

Abstract
Background
Spindle cell rhabdomyosarcoma (RMS) is a rare variant of RMS accounting for up to 10% of cases in infants. In older children and adults, spindle cell RMS is associated with MYOD1 mutations and a poor prognosis. In infants, it is associated with recurring fusions involving NCOA2 and VGLL2. Reports in the literature suggest a favorable prognosis for this subset, however, little is known about treatment and outcome data of infants with spindle cell RMS.
Methods
Characteristics, treatment, and outcome of an international cohort of 40 patients aged ≤12 months with spindle cell RMS treated from 1997–2018 were evaluated.
Results
Localized disease (LD) was diagnosed in 39 patients. The median age at diagnosis was 2.5 months (range 0–12 months). Expert pathologic review confirmed the diagnosis of spindle cell RMS in all patients. Among 26 tumors that had molecular evaluation, 13 had rearrangements of NCOA and/or VGLL. Multimodal treatment of infants with LD included conventional (age adjusted) chemotherapy (n=37), resection (n=31) and radiotherapy (RT) (n=5, brachytherapy in 3). Complete remission was achieved in 37/39 patients. Progressive disease occurred in two infants, relapsed disease in three. Microscopically complete surgical resection was associated with 5-year event-free survival (EFS) and overall survival (OS) of 100%. Two patients with tumors ≤ 5cm were treated with microscopically complete resection only and were alive 1 and 4.2 years after diagnosis. The 5-year EFS and OS for infants with LD were 86 % (±11; CI 95%) and 91% (±9; CI 95%), respectively. One patient had metastatic disease (NCOA fusion positive) with primary tumor in head and neck and brain metastases. This patient died despite chemotherapy and delayed resection of the primary tumor due to respiratory failure secondary to cytomegalovirus infection 1.2 years after diagnosis.
Conclusion
Infants with spindle cell RMS have an excellent prognosis. Multimodal treatment including microscopically complete resection of the tumor is strongly recommended.
Keywords: spindle cell rhabdomyosarcoma, NCOA, VGLL, infants, localized disease
Table of contents:
The treatment and outcome of 39 infant patients with localized spindle cell rhabdomyosarcoma and a single patient with metastatic disease enrolled in international trials and registries (1997–2018) were analyzed.
INTRODUCTION:
Although the majority of rhabdomyosarcoma (RMS) is diagnosed in children under 6 years, the disease is uncommon in infants[1]. Historically, age ≤ 12 months has been reported as a poor prognostic factor [2, 3], in part because of high rates of local failure due to the difficulty in delivering aggressive local treatment, especially radiotherapy, in such young children[2, 4, 5]. More recent studies, however, have not shown this discrepancy in outcomes for infants, with infants demonstrating either no difference or improved overall survival (OS) compared to children 12–36 months [5, 6]. Spindle cell and sclerosing RMS is a rare variant which accounts for between 5 to 10% of all RMS. In infants, spindle cell RMS accounts for 10% of RMS cases [6]). In older children and adults, spindle cell and sclerosing RMS may be associated with MYOD1 mutations and a poor prognosis[7, 8]. In contrast, infantile spindle cell RMS has been associated with recurring fusions involving VGLL2 or NCOA2[9]. The majority of cases reported in the literature suggest that molecularly defined spindle cell RMS in infants may be a biologically distinct entity with a favorable prognosis and may not require the aggressive multimodal treatment used for other subtypes of RMS, with behavior more closely reminiscent of ETV6-NTRK3-positive infantile fibrosarcomas [9–11]. However, a recent publication described 4 infants with unresectable tumors harboring VGLL fusions with poor outcomes, calling into question the previous suggestion of favorable outcome[12]. We sought to describe the clinical characteristics, outcomes and prognostic factors of an international cohort of infants diagnosed with spindle cell RMS between 1997–2018.
METHODS:
Patients aged ≤12 months at the time of diagnosis of spindle cell RMS were identified from the Children’s Oncology Group (COG), Cooperative Weichteilsarkom Studiengruppe (CWS), European paediatric Soft tissue sarcoma Study Group (EpSSG) and Italian Soft tissue Sarcoma Committee (STSC), clinical trial and registry databases as well as the Texas Children’s Hospital (TCH) pathology archives. Patients diagnosed between 1997 and 2018 were included. Guardians of patients who were enrolled in cooperative group clinical trials or registries had previously consented to data collection and retrospective chart review was performed per the requirements of the declaration of Helsinki and in accordance with the regulations of the respective ethical committee. Expert pathology review was performed either by the treating center or by central review for those enrolled on clinical trials. Gene fusions involving VGLL2 and NCOA2 were analyzed by fluorescence in situ hybridization (FISH) or reverse transcription polymerase chain reaction (RT)-PCR[10]. Fusion data were not available for all cases.
Definition of terms:
Initial staging procedures and assessment included imaging of the primary tumour and metastases by magnetic resonance imaging (MRI) or computed tomography (CT) with additional recommendation for whole body imaging with radionuclide bone scan or 18F-fluorodeoxyglucose positron emission tomography, and bone marrow aspirate/biopsy dependent on the extent of the primary tumor. The TNM classification was used and differentiated pre-treatment TNM and postsurgical TNM stages [13–15]. The clinical grouping system adapted from the International Rhabdomyosarcoma Study Group (IRS) was used [16, 17]. Margins were defined at the time of pathological assessment. Resection was classified as microscopically complete (R0), microscopically incomplete (R1) and macroscopically incomplete (R2). Delayed surgical resection was defined as occurring after initiation of chemotherapy. “Extent of resection” was defined as the best surgical result in the sum of surgeries performed in an individual during primary treatment. Response was assessed after 3–4 courses of chemotherapy: Complete response (CR), partial response (PR), and stable disease (SD). Progressive disease (PD) as first event was defined as any increase in tumor volume in patients who did not achieve CR[18]. Response was not assessable after up front R0/R1 resection. “Best response” was the most available data on response without standardized timepoint after start chemotherapy. The interval between pathologic diagnosis and detection of relapse or progression was defined as time to event.
