

Abstract
Background:
Elexacaftor-Tezacaftor-Ivacaftor (ETI) therapy is showing promising efficacy for treatment of cystic fibrosis (CF) and is becoming more widely available since recent FDA approval. However, little is known about how these drugs will affect lung infections, which are the leading cause of morbidity and mortality among people with CF (pwCF).
Methods:
We analyzed sputum microbiome and metabolome data from pwCF (n=24) before and after ETI therapy using 16S rRNA gene sequencing and untargeted metabolomics.
Results:
The sputum microbiome diversity, particularly its evenness, was increased (p=0.036) and the microbiome profiles were different between individuals before and after therapy (PERMANOVA F=1.92, p=0.044). Despite these changes, the microbiomes remained more similar within an individual than across the sampled population. No specific microbial taxa differed in relative abundance before and after therapy, but the collective log-ratio of classic CF pathogens to anaerobes significantly decreased (p=0.013). The sputum metabolome also showed changes associated with ETI (PERMANOVA F=4.22, p=0.002) and was characterized by greater variation across subjects while on treatment. Changes in the metabolome were driven by a decrease in peptides, amino acids, and metabolites from the kynurenine pathway, which were associated with a decrease in CF pathogens. Metabolism of the three small molecules that make up ETI was extensive, including previously uncharacterized structural modifications.
Conclusions:
ETI therapy is associated with a changing microbiome and metabolome in airway mucus. This effect was stronger on sputum biochemistry, which may reflect changing niche space for microbial residency in lung mucus as the drug’s effects take hold.
Keywords: Cystic Fibrosis, Sputum, Microbiome, Metabolome, Trikafta
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR is a cAMP-regulated ion channel used for the transport of anions across epithelial cells [1]. Mutations in this gene result in a thickening of mucosal secretions, primarily in the respiratory and gastrointestinal systems [1]. Common clinical manifestations of this disease include, but are not limited to, chronic polymicrobial sino-pulmonary infections, male infertility, decreased lung function, and pancreatic insufficiency [1]. Pancreatic sufficiency is closely linked to the specific mutation class, but other aspects of CF pathology have unclear links to genotype [2,3]. Those with severe disease, especially the F508del mutation, are plagued by chronic lung infection throughout their lifetime [4].
The lung microbiome of people with CF (pwCF) has been well characterized and includes bacteria, viruses, and fungi [5–7]. Studies of sputum expectorated from the airways have demonstrated that the CF lung microbiome diversity decreases as the disease progresses over time, becoming dominated by opportunistic pathogens, such as Pseudomonas aeruginosa [4,8]. Thickened mucus within the lungs allows for these pathogens to form a biofilm and thrive [9]. The chemical composition of this matrix has been shown to mainly include DNA, amino acids, peptides, antibiotics, inflammatory lipids, and a myriad of small molecules from host, microbial, and xenobiotic sources [10–14].
In November 2019, a new triple therapy, comprised of three compounds Elexacaftor-Tezacaftor-Ivacaftor (ETI, Trikafta®), was approved by the United States Food and Drug Administration (FDA) for the treatment of CF [15,16]. People with at least one copy of the F508del mutation, the most common across CF patients, are eligible to take ETI. Studies from clinical trials and data available since approval have shown that the treatment is providing remarkable improvements in lung function and other disease symptoms [17,18]. However, little is known about how this new therapy will affect the CF lung microbiome and metabolome.
In this study, paired sputum samples from pwCF (n=24) were collected before and after therapy (within one year of FDA approval) and analyzed using an integrated multi-omics approach including 16S rRNA amplicon sequencing and LC-MS/MS untargeted metabolomics. We hypothesized that the sputum microbiome and metabolome profiles would be altered before and after taking ETI. We measured alpha-diversity (the number and relative abundances of features in the samples), and beta-diversity (the relative similarity of overall profiles between samples) and found that the microbiome and metabolome were significantly different after ETI. These changes indicated a shift in the niche space of airway mucus and its microbial occupancy associated with ETI therapy.
2. Methods
2.1. Further detail available in supplemental methods
2.1.1. Sample Collection
Sputum samples were collected during routine clinical visits from adult pwCF (>18 years) at two separate CF clinics (patient details table S1). Inclusion criteria for the study included adult subjects that could produce sputum and had an initial sample collected within one year of ETI administration and a paired sputum sample within one year after. Samples were obtained from the most recent clinical visit prior to ETI administration and the most recent visit after ETI administration if sputum production was possible. Thus, subjects had different intervals between paired samples (mean 202 days +/− 108, Table 1, S1). Ethical approval for the collections at the University of California San Diego adult CF clinic was obtained from the UCSD Human Research Protections Program Institutional Review Board under protocol #160078. Institutional review board approval was also provided for the collections at the Spectrum Health adult CF clinic in Grand Rapids, MI by the Spectrum Health Human Research Protection Program Office of the Institutional Review Board under IRB #2018–438.
Table 1.
Clinical, microbiological and sampling data on pwCF in this study prior to and after ETI therapy. Numbers are reported means. NA=not applicable, +/− = Standard Deviation of the mean.
| Prior to ETI | Post ETI | Delta | |
|---|---|---|---|
| Number of subjects | 24 | 24 | NA |
| % Male | NA | 0.54 | NA |
| Average age (from range) | 32.50 | 33.00 | 0.50 |
| FEV1%-predicted | 50.21 (+/−17.8) | 65.86 (+/−18.73) | 15.65 |
| FVC | 71.83 (+/−20.0) | 81.95 (+/−18.36) | 10.12 |
| BMI | 22.14 (+/−3.44) | 23.63 (+/−3.31) | 1.49 |
| Height (cm) | 166.58 | 166.39 | −0.19 |
| # Days on ETI | NA | 149.71 (+/−81.17) | NA |
| # Days between samples | NA | 201.79 (+/−107.97) | NA |
| % Pseudomonas by culture | 70.00 | 84.00 | 14.00 |
| Exacerbations per year | 2.63 | 2.45 | −0.17 |
| % Pancreatic sufficient | NA | 0.04 | NA |
To serve as a control group for comparison of changes related to ETI therapy we mined publicly available microbiome and metabolome data from a previous study with a similar time of colleciton between paired samples but prior to ETI approval (mean 157 days +/− 119, mean FEV1%-predicted = 67.77%, n=10 pwCF, Table S1) [13]. These samples were collected under the same UCSD IRB protocol with the same procedures.
2.1.2. DNA Extraction, qPCR and 16S rRNA single amplicon sequencing
A Qiagen® PowerSoil® DNA extraction kit was used to extract DNA from the sputum samples following standard protocol. PCR amplification was then performed using 27F and 1492R primers targeting the bacterial 16S rRNA gene to test for DNA amplification quality. If amplifiable, bacterial 16S rRNA V4 amplicon sequencing was performed with primers 515f/806r on an Illumina® MiSeq® at the Michigan State University Sequencing Core. The raw sequences were processed using QIITA (qiita.ucsd.edu [19]), which is driven by QIIME2 algorithms [20], and quality filtered to generate amplicon sequence variants (ASVs) through the Deblur method [21]. ASVs in the microbiome data were classified as ‘classic CF pathogens’ or ‘anaerobes’ based on the methods of Raghuvanshi et al. (2020) and Carmody et al. (2018) [22,23]. The specific ASVs and their classifications are available in table S2. Quantitative PCR (qPCR) was performed using universal 16S primers [43] and Applied Biosystems SYBR Green PCR Master Mix with three technical replicates for each sample. The microbiome data is publicly available at the Qiita repository under study #13507. Microbiome data for the follow up control cohort was generated with the same extraction methods as described in [13].
