

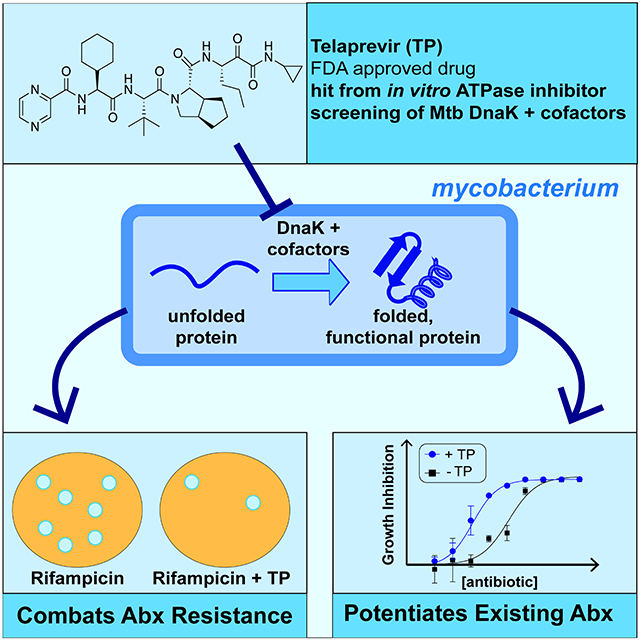
Summary:
DnaK is the bacterial homolog of Hsp70, an ATP-dependent chaperone that assists cofactor proteins to catalyze nascent protein folding and salvage misfolded proteins. In the pathogen Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), DnaK and its cofactors are proposed antimycobacterial targets, yet few small molecule inhibitors or probes exist for these classes of proteins. Here, we describe the repurposing of a drug called telaprevir that is able to allosterically inhibit the ATPase activity of DnaK, and prevents chaperone function by mimicking peptide substrates. In mycobacterial cells, telaprevir disrupts DnaK/cofactor-mediated cellular proteostasis, resulting in enhanced efficacy of aminoglycoside antibiotics and reduced resistance to the frontline TB drug rifampin. Hence, this work contributes to a small but growing collection of protein chaperone inhibitors, and demonstrates that these molecules disrupt bacterial mechanisms of survival in the presence of different antibiotic classes.
Keywords: chaperones, cofactors, DnaK, Hsp70, DnaJ, proteostasis, antibiotic adjuvants, resistance, mycobacteria, tuberculosis
Graphical Abstract
eTOC:
Hosfelt and Richards et al. discover that an FDA-approved antiviral drug called telaprevir inhibits chaperone activation by cofactors in bacteria, and in doing so can increase bacterial sensitivity to antibiotics and reduce the likelihood of drug resistance.
Introduction:
The molecular chaperone heat shock protein 70 (Hsp70) is conserved throughout all domains of life, and is intimately involved in regulating protein homeostasis for cell survival (Calloni et al., 2012; Zuiderweg et al., 2013; Kim et al., 2013; Balchin, Hayer-Hartl and Hartl, 2020). Hsp70s have been the targets of ongoing drug discovery campaigns due to their link to protein misfolding-related diseases and certain cancers (Chang et al., 2011; Li et al., 2013; Chang et al., 2008; Cesa et al., 2013; Shao et al., 2018; Rodina et al., 2013; Taldone et al., 2014; Kang et al., 2014). Eukaryotes contain several Hsp70 isoforms (Sharma and Masison, 2009); however bacteria typically encode one major protein folding isoform called DnaK (Skowyra, Georgopoulos and Zylicz, 1990). In the human pathogen Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), dnaK is predicted to be essential as shown in the related non-pathogenic bacterium M. smegmatis (Msm) (Sassetti, Boyd and Rubin, 2003; Griffin et al., 2011; Fay and Glickman, 2014; Bosch et al., 2021). Despite their importance, we have relatively few selective small molecule probes or inhibitors for chaperones, especially when compared to other enzymes such as kinases (Gestwicki and Shao, 2019; Knight and Shokat, 2005; Evans, Chang and Gestwicki, 2010; Ambrose and Chapman, 2021). While some existing eukaryotic Hsp70 inhibitors show cross-reactivity against bacterial DnaKs (Taylor et al., 2020), our goal was to discover chemical scaffolds that directly inhibit Mtb DnaK, since it represents a potential antibiotic target and new TB drugs are in much demand (Lupoli et al., 2018; Rock, 2019; Patel, Li and Seeliger, 2019).
Hsp70s function as a part of an expansive network of protein-protein interactions (PPIs), which include cofactor proteins. The prototypical Hsp70, Escherichia coli DnaK, is known to function alongside the cofactors DnaJ and GrpE, a nucleotide exchange factor (NEF) (Figure 1) (Calloni et al., 2012; Balchin, Hayer-Hartl and Hartl, 2016; Balchin, Hayer-Hartl and Hartl, 2020; Wu et al., 2020). DnaK is highly allosteric and able to assume a wide range of conformations due to the flexible nature of a linker that connects the N-terminal, or nucleotide binding domain (NBD), to the C-terminal substrate binding domain (SBD) (Figure 1A). When bound to ATP, DnaK occupies a more compact state (Kityk et al., 2012). Dimeric DnaJ delivers unfolded protein substrates to DnaK-ATP (Figure 1B), which leads to the hydrolysis of ATP to ADP and results in a conformational change to an extended state. Interaction of DnaK with GrpE leads to an exchange of ADP for ATP, and restarts the catalytic cycle of substrate bind and release.
Figure 1. DnaK plays a crucial role in protein folding and reactivation following stress.

(A) Cartoon representation of E. coli DnaK bound to ATP (PDB ID: 5NRO) and ADP (2KHO). DnaK is composed of an N-terminal nucleotide binding domain (NBD) and C-terminal substrate binding domain (SBD). (B) Non-native proteins are folded by mycobacterial DnaK assisted by cofactor proteins DnaJ1 and DnaJ2 (DnaJ proteins), which deliver protein substrates, and the nucleotide exchange factor GrpE (Balchin, Hayer-Hartl and Hartl, 2020; Lupoli et al., 2018). Any step of the catalytic cycle, shown above, might be inhibited to disable this essential chaperone network in mycobacteria. A representative protein structure is shown (PDB ID: 3IEP).
DnaK is involved in both folding of newly synthesized proteins, as well as transport and re-folding of aggregated proteins (Figure 1B). We and others have shown that Mtb DnaK works in conjunction with two DnaJ proteins (DnaJ1 and DnaJ2) and GrpE to fold proteins in vitro, and that disrupting this chaperone network through rational point mutations leads to cell death in mycobacteria (Lupoli et al., 2016; Fay and Glickman, 2014). Based on this observation, we sought to discover a small molecule that could phenocopy genetic disruption of DnaK-mediated protein folding. Similar to other chaperones (Lukačišinová, Fernando and Bollenbach, 2020; Cowen and Lindquist, 2005), mycobacterial DnaK and its cofactors have been implicated in resistance mechanisms to antibiotics (Fay et al., 2021); hence, we hypothesized that mycobacterial DnaK-cofactor inhibitors may have broad applications that span from fundamental to combating avenues of drug resistance.
We performed a high-throughput multiprotein screen to identify inhibitors of the mycobacterial DnaK chaperone network. We emphasized discovery of allosteric over ATP competitive inhibitors, as the latter might exhibit promiscuity in the cell (Massey, 2010; Terrab and Wipf, 2020; Gestwicki and Shao, 2019). In doing so, we identify two clinical drugs that are able to modulate Mtb DnaK activity, one of which has activity against mycobacterial cells and enhances the potency of aminoglycosides, while reducing the evolution of resistance against a frontline TB drug. Our work contributes to a small but growing list of chemical scaffolds that have activity against protein chaperones, and demonstrates that DnaK inhibitors can be used to disrupt bacterial stress response mechanisms.
Results:
Small molecule screen reveals clinical drugs that disrupt Mtb DnaK.
In order to identify small molecules capable of modulating the DnaK-mediated proteostasis network, we screened for inhibitors of DnaK in the presence of cofactor proteins. We have shown that the ATPase activity of DnaK is enhanced by the addition of physiologically-relevant sub-stoichiometric concentrations of cofactor proteins DnaJ2 and GrpE (Figure 2A) (Lupoli et al., 2016), which enables an in vitro chaperone network assay. We sought to enlist a screening approach that offered improved sensitivity over kinase-based ATP consumption assays. Using a luciferase-coupled assay to detect the production of ADP (Figure S1A), we optimized the ratio of DnaK to cofactors (10:1:1, DnaK:DnaJ2:GrpE) and validated a high-throughput set-up with a small pilot library, which gave a favorable Z’ score (>0.8) and showed high reproducibility (Figure S1B). We then screened two collections of small molecules (Figure 2A-B). The first collection (called drug repurposing library) consisted of ~1300 approved drugs, and ~3700 compounds from peer-reviewed research and patents. The second collection (called drug-like) consisted of ~20,000 small molecules that were ranked based on quantitative estimate of drug-likeness (Bickerton et al., 2012). Compounds were screened near the KM (~20 μM) of Mtb DnaK for ATP and its affinity for DnaJ2 (~20 μM) (Lupoli et al., 2016) to uncover compounds that might disrupt DnaK activation, as well as controls that might compete with ATP.
Figure 2. High-throughput screen leads to the identification of small molecules that attenuate the ATPase activity of Mtb DnaK-DnaJ2-GrpE.

(A) Schematic of small molecule screen employed to discover allosteric inhibitors of the mycobacterial chaperone network. (B) Scatter plots for the normalized percent inhibition (NPI) of ATP hydrolysis by Mtb DnaK-DnaJ2-GrpE for each compound tested. (C) Chemical structures for selected hit compounds, both from the drug repurposing collection. (D) Dose-response curves for compounds from part B in the presence of Mtb DnaK-DnaJ2-GrpE. (E) Chemical structures of additional small molecules that inhibit Hepatitis C Virus (HCV) protease, similar to Telaprevir (TP). (F) Dose-response assay of additional HCV inhibitors against Mtb DnaK-DnaJ2-GrpE. For dose-response curves, error bars represent standard deviation (SD) (n=3). See also Figure S1, Tables S4 and S5.
The effect of the compounds was judged by a normalized percentage of inhibition (NPI) of ATPase activity relative to control reactions of DnaK with and without cofactors. Based on a cutoff of ≥ 40 NPI (Figure 2B), eighteen molecules were identified as hits with confirmed half-maximal inhibition (IC50) values ≤ 20 μM against DnaK activated by cofactors (Tables S4-S5). Out of these hits, sixteen exhibited inhibitory activity against luciferase and were classified as false positives (Figure S1C-E), or were largely hydrophobic and expected to exhibit off-target effects. Two molecules from the drug repurposing collection, telaprevir (TP) and 2Cl-IB-MECA (MECA) (Figure 2C), were confirmed to have IC50 values of ~ 4.0 μM and 4.5 μM, respectively, against Mtb DnaK-cofactor ATPase activity (Figure 2D) and no activity against luciferase alone (Figure S1C). The structural disparity of the two molecules hinted that each inhibited ATPase activity by a different mechanism, and they were chosen for further analysis.
Drugs similar to TP do not inhibit the Mtb chaperone network in vitro.
MECA is a nucleoside analog A3 adenosine receptor agonist (Kim et al., 1994), and is currently in stage III clinical trials for the treatment of liver cancer. TP is the first of many FDA-approved Hepatitis C Virus (HCV) drugs that target an essential viral protease called nonstructural protein 3/4A (NS3/4A) (Grillot et al., 2011). As seen in Figure 2C, TP inhibited the chaperone’s ATPase activity more effectively than MECA, and so analogs of TP were explored further.
Both TP and another first-generation HCV drug, boceprevir (BC) (Figure 2C, E) possess a ketoamide warhead that forms a reversible covalent linkage to the protease active site. Along with the second-generation macrocyclic compounds grazoprevir and danoprevir, BC was present in the drug repurposing screening collection; however, none of these HCV drugs were selected as hits by the criteria described above. To better assess potency, dose-response assays were carried out with representative HCV protease inhibitors (BC, grazoprevir, simeprevir, danoprevir and paritaprevir), as well as an HCV RNA-binding protein inhibitor (velpatasvir) (Figure 2F and S2A). Only simeprevir showed measurable inhibition of ATPase activity, but with an IC50 value ~8-fold greater than that of TP and a steep slope about the inflection point that is indicative of compound aggregation (McGovern et al., 2003). These data suggest that TP does not inhibit mycobacterial DnaK through non-selective covalent labeling (since BC was inactive) and that potent DnaK inhibition is unique to the TP scaffold.
TP and MECA inhibit the stimulation of Mtb DnaK’s ATPase activity.
To obtain insight into TP’s mode of inhibition, we used an HPLC-based assay to directly measure ATP and ADP levels, in place of the coupled assay used for screening. We observed that addition of TP to DnaK and cofactor reactions returned the level of ADP to that of reactions containing DnaK only; while addition of TP to DnaK reactions did not significantly change the level of ADP produced (Figure 3A). We also measured DnaK’s ATPase activity over a time course with and without cofactors in the presence of TP and select HCV inhibitors, which further validated that TP inhibits activation of DnaK (Figure S2B). To tease out the effect of TP on the function of individual cofactor proteins, cofactor titration experiments were performed. In these experiments, the concentration of DnaJ2 or GrpE was varied in reactions containing DnaK and a sub-stoichiometric concentration of the other cofactor, and ATPase rates were measured in the presence and absence of excess HCV inhibitors (Figure 3B). We found that TP and simeprevir significantly disrupted both DnaJ2- and GrpE-mediated activation of DnaK, while BC did not.
Figure 3. TP and MECA inhibit activation of Mtb DnaK by cofactors through different mechanisms.

(A) HPLC analysis (left) and quantification of ATP and ADP levels (right) in DnaK reactions with and without cofactor indicate that TP inhibits DnaK activation. (B) Titration of DnaK (4 μM) and one cofactor (GrpE or DnaJ2, 0.4 μM) with increasing concentrations of the other cofactor in the presence and absence of excess HCV inhibitors. (C) Cofactor titrations in the presence and absence of excess MECA. (D) Chemical structures of MECA analogs, AB-MECA and VER-155008, with moieties that differ from 2Cl-IB-MECA shown in red. (E) Percent ATP hydrolysis by equimolar DnaK:GrpE and sub-stoichiometric DnaJ2 show MECA inhibits, while analogs do not. (F) Proposed model for disruption of DnaK activation by TP and MECA. For all experiments, error bars represent SD (n=3), **p<0.05 (Welch’s t-test was used for comparison). See also Figure S2.
MECA is a nucleoside analog, and so we hypothesized that it might compete with ATP for binding to the nucleotide binding pocket of DnaK. However, kinetic analysis of ATP hydrolysis by DnaK-DnaJ2-GrpE with and without MECA indicated no competition with ATP (Figure S2C). In comparison, TP increased the KM of DnaK by ~2-fold. Since this result suggested that MECA might impede ATP hydrolysis indirectly, we performed cofactor titration experiments with and without added MECA (Figure 3C). While MECA hindered DnaJ2-mediated activation of DnaK’s ATPase activity, it nearly abrogated activation of DnaK by increasing concentrations of GrpE, most notably at 1:1 DnaK:GrpE with sub-stoichiometric DnaJ2 present. To further probe this observation, we utilized HPLC analysis to directly monitor the effect on ATP/ADP levels under these conditions. We compared the effect of MECA to the close structural analogs AB-MECA and VER-155008, a known Hsp70 inhibitor (Figure 3D) (Grillot et al., 2011; Olah et al., 1994). Neither of these analogs caused significant ATPase inhibition in the high-throughput screen as members of the drug repurposing library. Accordingly, out of the three compounds tested, only MECA showed inhibition of ATPase activation at 50 μM in the presence of equimolar GrpE (Figure 3E).
Since bacterial DnaK and GrpE have high affinity for one another (Melero et al., 2015), we examined the effect of MECA on DnaK-GrpE complex formation by native gel analysis. We observed that DnaK-GrpE complex formation is not affected by MECA, regardless of the presence or absence of nucleotide (Figure S3A-B). Isothermal titration calorimetry (ITC) analysis confirmed that MECA does not bind to DnaK alone (Figure S3C), supporting the observation that MECA interacts with DnaK through its association with cofactors. Taken together, these data suggested that TP and MECA inhibit ATP turnover by two distinct allosteric mechanisms, with TP primarily inhibiting DnaJ2-mediated activation and MECA disrupting GrpE-mediated nucleotide exchange that occurs after interactions between DnaK and DnaJ2 (Figure 3F). Since MECA demonstrated its most potent inhibition of DnaK only at equimolar concentrations of GrpE, which is non-physiologic in bacteria (Fay and Glickman, 2014), TP showed more promise as a direct inhibitor of Mtb DnaK.
