

Abstract
Background and Purpose:
Advances in the treatment of triple-negative breast and ovarian cancer remain challenging. In particular, resistance to the available therapy, by restoring or overexpressing the DNA repair machinery, has often been reported. New strategies to improve the therapeutic outcomes of these cancers are needed. Herein, we disclose the dregamine 5-bromo-pyridin-2-ylhydrazone (BBIT20), a natural monoterpene indole alkaloid derivative, as an inhibitor of homologous DNA repair.
Experimental Approach:
To unveil BBIT20 antitumour activity and underlying molecular mechanism of action, two-dimensional (2D) and three-dimensional (3D) cell cultures, patient-derived cell lines and xenograft mouse models were used.
Key Results:
BBIT20 disrupted the BRCA1-BARD1 interaction, triggering nuclearto-cytoplasmic BRCA1 translocation, cell cycle arrest and downregulation of homologous DNA repair-related genes and proteins, with subsequent enhancement of DNA damage, reactive oxygen species generation and apoptosis, in triple-negative breast and ovarian cancer cells. BBIT20 also displayed pronounced antitumour activity in patient-derived cells and xenograft mouse models of ovarian cancer, with low toxicity in non-malignant cells and undetectable side effects in mice. Additionally, it did not induce resistance in triple-negative breast and ovarian cancer and displayed marked synergistic effects with cisplatin and olaparib (a poly [ADP-ribose] polymerase inhibitor), on 2D and 3D models of these cancer cells.
Conclusion and Implications:
These findings add an inhibitor of the BRCA1-BARD1 interaction to the list of DNA-damaging agents. Importantly, either as a single agent or in combination therapy, BBIT20 reveals great potential in the personalized treatment of aggressive and resistant cancers, particularly triple-negative breast and advanced ovarian cancer.
Keywords: BRCA1, homologous recombination, indole alkaloids, targeted anticancer therapy
1 |. INTRODUCTION
Breast cancer type 1 susceptibility protein (BRCA1) coordinates multiple cellular processes, with particular relevance in regulation of DNA repair and cell cycle progression (Sato et al., 2012). BRCA1 plays these roles in association with its binding partner, the BRCA1-associated ring domain protein (BARD1), which stabilizes and confines BRCA1 to the nucleus (Tarsounas & Sung, 2020). In the nucleus, BRCA1BARD1 heterodimer functions as an E3 ubiquitin ligase, facilitating DNA double-strand break repair, mainly by homologous recombination (Tarsounas & Sung, 2020). BRCA1 also functions as a central hub of a complex molecular network, coordinating several DNA repair proteins (Tarsounas & Sung, 2020). Due to its key role in the maintenance of genomic integrity, a dysfunctional BRCA1 activity, either by mutation or low BRCA1 protein expression levels, is associated with an increased risk of developing hereditary and sporadic cancers, particularly breast and ovarian cancer (Drost & Jonkers, 2014; El-Deiry et al., 2019). Nevertheless, in established cancers, a full or residual BRCA1 function is associated with poor prognosis and therapeutic resistance (Abbotts et al., 2014).
BRCA1-deficient cancers, with a defective homologous DNA repair activity (Sato et al., 2012), are usually sensitive to DNA-damaging agents, as platinum drugs, and to poly(ADP)-ribose polymerase (PARP) inhibitors (Kamel et al., 2018). Particularly, different PARP inhibitors have recently been approved for the personalized treatment of homologous DNA repair-deficient cancers, including metastatic breast and ovarian cancer (Hengel et al., 2017; Murata et al., 2016; Robson et al., 2017; To et al., 2014; Washington et al., 2019). However, despite quite effective in homologous DNA repair-defective cancers, de novo resistance to these agents is commonly observed, particularly in triple-negative breast cancer (aggressive molecular breast cancer subtype) and ovarian cancer, due to a residual BRCA1 activity (as heterozygosity) (Hengel et al., 2017; Johnson et al., 2016; Yap et al., 2019). Thus, although the exciting benefit of PARP inhibitors, their clinical use has been highly limited due to inefficiency in homologous DNA repair-proficient cancers and recurrent acquired resistance in initially responding tumours. Therefore, therapeutic alternatives to impair homologous DNA repair pathway, sensitizing cancer cells to the effect of PARP inhibitors and other DNA-damaging agents, would have major relevance in cancer therapy.
This work discloses the natural monoterpene indole alkaloid derivative dregamine 5-bromo-pyridin-2-ylhydrazone (BBIT20) as a new homologous DNA repair inhibitor with encouraging anticancer activity, particularly towards triple-negative breast and ovarian cancer.
2 |. METHODS
2.1 |. Human cell lines and growth conditions
Detailed information on human cell lines is provided in Table S1, along with sources and culture conditions. MDA-MB-231 (RRID:CVCL_0062), HCC1937 (RRID:CVCL_0290) and IGROV-1 (RRID:CVCL_1304) cell lines were validated by short tandem repeat analysis. All cells were routinely tested for mycoplasma contamination.
2.2 |. Patient-derived ovarian cancer cells
Patient-derived ovarian cancer cells (#1, #9, #41, #49, #62) were obtained from ascitic effusions of patients bearing epithelial ovarian cancer (detailed information in Table S2). Patient-derived ovarian cancer cells were routinely maintained as serial xenotransplants as described (Indraccolo et al., 2006). The study was approved by IOV Institutional Review Board and Ethics Committee (EM 23/2017) and performed in accordance with declaration of Helsinki. Informed consent was obtained from patients who entered this study. Procedures involving animals were conformed to institutional guidelines that comply with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, 12 December 1987) and were authorized by the Italian Ministry of Health (Authorization No. 617/2016 PR). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
2.3 |. Next generation sequencing analysis of BRCA status
Targeted next generation sequencing (NGS) was performed on patient-derived ovarian cancer xenograft-derived cells. Genomic DNA was extracted with Easy DNA kit (Life Technologies), quantified with Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and subjected to quality control using agarose gel prior to library preparation. The samples were sequenced using a custom amplicon-based panel (Illumina, San Diego, CA) covering 66 genes of biological relevance in ovarian cancer on Miseq (Illumina) in paired-end mode. Mutations with allele frequencies of at least 10% and adequate coverage (>200 reads) in target regions were considered. Polymorphisms, synonymous or intronic mutations were excluded. The Catalogue of Somatic Mutations in Cancer (COSMIC) database was used to access the clinically relevant variants.
2.4 |. Cell viability and proliferation assays
MCF-7 (RRID:CVCL_0031), T47D (RRID:CVCL_0553) (4.0 × 103), MDA-MB-231, HCC1937, OVCAR-3 (RRID:CVCL_0465), SKOV-3 (RRID:CVCL_0532), IGROV-1 (5.0 × 103), SK-BR-3 (RRID:CVCL_0532), MDA-MB-468 (RRID:CVCL_0419), patient-derived ovarian cancer #1, #9, #41, #49, #62 (7.5 × 103) and MCF10A (RRID:CVCL_0598), HFF-1 (RRID:CVCL_3285) (1.0 × 104) cells/well were seeded in 96-well plates and allowed to adhere overnight, followed by treatment with serial compound dilutions, for 48 h. For patient-derived ovarian cancer cells, half-maximal inhibitory concentration (IC50) values were determined with the CellTiter96®Aqueous one solution cell proliferation assay (MTS assay; Promega, Italy) after 48 h of treatment; for the other human cells, IC50 values were measured by sulforhodamine B assay, as described (Raimundo et al., 2018). Briefly, 20 μl of reagent were added to each well at the end of treatment and allowed to react for 2 h at 37°C. IC50 values were determined using the GraphPad Prism software, Version 7.0 (La Jolla, CA, USA).
For the colony formation assay, 5 × 102 cells/well were seeded in six-well plates and treated with serial concentrations of BBIT20 for 8 (MDA-MB-231 and IGROV-1) or 16 (HCC1937) days. Colonies were fixed, stained and analysed as described (Raimundo et al., 2018).
2.5 |. Mammospheres generation
A total of 1.5 × 103 HCC1937 cells/well was seeded in 24-well plates covered with 1% agarose in DMEM:F12 supplemented with 20 ng ml−1 bFGF, 40 ng ml−1 EGF (Bio-techne, Citomed Lda, Lisboa, Portugal), 1 × B27 (Life Technologies, Porto, Portugal), 10 μg ml−1 insulin (Sigma-Aldrich, Sintra, Portugal) and 2 mM L-Glutamine (Sigma) and treated with BBIT20 at the seeding time (Bessa et al., 2018). Mammospheres also grown in compound-free medium for 3 followed by BBIT20 treatment (Bessa et al., 2018).
Using 3-day-old mammospheres, BBIT20 synergistic potential was evaluated through analysis of its effect, alone and in combination with cisplatin (CDDP) or olaparib, on spheroid growth for additional 14 days. New medium with the drugs (or vehicle only) was added to the wells at days 1, 3, 6, 9 and 12. Mammospheres were monitored using an inverted Nikon TE 2000-U microscope at 100X magnification, with DXM1200F digital camera and Nikon ACT-1 software (Raimundo et al., 2018). Spheroid diameters were quantified using Fiji Software (Schindelin et al., 2012).
2.6 |. RNA extraction and whole-transcriptome sequencing (RNA-seq)
HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 (2.25 × 105) cells/well were seeded in 6-well plates and allowed to adhere overnight, followed by treatment with BBIT20 for additional 48 h. Total RNA was extracted, using the IllustraTM RNAspin Mini RNA Isolation Kit (GEHealthcare) according to the manufacturer’s instructions and the concentration was measured using the Qubit™ RNA HS Assay Kit (Invitrogen, ThermoFisher Scientific, Portugal). Total RNA integrity was determined using the Agilent 2100 Bioanalyzer (Agilent, Portugal).