2.1.3. Metabolomics
Organic metabolite extraction was performed by adding twice the sample volume of chilled 100% methanol, vortexing briefly, and incubating at room temperature for 2 hours. Samples were then centrifuged at 10,000 x g for 10 minutes and the supernatant was collected. Methanolic extracts were analyzed on a Thermo Q-Exactive® Hybrid Quadrupole-Orbitrap mass spectrometer coupled to a Vanquish® ultra-high-performance liquid chromatography system. All raw files were converted to .mzXML format and then processed with MZmine 2.53 software [24], GNPS molecular networking [25] and SIRIUS [26]. MZmine 2 parameters are available in the supplementary information (Table S3). The network job is available at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=c700397169ff447490f764c34abb5abd and the mass spectrometry data were deposited on public repository massive.ucsd.edu under MassIVE ID MSV000087364. Metabolomics data for the follow up cohort was generated with the same extraction methods as described above and further detail is provided in [13].
2.2. Statistical Analysis
Statistical approaches for both the microbiome and metabolome data were similar, due to the inherent structural similarity of the multivariate data sets. Normality of the different quantitative measures was first tested using a Shapiro-Wilk (SW) test in order to determine the appropriate statistical methods. If the data were normally distributed, a paired dependent means t-test (DM t-test) was used, if not, a Wilcoxon signed-rank test (WSRT) was used. Alpha-diversity was calculated for both datasets using the Shannon index. Beta-diversity measures were calculated using the weighted UniFrac distance for the microbiome and Bray-Curtis distance for the metabolome. Beta-diversity was visualized for both datasets using principal coordinates analysis (PCoA) and the EMPeror software [27]. Beta-diversity clustering significance pre- and post-ETI were tested using a Permutational Multivariate Analysis of Variance (PERMANOVA) method with 999 permutations. Cross population beta-diversity comparisons were done between pwCF before and after ETI therapy, across the whole dataset, and within individuals pre- and post-therapy.
To identify metabolite and microbial drivers of the difference pre- and post-therapy, a random forest (RF) machine learning approach was used via the randomForest package in R [28]. The top 50 variables of importance were further explored. As all individual metabolite and microbiome abundance data were not considered normally distributed, statistical significance for individual microbial and metabolite changes before and after ETI therapy were calculated using the WRST. The p-values were adjusted for multiple comparisons using the Benjamini-Hochberg method.
Microbe and metabolite association vectors were calculated using mmvec [29]. Detail of the mmvec parameters and analysis are available in the online supplement.
3. Results
3.1. Patient Clinical Changes after ETI Therapy
Sputum was collected from twenty-four pwCF during routine clinical visits most recently before ETI therapy and most recently after (Table 1, Table S1). Subjects had varied time periods before and after treatment (mean 202 days +/−108 days between samples, mean 150 days +/−81 days since ETI prescription, Table 1). The FEV1%-predicted across the cohort significantly increased after ETI therapy (mean change = +15.6%, +/−11.8%, DM t-test p < 0.0001), as did the FVC measure (mean change = +10.1 ml, +/− 10.64 ml; DM t-test p < 0.0001) and BMI (Mean Change = +1.49, +/− 1.53; DM t-test p <0.0001, Table 1). The control cohort of successive paired samples from previously published data [13] (n = 10 pwCF, 24 paired samples, mean 157 days between samples +/− 119) did not show significant changes in lung function between samples (mean change = 1.19% +/−3.43, Table S1). Antibiotic administration before and after ETI was not significantly different (Chi-squared test: inhaled antibiotics p = 0.10, Oral and/or IV antibiotics p = 0.0857, any antibiotic p = 0.549), as many of the subjects continued their routine antibiotic regimen during ETI therapy. From available paired sample microbial culture data (n=16) 14 subjects that cultured Pseudomonas aeruginosa prior to ETI therapy cultured it in the sample collected after, while two subjects cultured this bacterium only when on ETI (Table S4). Other notable changes include the loss of Aspergillus sp. positive cultures in 4 of 6 subjects (Table S4).
3.2. Microbiome and Metabolome Diversity Changes after ETI Therapy
Diversity measures in this study included alpha-diversity, representing the number and relative abundances of features in the samples, and beta-diversity, the relative similarity of overall profiles between samples. Measures of microbiome alpha-diversity, both the Shannon index and Peilou evenness, showed a significant increase after ETI therapy (Shannon SW normality p=0.238, WSRT p=0.038; Peilou evenness SW normality p=0.074, WSRT p=0.036). The number of amplified sequence variants also increased, but did not reach statistical significance (ASVs SW normality p=0.0043, DM t-test p=0.12, Fig. 1a). The microbiome alpha-diversity of our control cohort did not show a significant difference in similarly paired sputum samples prior to ETI approval (Fig. S1). The metabolome did not show a significant change in Shannon index (SW p=0.0017, DM t-test, p=0.45, Fig. 1b) or evenness (Pielou evenness SW p=0.13, WRST p=0.30), but the number of molecular features in the metabolome did decrease significantly after ETI therapy (SW p=0.0027 DM t-test p=0.010). We also explored changes in microbiome alpha-diversity with other clinical parameters and drugs detected in the metabolome data to determine if these were significant confounders. The amount of antibiotics present in the metabolome data did not correlate with alpha-diversity measures and was not significantly different before or after ETI therapy (Fig. S2). The change in FEV1%-predicted also did not correlate with a changing microbiome diversity (Fig. S3). Collectively, this alpha-diversity analysis demonstrates that new microbial ASVs were not being introduced into the sputum microbiome after ETI therapy (ie. no change in richness), but rather the community became more even with previously present taxa. In the metabolome, only a decrease in the number of total molecules detected was observed (ie. loss of richness).
Fig. 1.
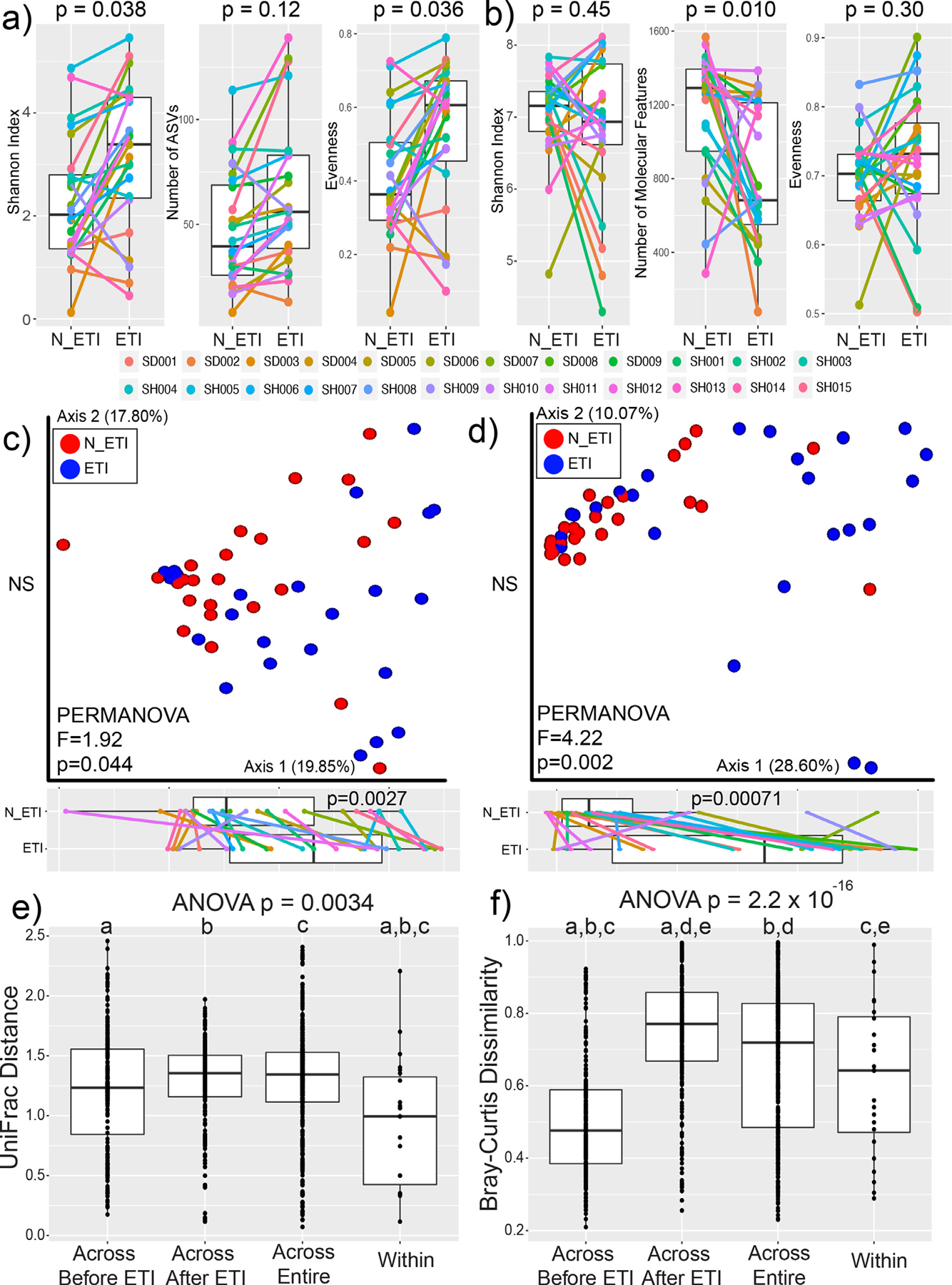
Alpha and Beta-diversity of lung microbiomes before and after ETI. Alpha-diversity measures of a) microbiome data and b) metabolome data before (N_ETI) and after ETI therapy. P-values shown are from either the DM t-test or WSRT after testing for normality. Principal coordinate analysis plots of beta-diversity data for c) microbiome data with significance calculated utilizing the weighted UniFrac distance and d) metabolome data with significance calculated utilizing the Bray-Curtis distance. PERMANOVA statistics and the percent of variance explained by each axis are shown. Boxplots of positions on the first principal coordinate are shown tested for significance with the DM t-test. Beta-diversity cross comparisons within the e) microbiome and f) metabolome data. Cross comparisons were done across subjects before and after ETI therapy, across the entire dataset, and within a subject’s paired samples (within). Statistical significance was first tested with an ANOVA followed by an ad-hoc Tukey’s test. Shared letters denote distributions that are significantly different from each other.