TP interacts with the C-terminus of DnaK and affects its conformational state.
Based on the results of dose-response assays, we initially hypothesized that TP binds to DnaK near the ATP site to attenuate turnover (Figure 4A, DnaKNBD). In order to determine if TP interacts directly with DnaK, we utilized a catalytically dead mutant, DnaK T175A (Lupoli et al., 2016; Barthel, Zhang and Walker, 2001; Chang et al., 2010) for ITC experiments following removal of bound nucleotide, as described by others for Hsp70 binding studies (Rauch and Gestwicki, 2014). ITC analysis of full-length apo DnaK (DnaKFL) with TP confirmed a direct binding interaction (KD ~ 590 nM) (Figure 4B, S3D); however, ITC analysis of TP with a DnaK truncation consisting of the NBD (DnaKNBD) indicated that there was no interaction (Figure 4C, S3E). Since TP bound to full-length DnaK but not DnaKNBD, we reasoned that TP instead interacts with the C-terminus of DnaK (DnaKSBD). The NBD and SBD of Hsp70s allosterically communicate through a hydrogen bond network that includes a universally conserved Pro residue, thought to act as a “molecular switch”, and an Arg residue that resides in the NBD near the interdomain linker (Vogel, Bukau and Mayer, 2006). Mutation of this Arg (Arg127Ala) in Mtb DnaK resulted in tolerance to excess TP, unlike the slight inhibition seen for wild-type DnaK (Figure S4A). Additionally, when the last ~110 residues (A513-K625) were removed from DnaK, which constitute most of the C-terminal α-helical lid, TP still disrupted ATPase activity (Figure S4B). These observations suggested that the SBD, containing the β-sandwich domain (P370-F450) involved in client binding, might directly interact with TP, which leads to inhibition of ATPase activity in the NBD through interdomain communication.
Figure 4. TP interacts with the SBD of DnaK and promotes a monomeric state.

(A) Schematic of DnaK with indicated binding site of NR peptide, a substrate mimic. (B) ITC measurements depict interaction of full-length DnaK (DnaKFL) with TP. KD represents the average of n=3 experiments with SD shown in parentheses; n: stoichiometry value; exp: experimental; teo: theoretical. (C) An N-terminal truncation (DnaKNBD) does not interact with TP under similar conditions to those used in B. (D) Fluorescence polarization (FP) assays demonstrate that TP and an analog (7) displace FL-NR from both DnaKFL (left) and an SBD truncation (DnaKSBD) (right). BC does not compete with FL-NR. 95% confidence intervals of the EC50 values are indicated (far right). (E) Limited proteolysis of apo DnaK +/− excess nucleotides show that samples with TP retained the greatest amount of full-length (FL) DnaK compared to the apo and NR samples. *Indicates partial cleavage of the C-terminal α-helical lid (~60 kDa fragment). (F) Native gel electrophoresis analysis and (G) quantification of replicate experiments (n=3, error bars represent SD) of apo DnaK incubated with a range of [TP] and excess NR indicates that TP shifts DnaK to a monomeric species while NR does not. Oligomeric states are approximated, where M = monomer; D = dimer and T = trimer. Gels and traces are all representative of n = 3 experiments. See also Figures S3 and S4.
A heptameric peptide, NRLLLTG (NR), has long been used as a mimic for protein substrates of eukaryotic Hsp70s and bacterial DnaKs (Figure 4A) (Zhu et al., 1996; Gragerov et al., 1994; Rüdiger, Buchberger and Bukau, 1997; Zhang et al., 2014). We sought to use NR as a probe to study interactions with the SBD. While excess NR is known to stimulate the ATPase activity of some Hsp70 homologs, (Meng et al., 2018) we found that while excess TP slightly inhibits DnaK ATPase activity, NR does not cause a significant effect (Figure S4C). For Hp70s such as human HspA1, NR is known to bind to the SBD, even though it does not stimulate ATPase activity. Hence, we hypothesized that NR could still be used as a probe for mycobacterial DnaK. To assess this assumption, we fluorescently tagged NR (FL-NR) to use in fluorescence polarization (FP) experiments with DnaKFL (Ricci and Williams, 2008). We confirmed that FL-NR bound to Mtb DnaK (Figure S4D), and then performed competition assays in the presence of increasing concentrations of untagged NR, TP or the HCV inhibitor BC. We found that TP disrupts binding of FL-NR at lower concentrations (half-maximal effective concentration (EC50) ~ 2.01 μM) than the unlabeled peptide (~ 12.9 μM), and that BC has no effect on binding of the probe, as expected (Figure 4D, left, S4E). To distinguish between TP binding directly to the SBD of DnaK or indirectly, causing allosteric changes in the C-terminus by binding elsewhere on the protein, we purified DnaK’s C-terminus (DnaKSBD, residues 360-625) and performed additional FP experiments (Figure 4D, right), which demonstrated that TP only requires the C-terminal domain of DnaK for competition with the probe.
Since TP and NR appeared to compete for binding to the same site of DnaK but only TP functions as an inhibitor of ATPase activity, we sought to assess how each peptide affects DnaK structure. Hsp70s/DnaKs are known to populate different overall conformations in apo, ATP- and ADP-bound states (Figure 1A) (Kityk et al., 2012; Bertelsen et al., 2009). A limited proteolysis approach has been used to distinguish these conformations, since the flexible linker region is more readily cleaved when DnaK is extended, which typically occurs in the ADP-bound and/or apo state (see Figure 4A), depending on the organism (Meng et al., 2018; Kassenbrock and Kelly, 1989; Wei, Gaut and Hendershot, 1995; Kamath-Loeb et al., 1995; Buchberger et al., 1995; Preissler et al., 2017). TP or NR was added to apo, ATP-bound or ADP-bound DnaK T175A (Rauch and Gestwicki, 2014), prior to brief treatment with proteinase K. Since TP is known to inhibit another protease (NS3/4A), we selected a concentration of TP (50 μM) that did not affect cleavage by proteinase K (Figure S4F), and was still in excess to the KD of TP for DnaK. Similar to reports on the digestion of apo and ADP-bound E. coli DnaK, SDS-PAGE analysis (Figure 4E) indicated that addition of proteinase K to the apo, ATP- and ADP-bound states leads to cleavage of the C-terminal α-helical “lid” region (Figure 4A, dark gray) resulting in a truncated product (*), which is further cleaved to the corresponding NBD and SBD fragments (compare lane 1 to lanes 2, 5, and 8). Addition of TP protects full-length DnaK from cleavage (lanes 4, 7 and 10), while NR-treated DnaK was digested in a similar manner to the control, regardless of the addition of nucleotide (compare lanes 2 and 3, 5 and 6, 8 and 9). The linker region might also be occluded when DnaK is bound to TP in the apo and nucleotide-bound states, since the NBD band appears to be shifted, which is not expected to change due to lid cleavage (compare lanes 2 and 4, 5 and 7, 8 and 10). Hence, TP might lock DnaK into a more compact state (see Figure 1A), unique from that of substrate-bound DnaK (Zhu et al., 1996).
DnaK is also known to form dimers and higher order oligomers through intermolecular interactions between the SBD and the interdomain linker. Analysis of the ITC data above suggested that TP and DnaK interact according to a 1:2 stoichiometry (TP: DnaK ~ 0.5, Figure 4B), suggesting that TP may influence the oligomeric state of DnaK. To test this hypothesis, we performed native gel electrophoresis analysis with increasing concentrations of TP added to apo DnaK (Figure 4F). We found that addition of excess TP inhibited formation of dimers and higher order oligomers of DnaK (compare lanes 1-3), while addition of excess NR did not affect the oligomeric state (Figure 4G). Hence, these data suggest that while TP may compete with NR for binding to the SBD of DnaK, the two peptides induce distinct structural changes.
A photoactivatable probe indicates TP binds the peptide binding cleft.
While TP is the result of a rational inhibitor design strategy (Grillot et al., 2011), there are no solved structures of full-length Mtb DnaK (Kelley et al., 2015). Hence, we decided to take an unbiased approach to determine the binding cleft of TP using a small-scale structure-activity relationship (SAR) study.
TP is a tetrapeptide composed of unnatural amino acids with caps on both the N- and C-termini (Figure 5A, top left). We reasoned that the bicyclic carbocycle in TP could be replaced with a more accessible Pro residue, which was maintained in all analogs since a strained secondary amide is likely structurally important and known to mitigate loss in entropy upon target binding (Zahn et al., 2013) (Figure 5A, top right). Similarly, the N-terminal pyrazine cap was maintained in initial scaffolds because it might participate in pi stacking and/or van der Waal interactions with DnaK. We varied the remaining positions with commercially available amino acids that are structural analogs of the unnatural residues found in TP, and performed DnaK-J2-GrpE ATPase dose-response experiments. Several of these analogs were inactive even at high compound concentration (Figure 5A, table). Val in position R2 led to greater inhibition than the original cyclohexyl side chain (compare 2 vs 1 and 4). Methylation of the C-terminus of peptide 2 to give 5 led to a >2-fold increase in potency (IC50 ~ 192 μM versus 76.0 μM, respectively). Based on the structure of 5, replacement of the tert-butyl side chain at R3 with Val was also tolerated, along with Leu at R4. We concluded that active peptide analogs, like TP, interacted with a largely hydrophobic binding pocket of DnaK.
Figure 5. A photoactivatable analog of TP covalently crosslinks to the peptide binding site of Mtb DnaK.

(A) SAR study of peptide analogs of TP with indicated IC50 values in DnaK-DnaJ2-GrpE ATPase assays. (B) Schematic of Mtb DnaK model (Kelley et al., 2015) with photoactivatable probe 7 (top). Mass spectrometry (MS) analysis indicates sites of covalent modification of the SBD by 7 based on crosslink spectral matches (CSMs) (bottom). Non-modified residues that were mutated are indicated in blue. (C) Higher-Energy Collision Dissociation (HCD) MS/MS spectrum of one of the most highly modified DnaK SBD peptides by probe 7. N-terminal fragment ions (b-type ions) are indicated by ⌋ and C-terminal fragment ions (y-type ions) are indicated by ⌈. The red character indicates the likely placement of the probe modification. PB2 and PB3 indicate fragment ions of 7 (206.093 and 305.161 Da respectively). (D) ATPase activity (left) and FP assays (right) of DnaK mutants support that TP interacts with the peptide binding site of DnaK. F400A (boxed) was used at higher concentrations than WT because of weakened binding affinity for FL-NR. Error bars represent SD (n=3). 95% confidence intervals of EC50 values are shown. See also Figures S4 and S5, Table S6.
Since 5 showed reasonable inhibitory activity, we used this scaffold for further modification to create a UV-activatable probe that would enable identification of the protein binding site. When the readily accessible photoreactive moiety para-benzoyl-phenylalanine replaced the N-terminal pyrazine cap, the resulting peptide showed no activity (6, similar to the free acid 3), likely due to its bulky nature. In order to minimize structural changes, we then substituted Leu at R4 with the UV-activatable residue photoleucine (7), and saw an improvement in inhibitory activity (IC50 = 52.5 μM).
To confirm that the photoreactive probe (7) bound to the same region of DnaK as TP, we performed FP-based competition experiments. We found that 7 competed with FL-NR for binding to DnaK, as seen with TP (Figure 4D). To identify the exact side of binding, we incubated DnaKSBD with excess 7, irradiated with UV-light (Figure 5B, top) over a time course and selected an optimal time to obtain a single cross-linked species as indicated by MALDI-TOF analysis (Figure S5A,B). The resulting mixture of native and modified DnaK was digested with trypsin, and resulting peptides were analyzed by LC MS/MS. Amino acids identified as sites of attachment to probe 7 are indicated in the structural model of Mtb DnaK (Kelley et al., 2015) (Figures 5B and S5C, Table S6, Data S1). Since the peptide 7 was anticipated to fragment during MS analysis, crosslinked peptides were identified by MS/MS filtering that required two fragments of the probe to be present in an MS/MS spectrum, as exemplified by the spectra shown in Figures 5C and S5D. Based on crosslink spectral matches (CSMs), 80% of the modified peptides were in the β-sandwich domain spanning from Glu398 to Ser409. The most highly represented modified residue was Asn406, followed by Glu398 and Asp405, all of which are present in the peptide binding cleft. Since Glu398 and Asn406, which are on opposite sides of the protein (see rotated inset in Figure 5B), interact with the side chain R4 of 7, these results suggest the probe can bind in two different orientations or near a DnaK dimer interface. In accordance with the former scenario, similar peptides are known to bind bacterial DnaKs in both “forward” and “reverse” modes, especially those containing branched amino acid-Pro motifs (Zahn et al., 2013; Tapley, Cupp-Vickery and Vickery, 2005). Although residues in the α-helical lid were also crosslinked, albeit with lower CSMs (Figure S5C, Table S6), we found that the lid is not important for inhibition (Figure S4B); hence, interactions with the lid are likely non-specific and due to the use of excess probe.
Mutagenesis of select residues (Figure 5B, red and blue) in and around the peptide binding cleft was performed to confirm the binding site of TP. Among the mutants that exhibited ATPase activity, we observed a 1.4-fold increase in the IC50 value for TP when Glu514 was mutated to Ala compared to wild-type (Figure 5D, left); while mutation of Gly398 or Asp404 to Ala led to slight enhancements in the inhibitory activity of TP (~0.5-0.75 reduction in IC50 compared to wild-type). Mtb DnaK Glu514 is analogous to E. coli DnaK Asp540, known to form a salt bridge that stabilizes a “closed” interaction between the α-helical lid to the β-sandwich subdomain in the SBD (Sarbeng et al., 2015; Mayer et al., 2000); hence, our results suggest that TP interacts with both subdomains. For ATPase-dead mutants Phe400Ala and Thr401Ala, we used FP analysis with FL-NR (Figure 5D, right, S4E) and found that Phe400Ala had decreased affinity for FL-NR (~40-fold higher EC50 than WT), since Phe400 is likely involved in binding peptide substrates. As a result, a higher concentration of Phe400Ala was used in displacement assays to achieve binding to the probe, and we observed a dramatic increase in the concentration of TP required for competition. Mutation of the adjacent residue, Thr401, did not significantly affect TP binding relative to the wild-type (compare triangles to circles), perhaps because the Thr side chain is oriented outside of the pocket. Taken together, these experiments indicate that TP likely competes with peptide substrates in the SBD through interactions with Phe400.
TP is a pan inhibitor of Hsp70s.
Hsp70s and DnaKs show high sequence conservation throughout all domains of life; E. coli DnaK shares 52.8% sequence identity with Mtb DnaK. To address if TP bound to other bacterial DnaKs, we performed E. coli DnaK, DnaJ and GrpE ATPase-based dose-response experiments with select HCV analogs. Both TP and simeprevir inhibited E. coli DnaK-mediated ATP hydrolysis with similar IC50 values (3.12 μM for TP; >100 μM for simeprevir) observed for the Mtb DnaK chaperone components (compare Figures 6A and 2D, F).
Figure 6. TP is a pan inhibitor of Hsp70s.

(A) Dose-response curves of HCV protease inhibitors demonstrate disruption of E. coli DnaK-DnaJ-GrpE ATPase activity. (B) Denatured luciferase reactivation by human chaperones Hsc70 and DnaJA2 +/− TP (100 μM) over a time course (left) and a concentration gradient of TP (right) demonstrates dose-dependent inhibition of refolding, unlike excess BC. (C) Superposition of the SBD crystal structures of E. coli DnaK, human Hsp72 (representative for Hsc70) and a model of Mtb DnaK (Kelley et al., 2015) with NR from indicated structures. Red residues in the peptide binding cleft are conserved across all three species. Error bars represent SD (n=3). See also Figure S6.