RNA sequencing was used to detect changes in expression levels of over 20 800 human RefSeq genes (18 574 coding genes and 2228 non-coding genes based on UCSC hg19 annotation). Briefly, 10 ng of RNA were used for cDNA amplification using the SuperScript® VILO™ cDNA Synthesis Kit (ThermoFisher Scientific). Libraries were constructed using Ion AmpliSeq™ Transcriptome Human Gene Expression Kit and multiple samples pooled after ligation of unique barcodes. The pooled libraries were processed on Ion Chef™ System and the loaded Ion 550™ chip was sequenced on Ion S5™ XL System (ThermoFisher Scientific). Sequencing quality was assessed through the plug-in coverage analysis and the samples were analysed on the torrent Suite™ Software using the Ion AmpliSeq™ RNA plug-in v.5.12 and target region hg19_AmpliSeq_Transcriptome_21K_v1) within the Ion Reporter v5.6 server (Ion Torrent, Waltham, MA, USA). Samples with a number of reads <100 000 and/or the average base coverage <500× were considered inadequate for analysis. Differentially expressed genes in BBIT20-treated cells, compared to control cells, were evaluated in the Transcriptome Analysis Console (TAC; RRID: SCR_018718) software v4.0.2 (ThermoFisher Scientific) using a double threshold based on fold change (<1.5 and >1.5) and statistical significance of the change with P < 0.05 (ANOVA). Metascape webtool and Ingenuity Pathway Analysis software (RRID:SCR_008653) were used to perform Gene Ontology enrichment analysis and comparison of enriched pathways and molecular functions of the transcriptome of the differentially expressed genes list; in Metascape, the pathway and enrichment process analysis has been carried out with the following ontology sources: KEGG Pathway, Gene Ontology Molecular Functions, Gene Ontology Biological Processes, Reactome Gene Sets, Hallmark Gene Sets, Canonical Pathways.
2.7 |. Cell cycle and apoptosis analyses
HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 and patient-derived ovarian cancer #1 (2.25 × 105) cells/well were seeded in six-well plates and treated in the next day with BBIT20 for 48 h. Cell cycle and apoptosis analysis were performed as described (Raimundo et al., 2018). For patient-derived ovarian cancer #1, apoptosis analysis was performed according to the manufacturer’s instructions, using the Annexin-V-Fluos staining kit (Roche, Sigma-Aldrich, Italy). The BD LSR II flow cytometer, with the BDF FACS Diva software version 6.1.3 (BD Biosciences, Italy), was used. FlowJo v10.0.7 (Treestar, Ashland, OR, USA) was used for quantification of cell cycle phases.
2.8 |. Western blot
HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 (2.25 × 105) cells/well were seeded in six-well plates, allowed to adhere overnight and treated with BBIT20. Samples preparation and western blot were performed as described (Raimundo et al., 2018). Band intensities were quantified using Image Lab software (version 5.2.1; Bio-Rad laboratories, Amadora, Portugal). Specific antibodies are described in Table S3.
2.9 |. Comet assay
Cellular DNA damage was detected at a single-cell level using the alkaline comet assay, basically as described (Marabini et al., 2019). HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 (2.25 × 105) cells/well were seeded in six-well plates and allowed to adhere overnight, followed by 48 h of treatment with BBIT20. Cells were then harvested, resuspended in 150 μl of 1% low melting-point agarose gel and spread on pre-coated slides with 1% agarose. Slides were then stored at 4°C for 10 min to allow solidification. Slides with cells were immersed in lysis buffer, electrophoresis (buffer: 0.3 M NaOH, 1 mM Na-EDTA, pH 13) was carried out; cells were then fixed with ethanol, stained with propidium iodide (PI; 20 μg ml−1) and photographed using a Nikon DS-5Mc camera and a Nikon Eclipse E400 fluorescence microscope and images processed with Nikon ACT-2 U software (Izasa). For each sample, 100 randomly selected nucleoids were analysed and quantified using TriTek Comet Score Imaging Software V2.0. DNA damage was measured by quantification of the tail DNA (percentage of comet-positive cells with more than 5% of DNA in the tail) and the tail moment (product of the tail length and % of DNA in the tail).
2.10 |. Cell-based homologous DNA repair assays
Homologous DNA repair assays were performed using MCF7 DR-GFP cells as previously described (Kaplan et al., 2019). Briefly, cells were seeded and pre-treated with BBIT20 for 48 h. Site-specific DNA double-strand breaks were introduced using electroporation with Amaxa Nucleofector II and Nucleofector Kit V (Lonza) to deliver 10 ng of a plasmid encoding the restriction enzyme I-Sce I. After 48 h transfection, the medium was replaced by fresh one without compound. After 72 h of DNA double-strand break induction, MCF7 DR-GFP cells were analysed by flow cytometry to quantify the percentage of GFP-positive cells.
2.11 |. Immunofluorescence staining
HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 (2.25 × 105) cells/well were seeded in culture slides (Corning, ThermoFisher Scientific, Portugal) and allowed to adhere overnight, followed by 48 h of treatment with BBIT20. Immunofluorescence was performed basically as described (Vazquez et al., 2017). Specific antibodies are described in Table S3. Images were acquired with a Nikon eclipse Ts2R-C-AL microscope with a Nikon LV-TV camera and NIS Elements BR-5.20 software (Nikon Corporation). Representative images were generated using Fiji software (RRID:SCR_002285), also used for quantification of foci formation (Schindelin et al., 2012).
2.12 |. Reactive oxygen species generation
HCC1937 (1.5 × 105), IGROV-1 (2.0 × 105) and MDA-MB-231 (2.25 × 105) cells/well were seeded in six-well plates and allowed to adhere overnight, followed by treatment with BBIT20 for 48 h. Reactive oxygen species (ROS) analysis was performed using the 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA; ThermoFisher Scientific), for 30 min at 37°C, as described (Soares et al., 2015).
2.13 |. Co-immunoprecipitation assay
Co-immunoprecipitation was performed using the Pierce Classic Magnetic IP and CO-IP Kit (Thermo Scientific, Dagma, Carcavelos, Portugal) as described (Raimundo et al., 2018). The anti-BRCA1 (IP:BRCA1) and anti-immunoglobulin G (IgG) antibodies were used after 18 h (MDA-MB-231 and HCC1937) and 24 h (IGROV-1) treatments with BBIT20, followed by western blot detection of BRCA1, BARD1 and GAPDH (Table S3).
2.14 |. Combination therapy assays
Cells were treated with BBIT20 (at a concentration with no significant effect on cell growth) and/or increasing concentrations of cisplatin and olaparib, for 48 h. The effect of combined treatments was analysed by MTS (for patient-derived ovarian cancer cells) or sulforhodamine B assay and in 3D-spheroid model. Combination index and dose reduction index were calculated and analysed as described (Raimundo et al., 2018).
2.15 |. Acquired resistance studies
MDA-MB-231, HCC1937 and IGROV-1 cells were exposed to five rounds of selection with increasing concentrations of BBIT20, which were added to the culture medium for 24 h, followed by a recovery time of 2 to 3 days. Cells were harvested, seeded and treated three times with each concentration. For each round, IC50 values were determined as described (Raimundo et al., 2018).
2.16 |. Antitumour assays in heterotopic xenograft mouse models
In vivo studies were performed using the C57BL/6-Rag2−/−IL2rg−/− mice model, generously provided by Prof. James Di Santo (Institute Pasteur, Paris, France). A total of 2.5 × 106 IGROV-1 cells (expressing heterozygous mutBRCA1) were implanted subcutaneously (in PBS/Matrigel 1:1; Corning, Enzifarma, Porto, Portugal) in the right flank of female mice with 6 to 8 weeks. Seven intraperitoneal injections, three times a week, of BBIT20 (2 mgkg−1), olaparib (50 mgkg−1) or vehicle were performed for tumours with approximately 100 mm3 (8 days after implantation). Tumour volume was measured three times a week using calliper and the formula (axb2)/2 (a and b represent the longest and shortest tumour axes, respectively). Tumour growth studies were performed with n = 12 mice for BBIT20 and vehicle group or n = 8 mice for olaparib experimental group, which have a power around 80% to detect difference of 1.25 SDs between two groups at a 5% significance level. Female mice were randomly assigned to the different treatment groups. Experiments were not blinded. At the end of treatment, blood samples were collected for toxicological analysis, and animals were killed by cervical dislocation. All animals were housed in polycarbonate cages (two to six per cage) and kept on a 12 h light/dark cycle. Food and water were given ad libitum. Studies were reviewed by the Animal Ethics Committee and Animal Welfare Body of the i3S (reference 2016/22), authorized by the national authority Direção Geral de Alimentação e Veterinária (DGAV reference 0421/000/000/2017). Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology.
2.17 |. Immunohistochemistry analysis
Immunohistochemistry analysis of xenograft tumour tissues was performed as described (Soares et al., 2016), for staining with H&E or antibodies, for assessment of TUNEL-positive cells, and evaluation of 3,3-diaminobenzidine (DAB) intensity. Images were analysed using Fiji software (Schindelin et al., 2012). Antibodies are listed in Table S3.
2.18 |. Randomization and blinding
For in vivo experiments, the animals were randomized to each treatment group, 12 or 8 animals by group. Tumours were removed for histological analyses. The slides obtained from paraffin-embedded tumours were labelled with numbers and the analysis was performed under blinded conditions. For in vitro studies, blinded analysis was not performed due to the nature of the assays.
2.19 |. Statistical analysis
Data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Data were plotted in bar charts, except for the cases of dose–response curves, considering that a scatter plot or before–after charts did not reveal unusual or interesting aspects of the data not obvious from the bar chart. Data are presented as mean ± SD or ± SEM of n samples, where n refers to independent experiments, not replicates. Values of n and number of technical replicates, if performed, are given in figure legends. Where replicates were used, their values were averaged to provide a single value to the dataset. Data analyses was carried out in GraphPad Prism (RRID:SCR_002798) Software Version 8.4.0 (La Jolla). All assays with five or more independent experiments were subjected to statistical analysis. Normalization was made for controlling unwanted sources of variation and data analysis was performed setting controls (DMSO or non-treated cells) as 100% or as one for comparison purposes. Different statistical tests were used depending on dataset; for comparison of two groups, unpaired Student’s t-test was used; for comparison of multiple groups, the oneway or the two-way ANOVA followed by multiple comparison tests (post hoc Tukey’s, Sidak’s or Dunnet’s multiple comparison tests) were used relative to controls. Statistical significance was set as *P <0.05. Post hoc tests were run only if F achieved P <0.05 and when no significant variance in homogeneity was observed.
