

Abstract
Molibresib (GSK525762) is an investigational orally bioavailable small‐molecule bromodomain and extraterminal (BET) protein inhibitor for the treatment of advanced solid tumors. In the first‐time‐in‐human BET115521 study of molibresib in patients with solid tumors, thrombocytopenia was the most frequent treatment‐related adverse event (AE), QT prolongation was an AE of special interest based on preclinical signals, and gastrointestinal (GI) AEs (nausea, vomiting, diarrhea, and dysgeusia) were often observed. The aims of this analysis were the following: (i) develop a population pharmacokinetic (PK)/pharmacodynamic (PD) model capable of predicting platelet time courses in individual patients after administration of molibresib and identify covariates of clinical interest; (ii) evaluate the effects of molibresib (and/or its two active metabolites [GSK3529246]) exposure on cardiac repolarization by applying a systematic modeling approach using high‐quality, intensive, PK time‐matched 12‐lead electrocardiogram measurements; (iii) evaluate the exposure–response (ER) relationship between molibresib and/or GSK3529246 exposures and the occurrence of Grade 2 or higher GI AEs. Overall, the PK/PD model (including a maximal drug effect model and molibresib concentration) adequately described platelet counts following molibresib treatment and was used to simulate the impact of molibresib dosing on thrombocytopenia at different doses and regimens. ER analyses showed no clinically meaningful QT interval prolongation with molibresib at up to 100 mg q.d., and no strong correlation between molibresib exposure and the occurrence of Grade 2 or higher GI AEs. The models described here can aid dosing/schedule and drug combination strategies and may support a thorough QT study waiver request for molibresib.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
In a first‐time‐in‐human study of molibresib in advanced solid tumors, thrombocytopenia and gastrointestinal (GI) adverse events (AEs) were commonly observed. As QT prolongation was observed in dogs, pharmacokinetic (PK) time‐matched 12‐lead electrocardiogram measurements were obtained. A population PK/pharmacodynamic (PD) model of molibresib and its active metabolite composite (GSK3529246) is also available.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study evaluates relationship between molibresib, GSK3529246, or total active moiety (TAM) exposure and thrombocytopenia, QTc prolongation, or GI AEs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Semimechanistic PK/PD and exposure–response models adequately described the relationship between TAM exposures and thrombocytopenia as well as GSK3529246 exposures and QTc prolongation. QTc analysis demonstrated that molibresib does not affect QTc prolongation at the clinically relevant dose. No exposure measures were associated with the occurrence of Grade 2 or higher GI AEs.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
These models can be used to optimize molibresib dosing/schedules and design combination studies to minimize the risk of thrombocytopenia. These analyses extend the knowledge of modeling for drugs with active metabolites.
INTRODUCTION
Molibresib (GSK525762) is an orally bioavailable, small‐molecule bromodomain and extraterminal (BET) protein inhibitor 1 , 2 investigated for the treatment of advanced cancers. The recently completed two‐part, first‐time‐in‐human (FTIH) phase I/II study in patients with nuclear protein in testis carcinoma (NC) 3 and other solid tumors evaluated the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of molibresib and showed preliminary antitumor activity with this agent in NC (NCT01587703; BET115521; S Cousins, J‐Y Blay, IB Garcia, et al., unpublished data, 2021). 4 Molibresib is eliminated rapidly with an average terminal phase half‐life of 3–6 h and is metabolized by cytochrome P450 3A4 enzymes to produce two major active metabolites that are equipotent to the parent molecule. 4 The metabolites are measured together after full conversion of one to the other and reported as an active metabolite composite (GSK3529246). 5 A semi‐mechanistic liver‐compartment population PK model including the autoinduction of molibresib clearance was recently developed using the PK data from the FTIH study, which adequately described the PK of both molibresib and GSK3529246. 5
In the FTIH study, thrombocytopenia (TCP) was the most common treatment‐related adverse event (AE) and the most common reason for treatment interruptions, dose reductions, and discontinuations. 4 TCP is an established clinical effect of BET inhibition 4 and a common challenge with many chemotherapies and radiotherapies. 6 Therefore, a better understanding of platelet kinetics in patients receiving molibresib is needed to evaluate potential ways to reduce the risk of this AE, such as alternate dosing or individualized dosing regimens. Development of a PK/PD model of platelet kinetics following molibresib treatment could achieve this goal and thereby help to improve patient outcomes. A semi‐mechanistic mathematical model developed by Friberg et al. is often used to describe platelet kinetics after treatment with chemotherapy and has been applied to various chemotherapeutic agents. 7 , 8 , 9 The Friberg model provided an appropriate initial structure for the development of such a PK/PD model.
The International Conference on Harmonization (ICH) issued guidance (E14) in 2005 specifying that all experimental drugs must undergo a dedicated “thorough QTc” (TQT) study to assess drug‐induced QTc prolongation. 10 , 11 As a result, phase I studies are now being designed to integrate high‐quality, intensive, PK time‐matched 12‐lead electrocardiogram (ECG) measurements using exposure–response (ER) modeling to detect QTc liability early. 11 , 12 , 13 , 14 , 15 , 16 , 17 The use of drug concentration‐QTc (C‐QTc) modeling at different phases of drug development can influence the need for and/or timing of a TQT study. 18 QT prolongation was observed for molibresib (unpublished results) in dogs after a single oral dose of 30 mg/kg or daily repeat oral and intravenous dosing at ≥1 mg/kg/day, with a maximum prolongation of 41 milliseconds following 12 doses of 3 mg/kg/day orally (mean pre‐ and post‐treatment difference). Given the risks of QTc prolongation, patients in the FTIH study were monitored closely for any changes in QTc with triplicate 12‐lead ECG measurements and time‐matched exposures of molibresib, GSK3529246, and total active moiety (TAM; molibresib + GSK3529246) as well as change from baseline in QT interval corrected for heart rate by Fridericia's formula (dQTcF). The C‐QTc analysis was performed for a wide range of doses to account for changes in drug exposures attributed to drug–drug interactions (DDIs), organ impairment, or other special population effects. The following differences specific to oncology therapies were also taken into account: (i) based on recent publications or guidance, a QT prolongation threshold of a <20 millisecond change is considered appropriate for oncology drugs, 19 , 20 , 21 , 22 and (ii) although regulatory authorities typically request an investigation of three times the therapeutic dose to assess any effect on QT interval, for many cancer drugs this is not possible as the therapeutic dose may be similar to the maximum tolerated dose.
