

Abstract
Helicobacter pylori infection (Hp-I) represents a typical microbial agent intervening in the complex mechanisms of gastric homeostasis by disturbing the balance between the host gastric microbiota and mucosa-related factors, leading to inflammatory changes, dysbiosis and eventually gastric cancer. The normal gastric microbiota shows diversity, with Proteobacteria [Helicobacter pylori (H. pylori) belongs to this family], Firmicutes, Actinobacteria, Bacteroides and Fusobacteria being the most abundant phyla. Most studies indicate that H. pylori has inhibitory effects on the colonization of other bacteria, harboring a lower diversity of them in the stomach. When comparing the healthy with the diseased stomach, there is a change in the composition of the gastric microbiome with increasing abundance of H. pylori (where present) in the gastritis stage, while as the gastric carcinogenesis cascade progresses to gastric cancer, the oral and intestinal-type pathogenic microbial strains predominate. Hp-I creates a premalignant environment of atrophy and intestinal metaplasia and the subsequent alteration in gastric microbiota seems to play a crucial role in gastric tumorigenesis itself. Successful H. pylori eradication is suggested to restore gastric microbiota, at least in primary stages. It is more than clear that Hp-I, gastric microbiota and gastric cancer constitute a challenging tangle and the strong interaction between them makes it difficult to unroll. Future studies are considered of crucial importance to test the complex interaction on the modulation of the gastric microbiota by H. pylori as well as on the relationships between the gastric microbiota and gastric carcinogenesis.
Keywords: Helicobacter pylori infection, Gastric microbiota, Gastric cancer, Oncogenesis, Dysbiosis, Helicobacter pylori eradication
Core Tip: Gastric adenocarcinoma is a leading cause of cancer-related death in the world. Chronic gastric infection caused by Helicobacter pylori (H. pylori) is the strongest identified risk factor for gastric adenocarcinoma, prompting the World Health Organization to classify it as a class I carcinogen. It has been shown that in H. pylori-colonized patients, this pathogen accounts for more than 90% of all gastric microbiota modifying healthy microbiota and reducing its overall diversity. In this review, we tackle the complicated relationship between H. pylori, gastric microbiota and gastric cancer in an effort to unroll this tangle.
INTRODUCTION
Gastric cancer (GC) has been recognized as a global health concern; it is still the fifth most frequent global malignancy and one of the main causes of cancer-related death[1]. Likewise, Helicobacter pylori infection (Hp-I), an important public health burden affecting more than half of the global population[2], is related with the majority of GC, with an estimate between 74.7% to more than 90% of the new non-cardia GC cases[1,3].
Regarding the interaction between Hp-I and GC, relevant mechanisms known for many years have been studied and are constantly being enriched with new data (Figure 1)[4-17]. In this regard, arising evidence indicates that Helicobacter pylori (H. pylori), as the most important member of abnormal gastric microbiota (GM), might induce gastric microbiome modifications[11] thereby possibly leading to gastric oncogenesis. The gastric flora may be involved in the H. pylori-related oncogenicity, and the variations in the GM composition of patients with GC, intestinal metaplasia (IM) and chronic gastritis are defined[18]. For instance, Campylobacter is among the most influential genera in H. pylori-associated atrophic gastritis and gastric atrophy-induced alterations of the GM, namely gastric dysbiosis, might contribute to gastric tumorigenic effect[1]. Moreover, H. pylori-related metabolic syndrome induces dysbiosis of gastrointestinal tract (GIT) microbiota, thereby contributing to lower and upper GIT carcinogenesis including GC[19-21]. However, the interaction between the host, microbiota and H. pylori in the pathogenesis of GC still has to be fully elucidated[22].
Figure 1.

Possible mechanisms involved (A) in the etiology of non-cardiac gastric cancer (intestinal type) resulting in the classical cascade of Correa histopathological precancerous lesions (B) as seen in an upper gastrointestinal endoscopy. Hp-I: Helicobacter pylori infection; GC: Gastric cancer; PPIs: Proton pump inhibitors; CagA: Cytotoxin-associated gene A; VacA: Vacuolating cytotoxin A; GGT: γ-glutamyl transpeptidase; BabA: Blood-group-antigen-binding adhesin; SabA: Sialic acid-binding adhesin; OipA: Outer inflammatory protein; NapA: Neutrophil activation protein A; EMT: Epithelial-mesenchymal transition; ROS/RNS: Reactive oxygen species/Reactive nitrogen species; EGFR: Epidermal growth factor receptor; SPEM: Spasmolytic polypeptide-expressing metaplasia; CSC: Cancer stem cell; BMDSCs: Bone marrow-derived stem cells; IEN: Intraepithelial neoplasia.
