomic units (OTUs) using a 97% similarity cutoff with the average neighbor clustering algorithm. The number of sequences in each sample was rarefied to the sample with the lowest number to minimize the effects of uneven sampling. Mothur-generated output data were analyzed using R software with the following packages: ape, dplyr, ggplot2, gplots, lme4, phnagorn, plotly, tidyr, vegan, phyloseq – and confirmed using Microbiome Analyst web-based platform (Dhariwal et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample or experiment sizes were determined empirically for statistical power. No statistical tests were used to predetermine the size of experiments. Prism 8.3.0–8.3.4 (GraphPad Software, Inc.) software was used for statistical analysis. Statistical differences for two groups comparison were determined by unpaired two-tailed Student’s t-test. More than two groups were compared using one-way analysis of variance (ANOVA) followed by Tukey test. A P value of ≤ 0.05 was considered statistically different. Data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Supplementary Material
Acknowledgments
We thank Dr. B. Kazmerciak for providing P. aeruginosa gDNA. We would like to thank all current and former members of Medzhitov lab for helpful discussions. ΔLasB and ΔLasR PA01 strains were obtained from PA01 Mutant Library (NIH P30 DK089507). Work in the R. M. lab was supported by the Howard Hughes Medical Institute, the Blavatnik Family Foundation, the Food Allergy Science Initiative, and a grant from NIH (AI144152–01). C.S.D.C. is supported by NIH HL126094, Department of Defense Grant and Veterans Affairs Merit Grant. L. S. is supported by Francis B Parker Fellowship and American Lung Association Catalyst Award. R.C.H. is supported by NIH HL136919. K. A. was supported by the Yale Brown-Coxe Post-Doctoral Fellowship and is Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. Images were Created with BioRender.com.
Footnotes
Declaration of interests
All authors declare no competing interests. B. V. is currently an employee of EMD Serono.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure sex balance in the selection of non-human subjects.
References
- Abebe F (2019). Synergy between Th1 and Th2 responses during Mycobacterium tuberculosis infection: A review of current understanding. Int Rev Immunol 38, 172–179. [DOI] [PubMed] [Google Scholar]
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gruning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic acids research 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JE, and Wynn TA (2011). Evolution of Th2 Immunity: A Rapid Repair Response to Tissue Destructive Pathogens. PLoS pathogens 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato F, Romagnani C, and Romagnani S (2015). The 3 major types of innate and adaptive cell-mediated effector immunity. The Journal of allergy and clinical immunology 135, 626–635. [DOI] [PubMed] [Google Scholar]
- Arabyan N, Park D, Foutouhi S, Weis AM, Huang BHC, Williams CC, Desai P, Shah J, Jeannotte R, Kong N, et al. (2016). Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling. Scientific reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristoteli LP, and Willcox MD (2003). Mucin degradation mechanisms by distinct Pseudomonas aeruginosa isolates in vitro. Infection and immunity 71, 5565–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan SP, Treuting PM, and Rudensky AY (2015). A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerzoug S, Bounab B, Rose S, Gosset D, Biet F, Cochard T, Xavier A, Rouxel N, Fauconnier L, Horsnell WGC, et al. (2019). Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity. Cell reports 27, 2649–2664 e2645. [DOI] [PubMed] [Google Scholar]
- Berasain C, and Avila MA (2014). Amphiregulin. Seminars in cell & developmental biology 28, 31–41. [DOI] [PubMed] [Google Scholar]
- Bever RA, and Iglewski BH (1988). Molecular characterization and nucleotide sequence of the Pseudomonas aeruginosa elastase structural gene. Journal of bacteriology 170, 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel CP (2005). ADAMs: key components in EGFR signalling and development. Nature reviews Molecular cell biology 6, 32–43. [DOI] [PubMed] [Google Scholar]
- Borah K, Beyss M, Theorell A, Wu HH, Basu P, Mendum TA, Noh K, Beste DJV, and McFadden J (2019). Intracellular Mycobacterium tuberculosis Exploits Multiple Host Nitrogen Sources during Growth in Human Macrophages. Cell reports 29, 3580-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broaddus VC, Mason RJ, Ernst JD, King JTE, Murray JF, and Nadel JA Murray and Nadel’s Textbook of Respiratory Medicine (Sixth Edition) (W.B. Saunders.). [Google Scholar]
