

SUMMARY
The Epidemiologic Study of Cystic Fibrosis (ESCF) was a prospective observational study of over 32,000 people with cystic fibrosis (CF) from 250 clinical care sites in North America from 1994 to 2005. Begun as a pharmacovigilance study in connection with the approval of dornase alfa in 1993, ESCF was open to all people with CF treated at any participating site in the United States or Canada. In addition to obtaining safety and effectiveness data on dornase alfa, ESCF collected encounter-based data to characterize the natural history and management of CF with a special focus on lung disease. During the study, 32,178 patients reported at least one encounter, contributing 869,136 encounters, 622,592 pulmonary function tests, 432,896 cultures, and 118,563 pulmonary exacerbations treated with intravenous antibiotics. Although ESCF data collection concluded in 2005, through a collaboration with the U.S. Cystic Fibrosis Foundation Patient Registry, additional follow-up data through 2017 was available for two-thirds of patients. This allowed for updating of CF genotype and survival information. Fifty-six peer-reviewed publications (cited over 3600 times) resulted from this study. In this manuscript we summarize the published ESCF manuscripts in thematic groups with key study findings and brief comments, and speculate on how ESCF findings will inform future data registries and patient care practices.
Keywords: cystic fibrosis, epidemiology, lung function, pulmonary exacerbation
INTRODUCTION
The Epidemiologic Study of Cystic Fibrosis (ESCF) was an observational study begun in 1993 and ultimately spanning more than 10 years of data collection. There have been 56 peer-reviewed publications [1-56] that have been cited over 3600 times (mean 65, median 43). ESCF was initiated in response to an FDA request for post marketing pharmacovigilance on the newly approved therapy dornase alfa (Pulmozyme®) and was sponsored by Genentech, Inc. The study was designed to obtain safety and effectiveness data on dornase alfa and to use encounter-based data collection to characterize the natural history and management of CF with a special focus on lung disease [1].
Although begun in connection with the approval of dornase alfa, ESCF was open to all patients with CF in the United States or Canada; by 1996, 20,000 patients were enrolled. A total of 250 sites and 28,909 patients had 675,117 encounters recorded in the first phase of ESCF, which used paper case report forms (CRFs). In 2003, data collection was changed from paper-based to electronic and sites and patients were re-enrolled. There were 184 sites and 17,963 patients who contributed 194,019 encounters using the electronic CRF. Data collection concluded 12/21/2005. Throughout the study a total of 32,178 patients reported at least one encounter, contributing 869,136 encounters, 622,592 pulmonary function tests, 432,896 cultures, and 118,563 pulmonary exacerbations treated with intravenous (IV) antibiotics (IV exacerbations).
Although ESCF data collection concluded in 2005, through a collaboration with the U.S. Cystic Fibrosis Foundation Patient Registry (CFFPR) additional follow-up data has been available for the two-thirds of patients who provided CFFPR patient identification numbers [57]. This allowed for updating of CF genotype and survival information.
In this manuscript we summarize the published ESCF manuscripts in thematic groups with key study findings and brief comments. We then speculate on how ESCF findings will inform future data registries and patient care practices.
PATIENT STATUS, PRACTICE PATTERNS, AND CHANGE OVER TIME
Over the 12 years of ESCF, there were striking changes in therapeutic practice patterns (including several new therapies) and improved patient outcomes that were captured in the encounter-based data collection.
Practice patterns [2,3].
The long-term nature of ESCF allowed for both cross-sectional characterization and longitudinal evaluation of patient status, practice patterns, and their relationships. Adherence to the 1990 CF practice guidelines [58] varied: 57.5% had ≥4 visits (but only 27.4% had ≥4 routine visits), 75.8% had ≥2 spirometries, 79.3% had ≥1 respiratory culture, and 68.3% had ≥1 radiograph. Use of therapies showed indication bias (sicker patients were more likely to be treated).
Center-based analysis of practice patterns [7].
Variation in care patterns across CF centers provides an opportunity to address indication bias by comparing outcomes at different sites. Practice patterns in 1995-1996 were compared between top and bottom quartile sites based on median lung function as percent predicted forced expiratory volume in 1 second (ppFEV1) by age group (6-12, 13-17, and 18+ years). Patients at top quartile sites had more frequent monitoring of clinical status, spirometry, and respiratory cultures and generally received more interventions, especially IV antibiotic treatments.
Practice patterns for young children [12].
A similar approach was used to compare care patterns for young children (age 0 to 3 years) based on each site’s ppFEV1 for 6- to 12-year-olds. Top quartile sites had more CF infants diagnosed by family history or newborn screening and correspondingly fewer infants diagnosed because of symptoms. Infants at top quartile sites had more visits; more respiratory tract cultures; and more frequent use of IV antibiotics, oral corticosteroids, mast cell stabilizers, and mucolytics; but they received less chest physiotherapy, inhaled bronchodilators, oral nutritional supplements, and pancreatic enzymes.
Trends in inhaled antibiotic use [15].
An increase in chronic inhaled antibiotic use from 1996 through 2005 was accompanied by a decrease in acute use to treat exacerbations.
Trends in pulmonary function and respiratory signs and symptoms [16].
From 1995-2005, pulmonary function improved and the percentages of patients reporting cough or sputum production or having crackles or wheeze decreased. Changes in pulmonary function were not consistently mirrored by changes in symptoms.
Trends in use of routine therapies [23].
From 1995 to 2005, use of airway clearance, inhaled bronchodilators, dornase alfa, inhaled corticosteroids, inhaled antibiotics, oral nutritional supplements, and insulin/oral hypoglycemic agents increased substantially, while use of mast cell stabilizers and oral bronchodilators decreased. Three therapies not tracked in 1995, oral macrolide antibiotics, leukotriene inhibitors/antagonists, and inhaled hypertonic saline, had notable use by 2005.
Choice of dornase alfa regimen [52].
Dornase alfa was approved with two regimens: 2.5 mg inhaled once daily (QD) or twice daily (BID), with little guidance as to when to use each regimen. Patients beginning BID regimens 1994-2005 had worse lung function and more frequent IV-treated exacerbations than those beginning QD regimens. At the time of regimen switch, patients switching from QD to BID had more exacerbations and more signs and symptoms, whereas patients switching from BID to QD did not. Switching from QD to BID was associated with increased exacerbations; deterioration in lung function did not appear to be a driver for this switch. Patients switching from BID to QD were clinically stable, on average, suggesting that treatment burden and cost may have been drivers of the decision to switch regimens.
Socioeconomic status (SES) and treatment [19,27].
Starting in 2003, median household income by zip code, maternal educational attainment, and state insurance coverage were collected. There were no differences in chronic therapy use related to SES. Similarly, SES had little effect on treatment for exacerbation with any antibiotic. However, IV antibiotics were prescribed more frequently for patients with lower SES.
Comment:
Through quality improvement programs and studies that have shown the effectiveness of a range of therapies, there has been an increase in the use of therapies and a concomitant improvement in health outcomes. The disparities in health outcomes associated with low SES need further study.
RISK FACTORS IN EARLY CHILDHOOD
Early diagnosis and management of children with CF can have a profound effect on their health later in life. Beyond newborn screening, recognition of risk factors that manifest in early childhood may improve outcomes.
Risk factors for poor lung function at age 6 years [8].
Growth/nutritional indexes (weight for age (WFA), height for age (HFA), and percent ideal body weight) and pulmonary signs and symptoms (cough, sputum, clubbing, crackles), and presence of Pseudomonas aeruginosa (Pa) at age 3 were strongly associated with ppFEV1 at age 6 years (Table 1). Analysis of forced vital capacity (FVC) and FEV1/FVC ratios indicated that deficits in lung function at age 6 years were largely due to smaller lungs and not obstructive in nature.
Table 1:
Signs and symptoms of lung disease at age 3 years and pp FEV1 at age 6 years (mean ± SD)
| Characteristic | ppFEV1 With | ppFEV1 Without | P value |
|---|---|---|---|
| Cough | 95 ± 21 | 100 ± 19 | < 0.001 |
| Sputum | 93 ± 22 | 98 ± 20 | 0.001 |
| Clubbing | 94 ± 22 | 97 ± 20 | 0.035 |
| Crackles | 88 ± 22 | 97 ± 20 | < 0.001 |
| Pa | 92 ± 21 | 100 ± 19 | < 0.001 |
Pa, Pseudomonas aeruginosa. Adapted from [8] Table 4 p. 628
Risk factors for persistence of respiratory signs and symptoms [31].
Persistence of signs and symptoms (cough, sputum, clubbing, crackles, and wheeze) was studied over time in patients <4 years of age at enrollment. Earlier onset was associated with pancreatic enzyme use, presence of Pa, and prior diagnosis of asthma. Signs and symptoms of lung disease begin early and especially for cough and crackles a majority of those eventually affected have symptoms by age 2 years (Figure 1).
Figure 1.
