

Abstract
DNA helicases represent a specialized class of enzymes that play crucial roles in the DNA damage response. Using the energy of nucleoside triphosphate binding and hydrolysis, helicases behave as molecular motors capable of efficiently disrupting the many noncovalent hydrogen bonds that stabilize DNA molecules with secondary structure. In addition to their importance in DNA damage sensing and signaling, DNA helicases facilitate specific steps in DNA repair mechanisms that require polynucleotide tract unwinding or resolution. Because they play fundamental roles in the DNA damage response and DNA repair, defects in helicases disrupt cellular homeostasis. Thus, helicase deficiency or inhibition may result in reduced cell proliferation and survival, apoptosis, DNA damage induction, defective localization of repair proteins to sites of genomic DNA damage, chromosomal instability, and defective DNA repair pathways such as homologous recombination of double-strand breaks. In this chapter, we will describe step-by-step protocols to assay the functional importance of human DNA repair helicases in genome stability and cellular homeostasis.
Keywords: Helicase, DNA repair, Genomic stability, DNA damage, Replication stress, Genetic disease
1. Introduction
DNA helicases represent a large class of enzymes that play integral roles in the maintenance of cellular homeostasis and chromosomal stability. Inherited genetic defects in DNA repair helicases can lead to various genetic disorders including (but not limited to) Werner syndrome, Bloom’s syndrome, Rothmund–Thomson syndrome, Baller–Gerold syndrome, RAPADILINO, Fanconi anemia (FA), Trichothiodystrophy, Xeroderma pigmentosum, and Cockayne’s syndrome [1–3]. In a nucleoside triphosphate-dependent manner, helicases catalytically unwind double helical nucleic acid molecules (DNA–DNA, DNA–RNA, and/or RNA–RNA) that are stabilized by many noncovalent hydrogen bonds between the bases of the two strands [4–7]. In addition to conventional duplex DNA substrates, certain DNA helicases unwind specialized non-B form DNA structures such as triplex DNA [8, 9], G-quadruplex DNA [10–12], and Z-DNA [13]. Such alternatively arranged DNA structures are believed to interfere with normal cellular DNA transactions, but the mechanisms are still being investigated. Additionally, specialized DNA helicases resolve more complex duplex DNA structures which represent replication and repair intermediates including stalled or regressed replication forks, four-stranded Holliday junctions, and three-stranded displacement (D)-loop or flap structures. A subset of DNA helicases, particularly those of the RecQ family, stimulate annealing of complementary single-stranded DNA [1, 14, 15], but the physiological importance of this biochemical activity has not yet been revealed. Altogether, DNA helicases are crucial enzymes in numerous cellular processes including DNA repair, replication, and transcription.
DNA helicases are upregulated in various cancer cell lines, including breast, colorectal, hypopharyngeal, ovarian, and lung cancers [16, 17]. Increased expression of a given DNA helicase in cancer cells has been associated with increased proliferation and resistance to chemotherapeutic drugs [16]; conversely, inhibition of DNA helicase activity either by its depletion through RNA interference (RNAi) [18, 19] or pharmacological inhibition of helicase-catalyzed DNA unwinding [20–23] has been shown to lead to reduced cell proliferation, accumulation of DNA damage, and increased cellular apoptosis. Thus, DNA repair helicases are proposed to represent a novel class of enzymes which may serve as therapeutic targets for increasing the efficacy of chemotherapeutic agents [20, 24, 25]. In this protocol chapter, we will describe the experimental procedures by which we study the biological importance of human DNA repair helicases in response to endogenous or exogenously induced stress in terms of cellular proliferation and survival, apoptosis, and DNA damage induction as marked by phosphorylation of the histone protein H2AX.
Stalled replication forks can lead to double strand breaks (DSBs) due to broken replication forks, whereas ionizing radiation or certain DNA damaging agents (e.g., bleomycin) can directly introduce DSBs. DSBs introduced by either mechanism require efficient correction by nonhomologous end-joining or homologous recombination (HR) repair. Disruption of these DSB repair pathways can lead to chromosomal instability, age-related diseases, and cancer [26]. Thus, a quantitative method to measure each of these pathways in vivo can provide novel insights into the role of a given DNA repair protein in regulating these pathways. We use a strategy developed originally by the Jasin lab to examine the effect of helicase deficiency on HR repair in vivo [27] (elaborated below). Essentially, U2OS cells with a stably integrated recombination susceptible DNA sequence characterized by a disrupted gene encoding Green Fluorescence Protein (GFP) are scored for HR by restoration of the intact GFP, resulting in cellular immunofluorescence which can be detected by flow cytometry.
Altogether the combined approaches to measure cell proliferation, colony survival, apoptosis, immunofluorescent accumulation of DNA damage markers or DNA repair proteins to sites of damage, cytogenetic analysis for chromosomal stability, and HR repair using an in vivo reporter assay provide a comprehensive battery of techniques to begin to assess the effects of helicase deficiency or helicase inhibition in a biological setting. The techniques described in this book chapter provide a simple list of reagents, disposables, and equipment with step-by-step protocols to perform these assays.
We will begin by describing the laboratory procedures to measure total cell number by Coulter counting and cell proliferation assay by scoring metabolic activity of untreated cells compared to those exposed to the DNA damaging drug (Fig. 1a). Because cell proliferation assays are typically spectrophotometric, they can be easily set up in a high-throughput manner if so desired such as in the case of an RNAi screen. In addition, cell proliferation assays are typically conducted over a shorter period of time (such as 3–4 days), so these are more amenable to screens in which many treatments/conditions are being assessed for their effect on cell viability. For example, the assay can be adapted to use in a 96-well or 384-well plate to conduct large-scale screens to identify novel genetic or chemical interactions. Cell proliferation is measured using a tetrazolium salt such as sodium 5-(2,4-disulfophenyl)-2-(4-iodophenyl)-3-(4-nitrophenyl)-2H-tetrazolium inner salt (WST-1). The WST-1 assay is principally very similar to the MTT assay in which NADH produced by metabolically active cells reduces the positively charged tetrazolium salt 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl-2H-tetrazolium bromide (MTT) to form a purple-colored insoluble formazan dye [28]. Formazan that is produced by the cleavage of the tetrazole ring of the MTT is then solubilized, and the metabolic activity of the cells is measured by colorimetric quantification. Reduction of WST-1 by the NADH produced in proliferating cells leads to the formation of soluble yellow or orange-colored formazan dye, thereby eliminating the need for solubilization of formazan as required in the MTT assay. Thus, the WST-1 assay is a more convenient and time-saving assay for the determination of cellular proliferation compared to the MTT assay.
Fig. 1.