- Bry L, Falk PG, Midtvedt T, and Gordon JI (1996). A model of host-microbial interactions in an open mammalian ecosystem. Science 273, 1380–1383. [DOI] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic acids research 44, D471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LE, and Locksley RM (2014). Allergic inflammation--innately homeostatic. Cold Spring Harbor perspectives in biology 7, a016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogen AL, Nizet V, and Gallo RL (2008). Skin microbiota: a source of disease or defence? The British journal of dermatology 158, 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic acids research 42, D633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, Mehra S, Oehlers SH, Gregory SG, Kaushal D, et al. (2021). A non-canonical type 2 immune response coordinates tuberculous granuloma formation and epithelialization. Cell 184, 1757–1774 e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MF, Peng RD, McCormack MC, and Matsui EC (2015). Staphylococcus aureus colonization is associated with wheeze and asthma among US children and young adults. The Journal of allergy and clinical immunology 135, 811–813 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delekta PC, Shook JC, Lydic TA, Mulks MH, and Hammer ND (2018). Staphylococcus aureus Utilizes Host-Derived Lipoprotein Particles as Sources of Fatty Acids. Journal of bacteriology 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353 e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhariwal A, Chong J, Habib S, King IL, Agellon LB, and Xia J (2017). MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic acids research 45, W180–W188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitrijeva M, Kahlert CR, Feigelman R, Kleiner RL, Nolte O, Albrich WC, Baty F, and von Mering C (2021). Strain-Resolved Dynamics of the Lung Microbiome in Patients with Cystic Fibrosis. mBio 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, and Knight R (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S, Schnappinger D, and Rhee KY (2018). Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nature reviews Microbiology 16, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto Y, Orihara K, Takamasu T, Matsuda A, Gon Y, Saito H, Ra C, and Okayama Y (2009). Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. J Allergy Clin Immun 124, 913–920. [DOI] [PubMed] [Google Scholar]
- Esther CR Jr., Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, Ramsey KA, Markovetz MR, Garbarine IC, Forest MG, et al. (2019). Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Science translational medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer NL, Naseer N, Shin S, and Brodsky IE (2020). Effector-triggered immunity and pathogen sensing in metazoans. Nature microbiology 5, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florsheim EB, Sullivan ZA, Khoury-Hanold W, and Medzhitov R (2021). Food allergy as a biological food quality control system. Cell. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Niccum D, Dunitz JM, and Hunter RC (2016). Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS pathogens 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Phan C, and Hunter RC (2017). Genome-Wide Survey of Pseudomonas aeruginosa PA14 Reveals a Role for the Glyoxylate Pathway and Extracellular Proteases in the Utilization of Mucin. Infection and immunity 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, and Molin S (2012). Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nature reviews Microbiology 10, 841–851. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nature methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambello MJ, and Iglewski BH (1991). Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. Journal of bacteriology 173, 3000–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge ZM, Sheh A, Feng Y, Muthupalani S, Ge LL, Wang CW, Kurnick S, Mannion A, Whary MT, and Fox JG (2018). Helicobacter pylori-infected C57BL/6 mice with different gastrointestinal microbiota have contrasting gastric pathology, microbial and host immune responses. Scientific reports 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan JA, Irvine AD, and Foster TJ (2018). Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends in microbiology 26, 484–497. [DOI] [PubMed] [Google Scholar]
- Giacomin P, Zakrzewski M, Jenkins TP, Su X, Al-Hallaf R, Croese J, de Vries S, Grant A, Mitreva M, Loukas A, et al. (2016). Changes in duodenal tissue-associated microbiota following hookworm infection and consecutive gluten challenges in humans with coeliac disease. Scientific reports 6, 36797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayczyk JP, Harvey CJ, Laczkovich I, and Alonzo F (2017). A Lipoylated Metabolic Protein Released by Staphylococcus aureus Suppresses Macrophage Activation. Cell host & microbe 22, 678-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D, Griese M, Kappler M, Zissel G, Reinhardt D, Rebhan C, Schendel DJ, and Krauss-Etschmann S (2006). Pulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. The Journal of allergy and clinical immunology 117, 204–211. [DOI] [PubMed] [Google Scholar]
- Hoffman CL, Lalsiamthara J, and Aballay A (2020). Host Mucin Is Exploited by Pseudomonas aeruginosa To Provide Monosaccharides Required for a Successful Infection. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard NC, and Khader SA (2020). Immunometabolism during Mycobacterium tuberculosis Infection. Trends in microbiology 28, 832–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur GY, Lee SY, Lee SH, Kim SJ, Lee KJ, Jung JY, Lee EJ, Kang EH, Jung KH, Lee SY, et al. (2007). Potential use of an anticancer drug gefinitib, an EGFR inhibitor, on allergic airway inflammation. Experimental and Molecular Medicine 39, 367–375. [DOI] [PubMed] [Google Scholar]
- Jaramillo AM, Azzegagh Z, Tuvim MJ, and Dickey BF (2018). Airway Mucin Secretion. Ann Am Thorac Soc 15, S164–S170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PO, Givskov M, Bjarnsholt T, and Moser C (2010). The immune system vs. Pseudomonas aeruginosa biofilms. FEMS immunology and medical microbiology 59, 292–305. [DOI] [PubMed] [Google Scholar]