Additionally, gastrointestinal (GI) AEs such as nausea, dysgeusia, diarrhea, and vomiting were among the most frequent treatment‐related AEs during the FTIH study, in some cases leading to dose reductions and/or interruptions. 4 The development of models describing the PK/PD relationships between TCP and molibresib treatment, as well as the ER relationships between molibresib and/or GSK3529246 and the occurrence of QT prolongation or GI AEs, will be supportive for risk mitigation and dose‐optimization strategies.
The three main objectives of these analyses were to develop a semi‐mechanistic population PK/PD platelet model to assess the occurrence of TCP in patients with solid tumors treated with molibresib and identify important covariates associated with the occurrence of TCP to guide molibresib dosing strategies, to develop ER models for the relationship between molibresib/GSK3529246 exposure and change in corrected QT interval by Frederica (QTcF), and evaluate the ER relationship between molibresib/GSK3529246 exposure and the occurrence of selected Grade 2 or higher (Grade 2+) GI AEs (nausea, dysgeusia, diarrhea, and vomiting).
METHODS
Patients and study design
This analysis was performed using clinical data obtained from the multicenter, open‐label, two‐part, phase I/II FTIH study BET115521 investigating the use of molibresib in patients with NC and other solid tumors. Details on the design and methodology for the FTIH study have been published previously. 4 Briefly, Part 1 of the study was a dose‐escalation phase involving single‐dose and repeat‐dose oral administration of 2–100 mg molibresib as an amorphous free‐base formulation to evaluate the safety, PK, and PD of molibresib and to determine the recommended phase II dose (RP2D). 4 Part 2 of the study assessed the efficacy, safety, PK, and PD of molibresib as a besylate salt formulation at the RP2D of 75 mg once daily (S Cousins, J‐Y Blay, IB Garcia, et al., unpublished data, 2021). The study was conducted in accordance with ICH Good Clinical Practice and applicable country‐specific regulatory requirements as well as the ethical principles outlined in the 2008 Declaration of Helsinki. All patients provided written, informed consent prior to study enrollment.
Population PK model
Recently, a semimechanistic liver‐compartment population PK model including autoinduction of molibresib clearance was developed that adequately describes the PK of both molibresib and GSK3529246. 5 Covariate analysis indicated that body weight had a significant effect on the volume of distribution of molibresib and GSK3529246 and that higher levels of aspartate aminotransferase (AST) resulted in lower clearance of GSK3529246. For all patients in the analyses presented here, the individual post hoc PK parameter estimates (maximum plasma concentration [Cmax], trough plasma concentration [Cmin], and area under the concentration‐time curve [AUC0–24 h]) for molibresib, GSK3529246, and TAM were derived using this population PK model taking into account individual dosing history and clinical covariates of interest (baseline body weight, time‐varying AST). The population PK model was used to obtain individual molibresib, GSK3529246, and TAM plasma concentrations for timepoints where ECG and platelet data were also obtained.
Modeling software
The platelet analysis was performed using a nonlinear mixed‐effect modeling approach using NONMEM Version 7.4 (Icon Development Solutions). Model development was performed using first‐order conditional estimation with η–ϵ interaction (FOCE‐I). Model execution and visual predictive checks (VPCs) were performed using Perl‐speaks‐NONMEM Version 4.8.0. 23 Postprocessing of NONMEM analysis data was carried out in R Version 3.5.2, as was the analysis of QT interval prolongation and GI AEs. 24
Model diagnostics and qualification
During model development, a difference in objective function value (OFV) of 6.63 (equivalent to a p < 0.01; change in OFV [ΔOFV] of 10.8, nominal p < 0.001 for backwards elimination) was used to compare any two nested models that differed by one parameter. The accepted model was then determined on the basis of the lowest stable OFV, physiological plausibility of parameter values, successful numerical convergence, parameter precision (a relative standard error <50%), a low condition number (<1000), graphical goodness‐of‐fit (GOF) analyses, and acceptable prediction‐corrected VPC outcomes. 25 Graphical GOF analysis involved inspecting diagnostic plots of observed versus predicted values to determine any evidence of systemic lack of fit or bias in the error distributions. Prediction‐corrected VPCs were stratified by different covariates wherever applicable, with 90% confidence intervals (CIs).
Platelet modeling
Data assembly and handling
Blood samples for observed platelet count assessments were obtained at regular intervals throughout the FTIH study, with additional monitoring as clinically indicated. The platelets were assessed using data from both Part 1 and Part 2 of the study, including both once daily (q.d.) and twice daily (b.i.d.) dosing. Laboratory samples were obtained during Part 1 at screening and (Week x, Day x), W1D1, W1D6, W2D1, W2D6, W3D1, W4D1, W5D1, W7D1, W9D1, W11D1, and every 4 weeks on Day 1 (q4WD1) from Week 9, and at the end of treatment. In Part 2, samples were obtained at screening, W1D1, W2D1, W2D6, W3D1, W4D1, W5D1, W7D1, W9D1, W11D1, W13D1, q4WD1 from Week 9, and the end of treatment. The dosing was continued at the same dose level in case of Grade 1 and 2 TCP, with weekly or more frequent monitoring as necessary. In cases of Grade 3 TCP, the same dose, a reduced dose, or an alternate‐day dosing was recommended, with weekly or more frequent monitoring as necessary. In cases of Grade 4 TCP, temporary interruption of molibresib was recommended with monitoring every 2–3 days until the counts recovered to Grade 3 for at least two complete blood count reads at least 3 days apart. No imputations were performed for missing data; observations with missing platelet or time values were excluded from the analysis. PK parameter estimates were obtained as described in the ‘Population PK Model’ section.
Model development
Models were developed in increasing order of complexity using FOCE‐I. The established semi‐mechanistic Friberg myelosuppression model was used as the starting model, 7 with baseline platelet count, maturation rate constant (ktr), and feedback power term (γ) as the system‐related parameters and assuming treatment with molibresib resulted in a direct loss of megakaryocytes. The effects of molibresib, GSK3529246, and TAM on megakaryocyte proliferation were evaluated using linear and Maximal drug effect models. This was optimized in a stepwise fashion to adequately describe the platelet data from all parts of the FTIH study until further improvement in the platelet model fit was not supported by the data. Finally, random effects (residual variability and interindividual variability [IIV]) were evaluated.