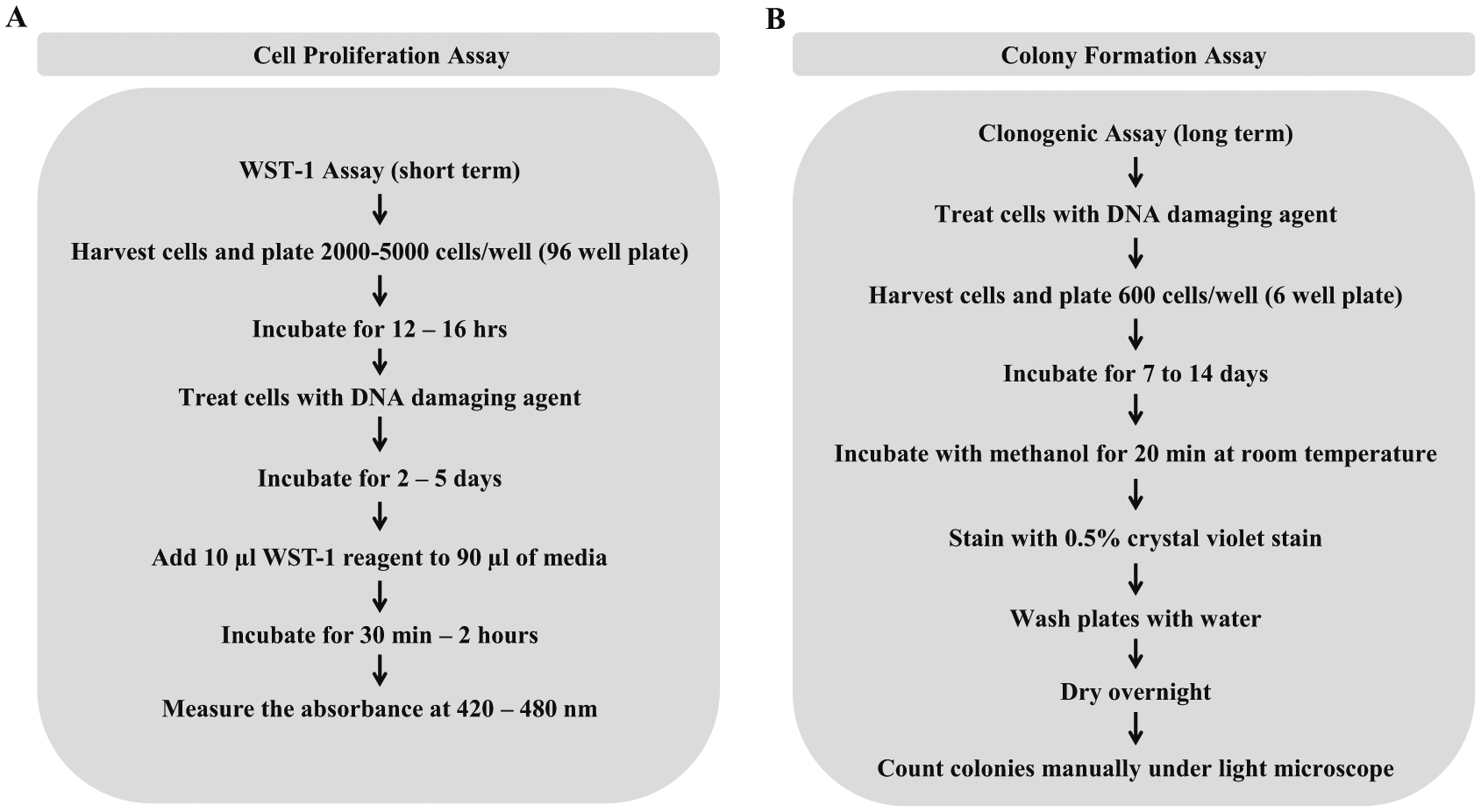
Flowchart representing assays to measure cell proliferation or colony formation. Panel (a) Protocol for determining cell proliferation using an MTT assay variant, WST-1 assay. NADP produced by metabolically active cells can produce a colored formazan dye by the reduction of WST-1. Colorimetric analysis of the soluble formazan dye is used to quantify cellular proliferation. Panel (b) Protocol for determining the potential of individual cells to proliferate by using colony formation assay. Change in proliferative ability of the cells before and after treatment with a DNA damaging drug can be studied by allowing individual cell to form colonies (50 cells or more). Further, quantification of colonies of cells with or without a helicase allows us to assess the role of the helicase in overcoming DNA damage as induced by the drug
Often researchers desire to assess the impact of helicase deficiency on cell growth and division, a truer measure of survival than the cell proliferation assay described above, which can be readily assessed by colony formation (Fig. 1b). The appropriate human wild-type and helicase mutant cell lines are treated with a specified compound that induces DNA damage or replication stress. Often, stably transfected cell lines exogenously expressing wild-type or mutant helicase proteins in a genetically null background are characterized for their sensitivity to various DNA damaging or replication stalling drugs. Alternatively, human cells depleted of a specific helicase by RNAi can be assayed for colony formation and compared to those cells that are transfected with a control (nontarget) siRNA. The simplest method to assess the number of cells before and after treatment with drug is by using a cell counting method utilizing a Coulter counter. This method can be used to determine the effect of treatment on a wide range of adherent and suspended cells. We also use a Coulter counter to seed the desired number of cells for subsequent analysis by WST-1, colony formation or apoptosis assay. However, one drawback of the Coulter counter method is that it fails to distinguish between live, dead and metabolically inactive cells. The colony forming assay can be used to measure the ability of human cells to survive and divide after they are treated with various DNA damaging agents. Using this assay, we can determine the capacity of every single cell in the selected sample to continuously divide and form colonies (which typically represent 50 cells or more). The colony formation assay is an accurate method to quantitatively assess the effect of drug treatment on the continued growth of the cells deficient or proficient for a given helicase.
As mentioned earlier, helicases such as FANCJ can resolve specialized non-B DNA structures such as G-quadruplexes thereby maintaining DNA replication and reducing DNA damage. Thus, FANCJ-depleted cells treated with a G-quadruplex stabilizer accumulate DNA damage and in-turn undergo apoptosis [29]. Like-wise, pharmacological inhibition of WRN helicase by a small molecule WRN-specific inhibitor in cancer cells leads to accumulation of double-strand DNA breaks and increased apoptosis [22]. To assess the biological effects of such agents, the apoptosis assay is a convenient and useful tool.
DNA fragmentation is one of the hallmarks of cell undergoing apoptosis and can be exploited for an accurate measurement of apoptosis. Various Ca2+/Mg2+-dependent endonucleases are activated during apoptosis, and they cleave the genomic DNA into mononucleosomes and multinucleosomes. These apoptotic nucleases cleave the accessible internucleosomal DNA to produce DNA–histone fragments or nucleosomes [30]. This principle has been utilized by several assays including TUNEL and ELISA to detect cytoplasmic DNA fragments indicative of apoptosis. In this chapter we will discuss ELISA to quantitate cytoplasmic nucleosomes as an indicator of apoptosis. This method relies on the fact that during apoptosis the chromosomal nucleosome units travel to the cytoplasm and can then be extracted from the dying cells and quantitated using a colorimetric method.
In our lab, we have used immunofluorescence confocal microscopy to detect various DNA damage markers in cells mutated for a given helicase, depleted of a target helicase, or inhibited pharmacologically by a helicase-specific small molecule (Table 1). Similarly, the formation of DNA repair and/or replication protein foci in helicase-deficient or helicase-inhibited cells exposed to a DNA damaging agent can be visualized by fluorescence microscopy (Table 2). Recruited proteins can act as DNA damage sensors, amplifiers, or downstream effectors at sites of DNA damage or stalled replication forks. We have also detected the colocalization of DNA repair proteins with DNA helicases in human cells exposed to agents that impose replication stress or DNA damage, suggesting their interactive functions or common pathways of DNA metabolism (Table 3). A representative example of microscopic imaging is shown in Fig. 2. Here, the immunofluorescence data suggest that nuclear FANCJ foci are larger and better colocalize with the single-stranded DNA binding protein RPA after ionizing radiation (IR) or mitomycin C (MMC) exposure to human fibroblasts that express FANCA, an upstream player in the FA pathway of interstrand cross-link repair.