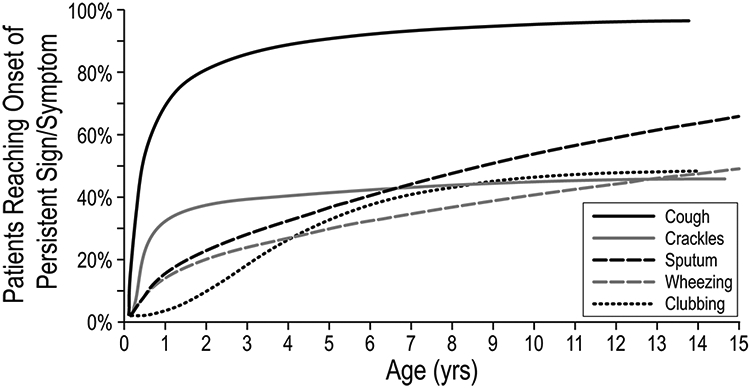
FIGURE 1Percentage of patients reaching onset of persistent sign/symptom by age. Adapted with permission from Pediatric Pulmonology
Wheeze in early childhood [42].
Any history of wheeze in the first 6 years of life (61% of patients) was associated with lower ppFEV1 at age 6 to 8 years (Table 2).
Table 2:
Wheezing phenotype and ppFEV1 at age 6y to <8y (adjusted mean ± SE)
| Wheezing | ppFEV1 at age 6y to <8y |
|---|---|
| No Wheeze (no wheeze by age 6y) | 104.0 ± 0.8 |
| Transient wheeze (before age 3y but not after age 3y) | 97.6 ± 0.9 |
| Late Wheeze (after age 3y) | 100.0 ± 1.6 |
| Persistent Wheeze (before age 3y and after age 3y) | 95.7 ± 1.0 |
Adapted from [42] Table 2 p. 748
BMI and BMI percentile in children [48].
Percent ideal body weight (%IBW), BMI, and BMI percentile have been used as composite measures of nutritional status, although %IBW has been discredited. However, BMI and BMI percentile fail to identify a substantial proportion of children 2-18 years with CF who are stunted or have potentially poor nutritional status as measured by WFA and/or HFA percentile.
Risk factors for early mortality [50].
CFFPR mortality data in 2013 linked to ESCF data 1994-2005 showed that 5.7% of patients died before age 18 years. Mortality risk factors included sign and symptoms, demographics, Pa, and WFA (Figure 2). Addition of ppFEV1 to the model had minimal effect on the other factors.
Figure 2.
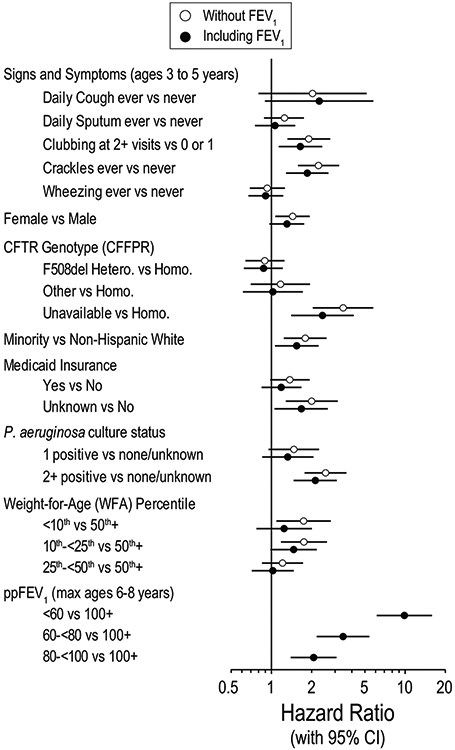
Risk factors for mortality before age 18 years. Hazard ratios for two survival models: one that does not include ppFEV1category as a predictor (open circles) and one that includes ppFEV1category as a predictor (filled circles). Adapted with permission from Pediatric Pulmonology.50CFFPR, Cystic Fibrosis Foundation Patient Registry; ppFEV1, percent predicted forced expiratory volume in 1 s
Comments:
These findings suggest that aggressive intervention early in life aimed at growth/nutrition status, and pulmonary signs/symptoms may lead to better pulmonary function later in childhood. The BMI finding points out the importance of not relying on a single measure for assessing growth and nutritional status. The predictive power of clubbing and crackles at age 3 to predict mortality even after including FEV1 as a predictor strongly suggest that they are important clinical findings that should inform care and be routinely collected in patient registries and observational studies of therapeutic effectiveness.
AIRWAY INFECTION AND LUNG DISEASE PROGRESSION
CF is characterized by complex chronic airway infections that contribute to inflammation and lung disease progression but demonstrating relationships between bacterial infections and outcomes is difficult, requiring at minimum comprehensive microbiologic information and extended observation periods.
Microbiology procedures [6].
In a site survey of pulmonary specimen collection and microbiology laboratory procedures, differences in culture frequency and conditions contribute to differences in species prevalence across sites.
Allergic bronchopulmonary aspergillosis (ABPA) [5].
Only 2% of patients had ABPA in 1994-1996 with substantial regional differences. In 2018, prevalence was 5.4% [57].
Methicillin resistant S. aureus (MRSA) [13].
Among the 7.5% of patients who had S. aureus but no other organisms detected in 2001, the 11% with MRSA had mean ppFEV1 lower by about 9 points than those with methicillin-sensitive S. aureus (MSSA). Likelihood of hospitalization and treatment with oral, inhaled, and IV antibiotics were all significantly higher in MRSA patients.
MRSA and change in FEV1 [18].
Compared to patients who remained MRSA-negative, the 12% of patients ≥6 years old with incident MRSA detection 2001-2003 had lower baseline ppFEV1, received more antibiotic and other therapies, and had a higher rate of ppFEV1 decline both before and after the incident culture. However, incident MRSA detection was not associated with a statistically significant change in rate of ppFEV1 decline.
Multiple antibiotic-resistant Pa (MARPA) and change in FEV1 [32].
Incident MARPA was defined as Pa (1) resistant to gentamicin and either tobramycin or amikacin, and (2) resistant to ≥1 antipseudomonal beta lactam after ≥2 years of Pa-positive but MARPA-negative respiratory cultures. The 25.5% MARPA-incident patients 1996-2003 had lower baseline ppFEV1 and received more oral and inhaled antibiotic therapies. Decline in ppFEV1 the two years before MARPA detection was similar to the decline two years after detection.
Treatment of newly detected Pa [36].
The EPIC (Early Pseudomonas Infection Control) study used ESCF data as historical controls. Protocol-based therapy for newly detected Pa resulted in lower Pa recurrence rates but comparable hospitalization rates compared to historical controls.
Comment:
These findings support the recommendation for a minimum of four respiratory cultures per year. MRSA and MARPA are more likely to be a marker of more severe disease and more intensive therapy, and less likely to be contributing independently to more rapid lung function decline.
DIAGNOSIS OF PULMONARY EXACERBATIONS
Although pulmonary exacerbations are important clinical events, there is no consensus as to what clinical presentations constitute exacerbation. Epidemiologic definitions of exacerbation often rely on antibiotic treatment and specifically IV treatment.
Clinical characteristics associated with exacerbations [9].
The four clinical characteristics most associated with exacerbation treatment (subsequently termed “Rabin criteria”) differed by age group (Table 3). Although Rabin criteria were likely to be present when exacerbations were diagnosed, the converse was not the case: presence of Rabin criteria was not a useful predictor of exacerbation diagnosis.
Table 3:
Clinical characteristics most associated with exacerbation treatment by age group
| Age <6 years | Age 6-12 years | Age 13-17 years | Age 18+ years |
|---|---|---|---|
| New crackles | Decline in ppFEV1 | Decline in ppFEV1 | Decline in ppFEV1 |
| Increased cough | Increased cough | Increased cough | New crackles |
| Decline in WFA | New crackles | New crackles | Hemoptysis |
| Increased sputum | New Pa | Hemoptysis | Increase cough |
Adapted from [9] Table 3 p. 403
Acute FEV1 drop [40]
Nearly one-third of children 6-12 years and one-fourth of adolescents 13-17 years with acute relative FEV1 drop ≥10% were neither hospitalized nor treated with a new antibiotic course, even though their mean drop was near 20%. (Figure 3A) Those with the best lung function were one-sixth as likely to receive IV antibiotics as those with the worst lung function, suggesting a missed opportunity to preserve lung function and possibly contributing to the more rapid decline observed in children with high FEV1.
Figure 3.