Gene expression data was Log2 transformed before analysis and all genes with a fold-change of at least 1.5 (<−1.5 and >1.5) and the adjusted Pa value (FDR) of less than 5%, were considered differentially expressed genes. A list of differentially expressed gene was generated using Transcriptome Analysis Console Software v4.0.2 (ThermoFisher Scientific).
2.20 |. Materials
BBIT20 was prepared by reaction of the monoterpene indole alkaloid dregamine, isolated from the alkaloid fraction of the methanol extract of the roots of Tabernaemontana elegans (Paterna et al., 2015), with 5-bromo-2-hydrazinopyridine (Sigma-Aldrich, Steinhelm, Germany), as previously described (Paterna et al., 2015). Its structure was assigned by spectroscopic methods, including 1D and 2D NMR experiments.
Cisplatin (Enzo Life Science, Taper, Sintra, Portugal) was dissolved in saline and Olaparib (AZD2281; Santa Cruz Biotechnologies, Frilabo, Portugal) and BBIT20 in DMSO (Sigma-Aldrich, Sintra, Portugal). The solvent (maximum 0.1%) was included as control.
2.21 |. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Fabbro, et al., 2019; Alexander, Kelly, et al., 2019).
3 |. RESULTS
3.1 |. BBIT20 inhibits the growth of human breast and ovarian cancer cells
In an attempt to search for new therapeutic options against breast and ovarian cancer, the anti-proliferative activity of a small library of monoterpene indole alkaloid derivatives was investigated in an array of human breast and ovarian cancer cells with different BRCA1 status: wild-type (wt), mutant (mut) and loss of heterozygosity (LOH; Table 1). These compounds were obtained, in previous work, by chemical modification of two epimeric indole alkaloids isolated from the alkaloid fraction of the methanol extract of the African medicinal plant Tabernaemontana elegans (Apocynaceae; Paterna et al., 2015). In accordance with the IC50 values obtained by sulforhodamine B assay, among the tested compounds, the dregamine 5-bromo-pyridin-2-ylhydrazone derivative (BBIT20; Figure 1a), resulting from condensation reaction of the ketone function of the natural indole alkaloid dregamine with 5-bromo-2-hydrazinopyridine, was found to be highly effective, particularly against triple negative breast cancer cells (Table 1). In fact, the IC50 values of BBIT20 ranged from 4–6 μM in BRCA1-deficient to 9–14 μM in BRCA1-proficient cancer cells. The effectiveness of BBIT20 against breast and ovarian cancer cells could be further evidenced when compared to cisplatin (clinically used in triple-negative breast and ovarian cancer patients) and olaparib (approved for mutBRCA1-related breast and ovarian cancer patients) (Table 1). Notably, conversely to cisplatin, the BBIT20 growth inhibitory effect markedly decreased in non-malignant breast (MCF10a) and fibroblast (HFF-1) cells (Table 1; Figure 1b), evidencing its selectivity to cancer cells. Importantly, BBIT20 showed to be much more effective than olaparib in all tested cancer cells, regardless of BRCA1 status (Table 1).
TABLE 1.
Antiproliferative effect of BBIT20, cisplatin and olaparib, in a panel of human breast cancer, ovarian cancer and non-malignant cells
| IC50 (μM) | ||||
|---|---|---|---|---|
|
|
||||
| Human cells | BBIT20 | Cisplatin | Olaparib | |
| wtBRCA1-expressing cells | T47D | 11.5 ± 3.0 | 5.9 ± 0.05 | 30.0 ± 2.4 |
| MCF-7 | 11.1 ± 1.6 | 11.9 ± 3.4 | 39.2 ± 2.8 | |
| OVCAR-3 | 13.9 ± 1.7 | 3.2 ± 0.05 | 42.0 ± 4.5 | |
| SKOV-3 | 8.9 ± 2.5 | 12.0 ± 1.1 | 98.0 ± 1.9 | |
| BRCA1 LOH cells | MDA-MB-231 | 5.6 ± 1.4 | 12.1 ± 1.1 | 50.9 ± 1.7 |
| MDA-MB-468 | 4.3 ± 1.2 | 2.3 ± 0.3 | 43.0 ± 5.9 | |
| SK-BR-3 | 5.7 ± 1.4 | 14.5 ± 2.6 | 27.5 ± 3.5 | |
| mutBRCA1-expressing cells | HCC1937 (5382insC) | 5.7 ± 1.3 | 8.6 ± 0.9 | 30.5 ± 1.3 |
| IGROV-1 (280delA) | 4.6 ± 1.5 | 5.8 ± 0.7 | 15.9 ± 2.1 | |
| Non-malignant cells | HFF-1 | 33.6 ± 3.2 | 9.3 ± 0.9 | 36.0 ± 1.1 |
| MCF10a | 29.5 ± 2.7 | 2.3 ± 0.35 | 86.0 ± 3.1 | |
| Patient-derived ovarian cancer (PD-OVCA) cells | ||||
| Pathogenic mutBRCA1 | PD-OVCA #1 | 2.68 ± 0.57 | 15.53 ± 1.41 | 31.92 ± 5.08 |
| PD-OVCA #9 | 5.11 ± 1.03 | 8.40 ± 1.38 | 15.64 ± 3.49 | |
| Benign mutBRCA1 | PD-OVCA #41 | 14.09 ± 0.88 | 4.38 ± 1.05 | 64.58 ± 4.45 |
| wtBRCA1-expressing cells | PD-OVCA #49 | 15.10 ± 1.42 | 8.31 ± 1.76 | 23.56 ± 1.93 |
| PD-OVCA #62 | 14.24 ± 2.85 | 6.52 ± 1.51 | 56.17 ± 5.22 | |
Note: IC50 values were determined by Sulforhodamine B (in immortalized cells) or CellTiter96®Aqueous one solution cell proliferation assay (MTS; in PD-OVCA cells) assay, after 48 h of treatment. Data are mean ± SD (n = 5, with two replicates each).
Abbreviations: LOH, loss of heterozygosity; mut, mutant; patient-derived ovarian cancer, PD-OVCA; wt, wild-type.
FIGURE 1.

Growth inhibitory effect of BBIT20 on 2D and 3D models of human triple-negative breast and ovarian cancer cells. (a) Chemical structure of BBIT20. (b) Dose–response curves for the growth inhibitory effect of BBIT20 on human non-malignant (MCF10a and HFF-1), triple-negative breast cancer (MDA-MB-231, HCC1937) and ovarian cancer (IGROV-1) cells, after 48 h of treatment; growth obtained with control was set as 100%; data are mean ± SD, n = 5 independent experiments (two replicates each); values obtained for non-malignant cells are significantly different from cancer cells: *P <0.05 (one-way ANOVA followed by Dunnett’s test). (c, d) Effect of BBIT20 on colony formation of cancer cells after 8 (MDA-MB-231 and IGROV-1) and 16 (HCC1937) days of treatment. In (c), representative experiments are shown. In (d), quantification of colony formation; growth obtained with DMSO was set as 100%; data are mean ± SD, n = 5 independent experiments; *P <0.05, significantly different from DMSO (two-way ANOVA followed by Sidak’s test). (e, f) Evaluation of HCC1937 mammosphere formation after 72 h of treatment with BBIT20; treatment was performed at seeding time of HCC1937 cells. (g, h) Effect of BBIT20 on 3-day-old HCC1937 mammospheres, for up to 11 days treatment. In (e) and (g), representative images are shown (scale bar = 50 μm, 100X magnification). In (f) and (h), determination of mammosphere area at the end of treatment; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (unpaired Student’s t-test)
BBIT20 antitumour activity was further studied in a panel of patient-derived ovarian cancer cells expressing wt or mutBRCA1 (Table 1; Figure S1A). By MTS assay, BBIT20 was found to reduce cell viability of all patient-derived ovarian cancer cells (Table 1; Figure S1B), even more effectively than olaparib (Table 1). The more pronounced growth inhibitory effect of BBIT20, associated with a notorious change in cell morphology (Figure S1C), was observed in patient-derived ovarian cancer cells #1 (Table 1; Figure S1B).
The pronounced growth inhibitory effect of BBIT20 in triple-negative breast and ovarian cancer cells was further confirmed by colony formation assay (Figure 1c,d). BBIT20 was also tested in a 3D-mammosphere model generated from HCC1937 cells. At 3 and 6 μM, BBIT20 significantly inhibited mammosphere formation, leading to a complete abolishment of spheroids formation at 6 μM, when added upon seeding (Figure 1e,f). Additionally, 6 and 12 μM BBIT20 markedly reduced mammosphere growth, in 3-day-old spheroids, triggering mammosphere disintegration at 12 μM (Figure 1g,h).