Based on recent data, this review attempts to unroll the tangle regarding the interaction between Hp-I, GM and GC.
GASTRIC MICROBIOTA COMPOSITION
The GIT (mainly intestine) is colonized by 1-4 × 1015 microorganisms, co-existing in a balanced relationship[22]; the GIT microbiota is estimated to be up to 2 kg and affects health and disease[23]. The majority of the bacteria found in the adults’ gut consists of Bacteroides and Parabacteroides[23]. The anaerobic environment of intestinal lumen does not facilitate aerobic pathogens colonization and development under normal conditions, though anaerobic and facultative pathogenic species can invade it and promote diseases. Each site of the GIT has a unique distribution of microflora; when compared with the stomach and duodenum, bacteria density increases in the jejunum/ileum and colon. To yield the optimal conditions for their common interaction and survival, host and microbes have developed specific mechanisms; the disruption of those mechanisms triggers an imbalance in microbial species abundance, termed dysbiosis, which is incriminated for gut barrier dysfunction and induction of inflammatory response. In this regard, the failure to regulate the composition (microbial diversity), probably occurs during the beginning and course of several diseases including malignancies, such as GC[24].
Until recently, the gastric environment was considered as sterile, probably due to increased acidity, and the microbiota was believed to be isolated in the small intestine and colon. Subsequently, identifying H. pylori focused the attention on the gastric microbiota as “an ecological niche for bacteria”[23]. Emerging data have revealed that there is a broad range of microorganisms in the stomach with a density of 101 to 103 colony forming units/g[25,26]. Gastric microbiome is composed of bacteria ingested mainly through the ororespiratory tract and secondary from the intestine by transpyloric biliary reflux[27,28]. Most of those microorganisms cannot resist indigenous gastric defensive mechanisms and there are data indicating which microorganisms permanently colonize the gastric mucosa, other than H. pylori. Relative reports suggested that the predominant phyla in the gastric mucosa consist of Streptococcus, Rothia, Lactobacillus, Veillonella, Prevotella, Neisseria and Hemophilus, counting more than one hundred sorts[28,18]. Specifically, H. pylori, represents the most important member of the GM family with the highest relative abundance. Additional GM includes Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and Fusobacteria being the 5 most abundant phyla[18], in children and adults[29]. In culture-based studies where cultures of gastric juice or mucosa biopsies were examined, numerous members of the Firmicutes, Proteobacteria, Actinobacteria and Fusobacteria phyla were identified, while yeasts were recognized in a relatively low abundance[30,31]. Laboratory molecular techniques with high sensitivity indicated that Streptococcus, Prevotella, Neisseria, Veillonella and Rothia represent the main bacterial populations in the gastric tissue, with Streptococcus being the most dominant genus[32-36]. Sung et al[37] revealed heterogeneity in the flora of gastric fluid and mucosa. Gastric mucosa has a greater flora richness while gastric juice has a greater flora diversity[37]. The presence of bacteria in gastric juice could be just transient as a result of their ingestion with food, drinks or saliva without colonizing the gastric mucosa so they create a fictional image of the real diversity[18].
More specifically, Bik et al[36] by introducing a small subunit 16S rDNA clone library approach, described a diverse population of 128 phylotypes (totally 1833 bacterial isolates obtained from gastric biopsies of 23 healthy adults) within gastric mucosal samples with the majority of bacteria belonging to the five abovementioned major groups- Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and Fusobacteria phyla[36]. A lot of similar studies confirmed the presence and proportion of these phyla[4,38-41]. Table 1 shows the taxonomy of most prevalent GM at phylum and genus level.
Table 1.
