

Abstract
Atherosclerosis is a chronic disorder of the vessel wall. One key regulator of disease progression is lipid handling in macrophages. However, the role of macrophage mitochondrial-dependent fatty acid β-oxidation (FAO) in atherosclerosis is not well defined. To address this, we focused on carnitine palmitoyltransferase (CPT) 1 and 2, which play an essential role in the transport of long chain fatty acids (FAs) into the mitochondria. Using conditional alleles of these mitochondrial enzymes, we have generated myeloid-specific Cpt1a and Cpt2 knockout mutants (CPT1a M-KO and CPT2 M-KO). In culture, macrophages derived from CPT1a and CPT2 M-KO mice have impaired FAO, enhanced expression of the CD36 scavenger receptor, increased uptake of oxidized low-density lipoprotein (oxLDL), and augmented transformation into cholesterol-rich foam cells. In line with these in vitro observations, in the atherosclerosis-susceptible apolipoprotein E (ApoE) KO background, CPT2 M-KO mice demonstrated augmented atherosclerosis, accompanied by increased accumulation of aortic macrophages with elevated CD36 expression. These data suggest that macrophage FAO is athero-protective and that augmenting FAO may potentially slow atherosclerotic progression.
1. Introduction
Mitochondrial-dependent fatty acid β-oxidation (FAO) is an essential step in the generation of energy from fatty acids (FAs). FAO, in conjunction with other substrates including glucose and amino acids, plays a crucial role in maintaining whole-body metabolic homeostasis by generating adenosine triphosphate (ATP). The regulation of the oxidation of long-chain FAs is in large part determined by the carnitine palmitoyltransferase (CPT) system, which involves two mitochondrial membrane enzymes, CPT1 and CPT2 (Supplemental Fig. 1A) [1]. This system allows for the transport of long-chain acylcarnitines (ACs), metabolites of long-chain FAs, across the outer and inner mitochondrial membranes. In mammals, depending on the cell type, outer mitochondrial membrane long-chain AC transport is achieved by either CPT1a or CPT1b (the muscle specific isoform). In contrast, transport of long-chain ACs across the inner mitochondrial membrane is achieved by CPT2, which is expressed ubiquitously in all cell types.
Lipids play a central role in atherosclerosis. In particular, the development and progression of this disease is catalyzed by the accumulation of lipoproteins, excessive (ester form) cholesterol, and macrophage-derived foam cells that can expand and proliferate within the vessel wall. Local production of cytokines by vessel wall macrophages, which are derived from circulating blood monocytes, result in chronic inflammation and also stimulate modified (e.g., oxidized) low-density lipoprotein (LDL)-induced foam cell formation via scavenger receptors such as CD36 [2,3]. CD36 also plays a central role in oxidized LDL (oxLDL)-induced NLRP3 inflammasome activation, which can also participate in atherogenesis [4,5]. Furthermore, inhibition of FA-induced endoplasmic reticulum (ER) stress in macrophages can protect against atherosclerosis [6].
Recent evidence has revealed that macrophage FAO may play a crucial role in the cellular phenotype of these specialized cells [3,7,8]. Importantly, initial reports using the CPT1a inhibitor etomoxir suggested that macrophage FAO promotes the maturation of M2 alternative activation and is essential for the anti-inflammatory effects of this cell type [9–12]. In line with these observations, using human (THP-1) or murine (RAW 264.7) macrophage cell lines, two recent studies demonstrated that FAO protects against FA (palmitate)-induced ER stress and inflammatory signaling [13,14]. These studies would imply that macrophage FAO might have an overall anti-inflammatory effect. In contrast, some studies have suggested that macrophage FAO seems to contribute to a pro-inflammatory and pro-atherogenic milieu. For instance, in J774.2 murine macrophages, inhibition of FAO by etomoxir alleviated bactericidal activity by reducing mitochondrial reactive oxygen species (ROS) production [15]. Increased FAO has been linked to the redox-dependent activation of the NLRP3 inflammasome [16,17] and in bone marrow-derived macrophages (BMDMs), knockdown of Cpt1a by short hairpin RNA or etomoxir treatment suppressed NLRP3 inflammasome activation [18]. Thus, there are potentially discordant results in the literature regarding whether FAO in macrophages would theoretically act to inhibit or promote atherosclerosis. However, it should be noted that these previous studies all reflect in vitro assessments, often with immortalized cell lines rather than primary macrophages. Indeed, no study to date, has directly assessed how macrophage substrate metabolism effects atherosclerosis. Moreover, many, if not all of these past studies, relied on pharmacological means to inhibit FAO. This is potentially problematic since our group using a genetic strategy has demonstrated that FAO is not essential for macrophage M2 activation and that concentrations of etomoxir that are often employed in the literature have significant off-target effects [19]. Additional examples of the off-target effects of etomoxir have also been described recently by other groups [20,21]. Thus, it would seem that to unequivocally assess the role of macrophage FAO in atherosclerosis in vivo genetic strategies are required.