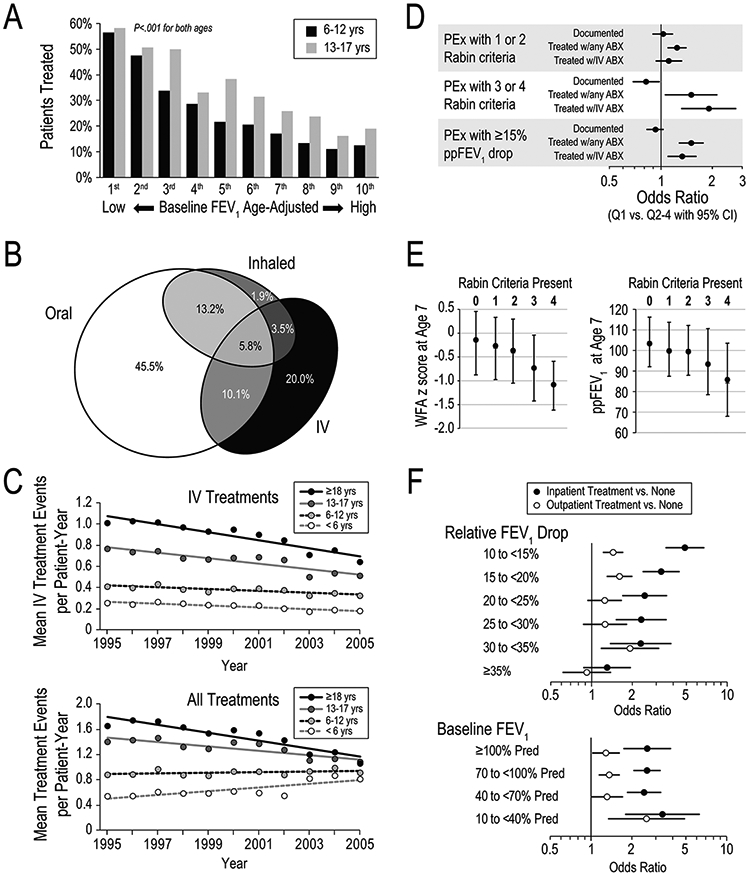
Treatment of PEx. Panel A. Percentage of children and adolescents treated with antibiotics after acute≥10% relative FEV1 drop stratified by ppFEV1 decile and age group. Adapted with permission from the Journal of Pediatrics.40Panel B. Area-proportional diagram of antibiotic treatments of clinician-identified exacerbations by route of administration. Adapted with permission from Pediatric Pulmonology.35Panel C. Mean antibiotic treatment events per patient‐year by IV route (upper panel) and by any route (lower panel). Adapted with permission from the Journal of cystic fibrosis.37Panel D. Documentation and antibiotic (ABX) treatment of PEx identified by Rabin Criteria or by≥15ppFEV1 drop. Odds ratios of treatment are shown for top quartile sites (Q1) versus other quartiles (Q2–Q4). Adapted with permission from Pediatric Pulmonology.44Panel E. Percentage of young children treated with antibiotics by number of Rabin Criteria present. Adapted with permission from Pediatric Pulmonology.34Panel F. Odds ratios for recovery of≥90% of baseline ppFEV1 after PEx for patients receiving in patient (filled circles) or outpatient (open circles) treatment as a function of ppFEV1 drop at diagnosis (upper panel) and Baseline ppFEV1(lower panel). Adapted with permission from the Annals of the American Thoracic Society.49IV, intravenous; PEx, pulmonary exacerbation; ppFEV1, percent predicted forced expiratory volume in 1 s
Acute FEV1 drop events and clinician-diagnosed exacerbation events [55]
Among ≥10% relative FEV1 drop events, 71% resulted in antibiotic treatment, of which two-thirds were IV. Of the clinician-diagnosed exacerbations, 32% had a drop ≥10%; 37% were treated intravenously. That is, a ≥10% relative FEV1 drop was not always considered an exacerbation and a lack of drop did not rule out an exacerbation diagnosis.
Comment:
The lack of treatment of substantial decline events was surprising and led to further investigations (see below).
TREATMENT OF PULMONARY EXACERBATIONS
Although ESCF collected information on treated exacerbations from inception, beginning in 2003 clinicians identified exacerbations which might or might not have been treated.
Exacerbation treatment and change in lung function [35].
Clinician-identified exacerbations were treated with oral (73%), inhaled (24%), and/or IV (39%) antibiotics (Figure 3B), implying that limiting analyses to IV exacerbations excludes over 60% of exacerbations. Mean ppFEV1 acutely improved 5.1 following IV and 2.0 following non-IV treatment, but regardless of route ppFEV1 declined 3.8 from the previous year as of 180 days after the exacerbation. Lung function loss was similar for patients with only one exacerbation and those with none.
Trends in exacerbation treatment [37].
Overall treatment incidence rate and IV incidence fell between 1995 and 2005, while non-IV incidence increased in children ≤12 years and decreased in older patients. (Figure 3C) Treatment thresholds (Rabin scores [9]) lowered over time, except for IV treatment in older patients. The overall decrease in treatment implies that lower treatment thresholds were more than offset by reduced treatment incidence (presumably resulting from improved health).
Exacerbation treatment by site [44].
High quartile sites (based on median FEV1) were more likely to treat exacerbations identified by Rabin criteria or by drop of ≥15 ppFEV1. (Figure 3D)
Exacerbation treatment outcomes in young children [34].
The number of Rabin criteria present in children <6 years old during a six-month evaluation predicted a higher hospitalization rate over the following year as well as lower WFA and lower ppFEV1 at age 7 years. Antibiotic treatment within 7 days before and 28 days after the encounter was associated with a lower proportion of children with crackles, cough, and Pa at follow-up (Figure 3E).
FEV1 recovery after exacerbation treatment [49].
All exacerbation treatments (hospitalizations, home IV antibiotics, new inhaled antibiotics, new oral quinolones, or other oral antibiotics) were associated with an increased likelihood of recovery to at least 90% of baseline lung function compared with no treatment, and inpatient treatment was better than outpatient treatment (Figure 3F). These findings were similar irrespective of baseline lung function.
Inpatient versus outpatient exacerbation treatment [53].
Employing three alternative techniques to control for indication bias (including instrumental variables), multiple response criteria, and multiple measures of inpatient versus outpatient administration, inpatient IV antibiotic treatment was consistently associated with higher likelihood of FEV1 recovery. Treatment response was not related to duration of IV therapy.
Evaluation of lung function recovery after exacerbation treatment [56].
Complete ppFEV1 recovery of baseline at follow-up was dependent on baseline definition, timing of assessment, and the ppFEV1 difference between baseline and treatment start. Lung function recovery as return to baseline is prone to artifact because of inherent FEV1 variability, sampling methodologies, and underlying disease progression.
Comment:
Research is needed to better understand how clinicians identify exacerbations and when and how they choose to treat (inpatient IV, outpatient IV, inhaled, or oral antibiotics). Special attention is needed for exacerbations treated only with oral or inhaled antibiotics in the outpatient setting. Acute drops of ppFEV1 are frequently not treated despite the evidence that treatment improves lung function irrespective of baseline level.
PREDICTING FUTURE LUNG FUNCTION
Chronic progressive lung disease is the most frequent cause of mortality in patients with CF.
Risk factors for lung function decline in children and adolescents [14] and in adults [33].
For children and adolescents, high baseline ppFEV1 and persistent crackles were significant risk factors for three-year ppFEV1 decline, and risk factors in some age groups female sex, presence of Pa, low WFA, sputum, wheezing, sinusitis, IV exacerbations, elevated liver function tests, and pancreatic insufficiency (Figure 4A). For adults 18-24 years, Burkholderia cepacia, pancreatic enzyme use, MARPA, and female sex predicted greater decline; low baseline ppFEV1 and sinusitis predicted less decline. For the ≥25y group, only pancreatic enzyme use predicted greater decline; low baseline ppFEV1 and sinusitis predicted less decline.
FIGURE 4.
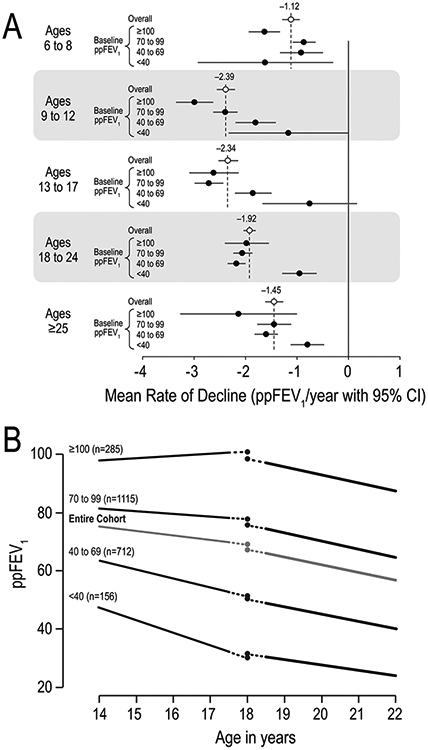
Lung function decline by age and baseline ppFEV1.Panel A. Mean rates of ppFEV1 decline by age group. Overall rates (open circles) and rates stratified by baseline ppFEV1 category (filled circles) adjusted for other risk factors are shown. Adapted with permission from the Journal of Pediatrics and the Journal of CysticFibrosis.14,33Panel B. Average changes in ppFEV1 around age 18years by ppFEV1 category adjusted for other risk factors. Adapted with permission from Pediatric Pulmonology.30ppFEV1, percent predicted forced expiratory volume in 1 s
Pulmonary outcome prediction (POP) tools [22].
