

Abstract
Cyclophilin A (CypA) is linked to diverse human diseases including viral infections. With the worldwide emergence of severe acute respiratory coronavirus 2 (SARS-CoV-2), drug repurposing has been highlighted as a strategy with the potential to speed up antiviral development. Because CypA acts as a proviral component in hepatitis C virus, coronavirus and HIV, its inhibitors have been suggested as potential treatments for these infections. Here, we review the structure of cyclosporin A and sanglifehrin A analogs as well as synthetic micromolecules inhibiting CypA; and we discuss their broad-spectrum antiviral efficacy in the context of the virus lifecycle.
Keywords: Cyclophilin A, Antiviral, Drug repurposing, Cyclosporin A, Debio-025
Introduction
Drug repurposing is an approach used to discover new medicinal uses for authorized or experimental medications that are not currently prescribed for such uses.1 This approach has several benefits in drug development. First, the toxicity of repurposed drugs is likely to be minimal when compared with newly developed drugs, because they have previously been proven to be sufficiently safe in preclinical or clinical Phase I studies. Thus, most tolerability research, such as safety evaluations, dosage optimization and route determination, can be bypassed or minimized.2, 3 Second, drug redevelopment investment can be lowered by using already established large-scale production processes and quality specifications or test methods for manufacturing processes. The absence of approved vaccines or virus-specific antivirals at the beginning of the currently circulating severe acute respiratory syndrome virus 2 (SARS-CoV-2) pandemic has focused attention on the potential value of successful drug repurposing strategies.
An example of the most recently developed, drug-repurposed antiviral agent is molnupiravir (also named MK-4482 or EIDD-2801) – a viral RNA-dependent RNA polymerase inhibitor for oral treatment of SARS-CoV-2 infection. The repurposing of molnupiravir was initiated by synthesizing an isopropylester prodrug of the ribonucleoside analog N4-hydroxycytidine (EIDD-1931) for development as a direct-acting, antiviral agent against influenza virus.4, 5 Being distinctive from the direct-acting antivirals, an alternative drug repurposing approach utilizes host-targeted antiviral agents. These include inhibitors of cyclophilin A (CypA), which is a ubiquitous, cytosolic protein with peptidyl–prolyl cis–trans isomerase (PPIase) activity first described in 1989, belonging to the immunophilin family.6 There is abundant experimental evidence suggesting that CypA is crucially involved in human diseases including not only cardiovascular diseases and cancers but also viral infections.7, 8, 9, 10, 11, 12, 13, 14, 15 However, in viral infections, CypA has converse roles: it supports viral replication in cells infected with hepatitis C virus (HCV), coronavirus (CoV), HIV or hepatitis B virus (HBV); whereas it destabilizes a viral protein or triggers host defense mechanisms in cells infected with influenza virus or rotavirus. To understand these contradictory biological functions (proviral and antiviral), this review focuses on the molecular interactions between CypA and different viral proteins or other cellular factors and the accompanying signaling pathways. It also summarizes progress in the development of CypA-targeting antiviral agents, based on the chemical structural diversity. We provide an insight into drug development strategies for broad-spectrum antivirals that target CypA and for the chemical modifications with improved antiviral efficacy but reduced immunosuppressive activity or undesirable side effects, such as nephrotoxicity, hypertension and dyslipidemia.16, 17, 18
CypA as a proviral host factor
CypA has been identified as a promising target for antiviral development because it works as a cofactor in viral genome replication, controls innate immune responses and stabilizes or structurally modulates viral proteins. Here, the proviral roles of CypA and its interaction with viral proteins or other cellular factors will be discussed by scrutinizing the lifecycles of different viruses, such as HCV, CoV, HIV and HBV.
HCV
HCV is a member of the family Flaviviridae and has a positive-sense ssRNA genome that encodes a single polyprotein of ∼ 3010 amino acids in length.19 It actively facilitates intracellular membrane rearrangement to maintain its cytoplasmic replication and exploits or modifies cellular proteins involved in cellular lipid metabolic pathways or immune responses.20, 21 Before the identification of CypA being one of the host proviral factors for HCV replication, cyclosporin A (also called ciclosporin A; CsA) was characterized as having antiviral activity against HCV in a cultured human hepatocyte cell line infected with HCV as well as in a viral replicon system.22, 23 At least three viral proteins: nonstructural protein 5B (NS5B; RNA-dependent RNA polymerase), NS5A and NS2, are known to bind directly to CypA.24, 25, 26 Another report suggested that NS5A–NS5B cleavage is controlled by CypA.12 The interactions of CypA with these proteins rely on the PPIase activity of CypA.27, 28 Indeed, when CypA is mutated at either arginine at position 55 or histidine at position 126, within the hydrophobic active pocket of the enzyme, viral replication is drastically impaired.24 This enzymatic activity of CypA is required for NS5B-mediated formation of double-membrane vesicles (DMVs), in which HCV RNA replication occurs (Fig. 1 , left panel). These intracellular compartments are also thought to contribute to preventing cytosolic pattern recognition receptors from sensing viral RNAs as non-self.29, 30 CsA, a natural cyclic polypeptide immunosuppressant, was reported to inhibit DMV formation efficiently in HCV-infected cells (Fig. 1, right panel).31
Figure 1.

Proviral functions of CypA in HCV replication. (Left panel) CypA roles in HCV replication. CypA recruits ISGylated NS5A to PKR, inhibiting the PKR signaling pathway and nuclear migration of IRF1. CypA participates in the genomic replication of HCV by interacting with viral proteins: NS5B and NS5A, within the double-membrane vesicles (DMVs) that provide an isolated compartment for HCV RNA-dependent RNA replication. (Right panel) Two ways of antiviral machinery of CsA – a CypA inhibitor. It interferes with CypA–NS5A interaction, resulting in activation of PKR-mediated antiviral response. Alternatively, CsA blocks CypA binding to NS5A and NS5B, the main components of the RNA-dependent RNA polymerase complex, finally resulting in disruption of DMV formation. Abbreviations: CypA, cyclophilin A; NS, nonstructural viral protein; ISG15, interferon-stimulated gene product 15; PKR, protein kinase R; IRF1, interferon regulatory factor 1; CsA, cyclosporin A.
Other studies demonstrated that the interaction of CypA with NS5A downregulates the activation of protein kinase R (PKR), which is the primary antiviral effector, and that this subsequently inhibits interferon regulatory factor-1 (IRF1) responses (Fig. 1, left panel).26, 32, 33 In detail, to promote CypA binding to NS5A, interferon (IFN)-stimulated gene product 15 (ISG15) is covalently attached at the CypA-binding site within NS5A (lysine residue 305) by the HERC E3-ubiquitin ligase.34 Like NS5B, NS5A is also involved in the maintenance of the membranous replication compartment by binding to CypA. Thus, CsA inhibits the binding of NS5A to CypA and eventually dissociates each component from the NS5A-NS5B-CypA complex (Fig. 1, right panel). Intrinsically, CsA is immunosuppressive by blocking enzymatic activity of calcineurin phosphatase, a key enzyme for interleukin (IL)-2 production.35, 36 Paradoxically, it participated in type I IFN stimulation by inhibiting CypA–NS5A interaction in HCV-infected cells.26 With understanding of these two contrasting functions of CsA in HCV infection, clinical trials of three CypA inhibitors [Debio-025 (alisporivir), SCY-635 and NIM811] have been scheduled.37 When compared with CsA, they are designed not to inhibit calcineurin activity and thus are not immunosuppressive, enhancing the pharmacological safety margin.38, 39, 40
CoV
CoVs are enveloped and positive-strand RNA members of the Coronaviridae family. The CoV genome encodes four structural proteins: spike (S), envelope (E), membrane (M) and nucleocapsid (N), and at least 16 nonstructural proteins (Nsps).41 CypA has been shown to support SARS-CoV and HCoV-NL63 infection by interacting with viral N protein and Nsp1.8, 14 SARS-CoV infection or Nsp1 overexpression stimulated the calcineurin/nuclear factor of activated T cells (NFAT) pathway and induced IL-2 expression through the activation of immune cells. It means that SARS-CoV activates the NFAT pathway via CypA-interacting Nsp1 for the cytokine dysregulation and immune-dependent pathogenesis. In the same context, single nucleotide polymorphisms near the active site of CypA or inhibition of its PPIase activity negatively affected HCoV-229E and MERS-CoV infections, emphasizing the importance of CypA enzymatic activity during CoV replication.42, 43 Based on these findings, it is not surprising that CsA inhibits a large population of coronaviruses, including HCoV-NL63, SARS-CoV, feline CoV and porcine CoV.14 As a pan-coronavirus antiviral agent, CsA inhibited SARS-CoV-2 infection of Vero E6 cells with an EC50 of 3.5 µM,44 whereas Debio-025 inhibited SARS-CoV-2 infection with an EC50 of 0.46 µM.45
Understanding the molecular basis of the cytokine storms induced by acute SARS-CoV-2 infection is of crucial importance for developing antivirals. Extracellular CypA has been suggested to interact with CD147 (basigin), which was also identified as a putative SARS-CoV-2 S-binding receptor together with angiotensin converting enzyme 2 (ACE2).46, 47 CD147 causes the activation of the mitogen-activated protein kinase (MAPK) pathway, resulting in the upregulation of cytokines and chemokines and promotion of a cytokine storm upon infection with SARS-CoV-2 and its variants.48 Moreover, the expression levels of CypA and CD147 are higher in SARS-CoV-2-infected patients than in uninfected people, and this might contribute to the prominent lymphocytopenia associated with COVID-19.49 However, it remains to be clearly addressed why both molecules, CsA and Debio-025, are effective against SARS-CoV-2 irrespective of immunosuppressive activity45 and whether antiviral activity and immunological safety of CsA can be guaranteed in SARS-CoV-2-infected patients undergoing a cytokine storm. Intuitively, immunosuppressive CypA inhibitors developed in the early generation, than nonimmunosuppressive ones (discussed below), could be more potent and clinically favorable against acute viruses, such as SARS-CoVs, that cause hyperinflammatory immune responses, but the community is awaiting the final results from clinical trials.
HIV
There are several reports exploring the multifunctions of CypA on retroviral infection or assembly.50, 51, 52 However, they exhibited converse experimental results in a cell-type- or viral-strain-dependent manner, in terms of favorable roles of CypA in viral infectivity. Because we have an interest in CypA as an antiviral target and, importantly, many studies agree with the fact that its genetic knockout suppresses most of retroviral infections,53 we will discuss its functions focusing on wild-type HIV type I (HIV-1) infection in human cells. HIV-1 is a member of the Retroviridae family with a positive-strand RNA genome. Its primary target cells are cluster of differentiation 4 (CD4)-positive T cells expressing C–C chemokine receptor 5 (CCR5) and/or C-X-C chemokine receptor type 4 (CXCR4) as a co-receptor and macrophages (Fig. 2 ).54 It was believed that HIV-1 uncoating occurs in the cytoplasm before reverse transcription.55 By contrast, recent studies suggest that nuclear import of the viral core proceeds reverse transcription and uncoating.56 After translation of viral mRNA in producer cells, the Gag polyprotein is cleaved by a viral protease to generate mature matrix (MA), capsid (CA) and nucleocapsid (NC) proteins. The host protein CypA selectively and directly binds to the N-terminal domain of CA and is packaged into the viral particle during virus assembly (Fig. 2, left).10, 11, 57 It catalyzes the isomerization of CA by binding to a single exposed loop containing Gly89 and Pro90 within CA.58, 59 These findings have raised an expectation that CypA incorporated within the viral particles could modulate uncoating and viral infectivity.11, 60 However, CypA-deficient virus produced from CypA null cells comparably infected Jurkat T cells as CypA-bearing HIV-1 particles did.53 Irrespective of CypA incorporation, their infectivity was reduced in CypA null target cells, indicating that the presence of CypA is required for incoming HIV infection only in target cells rather than in producer cells. In the same context, the inhibition of the CypA–CA interaction by CsA treatment suppressed HIV-1 infection in target cells, such as T cells, primary lymphocytes and macrophages.53, 61, 62, 63, 64, 65, 66.