PCoA plots were used to visualize beta-diversity of the two data types and PERMANOVA tests were used to determine if clustering based on ETI therapy was statistically significant (Fig. 1c, d). The overall microbiome profiles of the sputum samples changed after ETI therapy (PERMANOVA p=0.044). The metabolome profile also changed after treatment (p=0.002), with a stronger metric of difference in the metabolome compared to the microbiome (F-value=1.92 microbiome, F-value=3.12 metabolome, Fig. 1c, d). There was statistically significant movement along the first principal coordinate axis after ETI therapy for both the microbiome (SW test p=0.02, DM t-test p=0.0027) and the metabolome (SW p=0.0011, DM t-test p=0.00071). This indicates that the overall changes in the two data types occurred similarly, despite subjects having different initial profiles. To determine how variable the microbiome and metabolome profiles were overall, beta-diversity differences were compared before, between, and after ETI therapy across subjects and within subjects (SW normality beta-diversity microbiome p=1.4 × 10−15, metabolome p=2.2 × 10−16). The lowest microbiome variation was found within subjects before and after therapy, indicating that although the microbiome profiles change significantly (Fig. 1e), individuals were still more similar to themselves before and after therapy than across the cohort. The metabolome beta-diversity comparisons showed different trends than the microbiome. The largest beta-diversity variation in metabolite profiles was seen across patients in samples collected during therapy, signifying that the chemical makeup of sputum becomes far more varied across people once administered ETI (Fig. 1f). In contrast, the metabolomes were the most similar across subjects prior to ETI therapy, indicating that sputum metabolite profiles were relatively similar prior to ETI administration, but varied greatly across individuals after treatment.
3.3. Microbial Changes After ETI Therapy
A random forest machine learning classification was used to determine how well the microbiome data reflected the pre- or post-treatment groups and to rank the ASVs by their contribution to that classification. Overall, the random forest model poorly classified the microbiome data with an error rate of 44.7%. Veillonella parvula and Staphylococcus sp. were strong classifiers (Table S5), however, none of the ranked ASVs were significantly different between pre- and post-treatment samples after correction for false-discovery (Benjamini-Hochberg corrected, WSRT p>0.05). Similarly, at the family level, there was no significant difference before and after therapy following false discovery rate correction (Fig. 2a). The ASV representing Pseudomonas showed dynamic changes in some individuals, but it was not significantly different in the overall paired data (Fig. 2b). We therefore summed the abundance of all ‘CF pathogens’ and ‘anaerobes’ (as described by Raghuvanshi et al. (2020) [22], Table S2) and compared the log-ratio of pathogens/anaerobes. This ratio significantly decreased following ETI therapy (SW p = 0.233, WSRT p = 0.013, Fig. 2).
Fig. 2.
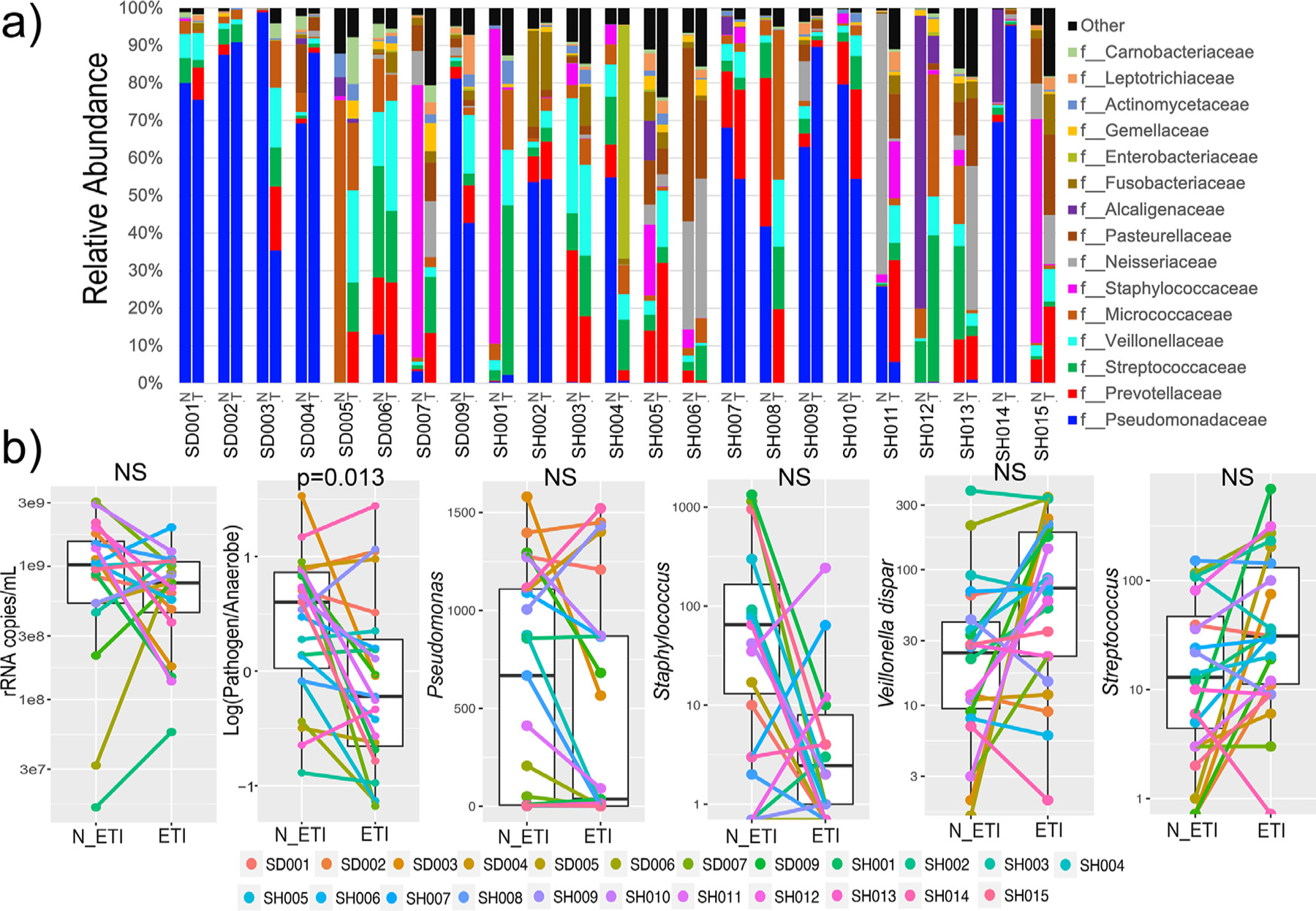
Microbiome changes throughout ETI therapy. a) Taxonomic dynamics at the family level of ASVs in each subject before (N) and after (T) ETI therapy. b) The rRNA copies/mL of sputum, log-ratio of pathogen:anaerobes and ASV dynamics before and after ETI therapy.