- Jia Z, Bao K, Wei P, Yu X, Zhang Y, Wang X, Wang X, Yao L, Li L, Wu P, et al. (2021). EGFR activation-induced decreases in claudin1 promote MUC5AC expression and exacerbate asthma in mice. Mucosal immunology 14, 125–134. [DOI] [PubMed] [Google Scholar]
- Kenanian G, Morvan C, Weckel A, Pathania A, Anba-Mondoloni J, Halpern D, Gaillard K, Solgadi A, Dupont L, Henry C, et al. (2019). Permissive Fatty Acid Incorporation Promotes Staphylococcal Adaptation to FASII Antibiotics in Host Environments. Cell reports 29, 3974-+. [DOI] [PubMed] [Google Scholar]
- Khokha R, Murthy A, and Weiss A (2013). Metalloproteinases and their natural inhibitors in inflammation and immunity. Nature reviews Immunology 13, 649–665. [DOI] [PubMed] [Google Scholar]
- Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, et al. (2015). A comprehensive transcriptional portrait of human cancer cell lines. Nature biotechnology 33, 306–312. [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noe L, and Touzet H (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. [DOI] [PubMed] [Google Scholar]
- Krall JA, Beyer EM, and MacBeath G (2011). High- and low-affinity epidermal growth factor receptorligand interactions activate distinct signaling pathways. PloS one 6, e15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, and Erle DJ (2002). Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nature medicine 8, 885–889. [DOI] [PubMed] [Google Scholar]
- Lan F, Zhang N, Holtappels G, De Ruyck N, Krysko O, Van Crombruggen K, Braun H, Johnston SL, Papadopoulos NG, Zhang L, et al. (2018). Staphylococcus aureus Induces a Mucosal Type 2 Immune Response via Epithelial Cell-derived Cytokines. Am J Respir Crit Care Med 198, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MS, Tan IS, Yan DH, Kang J, McCreary M, Modrusan Z, Austin CD, Xu M, and Brown EJ (2017). Host-derived fatty acids activate type VII secretion in Staphylococcus aureus. Proceedings of the National Academy of Sciences of the United States of America 114, 11223–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makki K, Deehan EC, Walter J, and Backhed F (2018). The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell host & microbe 23, 705–715. [DOI] [PubMed] [Google Scholar]
- Manzo ND, Foster WM, and Stripp BR (2012). Amphiregulin-Dependent Mucous Cell Metaplasia in a Model of Nonallergic Lung Injury. American journal of respiratory cell and molecular biology 47, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marichal T, Starkl P, Reber LL, Kalesnikoff J, Oettgen HC, Tsai M, Metz M, and Galli SJ (2013). A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity 39, 963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero J, Trujillo C, Rhee KY, and Ehrt S (2013). Glucose Phosphorylation Is Required for Mycobacterium tuberculosis Persistence in Mice. PLoS pathogens 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Neumann M, and Desai MS (2018). Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nature reviews Microbiology 16, 457–470. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. [Google Scholar]
- Medzhitov R (2001). Toll-like receptors and innate immunity. Nature reviews Immunology 1, 135–145. [DOI] [PubMed] [Google Scholar]
- Minutti CM, Drube S, Blair N, Schwartz C, McCrae JC, McKenzie AN, Kamradt T, Mokry M, Coffer PJ, Sibilia M, et al. (2017). Epidermal Growth Factor Receptor Expression Licenses Type-2 Helper T Cells to Function in a T Cell Receptor-Independent Fashion. Immunity 47, 710–722 e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrs M, Shinkai K, Mohrs K, and Locksley RM (2001). Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity 15, 303–311. [DOI] [PubMed] [Google Scholar]
- Monack DM, Mueller A, and Falkow S (2004). Persistent bacterial infections: the interface of the pathogen and the host immune system. Nature reviews Microbiology 2, 747–765. [DOI] [PubMed] [Google Scholar]
- Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, and Artis D (2015). IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proceedings of the National Academy of Sciences of the United States of America 112, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara K, and Tsuzuki H (1978). Phosphoramidon as an inhibitor of elastase from Pseudomonas aeruginosa. The Japanese journal of experimental medicine 48, 81–84. [PubMed] [Google Scholar]
- Moser C, Kjaergaard S, Pressler T, Kharazmi A, Koch C, and Hoiby N (2000). The immune response to chronic Pseudomonas aeruginosa lung infection in cystic fibrosis patients is predominantly of the Th2 type. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 108, 329–335. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, and Bossier BL (2019). Bacterial quorum sensing in complex and dynamically changing environments. Nature Reviews Microbiology 17, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G (2008). The ADAMs: signalling scissors in the tumour microenvironment. Nature reviews Cancer 8, 929–941. [DOI] [PubMed] [Google Scholar]