Statistical Methods:
Statistics were calculated using Statistica® version 6 (Statsoft) and IBM SPSS® 27 (Armonk, New York, U.S.). Graphs were created using R version 3.5.1. Overall survival (OS) and event-free survival (EFS), as well as post-relapse OS and EFS were calculated using the Kaplan-Meier estimator and confidence intervals (CI) stated at the 95% level [19]. For OS the time from diagnosis to death, either from therapy, disease, other reasons or last follow-up was calculated. For EFS the time from diagnosis to progression (any evidence of growth of a tumor which was not in clinical CR), first recurrence after CR, last follow-up, or death, was calculated. If there was no event the survival data was censored at last follow-up. For comparison of EFS levels the log-rank test was used in univariate analyses. P-values presented are not adjusted for multiplicity.
Treatment:
Patients were treated with a combination of therapies including chemotherapy, surgical resection, and radiation therapy (RT) according to their IRS stage and group. Therapy was determined either by the clinical trial of enrolment or at the discretion of the treating physician for those not enrolled on treatment studies. The initial chemotherapy combinations always included vincristine and dactinomycin (VA), often in combination with cyclophosphamide or ifosfamide (VAC/IVA) or doxorubicin (VAIA). Some patients also received maintenance chemotherapy with cyclophosphamide and vinorelbine[20]. Every protocol recommended that infants ≤12 months should receive dose reduced chemotherapy due to patient age and weight [21]. Resection was performed if a non-mutilating procedure was reasonable. RT was left to the discretion of the treating center.
RESULTS
Patients Characteristics and Demography
Since 1997, 39 patients with localised spindle cell RMS fulfilled the inclusion criteria to be eligible for the analysis. In addition, one patient with metastatic disease was identified. This patient was excluded from the overall analysis but is described below. Patientś characteristics are given in Tables 1 and 2. Median age at initial diagnosis was 2.5 months (0–12 months). The median follow up time was 5.3 years (0.6–12 years), the median overall EFS was 4.8 years (0.1–14.2 years) as of the data cut-off of December 2020. Among 26 tumors that had molecular evaluation, 13 (50%) contained a molecular rearrangement of VGLL2 or NCOA2 (Table 2).
Table 1.
Univariate analysis of characteristics and treatment of patients with localized spindle cell RMS
| Total1 (N=39) | 5-year EFS,% (95% CI) | p-value | 5-year OS, % (95% CI) | p-value | |
|---|---|---|---|---|---|
|
| |||||
| Sex | 39 | ||||
| female | 18 | 100 | 100 | ||
| male | 21 | 74±20 | 0.04 | 84±16 | 0.13 |
| Age | 39 | ||||
| ≤6 months | 30 | 84±14 | 92±11 | ||
| >6 months≤12 months | 9 | 89±21 | 0.75 | 89±21 | 0.79 |
| Age-2 | 39 | ||||
| ≤3 months | 22 | 83±14 | 100 | ||
| >3 months≤6 months | 8 | 70±36 | 69±36 | ||
| >6 months≤12 months | 9 | 89±21 | 0.39 | 89±21 | 0.05 |
| Patients Origin | 39 | ||||
| COG/Texas | 15 | 92±15 | 92±15 | ||
| CWS | 9 | 64±41 | 100 | ||
| EpSSG | 15 | 87±17 | 0.48 | 87±17 | 0.64 |
| Fusion status | 39 | ||||
| VGLL2 and/or NCOA2-positive | 13 | 90±19 | 100 | ||
| VGLL2 and/or NCOA2-negative | 12 | 75±25 | 0.25 | 82±23 | 0.15 |
| No fusion status available | 14 | 92±15 | 0.38 | 90±17 | 0.37 |
| Fusion status-2 | 13 | ||||
| VGLL2 positive | 6 | 83±30 | 100 | ||
| NCOA2 positive | 5 | 100 | 0.56 | 100 | - |
| VGLL2-NCOA2 positive | 2 | 100 | 0.72 | 100 | - |
| Tumor site | 39 | ||||
| favourable | 7 | 86±26 | 100 | ||
| unfavourable | 32 | 86±13 | 0.94 | 89±12 | 0.37 |
| Tumor location | 39 | ||||
| Extremities | 10 | 80±25 | 88±23 | ||
| Head and neck | 3 | 67±53 | 100 | ||
| GU | 8 | 86±26 | 88±23 | ||
| trunk | 18 | 93±13 | 0.58 | 93±14 | 0.90 |
| Initial tumor size | 39 | ||||
| ≤ 5 cm | 24 | 96±8 | 95±9 | ||
| >5 cm | 15 | 72±24 | 0.07 | 86±18 | 0.40 |
| Nodal status | 36 | ||||
| N0 | 35 | 87±12 | 93±9 | ||
| N1 | 1 | 0 | 0.002 | 0 | <0.001 |
| IRS group | 38 | ||||
| I | 12 | 100 | 100 | ||
| II | 6 | 75±43 | 80±35 | ||
| III | 20 | 80±17 | 0.34 | 90±14 | 0.44 |
| CHT | 34 | ||||
| VA | 6 | 75±43 | 100 | ||
| VAC/IVA | 14 | 92±16 | 92±16 | ||
| VAC/IVA+MT | 9 | 78±27 | 88±23 | ||
| VAIA | 5 | 75±43 | 0.79 | 80±35 | 0.76 |
| Best response to CHT | 19 | ||||
| CR | 10 | 100 | 100 | ||
| PR | 9 | 100 | - | 100 | - |
| RT | 35 | ||||
| yes | 5 | 100 | 100 | ||
| no | 30 | 85±14 | 0.37 | 92±10 | 0.52 |
| Surgical resection | 39 | ||||
| yes | 37 | 91±10 | 91±10 | ||
| no | 2 | 0 | <0.001 | 100 | 0.66 |
| Time of surgical resection | 35 | ||||
| upfront | 18 | 92±16 | 93±14 | ||