A qPCR assay using universal primers for the bacterial 16S rRNA gene [43] was used to calculate the total number of rRNA copies/mL of sputum pre- and post-therapy. The mean prior to therapy was 1.17 × 109 copies/mL and after therapy was 7.62 × 108 copies/mL. This difference was not statistically significant but did show a decreasing trend (SW p = 0.0027, DM t-test p = 0.061, Fig. 2).
3.4. Metabolite Changes After ETI Therapy
Grouping known metabolites into molecular families showed that the strongest metabolomic signature due to ETI therapy was a decrease in peptides and amino acids. Comparatively, phosphocholine and phosphoethanolamine molecular families did not change with ETI (Fig. 3a). A random forest machine learning classification was used to assess how well the complete metabolomic data reflected changes after ETI therapy. The out-of-bag error rate of the classification was 22.92% indicating that there was a metabolomic signal for ETI therapy, but not all samples were correctly classified as pre- or post-treatment, likely due to personalization. Of the 50 most important classifiers (Table S6), 13 had matches in the GNPS database, and of those, 10 were amino acids or peptides. These were primarily dipeptides, including Phe-Glu, Ile-Leu, Glu-Val, and Ser-Phe, as well as the amino acid tryptophan; all of which significantly decreased after ETI therapy (Fig 1b). Molecular network analysis (Fig. 3c) showed a diverse set of peptides that were more abundant prior to ETI. Comparatively, tryptophan and total peptides in our control cohort did not show a significant difference in paired samples across a similar timeframe prior to ETI approval (Fig. S1), implicating ETI in the changes observed here. Metabolites from the kynurenine pathway (which includes tryptophan) were also identified as strong classifiers in the model. Kynurenine, formylkynurenine, and indole abundances significantly decreased after ETI therapy (Fig. 3b). The P. aeruginosa siderophore pyochelin was detected in 10 of the 24 patients, and within those individuals, it also decreased (Fig. 3b). Though commonly detected in CF sputum with the metabolomics methods used here, other P. aeruginosa specialized metabolites were not detected in this study, except for one quinolone (NHQ) that was detected in 6 samples.
Fig. 3.
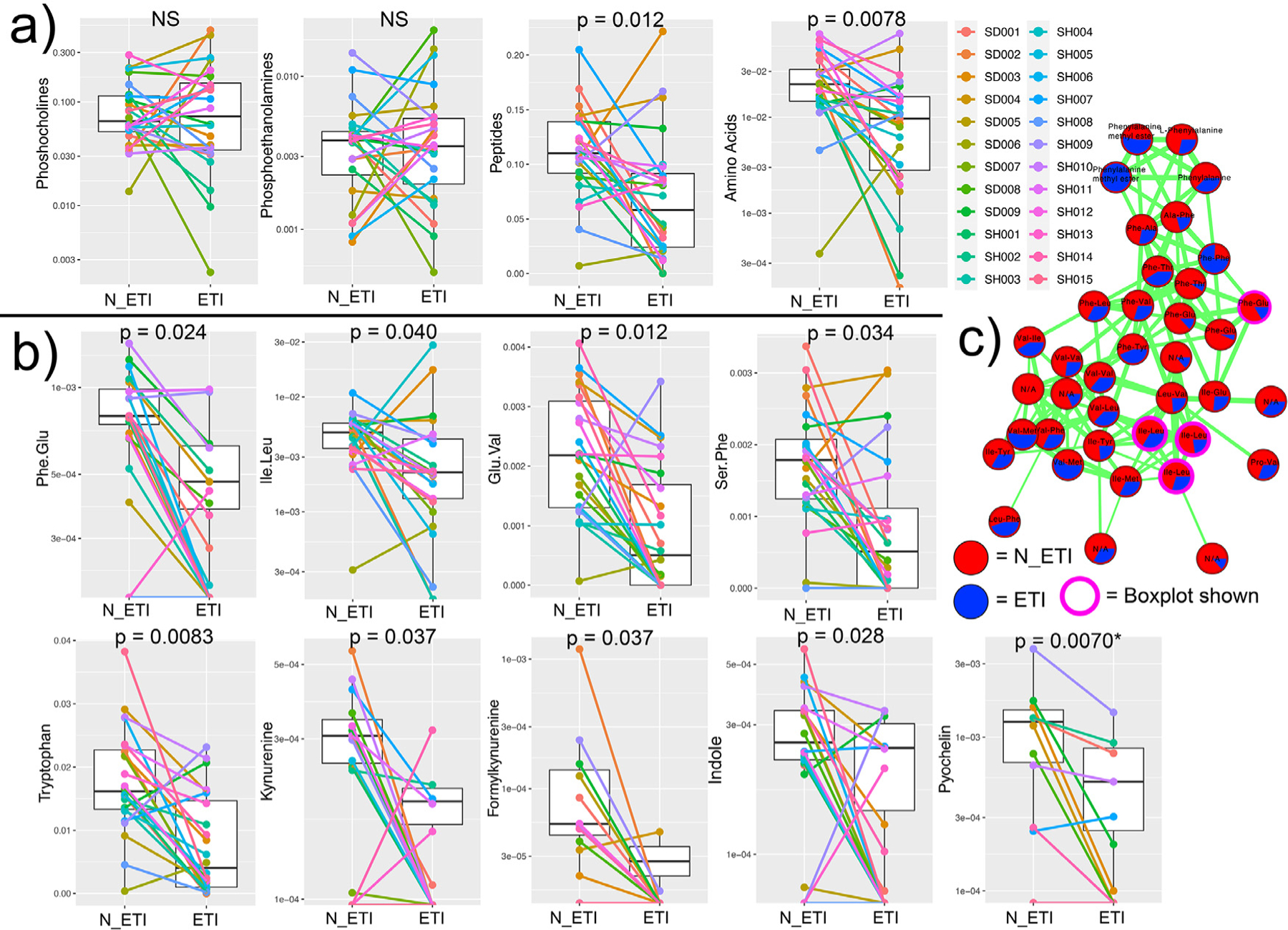
Molecular families and metabolite changes before and after ETI therapy. etabolite network of peptides and other metabolite changes. a) Molecular family metabolite abundance changes pre- and post-therapy. b) Individual metabolite changes pre- and post-therapy. c) Molecular network of peptides identified by GNPS library searching. Each node represents a unique MS/MS spectrum (putative metabolite), connections between the nodes are determined and width-scaled by the cosine score from MS/MS alignment. Pie charts are the total feature abundance colored according to the legend.