Taxonomy of the most prevalent gastric microbiota at phylum and genus level
|
Phylum
|
Genus
|
| Proteobacteria | Helicobacter, Enterobacteriaceae unknown, Acinetobacter, Pseudomonas, Haemophilus, Agrobacterium, Halomonas, Shewanella, Sphingomonas, Methylobacterium, Aquabacterium |
| Bacteroidetes | Prevotella, Chryseobacterium |
| Firmicutes | Streptococcus, Clostridium, Lactobacillus, Staphylococcus, Faecalibacterium, Veillonella, Bacillus, Peptostreptococcus, Selenomonas, Phascolarctobacterium, Gemella, Roseburia, Megamonas, Gemmiger, Lactococcus, Granulicatera, Dialister, Alcaliphylus, Ruminococcus, Blautia |
| Fusobacteria | Fusobacterium, Leptotrichia |
| Actinobacteria | Propionobacterium, Corynebacterium, Arthrobacter |
| Sprirochaetes | Bacteroeides |
| Acidobacteria | Streptophyta, Sphingobacterium, Pedobacter |
IMPACT OF HP-I ON GASTRIC MICROBIOTA COMPOSITION
Regarding Hp-I, its impact on the GM remains to be clarified. While Bik et al[36] did not depict an impact of the occurrence of H. pylori in gastric biopsies on the composition of GM, several subsequent studies characterize H. pylori as the regulator of the GM community. Andersson et al[42] revealed that H. pylori was the dominant bacterium whenever isolated, though its absence was associated with a diverse microbiota. Analytically, in samples from H. pylori(+) individuals, H. pylori was the mainstay species (ninety percent) of the samples examined by 454 pyro-sequencing. Thirty-three phylotypes were recognized solely, 229 less when compared with H. pylori(-) individuals[42]. The abovementioned signifies that H. pylori has inhibitory effects on the colonization of other bacteria harboring a significantly lower diversity of them in the stomach. The GM in H. pylori negative patients was mainly dominated by the same phyla, though with diverse percent abundances: 52.6% Proteobacteria, 26.4% Firmicutes, 12% Bacteroidetes and 6.4% Actinobacteria[43]. The common genera observed in H. pylori negative individuals included Gemella, Prevotella and Streptococcus[42].
In another study which introduced DNA microarrays to characterize the GM in 12 corpus biopsy samples (eight H. pylori positive), Maldonado-Contreras et al[44] isolated 44 phyla with four dominant Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Hp-I augmented the relative abundance of non-H. pylori—Proteobacteria, Spirochaetes, and Acidobacteria whereas lessening the relative abundance of Actinobacteria, Bacteroidetes and Firmicutes, compared to uninfected stomachs[44]. An additional study from Mongolia showed that patients infected with H. pylori exhibited a significantly lesser bacterial richness and Shannon and Simpson indices[45,46] compared with H. pylori negative arms. Moreover, enrichment of Firmicutes, Fusobacteria, Bacteroidetes and Actinobacteria at phylum level was shown in patients with H. pylori negative gastritis by the linear discriminant analysis effect size analysis[47].
Miao et al[48] studied the effect of H. pylori eradication in microbiota composition and found that GM profiles between H. pylori negative groups and previously H. pylori positive groups four months after successful eradication therapy were almost the same[48].
Table 2 shows the relative abundance of GM at phylum level among H. pylori positive and H. pylori negative patient groups. In particular, we present the minimum and the maximum values across the studies[36,42,43,47,48]. Also, we calculated the pooled percentages and the relative 95% confidence intervals. Among H. pylori positive patient groups, proteobacteria were more frequent, while among H. pylori negative patient groups, firmicutes and proteobacteria were more frequent.
Table 2.
Relative abundance of gastric microbiota at phylum level among Helicobacter pylori positive and Helicobacter pylori negative patient groups
| Phylum |
H. pylori
-positive
|
H. pylori
-negative
|
||||
|
Minimum
|
Maximum
|
Pooled (95%CI)
|
Minimum
|
Maximum
|
Pooled (95%CI)
|
|
| Proteobacteria | 68.7 | 96.7 | 88.4 (75.4-95.9) | 10.8 | 52.6 | 27.9 (12.7-43.9) |
| Bacteroeidetes | 0.8 | 8.3 | 3.1 (1.1-6.0) | 11.1 | 30.0 | 20.8 (12.7-28) |
| Firmicutes | 1.3 | 14.7 | 6.2 (1.8-12.9) | 16.3 | 29.9 | 31.1 (20.5-40.1) |
| Fusobacteria | 0.1 | 1.6 | 1.1 (0.2-2.3) | 1.1 | 6.1 | 3.5 (1.6-6.1) |
| Actinobacteria | 0.2 | 3.1 | 1.2 (0.4-2.5) | 2.8 | 46.8 | 16.7 (2.4-37.2) |
Values are expressed as percentages. CI: Confidence interval; H. pylori: Helicobacter pylori.
IMPACT OF FACTORS ON GASTRIC MICROBIOTA COMPOSITION BEYOND HP-I
Beyond H. pylori, the composition of GM could be modified by some other factors such as dietary habits, age, ethnicity, medication use and severity of gastric mucosa inflammation[18,27,49-53].