In this study, we characterize BMDMs from myeloid-specific Cpt1a and Cpt2 knockout mice (CPT1a M-KO and CPT2 M-KO, respectively). Here, we demonstrate that inhibition of FAO in myeloid cells augments foam cell formation and CD36 expression. Furthermore, the absence of FAO activity in macrophages exacerbates in vivo atherosclerosis in the setting of the apolipoprotein E (ApoE) knockout atherosclerosis-prone mouse. Taken together, our work demonstrates that manipulating macrophage metabolism may provide a novel strategy to reduce atherogenesis.
2. Methods
Detail methods are available in the online supplement.
3. Results and discussion
We first sought to characterize the components of the CPT system that were operational in BMDMs (Supplemental Fig. 1A). Previously, we had demonstrated that the inner mitochondrial membrane protein CPT2 is expressed in BMDMs and deletion of Cpt2 inhibits FAO [19,22]. As expected, expression analysis confirmed that BMDM also express the outer mitochondrial membrane Cpt1a isoform, and not the muscle-specific Cpt1b isoform (Supplemental Fig. 1B). To further understand how FAO might regulate macrophage function, we took advantage of a myeloid-specific deletion of Cpt2 (Cpt2fl/flLyz2-Cre; called ‘CPT2 M-KO’ here), which have been described in previous studies [19,20,22]. To complement and extend these studies, we also generated the Cpt1afl/fl mouse strain using a purchased embryonic stem (ES) cell line followed by CRISPR/Cas9 modification (see Methods). These Cpt1afl/fl mice were subsequently crossed with Lyz2-Cre mice [23] to generate mice with myeloid-specific deletion of Cpt1a (Cpt1afl/flLyz2-Cre; called ‘CPT1a M-KO’ here). Quantitative PCR and Western blot analyses confirmed the faithful deletion of CPT1a in BMDMs (Supplemental Fig. 1C, D). Consistent with the observation that Cpt1a but not Cpt2 is often regulated transcriptionally [24], we observed that knockout of Cpt1a did not alter downstream Cpt2 expression, while knockout of Cpt2 modestly increased upstream Cpt1a expression in the presence of oleic acid (Supplemental Fig. 1E).
While deletion of either Cpt1a or Cpt2 would each be expected to abrogate FAO, these genetic manipulations are known to have opposite effects on long-chain AC levels. In particular, based on their known enzymatic activity, deletion of Cpt1a should reduce long-chain AC formation, while Cpt2 deletion should elevate levels of these intermediaries (see Supplemental Fig. 1A). This biochemical difference is the clinical basis for discriminating whether patients with inherited disorders of FAO have mutations in either CPT1 or CPT2 [1,24]. Metabolomic analysis of ACs revealed the expected alterations in long-chain AC levels in CPT1a M-KO and CPT2 M-KO derived macrophages and both Cpt1a and Cpt2 deletion reduced short/medium-chain ACs levels (Supplemental Fig. 1F).
In order to assess the metabolic consequences of disrupting FAO, we next assessed basal and maximal respiration in control (wild type), CPT1a M-KO and CPT2 M-KO derived macrophages. In the presence of media containing high glucose (25 mM) and fetal bovine serum (which contains some FAs; denoted as ‘Glu’ in figures), there was a non-significant reduction in both basal and maximal oxygen consumptions rates (OCR) in macrophages lacking either Cpt1a or Cpt2 (Supplemental Fig. 1G). These energetic deficits were, however, much more apparent when macrophages were supplemented with a medium containing physiological levels of carnitine and oleic acid (150 μM), the most abundant FA found within atheroma [25]. Indeed, when BMDMs were given oleic acid as a primary substrate (denoted as ‘Ole’ in figures), both CPT1a M-KO and CPT2 M-KO derived macrophages exhibited a reduction in basal and maximal mitochondrial respiration (Supplemental Fig. 1G). We also observed similar FA-dependent metabolic differences when the respiration of CPT1a M-KO macrophages were assessed in the presence of bovine serum albumin (BSA) alone or in the presence of palmitate-BSA (Supplemental Fig. 1H). Consistent with this impaired ability to increase mitochondrial respiration in the setting of a FA substrate, CPT1a M-KO and CPT2 M-KO derived macrophages had reduced ATP levels when given oleic acid as the primary substrate (Supplemental Fig. 1I). In previous studies, hepatic-specific Cpt2 knockouts and endothelial-specific Cpt1a knockouts revealed evidence of oxidative stress [26,27]. Interestingly, levels of mitochondrial ROS were also elevated in CPT1a M-KO and CPT2 M-KO macrophages, particularly when macrophage ROS levels were assessed in the presence of exogenous oleic acid (Supplemental Fig. 1J).