3.2 |. BBIT20 decreases homologous DNA repair and induces cell cycle arrest and apoptosis
To investigate further into BBIT20 molecular mechanism, the transcriptome analysis of triple-negative breast and ovarian cancer cells was performed, after 48 h of treatment with 12 μM BBIT20. The results showed broad changes in the gene expression profile of BBIT20-treated cancer cells (Figures 2a and S2A). In fact, differentially expressed gene analysis revealed that 1714 (in MDA-MB-231), 2200 (in HCC1937) and 3249 (in IGROV-1) genes were significantly altered by ±1.5-fold in BBIT20-treated cells when compared to control cells (Figure S2B). For each cancer cell line, statistical assessment and pathway enrichment analysis of upregulated and downregulated genes were performed. To functionally characterize the differentially expressed gene, Metascape and Ingenuity Pathway Analysis were carried out. Using the Metascape webtool, the enrichment analysis applied to the list of genes upregulated by BBIT20 treatment was highly divergent among the three cancer cell lines, only converging in the enrichment of the autophagy mechanism (Figure S2C). Conversely, Metascape analysis deduced a significant enrichment of downregulated genes involved in cell proliferation, cell cycle progression, DNA repair and replication and MYC targets variant 1, in BBIT20-treated MDA-MB-231, HCC1937 and IGROV-1 cells (Figure 2b). Interestingly, the DNA repair GO biological process included enrichment of DNA double-strand breaks repair (Figure 2b), mainly through homologous recombination (Figure 2c), in all the three BBIT20-treated cancer cell lines. The Metascape results were corroborated by Ingenuity Pathway Analysis (Figures S3 and S4) that also identified signatures consistent with downregulation of cell proliferation, cell cycle progression, DNA replication, recombination and repair and upregulation of mechanisms involved in cell death (Figure S3). Moreover, the analysis of top enriched canonical pathways indicated that BBIT20-treated cells features a transcriptional change of genes mostly involved in cell cycle control and DNA damage (Figure S4). In fact, the “Role of BRCA1 in DNA damage and response” was one of the top enriched canonical pathways (Figure S4).
FIGURE 2.

Transcriptome enrichment analysis upon BBIT20 treatment in triple-negative breast and ovarian cancer cells. (a) Heatmap of differentially expressed genes between BBIT20- and DMSO-treated cancer cells. (b) Effect of 12 μM BBIT20 in the transcriptome of cancer cells, after 48 h of treatment; Metascape was used to perform gene set enrichment analysis of downregulated genes in cancer cells. (c) Analysis of enriched terms in DNA repair cluster of downregulated genes in cancer cells. In (a)–(c), data are mean ± SEM, n = 3 independent experiments; fold change, <1.5 to >1.5; FDR<0.05
Consistently, several genes involved in DNA damage repair, particularly through homologous recombination, were found downregulated such as RAD51, RAD51AP1, FANCD2, EME1, POLQ (in MDA-MB-231, HCC1937, IGROV-1 cells), BRCA1, MSH2, BLM, EXO1 (in MDA-MB-231 and HCC1937 cells), BRCA2, BARD1, RAD52, MRE11A, MSH6 and PCNA (in MDA-MB-231 and IGROV-1 cells; Figure 3a). Accordingly, a significant decrease in expression levels of homologous DNA repair-related proteins was observed in all BBIT20-treated cancer cells (Figure 3b,c). Interesting changes could also be observed in the protein levels of ATM/pATM and ATR/pATR, which are key DNA damage repair upstream mediators (Marechal & Zou, 2013). In fact, a significant decrease in the ATM protein levels was observed in MDA-MB-231 and IGROV-1 cells. Although HCC1937 cells have not shown any change in ATM or pATM levels, a significant reduction in the pATR protein levels could be observed (Figure 3b,c). Of note, for some homologous DNA repair-related proteins, a reduction in the mRNA levels could not be observed (Figure 3a), which can be related to the timepoint selected for the analysis.
FIGURE 3.

BBIT20 regulates the expression levels of genes and proteins involved in homologous DNA repair, in triple-negative breast and ovarian cancer cells. (a) Effect of 12 μM BBIT20 on mRNA expression levels of genes involved in homologous DNA repair, after 48 h of treatment; data are mean ± SEM, n = 5 independent experiments; fold change <1.5 to >1.5; *P <0.05 (ANOVA). (b, c) Effect of 12 μM BBIT20 on expression levels of proteins involved in homologous DNA repair after 48 h of treatment. In (b), representative immunoblots are shown; GAPDH was used as loading control. In (c), quantification of protein expression levels relative to DMSO; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (unpaired Student’s t-test)
Among the downregulated genes mainly involved in cell cycle progression and therapeutic response were PLK1, FOXM1, CDC25C, CDC20, CCNA2, CCNB1, CDK1, DNA2, BIRC5/Survivin (in MDA-MB231, HCC1937 and IGROV-1 cells), CDC25B (in MDA-MB-231 and HCC1937), CDK2 (in HCC1937 and IGROV-1 cells) and POLE2 (in MDA-MB-231 and IGROV-1; Figure 4a). In line with this, BBIT20 led to a significant reduction of CDK1, CDK2, Cdc20 and survivin (baculoviral IAP repeat containing 5) protein expression levels (Figure 4b,c). Despite their roles in cell cycle progression and therapeutic resistance, these proteins are also associated with enhanced DNA damage repair activity. Hence, the downregulated genes signature of BBIT20-treated cells is clearly related to DNA repair, particularly through homologous recombination and cell cycle regulation.
FIGURE 4.

BBIT20 regulates the expression levels of proliferation and therapeutic resistance factors, inducing cell cycle arrest, apoptosis and ROS generation, in triple-negative breast and ovarian cancer cells. (a) Effect of 12 μM BBIT20 on mRNA expression levels of genes involved in cell cycle progression and therapeutic response, after 48 h of treatment; data are mean ± SEM, n = 5 independent experiments; fold change <1.5 to >1.5; *P <0.05 (ANOVA). (b, c) Effect of 12 μM BBIT20 on protein expression levels after 48 h of treatment. In (b), representative immunoblots are shown; GAPDH was used as loading control. In (c), quantification of protein expression levels relative to DMSO; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (unpaired Student’s t-test). Effect of 6 and 12 μM BBIT20 on cell cycle progression (d), apoptosis (e) and ROS production (f), in triple-negative breast and ovarian cancer cells, after 48 h of treatment; data are mean ± SD, n = 5 independent experiments; values significantly different from DMSO: *P <0.05 (two-way ANOVA followed by Dunnett’s test)
In accordance with the enrichment of downregulated genes involved in cell cycle progression, an increase of CDKN1A/p21 mRNA (Figure 4a) and protein (Figure 4b,c) expression levels was observed in all cancer cells. In fact, 6 and 12 μM BBIT20 induced cell cycle arrest at G0/G1 (in MDA-MB-231 cells), G2/M (in HCC1937 cells) and S (in IGROV-1 cells) phases, after 48 h of treatment (Figure 4d). Consistently, 6 μM BBIT20 induced G0/G1 phase cell cycle arrest in patient-derived ovarian cancer cells #1 (Figure S1D).
BBIT20 also upregulated mRNA expression levels of BBC3/PUMA (in MDA-MB-231 and IGROV-1 cells), while downregulating PARP1 (in MDA-MB-231 and HCC1937 cells) (Figure 4a). Moreover, Ingenuity Pathway analysis of differentially expressed genes revealed that some of the top enriched biological pathways are related to cancer cell death by apoptotic mechanisms (Figure S3). The induction of apoptosis by BBIT20 was further supported by increased PUMA and cleaved PARP protein levels (at 12 μM; Figure 4b,c) and Annexin-Vpositive cells (at 6 and 12 μM; Figure 4e), in MDA-MB-231, HCC1937 and IGROV-1 cells. Of note, 6 μM BBIT20 also increased Annexin-V-positive cells, patient-derived ovarian cancer cells #1 (Figure S1E). Interestingly, a dose-dependent increase in ROS generation was also observed in 6 and 12 μM BBIT20-treated cancer cells (Figure 4f).
3.3 |. BBIT20 decreases homologous DNA repair with disruption of the BRCA1-BARD1 interaction
The above results indicated a marked inhibition of DNA repair by BBIT20. To further corroborate these data, the interference of BBIT20 in DNA damage was evaluated by measuring DNA double-strand break in cancer cells, after 48 h of treatment, using the alkaline comet assay. In MDA-MB-231, HCC1937 and IGROV-1 cells, 6 and 12 μM BBIT20 significantly increased the percentage of comet-positive cells, particularly of tail DNA (Figure 5a,b) and tail moment (Figure 5a,c).
FIGURE 5.

BBIT20 enhances DNA damage by inhibiting homologous DNA repair, in triple-negative breast and ovarian cancer cells. (a–c) Measurement of DNA damage in cancer cells, after 48 h of treatment with 6 and 12 μM BBIT20, was measured by comet assay. In (a), representative images are shown (scale bar = 20 μm; 200X magnification). In (b), quantification of tail DNA percentage; in (c), quantification of tail moment; data are mean ± SD, n = 5 independent experiments (200 cells per sample); *P <0.05 significantly different from DMSO (two-way ANOVA followed by Dunnett’s test). (d) Homologous DNA repair activity was determined in MCF7 DR-GFP cells treated with 6 and 12 μM BBIT20, using the chromosomal DR-GFP assay; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (one-way ANOVA followed by Dunnett’s test). (e, f) Analysis of γH2AX expression levels in cancer cells, after 48 h of treatment with 12 μM BBIT20. In (e), representative immunoblots are shown; GAPDH was used as loading control. In (f), quantification of protein expression levels relative to DMSO; data are mean ± SD, n = 5 independent experiments; RLU (relative light unit). (g) γH2AX, RAD51 and BRCA1 foci formation, and BRCA1 cellular localization after 48 h of treatment with 12 μM BBIT20; representative images (scale bar = 100 μm; 400X magnification) are shown. Quantification of γH2AX (h), RAD51 (i) and BRCA1 (j) foci formation; data are mean ± SD, n = 5 independent experiments (100 cells per sample); *P <0.05 significantly different from DMSO (unpaired Student’s t-test)
To investigate the effect of BBIT20 on homologous recombination, a chromosomally based homologous DNA repair reporter cell line (MCF7 DR-GFP), carrying site-specific DNA double-strand breaks, was used to monitor homologous DNA repair function. As expected, a marked reduction in homologous DNA repair capacity was observed with 2 and 6 μM BBIT20 (Figure 5d). Consistently, 12 μM BBIT20 increased the amount of phosphorylated (Ser139) histone H2AX (γH2AX) (Figure 5e,f) and the number of γH2AX-positive foci formed in MDAMB-231, HCC1937 and IGROV-1 cells (Figure 5g,h). Corroborating the decline of homologous DNA repair activity, a pronounced reduction in RAD51-foci formation could also be observed by immunofluorescence analysis, in BBIT20-treated cancer cells (Figure 5g,i). Furthermore, 12 μM BBIT20 triggered nuclear-to-cytoplasmic BRCA1 translocation, in MDA-MB-231, HCC1937 and IGROV-1 cells (Figure 5g,j).