3.5. ETI Metabolism in CF Mucus
Ivacaftor, Elexacaftor, and Tezacaftor were all identified in the sputum metabolome by MS/MS analysis with similar fragmentation behavior to that described by Reyes-Ortega et al. (2020) [30]. This included the known and unknown metabolized products of the parent drugs with related MS/MS spectra (Fig. 4). Ivacaftor had extensive metabolism revealed by molecular networking with the parent drug having six related nodes with unique retention times. Two of these are the known M1 and M6 metabolites, representing hydroxymethyl ivacaftor and ivacaftor carboxylate, respectively (level 2 matches according to [31]). Other modifications of the compounds were also seen, including a hydroxylated quinolone ring (m/z 425.2067, C24H29N2O5+H+) and further hydroxylations and carboxylations on two trimethyl groups, but the exact the location of these modifications cannot be discerned (level 3 annotation [31], Fig. S4). Tezacaftor metabolism was also identified including a known dehydrogenation (metabolite M1, m/z 519.1400, C26H26F3N2O6+H+), a known glucuronate (metabolite M3, Fig. 4a) and a phosphorylated metabolite (m/z 599.1401, C26H27F3N2O9P+H+). Elexacaftor exhibited only one metabolic transformation – the loss of a methyl group, but its location could not be determined by MS/MS analysis (level 4 match, Fig. 4). Newly annotated metabolites are only putative and require further analytical analysis to validate proposed structures.
Fig. 4.
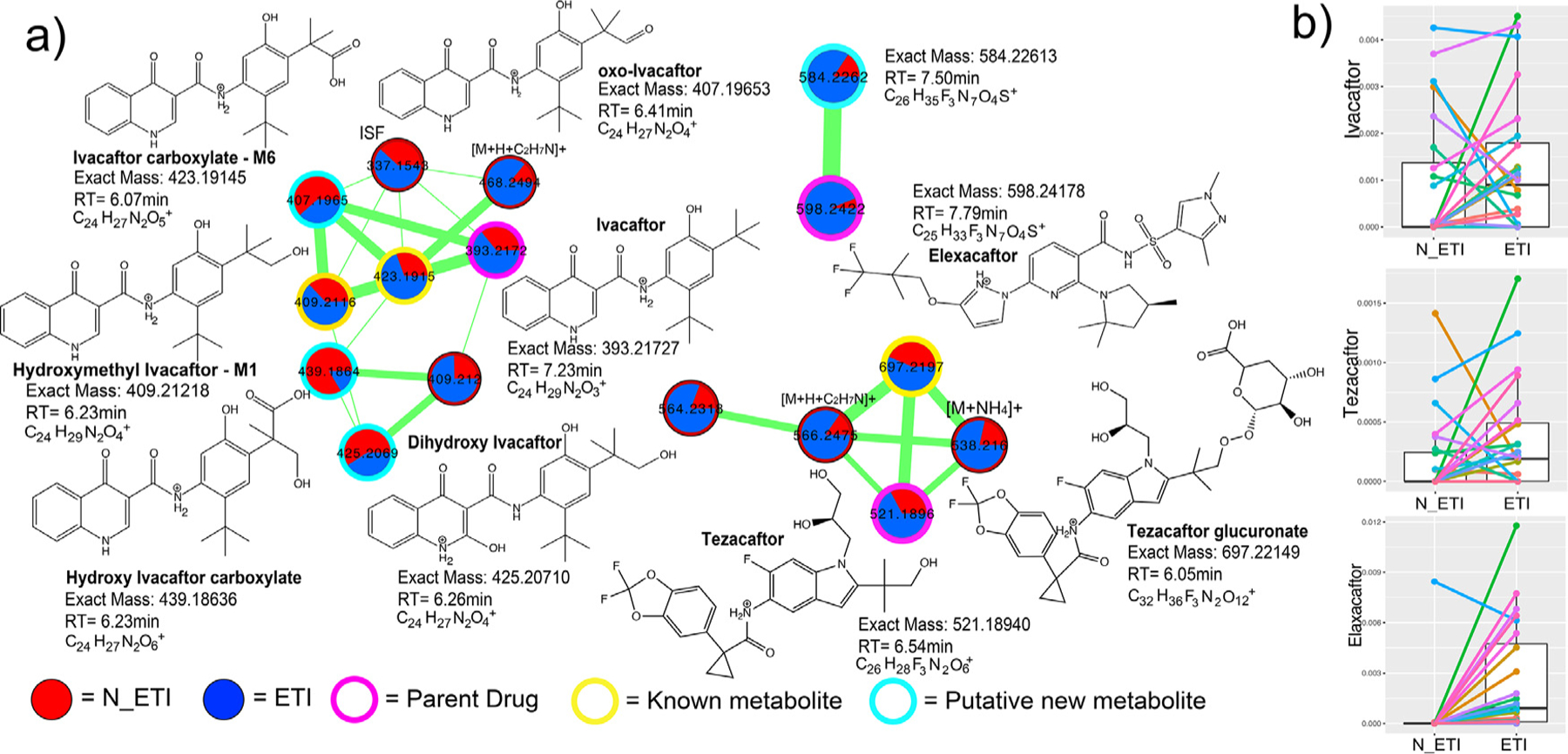
ETI metabolism detected in sputum metabolomic data. a) Three separate molecular networks are shown for Ivacaftor, Tezacaftor, or Elexacaftor and their related metabolic products as identified by MS/MS spectral alignments. Each node in a network represents a unique MS/MS spectrum and connections between the nodes indicate spectral similarity as identified by the cosine score. The width of the edges are scaled to the cosine score and the pie chart inside nodes represent the sum of the area-under-curve abundance of that molecule in either pre- (red) or post-treatment (blue) sputum samples. The nodes are highlighted by whether they represent parent drug, known metabolized product, or putative unknown metabolized product. Putative structures of the metabolites are shown with their molecular formulas, retention times, and exact masses. Note that the stereochemistry of some of the metabolized products cannot be discerned with this level of MS/MS annotation. ISF= in source fragment. b) Boxplots of the area-under-curve abundance of the three parent drugs in pre- and post-ETI samples.
Similar to the ability to detect antibiotics directly in the sputum metabolome, Ivacaftor and Tezacaftor were also detected both prior to and after ETI administration. These two compounds were released as therapies in prior formulations of CFTR modulators, likely explaining their presence. In light of this detection, we investigated whether or not the presence of previously approved correctors/potentiators in sputum prior to administration of ETI may have buffered the microbiome dynamics observed. Ivacaftor (a component of Kalydeco®, Orkambi®, and Symdeko®) was found in 11 of the 24 patients prior to ETI therapy. There was no significant difference in the alpha or beta-diversity changes between subjects that had Ivacaftor in their sputum prior to ETI and those that did not (p>0.05, Fig. S5). Thus, prior CFTR corrector/potentiator therapy did not contribute significantly to the overall changes seen with ETI, allowing these changes to be more definitively attributed to the triple therapy. Elexacaftor, the next-generation corrector unique to ETI, was present in sputum after its prescription as expected. However, one subject unexpectedly had Elexacaftor present in their lung sputum prior to knowledge of clinical administration of ETI.
3.6. Microbiome/Metabolite Associations Through ETI Therapy
We employed the novel neural network algorithm mmvec [29] to integrate the microbiome and metabolome data and provide a picture of collective changes with ETI. Mmvec calculates conditional probabilities of the association between all ASVs in the microbiome data with all metabolite features. The overall neural network showed a strong association between a changing microbiome and metabolome (Fig. S6). The biplot of the mmvec algorithm enabled visualization of the microbiome vectors associated with the metabolomic changes. There was a clear separation in vector directionality between pathogen and anaerobe associations with the metabolome in the mmvec biplot. This indicates that the metabolite changes associated with classic pathogens are not the same metabolites associated with changing anaerobes. Drivers of the metabolite changes associated with classic pathogens were mostly peptides (Fig. 5a); those same peptides shown to be decreasing after ETI therapy. Plotting the conditional probabilities of each peptide with the mean of all pathogens and all anaerobes in the dataset showed that the peptides were significantly associated with classic pathogens (WSRT p<0.001) (Fig. 5b). Kynurenine, another metabolite found to decrease with ETI therapy, was also strongly associated with pathogens. This analysis indicates the decrease of peptides and kynurenine in sputum samples associated with ETI corresponds to a decrease in the relative abundance of classic pathogens (Fig. 5c).