| delayed | 17 | 94±11 | 0.99 | 94±12 | 0.91 |
| Extend of resection | 37 | ||||
| R0 | 17 | 100 | 100 | ||
| R1 | 14 | 92±16 | 93±15 | ||
| R2 | 6 | 67±38 | 0.03 | 83±30 | 0.39 |
Abbreviations: EFS event free survival; OS overall survival; CHT chemotherapy; CR complete response; IVA Vincristine, actinomycin-D, ifosfamide; PR partial response; R0 complete resection; R1 microscopic incomplete resection; R2 macroscopic incomplete resection; RT radiotherapy; VA vincristine, actinomycin-D; VAC vincristine, actinomycin-D, cyclophosphamide; VAIA Vincristine, actinomycin-D, ifosfamide, doxorubicin; y years.
Variables displayed may have missing values. For each variable the available cases are used and therefore subcategories do not always sum up to N=39.
Table 2.
Characteristics and treatment of 39 patients with localized disease according to fusion status
| Fusion | N | Age (mo) | Tumor location | Initial tumor size | N-status | IRS Group | CHT | Extend of resection | RT | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| NCOA2-VGLL2 | 2 | 0.24 and 1.6 | extremity (n=1) trunk (n=1) |
≤5cm (n=1) >5 (n=1) |
N0 | III (n=2) | VA(C) (n=1) VAIA (n=1) |
R1 (n=1) R2 (n=1) |
no (n=2) | A1.CR (n=2) |
| NCOA2- | 51 | 1.3 (0.3–8) | extremity (n=3) trunc (n=2) |
≤5cm (n=3) >5 (n=2) |
N0 | I (n=2) II (n=1) III (n=2) |
VA(C) (n=4) IVA (n=1) |
R0 (n=3) R1 (n=1) R2 (n=1) |
no (n=5) | A1.CR (n=5) |
| VGLL2- | 62 | 0.4 (0.2–3.0) | extremity (n=1) trunc (n=5) head and neck (n=1) |
≤5cm (n=1) >5 (n=6) |
N0 | II (n=1) III (n=5) n.a. (n=1) |
VA(C)/IVA(n=2) IVA+MT (n=3) VAIA (n=2) |
R0 (n=1) R1 (n=5) R2 (n=1) |
no (n=6= yes (n=1) |
A1.CR (n=6) relapse and A2.CR (n=1) |
| No fusion identified | 123 | 4.3 (0–12) | extremity (n=4) trunc (n=3) head and neck (n=1) GU (n=4) |
≤5cm (n=8) >5 (n=4) |
N0 (n=11) N1 (n=1) |
I (n=5) II (n=7) |
VA(C)/IVA(n=6) IVA+MT (n=2) VAIA (n=1) n.a. (n=2) |
R0 (n=6) R1 (n=2) R2 (n=3) n.a. (n=1) |
no (n=11) yes (n=2) |
A1-CR (n=5) DOD (n=2) |
| Not tested/Insufficient sample | 14 | 3.9 (0.2–12.4) | extremity (n=1) trunc (n=8) head and neck (n=1) GU (n=4) |
≤5cm (n=11) >5 (n=3) |
N0 (n=13) n.a. (n=1) |
I (n=5) II (n=4) III (n=5) |
VA(C)/IVA(n=7) IVA+MT (n=3) VAIA (n=2) No CHT (n=2) n.a. (n=1) |
R0 (n=7) R1 (n=6) n.a. (n=1) |
no (n=12) yes (n=2) |
A1.CR (n=13) DOD (n=1) |
Abbreviations: A1.CR alive in 1st complete remission; A2. CR alive in 2nd complete remisison; CHT chemotherapy; CR complete response; DOD dead of disease; GU genitourinary tract; IVA Vincristine, actinomycin-D, ifosfamide; n.a. not available; PR partial response; R0 complete resection; R1 microscopic incomplete resection; R2 macroscopic incomplete resection; RT radiotherapy; VAC vincristine, actinomycin-D, cyclophosphamide; VAIA Vincristine, actinomycin-D, ifosfamide, doxorubicin
TEAD1-NCOA2 n=4, partner unknown n=1
VGLL2-CITED2 n=2, partner unknown n=4
One sample only tested for NCOA2 and 2 samples only tested for VGLL2 containing fusions
Patients with localized disease
Thirty-nine patients had localized disease: IRS I (n=12), IRS II (n=6) and IRS III (n=20); n.a. (n=1). Thirty patients were ≤ 6 months old at diagnosis, with 22 of them <3 months. Data for primary tumor size, chemotherapy regimens and response to chemotherapy are given in Table 1. One infant had regional lymph node involvement (primary in the extremity without molecular rearrangement). In 2 patients, no chemotherapy was given at the discretion of the treating center or parental refusal. Both patients achieved CR after upfront R0 resection (localisation was paratesticular and trunk, both less than 5 cm). Both are alive in CR 4.2 and 1 year after diagnosis. After initial chemotherapy, delayed primary resection was performed in 17 patients with IRS III disease. The extent of resections resulted in R0 (n=17, 44%), R1 (n=14, 36%) or R2 (n=6, 15%). RT was used in 5/39 patients, all of whom had IRS III disease and delayed resection resulted in a positive margin: R1 margin in 4 patients and R2 in 1 patient. Overall, 38/39 patients achieved CR, including 5/6 patients after R2 resection (one patient received additionally RT after R2 resection). Thirty-five patients had no relapse (92% of patients in CR) and 36 were alive in clinical remission at last follow up. One patient died of progressive disease despite chemotherapy after R2 resection and two of recurrent disease (one patient with N1 disease died from metastatic relapse, and one patient died with unknown site of recurrence). Of the three patients who died from disease, no molecular rearrangement could be found (n=2) or was not tested (n=1). The 5-year EFS was 86 % (±11; CI 95%), the 5-year OS was 91% (±9; CI 95%) (Figure 1).