Eukaryotic Hsp70s show ~50% sequence identity with prokaryotic DnaKs (Daugaard, Rohde and Jäättelä, 2007), although identity among SBDs is lower than that of NBDs (Schlecht et al., 2013; Chiappori et al., 2015). The most enriched Hsp70 in the cytoplasm of human cells is the constitutively expressed Hsc70 (HspA8), which has been shown to refold denatured model proteins with the assistance of a single cofactor (DnaJA2) in vitro (Tzankov et al., 2008; Baaklini et al., 2012; Taylor et al., 2018; Rodina et al., 2013). We tested the effect of TP and BC using well-described denatured luciferase refolding assays (Lupoli et al., 2016; Nillegoda and Bukau, 2015), and found that TP inhibits human Hsc70 and DnaJA2 folding over a time course (Figure 6B, left) and in a concentration-dependent manner, resulting in only 35% of refolding activity with 100 μM TP compared to DMSO alone (Figure 6B, right). As expected, excess BC does not inhibit human chaperones under these conditions. Structural alignment of the SBDs of E. coli DnaK, a human Hsp70 and a structural model of Mtb DnaK (Kelley et al., 2015) illustrates the similarity in the structure of this domain across organisms (Figure 6C). NR binds to both human (yellow) and E. coli (green) Hsc70/DnaK at this site, although in slightly different conformations. The peptide binding region of Mtb DnaK, which has some overlap with the proposed binding site of TP (compare to Figure 5B), has several residues that are conserved across E. coli, Mtb and humans (Figure 6C, box, residues indicated in red, S6). Most notably, Phe400, which likely directly interacts with TP, is found across bacterial and eukaryotic species. Hence, we postulate that TP shows pan activity against Hsp70s from diverse organisms because residues within this pocket are conserved. While TP did not show species selectivity in vitro, we anticipated it could still be used to study mycobacterial chaperone function in vivo.
TP disrupts bacterial proteostasis leading to enhanced sensitivity to proteotoxic stress.
Our previous work suggested that disruption of cofactor-mediated activation of DnaK can lead to mycobacterial cell death (Lupoli et al., 2016). Msm requires dnaK, grpE, and at least one dnaJ for growth (Fay and Glickman, 2014; Lupoli et al., 2016). We have shown that Mtb DnaJ proteins can replace native DnaJs in Msm cells, and have used Msm to study Mtb chaperone function in vivo (Lupoli et al., 2016). Since TP inhibited the activation of DnaK by cofactors in vitro, we hypothesized that addition of TP to mycobacterial cells might lead to growth defects. To assess the effect of TP on both Msm and Mtb DnaK in vivo, cellular dose-response assays were performed with wild-type Msm, as well as Msm ΔdnaK cells that expressed Mtb DnaK. Both strains behaved similarly in the presence of the antibiotic kanamycin and TP, which caused growth inhibition only at high concentrations (>100 μM, Figure S7A-C). This result indicated that TP is a broad inhibitor of mycobacterial DnaKs, as expected. Since the IC50 of TP is over an order of magnitude lower than the predicted MIC90 value, we reasoned that TP might not be able to permeate mycobacterial cells under standard growth conditions (Nikaido and Jarlier, 1991), is diluted by off-target effects, or might require other cellular stresses to demonstrate activity in cells.
Many chaperones are annotated as heat shock proteins (Hsps) due to their role in the global reactivation of proteins that aggregate following heat stress (Balchin, Hayer-Hartl and Hartl, 2016). Cells with chaperone defects that are heat shocked often show poor recovery (Jastrab et al., 2017; Sell et al., 1990), and so we sought to evaluate the effect of TP on heat-stressed mycobacterial cells (Jastrab et al., 2017; Harnagel et al., 2021; Fay and Glickman, 2014). We tested wild-type and dnaJ deletion strains because we reasoned that cofactor-deficient cells might be more sensitive to DnaK inhibition. Msm cells with and without dnaJ1 or dnaJ2 were heat shocked prior to recovery on plates +/− TP (100 μM) at normal (37 °C) versus elevated (40 °C) temperatures. As seen in Figures 7A and S7D, only cells lacking dnaJ1 consistently show a slight (~1-log10) heat recovery defect in plates lacking TP incubated at elevated temperatures (compare input (in) and heat-shocked (Δ) cells for ΔJ1 in plate 2, Figure 7A). This defect was exacerbated in the presence of TP with a >2-log10 decrease in growth of ΔdnaJ1 input and heat-shocked cells relative to respective wild-type samples (plate 4). Incubation of Msm at 37 °C partially relieved the growth defects in Δdnaj1 cells without and with drug (plates 1 and 3, respectively) compared to elevated temperatures (plates 2 and 4, respectively). Wild-type Msm and ΔdnaJ2 cells behaved similarly under all conditions tested, suggesting that DnaJ2 does not play a major role in the heat-shock response. Time course analysis indicated that prolonged heat treatment sensitized Msm wild-type cells to TP (Figure S7E), which likely also aided in compound uptake through the waxy mycobacterial membrane (Touchette and Seeliger, 2017; Tsuchido and Takano, 1988). These data indicated that TP can inhibit the Msm DnaK-cofactor network in vivo.
Figure 7. TP disrupts protein folding in mycobacterial cells, leading to increased sensitivity to stress and reduced frequencies of drug resistance.

(A) Recovery of indicated Msm strains following heat shock prior to growth +/− TP at 37 °C or 40 °C indicates that cells lacking dnaJ1 are the most sensitive to TP following global protein denaturation. WT: Msm; ΔJ1: Msm ΔdnaJ1; ΔJ2: Msm ΔdnaJ2; Δ: heat shock; in: input. (B) Time course of denatured luciferase reactivation by indicated chaperone mixtures demonstrates DnaJ2 reactions are more sensitive to TP than DnaJ1 reactions. (C) TP exhibits a dose-dependent inhibition of chaperone network, whereas BC shows negligible inhibition. (D) Minimum inhibitory concentration (MIC) experiments in Msm show that addition of non-lethal concentrations of TP enhance the potency of aminoglycoside antibiotics. (E) While TP does not affect the activity of rifampicin (RIF) (left), addition of TP lowers the frequency of resistance (FOR) of Msm exposed to high levels of RIF (n = 6, lines indicate the median). ****p<0.0001 (Paired t-test was used for comparison). Error bars represent SD (typically n=3, except where otherwise indicated). See also Figure S7 and Table S7.
The observation that heat-shocked cells lacking either DnaJ1 or DnaJ2 responded differently to TP suggested that the function of each J-protein in folding is affected differently by TP. Hence, we examined denatured luciferase refolding by mycobacterial DnaK and GrpE with added DnaJ1 or DnaJ2 with and without TP added (Figure 7B). Chaperone mixtures with either DnaJ1 or DnaJ2 could each restore the activity of denatured luciferase to ~75% of its native level over the indicated time course (black circles or triangles, respectively). When excess TP was added to reactions containing DnaJ2 (red triangles), reactivation of luciferase was only ~30% relative to the level detected without drug, compared to the >60% relative reactivation that occurred with DnaJ1 in the presence of TP (red circles). Inhibition of DnaJ2-mediated folding by TP was concentration-dependent (Figure 7C). When excess BC was added to these reactions, only a slight effect was observed. Hence, protein folding reactions mediated by DnaJ2 exhibit greater sensitivity to TP, similar to the effect observed in heat-treated cells that contain dnaJ2 but lack dnaj1.
Due to the influence of chaperones on cellular proteostasis and fitness (Aguilar-Rodríguez et al., 2016), we reasoned that TP might synergize with molecules that could also disrupt proteostasis, such as the aminoglycoside antibiotics kanamycin and streptomycin, which inhibit bacterial protein synthesis; the latter of which is still a commonly prescribed TB drug. Interestingly, addition of a nontoxic concentration of TP (100 μM) in Msm dose-response assays with kanamycin and streptomycin resulted in a ~2- to 4-fold improvement in the minimum inhibitory concentrations to inhibit 50% of bacterial cell growth (MIC50’s) of these drugs, respectively (Figure 7D). As we have shown previously (Lupoli et al., 2016), Msm ΔdnaJ1 and ΔdnaJ2 show similar MIC50 values for kanamycin as wild-type, which we confirmed (Table S7). Similar to wild-type, cells lacking dnaJ2 demonstrated lower tolerance for kanamycin and streptomycin in the presence of TP (Table S7), while ΔdnaJ1 shows growth defects in the presence of 100 μM TP (Figure S7F). These data are in line with our observations that mycobacterial cells lacking dnaJ1 are more sensitive to growth inhibition in the presence of TP than wild-type and ΔdnaJ2 cells, especially when exposed to stress.
A recent report has shown that DnaK and cofactors, in particular DnaJ2, are required for the evolution of resistance of mycobacteria to the frontline TB drug rifampicin (RIF), as chaperones are known to stabilize mutants of the target, RNA polymerase, that confer drug tolerance (Fay et al., 2021). Since TP disrupts DnaK-DnaJ2 chaperone activity in protein renaturation experiments, we assessed the effect of TP on the frequency of resistance (FOR) of Msm to high concentrations of RIF (Figure 7E). While TP does not affect the growth of wild-type cells on plates (Figure 7A, plate 3 compared to plate 1), addition of TP indeed lowered the FOR of RIF by about 1-log10, demonstrating that chaperone inhibitors can be used to interfere with antibiotic resistance mechanisms.
Discussion:
Molecules that can inhibit the function of a mycobacterial chaperones represent potential TB therapeutics (Lupoli et al., 2018), and/or antibiotic adjuvants (Wright, 2016; Cheng and Wuest, 2019; Mattingly et al., 2020). We used a small molecule screening approach to discover mycobacterial chaperone inhibitors, similar to other endeavors that aimed to disrupt key PPIs involved in chaperone networks (Chang et al., 2011; Cesa et al., 2013; Chang et al., 2008). Our results validated that known drugs TP and MECA allosterically inhibit DnaK, while only TP appears to cause growth inhibition of mycobacterial cells at high concentrations (Figure S7B-C).
TP is a first-in-class HCV protease inhibitor with a Ki of 44 nM (Grillot et al., 2011), and shows micromolar activity against bacterial and human Hsp70s. We did not observe similar behavior for the structural analog BC, which also inhibits HCV protease through a reversible covalent mechanism. Simeprevir showed weak DnaK-cofactor inhibitory activity in vitro, but did not affect Msm growth in vivo (Figure S7B-C). TP now joins a collection of other peptide-based inhibitors that disrupt DnaK activity by binding to the SBD, which typically occurs alongside other chaperone targets in the cell (Chiappori et al., 2015; Shah et al., 2016; Knappe et al., 2010; Scocchi, Tossi and Gennaro, 2011; Kragol et al., 2002; Kragol et al., 2001; Chesnokova, Slepenkov and Witt, 2004; Zahn et al., 2013; Welch et al., 2020). Most known DnaK peptide ligands contain Pro-rich sequences and/or a hydrophobic stretch of five amino acids bordered by positively charged residues, such as Arg (Chiappori et al., 2015; Rüdiger et al., 1997). TP is a tetrapeptide analog that contains a Pro-like residue, but noticeably lacks any positively charged side chains, and interacts with bacterial DnaKs with similar affinity to longer antimicrobial peptides (18-22 residues) that bind in the low micromolar-high nanomolar range (Chiappori et al., 2015; Dalphin et al., 2020). Like other re-purposed drugs, TP has already been through a vigorous FDA approval process, while most antimicrobial DnaK peptides show metabolic instability and low specificity (Craik et al., 2013). Since a wealth of other HCV protease inhibitors exist, we are now poised to explore related scaffolds as putative chaperone modulators.
Our proteolysis data suggests that DnaK bound to TP assumes a compact conformation (Figure 1A), which protects the α-helical lid of the SBD from cleavage. Previous work has demonstrated that the NBD and SBD communicate through conformational changes in the interdomain linker (Zhuravleva and Gierasch, 2015). TP bound to the SBD likely allosterically modulates ATP-turnover in the NBD by locking DnaK in an inactive conformation with the linker occluded, unique from the conformation assumed by DnaK bound to the substrate analog NR (Gragerov et al., 1994; Bertelsen et al., 2009). It has been shown previously that E. coli DnaK functions as a dimer in the active state (Sarbeng et al., 2015; Li, Qian and Sha, 2009), and that mutation of E. coli DnaK Asp540 (Mtb DnaK Glu514) leads to loss of dimer formation in DnaK (Sarbeng et al., 2015). Accordingly, mutation of Mtb DnaK Glu514 to Ala was shown to reduce TP inhibitory activity, and Mtb DnaK bound to TP is predominantly monomeric, suggesting that TP binds residues that mediate oligomerization. Just as studies on DnaK bound to substrates have informed our understanding of the allosteric mechanism of ATPase activation (Swain, Schulz and Gierasch, 2006; Zhuravleva and Gierasch, 2015), future structural studies on DnaK from various organisms co-complexed with TP will provide a better understanding of this inactive conformation.
Key to its role in protein folding, DnaK’s SBD can interact with various peptide sequences within the same pocket. A recent computational study identifies certain “hot spot” residues within the SBD of bacterial DnaKs that commonly interact with peptide ligands (Chiappori et al., 2015). E. coli DnaK residues Thr409 (Mtb DnaK Thr383), Phe426 (Mtb DnaK Phe400), and Gln538 (Mtb DnaK Gln512) are predicted to make key contacts with NRLLLTG and several antimicrobial peptides (Figure 6C). Our data indicate that TP and the synthetic peptide analog 7 interact with a similar surface area. While there are unique residues in mycobacterial DnaK compared to eukaryotic Hsp70s (Figure S6) that may facilitate species specific-targeting, this goal remains a challenge (Evans, Chang and Gestwicki, 2010).
Our follow-up studies demonstrated that the use of DnaJ2 in the initial small molecule screen resulted in the identification of a compound that specifically modulates DnaK-DnaJ2 function in cells. As indicated by our biochemical results, TP primarily disrupts DnaK-DnaJ2-GrpE-mediated protein folding; hence, TP is not toxic to wild-type mycobacteria containing both J-proteins, since DnaJ1 can compensate for the disruption of DnaK-DnaJ2 function by TP. Accordingly, TP is most potent in cells without dnaJ1, especially after proteotoxic stress, since DnaK lacks a functional DnaJ partner for protein reactivation in these cells. Accordingly, TP also potentiates aminoglycosides known to both cause proteome damage and promote small molecule uptake into cells, which may enhance access of both TP and antibiotics to cellular targets (Busse, Wöstmann and Bakker, 1992; Touchette and Seeliger, 2017; Tsuchido and Takano, 1988). It is important to note that there is no evidence TP inhibits DnaJ2 directly, and so we do not expect Msm cells lacking dnaJ2 to phenocopy defects in cells exposed to TP. While we examined the effect of TP in non-pathogenic Msm in place of Mtb, we showed that Msm cells expressing Mtb DnaK respond similarly to wild-type to TP. A recent whole genome depletion study has indicated that dnaK, dnaJ1 and dnaJ2 are each essential in pathogenic Mtb (Bosch et al., 2021), suggesting that interference of this chaperone network would also be detrimental to Mtb cells under stress, even though other proteostasis machinery differs among mycobacteria (Knipfer et al., 1999).
Milestone work has illustrated that chaperone inhibition reduces drug resistance in cancer cells (Wang et al., 2016; Whitesell et al., 2014) and fungi (Cowen and Lindquist, 2005; Vincent et al., 2013), primarily through disruption of the eukaryotic chaperone Hsp90. The bacterial homolog of Hsp90, HtpG, (Genest et al., 2011; Harnagel et al., 2021) has not yet been linked to bacterial drug resistance mechanisms (Taipale, Jarosz and Lindquist, 2010). In bacteria, dnaK and dnaJ genes have recently been implicated in resistance to RIF (Fay et al., 2021), as well as tetracycline and chloramphenicol (Lukačišinová, Fernando and Bollenbach, 2020). We found that the FOR of Msm for the TB drug RIF is reduced from ~10−7 to ~10−8 with TP added, while Msm ΔdnaJ2 cells demonstrated an FOR value below the limit of detection (<10−10) (Fay et al., 2021). Since addition of TP does not fully inhibit DnaJ2, we do not expect to eliminate mycobacterial resistance to RIF with this small molecule. However, to our knowledge, the work described here provides the first direct evidence that a small molecule DnaK inhibitor can mitigate antibiotic resistance in bacteria. We anticipate this chemical strategy will be applied to modulating drug resistance in other species, as TP can inhibit DnaKs/Hsp70s from bacteria and humans. Since the distinct cellular roles of DnaJ1 and DnaJ2 remain unclear, we envision that TP will also be valuable in discriminating the biochemical function of J-proteins in other species, including pathogenic Mtb, which might provide insight into the diverse functions of J-protein-mediated Hsp70 activation in other cell types.