We reasoned that the inability to metabolize FA might lead to the accumulation of lipid substrates within CPT1a M-KO and CPT2 M-KO macrophages. Consistent with this, in CPT1a M-KO and CPT2 M-KO macrophages, lipid staining by boron-dipyrromethene (BODIPY) 493/503 was modestly elevated in normal medium, but markedly elevated in the presence of oleic acid (Fig. 1A, B). Previous analysis has demonstrated that the uptake of modified LDL is achieved in part through the CD36 receptor [2]. This scavenger receptor is regulated by a series of FA-dependent transcription factors including peroxisome proliferator-activated receptor gamma (PPARγ) [28,29]. We hypothesized that disruption of FAO within BMDMs might therefore alter the lipid-dependent regulation of CD36 expression. Consistent with this notion, we noted that particularly in the presence of oleic acid, levels of CD36 messenger RNA (mRNA) and protein were elevated in CPT1a M-KO and CPT2 M-KO macrophages (Fig. 1C, D, and Supplemental Fig. 2A), while other scavenger receptors, such as scavenger receptor A (SR-A) (encoded by Msr1) or Olr1 (which is only expressed in LPS-activated cells and encodes lectin-like oxidized LDL receptor 1 (LOX1)) were not altered (Supplemental Fig. 2B–D). To understand the physiological and functional effects of such dysregulation, we next assessed oxLDL-induced foam cell formation in control, CPT1a M-KO or CPT2 M-KO macrophages. Cells were first exposed to oleic acid for 24 h (to induce CD36 expression) followed by incubation with oxLDL for 24 h. As noted, lipid accumulation, as measured by BODIPY staining and total cholesterol accumulation, were markedly increased in FAO-deficient macrophages, while levels of free FAs were not affected (Fig. 1E–G, and Supplemental Fig. 2E). This increase was inhibited by treating cells with sulfosuccinimidyl oleate (SSO), a specific CD36 inhibitor (Supplemental Fig. 2F, G). Recent observations have demonstrated that low concentrations of etomoxir (3 μM), a CPT1a inhibitor, effectively inhibits FAO without producing significant off-target effects such as reducing M2 polarization of BMDMs [20]. As expected, etomoxir treatment of wild type macrophages mimics the effects of genetic disruption of Cpt1a or Cpt2 deletion and significantly increased CD36 expression, oxLDL uptake and foam cell formation (Supplemental Fig. 3A–D). Consistent with the notion that CPT1A and CPT2 work sequentially in the import of FAs and that both proteins are required for macrophage FAO, we observed that in CPT2M-KO macrophages knocking down Cpt1a did not further augment CD36 message or protein expression or change the magnitude of lipid accumulation seen with deletion of Cpt2 alone (Supplemental Fig. 3E–G).
Fig. 1.
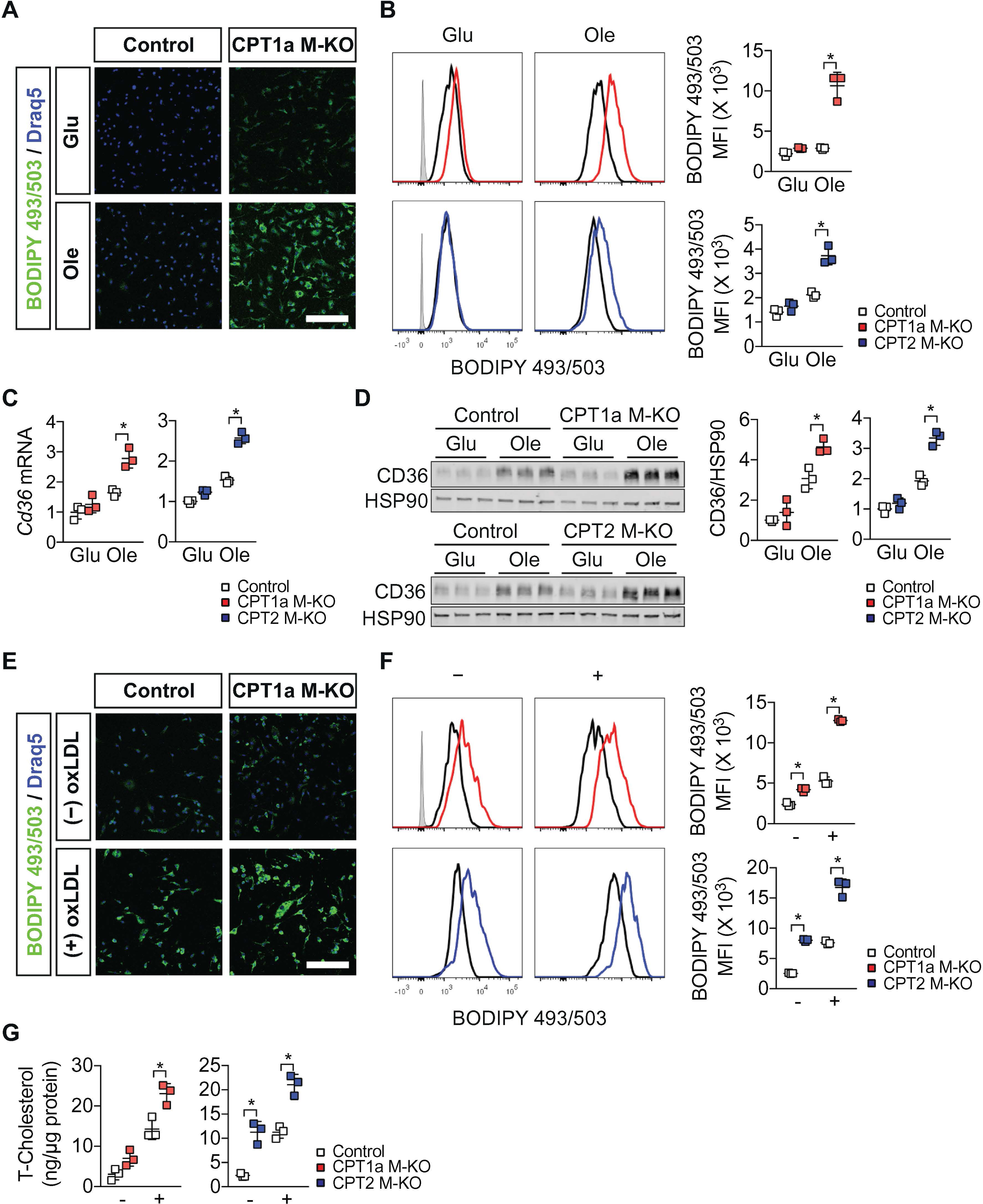
Inhibition of macrophage FAO increases CD36 expression and cholesterol accumulation. (A) Representative images of the BODIPY 493/503 (lipids; green) and Draq5 (nuclei; blue) staining in control and CPT1a BMDMs (cells obtained from n = 3 mice per group). Analysis was performed in standard high glucose media (Glu) or in the presence of a FA substrate (Ole) for 48 h. Scale bar, 100 μm. (B) Representative and quantification of flow cytometry analyzing BODIPY 493/503 staining in control, CPT1a M-KO or CPT2 M-KO BMDMs (cells obtained from n = 3 mice per group). Analysis was performed in standard high glucose media (Glu) or in the presence of a FA substrate (Ole) for 48 h. Data are representative of three independent experiments. (C) Quantitative PCR analysis of Cd36 mRNA in control, CPT1a M-KO or CPT2 M-KO BMDMs (cells obtained from n = 3 mice per group). Analysis was performed