Simple integer-based pulmonary outcome prediction (POP) tools to estimate lung function at age 6 in patients aged 2–5 years (POP2–5) and lung function change over a 4-year period in patients aged 6–17 years (POP6–17) were developed using variables available at clinical visits. Both tools included WFA, clubbing, crackles, wheeze, number of exacerbations, and presence of Pa. POP2-5 added daily cough; POP6-17 added age, sex, and ppFEV1.
Risk factors for lung function decline in young adulthood [30].
Rate of ppFEV1 decline was greater in young adulthood (18.5 to 22.0 years) than in adolescence (14.0–17.5 years). Factors during adolescence that predicted substantial decline (≥5 ppFEV1 per year) in young adulthood were: slower rate of FEV1 decline, greater FEV1 variability, faster BMI decline, male sex, chronic inhaled antibiotics, presence of Haemophilus influenzae, and absence of MARPA. Additional risk factors were lower than expected FEV1 and BMI around age 18 (Figure 4B).
Year-to-year changes in FEV1 [21].
The year-to-year change in ppFEV1 for individuals (using the highest ppFEV1 for each year) was compared to the change in the population median for the corresponding ages. Individual ppFEV1 decreased 1–3 points per year, with maximal decreases in 14–15 year olds. Population changes agreed with individual changes up to age 15. After age 30, population change approximated zero while individual ppFEV1 decreases were 1–2 points per year, indicating loss of FEV1 is a persistent risk in 6–45 year old CF patients (Figure 5).
FIGURE 5.
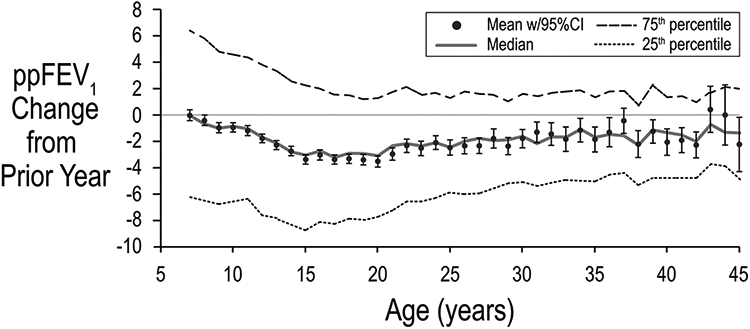
Change in lung function by age. mean, median, and quartile changes in best recorded ppFEV1 from the prior year are shown as a function of age. Bars represent 95% CI. Adapted with permission from the Journal of Cystic Fibrosis. 95% CI, 95% confidence interval; ppFEV1, percent predicted forced expiratory volume in 1 s
Using decline in lung function to predict future decline [46].
For each year of age, the best ppFEV1 and associated ppFVC and ppFEF25–75, were used to calculate 2-year slopes. Slopes were most negative among those 13–17 years old, especially for FEF25–75. Rate of lung function decline had a moderate correlation with later level of lung function, but contrary to expectations did not predict future rate of decline 3-8 years later either within or across spirometric variables.
Lung function variability [47].
Five different ppFEV1 variability measures from a 2-year baseline period were predictive of the best ppFEV1 in the subsequent 2 years both alone and after controlling for baseline demographic and clinical factors and the slope and level of ppFEV1. The easy-to-calculate median deviation from the best ppFEV1 in the baseline period had the highest contribution to explanatory power and exceeded the total contribution of all other factors excluding the rate of ppFEV1 decline (Figure 6). Additional analyses revealed that number of exacerbations did not explain much of the variability [59,60].
FIGURE 6.
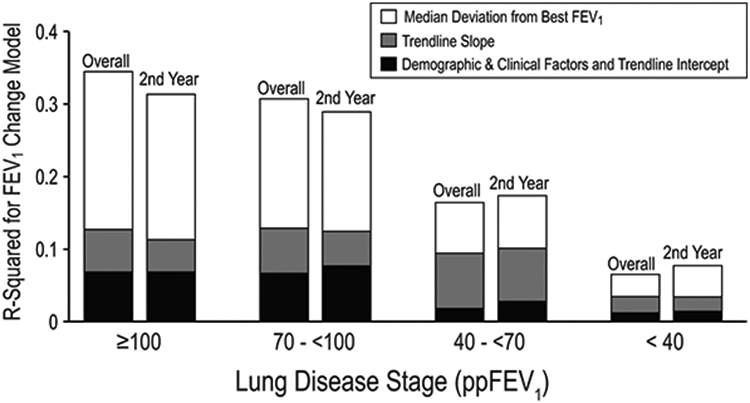
Prediction of best ppFEV1 in subsequent 2 years after2-year baseline. Contribution to percentage of variance explained(R2) in a regression model of ppFEV1 variability (as measured by median deviation from best ppFEV1; open bars), trendline slope (rate of ppFEV1 decline; gray bars), and all other factors (demographics, clinical factors, and trendline intercept; black bars). Adapted with permission from the Journal of Pediatrics. ppFEV1, percent predicted forced expiratory volume in 1 s
Comment:
Clinicians should not be reassured by high lung function, particularly in young children, because it is associated with more rapid decline in ppFEV1. This is possibly due to failure to treat acute drops in lung function in patients with high ppFEV1. Crackles predict future lung function decline in all ages and warrant attention in clinical care as well as research studies and are now captured in the CFFPR. Variability in ppFEV1 is getting increased attention and may play an important role not only as a predictor but also as a potential surrogate variable for lung function decline.
DIABETES, PREGNANCY, AND LIVER DISEASE
Cystic fibrosis-related diabetes (CFRD) [10].
Patients with CFRD (defined as using insulin or an oral hypoglycemic agent) had more liver disease, more advanced lung disease, more exacerbations, and worse nutritional status. Previously reported CFRD risk factors of older age, female sex, and pancreatic insufficiency were confirmed.
Relationship between pregnancy, therapies and health outcomes [11].
Pregnant women (n=216) were compared with a group of never-pregnant women matched using propensity scores. Baseline ppFEV1 was higher for the pregnant women but declines from baseline to follow-up were not different. Compared to the nonpregnant group, outpatient visits for pregnant patients were more frequent before and increased during pregnancy. Exacerbations and hospitalizations were similar between the groups at baseline but increased during pregnancy. Treatment for diabetes rose from 9.3% to 20.6% during pregnancy and then dropped to 14.4%. Although pregnant women experienced similar respiratory and health trends, they used a greater number of therapies and received more intense monitoring than nonpregnant women.
Pregnancy and motherhood [38].
A group of 119 women reporting a pregnancy and followed a median of 6.0 years from the pregnancy were propensity-score matched with never-pregnant women. No group differences were found in change from baseline for FEV1 or BMI, in respiratory signs and symptoms, or in prescribed chronic therapies. Women who had been pregnant had lower health-related quality of life (HRQOL) scores, more exacerbations and more illness-related visits but showed no increase in chronic therapies.
Liver disease in Hispanics [43].
Abnormal liver findings were present in 20.8% of Hispanic patients compared with 16.0% of non-Hispanic white patients. This higher prevalence of liver involvement persisted after adjusting for demographics and meconium ileus and was most pronounced in the first year of life. Follow up of infants with elevated liver enzymes found that 10% progressed to more severe liver disease.
Comment:
Research to understand what occurs in patients before diagnosis with CFRD. With improvements in survival, CFRD is likely to become a more important problem. Women with CF can successfully navigate pregnancy and motherhood. Disease outcomes and associated risk factors for Hispanics and other minorities with CF need further investigation.
HEALTH-RELATED QUALITY OF LIFE (HRQOL)
Patient-reported outcomes are increasingly used in clinical trials to assess the natural history of chronic diseases and the efficacy of new treatments. Beginning in 2003, there were 14,258 Cystic Fibrosis Questionnaire-Revised (CFQ-R) forms completed by 7,763 patients and/or their parents from 160 sites.
Socioeconomic status (SES) and HRQOL [20].
Low SES was associated with lower CFQ-R scores on a majority of questionnaire domains. Even after controlling for lung function and SES, African American and Hispanic patients reported worse emotional and social functioning.
Change in CFQ-R [24].
Increased respiratory signs/symptoms were associated with worse CFQ-R Respiratory Symptom scores and declining weight was associated with worse scores on nutritional health domains. A Treatment Complexity Score (TCS) based on 37 chronic therapies was developed. For parents, increases in TCS were associated with worsened CFQ-R Treatment Burden scores.
Treatment complexity [39].
Treatment complexity as measured by the TCS were highest in adults and increased over the three years 2003-2005 for all ages. Sites were stratified by quartiles based on ppFEV1, BMI, or CFQ-R Treatment Burden score. TCS did not differ by ppFEV1 quartile; was higher in the highest BMI quartile; and was lower in the CFQ-R Treatment Burden quartile with lowest burden.
CFQ-R psychometrics [29].