Table 1.
Immunofluorescent detection of endogenous or exogenously induced DNA damage in helicase-compromised cells
| Helicase deficiency/inhibitor | Agent | Markera | DNA structure | Reference |
|---|---|---|---|---|
| FANCJ-RNAi, U2 OS cells | Telomestatin | γ-H2AX | Double-strand break | [36] |
| FANCJ mutant DT40 cells | Telomestatin | G4-DNA antibody | G-quadruplex | [37] |
| FANCJ mutant fibroblasts | Mitomycin C | γ-H2AX | Double-strand break | [38] |
| RECQL1-RNAi, HeLa cells | Camptothecin | 53BP1 | Double-strand break | [39] |
| WRNi NSC 617145b, HeLa cells | None | γ-H2AX | Double-strand break | [22] |
| PCNA | Stalled replication fork |
DNA damage marker or protein foci are elevated upon treatment with agent in helicase-compromised cells
WRNi NSC 617145 is a WRN-specific small molecule helicase inhibitor
Table 2.
Immunofluorescent detection of DNA repair proteins in helicase-compromised cells challenged with a DNA damaging agent
| Helicase deficiency/inhibitor | Agent(s) | Repair protein foci | Function | Reference |
|---|---|---|---|---|
| WRN inhibitor NSC 617145 | MMC | DNA-PKcs pS2056 | Nonhomologous end-joining | [21] |
| ATM | DNA damage sensing and signaling | [21] | ||
| Rad51 | Strand exchange, HR repair | [21] | ||
| RECQL1-RNAi | Camptothecin | RPAa | Single-stranded DNA binding | [39] |
| Rad51a | Strand exchange, HR repair | [39] | ||
| Mre11 | Nucleolytic trimming, HR repair | [39] |
Unless stated otherwise, protein foci are elevated upon treatment with agent in helicase-compromised cells. However, in the case of RPA or RAD51, foci are decreased in RECQL1-RNAi depleted cells exposed to camptothecin
Table 3.
Immunofluorescent detection of DNA repair proteins colocalizing with human DNA
| Helicases | Agent | Colocalizing protein | Function | Reference |
|---|---|---|---|---|
| WRN | Hydroxyurea | RPA | Single-stranded DNA binding | [40] |
| WRN | Mitomycin C 4-nitroquinoline-1-oxide Methylmethanesulfonate |
FEN-1 | Processing of DNA replication and repair intermediates | [41] |
| BLM | Hydroxyurea | FANCJ | ICL repair, replication stress response | [42] |
| FANCJ | MMC Ionizing radiation Hydroxyurea | RPA | Single-stranded DNA binding | [43] |
Fig. 2.
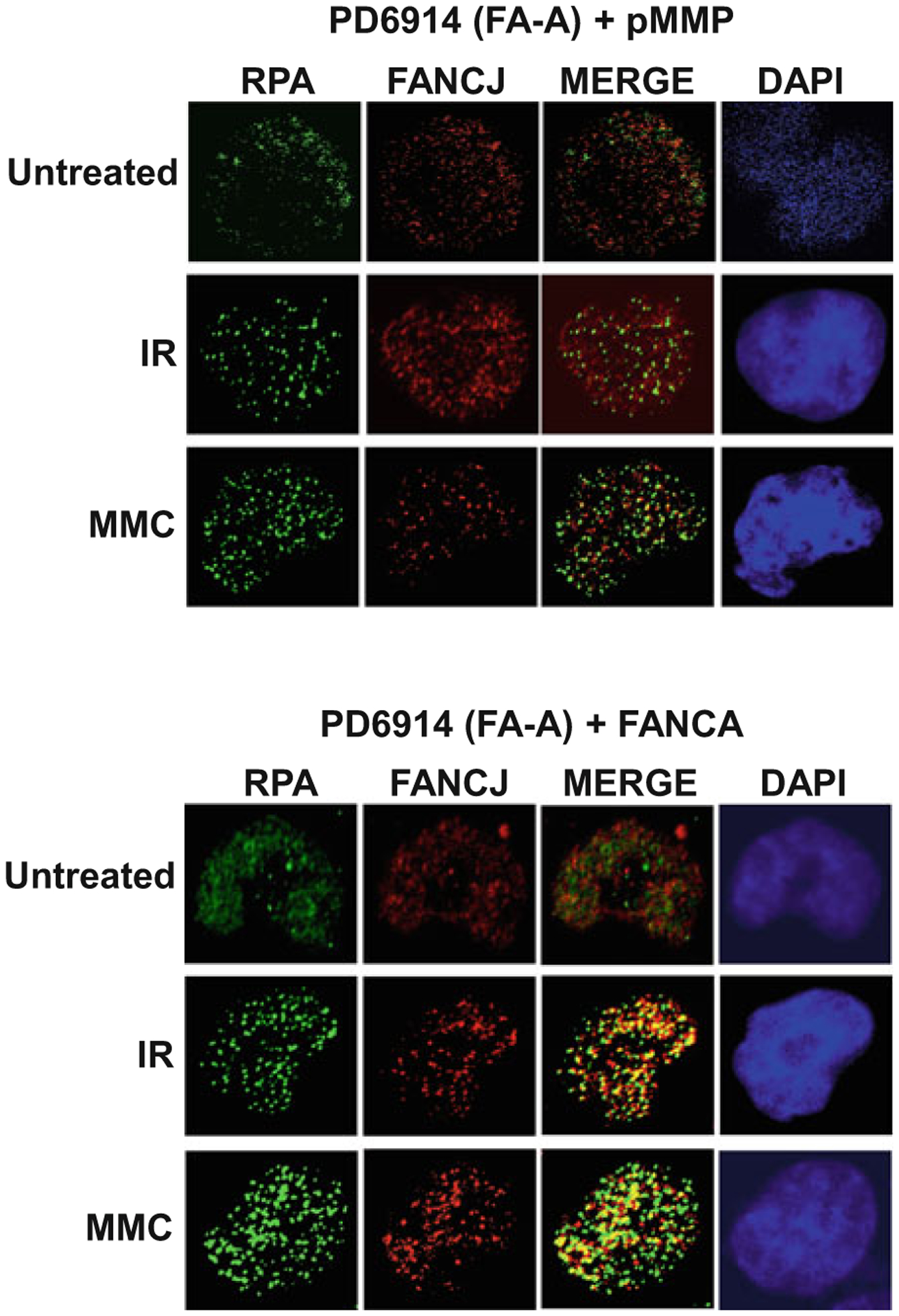
Colocalization of FANCJ helicase and RPA is dependent on FANCA expression in cells exposed to DNA damaging agents. FA-A fibroblasts (PD6914 + pMMP vector) and corrected fibroblasts (PD6914 + FANCA) [35] were treated with the DNA-damaging agent MMC (500 ng/mL) for 16 h or exposed to 10 Gy IR followed by 6 h recovery. Cells were fixed (3.7% formaldehyde), permeabilized, and incubated with mouse anti-RPA34 Ab-2 (Millipore Sigma) and rabbit anti-FANCJ (Millipore Sigma) antibodies. For immunofluorescent detection, cells were incubated with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 568 goat anti-rabbit IgG (Thermo Fisher Scientific) followed by ProLong Gold mountant with DAPI (Thermo Fisher Scientific). Immunofluorescence was imaged on a Zeiss LSM 510 META inverted Axiovert 200M laser scan microscope (Carl Zeiss, Jena, Germany) with a Plan-Apochromat 63×/1.4 oil DIC objective. After treatment with MMC or IR, RPA (green) localizes in nuclear foci that coincide with FANCJ (red) foci as shown in the overlapped images (yellow) in the presence of FANCA protein. In the absence of FANCA, RPA and FANCJ staining does not overlap. DAPI staining of the nucleus in each cell is indicated by the color blue
Immunofluorescence imaging of DNA repair proteins has provided novel insights into DNA damage signaling and the repair process [31]. For example, in response to replication stress ATR acts as one of the early sensors and is required for the formation of RPA and Rad9 foci which act as amplifiers. Formation of RPA and Rad9 foci lead to the activation of Chk1 which acts as the effector molecule that is known to cause cell cycle arrest [32]. Immunofluorescence microscopy is a very powerful technique that helps to identify and monitor nuclear relocalization of these proteins, protein–protein interactions, and histone modifications. Some DNA repair proteins, like ATR, ATRIP, Rad9, WRN, MRE11, BRCA1, and RPA, do not form detectable foci until the fraction of proteins that is not bound to the chromatin is washed away. In situ fractionation can be used to remove the excess of the nucleoplasmic proteins not bound to the chromatin to enhance the detection of the proteins bound to the chromatin. We have adapted and modified a technique originally described by Mirzoeva and Petrini [33] to enhance image quality, and this is described in the Methods section.