FIGURE 1.
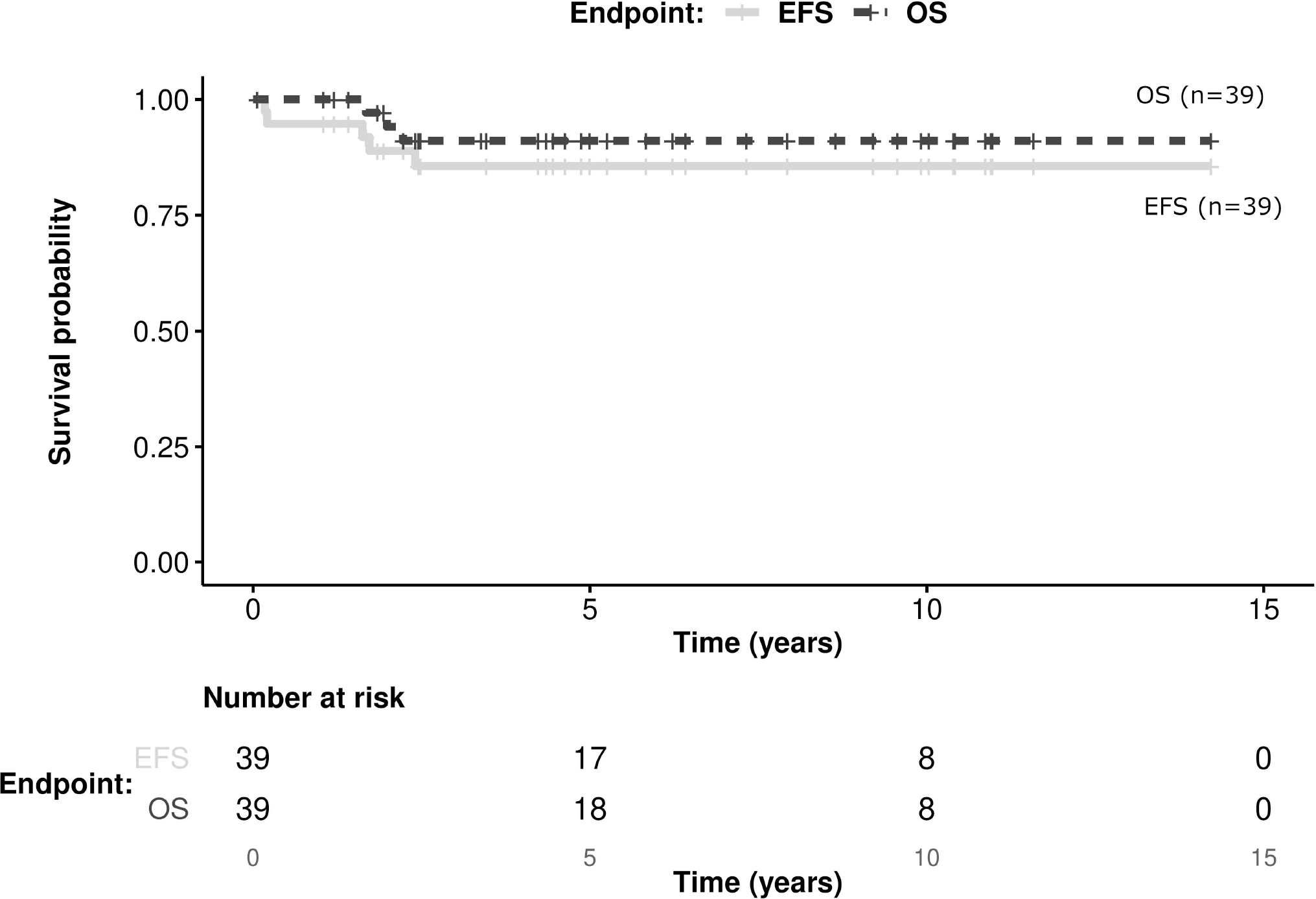
Kaplan-Meier estimates presenting EFS and OS of 39 patients with localized disease. Total number of events: 5 in EFS, 3 in OS.