Significance:
Hsp70s are ubiquitous ATP-binding proteins that work in concert with co-chaperones (cofactors) to fold nascent proteins and salvage aggregated proteins following periods of stress. In eukaryotes, several isoforms of Hsp70 exist in each cell type; in most bacteria, there is a single general Hsp70 called DnaK. In the pathogen Mycobacterium tuberculosis. the causative agent of tuberculosis (TB), DnaK and its cofactor proteins DnaJ1, DnaJ2 and GrpE are proposed drug targets. Despite the importance of chaperone function in human cancers and infectious disease, there are limited chemical probes or inhibitors that enable in vivo studies on the function of individual chaperones/cofactors. Here, we discover small molecule inhibitors of mycobacterial DnaK from a re-purposed drug library, most notably a peptidomimetic called telaprevir, which is able to inhibit chaperone function through interactions with the peptide-binding cleft of DnaK. Binding of telaprevir leads to allosteric conformational changes that prevent ATP hydrolysis in a distal domain. We find that telaprevir also inhibits E. coli DnaK and human Hsc70 chaperones due to high conservation of Hsp70 sequences. Using in vitro and in vivo chaperone assays, we demonstrate that telaprevir modulates the function of mycobacterial DnaK and its cofactor protein DnaJ2 in cells, which disrupts cellular proteostasis. Co-treatment of mycobacteria with telaprevir and aminoglycosides, which further stress the proteome, enhances the potency of these antibiotics. In addition, telaprevir combats mycobacterial resistance to the frontline TB drug rifampin, as DnaK-DnaJ2 function is required for stabilization of protein mutants that confer drug tolerance. This work sets the stage for telaprevir and related scaffolds to be employed for multifaceted applications that span from potentiators of known antibiotics and chemical probes for chaperone-cofactor interactions.
STAR Methods:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tania Lupoli (tjl229@nyu.edu).
Materials availability
Plasmids generated in this study are available from the lead contact upon request and upon completion of an MTA.
Data and code availability
Small molecule screening data have been deposited in OSF and are publicly available as of the date of publication. Link is listed in the key resources table. Mass spectrometry-based proteomics data have been deposited in MassIVE and are publicly available as of the date of publication. The accession number is listed in the key resources table. Analyses of mass spectrometry data is present in a Supplemental data file.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli Mach1 | ThermoFisher | Cat# C862003 |
| Escherichia coli Rosetta2 (DE3) | Millipore Sigma/Novagen | Cat# 71400-3 |
| Escherichia coli Rosetta2 (DE3) pLysS | Millipore Sigma/Novagen | Cat# 71403-3 |
| Mycobacterium smegmatis wild-type (mc2155) | Glickman Lab (Memorial Sloan Kettering) | N/A |
| Mycobacterium smegmatis Δdnaj1 | Lupoli et al., 2016 | N/A |
| Mycobacterium smegmatis Δdnaj2 | Lupoli et al., 2016 | N/A |
| Mycobacterium smegmatis ΔdnaK | Fay & Glickman, 2014 | N/A |
| M. smegmatis ΔdnaK attB::Mtb dnaK | This work | N/A |
| Escherichia coli strains for protein overexpression, see Table S3 | Various | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fmoc-protected natural amino acids | ChemImpex | N/A |
| Fmoc-L-Photo-Leu (Fmoc- photoLeu-OH) | Sigma-Aldrich | CAS: 1360651-24-6 |
| Fmoc-L-tert-leucine (Fmoc-Tle-OH) | ChemImpex | CAS: 132684-60-7 |
| Fmoc-L-cyclohexyl-glycine (Fmoc-Chg-OH) | ChemImpex | CAS: 161321-36-4 |
| Fmoc-L-β-alanine (Fmoc-β-Ala-OH/Fmoc-βA-OH) | ChemImpex | CAS: 35737-10-1 |
| Pyrazine-2-carboxylic acid (Pz) | Sigma-Aldrich | CAS: 98-97-5 |
| Fluorescein isothiocyanate isomer 1 (FL-NCS) | ChemImpex | CAS: 3326-32-7 |
| 4-Benzoylbenzoic Acid (4-BBA) | Sigma-Aldrich | CAS: 611-95-0 |
| Telaprevir | Cayman Chemical | CAS: 402957-28-2 |
| Simeprevir | Cayman Chemical | CAS: 1241946-89-3 |
| Boceprevir | Cayman Chemical | CAS: 394730-60-0 |
| Grazoprevir | Cayman Chemical | CAS: 1350514-68-9 |
| Velpatasvir | Cayman Chemical | CAS: 1377049-84-7 |
| Danoprevir | Celia Schiffer (UMass Medical School) | CAS: 850876-88-9 |
| Paritaprevir | Celia Schiffer (UMass Medical School) | CAS: 1216941-48-8 |
| 2-Cl-IB-MECA | Cayman Chemical | CAS: 163042-96-4 |
| AB-MECA | Cayman Chemical | CAS: 152918-26-8 |
| VER 155008 | Cayman Chemical | CAS: 1134156-31-2 |
| NH2-Asn-Arg-Leu-Leu-Leu-Thr-Gly-OH | Biomatik | N/A |
| FL-β-Ala-Asn-Arg-Leu-Leu-Leu-Thr-Gly-OH | This work | N/A |
| Pz-Chg-Tle-Pro-Leu-OH (1) | This work | N/A |
| Pz-Val-Val-Pro-Leu-OH (2) | This work | N/A |
| 4BB-Val-Val-Pro-Leu-OH (3) | This work | N/A |
| Pz-Chg-Tle-Pro-Leu-OMe (4) | This work | N/A |
| Pz-Val-Val-Pro-Leu-OMe (5) | This work | N/A |
| 4BB-Val-Val-Pro-Leu-OMe (6) | This work | N/A |
| Pz-Val-Val-Pro-photoLeu-OMe (7) | This work | N/A |
| Firefly Luciferase: QuantiLum Recombinant Luciferase | Promega | Cat# E1701 |
| Critical commercial assays | ||
| Luciferase Assay System | Promega | Cat# E1500 |
| ADP-Glo Kinase System | Promega | Cat# V6930 |
| Kinase-Glo Luminescent Kinase Assay | Promega | Cat# V6711 |
| Deposited data | ||
| Raw small molecule screening data from pilot and primary screens | This paper; OSF | https://osf.io/cmqxd/?view_only=a2e3d69e9d404bd598f9f6475b042a77 |
| Analysis of mass spectrometry data of proteolyzed DnaK crosslinked to probe | This paper; SI file | N/A |
| Mass spectrometry raw files of proteolyzed DnaK crosslinked to probe | MassIVE | https://massive.ucsd.edu; MassIVE ID: MSV000088127 |
| Oligonucleotides | ||
| Primers for cloning bacterial and human chaperones into overexpression vectors, see Table S1 | This paper | N/A |
| Primers for inserting mutations into DnaK, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pETHisSUMO | Gift from Scott Gradia, University of California, Berkeley | Addgene Plasmid #29711 |
| Plasmid: pHYRS52: overexpression vector for Ulp1 | Gift from Hideo Iwai, University of Helsinki; Muona, M. et al. 2008 | Addgene Plasmid #31122 |
| Plasmid: pET-47b(+) | Millipore Sigma/Novagen | Cat# 71461-3 |
| Plasmid: pcDNA5/FRT/TO HIS HSPA8: contains gene encoding Hsc70 | Gift from Harm Kampinga, University of Groningen; Hageman, J. et al. 2009 | Addgene Plasmid #19541 |
| Plasmid: pcDNA5/FRT/TO DNAJA2: contains gene encoding DnaJA2 | Gift from Harm Kampinga, University of Groningen; Hageman, J. et al. 2009 | Addgene Plasmid #19465 |
| Plasmid: pAJF465: M. tuberculosis dnaK expression in mycobacteria | This work | N/A |
| Plasmids for overexpression of bacterial and human chaperones and mutants in E. coli, see Table S2 | This work | N/A |
| Software and algorithms | ||
| Prism 9 | Mathpad | https://www.graphpad.com/scientific-software/prism/ |
| AFFINImeter | AFFINImeter | https://www.affinimeter.com/site/ |
| Chromeleon 7 | ThermoFisher Scientific | https://www.thermofisher.com/order/catalog/product/CHROMELEON7#/CHROMELEON7 |
| PyMOL 2.0 | Schrodinger | https://www.schrodinger.com/products/pymol |
| Image Lab | Biorad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Byonic | Protein Metrics | https://proteinmetrics.com/resources/byonic-advanced-peptide-and-protein-identification-software/ |
| Other | ||
| Compound libraries used for small molecule screens | Various sources; assembled by The Rockefeller University High Throughput and Spectroscopy Center | https://www.rockefeller.edu/htsrc/libraries/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
Escherichia coli strains were grown at 37°C in Luria-Bertani (LB) medium (BD Biosciences), unless otherwise noted, with appropriate antibiotic (carbenicillin 50 μg/mL; chloramphenicol 30 μg/mL) for resistance selection during molecular cloning and recombinant protein overexpression. LB Agar (BD Difco) was used for growth on plates. Mycobacterium smegmatis (Msm) strains were derivatives of mc2155. Msm was grown at 37°C in Middlebrook 7H9 medium (BD Difco), unless otherwise noted, supplemented with 0.2% glycerol, 0.05% Tween-80, 0.5% bovine serum albumin (Roche), 0.2% dextrose, and 0.085% NaCl (called 7H9 complete/tween). Appropriate antibiotics were used for selection (streptomycin 20 μg/mL). Middelbrook 7H10 Agar was used for plates prepared according to manufacturer’s instructions (BD Difco) with Middlebrook OADC Enrichment (BD Biosciences) added unless otherwise noted. Please note that these cell lines were not subjected to separate authentication by whole genome sequencing.
METHOD DETAILS
General Reagents and Equipment.
All reagents were purchased from Sigma-Aldrich unless otherwise specified. FPLC separations were performed using an AKTA pure 15 L instrument (GE Healthcare). HPLC purification and analysis was performed on a Dionex UltiMate 3000 UHPLC+ (Thermo Scientific). For ATPase assays, ATP (Sigma Aldrich) and ADP (Promega) used in these assays were each 99% pure. Compound mass spectrometry analysis was performed on an Agilent 6224 TOF LC/MS, unless otherwise noted.
Plasmid construction.
All primers are listed in Table S1; plasmids and details on construction are shown in Table S2; strains are listed in Table S3. pET His6 Sumo TEV LIC cloning vector (2S-T) was a gift from Scott Gradia (Addgene plasmid # 29711 ; http://n2t.net/addgene:29711 ; RRID:Addgene_29711). pHYRS52 was a gift from Hideo Iwai (Addgene plasmid # 31122; http://n2t.net/addgene:31122; RRID:Addgene_31122) (Muona, Aranko and Iwai, 2008). pcDNA5/FRT/TO HIS HSPA8 was a gift from Harm Kampinga (Addgene plasmid # 19541;http://n2t.net/addgene:19541; RRID:Addgene_19541) (Hageman and Kampinga, 2009). pcDNA5/FRT/TO DNAJA2 was a gift from Harm Kampinga (Addgene plasmid # 19465; http://n2t.net/addgene:19465 ; RRID:Addgene_19465) (Hageman and Kampinga, 2009). Primers were purchased from Invitrogen. Overexpression plasmids Phusion™ DNA polymerase was from ThermoFisher, and restriction endonucleases were purchased from NEB (Table S2) and were constructed by using overlap extension PCR cloning techniques with indicated plasmid and primer pairs (Tables S1 and S2).(Bryksin and Matsumura, 2010) Phusion™ DNA polymerase was used for PCR reactions. Following PCR steps and DpnI treatment (New England Biolabs), DNA samples were purified using a PCR purification kit (Qiagen) and transformed into Escherichia coli Mach1 competent cells (Invitrogen). For point mutagenesis, QuikChange Site-Directed Mutagenesis Kit, (Stratagene) along with the indicated primers (Table S1), were used. Gene insertion was confirmed by DNA sequencing (Genewiz). For protein overexpression strains, plasmids were transformed into Escherichia coli Rosetta 2 (DE3) or Rosetta 2 (DE3) pLysS using standard techniques resulting in strains described in Table S3. For pAJ465 (Table S2), this plasmid was constructed using standard endonuclease cloning. Mycobacterium bovis strain BCG encodes dnaK with 100 percent identity to that of Mtb. M. bovis BCG Pasteur genomic DNA (gDNA) was PCR amplified with primers 29 and 30 (Table S1), digested with XbaI/EcoRI (NEB) and ligated (T4 ligase, NEB) to pDB60 digested with same endonucleases. Plasmid were transformed into mycobacteria used standard protocols.(Hatfull and Jacobs, 2014)
Purification of bacterial chaperones.
M. tuberculosis (Mtb) chaperones (DnaK, DnaJ2, GrpE and DnaJ1) were expressed and purified by a similar method to that described previously,(Lupoli et al., 2016) with slight changes described below. Mtb DnaK, DnaJ2, and GrpE were overexpressed in E. coli Rosetta2 (Table S3). Overnight cultures in 5 mL of LB (carbenicillin 50 mg/mL, 0.1% glucose (v/v)) were inoculated into 500 mL LB containing carbenicillin (50 mg/mL), and grown with shaking (37°C, 200 rpm) to log-phase as measured by OD600 (optical density at 600 nm), following induction for 3 to 4 hours with 1 mM IPTG (isopropyl β-D-thiogalactopyranoside) at 25 °C.
DnaK and GrpE cell pellets (each from 500 mL culture) were each resuspended in Buffer A (25 mM Tris(hydroxymethyl)aminomethane (Tris), pH 8.0, 400 mM NaCl, 10% glycerol (v/v)) containing lysozyme (0.1 mg/mL) and DNaseI (6.6 μg/mL), rocked for 1 hr at 4 °C, and lysed by sonication (Fisherbrand, 5 min, 30 sec on/30 sec off, 50% amplitude, twice with cooling between runs). Cellular debris was cleared by ultracentrifugation (Beckman Coulter Allegra X-15R, 10956 x g, 30 min, 4 °C). Supernatant was added to a pre-washed equilibrated 1 mL Ni-NTA (Qiagen) column containing 2 mM imidazole and incubated for 30 min. The protein was washed and eluted with imidazole in Buffer A (30 mM and 200 mM imidazole, respectively). SUMO protease His-Ulp1 (700 μg) was added to the elution fraction, as has been described,(Uehara et al., 2010) and dialyzed overnight using SnakeSkin Dialysis Tubing (Thermo Scientific, 10K MWCO, 35 mm) in 2 L Buffer A at 4 °C with stirring. Following dialysis, the sample was then incubated in a prewashed equilibrated Ni-NTA column (2 mL) for 30 min at 4 °C with rocking, collected and washed with Buffer A (10 mL), and concentrated using an Amicon centrifugal unit (MilliporeSigma, 10K MWCO, 1857 x g, 4 °C).
DnaJ1 cell pellet (from 500 mL culture) was resuspended in Buffer B (25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5, 400 mM NaCl, 10% glycerol (v/v)) containing lysozyme (0.1 mg/mL) and DNaseI (6.6 μg/mL), and rocked for 30 min before lysing using a cell homogenizer (Avestin Emulsiflex-C5) at 10,000 to 15,000 psi for 10 min. Cellular debris was cleared by ultracentrifugation (1857 x g, 30 minutes, 4°C). Supernatant was added to pre-washed equilibrated 2 mL His-Pur Cobalt resin (ThermoFisher) and incubated for 15 min at 4°C with rocking. Protein was washed and eluted with 30 and 200 mM imidazole respectively. SUMO protease His-Ulp1 (700 μg) was added to the elution fraction and dialyzed overnight using SnakeSkin Dialysis Tubing (Thermo Scientific, 10K MWCO, 35 mm) in 2L Buffer B at 4 °C with stirring. Following dialysis, the sample was then incubated in a prewashed equilibrated Ni-NTA column (2 mL, 15 min), collected and washed with Buffer B and Buffer B containing 30 mM imidazole. Collected fractions were concentrated using an Amicon 10kD MWCO ultra-centrifugal unit (MilliporeSigma, 1857 x g, 4 °C) along with subsequent 2 mL washes of Buffer B. Protein was further purified by size exclusion chromatography (Superdex 200 Increase 10/300 GL) using Buffer B. Major peaks were collected and concentrated using an Amicon centrifugal unit (10K MWCO, 1857 x g, 4 °C).