Figure 2.

Proviral functions of CypA in HIV-1 infection. (Left panel) CypA association with HIV-1 CA in producer cells. HIV-1 enters T cells by sensing CD4 as a receptor and either CCR5 or CXCR4 as a co-receptor. Nuclear viral mRNA is exported to the cytoplasm to express viral proteins. These viral proteins are self-assembled and complexed with CypA, for which the viral RNA genome is packaged inside. CypA-bearing progeny virions are secreted from the producer cells. (Right panel) Host factors affected by CA–CypA interaction in target cells. CypA null and CypA-bearing HIV-1 particles can enter CD4-expressing target cells. Host restriction factor TRIM5α facilitates degradation of CA. CypA inhibits TRIM5α binding to CA in a competitive manner. CPSF6 interacts with CA and traffics the viral core on microtubules, to accomplish HIV-1 capsid uncoating. CPSF6 recruits HIV-1 core to SC35 nuclear speckles for reverse transcription with help of inner nuclear envelope protein SUN2. CypA blocks these two steps by competing with CPSF6 for binding to CA. Abbreviations: CD4, cluster of differentiation 4; CCR5, C–C chemokine receptor 5; CXCR4, C-X-C chemokine receptor type 4; gp120, envelope glycoprotein 120; TRIM5α, tripartite-containing motif 5α; CPSF6, cleavage and polyadenylation specific factor 6; SUN2, inner nuclear membrane protein.
For modulation of HIV-1 infectivity, diverse intracellular proteins interact with CA in a CypA-dependent manner particularly at the early stage of virus infection in target cells. For example, antiviral tripartite-containing motif 5α (TRIM5α) competes with CypA for binding to HIV-1 CA, whereas cleavage and polyadenylation-specific factor 6 (CPSF6), SUN2 (UNC84B) and nucleoporins are recruited to CA depending on the presence of CypA for viral fusion, uncoating or nuclear localization and even reverse transcription.50, 56, 67, 68, 69 Of these, functions of TRIM5α and CPSF6 in CA–CypA interactions have been most actively investigated to address questions about how they regulate HIV-1 infection (Fig. 2, right). A retrovirus resistance factor in humans, named restriction factor 1 (Ref1), has been identified as TRIM5α.70 It blocks HIV-1 infection by accelerating degradation of cytosolic CA.71, 72 As a counter molecule, CypA protects HIV-1 CA from this CA-specific restriction factor TRIM5α.68, 73 Meanwhile, among the essential factors for HIV-1 infection, CPSF6 is also complexed with the CA protein. CPSF6 traffics HIV-1 core on microtubules for viral uncoating and recruits it to SC35 nuclear speckles for reverse transcription.56, 69 Disruption of CypA–CA binding after treatment with CsA increases the CA–CPSF6 interaction and prominently guides capsid trafficking to microtubules in HeLa cells and primary peripheral blood mononuclear cells (PBMCs). These findings inform that CypA interacts with CA to prevent CPSF6–CA interaction and affects cytoplasmic trafficking and uncoating of the viral nucleocapsid at the post-entry step.69 However, there is little evidence explaining whether stimulation of this proviral role of CPSF6 by blocking CypA could be directly induced by CsA in HIV-1-infected cells.
HBV
HBV is a member of the Hepadnaviridae family and is a small enveloped virus with a partially double-stranded circular DNA genome. HBV has abundant hepatitis B surface antigen (HBsAg), which affects pathogenesis during viral infection. HBsAg is composed of large HBV (LHB), middle HBV (MHB) and small HBV (SHB) surface antigens.74 Induction of CypA is accompanied by increased secretion of SHB into the sera of HBV-expressing mice.75 The secretion of SHB is reduced by treatment with CypA inhibitors CRV431 and Debio-025.76, 77 CypA acts as a proviral factor for HBV infection during virus entry, assembly and release. Interestingly, CsA inhibits the entry of HBV by directly interacting with its entry receptor sodium taurocholate co-transporting polypeptide (NTCP) receptor, potentially independently of CypA.78, 79, 80
CypA as an antiviral host factor
In contrast to the proviral effects discussed above, CypA inhibits virus replication either by inducing the degradation of a viral protein that is crucial for nuclear export of the viral genome during the virus assembly step (e.g., in influenza virus-infected cells) or by stimulating cellular antiviral responses (e.g., in rotavirus-, influenza- or Sendai-virus-infected cells).81, 82 In these cases, CypA inhibitors should be applied more carefully, because suppression of CypA can result in minimal antiviral effect or even undesirable virus amplification.
Influenza virus
Influenza viruses are negative-sense single-stranded enveloped RNA viruses comprising eight segments, belonging to the Orthomyxoviridae family. They are among the major human respiratory pathogens and encode at least ten distinct viral proteins.83, 84 At the entry step of the virus life cycle, low endosomal pH not only stimulates membrane fusion, mediated by hemagglutinin 2 protein (HA2), but also opens the proton channel of matrix protein 2 (M2), driving cytoplasmic release of viral ribonucleoproteins (vRNP) from the inner shell composed of the viral matrix protein 1 (M1).85, 86 In the nucleus, where RNA-dependent RNA replication occurs, newly synthesized vRNP again interacts with M1, recruiting nonstructural protein 2 (NS2; also named nuclear export protein, NEP), which can bind to the nuclear export receptor: chromosomal maintenance 1 (CRM1) (Fig. 3 ).87, 88, 89, 90, 91 This series of interactions is essential for nuclear export of vRNPs as well as their migration to the plasma membrane for production of viral progeny.
Figure 3.

Antiviral roles of CypA in influenza virus infection. After receptor-mediated endocytosis, eight-segmented influenza vRNP complexes migrate into the nucleus for triggering RNA-dependent RNA replication. M1 plays a crucial part in nuclear export of newly synthesized vRNPs by recruiting NS2 (also named NEP), which has an ability to bind the nuclear export receptor CRM1. CypA blocks nuclear import of M1 and induces M1 degradation by recruiting AIP4 – an E3 ubiquitin ligase. Abbreviations: HA, hemagglutinin; NA, neuraminidase; PB1, polymerase basic protein 1; PB2, polymerase basic protein 2; PA, polymerase acidic protein; NP, nucleoprotein; M1, matrix protein 1; M2, matrix protein 2; NS2, nonstructural protein 2; CRM1, chromosomal maintenance 1; AIP4, atrophin-interacting protein 4; CypA, cyclophilin A.
As described for HIV-1 above, CypA is incorporated into influenza virus particles.92 Yeast two-hybrid assays and mammalian cell-based assays have shown that CypA directly interacts with the M1 protein, which negatively affects virus replication at two steps: the localization of M1 protein to the nucleus and M1-mediated nuclear export of vRNP complexes (Fig. 3).13 With regard to the latter mechanism, CypA accelerates M1 degradation by summoning the E3 ubiquitin ligase atrophin-interacting protein 4 (AIP4) to M1, thereby blocking nuclear escape of vRNP.81, 93 Crucially, the findings, showing increased influenza viral infectivity in the absence of CypA using a CypA-knockout cell line but strong disease resistance to viral infection in human CypA-expressing transgenic mice, prove that CypA is an antiviral factor restricting influenza viral infection in vitro and in vivo. 81, 94 Liu and colleagues suggested that CsA inhibits influenza virus replication with reduction of the viral protein level but failed to exhibit inhibition of influenza viral genome replication or transcription.95 Mysteriously, CsA impairs the nuclear export of viral mRNA in CypA-depleted cells, which was independent of calcineurin signaling, meanwhile it enhanced CypA–M1 interaction in CypA-expressing cells. The isomerase activity of CypA is not even necessary for influenza viral replication in cells.13 As they reviewed, this incomprehensible antiviral mechanism of CsA might be induced via complicated, CypA-dependent and -independent pathways.95 A limited number of reports proposed that CsA and its analogs block influenza viral infection, thus further accumulating evidence is required for ensuring feasibility of CypA as an anti-influenza viral target and for understanding its CypA-dependent or -independent modes of antiviral action.
Rotavirus
Human rotaviruses are members of the Reoviridae family and are non-enveloped viruses. Rotavirus genomes consist of 10–12 segmented, double-stranded DNA molecules that encode the λ, μ and σ proteins. Rotaviruses are gastrointestinal pathogens that cause acute diarrheal disease.96 CypA is induced in the early stage of rotavirus infection and inhibits rotavirus replication. Although the Nsp1 of rotavirus inhibits IRF3, IRF5, IRF7 and nuclear factor (NF)-κB to sequentially downregulate the type I IFN signaling pathway, the production of IFN-β is still induced after rotavirus infection via activation of CypA. Interestingly, this event occurs depending on c-JUN N-terminal kinase (JNK) activation rather than the PPIase activity of CypA.97 Consistent with this, CsA treatment marginally affected the growth of rotavirus.98 In addition, the elevated expression of CypA in enterocytes prevented acute dehydrating diarrhea, following rotavirus infection, in BALB/c mice.99 Furthermore, CypA is recruited to within rotavirus particles via its interaction with the structural protein VP2, which reduces rotavirus infectivity. It is unclear why the virus allows incorporation of the antiviral CypA protein into progeny virus particles at the assembly step.
CypA inhibitors
Among the CypA inhibitors, CsA and sanglifehrin A are naturally existing peptidic macromolecules and historically presentative ones. Both molecules are immunosuppressive and can inhibit PPIase activity of a class of cyclophilins.100, 101 These PPIase enzymes catalyze the interconversion of the cis–trans peptide bond of proline residues. Because the cis conformation of peptidyl–prolyl bonds is found in > 30% of newly synthesized proteins in cells and needs to be converted to the trans conformation for those proteins to have biological activity, it follows that cyclophilins are essential in the post-translational process.102 In this section, the chemical structures, antiviral efficacy and immunosuppressive effect of CypA inhibitors will be discussed.
CsA analogs
CsA, a commonly used immunosuppressant medication, binds to CypA and this complex then interacts with a third partner: calcineurin. Binding of CsA–CypA to calcineurin inhibits its calcium-dependent phosphatase activity, which plays a vital part in the nuclear translocation of NFAT.103 Natural CsA (structure 1,Fig. 4 ) is a cyclic peptide with multiple methylated amide bonds, of which molecular weight is > 1200 Da.104 The high molecular weight, cyclic structure and conformational flexibility of CsA contribute to its cellular permeability and oral bioavailability.105 The numbering pattern of CsA is shown in Fig. 4. The binding affinity of CsA for CypA mainly relates to the side chains P1, P2, P9 and P10.106 The rest of the CsA molecule has relatively little impact on CypA binding, based on the structural analysis of CsA–CypA co-crystals. The immunosuppressive effect of CsA is due to its ability to bind to calcineurin, mainly through the P4, P5 and P6 side chains of CsA (see Figure S1 in supplementary material online).107 Consequently, to generate a CsA derivative with more-potent CypA inhibition but a lesser immunosuppressive effect, a modified CsA has been designed with substitutions in the side chains P4, P5 and P6 and a change to the P3 side chain to increase its binding to CypA.