The CFQ-R showed strong internal consistency reliability, few floor or ceiling effects, good discrimination between clinically defined groups, strong parent-child agreement on respiratory symptoms, and convergence between CFQ-R scales and health outcomes. Females reported better HRQOL than males on scales related to body image and weight and worse HRQOL on other scales.
Comment:
Health-related quality of life measures such as those from the CFQ-R are increasingly used in clinical care as well as in research.
DESIGN OF PROSPECTIVE STUDIES
When designing a clinical trial, study duration, outcome measures, and inclusion/exclusion criteria need to be determined. Observational studies can inform these decisions.
FEV1 decline as endpoint [25].
Increased study duration and exclusion of lower risk patients both substantially reduced sample size requirements but increasing visit frequency had only modest impact. Studies of 1.5 years in duration appeared feasible, but shorter studies are unlikely to be adequately powered, irrespective of visit frequency.
Exacerbation rates as endpoint [28].
Rates of clinician-identified exacerbations (those treated with any antibiotic or only those treated with IV antibiotics) were estimated for durations from 3 to 12 months for subpopulations stratified by age, FEV1, sex, WFA percentile, respiratory signs and symptoms, and history of exacerbations and bacterial culture. Prospective studies including patients with lower ppFEV1, older age, female sex, recent history of exacerbations, and presence of Pa required lower sample sizes.
Pulmonary function outcomes [45].
Six potential outcome variables (best FVC, FEV1, and FEF25–75 in 2002 and rate of decline for each variable from 2000 to 2002) were used to rank sites and associations with practice patterns and follow-up pulmonary function assessed. Top quartile sites had more frequent monitoring, treatment of exacerbations, and use of chronic therapies and oral corticosteroids, but follow-up rates of FEV1 decline were not predicted by site ranking. Annual FEV1 was at least as good as any other pulmonary function measure. The current site ranking only moderately predicted future ranking.
Choice of FEV1 reference equation [51].
The dornase alfa rate of decline study [26] was reanalyzed to compare the effect of using different reference equations: Knudson [61], Wang [62] & Hankinson [63], Stanojevic [64], and GLI [65]. The study conclusions about acute change and change in rate of decline were minimally affected. Based on this analysis, the recent GLI equations were recommended for future studies, but prior results based on older equations should be accepted as reliable.
Comment:
Observational studies such as ESCF can provide guidance for clinical trial design. While choice of ppFEV1 reference equations matters for individual care decisions, intervention evaluations are not greatly affected.
COMPARATIVE EFFECTIVENESS
In contrast to randomized clinical trials, which can determine the efficacy of an intervention, observational studies such as ESCF are ideal for determining the real-world effectiveness of interventions. Although limited by various types of bias, especially indication bias, observational studies have the strengths of large sample sizes, broad inclusion criteria, and extended observation periods. These strengths allow the evaluation of long-term outcomes such as long-term lung function decline or survival that are not practical in clinical trials.
Dornase alfa [4,26].
The randomized controlled trial for dornase alfa demonstrated a 5.8 ppFEV1 sustained improvement in ppFEV1 [66]. In a 1997 analysis, dornase alfa patients in ESCF age ≥6 years with baseline ppFEV1 ≥40 had lower pulmonary function, more bacterial infections, and more exacerbations at baseline than untreated comparators [4]. Treated patients had a 3.9 ppFEV1 increase compared with a 1.6 ppFEV1 decrease for untreated patients that was sustained for a year.
In a later study using data through 2005, a change-point model was used to relate initiation of dornase alfa to acute ppFEV1 change and subsequent change in rate of ppFEV1 decline, an endpoint not previously explored [26]. Change-point analysis of lung function looks before and after an event (such as the initiation of a new therapy) to see if there is an acute change (change in intercept) or a trend change (change in slope). This analysis introduced innovative methods, described in some detail in the online supplement [26]. Age-specific ppFEV1 deciles accounted for the fact that patients with relatively high lung function have a shallower decline before and a steeper decline after an arbitrary point in time and the opposite for those with relatively low lung function. In addition, patients could contribute multiple four-year time spans to the comparator group (as was done for the inhaled corticosteroid analysis) but in addition dornase alfa patients could be included in the comparator group up to the time they received dornase alfa. Sensitivity analyses showed that this approach had greater power than using just one four-year time interval chosen at random and was additionally less biased than using the first or last possible interval.
Patients treated with dornase alfa had lower ppFEV1 at treatment initiation and a more rapid decline prior to treatment relative to comparators. There was an acute improvement and the rate of ppFEV1 decline was reduced by 32.1% among children (95% CI 15.1% to 46.8%) and by 28.8% among adults (−1.7% to 51.9; Figure 7A). For the comparators, there was no material change in intercept and the rate of decline worsened in children and did not change in adults.
FIGURE 7.
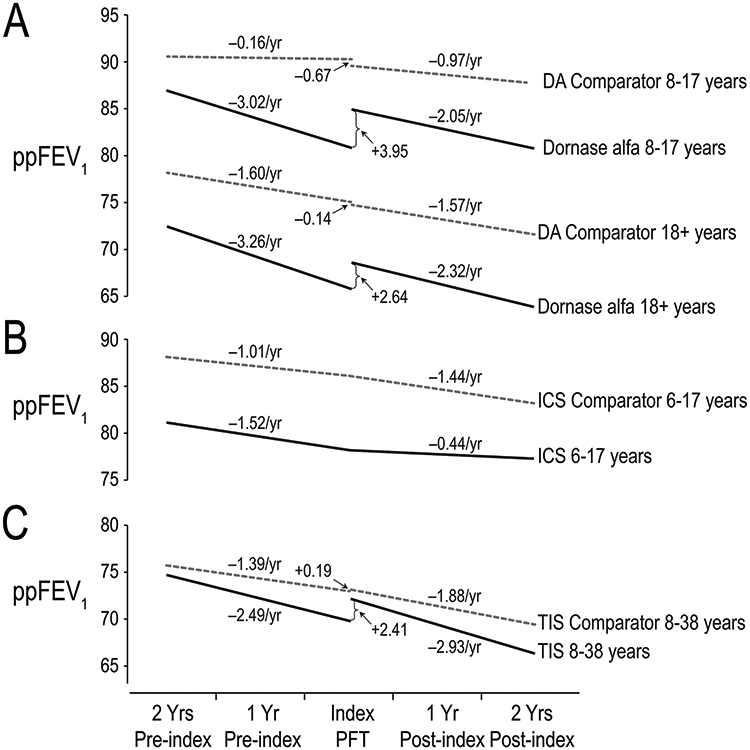
Change-point models of ppFEV1 before and after initiation of chronic treatments. Panel A. Change in ppFEV1 among children (upper lines) and adults (lower lines) receiving chronic dornase alfa treatment (DA; solid lines) versus an untreated comparator group (dashed lines). Mean ppFEV1 decline rates (slopes) are shown above lines; differences in intercept between lines are shown with arrows. Adapted with permission from Pediatric Pulmonology.26Panel B. Change in ppFEV1 among children receiving chronic ICS (solid lines) versus an untreated comparator group (dashed lines). Derived from previously published data.17Panel C. Change in ppFEV1 among patients ages 8 to38 years receiving chronic TIS (solid lines) versus an untreated comparator group (dashed lines). Mean ppFEV1 decline rates (slopes) are shown next to lines; differences in intercept between lines are shown with arrows. Derived from previously published data.41ICS, inhaled corticosteroids; ppFEV1, percent predicted forced expiratory volume in 1 s; TIS, tobramycin inhalation solution
Chronic inhaled corticosteroid (ICS) [17].
The change-point model for ICS allowed for a change in rate of ppFEV1 decline at initiation of therapy but no acute change. For ICS patients, rate of ppFEV1 decline was reduced by 71% (95% CI 40% to 98%), whereas the rate of ppFEV1 decline in the comparator population increased 43% (Figure 7B). ICS use was associated with decreased HFA and increased insulin/oral hypoglycemic use.
Tobramycin inhalation solution (TIS) [41].
Using the same methods as the dornase alfa change-point investigation, acute ppFEV1 change and subsequent change in rate of ppFEV1 decline associated with initiation of chronic TIS were evaluated; comparators used TIS<10% of the time. TIS patients improved at initiation versus no change for comparators, whereas the rate of ppFEV1 decline worsened in both groups by similar amounts (Figure 7C).
High-dose ibuprofen (HDI) [54].
Although treatment-associated reduction in rate of lung function loss should theoretically improve survival, no such relationship had been shown for a chronic CF therapy. In part, this is because the ages of most rapid lung function decline [14,21] -- early adolescence – occur more than a decade before the median age of death. HDI had previously been shown to reduce rate of ppFEV1 decline without a corresponding acute improvement, but no survival benefit had been published. Children age 6-17 years receiving HDI for 2 years were matched 1:5 with comparator children not treated with HDI. HDI patients had a 37.5% reduction in rate of ppFEV1 decline during 2 years of treatment and an 18% reduction in mortality (in ESCF and subsequently up to 16 years in the U.S. CFFPR) (Figure 8).