in standard high glucose media (Glu) or in the presence of a FA substrate (Ole) for 48 h. Data are representative of three independent experiments. (D) Immunoblot analysis and the quantitative densitometry of CD36 protein in total lysate of control, CPT1a M-KO or CPT2 M-KO BMDMs (cells obtained from n = 3 mice per group). HSP90 is shown as a loading control. Analysis was performed in standard high glucose media (Glu) or in the presence of a FA substrate (Ole) for 48 h. Data are representative of three independent experiments. (E) Representative images of BODIPY 493/503 (lipid) and Draq5 (nuclei) staining in control and CPT1a BMDMs after incubation of media containing a FA substrate for 24 h, followed by media with or without oxLDL (50 μg/ml) for 24 h (cells obtained from n = 3 mice per group). Scale bar, 100 μm. (F) Representative and quantification of flow cytometry analyzing the BODIPY 493/503 staining in control, CPT1a M-KO or CPT2 M-KO BMDMs after incubation of media containing a FA substrate for 24 h, followed by media with (+) or without (−) oxLDL (50 μg/ml) for 24 h (cells obtained from n = 3 mice per group). Data are representative of two independent experiments. (G) Total cellular cholesterol (T-Cholesterol) content in control, CPT1a M-KO or CPT2 M-KO BMDMs after incubation of media containing a FA substrate for 24 h, followed by media with (+) or without (−) oxLDL (50 μg/ml) for 24 h (cells obtained from n = 3 mice per group with each biological value representing a technical triplicate). Data are representative of two independent experiments. Results represent mean ± s.d. *P < .05, by one-way ANOVA with the Bonferroni correction.
Our in vitro data suggests that inhibition of macrophage FAO may increase foam cell formation and thereby exacerbate atherosclerosis. To further investigate this hypothesis, we crossed CPT2 M-KO mice to atherosclerosis-susceptible Apoe knockout mice [30] and established Cpt2+/+Lyz2-CreApoe−/− (described as “ApoE KO”) and Cpt2fl/flLyz2-CreApoe−/− (described as “ApoE CPT2 M-KO”) mice. Two separate cohorts of mice were exposed to an atherosclerosis-inducible Western diet (WD; 42% calorie from fat) or normal chow (NC) for 17 weeks (first cohort) or 20 weeks (second cohort). In the setting of Apoe deficiency, conditional deletion of Cpt2 did not alter body weight (Fig. 2A), body composition (Fig. 2B), blood glucose and insulin (Fig. 2C) or cholesterol levels (Fig. 2D). When these mice were fed a WD, we observed a significant increase in vascular lesion formation in both the thoraco-aorta and the aortic root of ApoE CPT2 M-KO mice (Fig. 2E, F). These increases were roughly 20% and 50%, respectively. In ApoE CPT2 M-KO, CD68-positive macrophages appeared to compose a larger percentage of the atherosclerotic lesion (Fig. 2G). In contrast, the ApoE CPT2 M-KO atherosclerotic lesions had similar levels of smooth muscle cells (stained by alpha smooth muscle actin (αSMA)), collagen (stained by Sirius red), and ROS (stained by dihydroethidium (DHE)) (Fig. 2G). Moreover, consistent with our in vitro observations, mRNA expression of Cd36, but not other cholesterol scavenger receptors (Msr1 and Olr1), were increased in ApoE CPT2 M-KO mice (Fig. 2H, J). A similar increase in aortic CD36 protein levels was also observed (Fig. 2I). Further analysis of other potential markers within the vessel wall (Fig. 2J) demonstrated that the aortas of ApoE CPT2 M-KO exhibited reduced Cpt2 expression, in conjunction with elevated levels of various macrophage markers (Mrc1, Clec10a, Mgl2) and increased expression of key lipid-regulated transcription (co)factors and chaperones (Pparg, Ppargc1b, Fabp4).