Figure 4.

Chemical structure of CsA (compound 1) and its derivatives as CypA inhibitors. The analogs, compounds 2 and 3 are modified at positions 3 (P3) and 6 (P6), respectively. The analogs 4 to 14 are modified at position 3 (P3) and/or 4 (P4) of CsA. Modification sites are highlighted in red.
In addition to its potential CypA inhibition activity and the related immunosuppressive effect, CsA was also found to remarkably inhibit several endogenous transporters, including organic anion transporting receptors (OATP1B1 and OATP1B3),108 bile salt export pump, multidrug resistance protein 1 (MDR1; also named ABCB1 or P-gp)109 and MDR2.110 From 1984 to 1997, CsA derivatives were chemically synthesized and evaluated for their ability to inhibit CypA, and were subjected to detailed SAR analyses by several research groups. As shown in Fig. 4, introducing a benzyl group as a P3 side chain into CsA to obtain derivative 2 does not affect the ability of CsA to inhibit CypA but dramatically decreases its immunosuppressive effect.111 Introducing a bulky group into P3 resulted in the first identified, non-immunosuppressive CypA inhibitor. By switching an isopropyl to a methyl group as the P6 side chain the resulting CsA derivative (3) has ∼ 50% of the CypA binding affinity of natural CsA. Surprisingly, it also reduces immunosuppression to 0.4% of that of CsA.112
Replacement of the P4 side chain in 1 to produce derivatives 4 or 5 can preserve or improve CypA binding while sharply reducing its activity in immunosuppression assays. Based on these results, the authors concluded that calcineurin has a unique pocket for P4. This hypothesis was later confirmed by crystallization experiments.113 Derivatives 6 and 7 (NIM811) with changes in the P4 side chain inhibited replication of HIV in cellular assays while significantly impairing the immunosuppressive effect. Intriguingly, derivative 6 was 4-times more effective than derivative 7 in binding assays but, in the anti-HIV cellular assay, 6 was less potent than 7.114, 115 In these preliminary tests, derivative 7 exhibited a stronger affinity for CypA than natural CsA.116 This inhibitor has been tested in animal and cell models of numerous diseases, as well as in clinical trials as an HCV treatment.117 Following the finding that it inhibits HCV replication, further research involving the derivative 7 was carried out to examine whether it could be employed as a therapy for HCV infection.118 Non-immunosuppressive CypA inhibitors have substantially high genetic barriers to the evolution of viral resistance, according to this research, which is also predicted to be the case for host-targeted therapies.
CsA derivative 8 (Debio-025 or alisporivir) has a potency of 5–10-times that of CsA in a cellular subgenomic HCV replication system.119 This inhibitor was more effective than derivative 7 in cell-culture-based antiviral assays.120 The antiviral activity of derivative 8 was higher than that of earlier CsA-like inhibitors, which might be attributable to its altered P3 groups, conferring higher affinity for CypA. The progression of HCV resistance to derivative 8 is a protracted one. Cells swiftly remove the replicon when this molecule is coupled with pegylated (peg)IFN-α and ribavirin.120 Moreover, this CypA inhibitor has been reported to be very effective for the treatment of genotype 2 and 3 infections of HCV.121 Originally, the modified CsA derivative 8 was developed as a therapy against HIV infection. The anti-HIV action of derivative 8 stems from its ability to prevent CypA from attaching to the HIV-p24 Gag protein.122 Once-daily treatment resulted in a moderate decrease in HIV-1 plasma RNA in an initial 10-day investigation in infected individuals.
Derivative 9 was found in a pharmaceutical investigation aimed at developing a novel type of CsA inhibitor for the treatment of HIV infections. It improved anti-HIV efficiency while simultaneously lowering in vitro immunosuppressive activity.123 This chemical inhibited CypA isomerase activity more effectively than native CsA 1.124 The decrease in HCV replication in vitro was time-dependent, with total replication suppression not occurring until 72 h.125 In comparison with other CypA inhibitors, derivative 9 was a less effective transporter protein inhibitor. P-glycoprotein efflux activity was totally inhibited at 15 µM, although multidrug resistance 2 (MRP2)-mediated transport was only moderately affected 150 µM.
A series of novel CsA analogs with a variety of P3 and P4 residues have been further synthesized and analyzed. The most representative compound is derivative 10, an analog with a morpholine moiety at the P4 residue. Surface plasmon resonance (SPR) assays revealed that it had a binding affinity of 1.1 nM. It inhibited HCV replication with an EC50 of 36 nM and was eightfold less effective in inhibiting the OATP1B1 transporter.126
EDP-546 (a structurally unknown CypA inhibitor) is similar to derivative 11. In cellular passaging assays with EDP-546 using HCV genotype 1a and 1b replicon systems, selection for the D320E mutation within the NS5A protein resulted in moderately (two-to-fivefold) higher resistance. The antiviral activity of combinations of HCV protease inhibitors and HCV NS5A inhibitors was synergistic, and co-incubation of EDP-546 with these antiviral agents reduced viral resistance to these agents. The cellular activity of the inhibitor was unaffected by the addition of 40% human serum to the growth medium, and EDP-546 was more stable in human microsomes than derivative 8. EDP-546 exhibited limited blood clearance and substantial distribution in the liver in preclinical pharmacokinetic investigations. In aqueous buffer solutions, good solubility was reported, especially at pH values less than 7. Furthermore, EDP-546 is a significantly weaker inhibitor of bilirubin and drug transporters MRP2 (about 100-fold less) and OATP1B1 (roughly threefold less) than derivative 8.127
Derivative 12, an example as a 3- and 4-modified CsA analog, showed potent anti-HCV activity in a cellular replicon assay. Derivative 13 differs from derivative 12 in having a hydroxyl group at the P4 chain and differs from CsA in having a bulky thioether group at P3 (1). Both derivatives exhibited similar CypA inhibition in an in vitro enzyme assay but lower inhibition of immune responses, with 50-fold lower immunosuppression than that of CsA in a mixed lymphocyte reaction.128
Derivative 14 (STG-175) is a CsA analog with a thioether and a hydroxyl butyl group in the P3 side chain. It inhibits CypA isomerase activity with an IC50 of 0.6 nM. Its immunosuppressive effect was ∼ 1000-fold less than that of CsA in an IL-2 release assay with the Jurkat cell line. The anti-HCV activity EC50 values of derivative 14 on different HCV replicon genotypes, including GT1a, GT1b, GT2a, GT3a and GT4a, ranged from 11.5 nM to 38.9 nM. It could greatly enforce the HCV resistance barrier when combined with direct-acting antivirals without any cross-resistance.129
A series of modifications at P1 has been challenged (Fig. 5 ). Derivative 15 differs from CsA in having an extended P1 and an additional methyl group on P3. It was 15-fold more potent in inhibiting CypA PPIase activity than CsA but dramatically decreased immunosuppression.130
Figure 5.

Chemical structure of CsA derivatives, including compounds 15 to 20. In compounds 15 and 16 P1 and P3 are modified; in compounds 17 and 18 P1 is modified; in compound 19 P3 and P5 are modified; and in compound 20 P8 is modified compared with CsA (1) (Fig. 4). Modification sites are highlighted in red.
Derivative 16 (CPI-431–32 or CRV-431) efficiently inhibited HIV-1 and HCV growth individually and during co-infection. Derivative 16 inhibited CypA–HCV-NS5A and CypA–HIV-1-CA interactions with IC50 values of 0.18 µM and 0.21 µM, respectively. In addition, it inhibited CypA isomerase activity with an IC50 of 1.8 nM, indicating that it was almost ninefold more potent than CsA. Derivative 16 suppressed HIV-1 reverse transcription and nuclear import.131, 132
Derivative 17, with a benzimidazole group in P1, was developed as an extracellular CypA inhibitor to suppress leukocyte trafficking. The binding affinity of derivative 17 was similar to that of CsA in a PPIase binding assay. Because cellular CypA is crucial, analysis using fluorescently labeled CsA derivatives showed that derivative 15 was at least 50-fold less permeable than CsA. Despite its reduced cell permeability, derivative 15 hindered leukocyte migration to CypA. In a mouse model of allergic contact hypersensitivity, this inhibitor also decreased leukocyte activation.115
A recent study showed that a P5-modified CsA analog acts as an NTCP inhibitor and reduces bile acid uptake. The markedly potent and specific inhibitor, derivative 18, exhibited broad antiviral activity against genomic HBV and hepatitis D virus infection in vitro with excellent pharmacokinetics and oral bioavailability. However, it had lower immunosuppressive potency than CsA.133
Through another approach, the P3- and P5-modified CsA derivative, 19, was identified as a CypA and CypD inhibitor with a dissociation constant (K d) of 60 nM and an EC50 < 200 nM in an HCV replicon assay. Derivative 19 was ∼ 40-fold more potent in inhibiting IL-2 release in a stimulated T cell assay than CsA.134 Changing the side chain at the P8 position of CsA, resulting in derivative 20, improved its anti-HCV activity but reduced the immunosuppressive activity.135
Sanglifehrin A derivatives
Sanglifehrin A (compound 21; Fig. 6 ) is the most extensively studied natural product of its family and binds to CypA 60-times more strongly than CsA. The immunosuppressive effect of sanglifehrin A is not attributable to a reduction in its calcium-regulated phosphatase activity.101 The 3D co-crystalized structure with CypA revealed detailed information regarding this interaction (see Figure S2 in supplementary material online).136 Modifications to the spatial domain substitution pattern (regions C13–C26 on sanglifehrin A) consistently reduced potency. For example, in a procycling binding experiment, synthetic macrolide derivative 23 was ∼ 600-times less effective than derivative 22.101
Figure 6.

Chemical structure of sanglifehrin A (21) and its simplified derivatives, compounds 22 to 28, as CypA inhibitors. Modification sites are highlighted in different colors depending on their positions.