Proton pump inhibitor (PPI) raises the pH in the stomach thereby altering the GM. Likewise, PPIs-driven gastric hypo-chlorhydria can cause substantial changes in gut microbiota composition[54,55]. Two possible mechanisms by which the mentioned PPIs can influence the GM composition have been proposed: (1) By targeting directly bacterial and fungal proton pumps; and (2) By disturbing the natural gastric microenvironment through the gastric pH alkalization[56]. More specifically, GM of patients on PPIs therapy has more abundant bacteria compared to patients on H2RAs and untreated control. The composition of microbiota was quite similar to that of oropharyngeal or fecal bacteria[26]. Paroni Sterbini et al[57] showed a significant increase in the relative abundance of Streptococcus in patients taking PPIs irrespective of H. pylori status; they revealed that Streptococcus can be an independent indicator of the gastric microbiome changes in dyspeptic patients secondary to the use of PPIs[57]. On the other hand, Parsons et al[40] by using 16S rRNA sequencing in gastric samples, showed that patients receiving PPIs had relatively few changes in the GM compared to healthy controls[39]. Besides, numerous reports indicated that the H. pylori moving from the antrum to body and fundus of the stomach is recorded particularly by long-term PPIs usage[58]. Thus, Hp-I eradication is proposed for patients who received long-term PPI usage in order to prevent the proinflammatory trigger and thereby decreasing GC potential. Antibiotic ingestion also effects gastrointestinal microflora. Mason et al[59] revealed that treatment with cefoperazone caused changes in GM with an overgrowth of Enterococci and a decrease of Lactobacilli[59].
Attempting to correlate gastric mucosal inflammation with GM, a rise in Streptococcus and a reduction in Prevotella was found in patients with atrophic gastritis vs healthy subjects[36]. Patients with autoimmune atrophic gastritis exhibited a larger concentration of Firmicutes than patients with chronic atrophic gastritis (CAG) and a greater variety of microbial species than H. pylori-induced atrophic gastritis. This might be due to the differences in gastric acidity between the two conditions or additional factors such as their different immune profiles[39]. Researchers from Mexico obtained gastric tissue from patients with non-atrophic gastritis (NAG), IM and intestinal type GC through extraction of DNA for microbiota analyses using microarray methods and showed that bacterial diversity steadily decreased from NAG to IM to GC[59].
THE INTERACTION BETWEEN GASTRIC MICROBIOTA AND GASTRIC CANCER
The existence of multiple homeostasis mechanisms that take place in the human stomach is a well-recognized phenomenon contributing to health maintenance by balancing the interaction between host gastric microbial diversity and mucosa-related factors[60,61]. When this balance is interrupted, a cascade of events occurs resulting in the emergence of inflammatory changes, dysbiosis and consequently, diseases including GC[36].
The mentioned hypochlorhydria appears to promote a decrease in microbial heterogeneity as well as the development of microorganisms which exhibit genotoxic changes, and raising the ratio of nitrate to nitrite reductase microbe capacities implicated in gastric oncogenesis. Furthermore, the bacterial balance differentiates by raising the stomach pH, giving growth mostly of oral bacteria, such as Streptococcus anginosus, Peptostreptococcus stomatis, Slackia exigua and Parvimonas micra as well as Dialister pneumosintes. Such bacteria might play a role in GC progression via the induction of various metabolic pathways[62]. Thus, to improve the understanding of the influence of promoting the survival and spread of potentially genotoxic bacteria in the stomach and other GIΤ locations, it will be critical to describe the properties of the mentioned PPIs in GM composition. Nevertheless, no consensus exists regarding the role of PPIs in GC development. Based on a number of metanalyses and studies, there is an increased GC risk in patients using PPIs for a long time period[63] (approximately 2.4 times more than non-users), despite H. pylori eradication[4,64,65].
Hp-I is a precise paradigm of the GM homeostasis disturbance sequelae[66]. The H. pylori-related inflammatory effects primarily act on the mucosal surface of the stomach variably affecting the production of mucin[67]. Differentiations of the latter seem to play a crucial role regarding the gastric carcinogenesis pathway[9]. Nevertheless, it should be stated that studies on the H. pylori-related mucin production changes have not yet been able to sort out whether this GC sequelae results in dysbiosis in the stomach or, conversely, to microbial diversity. These effects could be the backbone of GC development, given the fact that at the last stage of gastric malignancy oral or intestinal-type bacteria are predominantly discovered, something not seen in premalignant conditions (chronic gastritis, atrophy and IM) where H. pylori abundancy is more than clear. Whether this phenomenon is due to tumor-related mucin type differentiation, possibly resulting in GC-related microbiota must be elucidated[68].