Fig. 2.
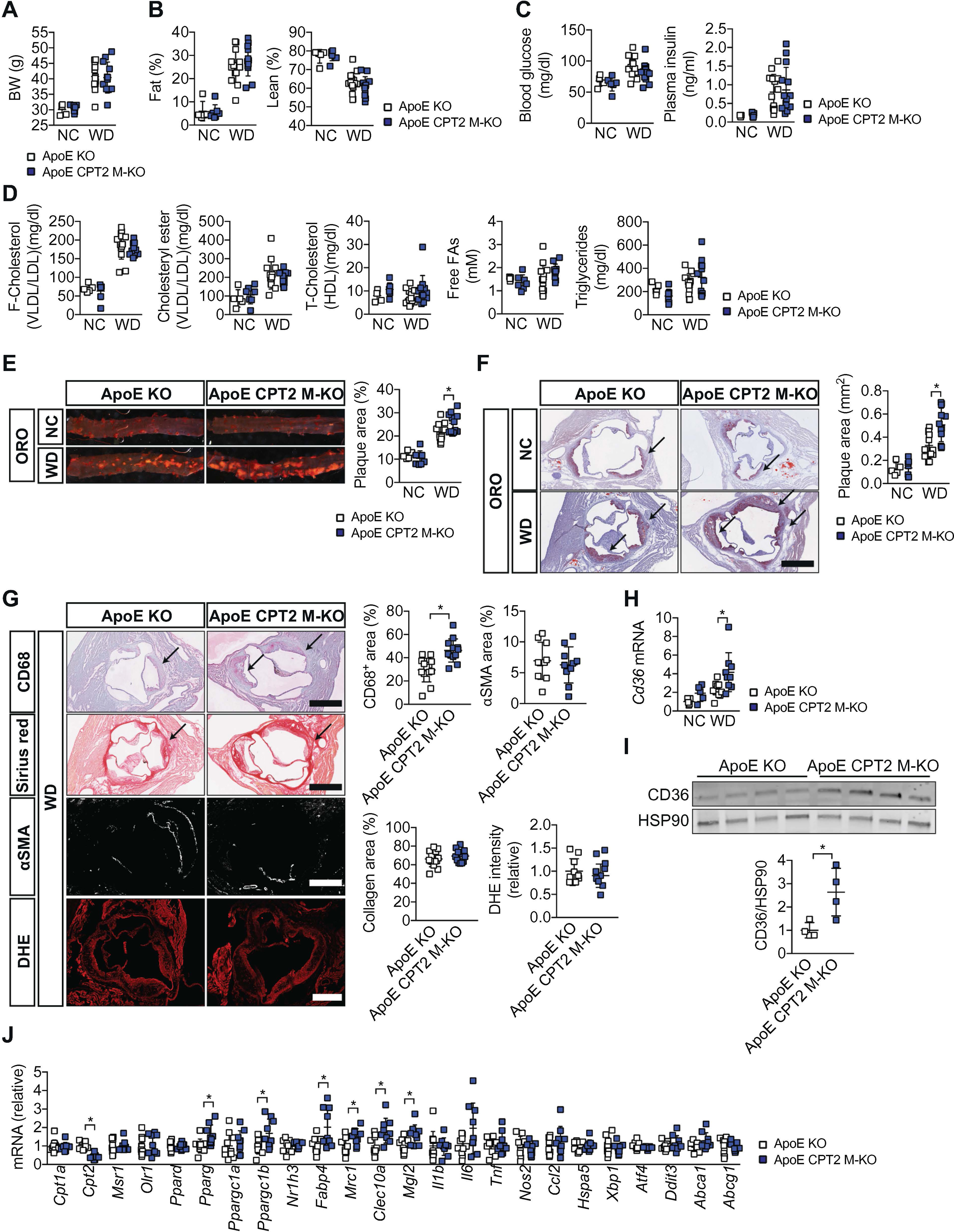
Inhibition of myeloid cell FAO exacerbates atherosclerosis. (A) Body weight of ApoE KO and ApoE CPT2 M-KO mice fed a normal show (NC) or Western diet (WD) for 17 weeks (n = 5–14 mice per group). (B) Fat and lean mass of ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 15 weeks (n = 5–14 mice per group). (C) Fasting (18 h) blood glucose and plasma insulin levels in ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 10 weeks (n = 5–14 mice per group). (D) Fasting (5 h) plasma lipid levels in ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 17 weeks (n = 5–14 mice per group). (E) Oil Red O (ORO) staining of the thoracoabdominal aorta and quantification of the plaque area of ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 17 weeks (n = 5–14 mice per group). (F) ORO staining of the aortic root and quantification of the plaque area of ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 17 weeks (n = 5–14 mice per group). Scale bar, 500 μm. (G) CD68 (macrophages), Sirius Red (collagen), alpha smooth muscle actin (αSMA), and dihydroethidium (DHE) (ROS) staining of the aortic root and quantification of the staining area of ApoE KO and ApoE CPT2 M-KO mice fed WD for 17 weeks (n = 10–14 mice per group). Scale bars, 500 μm. (H) Quantitative PCR analysis of Cd36 mRNA in the thraco-abdominal aorta of ApoE KO and ApoE CPT2 M-KO mice fed NC or WD for 20 weeks (n = 5–10 mice per group). (I) Immunoblot analysis and the quantitative densitometry of CD36 protein in total lysate of the thraco-abdominal aorta of ApoE KO and ApoE CPT2 M-KO mice fed WD for 20 weeks (n = 4 mice per group). (J) Quantitative PCR analysis of mRNA of Cpt1a, Cpt2, scavenger receptors (Msr1 and Olr1), lipid-regulated transcriptional (co) factors and chaperone (Ppard, Pparg, Ppargc1a, Ppargc1b, Nr1h3, Fabp4), macrophage markers (Mrc1, Clec10a, Mgl2), inflammatory markers (Il1b, Il6, Tnf, Nos2, Ccl2), ER stress markers (Hspa5, Xbp1, Atf4, Ddit3), and cholesterol efflux transporters (Abca1, Abcg1) in the thraco-abdominal aorta of ApoE KO and ApoE CPT2 M-KO mice fed WD for 20 weeks (n = 9–10 mice per group). Results represent mean ± s.d. *P < .05, by one-way ANOVA with the Bonferroni correction or unpaired two-tailed Studen’s t-test (G, I, and J).
Recently, several studies have demonstrated that FAO may play an important role in various macrophage functions including M2 polarization [9–12], ER stress [13,14], mitochondrial ROS production [15], and NLRP3 inflammasome activation [18]. The limitation of these studies, however, is that they have primarily used pharmaceutical approaches, or they have been limited to in vitro analysis, often using immortalized cell lines. Here, we have extended these efforts using a more rigorous genetic approach that allows for the in vivo physiological assessment following the manipulation of macrophage metabolism.