As shown in Fig. 6, the degradation product of sanglifehrin A, derivative 24 (AAE931), is a potent anti-HCV inhibitor with an EC50 of 130 nM in a replicon assay. The bioengineered product of sanglifehrin A, derivative 25 (NVP018), is a potential orally available CypA inhibitor for the treatment of HCV, HBV and HIV-1 infections. Derivative 26 (BC544) was developed through a combination of bioengineering and semi-synthesis. BC544 inhibits PPIase with an IC50 of 2.7 nM and the NS5A–CypA interaction with an IC50 of 0.36 µM, whereas it suppresses replication and replicons of the HCV genotype 1b in Huh5-2 cells with an EC50 of 125 nM, without exhibiting cytotoxicity at the maximum tested concentration of 100 µM.137, 138
With a pilot screen of a chemical library of ∼ 19,000 chemical compounds, hit compounds were not identified in a time-resolved fluorescence resonance energy transfer (FRET)-based binding assay.137 Chemical synthesis of sanglifehrin A simplified its structure and resulted in the synthesis of derivative 27 (corresponding to compound 3 from the original paper). Retention of the diene moiety and the reduction of the complexity of the molecule resulted in a CypA binding affinity (K d) of 25 nM and a K i value of 16 nM in a PPIase functional assay. It inhibited HCV genotype 1b replication with an EC50 of 600 nM.139 Switching the diene for a phenyl moiety led to a novel skeleton of sanglifehrin A, derivative 28, which had almost 17-fold more potency than 27 in inhibiting HCV genotype 1b replication and replicons. Determining the X-ray crystal structure of these new simplified sanglifehrin-A-based CypA inhibitors would be of significant value for the SAR analyses of related inhibitors and development of novel CypA inhibitors. The most representative analog was derivative 27 which possessed a quinolone unit. This derivative has a very high oral absorption rate in rats and dogs and is less effective in generating drug–drug interactions. The strong binding affinity (K d = 5 nM) and the availability of a systematic synthetic route establishes it as a candidate for HCV treatment.137
Micromolecular CypA inhibitors
Owing to the difficulty of synthesizing large molecules, and the possible side effects caused by their peptide skeletons, the synthesis of micromolecular CypA inhibitors has become the focus of extensive research efforts during the past two decades. Furthermore, the development of computer-aided drug design and virtual screening has nurtured robust growth in this area. Starting with a linear tetrapeptide: Suc-Ala-Gly-Pro-Phe-pNA, the macrocyclization of the template has led to the generation of a novel class of CypA inhibitors, such as derivative 29, that have a moderate K d of 10.5 µM for CypA, good aqueous solubility and a low rate of hepatic clearance (Table 1 ).140 This study provided the template for the subsequent development of diverse micromolecular CypA inhibitors.
Table 1.
Micromolecular CypA inhibitors.
| Compound no. | Chemical formula | Background and biological properties | Publication date |
|---|---|---|---|
| 29 | 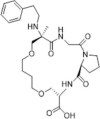 |
SPR analysis binding affinity, KD 7.09 µM; PPIase inhibition, IC50 = 0.41 µM; inhibition on the proliferation of spleen cells, IC50 = 4.32 µM; no obvious effect on cell viability at 10 µM concentration | 2016140 |
| 30 | 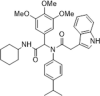 |
Identified by virtual screening and chemical modification, CC50 > 100 µM, EC50 = 5.2 µM; no effect on immune system; reducing the expression of NS5A and NS5B without cytotoxicity; Kd = 570 nM; synergistic effect when combined with IFN-α, ribavirin or telaprevir for HCV infection in mice | 2015141 |
| 31 | 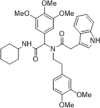 |
Anti-HCV activity, EC50 = 5.3 µM and CC50 > 100 µM; SPR analysis, KD = 3.66 µM | 2020143 |
| 32 | 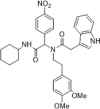 |
Anti-HCV activity, EC50 = 4.1 µM; inhibiting the expression of HCV core protein, KD = 4.6 µM determined by SPR | 2021142 |
| 33 | 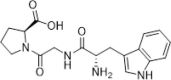 |
Identified by virtual screening focusing on peptide library; inhibition on the PPIase activity of CypA, IC50 = 33.11 nM; SPR assay, KD = 3.41 µM; exhibiting 75.5% inhibition of HIV-1 IIIB infection at 1 mM concentration | 2011144 |
| 34 | 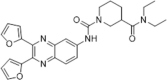 |
SPR assay binding affinity, KD = 7.09 µM; PPIase inhibition, IC50 = 0.41 µM; inhibition on the proliferation of spleen cells, IC50 = 4.32 µM; no obvious effect on cell viability at 10 µM concentration | 2006145 |
| 35 | 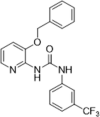 |
Virtual screening and pharmacophore-based drug design; PPIase isomerase inhibition, IC50 = 303 nM; inhibition on HIV-1 replication cycle on immortalized cells or human peripheral blood mononuclear cells | 2006146 |
| 36 | 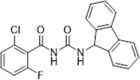 |
De novo drug design using the crystal structure of CypA with sanglifehrin A macrolide and pharmacophore-based design; CypA inhibition, IC50 = 1.52 nM | 2009147 |
| 37 | 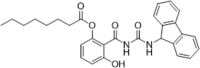 |
Chemical modification of existing hit compounds; inhibition of HCV activity replicon assay (isolate H77, genotype 1a), EC50 = 0.85 µM; inhibition JFH1 (genotype 2a), EC50 = 0.45 µM; inhibition of Con1 (genotype 1b), EC50 = 0.81 µM; CC50 > 16 µM; stable in pH 7.4 | 2015148 |
| 38 | 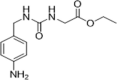 |
Fragment-based drug design using NMR and X-ray crystallography; CypA inhibition, IC50 = 13.1 µM | 2016149 |
| 39 | 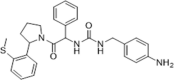 |
Inhibition of HCV genotype 1b replicon in Huh7 cells, EC50 = 0.4 µM, CC50 > 100 µM; inhibition of HCoV-229E in MRC5 cells, EC50 = 44.7 µM, CC50 > 100 µM; no calcineurin inhibition when treated with CypA; no effect on IL-2 production | 2016149 |
| 40 | 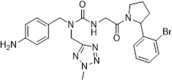 |
Through molecular dynamics simulation on CypA; SPR analysis, Kd = 0.6 µM; X-ray structure determined; dose-dependent growth inhibition at low micromolar concentrations (ranging from 10 nM to 10 µM) resulting in GI50 values twofold better than those for CsA; less toxic than CsA in MDA-MB-231 cells | 2019150 |
| 41 | 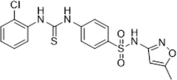 |
Identified through lead modification; PPIase activity assay, IC50 = 0.65 µM; dual inhibitor against HIV-1 capsid and human CypA | 2009151 |
| 42 | 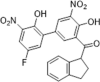 |
CypA-specific inhibitor, Ki = 0.52 µM; isothermal titration calorimetry, KD = 22.6 µM, inhibition on the chemotactic activity of CypA but not CypB | 2009152, 2011153 |
| 43 | 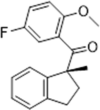 |
CypA-specific inhibitor, Ki = 7.5 µM | 2009152, 2011153 |
| 44 | 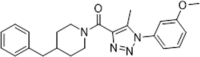 |
Identified by virtual screening; >30% inhibition of HIV-1 replication in MT4 and U87 cells at the concentration of 10 µM | 2013154 |
| 45 | 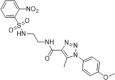 |
Identified by virtual screening; <30% inhibition of HIV-1 replication in MT4 and U87 cells at the concentration of 9 µM | 2013154 |
| 46 | 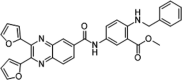 |
Identified by virtual screening; SPR analysis, Kd = 8.19 µM; PPIase inhibition, IC50 = 0.25 µM | 2006155 |
| 47 | 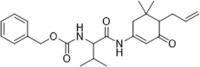 |
Identified by virtual screening; Kd = 22 µM; inhibition of fecundity and growth of Caenorhabditis elegans, IC50 = 190 µM | 2007156 |
| 48 | 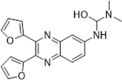 |
Identified by virtual screening; SPR assay, Kd = 0.218 µM; PPIase inhibition, IC50 = 3.47 µM; inhibition of HIV-1 replication by 65.2% at 10 µM; CC50 > 69.73 µM | 2007157 |
| 49 | 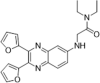 |
Identified by virtual screening; SPR assay, Kd = 5.12 µM; PPIase activity on human CypA; IC50 = 0.312 µM | 2006158 |
| 50 | 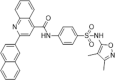 |
Identified by virtual screening; SPR assay, Kd = 10 µM; PPIase assay, IC50 = 2.84 µM; inhibition of the proliferation of mouse spleen cells at 10 µM | 2006159 |
| 51 | 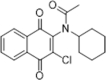 |
Identified by HTS; CypA PPIase inhibition, Kd = 2 µM determined by SUPREX (stability of unpurified proteins from rates of H/D exchange) analysis | 2010160 |
| 52 | 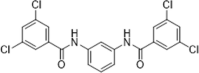 |
Identified by pharmacophore-based virtual screening; IC50 = 930 nM, promoting neurite outgrowth with EC50 around 100 to 1000 nM | 2003161 |
| 53 | 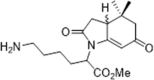 |
Native electron ionization mass spectrometry, Kd = 72.6 µM; direct stoichiometric analysis, Kd = 15.9 µM; PPIase assay, Ki = 6.8 µM; inhibition of normal growth and reproduction of C. elegans | 2011162 |
By structure-based screening a chemical library of > 200,000 compounds to identify hits with potential binding affinity for the CsA–CypA complex, compound 30 with a bisamide skeleton was identified as a promising hit compound. Compared with the synthetic schemes for CsA and sanglifehrin A, this bisamide was synthesized easily through a one-step Ugi reaction, which significantly reduced the difficulty in the preparation of macromolecular CypA inhibitors. Without causing cytotoxicity or immunosuppression, compound 30, when combined with IFN-α, ribavirin and telaprevir, showed excellent synergistic properties for the treatment of HCV infections.141 It had little or no immunosuppressive effect but, notably, inhibited HCV replication in vitro and in vivo. Its efficacy for the treatment of HCV infection in mice also increased synergistically when it was co-administrated with other direct-acting antivirals.141, 142, 143 SAR analysis of bisamides for antiviral activity has been extensively discussed in these reports. The more potent, optimized bisamide analogs as CypA inhibitors are compound 31 and 32, which inhibited HCV replicons with an EC50 of 5.3 µM and 4.1 µM, respectively, both with no cytotoxicity at a maximum concentration of 100 µM.
By dissection of the binding pocket of CsA on CypA, Pang and colleagues proposed that a peptide with three or four residues would be an effective CypA inhibitor. Virtual screening of a peptide library was utilized to identify compound 33 (WGP), which inhibited the PPIase activity of CypA with an EC50 of 33.11 nM. A SPR assay indicated that compound 33 has a K d value of 3.41 µM and inhibited HIV-1 replication by 75.5% at a concentration of 1 mM.144
One novel quinoxaline derivative, compound 34 (DC838), was designed and synthesized as a CypA inhibitor with a K d value of 7.1 µM for CypA, as measured by the SPR assay. Moreover, it suppressed PPIase activity with an IC50 of 0.41 µM and inhibited the replication of the HIV-1 cycle.145
Compound 35 (corresponding to compound 2 in the original paper), a CypA inhibitor with a molecular weight of < 400 Da, was identified using a computer-aided drug design approach with virtual screening and pharmacophore construction. This compound exhibited an IC50 value of ∼ 0.3 µM and substantially inhibited HIV infectivity.146
Two papers describing the identification of a small molecular CypA inhibitor were published by the same research group in China in 2009 and 2015.147, 148 The mother skeleton of the derivative series was carbonyl urea, which was discovered by combining virtual screening and pharmacophore-based drug design. Further lead optimization of these carbonyl urea CypA inhibitors was undertaken. Compound 36 (corresponding to analog 3i in the original paper) has an IC50 value of 1.52 nM. Compound 37 (corresponding to compound 25 in the original paper) inhibited different HCV genotypes with IC50 values ranging from 0.19 to 0.85 nM, including H77 (genotype 1a), JFH (genotype 2a) and Con1 (genotype 1b) in replicon assays and virus assays without causing toxicity at a maximum concentration of 16 µM.