Fig. 5.
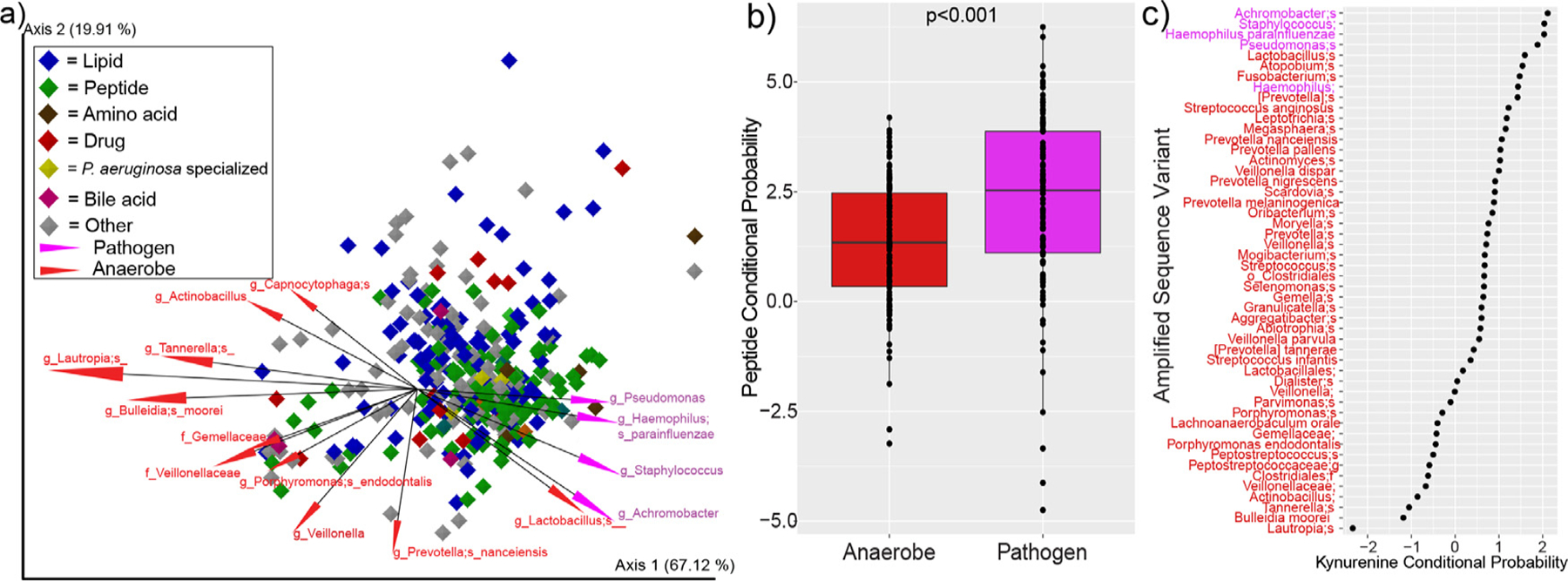
Mmvec analysis of sputum microbiomes and metabolomes from pwCF. a) Biplot of the metabolite and microbe vector associations. Each diamond represents a metabolite (only metabolites annotated within the GNPS library are shown) and they are colored by their molecular family. The vectors are the top 15 ASVs associated with the metabolomic dynamics and they are colored by whether or not they are considered clinical pathogens or anaerobes. b) Conditional probability distributions for the mean of all peptides identified in the dataset and their association with either anaerobes (red) or pathogens (purple, p-value from DM T-test). c) Rank abundance of the conditional probabilities of kynurenine with different anaerobes (red) and pathogens (purple) ASVs.
4. Discussion
This study assessed the multiomic changes in sputum from pwCF after administration of the novel CF triple therapy ETI. ETI has led to significant improvement in lung function and symptom measures of pwCF in clinical trials (and this study) with great potential to improve the lives of these individuals [15,16]. Promising as the treatment is, it is mostly unknown how the therapy will affect lung infections and the chemistry of sputum as CFTR function improves. This is of paramount importance, because if the microbial infections in the lungs of pwCF do not clear and/or change favorably, then the full benefits of the therapy may not be realized. Preliminary studies of other CFTR modulators, specifically Ivacaftor, which has received the most attention due to having the earliest FDA approval, have shown some changes in microbial diversity measures with treatment [32], specifically in the gut [33,34]. However, most studies find little change in the airway microbiome [34–37]. The addition of CFTR correctors, such as Lumacaftor, has shown an increase in microbial diversity in the CF airways [38], but other studies show less marked responses [39]. To our knowledge, this is the first study to report microbiome and metabolome changes resulting from ETI (which includes the new corrector Elexacaftor). Effects of the treatment were seen in both the microbiome and the metabolome. By beta-diversity measures, the effect was stronger in the metabolome, demonstrating that a change in the biochemical environment of CF mucus was associated with ETI therapy. Correspondingly, and similar to other studies [18], patients in our cohort showed improvements in clinical parameters, such as lung function and body mass index.
Microbial alpha-diversity increased after therapy, indicating that the microbiome in the lungs of pwCF became more complex. This increase was driven by a higher microbial evenness, a metric that contributes to the Shannon diversity, though the number of individual microbial sequences detected did not change. Therefore, the lung microbiome of pwCF was not necessarily gaining new or losing old members during therapy, but those present became more similar in their relative abundances. This was reflected in clinical culture data, where the major pathogens were mostly cultured before and after ETI. A similar change in alpha-diversity did not occur in our control cohort, supporting the notion that ETI is responsible for the changes observed. The overall profiles of the microbiome (beta-diversity) were changed significantly after ETI therapy and did so in a similar way across the study population, as shown by the homogeneous directional movement across the first principal coordinate axis. Despite these overall changes, no single organism was significantly altered from ETI therapy after multiple-comparisons correction. This is likely due to the widely known personalization in the CF microbiome, the phenomenon where individual pwCF have unique microbiome signatures which show some consistency over time for that individual [8,40]. Because individuals have very different microbial profiles, the start and endpoints from any pharmaceutical treatment may not be universal across subjects. This personalization was again observed here, as subjects were still more similar to themselves after ETI therapy than to other subjects. A larger sample size may have reached statistical significance for microbial taxa of interest because the trends for pathogens, such as P. aeruginosa and Staphylococcus, were showing reductions in relative abundance, while anaerobes were showing an increase. Accordingly, a collective comparison of the log-ratio of pathogen:anaerobe abundances did reach statistical significance. This ASV binning approach normalizes some of the personalized signatures, as not all subjects have the same bacteria, but when grouped in this clinically relevant manner, there is an overall reduction of classic pathogens relative to anaerobes. There was also a trend in the decreased bacterial load after ETI therapy, supporting that the increase in diversity seen from microbiome measures may be associated with a decrease in total bacteria in sputum, though this did not reach statistical significance. In summary, ETI therapy was associated with an altered lung microbiome, exemplified by an increased microbial evenness driven by a reduction in the relative abundance of pathogens in place of an increase in the relative abundance of anaerobes. This increase in anaerobes may be relevant for treating lung infections in the new era of highly effective CFTR modulators. The role of anaerobes in the CF lung is rather unclear however, as they are associated with better lung function [41], but also pulmonary exacerbations [22,23,42]. From a clinical standpoint, future studies need to examine if altering antimicrobial therapies for pulmonary exacerbations are needed as ETI and other highly effective modulators begin to reshape the lung microbiome.