Patient with primary metastatic disease
Only one patient with IRS group IV metastatic disease at diagnosis was identified. The patient was 1.2 months old at diagnosis and had a primary tumor located in the skull base measuring more than 5 cm, with metastatic disease in the brain. The tumor was found to have a NCOA2 fusion by FISH (fusion partner unknown). The patient was treated with chemotherapy (regimen unknown) and underwent a delayed R2 resection of the primary tumor. RT was not administered. Partial response was achieved, but the patient died from respiratory failure due to cytomegalovirus pneumonitis 1.2 years after diagnosis which was 3 months after completion of therapy.
Univariate Analysis and Prognostic Factors:
The 39 patients with localized disease were included in the univariate analysis. Patients with either a VGLL2 and/or NCOA2 fusion had a 5 year EFS of 90% (±19; CI 95%) and OS of 100%, while those with no detected fusion had 5 year EFS 75% (±25; CI 95%) and OS of 82% (±23; CI 95%). The presence of the VGLL2 and/or NCOA2 fusion was not a statistically significant prognostic factor (Figure 2). Extent of surgical resection R0 and R1 were statistically significant favorable prognostic factors for the 5-year EFS of localized spindle cell RMS patients diagnosed in infancy (Figure 3, Table 1). Use of RT was not a statistically significant prognostic factor, and no difference between the different chemotherapy regimens was detected (Table 1).
FIGURE 2.
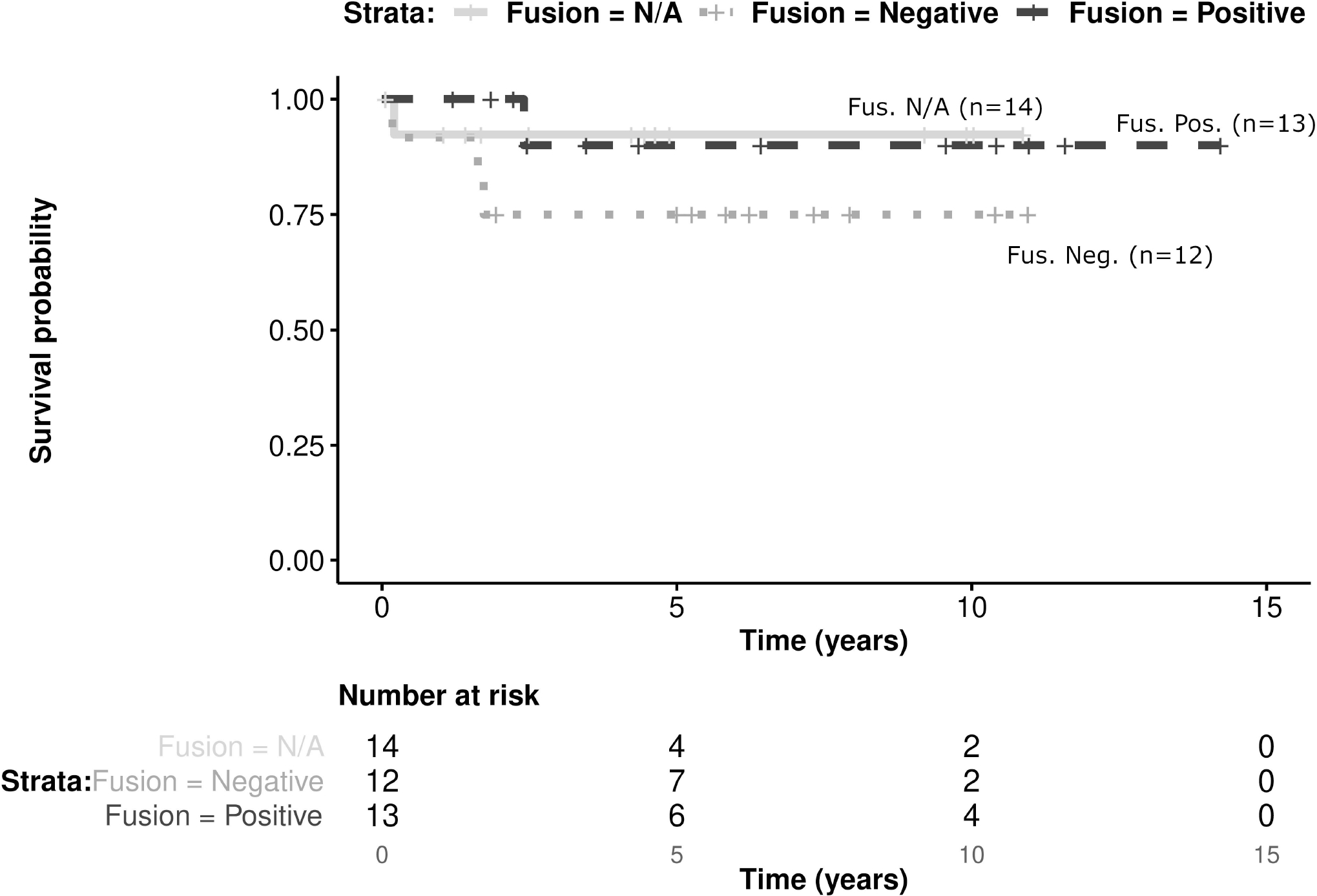
Kaplan-Meier estimates presenting EFS of 39 patients with localized disease according to fusion status (p=0.38)
FIGURE 3:
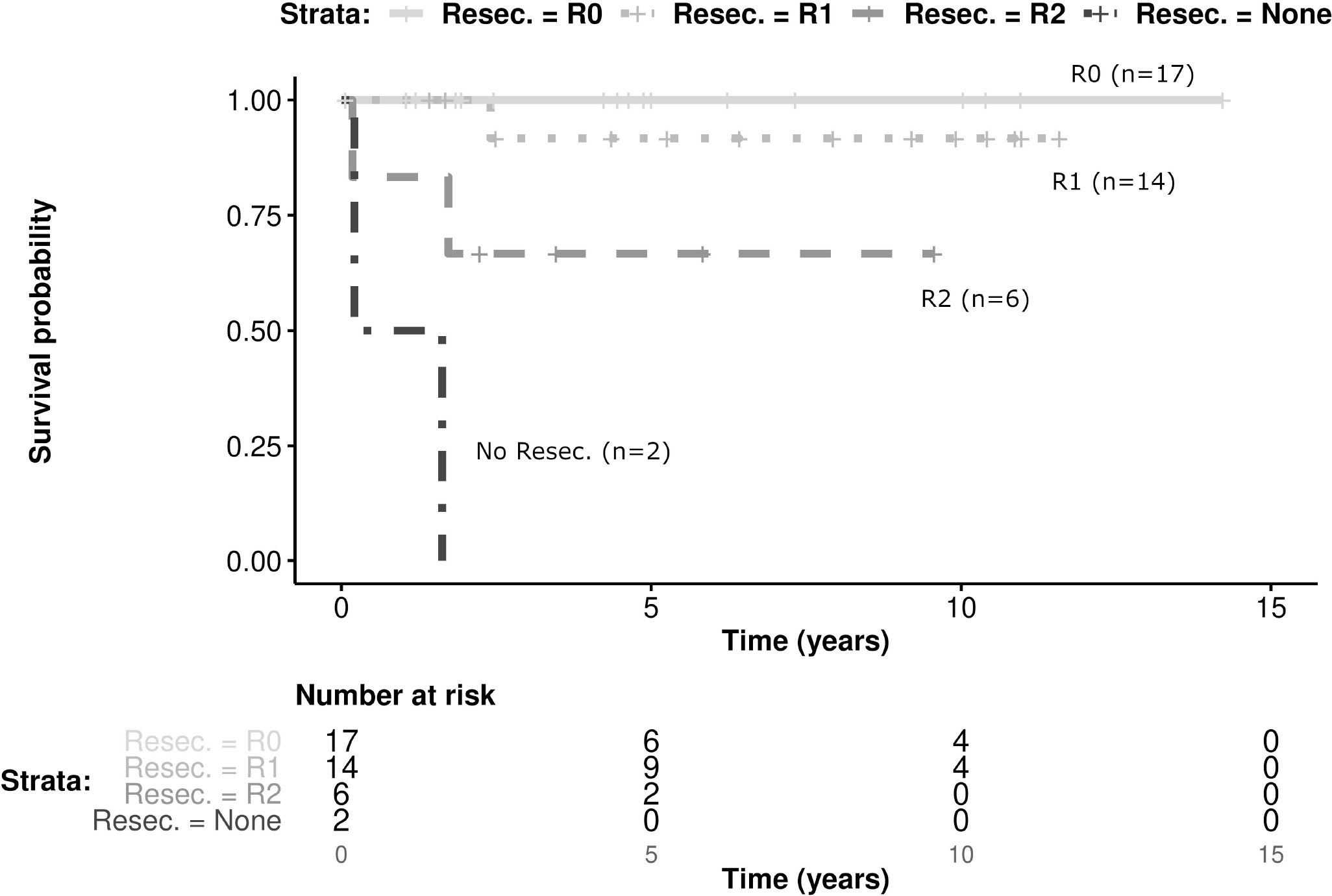
Kaplan-Meier estimates presenting EFS of 39 patients with localized disease according to extent of resection, including the no-resection group (p<0.001)
DISCUSSION:
Localized RMS diagnosed in the first year of life has high rates of relapse with 5-year failure free survival (FFS) rates ranging from 42% to 72% in reports from various international cooperative groups [2–4, 6, 21, 22]. The 5 year OS for RMS in children less than a year old ranges from 61–88% [2, 4, 21, 22], with more recent studies suggesting that OS in this age range is no be worse than older children[5, 6]. Within our international cohort the 5-year EFS and OS for infants with localized spindle cell RMS