Fragment-based drug discovery (FBDD) is based on the identification of small organic compounds (or fragments) that are then extended or combined to yield therapeutically beneficial drug lead compounds. A novel CypA inhibitor, which was structurally different from CsA and sanglifehrin A, was identified by using a combination of the FBDD method, NMR and X-ray crystallography. Compound 38 (hit compound 22 in the original paper) showed very potent inhibition of CypA with an IC50 of 13.1 µM. Structure-based lead optimization of this hit resulted in compound 39 (analog 33 in the original paper) being selected as a lead compound. It inhibited CypA, CypB and CypD more potently than compound 38 and showed broad-spectrum antiviral activities against HCV genotype 1b and human coronavirus HCoV-229E, without causing cytotoxicity or immunosuppressive effects.149
The CsA–CypA co-crystal X-ray structure identified Pro, Abu and three o’clock pockets, indicating that it could be used to develop a tri-vector CypA inhibitor that targets three pockets simultaneously. Molecular dynamics simulations designed for chemical optimization led to the design and synthesis of a series of linear CypA inhibitors with urea as the backbone. The most promising compound was 40 which bound to CypA, B and D with binding affinities of 0.07 to 0.6 µM. Furthermore, improved inhibition and reduced cytotoxicity were observed in cell assays, indicating that the FEB-based generation of this novel CypA inhibitor could provide a new approach for other structure-based drug designs.150
A CypA and HIV-1 CA dual inhibitor with thiourea was first described in 2009. The representative inhibitor, compound 41 (named D23 in the original paper), inhibited PPIase activity with an IC50 of 0.65 µM.151 The activities of CypA inhibitors with the aryl 1-indanylketone structure (compounds 42 and 43) have been discussed in many previous reports.152, 153
From 2003 to 2013, many reports described the use of virtual screening of different compound databases to identify potential CypA inhibitors. This resulted in the identification of compounds 44 to 50), the binding potentials of which were analyzed using SPR assays.154, 155, 156, 157, 158, 159 The promising CypA inhibitor was compound 48 (named FD-12), which had a quinoxaline-based backbone and a K d value of 0.218 µM. In addition, compound 51, a CypA inhibitor with a naphthalene-1,4-dione backbone and a K d value of 2 µM, determined by HTS, was described in a paper published in 2010.160
Similarly, CypA inhibitors with a N,N'-(1,3-phenylene)dibenzamide structure, such as compound 52, had IC50 values of ∼ 930 nM. Because such compounds can protect motor neurons and promote neurite growth at a concentration of 10 µM they have the potential to treat CNS diseases.161 Native-ESI-MS for high-throughput sampling and direct stoichiometric analysis was described by Dunsmore et al. and identified potent CypA inhibitors (including compound 53). Such an approach might provide an alternative route for the development of other anti-HCV agents.162
Concluding remarks
CypA, a ubiquitous, cellular protein with PPIase activity, is involved in the replication of chronic and acute viruses, such as HCV, CoV, HIV, HBV and influenza virus, with multiple roles in regulating uncoating, intracellular trafficking, viral RNA replication, and the stability of viral proteins and even in affecting host innate immune responses. In this review, we have discussed the opposing functions of CypA (i.e., its proviral and antiviral roles) and its modes of action in different viral infections. Clinical trials to evaluate antiviral potency and safety of CsA in humans have been challenged mainly against HCV and HIV (at https://www.clinicaltrials.gov). In 2008, a clinical trial to examine CsA in HCV clearance following liver transplantation finished (Phase IV; NCT00821587). However, the immunosuppressive property of CsA still raises concerns for its application as an antiviral.163 Long-term follow-up studies with a single treatment of non-immunosuppressive Debio-025 (Phase III; NCT02753699) or its combination with approved antivirals such as pegIFN-α2a and ribavirin (Phase II; NCT00537404) have completed against HCV infections. In parallel, Phase II studies to assess antiviral efficacy of NIM811 (NCT00983060) and SCY-6235 (NCT01265511) have been finished. To date, there is little progress report on clinical studies with Debio-25 or any other non-immunosuppressive CypA inhibitors against HIV.
The ongoing SARS-CoV-2 pandemic has generated significant interest in the feasibility of employing CypA inhibitors to combat infections caused by this highly variable RNA virus. The current urgent need for clinically efficacious antivirals has also focused attention on drug-repurposing strategies. To our knowledge, there are at least ten clinical trials of CsA, either alone or in combination with other drugs (NCT04412785, NCT04392531, NCT04540926, NCT04492891, NCT04979884, NCT04451239 and NCT04488081), and of Debio-025 (NCT04608214) and NIM-811 (NCT00983060) for the treatment of SARS-CoV-2 infection, as well as SARS-CoV-2-mediated pneumonia (at https://www.clinicaltrials.gov). In the case of SARS-CoV-2, immunosuppressive CypA inhibitors, such as CsA, could be employed additively to control severe hyperimmune responses acutely caused by this virus. By contrast, depending on host immune status, CypA inhibitors with no or weaker immunosuppression might be safer. Most of all, responding to these different clinical cases with the same virus, it is necessary to build a diverse antiviral candidate pool. For long-term development, the structural simplification is important not only for rendering them druggable by satisfying antiviral efficacy and pharmacokinetic parameters, such as immunotoxicity, oral bioavailability, membrane permeability, solubility and stability, but also for facilitating large-scale production. In line with this approach, minimizing the size of sanglifehrin-A-based derivatives has provided an important direction for the development of CypA inhibitors in recent years. Moreover, several small molecules, with excellent CypA inhibition in the nM range and reduced immunosuppression, have been discovered by HTS or computer-aided drug design methods. Basically, with understanding of biological roles of CypA, newly discovered CypA inhibitors should be reassessed in terms of their antiviral selectivity and broad genotypic or serotypic coverage.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (NRF-2018M3A9H4089601 to M.K. and NRF- 2018M3A9H4089602 to W.-J.C.) and by an intramural fund from KRICT (grant number SI2232-20 to M.K.).
Biographies

Jinhe Han received his PhD in pharmacy from Chonnam National University in 2020, supervised by Prof. Won-Jea Cho. He is now working as a post-doc on a fellowship in Prof. Cho’s lab and his current research interest focuses on the design, synthesis and evaluation of small organic compounds as potential antivirals and anti-neurodegenerative agents.

Won-Jea Cho has been a professor at Chonnam National University College of Pharmacy since 1995. After receiving his PhD from the Faculty of Pharmacy at Kanazawa University in Japan, he worked as a postdoctoral researcher at the University of Kansas and conducted research at Chugai Pharmaceutical, Japan. His research field is computer-aided drug design and synthesis of bioactive compounds in the field of anticancer, antiviral and dementia treatments.

Meehyein Kim received her PhD in chemistry in 2000 from Korea Advanced Institute of Science and Technology (KAIST). Since 2010, she has worked at the antiviral research group as a principal researcher in Korea Research Institute of Chemical Technology (KRICT). M. Kim has interests in discovery of antiviral compounds against influenza virus and coronavirus through investigation of their mode-of-action.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.drudis.2022.05.016.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 2.Breckenridge A., Jacob R. Overcoming the legal and regulatory barriers to drug repurposing. Nat Rev Drug Discov. 2019;18:1–2. doi: 10.1038/nrd.2018.92. [DOI] [PubMed] [Google Scholar]
- 3.Sirota M., Dudley J.T., Kim J., Chiang A.P., Morgan A.A., Sweet-Cordero A., et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3:96ra77 doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox R.M., Wolf J.D., Plemper R.K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat Microbiol. 2021;6:11–18. doi: 10.1038/s41564-020-00835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toots M., Yoon J.J., Cox R.M., Hart M., Sticher Z.M., Makhsous N., et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aax5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer G., Wittmann-Liebold B., Lang K., Kiefhaber T., Schmid F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 7.Bell R.D., Winkler E.A., Singh I., Sagare A.P., Deane R., Wu Z., et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carbajo-Lozoya J., Ma-Lauer Y., Malešević M., Theuerkorn M., Kahlert V., Prell E., et al. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res. 2014;184:44–53. doi: 10.1016/j.virusres.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davra V., Saleh T., Geng K., Kimani S., Mehta D., Kasikara C., et al. Cyclophilin A Inhibitor Debio-025 Targets Crk, Reduces Metastasis, and Induces Tumor Immunogenicity in Breast Cancer. Mol Cancer Res. 2020;18:1189–1201. doi: 10.1158/1541-7786.MCR-19-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorfman T., Weimann A., Borsetti A., Walsh C.T., Göttlinger H.G. Active-site residues of cyclophilin A are crucial for its incorporation into human immunodeficiency virus type 1 virions. J Virol. 1997;71:7110–7113. doi: 10.1128/jvi.71.9.7110-7113.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamble T.R., Vajdos F.F., Yoo S., Worthylake D.K., Houseweart M., Sundquist W.I., et al. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 12.Kaul A., Stauffer S., Berger C., Pertel T., Schmitt J., Kallis S., et al. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Sun L., Yu M., Wang Z., Xu C., Xue Q., et al. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol. 2009;11:730–741. doi: 10.1111/j.1462-5822.2009.01286.x. [DOI] [PubMed] [Google Scholar]
- 14.Pfefferle S., Schöpf J., Kögl M., Friedel C.C., Müller M.A., Carbajo-Lozoya J., et al. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vajdos F.F., Yoo S., Houseweart M., Sundquist W.I., Hill C.P. Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci. 1997;6:2297–2307. doi: 10.1002/pro.5560061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen S.Y., Wood C., Amin A.N., Papadimitriou J.C., Weir M.R., Coughlin T.R. Cyclosporine nephrotoxicity and dermal vascular alterations in renal transplants. Transplant Proc. 1989;21:1508–1510. [PubMed] [Google Scholar]
- 17.Iguro T., Okazaki H., Sato T., Jimbo M., Oguma S. The effect of donor age and sex on cyclosporine associated nephrotoxicity. Transplant Proc. 1989;21:1554–1555. [PubMed] [Google Scholar]
- 18.Burdick J.F., Colombani P.M., Pitt H.A., Perler B.A., Merritt W.T., Crandall B.C., et al. Overcoming early cyclosporine nephrotoxicity after liver transplantation. Transplant Proc. 1989;21:2236–2237. [PubMed] [Google Scholar]
- 19.Lindenbach B.D., Rice C.M. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 20.Heim M.H., Thimme R. Innate and adaptive immune responses in HCV infections. J Hepatol. 2014;61:S14–S25. doi: 10.1016/j.jhep.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 21.Paul D., Madan V., Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe. 2014;16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F., Robotham J.M., Nelson H.B., Irsigler A., Kenworthy R., Tang H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J Virol. 2008;82:5269–5278. doi: 10.1128/JVI.02614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watashi K., Hijikata M., Hosaka M., Yamaji M., Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 24.Chatterji U., Bobardt M., Selvarajah S., Yang F., Tang H., Sakamoto N., et al. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem. 2009;284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciesek S., Steinmann E., Wedemeyer H., Manns M.P., Neyts J., Tautz N., et al. Cyclosporine A inhibits hepatitis C virus nonstructural protein 2 through cyclophilin A. Hepatology. 2009;50:1638–1645. doi: 10.1002/hep.23281. [DOI] [PubMed] [Google Scholar]