DnaJ2 cell pellet (500 mL of culture) was resuspended in Buffer A containing SIGMAFAST EDTA-free protease inhibitor cocktail (1 tablet per 100 mL buffer), lysozyme (0.1 mg/mL), and DNase I (3.3 μg/mL) with rocking at 4 °C for 1 hour. The sample was lysed using a homogenizer (Emulsiflex-C5, 15,000 psi, at least 5 passages) and the cellular debris cleared by ultracentrifugation (10956 x g, 30 minutes, 4 °C). Supernatant was then incubated in a 0.75-1.5 mL pre-washed and equilibrated His-Pur Cobalt resin at 4 °C with rocking for 40 minutes. The column was washed with low imidazole in Buffer A (10, 20, 30 mM imidazole) and eluted with higher imidazole (50, 100, 200 mM imidazole). Elution fractions were combined and concentrated (1857 x g, 10 min, 4 °C) using an Amicon 10K MWCO ultra-centrifugal unit (MilliporeSigma) before addition of SUMO protease His-Ulp1 (700 μg). Samples were dialyzed overnight against Buffer A in Slide-A-Lyzer 10K MWCO dialysis cassettes (Thermo Fisher) or SnakeSkin 10K MWCO dialysis tubing. Following dialysis, the sample was incubated in a pre-washed 0.75 mL Ni-NTA column for 20 min or 1 hour with shaking at 4 °C. Cleaved DnaJ2 was collected as “flow-through” and the column was washed with Buffer A containing imidazole (30 mM imidazole). Flow-through and washes were concentrated using an Amicon 10kD MWCO ultra-centrifugal unit (1857 x g, 4 °C) and imidazole diluted out with intermittent Buffer A additions according to manufacterer’s instructions. Precipitated DnaJ2 was removed by centrifugation (Beckman Coulter Microfuge 20R, 10,000 x g, 2 min, 4 °C).
Overexpression and purification of Mtb DnaK R127A, T175A, E514A, and nucleotide binding domain (NBD) (Table S3) constructs were carried out as described for wild-type Mtb DnaK in Rosetta2 cells. Mtb DnaK E398A, F400A, T401A, D404A, and N406A, and the substrate binding domain (SBD) constructs (Table S3) were overexpressed in E. coli Rosetta2 pLysS and purified as described for wild-type Mtb DnaK.
For all experiments requiring apo DnaK (isothermal titration calorimetry (ITC) and limited proteolysis), DnaK T175A and DnaK T175A NBD were dialyzed over the course of two days into two different buffers based on previously described methods.(Moro, Fernández and Muga, 2003; Taneva et al., 2010) Following the purification for wild-type DnaK described above, samples were dialyzed in Buffer C (20 mM imidazole (pH 7.2), 2 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol) to remove metals and nucleotide from DnaK. Samples were then dialyzed against Buffer D (40 mM Tris-HCl (pH 7.4), 50 mM KCl, 10 mM MgCl2), which served as the ITC buffer.
All purification steps were performed at 4°C. Protein concentration was estimated by DC Protein Assay (Bio-Rad), using BSA (bovine serum albumin) as the standard. Following the purification, all proteins were frozen in N2(l) and stored at −80 °C until use. E. coli DnaK, DnaJ, and GrpE were expressed and purified using the same protocol as the corresponding Mtb chaperones or cofactors in Rosetta 2 cells (Table S3).
Purification of Homo sapiens chaperones.
Homo sapiens Hsc70 (HspA8) and DnaJA2 were overexpressed in E. coli Rosetta2 pLys cells (Table S3). Growth conditions were the same as Mtb chaperones except after growth to log-phase OD600 (37°C, 200 rpm), cells were induced overnight with 1 mM IPTG at 16 °C. Hsc70 was purified using the same protocol as Mtb DnaK, described above. DnaJA2 pellets (from 500 mL culture) were resuspended and incubated in Buffer A containing SIGMAFAST EDTA-free protease inhibitor cocktail), lysozyme (0.1 mg/mL), and DNase I (3.3 μg/mL) with shaking for 1 hour at 4 °C. The sample was lysed using a homogenizer (15,000 psi) and the cellular debris cleared by ultracentrifugation (10956 x g, 30 minutes, 4 °C). Supernatant was added to a pre-washed equilibrated 5 mL His-Pur Cobalt column and incubated for 30 min with rocking at 4 °C. The protein was washed with low concentrations of imidazole (10, 20, 30, and 50 mM imidazole) and eluted with 200 mM imidazole in Buffer A. SUMO protease His-Ulp1 (700 μg) was added to the pure elution fractions as analyzed by SDS-PAGE, and dialyzed overnight using SnakeSkin Dialysis Tubing (10K MWCO) in 2 L of Buffer A. Following dialysis, the sample was then incubated in a pre-washed equilibrated Ni-NTA column (5 mL, 10 min), collected and washed with Buffer A (10 mL), and concentrated using an Amicon centrifugal unit (10K MWCO, 1857 x g, 4 °C). Protein quantification, freezing and storage was carried out as described above.
High-throughput screen for inhibitors of chaperones.
Primary screen reactions were carried out in 15 μL volume in 384-well flat bottom polystyrene microplates (Lumitrac). After any addition of reagents, the plates were centrifuged at 180 x g for 30 sec to collect all the liquid at the bottom of the plate. The following final concentrations of each were used for screening: DnaK (2 μM), DnaJ2 (0.2 μM), GrpE (0.2 μM), and ATP (100 μM), which were determined following optimization in plate-based setup. 10 μL of ATP (150 μM) dissolved in the reaction buffer containing phosphate-buffered saline (PBS) pH 7.4 (Gibco), 5 mM MgCl2, and 0.01 % tween-20 (detergent was included to prevent compound aggregation) were added into plates using a Thermo Multidrop Combi dispenser (Thermo Scientific). 0.05 μL of screening compounds (5 mM in DMSO) were dispensed into the plate using a pintool (V&P Scientific) attached to an MDT Janus (Perking Elmer). The final concentration of each compound was 17 μM with 0.3% (v/v) DMSO final. A mixture of DnaK (6 μM), DnaJ2 (0.6 μM) and GrpE (0.6 μM) in reaction buffer was incubated for 15 min at room temperature and then 5 μL of this mixture were added to columns 1-23 using a Thermo Multidrop Combi dispenser (Thermo Scientific) and 5 μL of a mixture of DnaJ2 (0.6 μM) and GrpE (0.6 μM) pre- incubated at room temperature for 15 min were added to column 24. The plates were sealed with a hot sealer Plate Loc Velocity 11 (Agilent Technologies) and incubated at 37 °C for 30 min. The reaction was quenched with 5 μL of ADP-Glo Reagent (Promega), the plates were then sealed and shaken at room temperature for 45 min. After that time, 10 μL of Kinase Detection Reagent was added to each well and the plates were shaken for another 45 min. Luminescence was measured in relative lights units (RLUs) using a Biotek Synergy Neo plate reader (Biotek). The amount of ADP produced was normalized against the positive control in column 24 and negative control in column 23 as % normalized product inhibition (NPI) = (RLUsample RLUaverage negative control) / (RLUaverage positive control – RLUaverage negative control) x 100. All active compounds were retested in concentration response experiments following purity analysis by HPLC. Compound stocks of 5 mM DMSO from library plates were cherry picked and serially diluted in DMSO; 0.1 μL of these dilutions were transferred using pintool (V&P Scientific) attached to an MDT Janus (Perking Elmer). Marvin Beans (ChemAxon) software was used for rendering and manipulating chemical structures for analysis of compound hits.
Coupled ATPase assays for titrations.
For compound dose-response experiments, reactions (15 μL) were prepared in 96-well half-area black polypropylene plates (Corning) in PBS buffer (pH = 7.4) containing 2 mM MgCl2 and 0.1% Tween-20 to prevent aggregation of drug. Compound suspended in DMSO was added to a mixture containing DnaK (4 μM), followed by addition of GrpE, DnaJ2, or both (0.4 μM each). Plates were pre-incubated (typically 15-20 min, 37°C) following each cofactor addition. 100 μM ATP final concentration was added at room temperature to initiate, followed by incubation (typically 30 min, 37°C). Using the ADP-Glo Kinase Assay system described above, ADP-Glo Reagent (5 μL) was added to wells and incubated for 45 min (rocking at 200 rpm at room temperature), followed by addition of Kinase Detection Reagent (10 μL) and incubation for an additional 45 min (rocking at 200 rpm at room temperature) followed by luminescence measurements on a Spark plate reader (Tecan, 1s integration). Production of ADP in each reaction was quantified by comparison of RLU obtained from addition of ADP-Glo reagents to ATP/ADP standards in 1x PBS, 2 mM MgCl2, 0.01% Tween-20, 10% DMSO (v/v), as described by the manufacturer’s instructions. For some assays, ATP remaining was calculated by quenching reactions with 15 μL of Kinase-Glo Max (Promega) prior to incubation for 10 min with rocking (200 rpm at room temperature). Luminescence was measured on a FlexStation 3 plate reader (Molecular Devices). ATP in each well was calculated by comparing measured RLU to that measured for ATP standards in the reaction buffer. In order to determine “percent inhibition of activation”, reactions containing DnaK, DnaJ2 and GrpE were normalized to controls containing DnaK alone (100%) and chaperone/cofactor mixtures with DMSO only (0%). In order to determine “percent inhibition of ATPase activity”, DnaK reactions were compared to reactions containing no protein (100%) and chaperones with DMSO only (0%). Unless otherwise indicated, ATPase activity was assessed using the assays described above. In order to calculate half-maximal inhibitory concentrations (IC50) of compounds for inhibition of ATPase activity or activation, data were analyzed in Prism using the equation logEC50 = logECF – (1/HillSlope)*log(F/100-F)) and Y = Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)). F was set to 50 to calculate 50% inhibition values.
For cofactor titrations, reactions (15 μL) were prepared in microcentrifuge tubes using the same conditions as dose-response experiments. Besides reactions with DnaK alone, each reaction contained 4 μM DnaK, 0.4 μM of one cofactor, and varying concentrations of the other cofactor (0, 0.4, 2, and 4 μM). Reactions were initiated with 100 μM ATP, carried out at 37°C, and quenched with 15 μL 0.05% formic acid. Reactions were analyzed as described above using ADP-Glo Kinase Assay.
HPLC-analysis of ATP hydrolysis by chaperones.
Indicated ATPase reactions were set-up as described above in microcentrifuge tubes. After completion of reactions, each tube was centrifuged at 10,000 x g for 2 minutes, and then 15 μL of supernatant was loaded onto a 96 well plate (Thermo Scientific), diluted with 35 μL of 50 mM potassium phosphate (pH 6.0) as running buffer. HPLC analysis was conducted with an autosampler using a 2.6 μm, 4.6 x 150 mm Accucore aQ C18 Polar Endcapped column (ThermoFisher Scientific). Each sample (15 μL) was eluted with an isocratic gradient of running buffer at a flow rate of 0.700 mL/min for 10 min at a column temperature of 30 °C. The presence of ATP and ADP was detected at a wavelength of 260 nm and quantified using Chromeleon 7 software (ThermoFisher Scientific) using ATP/ADP standards that were analyzed under the same conditions.
Isothermal titration calorimetry (ITC) analysis of chaperones.
ITC measurements were performed at 25 °C using a MicroCal auto-iTC200 calorimeter (Malvern Panalytical). Apo DnaK T175A and NBD T175A were purified as described, with a final dialysis step against 40 mM Tris-HCl (pH = 7.4), 50 mM KCl, 10 mM MgCl2 for 16 h at 4 °C. The concentration of the proteins after dialysis was measured using a DC protein assay. For the ITC assay, the dialyzed proteins were prediluted to 60 μM in the dialysis buffer, then diluted 1:1 with 2X buffer to a final concentration of 30 μM in a final buffer containing 40 mM Tris-HCl (pH = 7.4), 50 mM KCl, 10 mM MgCl2 supplemented with 1% DMSO and 0.01% tween-20. Then, 19 injections of 2 μL telaprevir or MECA (100 μM), dissolved in the aforementioned buffer, were injected into 0.2 mL of either DNAK175A or NBD in the chamber every 150 s. For one replicate, telaprevir was dissolved in 40 mM Tris-HCl (pH = 7.4), 50 mM KCl, 10 mM MgCl2 supplemented with 1.5% DMSO and 0.015% tween-20. Data for raw ITC and thermodynamic curves were downloaded for analysis using Affinimeter software and plotted using GraphPad Prism. Heat release from dilution of drug into buffer was subtracted from the thermodynamic data.
Native gel electrophoresis analysis of DnaK complexes.
Purified apo DnaK T175A (200 μM) and telaprevir (TP) or NRLLLTG or DMSO were diluted 1:10 into native gel buffer (20 mM HEPES (pH = 7.5), 50 mM KCl, 2.5 mM MgCl2) and incubated for 30 min. For each sample, an equal volume of 2X native gel sample buffer (62.5 mM Tris-HCl, pH 6.8, 40% glycerol, 0.01% bromophenol blue) was added and mixed, followed by loading a volume equivalent to 10 μg of DnaK into a 10% polyacrylamide native gel, which prepared according to Biorad’s insructions. The gel was run for 3 hr under constant voltage (120 V) at 4°C.
For analysis of MECA’s effect on DnaK-GrpE complex formation, purified DnaK T175A was diluted into native gel buffer (20 mM HEPES (pH = 7.5), 50 mM KCl, 2.5 mM MgCl2) to a final concentration of 20 μM, followed by addition of MECA (100 μM) and incubation at 37 °C for 30 minutes. GrpE (at a final concentration 31% to that of DnaK) was added and incubated at 37 °C for 30 minutes. Lastly, ATP and ADP were added to a final concentration of 100 μM; addition of native gel buffer was used as a “no nucleotide added” control. Mixtures were incubated at 37 °C for 30 minutes native gel anaysis as described above. The percentages of Mtb DnaK occupying the monomeric, dimeric, and trimeric states were quantified using Image Lab (Biorad). Within the software, lanes were fit and bands were selected, and the value “% Lane” was obtained for each condition. Oligomeric states are approximated based on a molecular weight ladder.
Limited proteolysis analysis of proteins with added small molecules.
To determine the effect of TP on proteinase K activity, a hemoglobin proteolysis control experiment was conducted following manufacturer’s instructions for proteinase K (Sigma-Aldrich). Hemoglobin (20 mg/mL) was denatured in a buffer containing 100 mM potassium phosphate, pH 7.0, 6 M urea for 1 hr at 37°C. Denatured hemoglobin (10 mg/mL) was then incubated in buffer (100 mM potassium phosphate, pH 7.0, 3 M urea) supplemented with varying concentrations of TP (5% DMSO as vehicle) for 30 minutes at room temperature. Proteolysis was initiated by adding proteinase K to a final concentration of 0.05 μM at room temperature following incubation for 15 minutes at room temperature. Reactions were quenched with 1 mM phenylmethylsulfonyl fluoride (PMSF) and analyzed by SDS-PAGE (15% acrylamide gel) followed by Coomassie staining. Cleavage was quantified by calculating the ratio of band intensity of cleaved product to uncleaved hemoglobin monomer using Image Lab software.
Limited proteolysis experiments were adapted from previous studies.(Meng et al., 2018; Preissler et al., 2017) Experiments were performed on Mtb apo DnaK T175A following the extensive dialysis described for ITC experiments. Apo DnaK T175A (10 μM) was incubated in buffer containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 5 mM CaCl2, 100 mM KCl, 5 mM DTT. 2 mM nucleotides were added for ADP and ATP-bound samples. Nucleotide were preincubated with DnaK for 1 hour at room temperature, followed by incubation with 50 μM compound (or 5% DMSO as control) for 30 min. Proteinase K was then added to a final concentration of 0.05 μM. The reaction was run at room temperature. Reactions were quenched with 1 mM PMSF at t = 15 min and 1 μg total protein per lane was analyzed by SDS-PAGE (12% acrylamide gel, Invitrogen) prior to Coomassie Brilliant Blue staining. The molecular weight of partially cleaved DnaK (*) was predicted to be ~60 kD based on comparison to the migration of protein ladder standards (Biorad) using Image Lab software. FL, NBD and SBD truncations resolve at approximately 67 kDa, 40 kDa, and 27 kDa, respectively
Fluorescence polarization (FP) assays with DnaK and peptides.