Because the interaction with BARD1 is primarily responsible for BRCA1 nuclear localization and its subsequent DNA repair activity, we hypothesized whether BBIT20 could be a potential inhibitor of the BRCA1-BARD1 interaction. With this premise, co-immunoprecipitation experiments were performed in MDA-MB-231 (Figure 6a,d), HCC1937 (Figure 6b,d) and IGROV-1 (Figure 6c,d) cells treated with 12 and 20 μM BBIT20. A visible decrease in the amount of BARD1 co-immunoprecipitated with BRCA1 was observed, particularly for 20 μM BBIT20, which indicated a disruption of the BRCA1-BARD1 interaction by BBIT20, in all tested cancer cells. Of note, for the experimental timepoints used in this assay (18 and 24 h), no significant reduction of BRCA1 and BARD1 protein levels was observed in the input.
FIGURE 6.

BBIT20 inhibits the BRCA1-BARD1 interaction in triple-negative breast and ovarian cancer cells. (a–c) Co-IP was performed in MDA-MB-231 (a), HCC1937 (b) and IGROV-1 (d) cells treated with 12 and 20 μM BBIT20 for 18 h (in MDA-MB-231 and HCC1937 cells) and 24 h (in IGROV-1 cells). In (a)–(c), representative immunoblots are shown; whole-cell lysate is represented (input). In (d), quantification of BARD1 relative to DMSO (set as 1); BRCA1 from IP was used as loading control; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (unpaired Student’s t-test)
3.4 |. BBIT20 sensitizes cancer cells to the effect of cisplatin and olaparib
The ability of BBIT20 to sensitize triple-negative breast and ovarian cancer cells to olaparib and cisplatin was also investigated. For that, cancer cells were treated with a single concentration of BBIT20 (with no significant effect on cancer cell growth, IC5–IC10) and a concentration range of cisplatin or olaparib. The results revealed that BBIT20 enhanced the growth inhibitory effect of platinum and PARP inhibitors in MDA-MB-231 (Figure 7a), HCC1937 (Figure 7b) and IGROV-1 (Figure 7c) cells. The combination index and dose reduction index values were thereafter determined by multiple drug-effect analysis for every combination, in each cancer cell line (Figure 7a–c).
FIGURE 7.

BBIT20 has synergistic effects with cisplatin (CDDP) and olaparib in triple-negative breast and ovarian cancer cells. (a–c) Effect of BBIT20, alone and in combination with a concentration range of cisplatin or olaparib, on proliferation of MDA-MB-231 (a), HCC1937 (b) and IGROV-1 (c) cells. (d–f) Effect of BBIT20, alone and in combination with olaparib or CDDP, on apoptosis induction in MDA-MB-231 (d), HCC1937 (e) and IGROV-1 (f) cells; data are mean ± SD, n = 5 independent experiments; *P <0.05 significantly different from DMSO (two-way ANOVA followed by Dunnett’s test)
This analysis revealed promising synergistic effects between BBIT20 and cisplatin or olaparib (combination index < 1), with a noticeable reduction in the effective dose of these chemotherapeutic agents (Figure 7a–c). Consistently, these synergistic effects were associated with a significant increase in apoptosis induction, in MDA-MB-231, HCC1937 and IGROV-1 cells (Figure 7d–f). It is worth noting the promising synergistic effects achieved combining BBIT20 and olaparib in MDA-MB-231 and HCC1937 cells, based on the high resistance of these cells to olaparib. The synergistic effects between BBIT20 and cisplatin or olaparib were further confirmed in a 3D-model of HCC1937-derived mammospheres. For that, 3-day-old mammospheres were treated with 2 or 3 μM BBIT20 alone and in combination with 1 μM cisplatin or 60 μM olaparib for up to 14 days (Figure 8a,b). A synergic reduction in mammosphere area was reached for both cisplatin (combined with 2 and 3 μM BBIT20) and olaparib (combined with 3 μM BBIT20). Most importantly, a complete disintegration of mammospheres was achieved for 1 μM cisplatin in combination with 3 μM BBIT20 (Figure 8a,b).
FIGURE 8.

BBIT20 synergises with cisplatin (CDDP) and olaparib, in 3D mammosphere models and patient-derived cells, and does not induce resistance in triple-negative breast and ovarian cancer cells. (a, b) Effect of BBIT20, alone and in combination with 1 μM CDDP or 60 μM olaparib, on 3-day-old HCC1937 spheroids, for up to 14 days of treatment. In (a), representative images are shown (scale bar = 50 μm, 100X magnification). In (b), determination of spheroids area at the end of treatment; data are mean ± SD, n = 5 independent experiments; combination index (CI) was determined considering spheroid area. (c) Effect of BBIT20, alone and in combination with a concentration range of CDDP or olaparib, on proliferation of patient-derived ovarian cancer #1 cells. Cell proliferation was measured after 48 h of treatment; growth obtained with DMSO was set as 100%; data are mean ± SD, n = 5 to 6 independent experiments; *P <0.05 significantly different from chemotherapeutic drugs alone (two-way ANOVA followed by Sidak’s test); CI and dose reduction index (DRI) values for each combined treatment were calculated using CompuSyn software (CI < 1, synergy; 1 < CI < 1.1, additive effect; CI > 1.1, antagonism); data were calculated using a mean value effect of five or six independent experiments. (d) Cancer cells were exposed to five rounds of treatment with increasing concentrations (6, 9, 13, 21 and 33 μM) of BBIT20. IC50 values of BBIT20 were determined at the end of each round, after 48 h of treatment; growth obtained with DMSO was set as 100%; data are mean ± SD, n = 5 independent experiments (two replicates each); values obtained for treated cells are not significantly different from parental cells (two-way ANOVA followed by Sidak’s test)
The ability of BBIT20 to sensitize patient-derived ovarian cancer cells #1 to olaparib and cisplatin was also evaluated (Figure 8c). For that, as with the other cancer cells, a single concentration of BBIT20 (IC5–IC10) was combined with a concentration range of cisplatin and olaparib. As expected, BBIT20 enhanced the growth inhibitory effect of cisplatin and olaparib, exhibiting promising synergistic effects (combination index < 1) and reducing the effective dose for both agents, in patient-derived ovarian cancer cells #1 (Figure 8c). Of note, patient-derived ovarian cancer cells #1 was derived from a patient firstly treated for breast cancer with radical surgery followed by radiotherapy and chemotherapy with taxotere/DOXO. At the end of adjuvant chemotherapy, the patient was diagnosed with ovarian cancer, ended up dying 25 months after diagnosis (Indraccolo et al., 2006). The patient received different treatments based on carboplatin, followed by gemcitabine and topotecan, and finally mitoxantrone (Indraccolo et al., 2006), having developed post-chemo platinum resistance. As such, these cells presented IC50 values higher than expected for both cisplatin and olaparib, whereas BBIT20 maintained a high anti-proliferative effect (Table 1). These results denoted that BBIT20 may not induce cross-resistance in ovarian cancer cells resistant to platinum drugs.
Due to the well-known predisposition of triple-negative breast and ovarian cancer cells to acquire resistance to therapy, we further checked whether BBIT20 could induce resistance in these cancer cells after several rounds of treatment. However, as demonstrated by the preservation of the IC50 value in successive generations (Figure 8d), neither triple-negative breast cancer nor ovarian cancer cells developed resistance to BBIT20.
3.5 |. BBIT20 displays potent antitumour activity in xenograft mouse models of ovarian cancer cells
The in vivo antitumour activity of BBIT20 was evaluated using xenograft mouse models of IGROV-1 cells. Tumours with approximately 100 mm3 were treated with 2 mgkg−1 BBIT20 or 50 mgkg−1 olaparib, by intraperitoneal administration, three times a week (for a total of seven administrations). A pronounced reduction of tumour volume was obtained in BBIT20-treated mice when compared to vehicle and olaparib (Figure 9a). Moreover, during the experiment, mice did not show significant variation of body weight (Figure 9b) or morbidity signs. In fact, data related to potential primary signs of toxicity showed no significant alterations on organs weight (spleen, liver, heart and kidneys) nor haematological and biochemical blood parameters variations in BBIT20-treated mice compared to vehicle (Table S4).
FIGURE 9.