We had previously shown that deletion of Cpt2 in macrophages does not alter the ability of these cells to polarize to an M2 phenotype [19,22]. These results were in contrast to what had been observed with the CPT1a inhibitor etomoxir, which was reported to block M2 induction [9,10,12]. Interestingly, recent studies have demonstrated that high concentration of etomoxir (200 μM) appears to inhibit M2 activation not through the inhibition of FAO, but rather via depletion of coenzyme A [20]. As we previously noted following Cpt2 deletion [19,22], we also observed that deletion of Cpt1a did not alter interleukin (IL)-4 stimulated M2 induction or LPS-induced inflammatory M1 genes (with or without IL-4 stimulation) (Supplemental Fig. 4A, B). In contrast to previous reports [13,14,18], CPT1a and CPT2 M-KO macrophages did not appear to differ in their response to palmitate-induced c-Jun N-terminal kinase (JNK) activation, inflammatory and ER stress response genes (Supplemental Fig. 4C, D) or to stimuli known to induce NLRP3 inflammasome activation (Supplemental Fig. 5A–C). Nonetheless, we did note that inhibition of FAO increased mitochondrial ROS production (Supplemental Fig. 1I). Again, we believe the differences with some of the previous literature largely reflects differences in experimental design, as none of these previous studies have characterized stable genetic knockouts of primary mouse macrophages.
Our results suggest that in macrophages, CD36 expression is modulated by FAO. Since we observed that ROS levels increased following inhibition of FAO (Supplemental Fig. 1I), based on some previous observations [31], we hypothesized that CD36 levels might be sensitive to the cellular redox state. However, we did not observe any significant effect following addition of the antioxidant N-acetyl-L-Cysteine (NAC) on either CD36 levels or foam cell formation in either control or Cpt1a-deficient macrophages (Supplemental Fig. 6A–D). We also assessed whether supplementation with acetate, which is converted to acetyl-CoA by acyl-CoA synthetase short chain family member 1 (ACSS1) [27], could rescue FAO-deficient macrophages. Although acetate supplementation rescued ATP production in Cpt1a-deficient cells (Supplemental Fig. 6E), CD36 expression and foam cell formation were not altered after acetate supplementation (Supplemental Fig. 6F–H).