FP assays were carried out by following published protocols with minor changes.(Rossi and Taylor, 2011; Ricci and Williams, 2008; Moerke, 2009) For direct binding assays, FL-βA-NRLLLTG (dissolved in DMSO) was titrated with increasing concentrations of DnaK (diluted in PBS, 2 mM MgCl2, 0.1% Pluronic-127) in a 96-well flat bottom black plate (Corning). The plate was sealed with aluminum foil and rocked (30 min, 200 RPM, room temperature), and then the plate was read using a DTX 880 Multimode Detector (Beckman) at 25° C, with excitation and emission wavelengths at 485 and 525 nm, respectively. For competition assays, FL-βA-NRLLLTG (3.75 μM) and DnaK (6.25 μM for WT, SBD, and T401A, 45 μM for F400A) were preincubated with rocking for 30 min at room temperature. Each were then titrated with unlabeled NRLLLTG, telaprevir, boceprevir, or 7 (dissolved in DMSO) at increasing concentrations, then incubated with rocking (45 min, 200 RPM, room temperature). The plate was read using the same detector and method as described for the direct binding assay. Note that increased concentration of F400A was used to achieve sufficient fluorescent polarization for displacement assays and that the EC50 value with TP was estimated to be >100 μM. In order to calculate half-maximal effective concentration (EC50) of compounds for displacement of probe, data were analyzed by Prism using the equation logEC50 = logECF − (1/HillSlope)*log(F/100-F)) and Y = Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)). F was set to 50 to calculate 50% effective concentration of ligand.
Mass spectrometry analysis of DnaK with probe 7.
Mtb DnaK SBD (360-625) (200 μM) and 7 (10 mM) were diluted together 1:10 in dH2O. Protein/ligand were incubated for 30 minutes (37°C) before irradiation with UV light (365 nm) at various time points. Analysis of data suggested t = 5 min irradiation was the optimal balance between crosslinking efficiency and specificity. Crosslinking efficiency was evaluated by diluting protein 1:10 into Super DHB matrix solution (50 mg/mL in 50:50 aceonitrile (ACN):H2O, 0.1% trifluoroacetic acid (TFA)) and analyzing 1 uL of the resulting mixture by MALDI-TOF (Bruker, UltrafleXtreme TOF/TOF, linear mode, suppressed up to 28000 Da).
Mass spectrometry analysis of crosslinking positions in DnaK.
The crosslinked DnaK SBD sample from above was resuspended in NuPAGE LDS Sample buffer (1X) (Invitrogen) and loaded onto a NuPAGE 4-12% Bis-Tris Gel (1.0 mm x 10 well, Invitrogen) for SDS-PAGE analysis. The gel was stained with GelCode Blue Stain Reagent (Thermo Scientific) and the DnaK band excised and destained with several rounds of incubation in 1:1 (v/v) methanol: 100 mM ammonium bicarbonate at 4°C with agitation. The destained gel pieces were dehydrated in a SpeedVac concentrator. Dried gel pieces were resuspended in 200 μL of 100 mM ammonium bicarbonate with 250 ng Promega trypsin for overnight digestion. A 200 μL solution of 5% formic acid and 0.2% TFA Poros R2 50 μm beads (Applied Biosystems) slurry in water was added to the gel pieces before returning the samples to the shaker for an additional 3 hr at 4°C. Beads were loaded onto 0.1% TFA equilibrated C18 ZipTips (Millipore) using a microcentrifuge for 30 seconds at 6,000 rpm. The beads were washed with 0.5% acetic acid. Peptides were eluted with 40% acetonitrile with 0.5% acetic acid followed by 80% acetonitrile with 0.5% acetic acid. The organic solvent was removed using a SpeedVac concentrator. The peptide mixture was reconstituted in 0.5% acetic acid. An aliquot of the peptide mixture was loaded onto a 75 μm × 50 cm Pepmap EasySpray column using the autosampler of an Easy nLC-1200 nano-HPLC coupled to an Orbitrap Eclipse Tribrid mass spectrometer using solvent A = 2% acetonitrile in 0.5% acetic acid (v/v) and solvent B = 80% acetonitrile in 0.5% acetic acid (v/v)). The sample was eluted using a flow rate of 0.2 μL/min with a gradient of 0–100% solvent B over 95 min, a spray voltage of 2.0 kV, and 3 sec cycle time. MS1 scans were acquired at 60,000 resolution (@ 400 m/z), 50 ms maximum injection time, AGC Target of 4E5, and scan range of 400 to 1500 m/z. MS2 HCD spectra were acquired on precursors that carry 2-5 charges and ETD on precursors that carry 4-10 charges using the following settings: 2 m/z isolation window, target value of 2e5 ions, 1 microscan, 200 ms maximum injection time, scan range of 100-2000 m/z, 30,000 resolution (@ 400 mlz), ETD using charge dependent reaction time, HCD using 27 % normalized collision energy.
Mass spectrometry data analysis of DnaK-7 crosslinking.
Raw files were searched using the search engine Byonic (Protein Metrics). Given that the probe (7) fragments upon MS2 (theoretical fragment ions: 107.025 Da, 206.093 Da, 305.161 Da for HCD and 122.0354 Da, 221.1038 Da, 320.1722 Da for ETD) which confounds the regular search mode, we implemented MS/MS filtering. We required two fragment ions of the probe (0.02 Da tolerance) for an MS/MS spectrum to be searched. The filtered spectra data was searched with a 20 Da mass tolerance for MS1 and 20 ppm for MS2. Amino acids K, R, T, S, I, L, H, A, V, N, D, E, and Y were allowed to be modified by the probe (+530.2853 Da). In addition, we also searched the data without MS/MS filtering allowing 20 ppm on the precursor and 0.02 Da on the MS/MS masses and addition of the probe to the protein N-Term, Protein C-Term, and amino acids A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, and Y. Note, that while our analysis method is able to identify the exact modified amino acids, it is not possible in all cases to unambiguously assign the site of modification within the peptide due to lack of peptide backbone fragmentation coverage. For some assignments, the actual site of modification might be +/− 2 residues within the reported site.
Luciferase refolding assays to assess chaperone activity.
Chemical protein denaturation and refolding assays were carried out as previously described,(Tzankov et al., 2008; Rodina et al., 2013) with some changes. Firefly luciferase (Promega) was denatured in a buffer containing 6 M guanidinium-HCl and 5 mM DTT for 1 hour. Refolding reactions were pre-assembled at room temperature containing 5 μM DnaK in Buffer-R (20 mM HEPES-KOH (pH 7.5), 100 mM KCl, and 2 mM Mg(OAC)2, 5 mM DTT) supplemented with compound in DMSO (2.5% DMSO final used as vehicle). After t = 30 min incubation at room temperature, 1 μM DnaJ2 and 2.5 μM GrpE are added and incubated for another t = 30 min at room temperature. Reactions were supplemented with 2 mM ATP and initiated with addition of denatured luciferase (1:100) to a final concentration of 100 nM. Control reactions containing 100 nM native luciferase were conducted in the same buffer conditions. At t = 30 min or indicated time point, 10 μL aliquots of each reaction was transferred to a 96-well half-area black polypropylene plate (Corning) and 25 μL of Luciferase Assay reagent (Promega) was added. Luciferase activity was immediately measured by luminescence on a Molecular Devices plate reader (FlexStation 3, 1s integration). Human chaperone experiments were performed as described above except luciferase was denatured for 10 min. Refolding reactions were pre-assembled on ice containing Hsc70 (4 μM) with DnaJA2 (6 μM) in Buffer-R supplemented with 2 mM ATP and compound (2.5% DMSO final (v/v)). Denatured luciferase was diluted 1:100 in reactions to a final concentration of 100 nM and incubated at 30°C. At t = 60 min or indicated time point, 10 μL aliquots were analyzed as described above.
Recovery of M. smegmatis cells following heat shock.
To assess the effect of heat shock on indicated strains, Msm cells were grown to late-log phase (OD580 = 1.0), centrifuged (3100 × g, 5 min) and resuspended in fresh 7H9 complete/tween prior to dilution in the same medium to OD580 = 0.08, as described previously.(Jastrab et al., 2017; Harnagel et al., 2021) Strains (250 μL) were placed in sealed 2.0 mL microtubes (Sarstedt) and incubated at 53 °C or 37 °C for 1 hr in a heating block. Cells were serially diluted (10-fold) in PBS/0.05% Tween-80. Cell survival for input and heat-treated cells was determined by spot assays in which 2.5 μL, beginning with the undiluted stock of each strain, were added to 7H10 agar plates with or without 100 μM TP and incubated for t = 3 days at 37 °C or 40 °C under standard conditions. In these experiments, the 7H10 agar plates contained the following additives: 0.05% dextrose and 0.05% glycerol. It is important to note that addition of TP to cell cultures during heat shock did not cause a significant change in growth recovery, and so cells were heat shocked in media lacking compound.
Mycobacterial growth inhibition assays.
M. smegmatis (Msm) strains (mc2155 and MGM6933 attB:MtbdnaK) were grown in 7H9 complete/tween. MGM6933 was grown in streptomycin 20 μg/mL. Strains were grown to late log to stationary phase (OD580 ~ 1). Cultures were centrifuged (1857 x g, 5 minutes at room temperature) and supernatants discarded. Msm cells were washed with 1X PBS containing .05% Tween-80 and pellets were re-suspended in 7H9 complete/tween. Single-cell suspensions were made by centrifuging cells (74 x g, 2 minutes with no brake at room temperature) and cultures were re-diluted in media multiple times to a final OD580 of .01 for use in minimum inhibitory concentration (MIC) assays. Compounds (100x stocks) were added to cultures (200 μL per well) in 96-well clear-bottom plates (Corning) with control wells containing only vehicle and cells. Plates were wrapped in foil and shaken at 250 rpm for at least 3 minutes on a plate shaker (Thermo Scientific) prior to incubation at 37 °C with shaking at 200 rpm or indicated amounts of time. Cultures were mixed prior to absorbance readings at 580 nm using a plate reader (FlexStation 3, Molecular Dynamics). Percent growth inhibition for each well was calculated by the following equation: ((OD580vehicle− OD580well)/(OD580no drug)) x 100, where OD580vehicle is the well that contains only vehicle for the condition tested. Resulting data were analyzed in Prism using the following equation: logEC50 = logECF – (1/HillSlope)*log(F/100-F)) and Y = Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)) where F = 50 to calculate MIC50 for 50% growth inhibition or F = 90 to calculate MIC90 for 90% growth inhibition.
For MIC assays performed to assess antibiotic synergy with TP, mid- to late-log phase cells (OD580 = 0.5-1) in 7H9 complete/tween were diluted to OD580 = 0.01 as described above. 200 μL of this final diluted culture was aliquoted to 96-well clear-bottom plates. 2 μL of 100x of each indicated antibiotic were added to each well, followed by addition of 2 μL of 10 mM TP ([TP]final = 100 μM) or DMSO as a vehicle control. Plates were mixed (2 min, 200 RPM, RT) and incubated at 37°C without shaking. OD580 was measured at t = 24 hr, 48 hr, 72 hr, at which time cells were resuspended manually using a multichannel pipette and OD580 was measured using a plate reader (iD5, Molecular Devices). Data shown for rifampicin was after t = 24 hr incubation, while kanamycin and streptomycin data is shown after t = 48 or 72 hr incubation. MIC data was analyzed as described above, where drug wells containing TP were compared to a vehicle well that contained 100 μM TP.
Frequency of resistance (FOR) determination for mycobacteria.
To quantify the frequency of RIF-resistant colonies for wild-type Msm, a published protocol was adjusted.(Fay et al., 2021) Cells were grown in 7H9 complete/tween (30 mL) to mid-log phase (OD850 = 0.6 to 0.8), centrifuged (3100 x g, 5 min), washed with PBS buffer containing 0.05% Tween-80, and then resuspended in 3 mL of fresh 7H9 complete/tween. Per experiment, 1 mL of cells were then added to 7H10 agar plates containing a final concentration of 100 μM TP or 240 μM rifampicin (RIF) or both compounds (100 μM TP + 240 μM RIF). 10 μL of the input cells were serial diluted (10-fold) in PBS/0.05% Tween-80 and plated on 7H10 square plates to calculate the average colony forming units (CFU)/mL as “input”. Plates were incubated at 37 °C with 5% CO2 for t = 3 days (input plates) and 5 days (compound plates). We observed a lawn of cells on 100 μM TP plates, which were not possible to count. FOR was calculated by dividing CFU of resistant cells on compound plates by the CFU determined from the input, accounting for the volume of cells plated. Compound plates were monitored for at least 10 days. Note that fresh 20% Tween-80 was filtered and used to make 7H9 complete/tween for each experiment.
Synthesis of peptide probes.
A standard solid-phase peptide synthesis protocol based on 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) promoted peptide coupling reaction was followed, as described briefly here. Fluorenylmethoxycarbonyl (Fmoc)-protected amino acid monomers were purchased from Chemimpex, except for Fmoc-L-Photo-Leu (Sigma-Aldrich). 2-Pyrazine acid and 4-benzoylbenzoic acid were purchased from Sigma-Aldrich and Fluorescein isothiocyanate was purchased from Chemimpex. Briefly, 0.1 mmol equivalent of 2-Cl-trityl resin was loaded into a plastic syringe with a frit column plate. The resin was swelled with 3 mL of dry dichloromethane (DCM) for t = 30 min. After filtering DCM out, the resin was swelled with a solution of 0.12 mmol of Fmoc protected C-terminal amino acid monomer and 0.12 mmol of diisopropylethylamine (DIPEA) in 2 mL of dry DCM overnight. After filtration, the unreacted trityl chloride functional group was swelled with 2 mL blocking solution (DCM)/methanol (MeOH)/DIPEA=17:2:1 v:v:v) for 5 minutes (3X). Then, Fmoc protection was removed by swelling resin in 2 mL of 20% piperidine/DMF for 20 minutes (deprotection step). The resulting resin, after washing with solvent, was treated with a 2 mL solution of 0.3 mmol of corresponding Fmoc-amino acid monomer and 0.036 mmol of DIPEA in dry DMF. After swelling for 1 hr (2 hrs swelling for the amino acids next to Pro), the solution was filtered out and washed with DMF (Coupling step). The Deprotection-Coupling cycle was performed likewise until the N-terminal was capped with 2-pyrazine acid/4-benzoylbenzoic acid (Compounds 1-7) following the coupling step protocol. Fluorescein isothiocyanate (FITC) was used for N-terminal capping to produce the peptide probe Fl-βAla-NRLLLTG with DIPEA in DMF. The resulting peptide was washed with DMF and DCM. After drying by air flow, the resulting peptides were euted from the column after swelling with 2 mL of hexafluoro-isopropanol (HFIP) for 2 hours. The methyl ester analogues were produced from free C-terminal peptides via stirring in 3 mL of dry MeOH with 100 μL of thionyl chloride. For 7, the reaction scale was reduced to 0.05 mmol equivalent; therefore, all the monomers and coupling reagents were used in half the amounts of mentioned above.
Characterization of synthetic peptides:
Following synthesis, peptides were purified by preparative HPLC using a Waters SunFire Prep C18 OBD 150 x 19 mm, 5 μm column. For analytical analysis of purity, a Phenomenex Aqua C18 150 x 4.6 mm, 5 μm column was used. A 10% to 90% linear gradient of solvent D (water:ACN = 5:95, 0.1 % of formic acid) in C (water:ACN = 90:10, 0.1% of formic acid) was used as the mobile phase in analytical HPLC runs. All peptides were obtained as white powders after freeze-drying.
Spectral Characterization:
Compound 1. HRMS (APCI+) m/z: [M+H]+calcd 587.3553, found 587.3542. Compound 2. HRMS (APCI+) m/z: [M + H]+ calcd 533.3062, found 533.3083. Compound 3. HRMS (ES+) m/z: [M+H]+ calcd 635.3441, found 635.3439. Compound 4. HRMS (ES+) m/z:[M+H]+ calcd 601.3710, found 601.3711. Compound 5. HRMS (ES+) m/z: [M+H]+ calcd 547.3221,found 547.3239. Compound 6. HRMS (APCI+) m/z: [M+Na]+ calcd 671.3411, found 671.3431.Compound 7. HRMS (ES+) m/z: [M+H]+ calcd 559.2968, found 559.2991. FL-βA-NRLLLTG. MALDI-TOF (R+) m/z: [M + H]+ calcd 1246.556, found 1246.544.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise noted, biochemical and cell growth curves, along with associated statistical analyses, were produced using Prism 9 (Graphpad). For ITC analysis, AFFINImeter software was used for analysis and the resulting data was plotted in Prism, with independent experiments shown. For gel-based analysis, Image Lab (Biorad) was used for analysis and the resulting data was plotted in Prism and further analyzed. Details of replicates (were n represents the number of individual replicates), and data analysis for specific experiments can be found in the figure legends. Data are typically reported as means, which equal the average of the values found for each of the replicates, and the reported standard deviation (SD) indicates the variance of replicate data. The equations used for each curve fit are described in the Method Details section.