were 86% and 91%, respectively, suggesting a favorable outcome for infants with spindle cell RMS, with lower rates of relapse and possibly death than combined histologic subtypes of infantile RMS. As previously reported, these tumors are frequently found in axial locations, and are almost always localized at presentation. Extent of resection was one prognostic factor resulting from univariate analysis, which has not been previously described [4]. Interestingly, while patients who had gross disease left behind surgically (R2 resection) had inferior EFS, no significant difference in OS could be shown. The small sample size limits the statistical power to show such an effect, especially considering that IRS III patients may have received additional intervention (resulting in microscopical complete resection) attenuating the impact on OS. We emphasize that from a statistical perspective a non-significant result in this small and exploratory study does not constitute proof that no relevant difference may exist between the examined groups [23].
Very few patients in our cohort received RT, and there was no statistically significant difference in outcomes based on the use of RT. RT in infants may lead to significant growth impairment, and is therefore generally avoided in this age group. Our data suggests the majority of these patients can be cured without RT. Chemotherapy regimens varied widely with respect to specific agents used as well as doses of agents including cyclophosphamide. Overall, the common VA(C) regimen was used in most patients. Note that not only the very limited number of patients prevent us from making any strong scientific conclusions about the therapy, but that also a selection bias may influence results here, given that the choice of therapy may in general be related to the clinical presentation. However, the overall good prognosis for those who received VAC or IVA suggests that these additional therapies may not be necessary for these patients, underlying the importance of microscopically complete resection: Two patients with tumors <5cm and microscopically complete resections did not receive chemotherapy. Even omission of chemotherapy might be an option in patients with these small tumors and R0 resection. However, numbers of patients treated with resection only are limited not allowing us to draw conclusions.