The metabolome showed stronger beta-diversity changes than the microbiome. The sputum metabolomes were relatively similar prior to therapy, but when on drug, they became highly diverse across subjects. These interesting chemical dynamics indicate that ETI therapy is associated with a sort of metabolomic turmoil within the airways of pwCF, where the lung sputum biochemistry changes significantly with a highly varied outcome across individuals. However, similar to the microbiome, the directionality of change had some uniformity, indicating a common metabolomic shift driven by ETI. Uniform changes included a decrease in peptides, amino acids and kynurenine metabolism. The latter was identified as an important pathway associated with P. aeruginosa dynamics from Lumacaftor/Ivacaftor therapy in a previous study [39], which supports the findings here, and may represent a universal consequence of CFTR modulator treatment. The decrease in the overall abundance of peptides, particularly dipeptides, links these metabolites to a previous study that associated their abundance with worsening lung function and neutrophil elastase activity [13]. Though not measured in this study, the decrease in peptides may be a proxy for decreased neutrophil proteolysis and inflammation in the lung. Importantly, we re-analyzed some data from that study as a control cohort and did not find a decrease in peptides in similarly paired samples, implicating ETI in the peptide and amino acid dynamics observed here. There may be a link between the decrease in kynurenine metabolism and amino acids/peptides, as it is a principal pathway for the metabolism of tryptophan in humans and bacteria. The reduction of peptides in sputum may reduce their availability for pathogens (particularly P. aeruginosa) to metabolize through the kynurenine pathway or others. Mmvec analysis further supported the changing relationship between peptides, kynurenine metabolism and pathogens. This approach, robust to the statistical challenges of cross-omics comparisons from compositional datasets [29,43], showed that the decrease in peptides and kynurenine was associated with a reduction in classic pathogens. In light of this finding, we propose the hypothesis that ETI therapy, and possibly other CFTR modulators [39], reshape microbiome niche space in CF mucus by reducing peptide and amino acid availability. This shift may begin to squeeze out some pathogens, such as P. aeruginosa, which is known to preferentially metabolize amino acids in the lung [44–47]. This changing niche space could have clinical consequences, as treatment for lung infection may need to be tailored to a shifting microbiome. How much the microbiome will change however, and whether there will be an altered steady state of the bacterial and viral community, will require more long-term studies of the effects of ETI.
Metabolomics of complex clinical samples often detects xenobiotics, such as drugs administered to patients, which can become confounders of studies of a particular treatment such as ETI [14]. For example, we detected four antibiotics in the sputum metabolome data, allowing for comparison of their abundances with microbiome measures in the same samples. There was no difference between the amount of antibiotics measured before or after therapy and no correlation between their abundance and microbiome alpha-diversity. This supports the notion that antibiotics were not a strong confounding factor in our study of ETI, however, we do not detect all drugs administered and it cannot be ruled out that the changes observed may, in part, be due to the impacts of antibiotics. Interestingly, prior CFTR modulators were also detected in the sputum metabolome data and there was diverse metabolism of these drugs, particularly Ivacaftor. This created a unique opportunity to determine whether or not the presence of a previously approved CFTR modulator therapy in a patient’s sputum affected the microbiome and metabolome dynamics of ETI. There was no difference in the microbial and metabolite dynamics while on ETI between those previously taking CFTR modulators and those not, evidence that the changes observed were associated with ETI therapy, perhaps Elexacftor itself, which is known to be a highly effective CFTR corrector. These results show promise that ETI therapy may have a particularly strong effect on the CF sputum microbiome and metabolome where other CFTR modulators have not [34–37].
There are several caveats to our study, perhaps most importantly, ETI therapy is known to reduce sputum production in pwCF. It is difficult to discern if the changes we identified here are due to sputum chemical and microbial dynamics or a change in the ability to produce sputum when on CFTR modulators. Instructions for expectoration were not varied before and after ETI to normalize the sampling approach, and all subjects in this study were able to produce sputum for collection both prior to and during ETI therapy. Furthermore, the relatively small sample size may have masked some specific changes, particularly with individual microbial ASVs. Larger studies of pwCF before and after ETI therapy are warranted, though with the wide availability of these CFTR modulators currently makes collecting ETI naïve samples difficult. Follow up studies on subjects taking ETI for longer periods are also needed.
In conclusion, the highly effective CF triple therapy ETI results in an overall reduction in pathogens compared to anaerobes and reduced amino acid availability and kynurenine metabolism. This change was associated with improved clinical parameters, most notably lung function. Changing chemistry within lung mucus associated with ETI will begin to reshape the niche space for its resident microbiome. A reduction of amino acids and peptides in lung mucus may be unfavorable to pathogens that preferentially metabolize these nutrients, leading a different future for the CF lung microbiome with widespread availability of ETI.
Supplementary Material
Acknowledgments
The authors would like to thank the Spectrum Health and UC San Diego clinical teams who assisted with sample and metadata collection for this study. We would like to acknowledge funding from the National Institutes of Allergy and Infectious Diseases under R01 grant R01AI145925 awarded to PI Quinn.
Funding:
This project was funded by a National Institute of Allergy and Infectious Disease Grant R01AI145925
✩. Sources of support:
NIH R01AI145925
Footnotes
Declaration of Competing Interest
The authors declare no conflicts of interest. Dr. Ryan Thomas served as a consultant for Vertex Pharmaceuticals in 2020, but this consultation did not affect the study’s design, results, interpretation or conclusions.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jcf.2021.11.003.
References
- [1].Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ 2007;335:1255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zielenski J. Genotype and Phenotype in Cystic Fibrosis. Respiration 2000;67:117–33. [DOI] [PubMed] [Google Scholar]
- [3].Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann. Hum. Genet 2003;67:471–85. [DOI] [PubMed] [Google Scholar]
- [4].Coburn B, Wang PW, Caballero JDiaz, Clark ST, Brahma V, Donaldson S, Zhang Y, Surendra A, Gong Y, Tullis DElizabeth, Yau YCW, Waters VJ, Hwang DM, Guttman DS. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep 2015;5:10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E, Sime-Ngando T, Chabé M, Viscogliosi E. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PLoS One 2012;7:e36313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rabin HR, Surette MG. The cystic fibrosis airway microbiome. Curr. Opin. Pulm. Med 2012;18:622–7. [DOI] [PubMed] [Google Scholar]
- [7].Lim YW, Schmieder R, Haynes M, Willner D, Furlan M, Youle M, Abbott K, Edwards R, Evangelista J, Conrad D, Rohwer F. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros 2013;12:154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao J, Schloss PD, Kalikin LM, a Carmody L, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, Young VB, LiPuma JJ. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc. Natl. Acad. Sci. U. S. A 2012;109:5809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hassett DJ, Sutton MD, Schurr MJ, Herr AB, Caldwell CC, Matu JO. Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol 2009;17:130–8. [DOI] [PubMed] [Google Scholar]
- [10].Manzenreiter R, Kienberger F, Marcos V, Schilcher K, Krautgartner WD, Obermayer A, Huml M, Stoiber W, Hector A, Griese M, Hannig M, Studnicka M, Vitkov L, Hartl D. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros 2012;11:84–92. [DOI] [PubMed] [Google Scholar]
- [11].Reid DW, Misso N, Aggarwal S, Thompson PJ, Walters EH. Oxidative stress and lipid-derived inflammatory mediators during acute exacerbations of cystic fibrosis. Respirology 2007;12:63–9. [DOI] [PubMed] [Google Scholar]