Covariate analysis
Once the structural platelet model had been established, a generalized additive model (GAM) approach was used to identify clinical covariates of interest. The covariates evaluated were selected based on prior experience and scientific plausibility, and included observed baseline platelet count, dose, dosing regimen, age, sex, weight, baseline body surface area, baseline body mass index (BMI), cancer type, baseline hemoglobin, baseline Eastern Cooperative Oncology Group status, prior platinum‐based chemotherapy, and prior taxane‐based chemotherapy on the model parameters baseline platelet count, ktr, and γ. Also, the covariates dose, dosing regimen, age, sex, race, weight, baseline body surface area, baseline BMI, baseline hemoglobin, prior platinum‐based chemotherapy, and prior taxane‐based chemotherapy were evaluated for their impact on the drug/metabolite concentration required to provide half the maximal response (EC50). All significant covariates (p < 0.01; decrease in Akaike information criterion [AIC]) identified by the GAM were subsequently included in the model in a manual forward inclusion step, followed by a backwards elimination step to identify the statistically significant (p < 0.001) covariates.
Simulation of Grade 3 or 4 TCP
The final model was used to simulate the impact of TAM concentration on platelet count profiles for molibresib doses of 20 mg q.d., 60 mg q.d., 80 mg q.d., and 100 mg q.d. In total, 100 studies with 400 virtual patients (100 at each dose) were simulated using the PRIOR MWPRI functionality in NONMEM to account for uncertainty in parameter estimates, and the predicted incidence of Grade 3 or Grade 4 TCP was presented.
dQTcF modeling
Data assembly and handling
In Part 1 of the FTIH study, ECGs were obtained at the same time as PK samples from standard 12‐lead ECGs in 94 patients (65 received q.d. molibresib dosing, 19 received b.i.d. dosing, 10 were included in the besylate substudy). Four triplicate ECGs were evaluated at each timepoint by a central cardiologist, and the mean values were calculated for dQTcF using a single baseline observation (Week 1, Day 1 pre‐dose) per individual. 26 PK parameter estimates were obtained as described in the ‘Population PK Model’ section. Individual predicted plasma concentrations, rather than observed plasma concentrations, were used in the analysis as PK samples were not collected for all timepoints where dQTcF data were available.
Model development and covariate analysis
Initially, dQTcF was modeled as an effect of molibresib, GSK3529246, or TAM concentrations using linear mixed‐effects models according to the process outlined by Garnett et al. 18 using a target model structure outlined in Figure S1a. The dQTcF model was then constructed by the stepwise inclusion of individual terms and selection of the most appropriate model at each stage based on ΔOFV, GOF plots, model stability, and clinical plausibility of the parameter estimates. Our approach differed from that used by Garnett et al. 18 in that the circadian effects on dQTcF were tested during the development of the structural model rather than as a categorical effect of time, and a treatment‐specific intercept (θ 1) was not estimated because there was no placebo arm in the study. Once the structural dQTcF model had been determined, various covariates were considered using GAM. Interoccasion variability (IOV) was considered in the model because of the long molibresib treatment period. The structure of the term(s) for IOV was determined by the results of exploratory analysis conducted before modeling, and the effects of disease type, weight, and sex were considered as motivated by corresponding random effects plots.
Simulation of dQTcF effects
The final dQTcF ER model was subsequently used to evaluate dQTcF prolongation at the mean Cmax for molibresib doses of 60, 80, and 100 mg by GSK3529246 concentration and dosing occasion (e.g., Week 1, Day 1). Predicted response and a two‐sided 90% prediction interval (PI; representing the parameter uncertainty for a typical population value) was determined for the Cmax in the 60, 80, or 100 mg treatment group. The PI was determined for each value of exposure as the 90 percentiles of 1000 responses calculated with 1000 multivariate‐normal samples of parameter estimates from the variance–covariance matrix of the final dQTcF model. For diagnostic purposes, mean (and 90% CI) of observed values were overlaid on mean (and 90% CI) model predictions, stratified by occasion.
GI AE modeling
For GI AEs (nausea, dysgeusia, diarrhea, and vomiting), the date of the first Grade 2+ event for each patient was extracted. Derived exposure metrics obtained as described in the ‘Population PK Model’ section were then merged with the AE data by treatment date and restricted to the first occurrence within patient of Grade 2+ GI AEs. Logistic regression (a generalized linear model for binomial family) was used to describe the relationship between the first occurrence of each Grade 2+ treatment‐related GI AE and the exposure metrics. Probability of AEs of Grade 2+ was modeled as a function of exposure associated with the date of the event or, for those patients not experiencing the event or experiencing a Grade 1 event, date of last instance of the highest administered dose.
RESULTS
Analysis population
Demographics, treatments, and clinical characteristics at baseline for patients included in the FTIH study are presented in Table S1. Overall, 193 patients included in the analysis had a median age of 58 years (range, 16–86 years) and were predominantly White (83%) with a median body weight of 69.6 kg (range, 34–120 kg).
Platelet modeling
Exploratory analysis
In total, 2526 platelet count observations from the 193 patients were included in the analysis for model development (Table 1). Exploratory analysis of platelet count versus time profiles revealed a large day‐to‐day within‐patient variability, often related to frequent dose interruptions during treatment. Overall, there appeared to be a clear trend showing a reduction in platelet count over time with molibresib treatment (Figure S2).
TABLE 1.
Platelet and dQTcF observations (FTIH study) included in the modeling analysis
| Study cohort | Dose | N | Platelet obs. | dQTcF (rich) obs. | dQTcF (sparse) obs. |
|---|---|---|---|---|---|
| Part 1 q.d. a | 2 mg | 3 | 23 | 21 | 55 |
| 4 mg | 4 | 31 | 37 | 65 | |
| 8 mg | 1 | 7 | 14 | 20 | |
| 16 mg | 3 | 17 | 29 | 72 | |
| 30 mg | 4 | 43 | 46 | 81 | |
| 60 mg | 9 | 87 | 103 | 160 | |
| 80 mg | 32 | 544 | 296 | 543 | |
| 100 mg | 9 | 169 | 78 | 191 | |
| Part 1 b.i.d. a | 20 mg | 4 | 33 | 78 | 70 |
| 30 mg | 10 | 132 | 174 | 210 | |
| 40 mg | 5 | 63 | 95 | 97 | |
| Besylate substudy b | 80 mg | 10 | 160 | 0 | 171 |
| Part 2 b | 75 mg | 99 c | 1217 | 0 | 0 |
| Total | 193 | 2526 | 971 | 1735 |
Molibresib amorphous free‐base formulation.
Molibresib besylate salt formulation.
The total population for Part 2 of the FTIH study was 102; three patients were excluded from this modeling analysis.