A number of transcription factor use FA ligands as cofactors, most notably the PPAR and liver X receptor (LXR) family of transcription factors [29,32]. Previous studies have demonstrated that the expression levels of hepatic Cd36 is also robustly increased in Cpt2 liver-specific knockout mice when fasted or placed on a ketogenic diet [33]. Similarly, in skeletal muscle, CD36 is also significantly increased in Cpt1b skeletal muscle-specific knockout mice [34–36]. Significant evidence suggests that macrophage CD36 is regulated by PPARγ [29]. Consistent with this, in these other models where FAO is genetically abrogated/ activated, activity of the PPAR family of transcription factors is induced/reduced, possibly by modulating the availability of endogenous FA ligands [33,37]. Interestingly, we also saw evidence of increased level of Pparg in the vessel wall of the ApoE CPT2 M-KO mice (Fig. 2J). However, a number of other observations argue against this pathway being substantially activated in our FAO-deficient cells. This includes the fact that the expression levels of the cholesterol efflux transporters and indirect (through LXR) PPARγ targets ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1) were reduced in knockout cells, as was ABCA1-dependent cholesterol efflux (Supplemental Fig. 7A–C). Moreover, the PPARγ inhibitors (SR202 and T0070907) did not reduce the elevated levels of CD36 observed in CPT1a M-KO or CPT2 M-KO cells (Supplemental Fig. 7D, E) and PPARγ expression was actually reduced in FAO-deficient BMDMs (Supplemental Fig. 7F, G). Thus, the precise mechanism through which a defect in macrophage FAO leads to an induction of CD36 expression will require additional studies.
In summary, our analysis suggests that inhibiting the ability of macrophages to use FA as a substrate results in increased foam cell formation. Moreover, in vivo, this metabolic defect in macrophages leads to increased atherogenesis. It is interesting to note that a recent report demonstrated that endothelial knockout of Cpt1a induced vascular leakiness and leukocyte infiltration [27], which suggests endothelial FAO may also be athero-protective. In general, rates of FAO decline as we age [38]. Whether or not this general decline in FA extends to specific cellular compartments such as macrophages is not known. Nonetheless, our results raise the possibility that therapies aimed at slowing or reversing this decline in FAO may have beneficial effects.
Supplementary Material
Acknowledgements
This research was supported by the National Institute of Health (Intramural Funds), Foundation Leducq (T.F.) and Japan Society for the Promotion of Science (M.N.). We are grateful to members of the Finkel and Mukoyama lab, and to the NHLBI Flow Cytometry Core.
Footnotes
Conflict of interest
None declared.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2019.01.003.
References
- [1].Houten SM, Violante S, Ventura FV, Wanders RJ, The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders, Annu. Rev. Physiol. 78 (2016) 23–44. [DOI] [PubMed] [Google Scholar]
- [2].Moore KJ, Sheedy FJ, Fisher EA, Macrophages in atherosclerosis: a dynamic balance, Nat Rev Immunol 13 (10) (2013) 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Koelwyn GJ, Corr EM, Erbay E, Moore KJ, Regulation of macrophage immunometabolism in atherosclerosis, Nat. Immunol. 19 (6) (2018) 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ, CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation, Nat. Immunol. 14 (8) (2013) 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E, NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals, Nature 464 (7293) (2010) 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, Hotamisligil GS, Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis, Nat. Med. 15 (12) (2009) 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Van den Bossche J, O’Neill LA, Menon D, Macrophage Immunometabolism: where are we (going)? Trends Immunol. 38 (6) (2017) 395–406. [DOI] [PubMed] [Google Scholar]
- [8].O’Neill LA, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists, Nat Rev Immunol 16 (9) (2016) 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A, Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation, Cell Metab. 4 (1) (2006) 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr., Artyomov MN, Pearce EL, Pearce EJ, Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages, Nat. Immunol. 15 (9) (2014) 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ, Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation, Immunity 45 (4) (2016) 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, Espinosa EC, Lamming D, Manning BD, Zhang Y, Blair IA, Horng T, Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation, Elife 5 (2016) e11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, Escote X, Gomez-Serrano M, Peral B, Salvado L, Fernandez-Veledo S, Casals N, Vazquez-Carrera M, Villarroya F, Vendrell JJ, Serra D, Herrero L, Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation, Am. J. Physiol. Endocrinol. Metab. 308 (9) (2015) E756–E769. [DOI] [PubMed] [Google Scholar]
- [14].Namgaladze D, Lips S, Leiker TJ, Murphy RC, Ekroos K, Ferreiros N, Geisslinger G, Brune B, Inhibition of macrophage fatty acid beta-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses, Diabetologia 57 (5) (2014) 1067–1077. [DOI] [PubMed] [Google Scholar]