- 26.Colpitts C.C., Ridewood S., Schneiderman B., Warne J., Tabata K., Ng C.F., et al. Hepatitis C virus exploits cyclophilin A to evade PKR. Elife. 2020;9 doi: 10.7554/eLife.52237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chatterji U., Lim P., Bobardt M.D., Wieland S., Cordek D.G., Vuagniaux G., et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol. 2010;53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorner M., Horwitz J.A., Donovan B.M., Labitt R.N., Budell W.C., Friling T., et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neufeldt C.J., Joyce M.A., Van Buuren N., Levin A., Kirkegaard K., Gale M., Jr, et al. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romero-Brey I., Merz A., Chiramel A., Lee J.Y., Chlanda P., Haselman U., et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterji U., Bobardt M., Tai A., Wood M., Gallay P.A. Cyclophilin and NS5A inhibitors, but not other anti-hepatitis C virus (HCV) agents, preclude HCV-mediated formation of double-membrane-vesicle viral factories. Antimicrob Agents Chemother. 2015;59:2496–2507. doi: 10.1128/AAC.04958-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gale M.J., Jr., Korth M.J., Tang N.M., Tan S.L., Hopkins D.A., Dever T.E., et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 33.Pflugheber J., Fredericksen B., Sumpter R., Jr., Wang C., Ware F., Sodora D.L., et al. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc Natl Acad Sci U S A. 2002;99:4650–4655. doi: 10.1073/pnas.062055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe T., Minami N., Bawono R.G., Matsui C., Deng L., Fukuhara T., et al. ISGylation of Hepatitis C Virus NS5A Protein Promotes Viral RNA Replication via Recruitment of Cyclophilin A. J Virol. 2020;94 doi: 10.1128/JVI.00532-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Albers M.W., Wandless T.J., Luan S., Alberg D.G., Belshaw P.J., et al. Inhibition of T cell signaling by immunophilin-ligand complexes correlates with loss of calcineurin phosphatase activity. Biochemistry. 1992;31:3896–3901. doi: 10.1021/bi00131a002. [DOI] [PubMed] [Google Scholar]
- 36.Fruman D.A., Klee C.B., Bierer B.E., Burakoff S.J. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flisiak R., Horban A., Gallay P., Bobardt M., Selvarajah S., Wiercinska-Drapalo A., et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- 38.Hopkins S., Bobardt M., Chatterji U., Garcia-Rivera J.A., Lim P., Gallay P.A. The cyclophilin inhibitor SCY-635 disrupts hepatitis C virus NS5A-cyclophilin A complexes. Antimicrob Agents Chemother. 2012;56:3888–3897. doi: 10.1128/AAC.00693-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zulian A., Rizzo E., Schiavone M., Palma E., Tagliavini F., Blaauw B., et al. NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum Mol Genet. 2014;23:5353–5363. doi: 10.1093/hmg/ddu254. [DOI] [PubMed] [Google Scholar]
- 40.Tiepolo T., Angelin A., Palma E., Sabatelli P., Merlini L., Nicolosi L., et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1-/- myopathic mice. Br J Pharmacol. 2009;157:1045–1052. doi: 10.1111/j.1476-5381.2009.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 42.Sauerhering L., Kupke A., Meier L., Dietzel E., Hoppe J., Gruber A.D., et al. Cyclophilin inhibitors restrict Middle East respiratory syndrome coronavirus via interferon-λ in vitro and in mice. Eur Respir J. 2020;56 doi: 10.1183/13993003.01826-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Brunn A., Ciesek S., von Brunn B., Carbajo-Lozoya J. Genetic deficiency and polymorphisms of cyclophilin A reveal its essential role for Human Coronavirus 229E replication. Curr Opin Virol. 2015;14:56–61. doi: 10.1016/j.coviro.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dittmar M., Lee J.S., Whig K., Segrist E., Li M., Kamalia B., et al. Drug repurposing screens reveal cell-type-specific entry pathways and FDA-approved drugs active against SARS-Cov-2. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.108959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Softic L., Brillet R., Berry F., Ahnou N., Nevers Q., Morin-Dewaele M., et al. Inhibition of SARS-CoV-2 Infection by the Cyclophilin Inhibitor Alisporivir (Debio 025) Antimicrob Agents Chemother. 2020;64:e00876–e920. doi: 10.1128/AAC.00876-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seizer P., Schonberger T., Schott M., Lang M.R., Langer H.F., Bigalke B., et al. EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis. 2010;209:51–57. doi: 10.1016/j.atherosclerosis.2009.08.029. [DOI] [PubMed] [Google Scholar]
- 47.Wang K., Chen W., Zhang Z., Deng Y., Lian J.Q., Du P., et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5:283. doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geng J., Chen L., Yuan Y., Wang K., Wang Y., Qin C., et al. CD147 antibody specifically and effectively inhibits infection and cytokine storm of SARS-CoV-2 and its variants delta, alpha, beta, and gamma. Signal Transduct Target Ther. 2021;6:347. doi: 10.1038/s41392-021-00760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su H., Wan C., Wang Z.D., Gao Y., Li Y.C., Tang F., et al. Expression of CD147 and Cyclophilin A in Kidneys of Patients with COVID-19. Clin J Am Soc Nephrol. 2021;16:618–619. doi: 10.2215/CJN.09440620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lahaye X., Satoh T., Gentili M., Cerboni S., Silvin A., Conrad C., et al. Nuclear Envelope Protein SUN2 Promotes Cyclophilin-A-Dependent Steps of HIV Replication. Cell Rep. 2016;15:879–892. doi: 10.1016/j.celrep.2016.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshikawa R., Izumi T., Nakano Y., Yamada E., Moriwaki M., Misawa N., et al. Small ruminant lentiviral Vif proteins commonly utilize cyclophilin A, an evolutionarily and structurally conserved protein, to degrade ovine and caprine APOBEC3 proteins. Microbiol Immunol. 2016;60:427–436. doi: 10.1111/1348-0421.12387. [DOI] [PubMed] [Google Scholar]
- 52.Berthoux L., Sebastian S., Sokolskaja E., Luban J. Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc Natl Acad Sci U S A. 2005;102:14849–14853. doi: 10.1073/pnas.0505659102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatziioannou T., Perez-Caballero D., Cowan S., Bieniasz P.D. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J Virol. 2005;79:176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baxter A.E., Russell R.A., Duncan C.J., Moore M.D., Willberg C.B., Pablos J.L., et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe. 2014;16:711–721. doi: 10.1016/j.chom.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Francis A.C., Melikyan G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe. 2018;23(536–48) doi: 10.1016/j.chom.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selyutina A., Persaud M., Lee K., KewalRamani V., Diaz-Griffero F. Nuclear Import of the HIV-1 Core Precedes Reverse Transcription and Uncoating. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ni T., Gerard S., Zhao G., Dent K., Ning J., Zhou J., et al. Intrinsic curvature of the HIV-1 CA hexamer underlies capsid topology and interaction with cyclophilin A. Nat Struct Mol Biol. 2020;27:855–862. doi: 10.1038/s41594-020-0467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoo S., Myszka D.G., Yeh C., McMurray M., Hill C.P., Sundquist W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J Mol Biol. 1997;269:780–795. doi: 10.1006/jmbi.1997.1051. [DOI] [PubMed] [Google Scholar]
- 59.Camilloni C., Sahakyan A.B., Holliday M.J., Isern N.G., Zhang F., Eisenmesser E.Z., et al. Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism. Proc Natl Acad Sci U S A. 2014;111:10203–10208. doi: 10.1073/pnas.1404220111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colgan J., Yuan H.E., Franke E.K., Luban J. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J Virol. 1996;70:4299–4310. doi: 10.1128/jvi.70.7.4299-4310.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braaten D., Franke E.K., Luban J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol. 1996;70:3551–3560. doi: 10.1128/jvi.70.6.3551-3560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Braaten D., Franke E.K., Luban J. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J Virol. 1996;70:4220–4227. doi: 10.1128/jvi.70.7.4220-4227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Towers G.J., Hatziioannou T., Cowan S., Goff S.P., Luban J., Bieniasz P.D. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med. 2003;9:1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- 64.Matsuoka S., Dam E., Lecossier D., Clavel F., Hance A.J. Modulation of HIV-1 infectivity and cyclophilin A-dependence by Gag sequence and target cell type. Retrovirology. 2009;6:21. doi: 10.1186/1742-4690-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saini M., Potash M.J. Novel activities of cyclophilin A and cyclosporin A during HIV-1 infection of primary lymphocytes and macrophages. J Immunol. 2006;177:443–449. doi: 10.4049/jimmunol.177.1.443. [DOI] [PubMed] [Google Scholar]
- 66.Sokolskaja E., Sayah D.M., Luban J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol. 2004;78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kane M., Rebensburg S.V., Takata M.A., Zang T.M., Yamashita M., Kvaratskhelia M., et al. Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. Elife. 2018;7 doi: 10.7554/eLife.35738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Selyutina A., Persaud M., Simons L.M., Bulnes-Ramos A., Buffone C., Martinez-Lopez A., et al. Cyclophilin A Prevents HIV-1 Restriction in Lymphocytes by Blocking Human TRIM5α Binding to the Viral Core. Cell Rep. 2020;30 doi: 10.1016/j.celrep.2020.02.100. 3766–77.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong Z., Ning J., Boggs E.A., Jang S., Wallace C., Telmer C., et al. Cytoplasmic CPSF6 Regulates HIV-1 Capsid Trafficking and Infection in a Cyclophilin A-Dependent Manner. mBio. 2021;12 doi: 10.1128/mBio.03142-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hatziioannou T., Perez-Caballero D., Yang A., Cowan S., Bieniasz P.D. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci U S A. 2004;101:10774–10779. doi: 10.1073/pnas.0402361101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chatterji U., Bobardt M.D., Gaskill P., Sheeter D., Fox H., Gallay P.A. Trim5alpha accelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J Biol Chem. 2006;281:37025–37033. doi: 10.1074/jbc.M606066200. [DOI] [PubMed] [Google Scholar]
- 72.Sebastian S., Luban J. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology. 2005;2:40. doi: 10.1186/1742-4690-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim K., Dauphin A., Komurlu S., McCauley S.M., Yurkovetskiy L., Carbone C., et al. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat Microbiol. 2019;4:2044–2051. doi: 10.1038/s41564-019-0592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfefferkorn M., Schott T., Böhm S., Deichsel D., Felkel C., Gerlich W.H., et al. Composition of HBsAg is predictive of HBsAg loss during treatment in patients with HBeAg-positive chronic hepatitis B. J Hepatol. 2021;74:283–292. doi: 10.1016/j.jhep.2020.08.039. [DOI] [PubMed] [Google Scholar]
- 75.Zhao C., Fang C.Y., Tian X.C., Wang L., Yang P.Y., Wen Y.M. Proteomic analysis of hepatitis B surface antigen positive transgenic mouse liver and decrease of cyclophilin A. J Med Virol. 2007;79:1478–1484. doi: 10.1002/jmv.20945. [DOI] [PubMed] [Google Scholar]