Abbreviations: b.i.d., twice daily; dQTcF, QT interval prolongation (corrected by Fridericia formula and by baseline); FTIH, first time in humans; obs., observations; q.d., once daily.
Structural model development
The key structural model building steps are summarized in Table 2a, the parameter estimates are presented in Table 3, and the model code is provided in Supplementary Text S1. The final structural model adequately described the time course of the platelet count and all parameters were estimated with acceptable precision. The IIV estimates derived from the final structural model for baseline platelet count, ktr, γ, and EC50 were 35.4%, 12.9%, 50.6%, and 60.2%, respectively (Table 3a). The residual variability was included as a proportional error model.
TABLE 2.
Summary of (a) key platelet structural model development steps and (b) key dQTcF model development steps
| (a) Key platelet structural model development steps | |||||
|---|---|---|---|---|---|
| Run | Ref | ΔOFV | Minimization | Description | Action |
| 201 | – | Successful | Flat baseline model, IIV on baseline platelet count | Accepted | |
| 203 | 201 | −2147.2 | Successful | Linear effect of TAM on the rate of megakaryocyte loss | Accepted |
| 204 | 203 | −940.1 | Successful | IIV on ktr, γ, and drug effect slope | Accepted |
| 205 | 204 | −39.1 | Successful | TAM effect moved to platelet proliferation rate | Accepted |
| 209 | 205 | −73.9 | Successful | Inclusion of Emax model (Emax fixed to 1) | Final |
| 211 | 209 | −21.6 | Successful | Estimation of Emax | Rejected a |
| 212 | 211 | −5.7 | Successful | Addition of omega block (γ, baseline platelet count, EC50) | Rejected |
| (b) Key dQTcF model development steps | |||||
|---|---|---|---|---|---|
| Run | Ref | ∆OFV | N parameters | Description | Action |
| 1 | – | – | 2 | IIV(intercept) | Accepted |
| 2 | 1 | −3.6 | 3 | + Intercept | Accepted |
| 3 | 2 | 825.3 | 4 | + Circadian: IIV(tmax) | Rejected |
| 4 | 2 | 640.9 | 4 | + Circadian: IIV(amplitude) | Rejected |
| 5 | 2 | −15.8 | 4 | + DMQTCF | Accepted |
| 6 | 5 | −16.1 | 5 | + Linear molibresib | Rejected |
| 7 | 5 | −28.9 | 5 | + linear TAM | Rejected |
| 8 | 5 | −43.9 | 5 | + linear GSK3529246 | Accepted |
| 9 | 8 | −0.1 | 6 | + Linear molibresib | Rejected |
| 10 | 8 | −2.6 | 7 | + Linear molibresib with interaction | Rejected |
| 11 | 8 | −0.1 | 6 | + linear TAM | Rejected |
| 12 | 8 | 12.8 | 5 | Emax GSK3529246 | Rejected |
| 13 | 6 | −0.7 | 5 | Emax molibresib | Rejected |
| 14 | 7 | −0.9 | 5 | Emax TAM | Rejected |
| 15 | 6 | −34 | 5 | Power(molibresib) | Rejected |
| 16 | 7 | −34.7 | 5 | Power(TAM) | Rejected |
| 17 | 8 | −38.7 | 5 | Power(GSK3529246) | Accepted |
| 18 | 17 | −32.2 | 6 | + Circadian(amplitude) | Rejected |
| 19 | 17 | −35.2 | 7 | + Circadian(tmax) | Rejected |
| 20 | 17 | −47.5 | 7 | IIV(GSK3529246) | Accepted |
| 21 | 20 | 0 | 8 | + IOV(GSK3529246) | Rejected |
| 22 | 20 | −449.7 | 8 | + IOV(intercept) | Accepted |
| 23 | 22 | −12.7 | 7 | + Occasion 2 power | Accepted |
| 24 | 23 | −8.9 | 8 | + Occasion 3 power | Final |
| 25 | 24 | 391 | 9 | + Intercept: occasion 2, occasion 3 | Rejected |
Rejected as correlation between EC50 and Emax was >95%.
Reference model numbers and ΔOFV values for accepted runs are shown in bold.
Abbreviations: ΔOFV, change in objective function compared with reference model; DMQTCF, the individual specific difference of baseline QTcF from population mean; dQTcF, QT interval prolongation corrected by Fridericia formula and by baseline; γ, feedback power term; EC50, drug concentration required to provide half the maximal response; Emax, maximal drug effect; ER, exposure–response; GSK3529246, molibresib active metabolite composite; IIV, inter‐individual variability; IOV inter‐occasion variability; ktr, maturation rate constant; PK, pharmacokinetic; Ref, Reference model; TAM, total active moiety (molibresib + GSK3529246); OFV, objective function value; TAM, total active moiety; tmax, time to maximum plasma concentration.
TABLE 3.
Platelet parameter estimates using the final structural platelet model (a) and the final platelet model (b)
| (a) Platelet parameter estimates – final structural model | |||||
|---|---|---|---|---|---|
| Description | Parameter (unit) | Estimate | RSE (%) | 95% CI | CV (%) |
| Baseline | BSLN (∙ 109/L) | 257 | – | (244–271) | – |
| Maturation rate | k tr (/h) | 0.0348 | – | (0.0331–0.0366) | – |
| Feedback power term | γ | 0.144 | – | (0.127–0.163) | – |
| Drug effect EC50 | EC50 (μM) | 4.82 | – | (4.22–5.51) | – |
| IIV on baseline | ω2 baseline | 0.125 | 12.4 | (0.0945–0.155) | 35.4 |
| IIV on ktr | ω2 ktr | 0.0167 | 28.2 | (0.00746–0.0259) | 12.9 |
| IIV on γ | ω2 γ | 0.256 | 24.1 | (0.135–0.376) | 50.6 |
| IIV on EC50 | ω2 EC50 | 0.363 | 19.8 | (0.222–0.504) | 60.2 |
| Proportional residual error | σ1 .1 | 0.0765 | 8.2 | (0.0641–0.0889) | – |
| (b) Platelet parameter estimates – final platelet model | |||||
|---|---|---|---|---|---|
| Description | Parameter (unit) | Estimate | RSE (%) | 95% CI | CV (%) |
| Baseline platelets | BSLN (∙ 109/L) | 257 | – | (244, 271) | – |
| Maturation rate | ktr(/h) | 0.0347 | – | (0.0331, 0.0365) | – |
| Feedback power term | γ | 0.157 | – | (0.137, 0.179) | – |
| Drug effect EC50 | EC50 (μM) | 4.85 | – | (4.22, 5.56) | – |
| Feedback GIST | γ GIST | 0.370 | 41.3 | (0.0707, 0.67) | – |
| Feedback NC, prostate | γ NC, prostate | ‐0.306 | 24.7 | (‐0.454, ‐0.158) | – |
| IIV on baseline platelets | ω2 baseline | 0.121 | 12.5 | (0.0914, 0.151) | 34.8 |
| IIV on ktr | ω2 ktr | 0.0171 | 27.6 | (0.00782, 0.0263) | 13.1 |
| IIV on γ | ω2 γ | 0.218 | 24.2 | (0.115, 0.322) | 46.7 |
| IIV on EC50 | ω2 EC50 | 0.383 | 19.5 | (0.237, 0.53) | 61.9 |
| Proportional residual error | σ1.1 | 0.0763 | 8.2 | (0.064, 0.0887) | – |
Abbreviations: γ, feedback power term; CI, confidence interval; CV, coefficient of variation (derived as 100 × √[ω2]); EC50, total active moiety plasma concentration (molibresib + active metabolite composite [GSK3529246]) that provides half the maximal response; GIST, gastrointestinal stromal tumor; IIV, inter‐individual variability; ktr, maturation rate constant; NC, nuclear protein in testis carcinoma; RSE, relative standard error.