Although immunofluorescent detection of DNA damage by γ-H2AX or 53BP1 foci to visualize DSBs is useful, often an analysis of chromosomal aberrations is warranted. In this chapter, we describe a very simple and useful technique to obtain high-quality chromosome spreads for subsequent microscopic analysis of chromosomal integrity. This technique can be used to identify aneuploidy, insertions, deletions, and genomic rearrangement within the same chromosome or between two different chromosomes.
Disruption or inhibition of a key DNA damage response protein leading to chromosomal instability requires further studies to delineate the precise pathway of DNA repair that is affected. Given that chromosomal instability may result from a defect in DSB repair, and HR is a preferred pathway of DSB repair during S/G2-phases of dividing cells, a quantitative assessment of HR repair is useful for the analysis of cell lines deficient in a DNA helicase, particularly those of the RecQ family but also the Fe-S helicase FANCJ. The DNA recombination substrate originally described by the Jasin lab [27] which we have used consists of a repeat sequence (SceGFP) that is nonfunctional due to the integration of the 18 bp recognition site for the I-SceI endonuclease. Downstream of the SceGFP gene is an internal GFP fragment (iGFP). Transfection of the cells with the plasmid expressing I-SceI endonuclease induces a double stranded break. HR using iGFP as a template restores the full-length functional GFP. GFP-positive cells are then detected by flow cytometry. We have used these reporter cell lines to study the effect of FANCJ helicase deficiency on the frequency of HR repair [34] (Fig. 3).
Fig. 3.
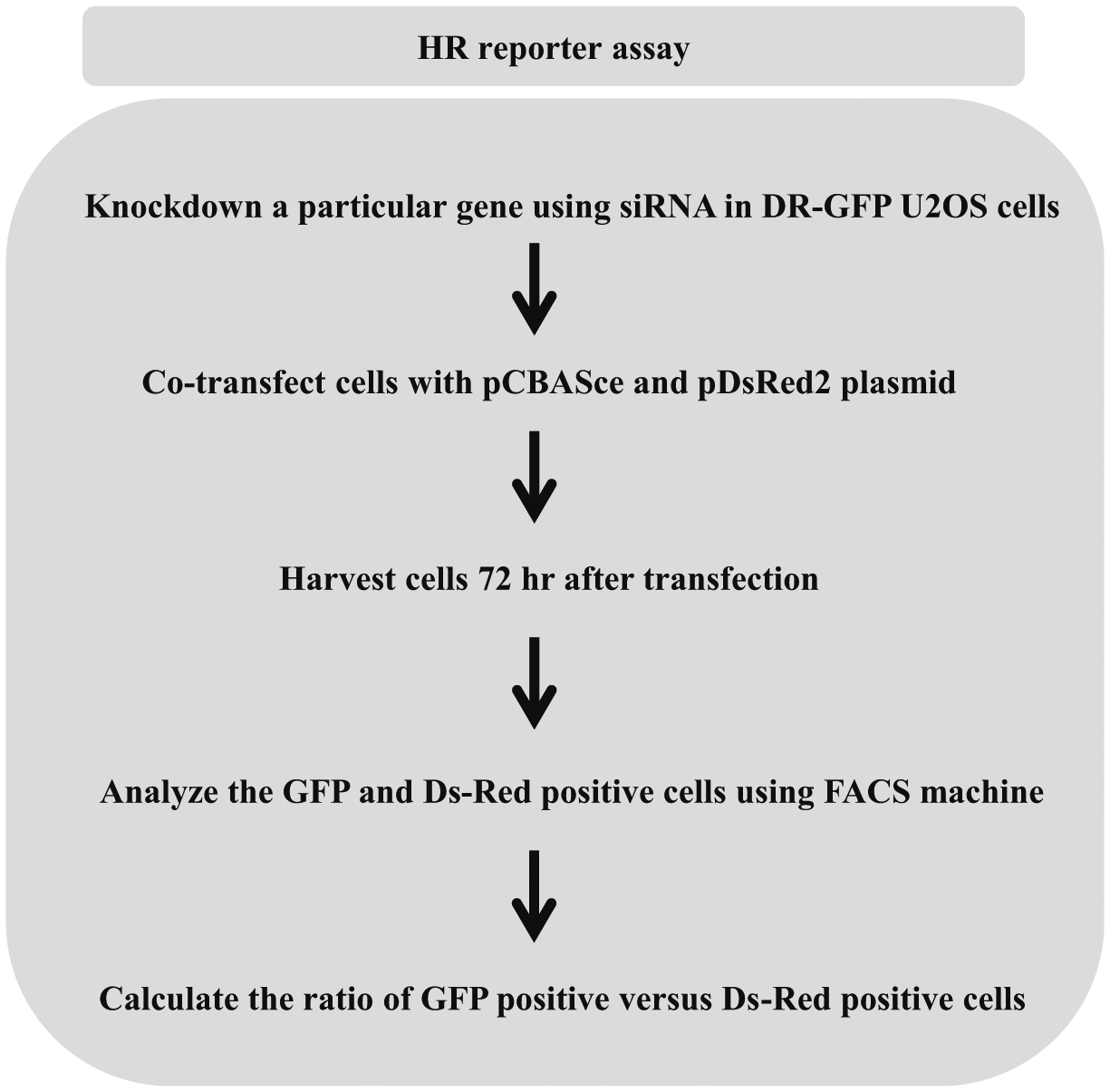
Flowchart representing homologous recombination repair assay. Cells are transfected with pCBASce and DsRed2 plasmids previously described in ref. [27]. I-SceI generates DSB that at integrated GFP sequence stimulates HR. GFP and Ds-Red signals are assayed after 3 days on an FACSCalibur flow cytometer
2. Materials
2.1. Cell Counting Using Coulter Counter
6-well tissue culture dish.
Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% Penicillin-Streptomycin.
DNA damaging agent.
Sterile phosphate buffered saline (PBS).
Trypsin–EDTA:0.25% trypsin solution with 1 mM EDTA.
Cell counter vials.
Coulter counter.
Isotonic diluent.
2.2. WST-1 Cell Proliferation Assay
96-well tissue culture plate.
Multichannel pipette.
Cell proliferation reagent WST-1.
Microplate spectrophotometer reader to measure the absorbance.
1% SDS solution (to quench reaction).
2.3. Colony Formation
Crystal violet powder. Dissolve 0.5 g of crystal violet powder in 80 mL distilled water and then add 20 mL methanol to make 0.5% crystal violet stain (see Note 1).
Distilled water.
100% methanol.
Bright-field microscope.
Transparent grid for colony counting.
2.4. Apoptosis Assay
Cell death detection ELISA kit.
8-well microplate module.
10× coating buffer.
Monoclonal anti-histone antibody.
1× incubation buffer.
10× washing buffer.
Monoclonal anti-DNA antibody, peroxidase conjugated.
5 mg tablets of ABTS or 2,2′-azino-bis(3-ethylbenzothiazo-line-6-sulfonic acid) substrate.
1× substrate buffer.
Adhesive foil to cover the microplate modules for incubation.
Microplate spectrophotometer reader to measure the absorbance.
2.5. Cellular Immunofluorescence
U2-OS Cells (ATCC).
Fetal bovine serum.
Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin–streptomycin.
Camptothecin.
Triton X-100.
Tween-20.
1 M HEPES pH 7.4.
1 M MgCl2.
5 M NaCl.
Sucrose.
Halt EDTA-free protease and phosphatase inhibitors.
Chamber slides.
PBS, pH 7.4.
Preextraction buffer 1: 1% Triton X-100, 10 mM HEPES pH 7.4, 10 mM NaCl, and 3 mM MgCl2 supplemented with 1× Halt EDTA-free protease and phosphatase inhibitors.
Preextraction buffer 2: 0.5% Triton X-100, 20 mM HEPES pH 7.4, 50 mM NaCl, 3 mM MgCl2, and 300 mM sucrose supplemented with 1× Halt EDTA-free protease and phosphatase inhibitors.
Fixing solution: Add 0.1% Triton X-100 to 4% Paraformaldehyde solution in 1× PBS. 100% ice-cold methanol can also be used for fixation.
Blocking Buffer: 5% normal horse serum in 1× PBS containing 0.3% Triton X-100. The normal serum is from the same species as the secondary antibody. SuperBlock Blocking buffer can also be used for blocking.
Antibody dilution buffer: 5% BSA in 1× PBS containing 0.1% Tween.
Primary antibody: ATR Antibody.
Secondary antibody: Fluorescein horse anti-mouse IgG antibody.
DAPI.
2.6. Chromosome Spreads
U2-OS cells.
Fetal bovine serum.
Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin–streptomycin.
Trypsin.
Methanol.
Glacial acetic acid.
Carnoy’s fixative → 3:1 ratio of methanol and glacial acetic acid.
KCl.
Giemsa stain.
Colcemid.
Mounting media.
2.7. Double-Strand Break HR Reporter Assay
U2-OS cells containing a stably integrated DR-GFP reporter made by the Jasin lab [27].
Fetal bovine serum.
Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin–streptomycin.
Amaxa® Cell Line Nucleofector® Kit V.
Nucleofector™ 2b Device.
2 μg of I-SceI expressing plasmid DNA (pCBASce) and 100 ng of ds-Red expressing plasmid DNA (pDsRed2-N1).
BD FACSCalibur Flow Cytometer Cell Analyzer.
3. Methods
3.1. Coulter Counter Assessment of Cell Number
For adherent cancer cell lines such as HeLa or U2-OS, plate ~15 × 104 cells are seeded per well in 1.5 mL of DMEM supplemented with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, and 100 U/mL penicillin (see Note 2).
Treat the cells with a predetermined concentration of DNA damaging agents and incubate for the appropriate duration (see Notes 3 and 4). This treatment can be done in duplicates to get more accurate and reliable data.
Remove media and wash the cells with PBS (see Note 5).
Add trypsin–EDTA solution to the well (cover the entire surface area of the well).
Incubate at 37 °C for 2–3 min to detach the cells.
Collect the cells in a tube by adding medium.
Centrifuge the tubes at 1000 × g for 5 min.
Aspirate out the medium and resuspend the pellet in appropriate volume of fresh medium.
Dilute 500 μL of cell suspension in 10 mL of the isotonic diluent in the cell counter vials. Resuspend well.
Place the cell counter vials on the Coulter counter lever. Make sure the tip of the Coulter counter with the aperture is completely immersed in the cell suspension (see Note 6).
Count the total number of cells using a dilution factor of 21 (see Note 7). To get accurate reading this process can be repeated several times and the average value can be used to determine cell number.
3.2. WST-1 Cell Proliferation Assay
The isogenic pair of wild-type and helicase mutant cell lines, or the target helicase-shRNA and conrol-shRNA treated cells are seeded in a 96-well plate. For cancer cell lines (including U2-OS and HeLa), plate 700–1000 cells per well in 100 μL in DMEM media (as mentioned above, see Note 2).
Treat the cells with appropriate concentration of DNA damaging agent (see step 2 in Subheading 3.1) 12–16 h after seeding, when the cells have attached (see Note 3). For control wells, treat the cells with the solvent (e.g., DMSO) that the drug is dissolved in (see Note 8). All the treatments and controls should be done in triplicates to obtain more accurate readings.
Incubate the cells for 2–5 days at 37 °C and 5% CO2 (see Note 9).
Equilibrate the temperature of WST-1 to room temperature (see Note 10). Add 10 μL WST-1 reagent to each well (see Note 11). Make sure the ratio of media to WST-1 is 9:1. For blank wells add 10 μL of WST-1 to 90 μL of culture medium (see Note 12). Tilt plate to mix or put it in a shaker for 30 s (see Note 13).
Incubate cells at 37 °C for 30 min to 2 h. To stop the assay 10 μL of 1% SDS may be added.
Measure the absorbance of the plate using a microplate reader or spectrophotometer at OD between 420 and 480 nm (see Note 14).
3.3. Colony Forming Assay
Treat the adherent cells with DNA damaging agents and harvest the cells after 24 h using trypsinization. For detailed procedure refer to the above protocol (see steps 1–7 in Subheading 3.1).
Resuspend the cells in fresh medium to the appropriate dilution, based on the kind of cell and treatment.
Count the cells using a Coulter counter (see steps 9–11 in Subheading 3.1) and plate around 500 cells in each well of a 6-well plate (see Note 15).
Incubate the plates for 7–14 days till the colonies are formed.
Carefully aspirate out the medium from the dish and gently add 100% methanol so as to not disturb the colonies. Make sure the entire plate is covered with methanol (see Note 16).
Incubate the dish at room temperature for 20 min. Keep the dish covered.
Rinse the cells with water and add 2 mL of 0.5% crystal violet stain. Incubate at room temperature 5–10 min.
Remove the crystal violet stain and wash the cells with water. Place the plates in a water bath or water filled sink to remove any excess dye (see Note 17).