- [12].Yang J, Eiserich JP, Cross CE, Morrissey BM, Hammock BD. Metabolomic profiling of regulatory lipid mediators in sputum from adult cystic fibrosis patients., Free Radic. Biol. Med 2012;53:160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quinn RA, Adem S, Mills RH, Comstock W, DeRight Goldasich L, Humphrey G, Aksenov AA, Melnik AV, da Silva R, Ackermann G, Bandeira N, Gonzalez DJ, Conrad D, O’Donoghue AJ, Knight R, Dorrestein PC. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019;7(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Quinn RA, Phelan VV, Whiteson KL, Garg N, Bailey BA, Lim YW, Conrad DJ, Dorrestein PC, Rohwer FL. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. ISME J 2015;10:1483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bear C A therapy for most with cystic fibrosis. Cell 2020;180:211. [DOI] [PubMed] [Google Scholar]
- [16].Ridley K, Condren M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther 2020;25:192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS. V.−445–103 T. Group, Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised. phase 3 trial. Lancet 2019;394:1940–8 London, England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, Marigowda G, McKee CM, Moskowitz SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Rowe SM, Jain R, Group V-445–102 S. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med 2019;381:1809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gonzalez A, Navas-Molina JA, Kosciolek T, McDonald D, Vázquez-Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, Sanders JG, Shorenstein J, Holste H, Petrus S, Robbins-Pianka A, Brislawn CJ, Wang M, Rideout JR, Bolyen E, Dillon M, Caporaso JG, Dorrestein PC, Knight R. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 2018;15:796–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al Ghalith GA, Alexander H, Alm EJ, Arumugam M, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Colin J, Brown CT, Callahan BJ, Mauricio A, Rodríguez C, Chase J, Cope E, Da Silva R, Dorrestein PC, Douglas GM, Duvallet C, Edwardson CF, Ernst M, Fouquier J, Gauglitz JM, Gibson DL, Gonzalez A, Huttley GA, Janssen S, Jarmusch AK, Kaehler BD, Bin Kang K, Keefe CR, Keim P, Kelley ST, Ley R, Loftfield E, Marotz C, Martin B, Mcdonald D, Mciver LJ, Alexey V, Metcalf JL, Morgan SC, Morton JT, Naimey AT. QIIME 2 : Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr 2018. doi: 10.7287/peerj.preprints.27295v2. [DOI] [PMC free article] [PubMed]
- [21].Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, Knight R. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017;2. available at http://msystems.asm.org/content/2/2/e00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Raghuvanshi R, Vasco K, Vázquez-Baeza Y, Jiang L, Morton JT, Li D, Gonzalez A, DeRight Goldasich L, Humphrey G, Ackermann G, Swafford AD, Conrad D, Knight R, Dorrestein PC, Quinn RA, Chia N. High-Resolution Longitudinal Dynamics of the Cystic Fibrosis Sputum Microbiome and Metabolome through Antibiotic Therapy. mSystems 2020;5:e00292 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carmody LA, Caverly LJ, Foster BK, Rogers MAM, Kalikin LM, Simon RH, VanDevanter DR, LiPuma JJ, Chotirmall SH. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS One 2018;13:e0194060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pluskal T, Castillo S, Villar-Briones A, Orešič M, Zmine M. 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010;11:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang M, Carver JJ, Phelan VV, Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu C-C, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw C-C, Yang Y-L, Humpf H-U, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, Boya CAP, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodríguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard P-M, Phapale P, Nothias L-F, Alexandrov T, Litaudon M, Wolfender J-L, Kyle JE, Metz TO, Peryea T, Nguyen D-T, VanLeer D, Shinn P, Jadhav A, Müller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BØ, Pogliano K, Linington RG, Gutiérrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 2016;34. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dührkop K, Fleischauer M, Ludwig M, Aksenov AA, Melnik AV, Meusel M, Dorrestein PC, Rousu J, Böcker S. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019;16:299–302. [DOI] [PubMed] [Google Scholar]
- [27].Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2013;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Breiman L Random Forests. Mach. Learn 2001;45:5–32. [Google Scholar]
- [29].Morton JT, Aksenov AA, Nothias LF, Foulds JR, Quinn RA, Badri MH, Swenson TL, Van Goethem MW, Northen TR, Vazquez-Baeza Y, Wang M, Bokulich NA, Watters A, Song SJ, Bonneau R, Dorrestein PC, Knight R. Learning representations of microbe–metabolite interactions. Nat. Methods 2019:1–9. [DOI] [PMC free article] [PubMed]
- [30].Reyes-Ortega F, Qiu F, Schneider-Futschik EK. Multiple Reaction Monitoring Mass Spectrometry for the Drug Monitoring of Ivacaftor, Tezacaftor, and Elexacaftor Treatment Response in Cystic Fibrosis: A High-Throughput Method. ACS Pharmacol. Transl. Sci 2020;3:987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol 2014;48:2097–8. [DOI] [PubMed] [Google Scholar]
- [32].Ronan NJ, Einarsson GG, Twomey M, Mooney D, Mullane D, NiChroinin M, O’Callaghan G, Shanahan F, Murphy DM, O’Connor OJ, Shortt CA, Tunney MM, Eustace JA, Maher MM, Elborn JS, Plant BJ. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores After CFTR Modulation With Ivacaftor. Chest 2018;153:395–403. [DOI] [PubMed] [Google Scholar]
- [33].Ooi CY, Syed SA, Rossi L, Garg M, Needham B, Avolio J, Young K, Surette MG, Gonska T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep 2018;8:17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kristensen MI, de Winter-de Groot KM, Berkers G, Chu MLJN, Arp K, Ghijsen S, Heijerman HGM, Arets HGM, Majoor CJ, Janssens HM, van der Meer R, Bogaert D, van der Ent CK. Individual and Group Response of Treatment with Ivacaftor on Airway and Gut Microbiota in People with CF and a S1251N Mutation. J. Pers. Med 2021;11. doi: 10.3390/jpm11050350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harris JK, Wagner BD, Zemanick ET, Robertson CE, Stevens MJ, Heltshe SL, Rowe SM, Sagel SD. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc 2019;17:212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Peleg AY, Choo JM, Langan KM, Edgeworth D, Keating D, Wilson J, Rogers GB, Kotsimbos T. Antibiotic exposure and interpersonal variance mask the effect of ivacaftor on respiratory microbiota composition. J. Cyst. Fibros 2018;17:50–6. [DOI] [PubMed] [Google Scholar]
- [37].Bernarde C, Keravec M, Mounier J, Gouriou S, Rault G, Férec C, Barbier G, Héry-Arnaud G. Impact of the CFTR-potentiator ivacaftor on airway microbiota in cystic fibrosis patients carrying a G551D mutation. PLoS One 2015;10:e0124124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Graeber SY, Boutin S, Wielpütz MO, Joachim C, Frey DL, Wege S, Sommerburg O, Kauczor H-U, Stahl M, Dalpke AH, Mall MA. Effects of Lumacaftor-Ivacaftor on Lung Clearance Index, Magnetic Resonance Imaging and Airway Microbiome in Phe508del Homozygous Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc 2021. doi: 10.1513/AnnalsATS.202008-1054OC. [DOI] [PubMed]
- [39].Neerincx AH, Whiteson K, Phan JL, Brinkman P, Abdel-Aziz MI, Weersink EJM, Altenburg J, Majoor CJ, Maitland-van der Zee AH, Bos LDJ. Lumacaftor/ivacaftor changes the lung microbiome and metabolome in cystic fibrosis patients. ERJ Open Res 2021;7:731–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lynch SV, Bruce KD. The cystic fibrosis airway microbiome., Cold Spring Harb. Perspect. Med 2013;3:a009738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lamoureux C, Guilloux C-A, Beauruelle C, Jolivet-Gougeon A, Héry-Arnaud G. Anaerobes in cystic fibrosis patients’ airways. Crit. Rev. Microbiol 2019;45:103–17. [DOI] [PubMed] [Google Scholar]
- [42].Quinn RA, Whiteson K, Lim Y-W, Salamon P, Bailey B, Mienardi S, Sanchez SE, Blake D, Conrad D, Rohwer F. A Winogradsky-based culture system shows an association between microbial fermentation and cystic fibrosis exacerbation. ISME J 2015;9. doi: 10.1038/ismej.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol 2017;8:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS Pathog 2016;12:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Thomas SR, Ray A, Hodson ME, Pitt TL. Increased sputum amino acid concentrations and auxotrophy of Pseudomonas aeruginosa in severe cystic fibrosis lung disease. Thorax 2000;55:795–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].BARTH AL, PITT TL. The high amino-acid content of sputum from cystic fibrosis patients promotes growth of auxotrophic Pseudomonas aeruginosa. J. Med. Microbiol 1996;45:110–19. [DOI] [PubMed] [Google Scholar]
- [47].Quinn RA, Lim YW, Maughan H, Conrad D, Rohwer F, Whiteson KL. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. MBio 2014;5. doi: 10.1128/mBio.00956-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.