Covariate analysis
The GAM analysis indicated that the observed baseline platelet count had an impact (p < 0.01) on ktr (AIC: −463.95), cancer type on γ (AIC: 180.2), prior platinum‐based chemotherapy on baseline platelet count (AIC: 99.3), and EC50 (AIC: 253.1) and prior taxane‐based chemotherapy (AIC: 256.9) and age (AIC: 245.5) on EC50. Forward inclusion steps resulted in addition of cancer type on γ (evaluated as either GI stromal tumor [GIST], NC, prostate cancer, or other cancers), observed baseline platelet count on ktr, and age on EC50. Backward elimination resulted in the removal of observed baseline platelet count on ktr and age on EC50 to produce the final model.
Final model
The final platelet model is illustrated in Figure 1a and presented mathematically in Figure 1b. Similar to the semi‐mechanistic Friberg myelosuppression model, 7 the final model consisted of a platelet proliferation compartment sensitive to drug effects, three transit compartments representing maturation, and a circulating platelet compartment. The model included an effect of TAM concentration on platelet proliferation rate (thus assumes that molibresib and GSK3529246 have equal potency) and cancer type (GIST, NC, prostate or other) on γ. Platelet parameter estimates using the final model are summarized in Table 3. Baseline platelet count was estimated to be 257 × 109/L, the mean transit time to be 115 h, and the EC50 for the effect of TAM on platelet proliferation rate to be 4.85 μM. Patients with GIST were predicted to have a 37% higher γ compared with other cancer types, resulting in greater feedback on platelet proliferation and a higher platelet proliferation rate. Conversely, patients with NC and prostate cancer were found to have a 31% lower γ, resulting in less feedback and a lower increase in the platelet proliferation rate. Platelet count simulations following administration of molibresib (75 mg q.d.) for different tumor types are presented in Figure S3.
FIGURE 1.
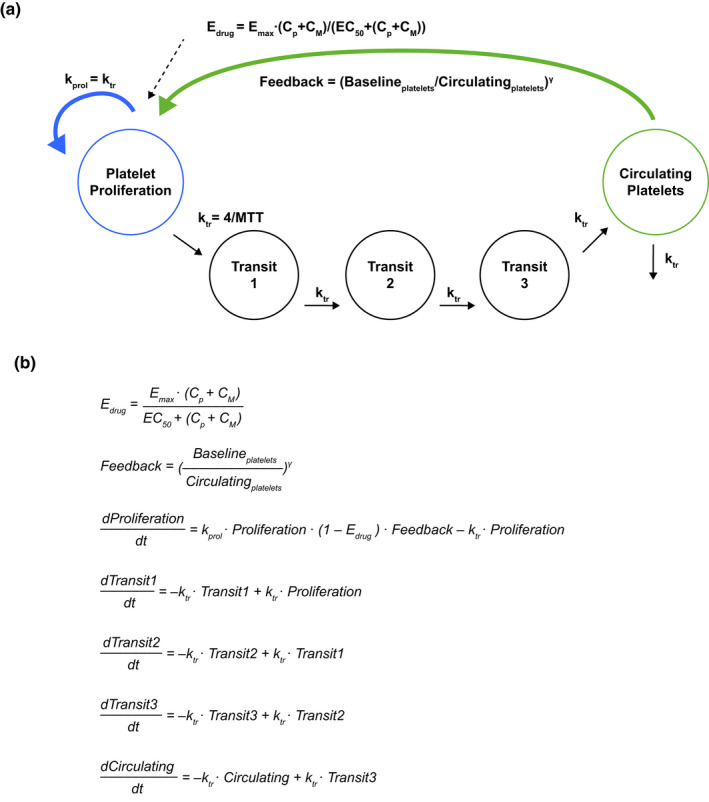
(a) Graphical representation of the final semi‐mechanistic PK/PD model for platelets after molibresib oral dosing, and (b) mathematical description of the final platelet model. Baselineplatelets, baseline platelet count; Circulatingplatelets, circulating platelet count; CM, active metabolite composite (GSK3529246) plasma concentration; Cp, molibresib plasma concentration; EC50, TAM plasma concentration that provides half the maximal response; Edrug, drug effect; Emax, maximal drug effect (fixed to 1); Feedback, feedback effect; γ, feedback power term; kprol, proliferation rate constant (equal to ktr); ktr, maturation rate constant; MTT, mean transit time; PK/PD, pharmacokinetic/pharmacodynamic; T, time; TAM, total active moiety
Model diagnostics and qualification
All platelet parameters were estimated (Table 3) with sufficient precision (based on the 95% CI values or a relative standard error <50%), and the condition number was low (20.2). The GOF plots for the final platelet model showed conditionally weighted residuals (CWRES) randomly scattered around the predicted range and across time (Figure S4). Overall, the diagnostic GOF plots showed that the final model demonstrated appropriate agreement between the predicted and observed data. The prediction‐corrected VPC showed that the final model adequately described the time course of platelet count data following molibresib treatment, although a slight underprediction of platelet count reduction is apparent at the highest doses (Figure 2).
FIGURE 2.