The plates can be left upright to air dry in room temperature alternatively they can be left inverted on tissue paper. Leave the plates overnight to ensure proper drying.
Count the colonies manually using a bright-field microscope and a transparent grid (see Note 18).
3.4. Apoptosis Assay
Seed the appropriate number of cells in a 6-well plate and treat with or without the DNA damaging agent for up to 72 h (see Note 19). Harvest the adherent cells using trypsinization method and resuspend in fresh medium.
Using the Coulter counter method, count the cells and transfer 5 × 104 cells into an eppendorf tube (see steps 9–11 in Subheading 3.1).
Pellet the cells by centrifuging the tubes at 1000–1500 × g for 5 min and resuspend the cell pellet well in 500 μL of incubation buffer.
Lyse the cells by incubating them for 30 min at room temperature.
Centrifuge the tube at 20,000 × g to separate the cell cytoplasmic fraction from the cell nuclear fraction.
Carefully aspirate out the supernatant into a fresh Eppendorf tube. The samples can now stored at −20 °C overnight.
Incubate the 8-well microplate module with 100 μL of the coating buffer with anti-histone antibody (see Notes 20 and 21) for 1 h at room temperature or overnight at 4 °C. Make sure that the microplate module is covered properly.
After removing the coating solution, add around 200 μL of incubation buffer to the microplate module.
Cover the microplate module carefully and incubate at room temperature for 30 min. This blocks the nonspecific binding sites on the surface of the coated microplate module.
Wash the microplate wells thrice using 300 μL of washing buffer (see Notes 22 and 23).
Dilute one part of the samples obtained in step 6 with nine parts of the incubation buffer. Add 100 μL of the diluted sample to the microplate module. Cover the microplate and incubate for 90 min at room temperature. To get “blank” absorbance values, add only incubation buffer without any sample lysate.
Repeat step 10.
Add 100 μL of freshly prepared incubation buffer with the anti-DNA-peroxidase conjugated antibody (9:1) to the microplate modules. Do not add this anti-DNA antibody for blank wells.
Cover the microplate module tightly and incubate for 90 min at room temperature.
Repeat step 10.
Add 100 μL of the ABTS substrate (see Note 24) to the microplate module.
Plate the microplate modules on a shaker incubator for 10–20 min at 250 rpm. The incubation time should be long enough for the development of color to perform the colorimetric quantification.
The contents of the microplate should be mixed thoroughly by tapping the sides of the microplate module and the absorbance can be read at 405 nm using a microplate spectrophotometer.
3.5. Cellular Immunofluorescence Detection
Maintain U2-OS cells in 5% CO2 at 37 °C in DMEM media. Split cells after every 3 days. Count cells using the cell counter and seed the cells onto the chamber slides. Seed 60,000 cells per well for four chamber slides or 30,000 cells per well for eight chamber slides.
Incubate cells in the 5% CO2 incubator and grow cells until they are 50–60% confluent.
Add 2 μM Camptothecin directly to the chamber slides for 2 h (see Note 25).
Add the preextraction buffer 1 (see Notes 26 and 27). Keep the slides at 4 °C for 5 min.
Wash with 1× PBS.
Add preextraction buffer 2 (see Notes 26 and 27). Keep the slides at 4 °C for 5 min.
Wash with 1× PBS.
Add 4% paraformaldehyde with 0.1% Triton X-100 at room temperature and make sure the cells are covered to a depth of ~3 mm. Cells can also be fixed with 100% methanol for 10 min at −20 °C.
After fixing the cells in paraformaldehyde for 15 min, wash the cells with 1× PBS three times.
Incubate cells in the Blocking buffer.
Dilute primary antibody (1:250) in PBS/T. Add the primary antibody on top of the cells in the chamber slides and incubate overnight at 4 °C.
Wash chamber slides three times for 5 min with PBS/T (see Note 28).
Dilute fluorescence labeled secondary antibody (1:500) in PBS/T. Add the secondary antibody on top of the cells and incubate for 3 h at room temperature in the dark.
Wash the chamber slides three times for 5 min with PBS/T in low lighting (see Note 28).
Aspirate off excess PBS and remove the chambers. Air dry the slides for 3 min in the dark.
Add a small drop of DAPI to each chamber and cover with glass coverslips. Gently tap the coverslips to remove the air bubbles and then seal the coverslips with a transparent nail polish. Store the slides at 4 °C in a dark box.
Images of the nuclei can be obtained by using a fluorescence microscope at 63× or 100 × magnification. If Z-sections are used, then all the sections should be merged so that all the foci are on a single visible plane.
Count at least 50 nuclei from three independent experiments and determine average number of foci per nucleus (see Note 29).
3.6. Cytogenetic Analysis of Chromosome Spreads
Maintain U2-OS cells in 5% CO2 atmosphere at 37 °C in DMEM media. Grow cells in a 10 cm dish and split cells after every 3 days.
When cells reach ~80% confluency, add colcemid to a final concentration of 200 ng/mL and incubate cells in the 37 °C incubator for 3 h (see Note 30).
Gently wash the cells with 1× PBS and aspirate off the PBS.
Add 1 mL of trypsin, swirl and keep the flask in 37 °C incubator for 3 min.
Tap the flask gently to detach the cells. Once the cells are detached, add 5 mL of media and transfer the cell suspension to a 15 mL conical tube.
Centrifuge at 180 × g for 5 min. Remove supernatant and resuspend pellet in 10 mL of 0.075 M KCl solution. Make sure that the KCl solution is prewarmed at 37 °C. Vortex the tube to mix the cells properly in the KCl solution (see Note 31).
Incubate cells in the 37 °C water bath for 10 min. Centrifuge at 180 × g for 5 min. Remove supernatant and resuspend pellet in 6 mL of fresh Carnoy’s Fixative (see Note 32).
Centrifuge at 180 × g for 5 min. Remove supernatant and resuspend pellet in 500 μL of Carnoy’s Fixative. Cells can be stored up to several months at 4 °C at this point.
Remove the top of the p1000 tip box and place the slide against one side of the box so that the slide is placed at an angle.
Hold a glass pasture pipet approximately 6 in. above the slide and release three drops of cell suspension on the glass slides at three different areas of the slide (see Note 33).
Place the slides in the chemical hood and allow it to dry (see Note 34).
Once the slide is dry, place the slides on a staining rack over the sink. Add the Giemsa staining solution on in the staining rack on top of the slides and stain for 5 min.
Wash the slides with distilled water, drain and allow the slides to air dry.
Add mounting media to the cover slips and place it on top of the slides. Put four drops of a clear nail polish on the corners of the coverslips and wait for 2 min to dry. Then add more nail polish on to the edges to seal the coverslip to the slides. Slides can be stored at −80 °C in slide box for up to 4 months.
Take pictures with a phase contrast microscope using 100× magnification. To obtain enough analyzable metaphase spread images, prepare at least eight glass slides of spreads for each data. Count at least 20 best metaphase spreads. Good spreads are the ones where chromosome spread is from a single cell. Calculate the individual chromosomes and ignore the ones where spreads from two chromosomes overlap. An individual chromosome is the one that does not overlap upon itself or onto the neighboring chromosomes. Both numerical and structural aberrations can be detected by this method.