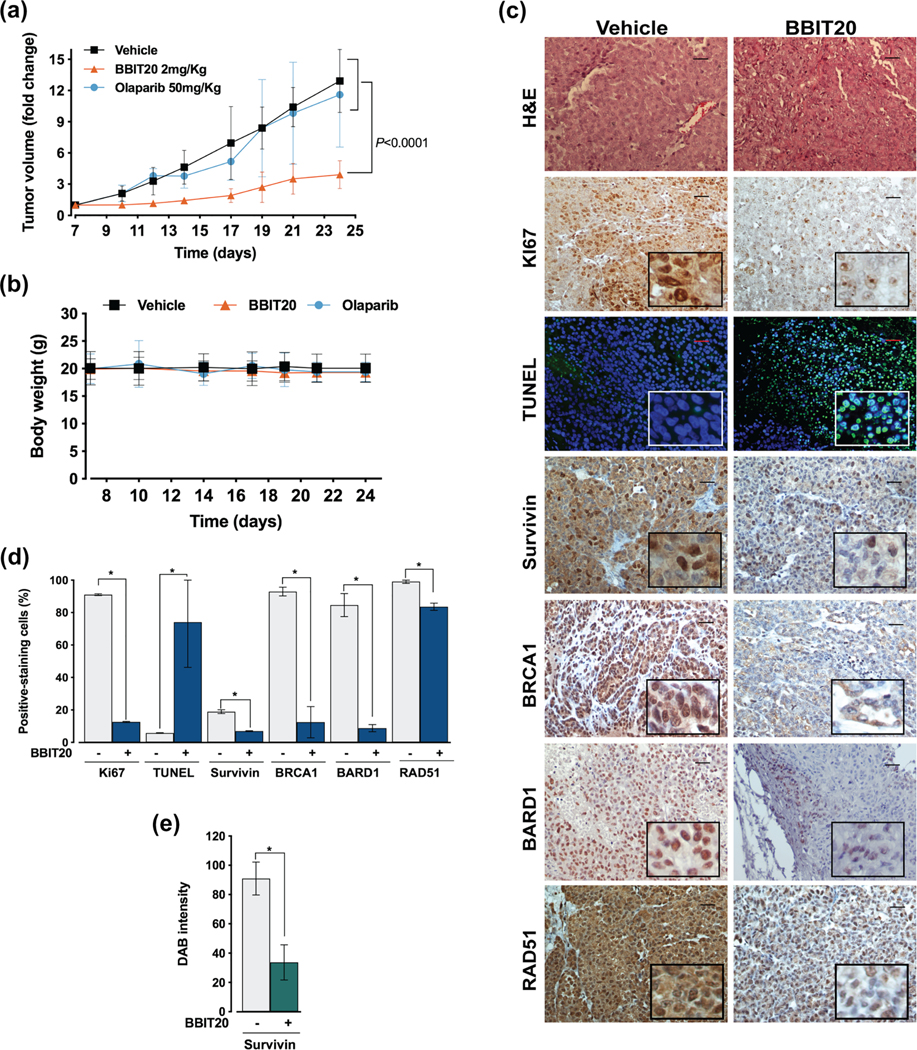
BBIT20 has pronounced antitumor activity in heterotopic xenograft mouse models of ovarian cancer cells. C57BL/6-Rag2−/− IL2rg−/− xenograft mice, carrying IGROV-1 cells implanted subcutaneously, were treated with 2 mgkg−1of BBIT20 or 50 mgkg−1 of olaparib by intraperitoneal injection three times per week (seven administrations in total). (a) Tumour volume curves of xenograft mice treated with BBIT20, olaparib or vehicle; values significantly different from vehicle: data are mean ± SD, n = 12 animals for BBIT20 and vehicle group and n = 8 for olaparib group; *P <0.05 (two-way ANOVA with Turkey’s multiple comparison test). (b) Mice body weight during treatment under each condition; values not significantly different from vehicle: n = 12 animals for BBIT20 and vehicle group and n = 8 for olaparib group; P > 0.05 (two-way ANOVA with Turkey’s multiple comparison test). (c–e) immunohistochemical staining of tumour tissues collected at the end of treatment from IGROV-1 xenograft mice. In (c), representative images of hematoxylin and eosin (H&E), Ki67, survivin, BRCA1, BARD1, RAD51 and TUNEL staining (scale bar = 20 μm; 200X magnification). In (d), quantification of Ki67, survivin, BRCA1, BARD1, RAD51 and TUNEL-positive nuclear staining. In (e), cytosolic survivin was quantified by evaluation of DAB intensity. In (d) and (e), data are mean ± SD, n = 5 independent staining’s (5 microphotographs per slide); values significantly different from vehicle: *P <0.05 (unpaired Student’s t-test)
Additionally, the immunohistochemical staining of tumour tissues from xenografts revealed that BBIT20 reduced Ki67 (decreasing proliferation), BRCA1, BARD1 and RAD51/DYRK4 (decreasing homologous DNA repair function/dual specificity tyrosine phosphorylation regulated kinase 4) and increased TUNEL-positive (DNA fragmentation) nuclear staining (Figure 9c,d), when compared to vehicle. A marked reduction of survivin staining could also be observed in both nucleus (Figure 9d) and cytoplasm (Figure 9e). These results unveiled a potent in vivo antitumour activity of BBIT20 through inhibition of cell proliferation and DNA repair and induction of an apoptotic cell death.
4 |. DISCUSSION
The discovery of effective therapeutic alternatives for the personalized therapy of triple-negative breast and high-grade ovarian cancer is still a challenge, with patients treatment mostly relying on chemotherapeutics as platinum drugs (Chalakur-Ramireddy & Pakala, 2018; Ellsworth et al., 2019; Mehanna et al., 2019). On the field of targeted therapies, PARP inhibitors are currently in the frontline for the treatment of breast and ovarian cancer with mutBRCA1 (Robson et al., 2017; Washington et al., 2019). However, once PARP inhibitors efficacy relates to homozygous mutBRCA1 and a complete loss of homologous DNA repair function, tumours with wt, heterozygous mutBRCA1 or loss of heterozygosity are commonly associated with PARP inhibitors resistance, due to a remaining homologous DNA repair activity (Johnson et al., 2016; Y. Kim et al., 2017; Noordermeer & van Attikum, 2019; Tarsounas & Sung, 2020). Furthermore, homozygous mutBRCA1 can also present resistance to PARP inhibitors and platinum drugs, mostly due to the restoration or residual homologous DNA repair activity (Johnson et al., 2016; Noordermeer & van Attikum, 2019). Based on the premise that cancer cells frequently become resistant to therapy by recovering its DNA repair machinery, inhibitors of homologous recombination have emerged as promising anticancer agents, particularly to (re)sensitize cancer cells to DNA-damaging agents (Abbotts et al., 2014; Trenner & Sartori, 2019).
This work reports the indole alkaloid derivative BBIT20 with marked growth inhibitory effect on triple-negative breast and ovarian cancer cells. Notably, the molecular mechanism underlying the efficient anticancer activity of BBIT20, in cancer cells with distinct BRCA1 status, showed to involve an inhibition of homologous recombination with disruption of the BRCA1–BARD1 interaction. The BRCA1–BARD1 heterodimer is known for its essential role in a myriad of homologous DNA repair steps, being the suppression of this complex a promising approach for inhibiting homologous DNA repair and therefore to overcome therapeutic resistance (Kawai et al., 2002). Interestingly, BBIT20 showed to be highly effective against the homozygous mutBRCA1 HCC1937 cells, initially described as having a complete loss of BRCA1 function and expression. However, recent data have shown residual BRCA1-mediated homologous DNA repair activity and BRCA1 interaction with BARD1 in these cells (Huen et al., 2010; Johnson et al., 2016; Liang et al., 2017). The potent BBIT20 antitumour activity was further validated in patient-derived and xenograft mouse models of ovarian cancer, confirming its superior activity to olaparib. Despite the existence of BRCA1-BARD1 heterodimers in non-malignant cells, BBIT20 revealed an evident selectivity for cancer cells. In fact, because non-malignant cells rely on intact DNA damage repair mechanisms, they are able to escape the cytotoxic effect of homologous DNA repair inhibitors (Gavande et al., 2016). Consistently, BBIT20 did not exhibit apparent side effects in mice.
BARD1 not only stabilizes BRCA1 but also plays a critical role in its translocation to the nucleus (Raimundo et al., 2020). Accordingly, disruption of the BRCA1-BARD1 heterodimer by BBIT20 resulted in an evident reduction in nuclear BRCA1 retention. In fact, BBIT20 led to BRCA1 cellular relocation and decreased its total protein expression levels, possibly by proteolytic degradation upon disruption of the BRCA1-BARD1 heterodimer (S. Kim et al., 2019; Nelson & Holt, 2010). These results were confirmed in tumour tissues, in which BBIT20 reduced BRCA1 levels and led to its translocation from the nucleus to the cytoplasm.
BRCA1 and BARD1 assemble into a stable heterodimer through the association of their RING domains (Tarsounas & Sung, 2020). As such, both RING domains of BRCA1 and BARD may represent potential binding sites for BBIT20. In fact, a number of cancer-causing mutations in the BRCA1 RING domain have been described by their ability to affect, although in different extensions, the BRCA1-BARD1 heterodimer formation (Densham & Morris, 2017; Hashizume et al., 2001). It was also shown that some proteins, like the BRCA1-associated protein 1 (BAP1), interact with the BARD1 RING domain, interfering with the BRCA1-BARD1 association (Nishikawa et al., 2009). That study also evidenced a possible binding pocket that could be applicable to BBIT20. Future studies capable of clarifying this issue will be of high relevance.
The decline in expression levels of several genes related to cell cycle progression and DNA repair corroborated the cell cycle arrest and inhibition of homologous recombination by BBIT20. These results were further supported in vivo by the evident reduction of proliferation (Ki67) and homologous DNA repair (BRCA1, BARD1 and RAD51) markers, in BBIT20-treated tumour tissues. Importantly, key players in therapeutic response were also downregulated by BBIT20 (Ha et al., 2014; Khongkow et al., 2014; Vequaud et al., 2016). In particular, FoxM1 and CDK2 overexpression are strictly correlated with triple-negative breast cancer low survival rates, increased epithelial-mesenchymal transition and chemoresistance (Chen et al., 2018; Tan, Wang, et al., 2019; Yao et al., 2018). Consistently, the inhibition of FoxM1 and CDK2 has been pointed out as an appealing anticancer therapeutic strategy (Tan, Wang, et al., 2019). RAD51 and RAD52 are also frequently overexpressed in mutBRCA1-related cancers to compensate for a deficient homologous recombination pathway, enhancing tumour progression and metastasis (Sullivan et al., 2016; Wiegmans et al., 2015). Compelling evidence has also established survivin upregulation as a crucial marker of chemoresistance and tumour aggressiveness. In fact, besides its association with an enhanced homologous recombination activity and cell cycle progression, survivin overexpression has also been related to enhanced drug export by cancer cells and subsequent therapeutic resistance (Singh et al., 2015; Vequaud et al., 2016). Interestingly, decreased survivin expression levels in both nucleus and cytoplasm was observed in tumour tissues collected from BBIT20-treated mice. Collectively, the reduction of key prognostic markers as survivin, FoxM1 and RAD51 by BBIT20 could greatly improve the therapeutic outcomes of cancer patients. Additionally, based on the inhibition of BRCA2 expression by BBIT20, it would also be interesting to study its effect on deficient BRCA2-related cancers, once BBIT20 led to a full disruption of the DNA damage repair pathway, thus sensitizing cancer cells to chemotherapy.