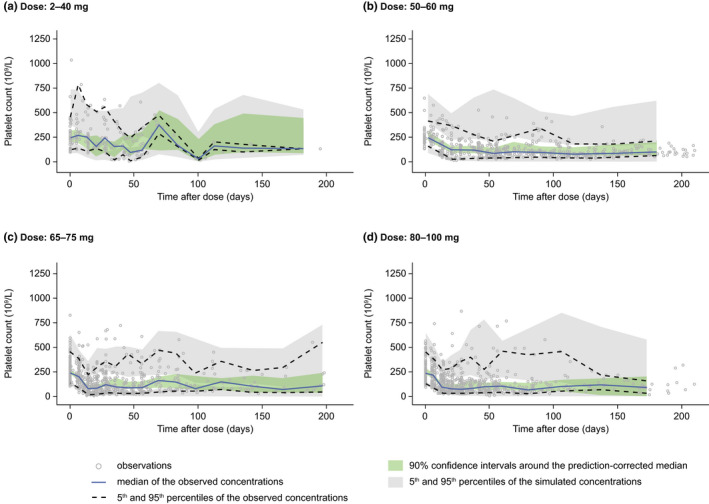
Visual predictive checks for the final platelet model simulated versus observed data by dose of molibresib: (a) 2–40 mg, (b) 50–60 mg, (c) 65–75 mg, and (d) 80–100 mg. All observations and predictions were adjusted using prediction correction 25
Simulations of Grades 3 and 4 TCP AEs
Platelet count time course simulations performed with the final model showed rates of Grade 3 or 4 TCP at 80 mg (46% predicted), similar to those reported in Part 2 of the FTIH study (43%) for molibresib 75 mg q.d. (besylate salt formulation), but greater than reported for the 60 mg q.d. dose (38% predicted versus 11% observed; Figure S5).
dQTcF modeling
Exploratory analysis
In total, 2706 dQTcF observations from 94 patients enrolled in Part 1 of the FTIH study were included in the analysis for model development (Table 1). Graphical exploratory analysis found no meaningful relationship between heart rate and molibresib or GSK3529246 exposure, and no compelling evidence of hysteresis (Figures S6 and S7).
Model development and qualification
The key structural model building steps are summarized in Table 2b. The final dQTcF model is described mathematically in Figure S1b; it included a population intercept, an effect of baseline dQTcF difference from the population mean, and an effect of GSK3529246 concentration on dQTcF described using a power model with separate power exponents for sampling occasions 2 (Week 1, Day 3 [W1D3]–predose Week 2, Day 1 [W2D1]) and 3 (W2D1–predose W3D1). The final model also included IIV and IOV on the intercept parameter.
The parameter estimates of the final dQTcF model and mean predicted dQTcF by occasion and dose are summarized in Table 4a,b. All model parameters were statistically significant (p ≤ 0.00011), and 95% CIs did not include zero. The GOF plots showed that residuals were symmetrically distributed and had negligible trends with predicted values and with time.
TABLE 4.
(a) Parameter estimates using the final dQTcF model and (b) mean predicted dQTcF (final dQTcF model) by occasion and dose
| (a) Parameter estimates – final dQTcF model | ||||
|---|---|---|---|---|
| Parameter | Estimate | Standard error | p value | 95% CI |
| DM | −0.22 | 0.05 | <0.0001 | −0.318 to −0.124 |
| INT | −4.00 | 1.03 | 0.0001095 | −6.02 to −1.98 |
| P145 | 0.25 | 0.02 | <0.0001 | 0.207–0.302 |
| P2 | 0.39 | 0.01 | <0.0001 | 0.37–0.419 |
| P3 | 0.33 | 0.02 | <0.0001 | 0.295–0.372 |
| IIV a | 7.84 | n/a | n/a | 6.54–9.42 |
| IOV a | 7.12 | n/a | n/a | 6.39–7.93 |
| RUV a | 9.18 | n/a | n/a | 8.92–9.45 |
| (b) Mean (90% CI) predicted dQTcF | |||
|---|---|---|---|
| Occasion | 60 mg (347 ng/ml) | 80 mg (380 ng/ml) | 100 mg (402 ng/ml) |
| W1D1–W1D3 predose | 1.76 (0.11–3.45) | 1.89 (0.236–3.6) | 1.98 (0.304–3.71) |
| W1D3–W2D1 predose | 10.6 (8.39–12.8) | 11.1 (8.82–13.4) | 11.4 (9.12–13.8) |
| W2D1–W3D1 predose | 5.74 (3.59–8.04) | 6.04 (3.82–8.41) | 6.23 (3.95–8.65) |
| W3D1–W4D1 predose | 1.76 (0.11–3.45) | 1.89 (0.236–3.6) | 1.98 (0.304–3.71) |
| W4 and above | 1.76 (0.11–3.45) | 1.89 (0.236–3.6) | 1.98 (0.304–3.71) |
Predictions (b) are presented as mean (90% CI) values for the mean maximum plasma concentration (in parentheses) across subjects at each dose level; doses represent q.d. dosing.
Random effects are expressed as standard deviations; standard errors and p values are not applicable.
Abbreviations: CI, confidence interval; D, day; DM, parameter describing the effect of the individual specific difference of baseline QTcF from population mean; dQTcF, QT interval prolongation corrected by Fridericia formula and by baseline; IIV, inter‐individual variability; INT, intercept; IOV, inter‐occasion variability; n/a, not applicable; P145, power estimate for occasions 1, 4, and 5; P2, power estimate for occasion 2; P3, power estimate for occasion 3; RUV, residual unexplained variability; W, Week.
Simulation of dQTcF using the final model
Observed and model‐predicted dQTcF by predicted GSK3529246 concentration and dosing occasion are summarized in Figure 3 and Table 4. The upper bounds of all 90% CIs for both observed and model‐predicted dQTcF were <20 milliseconds (and in most cases <10 milliseconds).
FIGURE 3.