3.7. Double-Strand Break HR Reporter Assay
Maintain U2-OS cells containing the stably integrated DR-GFP reporter in 5% CO2 atmosphere at 37 °C in a humid-ifier incubator in DMEM media. Grow cells in a T-25 flask and split cells after every 3 days.
When cells are around 70–80% confluent, transfect them using Amaxa Nucleofector per manufacturer’s instructions. In short, first trypsinize the cells and resuspend them in 100 μL of 1× transfection reaction solution. For each transfection add 18 μL of the supplement to 82 μL of Nucleofector Solution to make 100 μL of 1 transfection reaction solution. To the 1× transfection reaction add 2 μg of I-SceI expressing plasmid and 100 ng of ds-Red plasmid (see Note 35).
Transfer cell suspension into certified cuvette. All the sample must cover the bottom of the cuvette. Make sure there are no air bubbles. Close the cuvette with the cap.
Insert the cuvette with cell suspension into the Nucleofector cuvette holder and start the program number specific for U2-OS cells (X-001).
Take the cuvette out of the holder once the program is finished. Immediately add ~500 μL of warmed culture medium to the cuvette and gently transfer the sample into the 6-well plate containing 2 mL of DMEM media.
After 18–24 h, aspirate the media, wash cells with 1× PBS, and add fresh media. 72 h after transfection harvest the I-SceI and ds-Red cotransfected cells and the control cells. Control cells must include (1) cells transfected without I-SceI and ds-Red plasmids, (2) cells transfected with only I-SceI plasmid, and (3) cells transfected with only ds-Red plasmid.
Resuspend the cells in 300–500 μL of 1× PBS and transfer the mixture in the FACS tubes. Analyze the cells immediately after harvesting. Calibrate FACS by analyzing the cells transfected with only I-SceI plasmid, only ds-Red plasmid, and without I-SceI/ds-Red plasmids (see Note 36).
The percentage of GFP-positive cells represents the population of cells with accurate DSB repair and the ds-Red positive cells indicate the transfection efficiency. The relative efficiency of DSB repair is calculated as the ratio of the GFP positive cells to ds-Red positive cells (see Note 37).
4. Notes
Freshly made crystal violet stain solution can be stored in the dark at room temperature and used for up to 2 months.
The culture conditions for each cell line should be optimized as to provide the cells with sufficient surface area to grow without being confluent thereby avoiding contact mediated inhibition of growth.
To study the effect of DNA damaging agents on cellular proliferation, the culture conditions, including the concentration of the DNA damaging agents and duration of treatment, should be optimized according to cell type.
It is recommended to have “control” wells where the cells are only treated with the solvent that the DNA damaging drug is dissolved in. The number of cells in the “control” wells can be utilized to get the fold decrease or percentage decrease as a function of drug concentration.
Gentle washing of the cells with PBS is recommended. If cells are detached during washing, the PBS supernatant should be saved and centrifuged to recollect any detached cells.
The aperture should be free of any debris. To ensure this, the aperture should be washed with distilled water, “flushed” with the isotonic solution, and cleaned gently with a soft bristle brush before and in between measurements.
Since 500 μL of cell suspension was diluted with 10 mL of the isotonic diluent, a dilution factor of 21 is used. However, the cells can be further diluted and the dilution factor changed accordingly within the total volume ranging from 10 to 15 mL.
The absorbance value from the control cells should be used to quantify cell proliferation with drug treatment, as a fold change or percentage of the control value.
The duration of the incubation can be adjusted based on the seeding density, type of cell and kind of treatment.
Extended exposure of WST-1 to light should be avoided as it may cause increased reduction in WST-1 and increase the background absorbance readings.
The volume of media usually decreases around 10% over an incubation period of 3–5 days in a 96-well plate. Adjust the volume of WST-1 added according to the volume of medium in well.
The “blank” value is the background absorbance OD value and it generally increases with the incubation time. The blank value should be deducted from all readings while quantifying cell proliferation data.
Avoid forming bubbles while mixing the media. Introduction of air bubbles to the sample will lead to incorrect absorbance values.
Avoid exceeding the sensitivity of the microplate spectrophotometer reader (typically an optical density (OD) value range of 0–3). If the OD value exceeds the upper limit of the spectrophotometer, the number of cells seeded can be reduced and/or the incubation period minimized.
The number of cells can be adjusted according to the size of the plate and kind of treatment. Pilot experiments should be performed using serially diluted cells to determine the correct number of cells that allows the counting separate colonies without any overlap.
The washing and adding of methanol must be done carefully as colonies can detach from the plates. Tilting the tissue culture plate while washing and adding methanol can minimize the risk of dislodging the cells from the plate.
The colonies are now fixed and gently washing them with water will not dislodge the colonies.
Plates can be stored at room temperature for up to 50 weeks.
A cell culture control is recommended to measure the level of apoptosis in untreated cells that are seeded in a manner similar to those same cells exposed to a DNA damaging drug treatment. This control value will serve as a measurement of spontaneous apoptosis under the culture conditions used without treatment. Further, the “control” absorbance value can be used to normalize the absorbance values from cells treated with different concentrations of the drug.
Coating solution is prepared by diluting 10× Coating buffer to 1× with ddH2O and adding 1 mL of reconstituted anti-histone antibody to 9 mL of the 1× coating buffer. This should be prepared freshly before use.
The anti-histone antibody is biotin conjugated that in turn binds to the streptavidin coated microplate module.
Reconstitute the washing buffer from 10× to 1× by diluting with ddH2O at room temperature.
The wells should be washed carefully but thoroughly to avoid disturbing the coated surface of the microplate modules.
The ABTS substrate is prepared by dissolving 1 × 5 mg tablet in 5 mL of the substrate buffer at room temperature.
To obtain an optimal number of foci, we recommend standardizing the dose of DNA damaging agent and the treatment conditions by doing a pilot study prior to an actual experiment.
Some of the cell lines detach easily. We recommend adding the extraction and the washing buffers very gently and to check under microscope that the cells are not detaching.
Washes with the extraction buffer (steps 4 and 6) are required to obtain distinct ATR, ATRIP, Rad9, and RPA foci, and reduce background staining. This step is optional for some of the DNA Damage response proteins like γH2AX.
Longer duration of washes after the addition of the primary and secondary antibodies may help to reduce nonspecific signals.
This protocol can also be slightly modified to detect single stranded DNA by BrdU incorporation. Treat cells with 20 μM BrdU for 24 h. Repeat the steps exactly as mentioned above. Add anti-BrdU primary antibody (1:100, BD Pharmingen™) and goat anti-mouse secondary (1:1000 Invitrogen).
Incubation time with colcemid is crucial. Longer exposure time can result in shorter and thicker chromosomes and shorter exposure can result in fewer metaphase spreads.
Molarity of hypotonic solution is very important. It must be sufficient to swell the cells without lysing them.
Add the Carnoy’s Fixative solution to the pellet and vortex.
Drop the cells in the fixative agent onto the slides from a height of 6 in. and make sure that the slide is at a 45° angle.
Place the slides in 50% humidity and at 25 °C for quicker drying.
The quality of plasmid may affect the transfection efficiency. To obtain accuracy and consistency try to use same plasmid mix for one whole experiment.
Count at least 30,000 cells for each treatment group.
This protocol can be slightly modified to examine the effect of a gene product on HR repair. Cells can be treated with inhibitors/siRNA against a particular protein/gene before transfection with I-SceI and ds-Red plasmids.