- [15].Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, Ellett F, Lieschke GJ, Crosier KE, Crosier PS, Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ROS production, Cell Metab 18 (2) (2013) 265–278. [DOI] [PubMed] [Google Scholar]
- [16].Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation, Nature 469 (7329) (2011) 221–225. [DOI] [PubMed] [Google Scholar]
- [17].Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP, Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling, Nat. Immunol. 12 (5) (2011) 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabon MA, Rooney KT, Yoon JH, Ryter SW, Stout-Delgado H, Choi AM, NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages, Nat. Med. 22 (9) (2016) 1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [19].Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T, Fatty acid oxidation in macrophage polarization, Nat. Immunol. 17 (3) (2016) 216–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, Andreyev AY, Bowman CE, Caradonna K, Dranka BP, Ferrick DA, Liesa M, Stiles L, Rogers GW, Braas D, Ciaraldi TP, Wolfgang MJ, Sparwasser T, Berod L, Bensinger SJ, Murphy AN, Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis, Cell Metab. 28 (3) (2018) 490–503 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Van den Bossche J, van der Windt GJW, Fatty acid oxidation in macrophages and T cells: time for reassessment? Cell Metab. 28 (4) (2018) 538–540. [DOI] [PubMed] [Google Scholar]
- [22].Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, Wolfgang MJ, Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction, Am. J. Physiol. Endocrinol. Metab. 312 (5) (2017) E381–E393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I, Conditional gene targeting in macrophages and granulocytes using LysMcre mice, Transgenic Res. 8 (4) (1999) 265–277. [DOI] [PubMed] [Google Scholar]
- [24].Knottnerus SJG, Bleeker JC, Wust RCI, Ferdinandusse S, IJlst L, Wijburg FA, Wanders RJA, Visser G, Houtkooper RH, Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle, Rev. Endocr. Metab. Disord. 19 (1) (2018) 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, Kopf M, Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis, Nat. Immunol. 14 (10) (2013) 1045–1053. [DOI] [PubMed] [Google Scholar]
- [26].Lee J, Choi J, Selen Alpergin ES, Zhao L, Hartung T, Scafidi S, Riddle RC, Wolfgang MJ, Loss of hepatic mitochondrial long-chain fatty acid oxidation confers resistance to diet-induced obesity and glucose intolerance, Cell Rep. 20 (3) (2017) 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Bruning U, Scheinok S, Treps L, Cantelmo AR, Dubois C, de Zeeuw P, Goveia J, Zecchin A, Taverna F, Morales-Rodriguez F, Brajic A, Conradi LC, Schoors S, Harjes U, Vriens K, Pilz GA, Chen R, Cubbon R, Thienpont B, Cruys B, Wong BW, Ghesquiere B, Dewerchin M, De Bock K, Sagaert X, Jessberger S, Jones EAV, Gallez B, Lambrechts D, Mazzone M, Eelen G, Li X, Fendt SM, Carmeliet P, Quiescent endothelial cells upregulate fatty acid beta-oxidation for vasculoprotection via redox homeostasis, Cell Metab. 28 (6) (2018) 881–894 e13. [DOI] [PubMed] [Google Scholar]
- [28].Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM, PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL, Cell 93 (2) (1998) 241–252. [DOI] [PubMed] [Google Scholar]
- [29].Chawla A, Control of macrophage activation and function by PPARs, Circ. Res. 106 (10) (2010) 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL, Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells, Cell 71 (2) (1992) 343–353. [DOI] [PubMed] [Google Scholar]
- [31].Yang X, Yao H, Chen Y, Sun L, Li Y, Ma X, Duan S, Li X, Xiang R, Han J, Duan Y, Inhibition of glutathione production induces macrophage CD36 expression and enhances cellular-oxidized low density lipoprotein (oxLDL) Uptake, J. Biol. Chem. 290 (36) (2015) 21788–21799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang B, Tontonoz P, Liver X receptors in lipid signalling and membrane homeostasis, Nat. Rev. Endocrinol. 14 (8) (2018) 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee J, Choi J, Scafidi S, Wolfgang MJ, Hepatic fatty acid oxidation restrains systemic catabolism during starvation, Cell Rep. 16 (1) (2016) 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, Mynatt RL, Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism, Proc. Natl. Acad. Sci. U. S. A. 112 (25) (2015) E3300–E3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Warfel JD, Bermudez EM, Mendoza TM, Ghosh S, Zhang J, Elks CM, Mynatt R, Vandanmagsar B, Mitochondrial fat oxidation is essential for lipid-induced inflammation in skeletal muscle in mice, Sci. Rep. 6 (2016) 37941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Warfel JD, Vandanmagsar B, Wicks SE, Zhang J, Noland RC, Mynatt RL, A low fat diet ameliorates pathology but retains beneficial effects associated with CPT1b knockout in skeletal muscle, PLoS One 12 (12) (2017) e0188850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].van Weeghel M, Abdurrachim D, Nederlof R, Argmann CA, Houtkooper RH, Hagen J, Nabben M, Denis S, Ciapaite J, Kolwicz SC Jr., Lopaschuk GD, Auwerx J, Nicolay K, Des Rosiers C, Wanders RJ, Zuurbier CJ, Prompers JJ, Houten SM, Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B, Cardiovasc. Res. 114 (10) (2018) 1324–1334. [DOI] [PubMed] [Google Scholar]
- [38].Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, Auwerx J, The metabolic footprint of aging in mice, Sci. Rep. 1 (2011) 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.