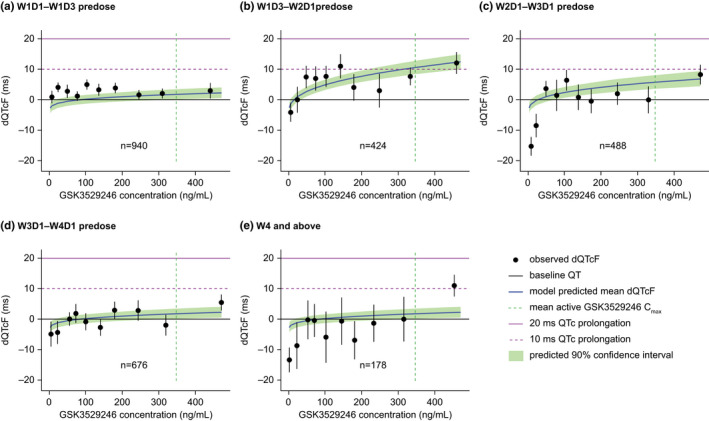
Predicted dQTcF prolongation (final dQTcF model) by active metabolite composite (GSK3529246) plasma concentration and dosing occasion. Filled black circles and vertical black lines represent means and 90% CI for deciles of observed dQTcF. Horizontal purple lines indicate dQTcF changes of 10 milliseconds (dashed) and 20 milliseconds (solid). Cmax, maximum plasma concentration; D, day; dQTcF, QT interval prolongation corrected by Fridericia formula and by baseline; q.d., once daily; W, Week
GI AE modeling
Exploratory analysis
In total, 101 first occurrences of Grade 2+ GI AEs among 193 patients were included in the analysis (Table S2). Correlation analyses are presented in Table S2 and showed that most pairwise combinations of GI AEs were weakly correlated, with the exception of nausea and vomiting, which were moderately correlated (coefficient, 0.57).
Model development
The models evaluated for nausea, dysgeusia, diarrhea, and vomiting are presented in Table S3 along with parameter estimates for the modeled probability of Grade 2+ nausea, dysgeusia, diarrhea, and vomiting events versus exposure metrics for molibresib, GSK3529246, and TAM. In all cases, models with exposure as a predictor performed better than models with dose as a predictor, which in turn performed better than models using intercept alone. For nausea, diarrhea, and vomiting, Cmin for molibresib provided the greatest improvement in statistical significance for the slope (p value), AIC, and OFV, although AUC for molibresib provided equivalent improvements in AIC and OFV for diarrhea. For dysgeusia, GSK3529246 Cmax provided the best improvements in AIC and OFV, while TAM Cmax provided the best improvement in the statistical significance of the slope.
Overall, however, the incidences of Grade 2+ nausea or dysgeusia were not associated with any exposure metric for molibresib, GSK3529246, or TAM. The incidences of Grade 2+ diarrhea and vomiting were found to be possibly related (i.e., a positive slope estimate and p < 0.05) to molibresib or TAM Cmin, but not associated with any Cmax metric (Figure S8). In addition, the occurrence of Grade 2+ diarrhea was possibly related to molibresib AUC.
DISCUSSION/CONCLUSIONS
Based on an established semi‐mechanistic mathematical model describing platelet kinetics following chemotherapy (Friberg et al. 7 ), we identified important covariates associated with the variability of molibresib and GSK3529246 exposure and TCP. We then used these to develop a PK/PD model that adequately described the time course of platelet count changes following treatment with molibresib, an approach that has been used successfully in previous modeling studies. 9 The final model consisted of a platelet proliferation compartment sensitive to drug effects, three transit compartments representing maturation, a circulating platelet compartment, and an effect of TAM concentration on the platelet proliferation rate. Interestingly, cancer type was shown to have a significant impact on the megakaryocyte proliferation rate (in response to the TCP effect of molibresib), with GIST showing an increase compared with other tumor types (NC and prostate cancer excluded), and NC and prostate cancer showing a decrease (as determined by a 37% increase and a 31% decrease in the model feedback power term γ, respectively). The reason for this is unclear, and patient numbers in the individual tumor data sets were low, so further investigation would be required to elucidate these observations and potentially provide guidance on the optimal dosing regimen for patients with different tumor types. Simulations using the final platelet model showed similar predicted and observed rates of Grades 3 and 4 TCP with 75 mg and 80 mg doses of molibresib, but higher predicted rates at a 60 mg q.d. dose (38% predicted vs. 11% observed). This may be the result of a small patient population in the 60 mg q.d. cohort (n = 9). Nevertheless, this analysis may still be used to predict doses and alternative dosing schedules for molibresib combination studies.
An ER model was also developed to explore the relationship between exposures to molibresib, GSK3529246, and TAM on change in the dQTcF interval. This analysis was initiated based on preclinical data suggesting a potential increased risk of arrhythmia and myocardial damage, and ECG data collected as part of this study were extensively analyzed to systematically assess this risk. A range of doses were also used in this analysis (2–100 mg). Overall, dQTcF was best described using a power model of GSK3529246 concentration; molibresib or TAM concentrations were not included in the final model. Model simulation from 60 mg q.d. (clinically relevant dose) to 100 mg q.d. (highest dose), allowing protection against possible inflated exposure in the patient population as a result of compromised renal/hepatic function or DDIs, showed that although a small prolongation of dQTcF occurred following molibresib treatment, this was <20 milliseconds in all cases, and usually <10 milliseconds. A threshold of <20 milliseconds change is considered appropriate for oncology drugs based on recent publications or guidance. 19 , 20 , 21 , 22 Providing an early assessment C‐QTc analysis may be used to justify a waiver for, or improve the design of, a resource‐intensive dedicated TQT study.
ER analyses of the relationship between molibresib, GSK3529246, and TAM concentrations and the occurrence of Grade 2+ GI AEs demonstrated that the occurrence of Grade 2+ nausea or dysgeusia was unrelated to any molibresib, GSK3529246, or TAM exposure metrices (Cmax, Cmin, or AUC). However, there was a trend showing a possible relationship between the occurrence of Grade 2+ diarrhea and molibresib Cmin, TAM Cmin, or molibresib AUC as well as between the occurrence of Grade 2+ vomiting and molibresib Cmin and TAM Cmin. The occurrences of GI Grade 2+ events were weakly correlated, except for nausea and vomiting, which were moderately correlated.
Limitations to be considered when interpreting the data from these analyses include the impact of the limited sample size for some doses and tumor cohorts and limited metabolite data from Part 1 individuals for the platelet analyses as well as the use of a single baseline observation for dQTcF correction. In addition, there was no placebo control for use in C‐QTc evaluation.
In conclusion, the analyses presented here demonstrate that a semi‐mechanistic population PK/PD model with transit compartments adequately describes platelet count changes over time following molibresib treatment. The covariates identified may be used prospectively to simulate the probability of TCP and optimize molibresib dosing/schedules. ER analyses showed no clinically meaningful prolongation of the dQTcF interval with molibresib up to a 100 mg supratherapeutic dose, and no strong correlation between molibresib treatment and the occurrence of Grade 2+ GI AEs (nausea, dysgeusia, diarrhea, and vomiting). The platelet model is being used to predict doses for a combination study where the combination drug also leads to TCP.