- 76.Gallay P., Ure D., Bobardt M., Chatterji U., Ou J., Trepanier D., et al. The cyclophilin inhibitor CRV431 inhibits liver HBV DNA and HBsAg in transgenic mice. PLoS ONE. 2019;14 doi: 10.1371/journal.pone.0217433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phillips S., Chokshi S., Chatterji U., Riva A., Bobardt M., Williams R., et al. Alisporivir inhibition of hepatocyte cyclophilins reduces HBV replication and hepatitis B surface antigen production. Gastroenterology. 2015;148(403–14) doi: 10.1053/j.gastro.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nkongolo S., Ni Y., Lempp F.A., Kaufman C., Lindner T., Esser-Nobis K., et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol. 2014;60:723–731. doi: 10.1016/j.jhep.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 79.Watashi K., Sluder A., Daito T., Matsunaga S., Ryo A., Nagamori S., et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP) Hepatology. 2014;59:1726–1737. doi: 10.1002/hep.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimura S., Watashi K., Fukano K., Peel M., Sluder A., Kawai F., et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J Hepatol. 2017;66:685–692. doi: 10.1016/j.jhep.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X., Zhao Z., Xu C., Sun L., Chen J., Zhang L., et al. Cyclophilin A restricts influenza A virus replication through degradation of the M1 protein. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0031063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu W., Li J., Zheng W., Shang Y., Zhao Z., Wang S., et al. Cyclophilin A-regulated ubiquitination is critical for RIG-I-mediated antiviral immune responses. Elife. 2017;6 doi: 10.7554/eLife.24425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tscherne D.M., Garcia-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest. 2011;121:6–13. doi: 10.1172/JCI44947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fukuyama S., Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol. 2011;23:481–486. doi: 10.1016/j.coi.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bron R., Kendal A.P., Klenk H.D., Wilschut J. Role of the M2 protein in influenza virus membrane fusion: effects of amantadine and monensin on fusion kinetics. Virology. 1993;195:808–811. doi: 10.1006/viro.1993.1435. [DOI] [PubMed] [Google Scholar]
- 86.Daniels R.S., Downie J.C., Hay A.J., Knossow M., Skehel J.J., Wang M.L., et al. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell. 1985;40:431–439. doi: 10.1016/0092-8674(85)90157-6. [DOI] [PubMed] [Google Scholar]
- 87.Brunotte L., Flies J., Bolte H., Reuther P., Vreede F., Schwemmle M. The nuclear export protein of H5N1 influenza A viruses recruits Matrix 1 (M1) protein to the viral ribonucleoprotein to mediate nuclear export. J Biol Chem. 2014;289:20067–20077. doi: 10.1074/jbc.M114.569178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bui M., Wills E.G., Helenius A., Whittaker G.R. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol. 2000;74:1781–1786. doi: 10.1128/jvi.74.4.1781-1786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cao S., Liu X., Yu M., Li J., Jia X., Bi Y., et al. A nuclear export signal in the matrix protein of Influenza A virus is required for efficient virus replication. J Virol. 2012;86:4883–4891. doi: 10.1128/JVI.06586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang S., Zhao Z., Bi Y., Sun L., Liu X., Liu W. Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1. J Virol. 2013;87:6182–6191. doi: 10.1128/JVI.03024-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ye Z., Robinson D., Wagner R.R. Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J Virol. 1995;69:1964–1970. doi: 10.1128/jvi.69.3.1964-1970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. Cellular proteins in influenza virus particles. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahesutihan M., Zheng W., Cui L., Li Y., Jiao P., Yang W., et al. CypA Regulates AIP4-Mediated M1 Ubiquitination of Influenza A Virus. Virol Sin. 2018;33:440–448. doi: 10.1007/s12250-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J., Chen C., Wong G., Dong W., Zheng W., Li Y., et al. Cyclophilin A protects mice against infection by influenza A virus. Sci Rep. 2016;6:28978. doi: 10.1038/srep28978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu X., Zhao Z., Li Z., Xu C., Sun L., Chen J., et al. Cyclosporin A inhibits the influenza virus replication through cyclophilin A-dependent and -independent pathways. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0037277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Santos-Ferreira N., Van Dycke J., Neyts J., Rocha-Pereira J. Current and Future Antiviral Strategies to Tackle Gastrointestinal Viral Infections. Microorganisms. 2021;9 doi: 10.3390/microorganisms9081599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.He H., Zhou D., Fan W., Fu X., Zhang J., Shen Z., et al. Cyclophilin A inhibits rotavirus replication by facilitating host IFN-I production. Biochem Biophys Res Commun. 2012;422:664–669. doi: 10.1016/j.bbrc.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 98.Yin Y., Wang Y., Dang W., Xu L., Su J., Zhou X., et al. Mycophenolic acid potently inhibits rotavirus infection with a high barrier to resistance development. Antiviral Res. 2016;133:41–49. doi: 10.1016/j.antiviral.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 99.He H., Mou Z., Li W., Fei L., Tang Y., Zhang J., et al. Proteomic methods reveal cyclophilin A function as a host restriction factor against rotavirus infection. Proteomics. 2013;13:1121–1132. doi: 10.1002/pmic.201100579. [DOI] [PubMed] [Google Scholar]
- 100.Heusler K., Pletscher A. The controversial early history of cyclosporin. Swiss medical weekly. 2001;131 doi: 10.4414/smw.2001.09702. [DOI] [PubMed] [Google Scholar]
- 101.Sedrani R., Kallen J., Martin Cabrejas L.M., Papageorgiou C.D., Senia F., Rohrbach S., et al. Sanglifehrin−Cyclophilin Interaction: Degradation Work, Synthetic Macrocyclic Analogues, X-ray Crystal Structure, and Binding Data. J Am Chem Soc. 2003;125:3849–3859. doi: 10.1021/ja021327y. [DOI] [PubMed] [Google Scholar]
- 102.Davis T.L., Walker J.R., Campagna-Slater V., Finerty P.J., Jr, Paramanathan R., Bernstein G., et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huai Q., Kim H.Y., Liu Y., Zhao Y., Mondragon A., Liu J.O., et al. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci U S A. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wenger R.M., France J., Bovermann G., Walliser L., Widmer A., Widmer H. The 3D structure of a cyclosporin analogue in water is nearly identical to the cyclophilin-bound cyclosporin conformation. FEBS Lett. 1994;340:255–259. doi: 10.1016/0014-5793(94)80149-5. [DOI] [PubMed] [Google Scholar]
- 105.Taylor P., Husi H., Kontopidis G., Walkinshaw M.D. Structures of cyclophilin-ligand complexes. Prog Biophys Mol Biol. 1997;67:155–181. doi: 10.1016/s0079-6107(97)00014-x. [DOI] [PubMed] [Google Scholar]
- 106.Ito K., Passioura T., Suga H. Technologies for the synthesis of mRNA-encoding libraries and discovery of bioactive natural product-inspired non-traditional macrocyclic peptides. Molecules. 2013;18:3502–3528. doi: 10.3390/molecules18033502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin L., Harrison S.C. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Amundsen R., Christensen H., Zabihyan B., Asberg A. Cyclosporine A, but not tacrolimus, shows relevant inhibition of organic anion-transporting protein 1B1-mediated transport of atorvastatin. Drug Metab Dispos. 2010;38:1499–1504. doi: 10.1124/dmd.110.032268. [DOI] [PubMed] [Google Scholar]
- 109.Ogimura E., Sekine S., Horie T. Bile salt export pump inhibitors are associated with bile acid-dependent drug-induced toxicity in sandwich-cultured hepatocytes. Biochem Biophys Res Commun. 2011;416:313–317. doi: 10.1016/j.bbrc.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 110.El-Sheikh A.A., Greupink R., Wortelboer H.M., van den Heuvel J.J., Schreurs M., Koenderink J.B., et al. Interaction of immunosuppressive drugs with human organic anion transporter (OAT) 1 and OAT3, and multidrug resistance-associated protein (MRP) 2 and MRP4. Transl Res. 2013;162:398–409. doi: 10.1016/j.trsl.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 111.Quesniaux V.F., Schreier M.H., Wenger R.M., Hiestand P.C., Harding M.W., Van Regenmortel M.H. Cyclophilin binds to the region of cyclosporine involved in its immunosuppressive activity. Eur J Immunol. 1987;17:1359–1365. doi: 10.1002/eji.1830170921. [DOI] [PubMed] [Google Scholar]
- 112.Sigal N.H., Dumont F., Durette P., Siekierka J.J., Peterson L., Rich D.H., et al. Is cyclophilin involved in the immunosuppressive and nephrotoxic mechanism of action of cyclosporin A? J Exp Med. 1991;173:619–628. doi: 10.1084/jem.173.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Papageorgiou C., Borer X., French R.R. Calcineurin has a very tight-binding pocket for the side chain of residue 4 of cyclosporin. Bioorg Med Chem Lett. 1994;4:267–272. [Google Scholar]
- 114.Steinkasserer A., Harrison R., Billich A., Hammerschmid F., Werner G., Wolff B., et al. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus type 1 (HIV-1): interference with early and late events in HIV-1 replication. J Virol. 1995;69:814–824. doi: 10.1128/jvi.69.2.814-824.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Malesevic M., Gutknecht D., Prell E., Klein C., Schumann M., Nowak R.A., et al. Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J Med Chem. 2013;56:7302–7311. doi: 10.1021/jm4007577. [DOI] [PubMed] [Google Scholar]
- 116.Lin K. Discovery of Cyclophilin Inhibitor NIM811 as a Novel Therapeutic Agent for HCV; 2011. p. 317–28.
- 117.Goto K., Watashi K., Murata T., Hishiki T., Hijikata M., Shimotohno K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 118.Puyang X., Poulin D.L., Mathy J.E., Anderson L.J., Ma S., Fang Z., et al. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob Agents Chemother. 2010;54:1981–1987. doi: 10.1128/AAC.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paeshuyse J., Kaul A., De Clercq E., Rosenwirth B., Dumont J.M., Scalfaro P., et al. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 120.Coelmont L., Hanoulle X., Chatterji U., Berger C., Snoeck J., Bobardt M., et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Flisiak R., Jaroszewicz J., Flisiak I., Łapiński T. Update on alisporivir in treatment of viral hepatitis C. Expert Opin Invest Drugs. 2012;21:375–382. doi: 10.1517/13543784.2012.658641. [DOI] [PubMed] [Google Scholar]
- 122.Chatterji U., Bobardt M.D., Stanfield R., Ptak R.G., Pallansch L.A., Ward P.A., et al. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J Biol Chem. 2005;280:40293–40300. doi: 10.1074/jbc.M506314200. [DOI] [PubMed] [Google Scholar]
- 123.Evers M., Barrière J.C., Bashiardes G., Bousseau A., Carry J.C., Dereu N., et al. Synthesis of non-immunosuppressive cyclophilin-Binding cyclosporin A derivatives as potential anti-HIV-1 drugs. Bioorg Med Chem Lett. 2003;13:4415–4419. doi: 10.1016/j.bmcl.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 124.Hopkins S., Scorneaux B., Huang Z., Murray M.G., Wring S., Smitley C., et al. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob Agents Chemother. 2010;54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wring S., Wille K., Rewerts C., Randolph R., Scribner A., Hopkins S. 676 IN VITRO MODELS FOR ASSESSING THE RELATIVE RISK OF HYPERBILIRUBINEMIA ASSOCIATED WITH CYCLOPHILIN INHIBITOR THERAPY. J Hepatol. 2010;52:S263. [Google Scholar]
- 126.Fu J., Tjandra M., Becker C., Bednarczyk D., Capparelli M., Elling R., et al. Potent nonimmunosuppressive cyclophilin inhibitors with improved pharmaceutical properties and decreased transporter inhibition. J Med Chem. 2014;57:8503–8516. doi: 10.1021/jm500862r. [DOI] [PubMed] [Google Scholar]
- 127.Owens C., Rhodin M.H.J., Polemeropoulos A., Long J., Wang G., Jiang L., et al. 1213 Cyclophilin inhibitor EDP-546 is a potential cornerstone drug for use in combination with ns5a and protease inhibitors due to its high barrier to hcv resistance. J Hepatol. 2013;58:S493. [Google Scholar]
- 128.Carling WR. Cyclosporin Analogs; 2011 [Vol. WO/2012/051193].