Under cell cycle arrest and accumulated unrepaired DNA damage, cell death mechanisms are activated. Accordingly, BBIT20 treatment triggered an in vitro and in vivo apoptotic cell death with elevated cellular levels of DNA double-strand break. Notably, the disruption of the BRCA1-BARD1 heterodimer with subsequent accumulation of BRCA1 at the cytoplasm, as observed upon BBIT20 treatment, has also been associated with induction of apoptosis (Takaoka & Miki, 2018).
Besides its potential in monotherapy, BBIT20 also showed great promising in combination therapy with PARP inhibitors and platinum drugs, acting as a second hit in a synthetic lethal approach, inhibiting the homologous DNA repair activity and enhancing the cytotoxic effect of DNA-damaging agents. Indeed, BBIT20 significantly reduced the effective dose of olaparib and cisplatin, increasing their apoptotic potential with concomitant reduction of toxic side effects commonly associated with these chemotherapeutic agents. Resistance to cisplatin is also a limiting factor to its clinical use (Tan, Xu, et al., 2019). The results obtained with BBIT20 in platinum resistant patient-derived ovarian cancer #1 cells are of particular interest. In fact, although cancer cells resistant to a particular treatment are often resistant to other drugs, patient-derived ovarian cancer #1 cells showed no cross resistance to BBIT20. Importantly, BBIT20 (re)sensitized triple-negative breast and ovarian cancer cells, including platinum resistant patient-derived ovarian cancer #1 cells, to the effect of cisplatin and olaparib, resulting in a pronounced reduction of their effective doses. It is also worth noting that despite triple-negative breast and ovarian cancer predisposition to acquire therapeutic resistance, these cells did not develop resistance to BBIT20. Interestingly, in a previous work, BBIT20 was described as a potent inhibitor of P-glycoprotein (ABCB1) in resistant cancer cells (Paterna et al., 2017), further strengthening its great potential on the reversion of therapeutic resistance.
In conclusion, this work discovers a new and effective inhibitor of homologous DNA repair capable of disrupting the BRCA1-BARD1 interaction. Indeed, despite the crucial role of BRCA1–BARD1 heterodimer in tumourigenesis, effective regulators of this interaction were still missing. These results support the promising application of BBIT20 in the treatment of resistant and aggressive cancers, particularly triple-negative breast and ovarian cancer, either as mono or combination therapy. Besides its contribution to the advance of personalized cancer therapy, BBIT20 may also represent the starting point for the development of new and improved pharmacological agents against hard-to-treat cancers that still lack effective therapeutic options.
Supplementary Material
What is already known
Triple-negative breast and ovarian cancers remain hard to treat often developing therapeutic resistance.
BRCA1 tumour suppressor protein is a key player in patients therapeutic response.
What this study adds
BBIT20 is a new and effective homologous DNA repair inhibitor.
BBIT20 displays promising in vitro/in vivo antitumour activity in triple-negative breast and ovarian cancers.
Clinical significance
BBIT20 reveals great potential in the personalized treatment of aggressive and resistant cancers.
ACKNOWLEDGEMENTS
This work received the financial support of FCT/MCTES (Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through UIDB/50006/2020, SAICTPAC/0019/2015 and PTDC/MED-QUI/30591/2017. We also thank for the financial support under the GenomePT project (POCI-01-0145-FEDER022184), supported by COMPETE 2020 – Operational Programme for Competitiveness and Internationalisation (POCI), Lisboa Portugal Regional Operational Programme (Lisboa2020), Algarve Portugal Regional Operational Programme (CRESC Algarve2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF), and by Fundação para a Ciência e a Tecnologia (FCT); FCT financial support through PhD fellowships SFRH/BD/117949/2016 (L. Raimundo), PD/BD/135 291/2017 (D. Cardoso) SFRH/BD/139760/2018 (J. Ribeiro), SFRH/ BD/115099/2016 (S.J. Neto), 2020.04613.BD (J. Calheiros) and the Programa Operacional Potencial Humano (POCH), specifically the BiotechHealth Programme (Doctoral Programme on Cellular and Molecular Biotechnology Applied to Health Sciences; PD/00016/2012). We also thank the AIRC IG 2020 - ID. 25179 project (S. Indraccolo). We thank Dr. Giorgia Nardo, Lidia Moserle and Simona Agata for their help with NGS analysis of BRCA status in patient-derived ovarian cancer samples.
Funding information
FCT, Grant/Award Numbers: PTDC/MEDQUI/30591/2017, SAICTPAC/0019/2015, UIDB/50006/2020, POCI-01-0145-FEDER-022184
Abbreviations:
- BARD1
BRCA1-associated ring domain protein
- BRCA1
Breast cancer type 1 susceptibility protein
- BBIT20
Dregamine 5-bromo-pyridin-2-ylhydrazone
- DMSO
Dimethyl sulfoxide
- H&E
Haematoxylin–Eosin
- MTS
CellTiter96®Aqueous one solution cell proliferation assay
- Mut
Mutant
- PARP
poly (ADP)-ribose polymerase
- RNA-seq
RNA sequencing wt, wild-type
Footnotes
CONFLICT OF INTEREST
A national patent request protecting the compound disclosed in this manuscript has been filed by the following authors: L.S., L.R., J.L.C., M.J.U.F., S.M., J.C. and A.P.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the British Journal of Pharmacology guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation and are commended by funding agencies and other organizations engaged with supporting research.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
All data associated with this study are present in the paper or in supporting information. Gene Expression Omnibus (GEO) will be provided upon revision request.
REFERENCES
- Abbotts R, Thompson N, & Madhusudan S. (2014). DNA repair in cancer: Emerging targets for personalized therapy. Cancer Management and Research, 6, 77–92. 10.2147/CMAR.S50497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & CGTP Collaborators. (2019). The concise guide to pharmacology 2019/20: Enzymes. British Journal of Pharmacology, 176(Suppl 1), S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Buneman OP, Cidlowski JA, Christopoulos A, Davenport AP, Fabbro D, Spedding M, Striessnig J, Davies JA, & CGTP Collaborators. (2019). The Concise Guide To Pharmacology 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessa C, Soares J, Raimundo L, Loureiro JB, Gomes C, Reis F, Soares ML, Santos D, Dureja C, Chaudhuri SR, Lopez-Haber C, Kazanietz MG, Gonçalves J, Simões MF, Rijo P, & Saraiva L. (2018). Discovery of a small-molecule protein kinase Cdelta-selective activator with promising application in colon cancer therapy. Cell Death & Disease, 9(2), 23–39. 10.1038/s41419-017-0154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalakur-Ramireddy NKR, & Pakala SB (2018). Combined drug therapeutic strategies for the effective treatment of triple negative breast cancer. Bioscience Reports, 38(1), 1–14. 10.1042/BSR20171357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, Carey JPW, Bui TN, Vijayaraghavan S, Evans KW, Yi M, Ellis DC, Cheung KL, Ellis IO, Fu S, Meric-Bernstam F, Hunt KK, & Keyomarsi K. (2018). Cyclin E overexpression sensitizes triple-negative breast cancer to Wee1 kinase inhibition. Clinical Cancer Research, 24(24), 6594–6610. 10.1158/1078-0432.CCR-18-1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, Hoyer D, Insel PA, Izzo AA, Ji Y, MacEwan DJ, Sobey CG, Stanford SC, Teixeira MM, Wonnacott S, & Ahluwalia A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Densham RM, & Morris JR (2017). The BRCA1 ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus, 8(2), 116–125. 10.1080/19491034.2016.1267092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost R, & Jonkers J. (2014). Opportunities and hurdles in the treatment of BRCA1-related breast cancer. Oncogene, 33(29), 3753–3763. 10.1038/onc.2013.329 [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Goldberg RM, Lenz HJ, Shields AF, Gibney GT, Tan AR, Brown J, Eisenberg B, Heath EI, Phuphanich S, Kim E, Brenner AJ, & Marshall JL (2019). The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA: A Cancer Journal for Clinicians, 69(4), 305–343. 10.3322/caac.21560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth DL, Turner CE, & Ellsworth RE (2019). A review of the hereditary component of triple negative breast cancer: High- and moderate-penetrance breast cancer genes, low-penetrance loci, and the role of nontraditional genetic elements. Journal of Oncology, 2019, 4382606. 10.1155/2019/4382606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, & Turchi JJ (2016). DNA repair targeted therapy: The past or future of cancer treatment? Pharmacology & Therapeutics, 160, 65–83. 10.1016/j.pharmthera.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, Hiebert S, & Bhalla KN (2014). Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget, 5(14), 5637–5650. 10.18632/oncotarget.2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, & Ohta T. (2001). The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. The Journal of Biological Chemistry, 276(18), 14537–14540. 10.1074/jbc.C000881200 [DOI] [PubMed] [Google Scholar]
- Hengel SR, Spies MA, & Spies M. (2017). Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chemical Biology, 24(9), 1101–1119. 10.1016/j.chembiol.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Sy SM, & Chen J. (2010). BRCA1 and its toolbox for the maintenance of genome integrity. Nature Reviews. Molecular Cell Biology, 11(2), 138–148. 10.1038/nrm2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indraccolo S, Tisato V, Agata S, Moserle L, Ferrari S, Callegaro M, Persano L, Palma MD, Scaini MC, Esposito G, Fassina A, Nicoletto O, Plebani M, Chieco-Bianchi L, Amadori A, D’Andrea E, & Montagna M. (2006). Establishment and characterization of xenografts and cancer cell cultures derived from BRCA1 / epithelial ovarian cancers. European Journal of Cancer, 42(10), 1475–1483. 10.1016/j.ejca.2006.01.057 [DOI] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, Lazaro JB, Kochupurakkal B, Sun H, Unitt C, Moreau LA, Sarosiek KA, Scaltriti M, Juric D, Baselga J, … Shapiro GI (2016). CDK12 inhibition reverses De novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Reports, 17(9), 2367–2381. 10.1016/j.celrep.2016.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel D, Gray C, Walia JS, & Kumar V. (2018). PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: An update of clinical trials. Current Drug Targets, 19(1), 21–37. 10.2174/1389450118666170711151518 [DOI] [PubMed] [Google Scholar]
- Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, & Glazer PM (2019). Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Science Translational Medicine, 11(492), 1–12. 10.1126/scitranslmed.aav4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Li H, Chun P, Avraham S, & Avraham HK (2002). Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene, 21(50), 7730–7739. 10.1038/sj.onc.1205971 [DOI] [PubMed] [Google Scholar]
- Khongkow P, Karunarathna U, Khongkow M, Gong C, Gomes AR, Yagüe E, Monteiro LJ, Kongsema M, Zona S, Man EP, Tsang JW, Coombes RC, Wu KJ, Khoo US, Medema RH, Freire R, & Lam EW (2014). FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene, 33(32), 4144–4155. 10.1038/onc.2013.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Jin H, Seo HR, Lee HJ, & Lee YS (2019). Regulating BRCA1 protein stability by cathepsin S-mediated ubiquitin degradation. Cell Death and Differentiation, 26(5), 812–825. 10.1038/s41418-018-0153-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim A, Sharip A, Sharip A, Jiang J, Yang Q, & Xie Y. (2017). Reverse the resistance to PARP inhibitors. International Journal of Biological Sciences, 13(2), 198–208. 10.7150/ijbs.17240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Dearnaley WJ, Varano AC, Winton CE, Gilmore BL, Alden NA, Sheng Z, & Kelly DF (2017). Structural analysis of BRCA1 reveals modification hotspot. Science Advances, 3(9), e1701386. 10.1126/sciadv.1701386 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lilley E, Stanford SC, Kendall DE, Alexander SP, Cirino G, Docherty JR, George CH, Insel PA, Izzo AA, Ji Y, Panettieri RA, Sobey CG, Stefanska B, Stephens G, Teixeira M, & Ahluwalia A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177, 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabini L, Galli CL, La Fauci P, & Marinovich M. (2019). Effect of plant extracts on the genotoxicity of 10-hydroxy alkenylbenzenes. Regulatory Toxicology and Pharmacology, 105, 36–41. 10.1016/j.yrtph.2019.03.017 [DOI] [PubMed] [Google Scholar]
- Marechal A, & Zou L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspectives in Biology, 5(9), a012716. 10.1101/cshperspect.a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehanna J, Haddad FG, Eid R, Lambertini M, & Kourie HR (2019). Triple-negative breast cancer: Current perspective on the evolving therapeutic landscape. International Journal of Women’s Health, 11, 431–437. 10.2147/IJWH.S178349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Zhang C, Finch N, Zhang K, Campo L, & Breuer EK (2016). Predictors and modulators of synthetic lethality: An update on PARP inhibitors and personalized medicine. BioMed Research International, 2016, 2346585. 10.1155/2016/2346585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AC, & Holt JT (2010). Impact of RING and BRCT domain mutations on BRCA1 protein stability, localization and recruitment to DNA damage. Radiation Research, 174(1), 1–13. 10.1667/RR1290.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, & Ohta T. (2009). BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Research, 69(1), 111–119. 10.1158/0008-5472.CAN-08-3355 [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, & van Attikum H. (2019). PARP inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends in Cell Biology, 29(10), 820–834. 10.1016/j.tcb.2019.07.008 [DOI] [PubMed] [Google Scholar]
- Paterna A, Borralho PM, Gomes SE, Mulhovo S, Rodrigues CM, & Ferreira MJU (2015). Monoterpene indole alkaloid hydrazone derivatives with apoptosis inducing activity in human HCT116 colon and HepG2 liver carcinoma cells. Bioorganic & Medicinal Chemistry Letters, 25(17), 3556–3559. 10.1016/j.bmcl.2015.06.084 [DOI] [PubMed] [Google Scholar]
- Paterna A, Kincses A, Spengler G, Mulhovo S, Molnar J, & Ferreira MJU (2017). Dregamine and tabernaemontanine derivatives as ABCB1 modulators on resistant cancer cells. European Journal of Medicinal Chemistry, 128, 247–257. 10.1016/j.ejmech.2017.01.044 [DOI] [PubMed] [Google Scholar]
- Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, … Würbel H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo L, Espadinha M, Soares J, Loureiro JB, Alves MG, Santos MMM, & Saraiva L. (2018). Improving anticancer activity towards colon cancer cells with a new p53-activating agent. British Journal of Pharmacology, 175(20), 3947–3962. 10.1111/bph.14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo L, Ramos H, Loureiro JB, Calheiros J, & Saraiva L. (2020). BRCA1/P53: Two strengths in cancer chemoprevention. Biochimica et Biophysica Acta. Reviews on Cancer, 1873, 188339. 10.1016/j.bbcan.2020.188339 [DOI] [PubMed] [Google Scholar]
- Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, & Conte P. (2017). Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. The New England Journal of Medicine, 377(6), 523–533. 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- Sato K, Sundaramoorthy E, Rajendra E, Hattori H, Jeyasekharan AD, Ayoub N, Schiess R, Aebersold R, Nishikawa H, Sedukhina AS, Wada H, Ohta T, & Venkitaraman AR (2012). A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Current Biology, 22(18), 1659–1666. 10.1016/j.cub.2012.07.034 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, & Cardona A. (2012). Fiji: An open-source platform for biological-image analysis. Nature Methods, 9(7), 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Krishnakumar S, Kanwar RK, Cheung CH, & Kanwar JR (2015). Clinical aspects for survivin: A crucial molecule for targeting drug-resistant cancers. Drug Discovery Today, 20(5), 578–587. 10.1016/j.drudis.2014.11.013 [DOI] [PubMed] [Google Scholar]
- Soares J, Raimundo L, Pereira NA, dos Santos DJ, Perez M, Queiroz G, Leão M, Santos MM, & Saraiva L. (2015). A tryptophanol-derived oxazolopiperidone lactam is cytotoxic against tumors via inhibition of p53 interaction with murine double minute proteins. Pharmacological Research, 95–96, 42–52. 10.1016/j.phrs.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Soares J, Raimundo L, Pereira NA, Monteiro A, Gomes S, Bessa C, Pereira C, Queiroz G, Bisio A, Fernandes J, Gomes C, Reis F, Gonçalves J, Inga A, Santos MM, & Saraiva L. (2016). Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53–1, a novel anticancer small-molecule. Oncotarget, 7(4), 4326–4343. 10.18632/oncotarget.6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, Cramer-Morales K, McElroy DL, Ostrov DA, Haas K, Childers W, Hromas R, & Skorski T. (2016). Identification of a small molecule inhibitor of RAD52 by structure-based selection. PLoS ONE, 11(1), e0147230. 10.1371/journal.pone.0147230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka M, & Miki Y. (2018). BRCA1 gene: Function and deficiency. International Journal of Clinical Oncology, 23(1), 36–44. 10.1007/s10147-017-1182-2 [DOI] [PubMed] [Google Scholar]
- Tan WX, Xu TM, Zhou ZL, Lv XJ, Liu J, Zhang WJ, & Cui MH (2019). TRP14 promotes resistance to cisplatin by inducing autophagy in ovarian cancer. Oncology Reports, 42(4), 1343–1354. 10.3892/or.2019.7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Wang Q, Xie Y, Qiao X, Zhang S, Wang Y, Yang Y, & Zhang B. (2019). Identification of FOXM1 as a specific marker for triplenegative breast cancer. International Journal of Oncology, 54(1), 87–97. 10.3892/ijo.2018.4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, & Sung P. (2020). The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nature Reviews. Molecular Cell Biology, 21, 284–299. 10.1038/s41580-0200218-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C, Kim EH, Royce DB, Williams CR, Collins RM, Kim EH, Royce DB, Williams CR, Collins RM, Risingsong R, Sporn MB, & Liby KT (2014). The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1-deficient mice. Cancer Prevention Research (Philadelphia, Pa.), 7(7), 698–707. 10.1158/1940-6207.CAPR-14-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenner A, & Sartori AA (2019). Harnessing DNA double-strand break repair for cancer treatment. Frontiers in Oncology, 9, 1388–1398. 10.3389/fonc.2019.01388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez C, Beachboard DC, & Horner SM (2017). Methods to visualize MAVS subcellular localization. Methods in Molecular Biology, 1656, 131–142. 10.1007/978-1-4939-7237-1_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vequaud E, Desplanques G, Jezequel P, Juin P, & Barille-Nion S. (2016). Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Research and Treatment, 155(1), 53–63. 10.1007/s10549-0153657-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington CR, Richardson DL, & Moore KN (2019). Olaparib in the treatment of ovarian cancer. Future Oncology, 15(30), 3435–3449. 10.2217/fon-2019-0271 [DOI] [PubMed] [Google Scholar]
- Wiegmans AP, Yap PY, Ward A, Lim YC, & Khanna KK (2015). Differences in expression of key DNA damage repair genes after epigenetic-induced BRCAness dictate synthetic lethality with PARP1 inhibition. Molecular Cancer Therapeutics, 14(10), 2321–2331. 10.1158/1535-7163.MCT-15-0374 [DOI] [PubMed] [Google Scholar]
- Yao S, Fan LY, & Lam EW (2018). The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance. Seminars in Cancer Biology, 50, 77–89. 10.1016/j.semcancer.2017.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap TA, Plummer R, Azad NS, & Helleday T. (2019). The DNA damaging revolution: PARP inhibitors and beyond. American Society of Clinical Oncology Educational Book, 39, 185–195. 10.1200/EDBK_238473 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or in supporting information. Gene Expression Omnibus (GEO) will be provided upon revision request.