- Newton JL, Jordan N, Oliver L, Strugala V, Pearson J, James OF, and Allen A (1998). Helicobacter pylori in vivo causes structural changes in the adherent gastric mucus layer but barrier thickness is not compromised. Gut 43, 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldak E, and Trafny EA (2005). Secretion of proteases by Pseudomonas aeruginosa biofilms exposed to ciprofloxacin. Antimicrobial agents and chemotherapy 49, 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, and Sperandio V (2012). Fucose sensing regulates bacterial intestinal colonization. Nature 492, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, and Medzhitov R (2012). Allergic host defences. Nature 484, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, and Medzhitov R (2013). Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity 39, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, and Bassler BL (2016). Quorum sensing signal-response systems in Gram-negative bacteria. Nature Reviews Microbiology 14, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, Subramanian C, and Rock CO (2014). Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proceedings of the National Academy of Sciences of the United States of America 111, 10532–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG, Turnbaugh PJ, et al. (2014). Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514, 638-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profet M (1991). The function of allergy: immunological defense against toxins. The Quarterly review of biology 66, 23–62. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glockner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research 41, D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinnell RJ, Pritchard DI, Raiko A, Brown AP, and Shaw MA (2004). Immune responses in human necatoriasis: association between interleukin-5 responses and resistance to reinfection. J Infect Dis 190, 430–438. [DOI] [PubMed] [Google Scholar]
- Ravimohan S, Kornfeld H, Weissman D, and Bisson GP (2018). Tuberculosis and lung damage: from epidemiology to pathophysiology. Eur Respir Rev 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, and Finn RD (2018). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic acids research 46, D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa BA, Supali T, Gankpala L, Djuardi Y, Sartono E, Zhou Y, Fischer K, Martin J, Tyagi R, Bolay FK, et al. (2018). Differential human gut microbiome assemblages during soil-transmitted helminth infections in Indonesia and Liberia. Microbiome 6, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford ST, and Bassler BL (2012). Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome biology 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe C, Thornton DJ, and Grencis RK (2018). A sticky end for gastrointestinal helminths; the role of the mucus barrier. Parasite Immunol 40, e12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, Davenport MT, Keith DB, and Musser JM (2008). The role of complex carbohydrate catabolism in the pathogenesis of invasive streptococci. Trends in microbiology 16, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LB, Salamon H, Eugenin EA, Pine R, Cooper A, and Gennaro ML (2015). Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Scientific reports 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, and Sonnenburg JL (2014). Starving our Microbial Self: The Deleterious Consequences of a Diet Deficient in Microbiota-Accessible Carbohydrates. Cell metabolism 20, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkl P, Watzenboeck ML, Popov LM, Zahalka S, Hladik A, Lakovits K, Radhouani M, Haschemi A, Marichal T, Reber LL, et al. (2020). IgE Effector Mechanisms, in Concert with Mast Cells, Contribute to Acquired Host Defense against Staphylococcusaureus. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, and UniProt C (2015). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Coureuil M, Charbit A, and Jamet A (2020). Multitasking Actors of Staphylococcus aureus Metabolism and Virulence. Trends in microbiology 28, 6–9. [DOI] [PubMed] [Google Scholar]
- Tropini C, Earle KA, Huang KC, and Sonnenburg JL (2017). The Gut Microbiome: Connecting Spatial Organization to Function. Cell host & microbe 21, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- UniProt C (2015). UniProt: a hub for protein information. Nucleic acids research 43, D204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, Gate RE, Haliburton GE, Ye CJ, Marson A, et al. (2016). A tissue checkpoint regulates type 2 immunity. Nature immunology 17, 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang HE, and Locksley RM (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, and Locksley RM (2014). I-L-C-2 it: type 2 immunity and group 2 innate lymphoid cells in homeostasis. Current opinion in immunology 31, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hayes BW, Phoenix C, Macias GS, Miao Y, Choi HW, Hughes FM Jr., Todd Purves J, Lee Reinhardt R, and Abraham SN (2020). A highly polarized TH2 bladder response to infection promotes epithelial repair at the expense of preventing new infections. Nature immunology 21, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, Gause WC, Osborne LC, and Artis D (2015). Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, and Mosmann TR (2006). Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314, 1746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting the findings of the article are available within the main text or supplementary materials. The published article includes datasets generated or used/analyzed during this study. Original data for the LC/MS/MS dataset are included in this manuscript in Table S1. Original shotgun metagenomics and 16S rRNA datasets used/analyzed in this study are available on ENA indicated in Key resources table.