FIGURE 8.
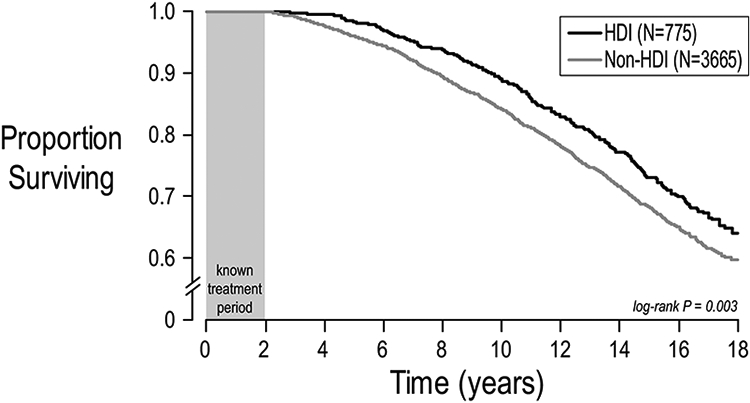
Survival after initiation of at least 2 years of chronic HDI in children (6–17 years). The 2-year HDI exposure period is shown as a gray box. Survival among patients receiving at least 2years of chronic HDI (N= 775) is represented as a black line and survival among propensity-matched comparator patients (N= 3665 )is shown in gray. Adapted with permission from the Annals of the American Thoracic Society.54HDI, high-dose ibuprofen
Comment:
Despite disease complexities and polypharmacy, observational studies can be used to evaluate comparative effectiveness of chronic therapies. Although an acute increase in ppFEV1 is desirable, it is more important to reduce the long-term rate of lung function decline which is not necessarily associated with an acute increase. The HDI results are consistent with the hypothesis that treatment-associated reduction of lung function decline in children with cystic fibrosis leads to improved survival. Although short term improvements in ppFEV1 can be assessed in clinical trials, only with long-term observational studies is it possible to evaluate lung function decline and survival.
IMPLICATIONS FOR FUTURE DATA REGISTRIES AND PATIENT CARE PRACTICES
ESCF was the first encounter-based patient registry for CF and has the informed the design of similar registries. In addition to the scientific publications, ESCF also provided site-specific reports with regional and national data for comparison, allowing for quality improvement initiatives. Although practice patterns will change dramatically with the introduction of highly effective modulator therapies (HEMT), patient registries such as the CFFPR remain critical to evaluate long-term outcomes including survival in relation to practice patterns. Furthermore, unlike the development of new therapeutic interventions, existing practices are unlikely to be evaluated formally outside of observational studies. Requirements of pharmacovigilance are no substitute for long-term registries that provide a comprehensive look at patients and their care over their lifetime.
As patients become healthier, we may become complacent or reliant on untested assumptions or analyses that are no longer relevant. As the natural history of CF changes, we need to update our understanding of risk factors for disease progression and poor outcomes so that we can continue to anticipate disease progression and deliver care accordingly. As survival improves, we will likely need to evaluate lung function much later in life than has been feasible in the CF population to date. This would allow the evaluation of risk factors for the development of progressive lung disease analogous to COPD and non-CF bronchiectasis.
ESCF and CFFPR has been an important source of information on practice patterns and also serve to provide historical control populations for new therapies. Until therapies are developed for the approximately 10% of patients not currently appropriate for HEMT, we will need registries to supply similar information for those patients.
Care summaries provided in connection with point of service data collection may allow clinicians to take action based on information such as ppFEV1 variability or acute drop. An electronic version of the POP tool or similar risk assessment tool could be used to guide care by highlighting patients at high risk of lung function decline.
Although attempts to develop more sensitive measures of lung disease (MBW, HRCT, MRI) are laudable, but greater attention to simple, available measures (chest exam, spirometry, height, and weight) is likely to improve outcomes.
Supplementary Material
Overall, 32,178 patients contributed 869,136 encounters.
Survival information was updated from the U.S. CFFPR for two-thirds of patients.
Sites with better lung function outcomes monitor and treat more aggressively [7,12]
Over the course of ESCF, there was an improvement in health outcomes and increase in the use of therapies [15,16,23]
Poorer health outcomes previously associated with low SES cannot be accounted for by decreased access to care or decreased diagnosis/treatment [19,27]
Respiratory signs and symptoms and nutritional indexes in young children predict lung function at age 6 years [8]
Persistent signs and symptoms of lung disease begin early, often by age 2 years [31]
Pediatric wheeze is an important predictor of reduced ppFEV1 at age 6 to 8 years [42]
BMI and BMI percentile fail to identify children who are stunted or have potentially poor nutritional status as measured by WFA and/or HFA percentile [48]
Signs and symptoms at an early age remain independent risk factors for mortality even after controlling for lung function [50]
Differences in respiratory culture frequency and conditions contribute to differences in apparent prevalence [6]
Likelihood of treatment was higher in MRSA patients [13] and MARPA patients [32], but neither incident MRSA nor incident MARPA was associated with a change in rate of ppFEV1 decline [18,32]
Limiting analyses to IV exacerbations excludes a majority of exacerbations [35]
Reduced exacerbation treatment would have been more pronounced if not for lower treatment thresholds [37]
Sites with highest lung function were more likely to treat exacerbations identified by Rabin criteria [44]
Antibiotic treatment of children presenting with at least one Rabin criterion was associated with a lower proportion with crackles, cough, or Pa at follow-up [34]
Patients with inpatient exacerbation treatment had better likelihood of lung function recovery than those with outpatient treatment which in turn is better than no treatment [49,53]
Lung function recovery from exacerbations as return to baseline is prone to artifact [56]
High ppFEV1 and persistent crackles are risk factors for lung function decline across all ages [14,22,33]
Variability in ppFEV1 is a major risk factor for lung function decline [30,47]
Decline in lung function is greatest in adolescents and although population decline is minimal after age 30, individual decline continues [21]
Past lung function decline does not predict future rate of decline ≥3 years later [46]
CFRD is a common complication associated with more severe disease [10]
Pregnancy and motherhood do not accelerate disease progression but are associated with more intense monitoring, more therapies, more illness-related visits and exacerbations, and a decrease in quality of life [11,38]
Higher prevalence of liver disease in Hispanics is not explained by demographics and prevalence of meconium ileus [43]
ESCF data has been used to quantify the effects of duration, measurement frequency, and inclusion/exclusion criteria on the statistical power of studies [25,28]
Annual FEV1 is at least as good a measure for distinguishing among site practice patterns as other spirometric parameters including slopes [45]
The GLI reference equations for ppFEV1 are recommended, but results based on older equations should be accepted [51]
Initiation of dornase alfa is associated with an acute ppFEV1 increase and then a slower rate of decline [4,26]
Initiation of ICS is associated with a slower rate of ppFEV1 decline without an acute increase [17]
Initiation of TIS is associated with an acute ppFEV1 without a change in rate of decline [41]
Initiation of HDI is associated with a slower rate of ppFEV1 decline and improved survival [54]
ACKNOWLEGEMENTS
The authors gratefully acknowledge the participation of the more 400 site investigators and coordinators of ESCF and thank the Cystic Fibrosis Foundation for the use of CFFPR genotype and mortality data. They also thank the patients and care providers throughout the United States and Canada for their contributions to ESCF and the CFFPR. The authors thank their coauthors and especially the North American Scientific Advisory Group members as well as the statistical analysts who have made these studies possible. Special thanks are due to Stefanie Millar from ICON Clinical Research for her more than 15 years of analytic contributions. Finally, the authors thank Genentech, Inc., who have generously supported ESCF from its inception.
Footnotes
The Epidemiologic Study of Cystic Fibrosis was funded by Genentech, Inc.
CONFLICT OF INTEREST
Michael Konstan, Donald VanDevanter, Jeffrey Wagener, and Wayne Morgan have received honoraria from Genentech from serving as members of the North American Scientific Advisory Group for ESCF and have served as consultants to Genentech. Jeffrey Wagener was previously an employee of Genentech. No compensation was provided to these authors in exchange for production of this manuscript. David Pasta was previously an employee of and consultant to ICON Clinical Research, which was paid by Genentech for providing analytical services for ESCF.