Acknowledgments
This work is supported by the National Institutes of Health, National Institute on Aging.
References
- 1.Estep KN, Brosh RM Jr (2018) RecQ and Fe-S helicases have unique roles in DNA metabolism dictated by their unwinding directionality, substrate specificity, and protein interactions. Biochem Soc Trans 46:77–95. 10.1042/bst20170044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suhasini AN, Brosh RM Jr (2013) Disease-causing missense mutations in human DNA helicase disorders. Mutat Res 752 (2):138–152. 10.1016/j.mrrev.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Brabant AJ, Stan R, Ellis NA (2000) DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet 1:409–459. 10.1146/annurev.genom.1.1.409 [DOI] [PubMed] [Google Scholar]
- 4.Byrd AK, Raney KD (2012) Superfamily 2 helicases. Front Biosci (Landmark Ed) 17:2070–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilman B, Tijerina P, Russell R (2017) Distinct RNA-unwinding mechanisms of DEAD-box and DEAH-box RNA helicase proteins in remodeling structured RNAs and RNPs. Biochem Soc Trans 45(6):1313–1321. 10.1042/bst20170095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raney KD, Byrd AK, Aarattuthodiyil S (2013) Structure and mechanisms of SF1 DNA helicases. Adv Exp Med Biol 767:17–46. 10.1007/978-1-4614-5037-5_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trakselis MA (2016) Structural mechanisms of hexameric helicase loading, assembly, and unwinding. F1000Res 5. F1000 Faculty Rev-111. 10.12688/f1000research.7509.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brosh RM Jr, Majumdar A, Desai S, Hickson ID, Bohr VA, Seidman MM (2001) Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J Biol Chem 276 (5):3024–3030. 10.1074/jbc.M006784200 [DOI] [PubMed] [Google Scholar]
- 9.Guo M, Hundseth K, Ding H, Vidhyasagar V, Inoue A, Nguyen CH, Zain R, Lee JS, Wu Y (2015) A distinct triplex DNA unwinding activity of ChlR1 helicase. J Biol Chem 290 (8):5174–5189. 10.1074/jbc.M114.634923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohaghegh P, Karow JK, Brosh RM Jr, Bohr VA, Hickson ID (2001) The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res 29 (13):2843–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Karow JK, Hickson ID, Maizels N (1998) The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem 273 (42):27587–27592 [DOI] [PubMed] [Google Scholar]
- 12.Vaughn JP, Creacy SD, Routh ED, Joyner-Butt C, Jenkins GS, Pauli S, Nagamine Y, Akman SA (2005) The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4-DNA resolving activity in HeLa cell lysates. J Biol Chem 280 (46):38117–38120. 10.1074/jbc.C500348200 [DOI] [PubMed] [Google Scholar]
- 13.Bacolla A, Wang G, Jain A, Chuzhanova NA, Cer RZ, Collins JR, Cooper DN, Bohr VA, Vasquez KM (2011) Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J Biol Chem 286 (12):10017–10026. 10.1074/jbc.M110.176636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma S, Doherty Kevin M, Brosh Robert M (2006) Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J 398 (Pt 3):319–337. 10.1042/BJ20060450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y (2012) Unwinding and rewinding: double faces of helicase? J Nucl Acids 2012:140601. 10.1155/2012/140601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brosh RM (2013) DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer 13(8):542–558. 10.1038/nrc3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma S (2014) An appraisal of RECQ1 expression in cancer progression. Front Genet 5:426. 10.3389/fgene.2014.00426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arai A, Chano T, Futami K, Furuichi Y, Ikebuchi K, Inui T, Tameno H, Ochi Y, Shimada T, Hisa Y, Okabe H (2011) RECQL1 and WRN proteins are potential therapeutic targets in head and neck squamous cell carcinoma. Cancer Res 71 (13):4598–4607. 10.1158/0008-5472.can-11-0320 [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Brosh RM Jr (2007) Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS One 2(12):e1297. 10.1371/journal.pone.0001297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal M, Banerjee T, Sommers JA, Brosh RM Jr (2013) Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 12(20):3329–3335. 10.4161/cc.26320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aggarwal M, Banerjee T, Sommers JA, Iannascoli C, Pichierri P, Shoemaker RH, Brosh RM Jr (2013) Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional Fanconi anemia pathway. Cancer Res 73(17):5497–5507. 10.1158/0008-5472.can-12-2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM Jr (2011) Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci U S A 108 (4):1525–1530. 10.1073/pnas.1006423108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, Bachrati CZ, Schultz L, Sakurai M, Savitsky P, Abu M, McHugh PJ, Bohr VA, Harris CC, Jadhav A, Gileadi O, Maloney DJ, Simeonov A, Hickson ID (2013) A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol 20(1):55–62. 10.1016/j.chembiol.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estep KN, Butler TJ, Ding J, Brosh RM Jr (2017) G4-interacting DNA helicases and polymerases: potential therapeutic targets. Curr Med Chem. 10.2174/0929867324666171116123345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hengel SR, Spies MA, Spies M (2017) Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chem Biol 24(9):1101–1119. 10.1016/j.chembiol.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brosh RM Jr, Bohr VA (2007) Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res 35(22):7527–7544. 10.1093/nar/gkm1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce AJ, Johnson RD, Thompson LH, Jasin M (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13(20):2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berridge MV, Herst PM, Tan AS (2005) Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 11:127–152. 10.1016/s1387-2656(05)11004-7 [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Shin-ya K, Brosh RM Jr (2008) FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol 28 (12):4116–4128. 10.1128/mcb.02210-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khodarev NN, Sokolova IA, Vaughan AT (1998) Mechanisms of induction of apoptotic DNA fragmentation. Int J Radiat Biol 73 (5):455–467 [DOI] [PubMed] [Google Scholar]
- 31.Bennett BT, Bewersdorf J, Knight KL (2009) Immunofluorescence imaging of DNA damage response proteins: optimizing protocols for super-resolution microscopy. Methods 48 (1):63–71. 10.1016/j.ymeth.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awasthi P, Foiani M, Kumar A (2015) ATM and ATR signaling at a glance. J Cell Sci 128 (23):4255–4262. 10.1242/jcs.169730 [DOI] [PubMed] [Google Scholar]
- 33.Mirzoeva OK, Petrini JH (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 21 (1):281–288. 10.1128/MCB.21.1.281-288.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson JY, Seidman MM, Brosh RM Jr (2013) Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol 33(11):2212–2227. 10.1128/mcb.01256-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8(3):255–265. 10.1016/j.ccr.2005.08.004 [DOI] [PubMed] [Google Scholar]
- 36.Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin-ya K, Teulade-Fichou MP, Kisker C, Brosh RM Jr (2013) Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J Biol Chem 288:28217–28229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henderson A, Wu Y, Huang YC, Chavez EA, Platt J, Johnson FB, Brosh RM Jr, Sen D, Lans-dorp PM (2014) Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res 42:860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-Ya K, Kitao H, Brosh RM Jr (2010) Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood 116:3780–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerjee T, Sommers JA, Huang J, Seidman MM, Brosh RM Jr (2015) Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr Biol 25:2830–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC (2000) Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep 1:80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, Kao HI, Bambara RA, Brosh RM Jr (2004) WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol Biol Cell 15:734–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM Jr (2011) Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J 30:692–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM Jr (2007) FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood 110:2390–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]