For the selection of compound hits in the high-throughput screen, the cutoff was set as >40 percent normalized product inhibition, as described in the Method Details section. The robustness of the primary screen is indicated by an average z’ of 0.861 with an SD of 0.868 over 78 plates. Hit rates were 0.5 % for the drug repurposing collection and 0.06 % for the drug-like collection.
For analysis of the proteomics data, the peptides were identified using the search engine Byonic (Protein Metrics) and peptides with a score of ≥300 were used for further analysis. The number of crosslinked peptide spectral matches to a specific modified peptide were used to identify the prevalent site of probe attachment as descried in the Method Details section.
Supplementary Material
Data S1. SI_Protein_MSMS_data: Data from described mass spectrometry analysis on Mtb DnaK crosslinked to peptide analog 7. Related to Figures 5 and S5, and Table S6.
Highlights.
The peptidomimetic drug telaprevir is a chaperone inhibitor
Telaprevir prevents activation of mycobacterial DnaK by cofactor DnaJ2
Addition of telaprevir sensitizes mycobacterial cells to other proteotoxic stresses
Inhibition of chaperone activity leads to lowered resistance to a key antibiotic
Acknowledgments:
The authors would like to thank Chloe Larson and Lavoisier Ramos-Espiritu (Rockefeller), Samantha Ciervo (NYU), Gideon Yawson (NYU) and Samuel Epstein (NYU) for experimental help. We acknowledge Jason Young (McGill) for sharing chaperone plasmids, Celia Schiffer (UMass Medical School) for contributing additional HCV inhibitors, Juan Sabin (Affinimeter) for data analysis advice, Chris Dashiell (Promega) for help with reagents, and Dr. Hermann Steller (Rockefeller) for supporting the purchase of the iTC200 through an HHMI grant. T.L. acknowledges NYU for funding. J.H. acknowledges NSF GRFP grant DGE1839302. B.U. acknowledges support for the mass spectrometric experiments by a shared instrumentation grant from the NIH, 1S10OD010582-01A1 for the purchase of an Orbitrap Eclipse Tribrid mass spectrometer.
Footnotes
Declaration of interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Aguilar-Rodríguez J, Sabater-Muñoz B, Montagud-Martínez R, Berlanga V, Alvarez-Ponce D, Wagner A and Fares MA (2016) 'The Molecular Chaperone DnaK Is a Source of Mutational Robustness', Genome Biol Evol, 8(9), pp. 2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose AJ and Chapman E (2021) 'Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones', J Med Chem., 64(11), pp. 7060–7082. [DOI] [PubMed] [Google Scholar]
- Baaklini I, Wong MJ, Hantouche C, Patel Y, Shrier A and Young JC (2012) 'The DNAJA2 substrate release mechanism is essential for chaperone-mediated folding', J Biol Chem, 287(50), pp. 41939–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M and Hartl FU (2016) 'In vivo aspects of protein folding and quality control', Science, 353(6294), pp. aac4354. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M and Hartl FU (2020) 'Recent advances in understanding catalysis of protein folding by molecular chaperones', FEBS Lett, 594(17), pp. 2770–2781. [DOI] [PubMed] [Google Scholar]
- Barthel TK, Zhang J and Walker GC (2001) 'ATPase-defective derivatives of Escherichia coli DnaK that behave differently with respect to ATP-induced conformational change and peptide release', J Bacteriol, 183(19), pp. 5482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen EB, Chang L, Gestwicki JE and Zuiderweg ER (2009) 'Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate', Proc Natl Acad Sci U S A, 106(21), pp. 8471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickerton GR, Paolini GV, Besnard J, Muresan S and Hopkins AL (2012) 'Quantifying the chemical beauty of drugs', Nat Chem, 4(2), pp. 90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B, DeJesus MA, Poulton NC, Zhang W, Engelhart CA, Zaveri A, Lavalette S, Ruecker N, Trujillo C, Wallach JB, Li S, Ehrt S, Chait BT, Schnappinger D and Rock JM (2021) 'Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis', Cell, 184(17), pp. 4579–4592.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV and Matsumura I (2010) 'Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids', Biotechniques, 48(6), pp. 463–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Theyssen H, Schröder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J and Bukau B (1995) 'Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication', J Biol Chem, 270(28), pp. 16903–10. [DOI] [PubMed] [Google Scholar]
- Busse HJ, Wöstmann C and Bakker EP (1992) 'The bactericidal action of streptomycin: membrane permeabilization caused by the insertion of mistranslated proteins into the cytoplasmic membrane of Escherichia coli and subsequent caging of the antibiotic inside the cells due to degradation of these proteins', J Gen Microbiol, 138(3), pp. 551–61. [DOI] [PubMed] [Google Scholar]
- Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, Tartaglia GG, Hayer-Hartl M and Hartl FU (2012) 'DnaK functions as a central hub in the E. coli chaperone network', Cell Rep, 1(3), pp. 251–64. [DOI] [PubMed] [Google Scholar]
- Cesa LC, Patury S, Komiyama T, Ahmad A, Zuiderweg ERP and Gestwicki JE (2013) 'Inhibitors of difficult protein-protein interactions identified by high-throughput screening of multiprotein complexes', ACS Chem Biol, 8(9), pp. 1988–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Bertelsen EB, Wisén S, Larsen EM, Zuiderweg ER and Gestwicki JE (2008) 'High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK', Anal Biochem, 372(2), pp. 167–76. [DOI] [PubMed] [Google Scholar]
- Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER and Gestwicki JE (2011) 'Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism', Chem Biol, 18(2), pp. 210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Thompson AD, Ung P, Carlson HA and Gestwicki JE (2010) 'Mutagenesis reveals the complex relationships between ATPase rate and the chaperone activities of Escherichia coli heat shock protein 70 (Hsp70/DnaK)', J Biol Chem, 285(28), pp. 21282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AV and Wuest WM (2019) 'Signed, Sealed, Delivered: Conjugate and Prodrug Strategies as Targeted Delivery Vectors for Antibiotics', ACS Infect Dis, 5(6), pp. 816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova LS, Slepenkov SV and Witt SN (2004) 'The insect antimicrobial peptide, L-pyrrhocoricin, binds to and stimulates the ATPase activity of both wild-type and lidless DnaK', FEBS Lett, 565(1-3), pp. 65–9. [DOI] [PubMed] [Google Scholar]
- Chiappori F, Fumian M, Milanesi L and Merelli I (2015) 'DnaK as Antibiotic Target: Hot Spot Residues Analysis for Differential Inhibition of the Bacterial Protein in Comparison with the Human HSP70', PLoS One, 10(4), pp. e0124563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE and Lindquist S (2005) 'Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi', Science, 309(5744), pp. 2185–9. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Fairlie DP, Liras S and Price D (2013) 'The future of peptide-based drugs', Chem Biol Drug Des, 81(1), pp. 136–47. [DOI] [PubMed] [Google Scholar]
- Dalphin MD, Stangl AJ, Liu Y and Cavagnero S (2020) 'KLR-70: A Novel Cationic Inhibitor of the Bacterial Hsp70 Chaperone', Biochemistry, 59(20), pp. 1946–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Rohde M and Jäättelä M (2007) 'The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions', FEBS Lett, 581(19), pp. 3702–10. [DOI] [PubMed] [Google Scholar]
- Evans CG, Chang L and Gestwicki JE (2010) 'Heat shock protein 70 (hsp70) as an emerging drug target', J Med Chem, 53(12), pp. 4585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A and Glickman MS (2014) 'An essential nonredundant role for mycobacterial DnaK in native protein folding', PLoS Genet, 10(7), pp. e1004516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A, Philip J, Saha P, Hendrickson RC, Glickman MS and Burns-Huang K (2021) 'The DnaK Chaperone System Buffers the Fitness Cost of Antibiotic Resistance Mutations in Mycobacteria', mBio, 12(2), pp. e00123–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genest O, Hoskins JR, Camberg JL, Doyle SM and Wickner S (2011) 'Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling', Proc Natl Acad Sci U S A, 108(20), pp. 8206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestwicki JE and Shao H (2019) 'Inhibitors and chemical probes for molecular chaperone networks', J Biol Chem, 294(6), pp. 2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragerov A, Zeng L, Zhao X, Burkholder W and Gottesman ME (1994) 'Specificity of DnaK-peptide binding', J Mol Biol, 235(3), pp. 848–54. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ and Sassetti CM (2011) 'High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism', PLoS Pathog, 7(9), pp. e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot AL, Farmer LJ, Rao BG, Taylor WP, Weisberg IS, Jacobson IM, Perni RB and Kwong AD (2011) 'Discovery and Development of Telaprevir', in Kazmierski WM (ed.) Antiviral Drugs: from Basic Discovery through Clinical Trials. Chichester: John Wiley & Sons, pp. 209–224. [Google Scholar]
- Hageman J and Kampinga HH (2009) 'Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library', Cell Stress Chaperones, 14(1), pp. 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnagel A, Lopez Quezada L, Park SW, Baranowski C, Kieser K, Jiang X, Roberts J, Vaubourgeix J, Yang A, Nelson B, Fay A, Rubin E, Ehrt S, Nathan C and Lupoli TJ (2021) 'Nonredundant functions of Mycobacterium tuberculosis chaperones promote survival under stress', Mol Microbiol, 115(2), pp. 272–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF and Jacobs WR (eds.) (2014) Molecular Genetics of Mycobacteria. 2nd edn. Washington, DC: American Society for Microbiology. [Google Scholar]
- Jastrab JB, Samanovic MI, Copin R, Shopsin B and Darwin KH (2017) 'Loss-of-Function Mutations in HspR Rescue the Growth Defect of a Mycobacterium tuberculosis Proteasome Accessory Factor E (pafE) Mutant', J Bacteriol, 199(7), pp. e00850–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath-Loeb AS, Lu CZ, Suh WC, Lonetto MA and Gross CA (1995) 'Analysis of three DnaK mutant proteins suggests that progression through the ATPase cycle requires conformational changes', J Biol Chem, 270(50), pp. 30051–9. [DOI] [PubMed] [Google Scholar]
- Kang Y, Taldone T, Patel HJ, Patel PD, Rodina A, Gozman A, Maharaj R, Clement CC, Patel MR, Brodsky JL, Young JC and Chiosis G (2014) 'Heat shock protein 70 inhibitors. 1. 2,5'-thiodipyrimidine and 5-(phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70', J Med Chem, 57(4), pp. 1188–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassenbrock CK and Kelly RB (1989) 'Interaction of heavy chain binding protein (BiP/GRP78) with adenine nucleotides', EMBO J, 8(5), pp. 1461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN and Sternberg MJ (2015) 'The Phyre2 web portal for protein modeling, prediction and analysis', Nat Protoc, 10(6), pp. 845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL and Jacobson KA (1994) '2-Substitution of N6-benzyladenosine-5'-uronamides enhances selectivity for A3 adenosine receptors', J Med Chem, 37(21), pp. 3614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M and Hartl FU (2013) 'Molecular chaperone functions in protein folding and proteostasis', Annu Rev Biochem, 82, pp. 323–55. [DOI] [PubMed] [Google Scholar]
- Kityk R, Kopp J, Sinning I and Mayer MP (2012) 'Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones', Mol Cell, 48(6), pp. 863–74. [DOI] [PubMed] [Google Scholar]
- Knappe D, Piantavigna S, Hansen A, Mechler A, Binas A, Nolte O, Martin LL and Hoffmann R (2010) 'Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens', J Med Chem, 53(14), pp. 5240–7. [DOI] [PubMed] [Google Scholar]
- Knight ZA and Shokat KM (2005) 'Features of selective kinase inhibitors', Chem Biol, 12(6), pp. 621–37. [DOI] [PubMed] [Google Scholar]
- Knipfer N, Seth A, Roudiak SG and Shrader TE (1999) 'Species variation in ATP-dependent protein degradation: protease profiles differ between mycobacteria and protease functions differ between Mycobacterium smegmatis and Escherichia coli', Gene, 231(1-2), pp. 95–104. [DOI] [PubMed] [Google Scholar]
- Kragol G, Hoffmann R, Chattergoon MA, Lovas S, Cudic M, Bulet P, Condie BA, Rosengren KJ, Montaner LJ and Otvos L (2002) 'Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin', Eur J Biochem, 269(17), pp. 4226–37. [DOI] [PubMed] [Google Scholar]
- Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R and Otvos L (2001) 'The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding', Biochemistry, 40(10), pp. 3016–26. [DOI] [PubMed] [Google Scholar]
- Li J, Qian X and Sha B (2009) 'Heat shock protein 40: structural studies and their functional implications', Protein Pept Lett, 16(6), pp. 606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ER, Sun D and Gestwicki JE (2013) 'Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents', ACS Med Chem Lett, 4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukačišinová M, Fernando B and Bollenbach T (2020) 'Highly parallel lab evolution reveals that epistasis can curb the evolution of antibiotic resistance', Nat Commun, 11(1), pp. 3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupoli TJ, Fay A, Adura C, Glickman MS and Nathan CF (2016) 'Reconstitution of a Mycobacterium tuberculosis proteostasis network highlights essential cofactor interactions with chaperone DnaK', Proc Natl Acad Sci U S A, 113(49), pp. E7947–E7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupoli TJ, Vaubourgeix J, Burns-Huang K and Gold B (2018) 'Targeting the Proteostasis Network for Mycobacterial Drug Discovery', ACS Infect Dis, 4(4), pp. 478–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey AJ (2010) 'ATPases as drug targets: insights from heat shock proteins 70 and 90', J Med Chem, 53(20), pp. 7280–6. [DOI] [PubMed] [Google Scholar]
- Mattingly AE, Cox KE, Smith R, Melander RJ, Ernst RK and Melander C (2020) 'Screening an Established Natural Product Library Identifies Secondary Metabolites That Potentiate Conventional Antibiotics', ACS Infect Dis, 6(10), pp. 2629–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Schröder H, Rüdiger S, Paal K, Laufen T and Bukau B (2000) 'Multistep mechanism of substrate binding determines chaperone activity of Hsp70', Nat Struct Biol, 7(7), pp. 586–93. [DOI] [PubMed] [Google Scholar]
- McGovern SL, Helfand BT, Feng B and Shoichet BK (2003) 'A specific mechanism of nonspecific inhibition', J Med Chem, 46(20), pp. 4265–72. [DOI] [PubMed] [Google Scholar]
- Melero R, Moro F, Pérez-Calvo M, Perales-Calvo J, Quintana-Gallardo L, Llorca O, Muga A and Valpuesta JM (2015) 'Modulation of the chaperone DnaK allosterism by the nucleotide exchange factor GrpE', J Biol Chem, 290(16), pp. 10083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W, Clerico EM, McArthur N and Gierasch LM (2018) 'Allosteric landscapes of eukaryotic cytoplasmic Hsp70s are shaped by evolutionary tuning of key interfaces', Proc Natl Acad Sci U S A, 115(47), pp. 11970–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerke NJ (2009) 'Fluorescence Polarization (FP) Assays for Monitoring Peptide-Protein or Nucleic Acid-Protein Binding', Curr Protoc Chem Biol, 1(1), pp. 1–15. [DOI] [PubMed] [Google Scholar]
- Moro F, Fernández V and Muga A (2003) 'Interdomain interaction through helices A and B of DnaK peptide binding domain', FEbS Lett, 533(1-3), pp. 119–23. [DOI] [PubMed] [Google Scholar]