REFERENCES
- 1.Morgan WJ, Butler SM, Johnson CA, Colin AA, FitzSimmons SC, Geller DE, Konstan MW, Light MJ, Rabin HR, Regelmann WE, Schidlow DV, Stokes DC, Wohl ME, Kaplowitz H, Wyatt MM, Stryker S. Epidemiologic study of cystic fibrosis: design and implementation of a prospective, multicenter, observational study of patients with cystic fibrosis in the U.S. and Canada. Pediatr Pulmonol 1999;28(4):231–241. [DOI] [PubMed] [Google Scholar]
- 2.Konstan MW, Butler SM, Schidlow DV, Morgan WJ, Julius JR, Johnson CA. Patterns of medical practice in cystic fibrosis: part I. Evaluation and monitoring of health status of patients. Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Pediatr Pulmonol 1999;28(4):242–247. [DOI] [PubMed] [Google Scholar]
- 3.Konstan MW, Butler SM, Schidlow DV, Morgan WJ, Julius JR, Johnson CA. Patterns of medical practice in cystic fibrosis: part II. Use of therapies. Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Pediatr Pulmonol 1999;28(4):248–254. [DOI] [PubMed] [Google Scholar]
- 4.Johnson CA, Butler SM, Konstan MW, Breen TJ, Morgan WJ. Estimating effectiveness in an observational study: a case study of dornase alfa in cystic fibrosis. The Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. J Pediatr 1999;134(6):734–739. [DOI] [PubMed] [Google Scholar]
- 5.Geller DE, Kaplowitz H, Light MJ, Colin AA. Allergic bronchopulmonary aspergillosis in cystic fibrosis: reported prevalence, regional distribution, and patient characteristics. Scientific Advisory Group, Investigators, and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Chest 1999;116(3):639–646. [DOI] [PubMed] [Google Scholar]
- 6.Shreve MR, Butler S, Kaplowitz HJ, Rabin HR, Stokes D, Light M, Regelmann WE. Impact of microbiology practice on cumulative prevalence of respiratory tract bacteria in patients with cystic fibrosis. J Clin Microbiol 1999;37(3):753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson C, Butler SM, Konstan MW, Morgan W, Wohl ME. Factors influencing outcomes in cystic fibrosis: a center-based analysis. Chest 2003;123(1):20–27. [DOI] [PubMed] [Google Scholar]
- 8.Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, Johnson CA, Morgan WJ; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr 2003;142(6):624–630. [DOI] [PubMed] [Google Scholar]
- 9.Rabin HR, Butler SM, Wohl ME, Geller DE, Colin AA, Schidlow DV, Johnson CA, Konstan MW, Regelmann WE; Epidemiologic Study of Cystic Fibrosis. Pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol 2004;37(5):400–406. [DOI] [PubMed] [Google Scholar]
- 10.Marshall BC, Butler SM, Stoddard M, Moran AM, Liou TG, Morgan WJ. Epidemiology of cystic fibrosis-related diabetes. J Pediatr 2005;146(5):681–687. [DOI] [PubMed] [Google Scholar]
- 11.McMullen AH, Pasta DJ, Frederick PD, Konstan MW, Morgan WJ, Schechter MS, Wagener JS. Impact of pregnancy on women with cystic fibrosis. Chest 2006;129(3):706–711. [DOI] [PubMed] [Google Scholar]
- 12.Padman R, McColley SA, Miller DP, Konstan MW, Morgan WJ, Schechter MS, Ren CL, Wagener JS; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Infant care patterns at epidemiologic study of cystic fibrosis sites that achieve superior childhood lung function. Pediatrics 2007;119(3):e531–537. [DOI] [PubMed] [Google Scholar]
- 13.Ren CL, Morgan WJ, Konstan MW, Schechter MS, Wagener JS, Fisher KA, Regelmann WE; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Presence of methicillin resistant Staphylococcus aureus in respiratory cultures from cystic fibrosis patients is associated with lower lung function. Pediatr Pulmonol 2007;42(6):513–518. [DOI] [PubMed] [Google Scholar]
- 14.Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, Stokes DC, Wohl ME, Wagener JS, Regelmann WE, Johnson CA; Scientific Advisory Group and the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr 2007;151(2):134–139. [DOI] [PubMed] [Google Scholar]
- 15.Moskowitz SM, Silva SJ, Mayer-Hamblett N, Pasta DJ, Mink DR, Mabie JA, Konstan MW, Wagener JS; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis (ESCF). Shifting patterns of inhaled antibiotic use in cystic fibrosis. Pediatr Pulmonol 2008;43(9):874–881. [DOI] [PubMed] [Google Scholar]
- 16.VanDevanter DR, Rasouliyan L, Murphy TM, Morgan WJ, Ren CL, Konstan MW, Wagener JS; Investigators, Coordinators of the Epidemiologic Study of Cystic Fibrosis. Trends in the clinical characteristics of the U.S. cystic fibrosis patient population from 1995 to 2005. Pediatr Pulmonol 2008;43(8):739–744. [DOI] [PubMed] [Google Scholar]
- 17.Ren CL, Pasta DJ, Rasouliyan L, Wagener JS, Konstan MW, Morgan WJ; Scientific Advisory Group and the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Relationship between inhaled corticosteroid therapy and rate of lung function decline in children with cystic fibrosis. J Pediatr 2008;153(6):746–751. [DOI] [PubMed] [Google Scholar]
- 18.Sawicki GS, Rasouliyan L, Pasta DJ, Regelmann WE, Wagener JS, Waltz DA, Ren CL; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. The impact of incident methicillin resistant Staphylococcus aureus detection on pulmonary function in cystic fibrosis. Pediatr Pulmonol 2008;43(11):1117–1123. [DOI] [PubMed] [Google Scholar]
- 19.Schechter MS, McColley SA, Silva S, Haselkorn T, Konstan MW, Wagener JS; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis; North American Scientific Advisory Group for ESCF. Association of socioeconomic status with the use of chronic therapies and healthcare utilization in children with cystic fibrosis. J Pediatr 2009;155(5):634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quittner AL, Schechter MS, Rasouliyan L, Haselkorn T, Pasta DJ, Wagener JS. Impact of socioeconomic status, race, and ethnicity on quality of life in patients with cystic fibrosis in the United States. Chest 2010;137(3):642–650. [DOI] [PubMed] [Google Scholar]
- 21.Liou TG, Elkin EP, Pasta DJ, Jacobs JR, Konstan MW, Morgan WJ, Wagener JS. Year-to-year changes in lung function in individuals with cystic fibrosis. J Cyst Fibros. 2010;9(4):250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.VanDevanter DR, Wagener JS, Pasta DJ, Elkin E, Jacobs JR, Morgan WJ, Konstan MW. Pulmonary outcome prediction (POP) tools for cystic fibrosis patients. Pediatr Pulmonol 2010;45(12):1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konstan MW, VanDevanter DR, Rasouliyan L, Pasta DJ, Yegin A, Morgan WJ, Wagener JS; Scientific Advisory Group; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Trends in the use of routine therapies in cystic fibrosis: 1995-2005. Pediatr Pulmonol 2010;45(12):1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawicki GS, Rasouliyan L, McMullen AH, Wagener JS, McColley SA, Pasta DJ, Quittner AL. Longitudinal assessment of health-related quality of life in an observational cohort of patients with cystic fibrosis. Pediatr Pulmonol 2011;46(1):36–44. [DOI] [PubMed] [Google Scholar]
- 25.Konstan MW, Wagener JS, Yegin A, Millar SJ, Pasta DJ, VanDevanter DR. Design and powering of cystic fibrosis clinical trials using rate of FEV(1) decline as an efficacy endpoint. J Cyst Fibros 2010;9(5):332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konstan MW, Wagener JS, Pasta DJ, Millar SJ, Jacobs JR, Yegin A, Morgan WJ; Scientific Advisory Group and Investigators and Coordinators of Epidemiologic Study of Cystic Fibrosis. Clinical use of dornase alpha is associated with a slower rate of FEV1 decline in cystic fibrosis. Pediatr Pulmonol 2011;46(6):545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schechter MS, McColley SA, Regelmann W, Millar SJ, Pasta DJ, Wagener JS, Konstan MW, Morgan WJ; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Socioeconomic status and the likelihood of antibiotic treatment for signs and symptoms of pulmonary exacerbation in children with cystic fibrosis. J Pediatr 2011;159(5):819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.VanDevanter DR, Yegin A, Morgan WJ, Millar SJ, Pasta DJ, Konstan MW. Design and powering of cystic fibrosis clinical trials using pulmonary exacerbation as an efficacy endpoint. J Cyst Fibros 2011;10(6):453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quittner AL, Sawicki GS, McMullen A, Rasouliyan L, Pasta DJ, Yegin A, Konstan MW. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Qual Life Res 2012;21(7):1267–1278. [DOI] [PubMed] [Google Scholar]; Corrected and republished as Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national, US sample in Qual Life Res. 2012. Sep;21(7):1279–1290. [DOI] [PubMed] [Google Scholar]