CONFLICT OF INTEREST
A.S.K. is an employee of GSK and hold stocks/shares in GSK. E.H. and T.B. are employees of qPharmetra LLC. A.D. has a family member employed by PRA Health Sciences, is an employee of GSK, and hold stocks/shares in GSK. M.P. is an employee of qPharmetra LLC. G.F‐B. is an employee of GSK and hold stocks/shares in GSK.
AUTHOR CONTRIBUTIONS
A.S.K., E.H., T.B., A.D., M.P., and G.F‐B. wrote the manuscript. A.S.K., A.D., M.P., and G.F‐B. designed the research. A.S.K., E.H., T.B., A.D., M.P., and G.F‐B. performed the research. A.S.K., E.H., T.B., A.D., M.P., and G.F‐B. analyzed the data.
Supporting information
Fig S1‐S8
Table S1‐S3
ACKNOWLEDGMENTS
Editorial support (in the form of writing assistance, including development of the initial draft based on author direction, assembling tables and figures, collating authors’ comments, grammatical editing, and referencing) was provided by Anna Polyakova, PhD, of Fishawack Indicia Ltd, UK, and was funded by GSK.
Krishnatry AS, Hanze E, Bergsma T, Dhar A, Prohn M, Ferron‐Brady G. Exposure–response analysis of adverse events associated with molibresib and its active metabolites in patients with solid tumors. CPT Pharmacometrics Syst Pharmacol. 2022;11:556–568. doi: 10.1002/psp4.12724
Funding information
This study was funded by GSK (115521; NCT01587703).
DATA AVAILABILITY STATEMENT
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
REFERENCES
- 1. Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao Y, Yang CY, Wang S. The making of I‐BET762, a BET bromodomain inhibitor now in clinical development. J Med Chem. 2013;56(19):7498‐7500. [DOI] [PubMed] [Google Scholar]
- 3. Wyce A, Soden P, Felitsky DJ, et al. Mechanism‐based combination strategies for BET inhibitors in NUT midline carcinoma. Can Res. 2016;76:4693. [Google Scholar]
- 4. Piha‐Paul SA, Hann CL, French CA, et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra‐terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Canc Spectr. 2019;4(2):pkz093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krishnatry AS, Voelkner A, Dhar A, Prohn M, Ferron‐Brady G. Population pharmacokinetic modeling of molibresib and its active metabolites in patients with solid tumors: a semimechanistic autoinduction model. CPT Pharmacometrics Syst Pharmacol. 2021;10(7):709‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuter DJ. Managing thrombocytopenia associated with cancer chemotherapy. Oncology (Williston Park). 2015;29(4):282‐294. [PubMed] [Google Scholar]
- 7. Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy‐induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20(24):4713‐4721. [DOI] [PubMed] [Google Scholar]
- 8. Kheifetz Y, Scholz M. Modeling individual time courses of thrombopoiesis during multi‐cyclic chemotherapy. PLoS Comput Biol. 2019;15(3):e1006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho YK, Irby DJ, Li J, et al. Pharmacokinetic‐pharmacodynamic model of neutropenia in patients with myeloma receiving high‐dose melphalan for autologous stem cell transplant. CPT Pharmacometrics Syst Pharmacol. 2018;7(11):748‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Food and Drug Administration . Guidance for industry. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. https://www.fda.gov/media/71372/download. Published 2005. Accessed December 15, 2020.
- 11. Salvi V, Karnad DR, Panicker GK, Kothari S. Update on the evaluation of a new drug for effects on cardiac repolarization in humans: issues in early drug development. Br J Pharmacol. 2010;159(1):34‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shah RR, Morganroth J. Early investigation of QTc liability: the role of multiple ascending dose (MAD) study. Drug Saf. 2012;35(9):695‐709. [DOI] [PubMed] [Google Scholar]
- 13. Darpo B, Garnett C. Early QT assessment–how can our confidence in the data be improved? Br J Clin Pharmacol. 2013;76(5):642‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Darpo B, Sarapa N, Garnett C, et al. The IQ‐CSRC prospective clinical Phase 1 study: "Can early QT assessment using exposure response analysis replace the thorough QT study?". Ann Noninvasive Electrocardiol. 2014;19(1):70‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mehrotra DV, Fan L, Liu F, Tsai K. Enabling robust assessment of QTc prolongation in early phase clinical trials. Pharm Stat. 2017;16(3):218‐227. [DOI] [PubMed] [Google Scholar]
- 16. Nelson CH, Wang L, Fang L, et al. A quantitative framework to evaluate proarrhythmic risk in a first‐in‐human study to support waiver of a thorough QT study. Clin Pharmacol Ther. 2015;98(6):630‐638. [DOI] [PubMed] [Google Scholar]
- 17. Zhu P, Hsu CH, Hu C, et al. Application of trial simulation in the design of a prospective study for concentration‐QTc analysis in support of a thorough QT study waiver. AAPS J. 2020;22(5):101. [DOI] [PubMed] [Google Scholar]
- 18. Garnett C, Bonate PL, Dang Q, et al. Scientific white paper on concentration‐QTc modeling. J Pharmacokinet Pharmacodyn. 2018;45(3):383‐397. [DOI] [PubMed] [Google Scholar]
- 19. Cohen‐Rabbie S, Berges AC, Rekić D, Parkinson J, Dota C, Tomkinson HK. QT prolongation risk assessment in oncology: lessons learned from small‐molecule new drug applications approved during 2011–2019. J Clin Pharmacol. 2021;61(8):1106‐1117. [DOI] [PubMed] [Google Scholar]
- 20. Bendell JC, Patel MR, Yoshida K, et al. Phase 1 study of cardiac safety of TAS‐102 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(6):1275‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lester RM, Paglialunga S, Johnson IA. QT Assessment in early drug development: the long and the short of it. Int J Mol Sci. 2019;20(6):1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. E14 Implementation Working Group . ICH E14 guideline: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. https://database.ich.org/sites/default/files/E14_Q%26As_R3_Q%26As.pdf. Published 2015. Accessed July 27, 2021.
- 23. Lindbom L, Pihlgren P, Jonsson EN. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241‐257. [DOI] [PubMed] [Google Scholar]
- 24. R Development Core Team . R: A Language and Environment for Statistical Computing. http://www.R‐project.org. Published 2008. Accessed July 2020. [Google Scholar]
- 25. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vandenberk B, Vandael E, Robyns T, et al. Which QT correction formulae to use for QT monitoring? J Am Heart Assoc. 2016;5(6):e003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8
Table S1‐S3
Data Availability Statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.