- 129.Gallay P.A., Chatterji U., Bobardt M.D., Long Z., Zhang S., Su Z. Characterization of the Anti-HCV Activities of the New Cyclophilin Inhibitor STG-175. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hegmans AF, BW, Trepanier DJ, Abel MD, Sugiyama S, Ure DR. Cyclosporine Analogue Molecules Modified at Amino Acid 1 and 3; 2012.
- 131.Liao Y., Luo D., Peng K., Zeng Y. Cyclophilin A: a key player for etiological agent infection. Appl Microbiol Biotechnol. 2021;105:1365–1377. doi: 10.1007/s00253-021-11115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gallay P.A., Bobardt M.D., Chatterji U., Trepanier D.J., Ure D., Ordonez C., et al. The Novel Cyclophilin Inhibitor CPI-431-32 Concurrently Blocks HCV and HIV-1 Infections via a Similar Mechanism of Action. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0134707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu Y., Ruan H., Li Y., Sun G., Liu X., He W., et al. Potent and Specific Inhibition of NTCP-Mediated HBV/HDV Infection and Substrate Transporting by a Novel. Oral-Available Cyclosporine A Analogue, J Med Chem. 2021;64:543–565. doi: 10.1021/acs.jmedchem.0c01484. [DOI] [PubMed] [Google Scholar]
- 134.Papageorgiou C., Kallen J., France J., French R. Conformational control of cyclosporin through substitution of the N-5 position. A new class of cyclosporin antagonists, Bioorganic & medicinal chemistry. 1997;5:187–192. doi: 10.1016/s0968-0896(96)00204-0. [DOI] [PubMed] [Google Scholar]
- 135.Scribner A., Houck D., Huang Z., Mosier S., Peel M., Scorneaux B. Synthesis and biological evaluation of [d-lysine]8cyclosporin A analogs as potential anti-HCV agents. Bioorg Med Chem Lett. 2010;20:6542–6546. doi: 10.1016/j.bmcl.2010.09.036. [DOI] [PubMed] [Google Scholar]
- 136.Kallen J., Sedrani R., Zenke G., Wagner J. Structure of human cyclophilin A in complex with the novel immunosuppressant sanglifehrin A at 1.6 A resolution. J Biol Chem. 2005;280:21965–21971. doi: 10.1074/jbc.M501623200. [DOI] [PubMed] [Google Scholar]
- 137.Steadman V.A., Pettit S.B., Poullennec K.G., Lazarides L., Keats A.J., Dean D.K., et al. Discovery of Potent Cyclophilin Inhibitors Based on the Structural Simplification of Sanglifehrin A. J Med Chem. 2017;60:1000–1017. doi: 10.1021/acs.jmedchem.6b01329. [DOI] [PubMed] [Google Scholar]
- 138.Hansson M.J., Moss S.J., Bobardt M., Chatterji U., Coates N., Garcia-Rivera J.A., et al. Bioengineering and semisynthesis of an optimized cyclophilin inhibitor for treatment of chronic viral infection. Chem Biol. 2015;22:285–292. doi: 10.1016/j.chembiol.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mackman R.L., Steadman V.A., Dean D.K., Jansa P., Poullennec K.G., Appleby T., et al. Discovery of a Potent and Orally Bioavailable Cyclophilin Inhibitor Derived from the Sanglifehrin Macrocycle. J Med Chem. 2018;61:9473–9499. doi: 10.1021/acs.jmedchem.8b00802. [DOI] [PubMed] [Google Scholar]
- 140.Granger B.A., Brown D.G. Design and synthesis of peptide-based macrocyclic cyclophilin inhibitors. Bioorg Med Chem Lett. 2016;26:5304–5307. doi: 10.1016/j.bmcl.2016.09.039. [DOI] [PubMed] [Google Scholar]
- 141.Yang S., K R.J., Lim S., Choi T.G., Kim J.H., Akter S., et al. Structure-Based Discovery of Novel Cyclophilin A Inhibitors for the Treatment of Hepatitis C Virus Infections. J Med Chem. 2015;58:9546–9561. doi: 10.1021/acs.jmedchem.5b01064. [DOI] [PubMed] [Google Scholar]
- 142.Li X., Han J., Lee H.W., Yoon Y.S., Jin Y., Khadka D.B., et al. SAR study of bisamides as cyclophilin a inhibitors for the development of host-targeting therapy for hepatitis C virus infection. Bioorg Med Chem. 2020;28 doi: 10.1016/j.bmc.2020.115679. [DOI] [PubMed] [Google Scholar]
- 143.Han J., Lee H.W., Jin Y., Khadka D.B., Yang S., Li X., et al. Molecular design, synthesis, and biological evaluation of bisamide derivatives as cyclophilin A inhibitors for HCV treatment. Eur J Med Chem. 2020;188 doi: 10.1016/j.ejmech.2019.112031. [DOI] [PubMed] [Google Scholar]
- 144.Pang X., Zhang M., Zhou L., Xie F., Lu H., He W., et al. Discovery of a potent peptidic cyclophilin A inhibitor Trp-Gly-Pro. Eur J Med Chem. 2011;46:1701–1705. doi: 10.1016/j.ejmech.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 145.Li J., Chen J., Zhang L., Wang F., Gui C., Zhang L., et al. One novel quinoxaline derivative as a potent human cyclophilin A inhibitor shows highly inhibitory activity against mouse spleen cell proliferation. Bioorg Med Chem. 2006;14:5527–5534. doi: 10.1016/j.bmc.2006.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Guichou J.-F., Viaud J., Mettling C., Subra G., Lin Y.-L., Chavanieu A. Structure-Based Design, Synthesis, and Biological Evaluation of Novel Inhibitors of Human Cyclophilin A. J Med Chem. 2006;49:900–910. doi: 10.1021/jm050716a. [DOI] [PubMed] [Google Scholar]
- 147.Ni S., Yuan Y., Huang J., Mao X., Lv M., Zhu J., et al. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J Med Chem. 2009;52:5295–5298. doi: 10.1021/jm9008295. [DOI] [PubMed] [Google Scholar]
- 148.Yan W., Qing J., Mei H., Mao F., Huang J., Zhu J., et al. Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization. Molecules. 2015;20:10342–10359. doi: 10.3390/molecules200610342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ahmed-Belkacem A., Colliandre L., Ahnou N., Nevers Q., Gelin M., Bessin Y., et al. Fragment-based discovery of a new family of non-peptidic small-molecule cyclophilin inhibitors with potent antiviral activities. Nat Commun. 2016;7:12777. doi: 10.1038/ncomms12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De Simone A., Georgiou C., Ioannidis H., Gupta A.A., Juarez-Jimenez J., Doughty-Shenton D., et al. A computationally designed binding mode flip leads to a novel class of potent tri-vector cyclophilin inhibitors. Chem Sci. 2019;10:542–547. doi: 10.1039/c8sc03831g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li J., Tan Z., Tang S., Hewlett I., Pang R., He M., et al. Discovery of dual inhibitors targeting both HIV-1 capsid and human cyclophilin A to inhibit the assembly and uncoating of the viral capsid. Bioorg Med Chem. 2009;17:3177–3188. doi: 10.1016/j.bmc.2009.02.051. [DOI] [PubMed] [Google Scholar]
- 152.Sambasivarao S.V., Acevedo O. Computational insight into small molecule inhibition of cyclophilins. J Chem Inf Model. 2011;51:475–482. doi: 10.1021/ci1004114. [DOI] [PubMed] [Google Scholar]
- 153.Daum S., Schumann M., Mathea S., Aumuller T., Balsley M.A., Constant S.L., et al. Isoform-specific inhibition of cyclophilins. Biochemistry. 2009;48:6268–6277. doi: 10.1021/bi9007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tian Y.S., Verathamjamras C., Kawashita N., Okamoto K., Yasunaga T., Ikuta K., et al. Discovery of novel low-molecular-weight HIV-1 inhibitors interacting with cyclophilin A using in silico screening and biological evaluations. J Mol Model. 2013;19:465–475. doi: 10.1007/s00894-012-1560-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li J., Zhang J., Chen J., Luo X., Zhu W., Shen J., et al. Strategy for Discovering Chemical Inhibitors of Human Cyclophilin A: Focused Library Design. Virtual Screening, Chemical Synthesis and Bioassay, Journal of Combinatorial Chemistry. 2006;8:326–337. doi: 10.1021/cc0501561. [DOI] [PubMed] [Google Scholar]
- 156.Yang Y., Moir E., Kontopidis G., Taylor P., Wear M.A., Malone K., et al. Structure-based discovery of a family of synthetic cyclophilin inhibitors showing a cyclosporin-A phenotype in Caenorhabditis elegans. Biochem Biophys Res Commun. 2007;363:1013–1019. doi: 10.1016/j.bbrc.2007.09.079. [DOI] [PubMed] [Google Scholar]
- 157.Chen S., Zhao X., Tan J., Lu H., Qi Z., Huang Q., et al. Structure-based identification of small molecule compounds targeting cell cyclophilin A with anti-HIV-1 activity. Eur J Pharmacol. 2007;565:54–59. doi: 10.1016/j.ejphar.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang F., Chen J., Liu X., Shen X., He X., Jiang H., et al. Synthesis and peptidyl-prolyl isomerase inhibitory activity of quinoxalines as ligands of cyclophilin A. Chem Pharm Bull (Tokyo) 2006;54:372–376. doi: 10.1248/cpb.54.372. [DOI] [PubMed] [Google Scholar]
- 159.Li J., Chen J., Gui C., Zhang L., Qin Y., Xu Q., et al. Discovering novel chemical inhibitors of human cyclophilin A: virtual screening, synthesis, and bioassay. Bioorg Med Chem. 2006;14:2209–2224. doi: 10.1016/j.bmc.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dearmond P.D., West G.M., Anbalagan V., Campa M.J., Patz E.F., Jr., Fitzgerald M.C. Discovery of novel cyclophilin A ligands using an H/D exchange- and mass spectrometry-based strategy. J Biomol Screen. 2010;15:1051–1062. doi: 10.1177/1087057110382775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wu Y.-Q., Belyakov S., Choi C., Limburg D., Thomas V.M., et al. Synthesis and Biological Evaluation of Non-Peptidic Cyclophilin Ligands. J Med Chem. 2003;46:1112–1115. doi: 10.1021/jm020409u. [DOI] [PubMed] [Google Scholar]
- 162.Dunsmore C.J., Malone K.J., Bailey K.R., Wear M.A., Florance H., Shirran S., et al. Design and synthesis of conformationally constrained cyclophilin inhibitors showing a cyclosporin-A phenotype in C. elegans. ChemBioChem. 2011;12:802–810. doi: 10.1002/cbic.201000413. [DOI] [PubMed] [Google Scholar]
- 163.Herrero J.I., de la Pena A., Quiroga J., Sangro B., Garcia N., Sola I., et al. Risk factors for recurrence of hepatitis C after liver transplantation. Liver Transpl Surg. 1998;4:265–270. doi: 10.1002/lt.500040406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.