- 30.Vandenbranden SL, McMullen A, Schechter MS, Pasta DJ, Michaelis RL, Konstan MW, Wagener JS, Morgan WJ, McColley SA; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Lung function decline from adolescence to young adulthood in cystic fibrosis. Pediatr Pulmonol 2012;47(2):135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McColley SA, Ren CL, Schechter MS, Regelmann WE, Pasta DJ, Konstan MW; Epidemiologic Study of Cystic Fibrosis. Risk factors for onset of persistent respiratory symptoms in children with cystic fibrosis. Pediatr Pulmonol 2012;47(10):966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren CL, Konstan MW, Yegin A, Rasouliyan L, Trzaskoma B, Morgan WJ, Regelmann W; Scientific Advisory Group, Investigators, and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J Cyst Fibros 2012;11(4):293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konstan MW, Wagener JS, VanDevanter DR, Pasta DJ, Yegin A, Rasouliyan L, Morgan WJ. Risk factors for rate of decline in FEV1 in adults with cystic fibrosis. J Cyst Fibros 2012;11(5):405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regelmann WE, Schechter MS, Wagener JS, Morgan WJ, Pasta DJ, Elkin EP, Konstan MW; Investigators of the Epidemiologic Study of Cystic Fibrosis. Pulmonary exacerbations in cystic fibrosis: young children with characteristic signs and symptoms. Pediatr Pulmonol 2013;48(7):649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagener JS, Rasouliyan L, VanDevanter DR, Pasta DJ, Regelmann WE, Morgan WJ, Konstan MW; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol 2013;48(7):666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayer-Hamblett N, Rosenfeld M, Treggiari MM, Konstan MW, Retsch-Bogart G, Morgan W, Wagener J, Gibson RL, Khan U, Emerson J, Thompson V, Elkin EP, Ramsey BW; EPIC, ESCF Investigators. Standard care versus protocol based therapy for new onset Pseudomonas aeruginosa in cystic fibrosis. Pediatr Pulmonol 2013;48(10):943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.VanDevanter DR, Elkin EP, Pasta DJ, Morgan WJ, Konstan MW; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Changing thresholds and incidence of antibiotic treatment of cystic fibrosis pulmonary exacerbations, 1995-2005. J Cyst Fibros 2013;12(4):332–337. [DOI] [PubMed] [Google Scholar]
- 38.Schechter MS, Quittner AL, Konstan MW, Millar SJ, Pasta DJ, McMullen A; Scientific Advisory Group; Investigators and Coordinators of Epidemiologic Study of Cystic Fibrosis. Long-term effects of pregnancy and motherhood on disease outcomes of women with cystic fibrosis. Ann Am Thorac Soc 2013;10(3):213–219. [DOI] [PubMed] [Google Scholar]
- 39.Sawicki GS, Ren CL, Konstan MW, Millar SJ, Pasta DJ, Quittner AL; Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Treatment complexity in cystic fibrosis: trends over time and associations with site-specific outcomes. J Cyst Fibros 2013;12(5):461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan WJ, Wagener JS, Yegin A, Pasta DJ, Millar SJ, Konstan MW; Scientific Advisory Group, investigators, and coordinators of the Epidemiologic Study of Cystic Fibrosis. Probability of Treatment Following Acute Decline in Lung Function in Children with Cystic Fibrosis is Related to Baseline Pulmonary Function. J Pediatr 2013;163(4):1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konstan MW, Wagener JS, Pasta DJ, Millar SJ, Morgan WJ. Clinical use of tobramycin inhalation solution (TOBI®) shows sustained improvement in FEV(1) in cystic fibrosis. Pediatr Pulmonol 2014; 49(6):529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren CL, Konstan MW, Rosenfeld M, Pasta DJ, Millar SJ, Morgan WJ; for the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Early childhood wheezing is associated with lower lung function in cystic fibrosis. Pediatr Pulmonol 2014; 49(8):745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagener JS, Woo MS, Pasta DJ, Konstan MW, Morgan WJ: Liver involvement in the Hispanic cystic fibrosis population of North America. J Pediatr Gastroenterol Nutr 2014;59(4):476–479. [DOI] [PubMed] [Google Scholar]
- 44.Schechter MS, Regelmann WE, Sawicki GS, Rasouliyan L, VanDevanter DR, Rosenfeld M, Pasta D, Morgan W, Konstan MW. Antibiotic treatment of signs and symptoms of pulmonary exacerbations: A comparison by care site. Pediatr Pulmonol 2015;50(5):431–440. [DOI] [PubMed] [Google Scholar]
- 45.Wagener JS, Elkin EP, Pasta DJ, Schechter MS, Konstan MW, Morgan WJ. Pulmonary function outcomes for assessing cystic fibrosis care. J Cyst Fibros 2015;14(3):376–383. [DOI] [PubMed] [Google Scholar]
- 46.Rosenfeld M, VanDevanter DR, Ren CL, Elkin EP, Pasta DJ, Konstan MW, Morgan WJ. Decline in lung function does not predict future decline in lung function in cystic fibrosis patients. Pediatr Pulmonol 2015;50(9):856–862. [DOI] [PubMed] [Google Scholar]
- 47.Morgan WJ, VanDevanter DR, Pasta DJ, Foreman AJ, Wagener JS, Konstan MW. Forced expiratory volume in 1 second variability helps identify patients with cystic fibrosis at risk of greater loss of lung function. J Pediatr 2016;169:116–121. [DOI] [PubMed] [Google Scholar]
- 48.Konstan MW, Pasta DJ, Wagener JS, VanDevanter DR, Morgan WJ. BMI fails to identify poor nutritional status in stunted children with CF. J Cyst Fibros 2017;16(1):158–160. [DOI] [PubMed] [Google Scholar]
- 49.Morgan WJ, Wagener JS, Pasta DJ, Millar SJ, VanDevanter DR, Konstan MW, Scientific Advisory Group, Investigators, and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Relationship of antibiotic treatment to recovery after acute FEV1 decline in children with cystic fibrosis. Ann Am Thorac Soc 2017;14(6):937–943. [DOI] [PubMed] [Google Scholar]
- 50.McColley SA, Schechter MS, Morgan WJ, Pasta DJ, Craib ML, Konstan MW. Risk factors for mortality before age 18 years in cystic fibrosis. Pediatr Pulmonol 2017;52(7):909–915. [DOI] [PubMed] [Google Scholar]
- 51.Konstan MW, Wagener JS, VanDevanter DR, Pasta DJ, Millar SJ, Morgan WJ, Scientific Advisory Group and the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Comparison of FEV1 reference equations for evaluating a cystic fibrosis therapeutic intervention. Pediatr Pulmonol 2017;52(8):1013–1019. [DOI] [PubMed] [Google Scholar]
- 52.VanDevanter DR, Craib ML, Pasta DJ, Millar SJ, Morgan WJ, Konstan MW. Cystic fibrosis clinical characteristics associated with dornase alfa treatment regimen change. Pediatr Pulmonol 2018;53(1):43–49. [DOI] [PubMed] [Google Scholar]
- 53.Schechter MS, VanDevanter DR, Pasta DJ, Short SA, Morgan WJ, Konstan MW. Treatment setting and outcomes of cystic fibrosis pulmonary exacerbations. Ann Am Thorac Soc 2018;15(2):225–233. [DOI] [PubMed] [Google Scholar]
- 54.Konstan MW, VanDevanter DR, Sawicki GS, Pasta DJ, Foreman AJ, Neiman EA, Morgan WJ. Association of high-dose ibuprofen use, lung function decline, and long-term survival in children with cystic fibrosis. Ann Am Thorac Soc 2018;15(4):485–493. [DOI] [PubMed] [Google Scholar]
- 55.Wagener JS, Williams MJ, Millar SJ, Morgan WJ, Pasta DJ, Konstan MW. Pulmonary exacerbations and acute decline in lung function in patients with cystic fibrosis. J Cyst Fibros 2018;17(4):496–502. [DOI] [PubMed] [Google Scholar]
- 56.Wagener JS, VanDevanter DR, Konstan MW, Pasta DJ, Millar SJ, Morgan WJ. Lung function changes before and after pulmonary exacerbation antimicrobial treatment in cystic fibrosis. Pediatric Pulmonology 2020:55(3):828–834. [DOI] [PubMed] [Google Scholar]
- 57.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2018 Annual Data Report, Bethesda, MD: 2019. Cystic Fibrosis Foundation [Google Scholar]
- 58.The Cystic Fibrosis Foundation Center Committee and Guidelines Subcommittee. Cystic Fibrosis Foundation Guidelines for Patient Services, Evaluation, and Monitoring in Cystic Fibrosis Centers. Am J Dis Child 1990;144(12):1311–1312. [DOI] [PubMed] [Google Scholar]
- 59.Morgan WJ, Millar S, Pasta D, Wagener J, Konstan MW. How much FEV1 variability can be explained by pulmonary exacerbations? Pediatr Pulmonol Suppl 50(S41):375–376 (488), 2015. [Google Scholar]
- 60.Morgan WJ, VanDevanter DR, Millar SJ, Pasta DJ, Konstan MW. What factors predict FEV1 variability in cystic fibrosis patients? Pediatr Pulmonol Suppl 51(S45):356 (428), 2016. [Google Scholar]
- 61.Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis 1983;127:725–734. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG Jr. Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol 1993;15:75–88. [DOI] [PubMed] [Google Scholar]
- 63.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999;159:179–187. [DOI] [PubMed] [Google Scholar]
- 64.Stanojevic S, Wade A, Stocks J et al. Reference ranges for spirometry across all ages: a new approach. Am J Respir Crit Care Med 2008;177:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quanjer PH, Stanojevic S, Cole TJ, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J 2012;40:1324–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994;331(10):637–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.