The subset of spindle cell RMS in infants with fusions involving VGLL2 or NCOA2 have previously been reported to have a favorable prognosis [10, 11], although a recent report of four infants with unresectable VGLL2 rearranged RMS who experienced local progression, metastatic disease, and 2 deaths from disease questioned these findings [12]. In that report, at initial diagnosis, 3 tumors were diagnosed as fibromatosis or infantile fibrosarcoma and initially managed as such, while 1 was a high-grade sarcoma. At relapse, 3 tumors showed high-grade morphology, while 1 retained a low-grade phenotype. These cases imply the importance of initial expert pathologic diagnosis, complete surgical resection, and suggest that RMS-type chemotherapy should be considered for unresectable low-grade tumors harboring these rearrangements, given the risk of high-grade transformation [12]. While our cohort did not demonstrate a significant difference in survival for those with VGLL2 or NCOA2 fusions, it is notable that no patients with a fusion died of their disease, though the one patient with metastatic disease and NCOA2 fusion died of infection. The ability to draw conclusions specific to NCOA2 or VGLL2 fusion status is limited given the information was only available for half of the cohort due to lack of suitable banked tissues for analysis in the others. Additionally, within those for whom tissue was available for testing, we identified a VGLL2 or NCOA2 containing fusion in less than 60%. It is possible that the use of FISH limited our ability to detect these fusions and that next generation sequencing methods may detect other fusions analogous to the known VGLL2 and NCOA2 containing fusions. Larger studies with comprehensive molecular analysis will be needed to truly define the incidence and prognostic implications of VGLL2 and NCOA2 fusions.
In addition to the lack of NCOA2 and VGLL2 fusion testing in half of our cohort, our study is also limited by its lack of complete genomic assessment of tumors including HRAS mutations which are frequently detected in FOXO1 fusion negative infantile RMS[24], or presence of MYOD1 mutations which are exceedingly rare in infants, but are associated with spindle cell RMS in older children and adults and carry important prognostic implications[24]. Knowledge about these and other genomic alterations would provide important contextual information related to prognosis in the patients with tumors lacking NCOA2 or VGLL2 fusions. In addition, the size of our study, potential selection bias, and a lack of complete treatment information for all patients made conclusions about best treatments for these patients difficult to determine. Nonetheless, this is the largest study of infants with spindle cell RMS and we internationally propose common first line treatment recommendations: R0 resection (if feasible without mutilation) and systemic treatment with risk-adapted therapy using the VA (or VAC/IVA) regimen; if no R0 resection seems feasible, start with VAC (IVA) with the aim of secondary microscopically complete resection (Figure 4). Anthracyclines and external beam RT can be avoided in the majority of patients. Further international studies are needed to answer the question if further reduction of treatment might be possible as reducing the cumulative dose of alkylating agents in this age after microscopically complete resection. Continued international collaboration and a prospective molecular analysis of spindle cell RMS is undoubtedly needed to further investigate a common treatment approach in this subgroup of patients.
Figure 4.

International consensus on initial treatment of infants with congenital spindle cell rhabdomyosarcoma
CHT chemotherapy; FISH fluorescence in situ hybridization; IVA Vincristine, actinomycin-D, ifosfamide; VAC vincristine, actinomycin-D, cyclophosphamide; RT radiotherapy; (RT)-PCR reverse transcription polymerase chain reaction;
Highlights:
international cohort of 40 patients aged ≤12 months with spindle cell rhabdomyosarcoma
Characteristics, treatment, and outcome analyzed in 39 patients with localized disease
Among 26 tumors that had molecular evaluation, 13 had rearrangements of NCOA2 and/or VGLL2
The 5-year EFS and OS for infants with localized disease were 86% and 91%, respectively.
ACKNOWLEDGEMENT:
We gratefully thank parents who allow sharing their children’s clinical data and are very thankful for the continuous cooperation of the contributing hospitals with EpSSG, COG, TCH and CWS. This work could not have been done without the excellent data management of all involved persons within EpSSG, COG, TCH and CWS.
Funding:
CWS-96, and -2002P trials were supported by grants from the German Cancer Aid Foundation, Bonn, Germany (CWS-96: T9/96/TrI, CWS-2002P: 50-2721-Tr2). The registry (SoTiSaR), was supported by the Deutsche Kinderkrebsstiftung, Bonn, Germany, grant no. A2007/13DKS2009.08 and by the Foerderkreis Krebskranke Kinder Stuttgart, Germany. Children’s Oncology Group trials and biobanking were supported by grants from the National Cancer Institute U10CA180886, U10CA180899, U10CA098543, U10CA098413, U24CA196173, and U24CA114766 and by the St. Baldrick’s Foundation. Drs. Barr and Pack were supported by the Intramural Research Program of the National Cancer Institute. The AIEOP STSC and EpSSG data collection has been supported by Fondazione Città della Speranza, Padova, Italy
Abbreviations
- AIEOP
Associazione Italiana di Ematologia e Oncologia Pediatrica
- CEVAIE
Carboplatin, epirubicin, vincristine, actinomycin-D, ifosfamide, etoposide
- CI
confidence interval
- CHT
chemotherapy
- COG
Children’s Oncology Group
- CR
complete remission
- CWS
Cooperative Weichteilsarkom Studiengruppe
- EFS
event free survival
- EpSSG
European Pediatric Soft Tissue Sarcoma Study Group
- EVAIA
etoposide, vincristine, actinomycin-D, ifosfamide, doxorubicine
- IRS
international rhabdomyosarcoma study group
- LD
localized disease
- MD
metastatic disease
- mPR
minor partial response
- MRI
magnetic resonance imaging
- OS
overall survival
- PFS
progression free survival
- PD
progressive disease
- PR
partial response
- RD
relapsed disease
- RMA
alveolar rhabdomyosarcoma
- RME
embryonal rhabdomyosarcoma
- RMS
habdomyosarcoma
- AIEOP STSC
AIEOP Soft Tissue Sarcoma Committee
- SD
stable disease
- TNM
Tumor-node-metastasis
- UICC
Union internationale contre le cancer
- VAC
vincristine, actinomycin-D, cyclophosphamide
- VACA
vincristine, actinomycin-D, cyclophosphamide, doxorubicine
- VAIA
vincristine, actinomycin-D, ifosfamide, doxorubicine
Footnotes
Declaration of Interest statement:
The authors declare that there is no conflict of interest.