- Muona M, Aranko AS and Iwai H (2008) 'Segmental isotopic labelling of a multidomain protein by protein ligation by protein trans-splicing', Chembiochem, 9(18), pp. 2958–61. [DOI] [PubMed] [Google Scholar]
- Nikaido H and Jarlier V (1991) 'Permeability of the mycobacterial cell wall', Res Microbiol, 142(4), pp. 437–43. [DOI] [PubMed] [Google Scholar]
- Nillegoda NB and Bukau B (2015) 'Metazoan Hsp70-based protein disaggregases: emergence and mechanisms', Front Mol Biosci, 2, pp. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitika, Blackman JS, Knighton LE, Takakuwa JE, Calderwood SK and Truman AW (2020) 'Chemogenomic screening identifies the Hsp70 co-chaperone DNAJA1 as a hub for anticancer drug resistance', Sci Rep, 10(1), pp. 13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah ME, Gallo-Rodriguez C, Jacobson KA and Stiles GL (1994) '125I-4-aminobenzyl-5'-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor', Mol Pharmacol, 45(5), pp. 978–82. [PMC free article] [PubMed] [Google Scholar]
- Patel HV, Li M and Seeliger JC (2019) 'Opportunities and Challenges in Activity-Based Protein Profiling of Mycobacteria', Curr Top Microbiol Immunol, 420, pp. 49–72. [DOI] [PubMed] [Google Scholar]
- Preissler S, Rohland L, Yan Y, Chen R, Read RJ and Ron D (2017) 'AMPylation targets the rate-limiting step of BiP's ATPase cycle for its functional inactivation', Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN and Gestwicki JE (2014) 'Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro', J Biol Chem, 289(3), pp. 1402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci L and Williams KP (2008) 'Development of fluorescence polarization assays for the molecular chaperone Hsp70 family members: Hsp72 and DnaK', Curr Chem Genomics, 2, pp. 90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock J (2019) 'Tuberculosis drug discovery in the CRISPR era', PLoS Pathog, 15(9), pp. e1007975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, Gozman A, Patel MR, Patel HJ, Chirico W, Erdjument-Bromage H, Talele TT, Young JC and Chiosis G (2013) 'Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein', Chem Biol, 20(12), pp. 1469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi AM and Taylor CW (2011) 'Analysis of protein-ligand interactions by fluorescence polarization', Nat Protoc, 6(3), pp. 365–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger S, Buchberger A and Bukau B (1997) 'Interaction of Hsp70 chaperones with substrates', Nat Struct Biol, 4(5), pp. 342–9. [DOI] [PubMed] [Google Scholar]
- Rüdiger S, Germeroth L, Schneider-Mergener J and Bukau B (1997) 'Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries', EMBO J, 16(7), pp. 1501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbeng EB, Liu Q, Tian X, Yang J, Li H, Wong JL and Zhou L (2015) 'A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein', J Biol Chem, 290(14), pp. 8849–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH and Rubin EJ (2003) 'Genes required for mycobacterial growth defined by high density mutagenesis', Mol Microbiol, 48(1), pp. 77–84. [DOI] [PubMed] [Google Scholar]
- Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, Bomke J, Eggenweiler HM, Mayer MP and Bukau B (2013) 'Functional analysis of Hsp70 inhibitors', PLoS One, 8(11), pp. e78443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scocchi M, Tossi A and Gennaro R (2011) 'Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action', Cell Mol Life Sci, 68(13), pp. 2317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sell SM, Eisen C, Ang D, Zylicz M and Georgopoulos C (1990) 'Isolation and characterization of dnaJ null mutants of Escherichia coli', J Bacteriol, 172(9), pp. 4827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Hsiao FS, Ho YH and Chen CS (2016) 'The proteome targets of intracellular targeting antimicrobial peptides', Proteomics, 16(8), pp. 1225–37. [DOI] [PubMed] [Google Scholar]
- Shao H, Li X, Moses MA, Gilbert LA, Kalyanaraman C, Young ZT, Chernova M, Journey SN, Weissman JS, Hann B, Jacobson MP, Neckers L and Gestwicki JE (2018) 'Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70)', J Med Chem, 61(14), pp. 6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D and Masison DC (2009) 'Hsp70 structure, function, regulation and influence on yeast prions', Protein Pept Lett, 16(6), pp. 571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Georgopoulos C and Zylicz M (1990) 'The E. coli dnaK gene product, the hsp70 homolog, can reactivate heat-inactivated RNA polymerase in an ATP hydrolysis-dependent manner', Cell, 62(5), pp. 939–44. [DOI] [PubMed] [Google Scholar]
- Swain JF, Schulz EG and Gierasch LM (2006) 'Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states', J Biol Chem, 281(3), pp. 1605–11. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF and Lindquist S (2010) 'HSP90 at the hub of protein homeostasis: emerging mechanistic insights', Nat Rev Mol Cell Biol, 11(7), pp. 515–28. [DOI] [PubMed] [Google Scholar]
- Taldone T, Kang Y, Patel HJ, Patel MR, Patel PD, Rodina A, Patel Y, Gozman A, Maharaj R, Clement CC, Lu A, Young JC and Chiosis G (2014) 'Heat shock protein 70 inhibitors. 2. 2,5'-thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70', J Med Chem, 57(4), pp. 1208–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneva SG, Moro F, Velázquez-Campoy A and Muga A (2010) 'Energetics of nucleotide-induced DnaK conformational states', Biochemistry, 49(6), pp. 1338–45. [DOI] [PubMed] [Google Scholar]
- Tapley TL, Cupp-Vickery JR and Vickery LE (2005) 'Sequence-dependent peptide binding orientation by the molecular chaperone DnaK', Biochemistry, 44(37), pp. 12307–15. [DOI] [PubMed] [Google Scholar]
- Taylor IR, Ahmad A, Wu T, Nordhues BA, Bhullar A, Gestwicki JE and Zuiderweg ERP (2018) 'The disorderly conduct of Hsc70 and its interaction with the Alzheimer's-related Tau protein', J Biol Chem, 293(27), pp. 10796–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IR, Assimon VA, Kuo SY, Rinaldi S, Li XK, Young ZT, Morra G, Green K, Nguyen D, Shao H, Garneau-Tsodikova S, Colombo G and Gestwicki JE (2020) 'Tryptophan scanning mutagenesis as a way to mimic the compound-bound state and probe the selectivity of allosteric inhibitors in cells', Chemical Science, 11(7), pp. 1892–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrab L and Wipf P (2020) 'Hsp70 and the Unfolded Protein Response as a Challenging Drug Target and an Inspiration for Probe Molecule Development', ACS Med Chem Lett, 11(3), pp. 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchette MH and Seeliger JC (2017) 'Transport of outer membrane lipids in mycobacteria', Biochim Biophys Acta Mol Cell Biol Lipids, 1862(11), pp. 1340–1354. [DOI] [PubMed] [Google Scholar]
- Tsuchido T and Takano M (1988) 'Sensitization by heat treatment of Escherichia coli K-12 cells to hydrophobic antibacterial compounds', Antimicrob Agents Chemother, 32(11), pp. 1680–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzankov S, Wong MJ, Shi K, Nassif C and Young JC (2008) 'Functional divergence between co-chaperones of Hsc70', J Biol Chem, 283(40), pp. 27100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Parzych KR, Dinh T and Bernhardt TG (2010) 'Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis', EMBO J, 29(8), pp. 1412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L and Lindquist S (2013) 'Fitness trade-offs restrict the evolution of resistance to amphotericin B', PLoS Biol, 11(10), pp. e1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M, Bukau B and Mayer MP (2006) 'Allosteric regulation of Hsp70 chaperones by a proline switch', Mol Cell, 21(3), pp. 359–67. [DOI] [PubMed] [Google Scholar]
- Wang M, Shen A, Zhang C, Song Z, Ai J, Liu H, Sun L, Ding J, Geng M and Zhang A (2016) 'Development of Heat Shock Protein (Hsp90) Inhibitors To Combat Resistance to Tyrosine Kinase Inhibitors through Hsp90-Kinase Interactions', J Med Chem, 59(12), pp. 5563–86. [DOI] [PubMed] [Google Scholar]
- Wei J, Gaut JR and Hendershot LM (1995) 'In vitro dissociation of BiP-peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis', J Biol Chem, 270(44), pp. 26677–82. [DOI] [PubMed] [Google Scholar]
- Welch NG, Li W, Hossain MA, Separovic F, O'Brien-Simpson NM and Wade JD (2020) '(Re)Defining the Proline-Rich Antimicrobial Peptide Family and the Identification of Putative New Members', Front Chem, 8, pp. 607769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Santagata S, Mendillo ML, Lin NU, Proia DA and Lindquist S (2014) 'HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models', Proc Natl Acad Sci U S A, 111(51), pp. 18297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD (2016) 'Antibiotic Adjuvants: Rescuing Antibiotics from Resistance', Trends Microbiol, 24(11), pp. 862–871. [DOI] [PubMed] [Google Scholar]
- Wu S, Hong L, Wang Y, Yu J, Yang J, Zhang H and Perrett S (2020) 'Kinetics of the conformational cycle of Hsp70 reveals the importance of the dynamic and heterogeneous nature of Hsp70 for its function', Proc Natl Acad Sci U S A, 117(14), pp. 7814–7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn M, Berthold N, Kieslich B, Knappe D, Hoffmann R and Sträter N (2013) 'Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK', J Mol Biol, 425(14), pp. 2463–79. [DOI] [PubMed] [Google Scholar]
- Zhang P, Leu JI, Murphy ME, George DL and Marmorstein R (2014) 'Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate', PLoS One, 9(7), pp. e103518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME and Hendrickson WA (1996) 'Structural analysis of substrate binding by the molecular chaperone DnaK', Science, 272(5268), pp. 1606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A and Gierasch LM (2015) 'Substrate-binding domain conformational dynamics mediate Hsp70 allostery', Proc Natl Acad Sci U S A, 112(22), pp. E2865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE and Ahmad A (2013) 'Allostery in the Hsp70 chaperone proteins', Top Curr Chem, 328, pp. 99–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. SI_Protein_MSMS_data: Data from described mass spectrometry analysis on Mtb DnaK crosslinked to peptide analog 7. Related to Figures 5 and S5, and Table S6.
Data Availability Statement
Small molecule screening data have been deposited in OSF and are publicly available as of the date of publication. Link is listed in the key resources table. Mass spectrometry-based proteomics data have been deposited in MassIVE and are publicly available as of the date of publication. The accession number is listed in the key resources table. Analyses of mass spectrometry data is present in a Supplemental data file.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli Mach1 | ThermoFisher | Cat# C862003 |
| Escherichia coli Rosetta2 (DE3) | Millipore Sigma/Novagen | Cat# 71400-3 |
| Escherichia coli Rosetta2 (DE3) pLysS | Millipore Sigma/Novagen | Cat# 71403-3 |
| Mycobacterium smegmatis wild-type (mc2155) | Glickman Lab (Memorial Sloan Kettering) | N/A |
| Mycobacterium smegmatis Δdnaj1 | Lupoli et al., 2016 | N/A |
| Mycobacterium smegmatis Δdnaj2 | Lupoli et al., 2016 | N/A |
| Mycobacterium smegmatis ΔdnaK | Fay & Glickman, 2014 | N/A |
| M. smegmatis ΔdnaK attB::Mtb dnaK | This work | N/A |
| Escherichia coli strains for protein overexpression, see Table S3 | Various | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fmoc-protected natural amino acids | ChemImpex | N/A |
| Fmoc-L-Photo-Leu (Fmoc- photoLeu-OH) | Sigma-Aldrich | CAS: 1360651-24-6 |
| Fmoc-L-tert-leucine (Fmoc-Tle-OH) | ChemImpex | CAS: 132684-60-7 |
| Fmoc-L-cyclohexyl-glycine (Fmoc-Chg-OH) | ChemImpex | CAS: 161321-36-4 |
| Fmoc-L-β-alanine (Fmoc-β-Ala-OH/Fmoc-βA-OH) | ChemImpex | CAS: 35737-10-1 |
| Pyrazine-2-carboxylic acid (Pz) | Sigma-Aldrich | CAS: 98-97-5 |
| Fluorescein isothiocyanate isomer 1 (FL-NCS) | ChemImpex | CAS: 3326-32-7 |
| 4-Benzoylbenzoic Acid (4-BBA) | Sigma-Aldrich | CAS: 611-95-0 |
| Telaprevir | Cayman Chemical | CAS: 402957-28-2 |
| Simeprevir | Cayman Chemical | CAS: 1241946-89-3 |
| Boceprevir | Cayman Chemical | CAS: 394730-60-0 |
| Grazoprevir | Cayman Chemical | CAS: 1350514-68-9 |
| Velpatasvir | Cayman Chemical | CAS: 1377049-84-7 |
| Danoprevir | Celia Schiffer (UMass Medical School) | CAS: 850876-88-9 |
| Paritaprevir | Celia Schiffer (UMass Medical School) | CAS: 1216941-48-8 |
| 2-Cl-IB-MECA | Cayman Chemical | CAS: 163042-96-4 |
| AB-MECA | Cayman Chemical | CAS: 152918-26-8 |
| VER 155008 | Cayman Chemical | CAS: 1134156-31-2 |
| NH2-Asn-Arg-Leu-Leu-Leu-Thr-Gly-OH | Biomatik | N/A |
| FL-β-Ala-Asn-Arg-Leu-Leu-Leu-Thr-Gly-OH | This work | N/A |
| Pz-Chg-Tle-Pro-Leu-OH (1) | This work | N/A |
| Pz-Val-Val-Pro-Leu-OH (2) | This work | N/A |
| 4BB-Val-Val-Pro-Leu-OH (3) | This work | N/A |
| Pz-Chg-Tle-Pro-Leu-OMe (4) | This work | N/A |
| Pz-Val-Val-Pro-Leu-OMe (5) | This work | N/A |
| 4BB-Val-Val-Pro-Leu-OMe (6) | This work | N/A |
| Pz-Val-Val-Pro-photoLeu-OMe (7) | This work | N/A |
| Firefly Luciferase: QuantiLum Recombinant Luciferase | Promega | Cat# E1701 |
| Critical commercial assays | ||
| Luciferase Assay System | Promega | Cat# E1500 |
| ADP-Glo Kinase System | Promega | Cat# V6930 |
| Kinase-Glo Luminescent Kinase Assay | Promega | Cat# V6711 |
| Deposited data | ||
| Raw small molecule screening data from pilot and primary screens | This paper; OSF | https://osf.io/cmqxd/?view_only=a2e3d69e9d404bd598f9f6475b042a77 |
| Analysis of mass spectrometry data of proteolyzed DnaK crosslinked to probe | This paper; SI file | N/A |
| Mass spectrometry raw files of proteolyzed DnaK crosslinked to probe | MassIVE | https://massive.ucsd.edu; MassIVE ID: MSV000088127 |
| Oligonucleotides | ||
| Primers for cloning bacterial and human chaperones into overexpression vectors, see Table S1 | This paper | N/A |
| Primers for inserting mutations into DnaK, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pETHisSUMO | Gift from Scott Gradia, University of California, Berkeley | Addgene Plasmid #29711 |
| Plasmid: pHYRS52: overexpression vector for Ulp1 | Gift from Hideo Iwai, University of Helsinki; Muona, M. et al. 2008 | Addgene Plasmid #31122 |
| Plasmid: pET-47b(+) | Millipore Sigma/Novagen | Cat# 71461-3 |
| Plasmid: pcDNA5/FRT/TO HIS HSPA8: contains gene encoding Hsc70 | Gift from Harm Kampinga, University of Groningen; Hageman, J. et al. 2009 | Addgene Plasmid #19541 |
| Plasmid: pcDNA5/FRT/TO DNAJA2: contains gene encoding DnaJA2 | Gift from Harm Kampinga, University of Groningen; Hageman, J. et al. 2009 | Addgene Plasmid #19465 |
| Plasmid: pAJF465: M. tuberculosis dnaK expression in mycobacteria | This work | N/A |
| Plasmids for overexpression of bacterial and human chaperones and mutants in E. coli, see Table S2 | This work | N/A |
| Software and algorithms | ||
| Prism 9 | Mathpad | https://www.graphpad.com/scientific-software/prism/ |
| AFFINImeter | AFFINImeter | https://www.affinimeter.com/site/ |
| Chromeleon 7 | ThermoFisher Scientific | https://www.thermofisher.com/order/catalog/product/CHROMELEON7#/CHROMELEON7 |
| PyMOL 2.0 | Schrodinger | https://www.schrodinger.com/products/pymol |
| Image Lab | Biorad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Byonic | Protein Metrics | https://proteinmetrics.com/resources/byonic-advanced-peptide-and-protein-identification-software/ |
| Other | ||
| Compound libraries used for small molecule screens | Various sources; assembled by The Rockefeller University High Throughput and Spectroscopy Center | https://www.rockefeller.edu/htsrc/libraries/ |