Consent statement: Informed consent has been obtained from all CWS, AIEOP STSC, EpSSG and COG participating patients and/or their parents/guardians according to the legal requirements.
Presented in part: Findings were included in part in the publication PMID: 33438323 & 31339226.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Steliarova-Foucher E, et al. , International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol, 2017. 18(6): p. 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrari A, et al. , Rhabdomyosarcoma in infants younger than one year old: a report from the Italian Cooperative Group. Cancer, 2003. 97(10): p. 2597–604. [DOI] [PubMed] [Google Scholar]
- 3.Joshi D, et al. , Age is an independent prognostic factor in rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Pediatr Blood Cancer, 2004. 42(1): p. 64–73. [DOI] [PubMed] [Google Scholar]
- 4.Malempati S, et al. , Rhabdomyosarcoma in infants younger than 1 year: a report from the Children’s Oncology Group. Cancer, 2011. 117(15): p. 3493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradley JA, et al. , Treatment Approach and Outcomes in Infants With Localized Rhabdomyosarcoma: A Report From the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Int J Radiat Oncol Biol Phys, 2019. 103(1): p. 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slater O, et al. , Localised rhabdomyosarcoma in infants (<12 months) and young children (12–36 months of age) treated on the EpSSG RMS 2005 study. Eur J Cancer, 2022. 160: p. 206–214. [DOI] [PubMed] [Google Scholar]
- 7.Agaram NP, et al. , MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol, 2019. 32(1): p. 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agaram NP, et al. , Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer, 2014. 53(9): p. 779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alaggio R, et al. , A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am J Surg Pathol, 2016. 40(2): p. 224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosquera JM, et al. , Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer, 2013. 52(6): p. 538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whittle SB, et al. , Congenital spindle cell rhabdomyosarcoma. Pediatr Blood Cancer, 2019. 66(11): p. e27935. [DOI] [PubMed] [Google Scholar]
- 12.Cyrta J, et al. , Infantile Rhabdomyosarcomas With VGLL2 Rearrangement Are Not Always an Indolent Disease: A Study of 4 Aggressive Cases With Clinical, Pathologic, Molecular, and Radiologic Findings. Am J Surg Pathol, 2021. 45(6): p. 854–867. [DOI] [PubMed] [Google Scholar]
- 13.Hermanek P, Sobin LH, and Wittekind C, How to improve the present TNM staging system. Cancer, 1999. 86(11): p. 2189–91. [PubMed] [Google Scholar]
- 14.Wittekind C, [New TNM classification of lung tumors]. Pathologe, 2014. 35(6): p. 578–85. [DOI] [PubMed] [Google Scholar]
- 15.Webber C, et al. , Improving the TNM classification: findings from a 10-year continuous literature review. Int J Cancer, 2014. 135(2): p. 371–8. [DOI] [PubMed] [Google Scholar]
- 16.Crist WM, et al. , Prognosis in children with rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma studies I and II. Intergroup Rhabdomyosarcoma Committee. J Clin Oncol, 1990. 8(3): p. 443–52. [DOI] [PubMed] [Google Scholar]
- 17.Crist W, et al. , The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol, 1995. 13(3): p. 610–30. [DOI] [PubMed] [Google Scholar]
- 18.Dantonello TM, et al. , Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol, 2009. 27(9): p. 1446–55. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan EL and Meyer P, Non-parametric estimation from incomplete observations. J Am Stat Assoc, 1958. 53: p. 457–481. [Google Scholar]
- 20.Bisogno G, et al. , Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol, 2019. 20(11): p. 1566–1575. [DOI] [PubMed] [Google Scholar]
- 21.Sparber-Sauer M, et al. , Rhabdomyosarcoma diagnosed in the first year of life: Localized, metastatic, and relapsed disease. Outcome data from five trials and one registry of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer, 2019. 66(6): p. e27652. [DOI] [PubMed] [Google Scholar]
- 22.Orbach D, et al. , Soft tissue sarcoma or malignant mesenchymal tumors in the first year of life: experience of the International Society of Pediatric Oncology (SIOP) Malignant Mesenchymal Tumor Committee. J Clin Oncol, 2005. 23(19): p. 4363–71. [DOI] [PubMed] [Google Scholar]
- 23.Altman DG and Bland JM, Absence of evidence is not evidence of absence. BMJ, 1995. 311(7003): p. 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shern JF, et al. , Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J Clin Oncol, 2021. 39(26): p. 2859–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]