As already stated, earlier studies have shown that H. pylori negative individuals exhibit a significant variability in microbiota composition which mainly consists of Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and Fusobacteria. On the contrary, the stomach of H. pylori positive patients is almost exclusively colonized by this infectious pathogen[42]. In line with this observation, it should be highlighted that from a specific point and beyond, the GC progress seems not to be related with H. pylori presence, since the gastric adenocarcinoma microbiota mainly consists of intestinal and oral bacterial genera, and in addition this progression can happen even after successful H. pylori treatment (Figure 2)[67]. Similar findings emerged from the study by Yu et al[27] who investigated 160 individuals with gastric malignancy residing in China and Mexico. They showed that in the non-cancerous gastric regions, the H. pylori presence was significantly high in contrast to the GC site with depletion even in the absence of H. pylori. The difference in microbiota diversity that patients with advanced malignant lesions exhibited was further verified in many studies which revealed a marked presence of Lactobacillus, Streptococcaceae, Staphylococcus, Clostridium and Fusobacterium among others, underlying the crucial role those intestinal microbes play[63,69]. Lastly, Robinson et al[70] showed, after utilizing an advanced computer-based search algorithm, that GC was the second most diversely abundant neoplasm in terms of bacterial DNA molecules with dominant species highly comprising Pseudomonas and not H. pylori.
Figure 2.
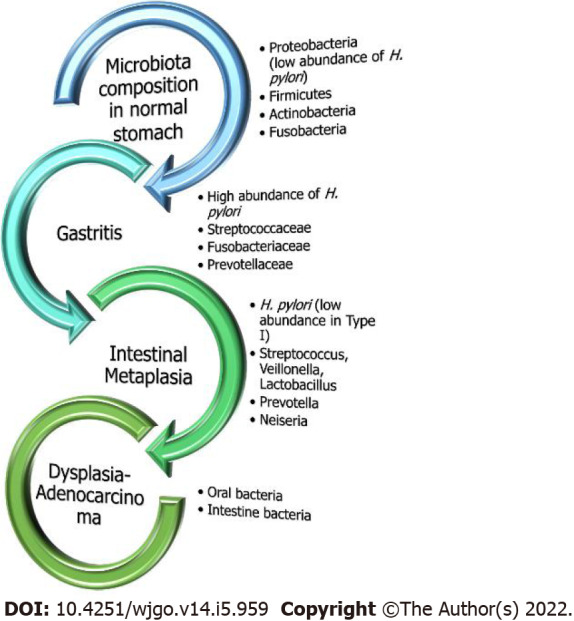
Gastric microbial composition in the healthy and diseased stomach. Under normal healthy conditions without evidence of excessive inflammation, Helicobacter pylori (H. pylori) exists in very low abundance. On the contrary, in chronic gastritis, H. pylori is the predominant bacteria with the presence of other microorganisms as well but at lower rates. However, as the sequalae of carcinogenesis moves towards malignancy, oral or intestinal-type pathogens exclusively predominate.
The above studies and their subsequent findings have been verified to an accountable level by well-designed animal model experiments, especially in C57BL/6 mice, where their stomach microbiota consisted of similar bacteria categories to those found in humans, namely Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria[71]. For instance, according to Lofgren et al[72], the H. pylori-related gastritis not only resulted in decreased GM variety (as seen in human individuals), but also significantly extended the interval to gastric malignancy emergence, especially when the only pathogen was H. pylori. The above interesting outcome was confirmed by the study of Lertpiriyapong et al[73], who showed that by adding even a small number of intestinal commensal pathogens to monocolonized by H. pylori germ-free insulin-gastrin (INS-GAS) transgenic mouse models’ stomach there was a progressive advancement to gastric neoplastic lesions.
Viewing the aforementioned data, while a role for H. pylori in gastric oncogenesis cannot be doubted, emerging data shows that additional bacteria in the GM also seem to be involved in the transformation of stomach epithelial cells[74]. Nevertheless, whether it is the Hp-I that stimulates growth of unwanted bacteria or vice versa warrants clarification.
In a survey, Jo et al[75] showed that in GC patients, the records of nitrosating/nitrate-reducing microbes other than H. pylori were no less than doubled in comparison with healthy controls exhibiting similar H. pylori status, albeit insignificantly. Thus, further basic research is necessary to illuminate whether GM alterations are crucial to GC development or are the result of alterations in the gastric setting.
Microbial infections have been incriminated for a variety of cancers by transforming host cells and triggering neoplastic characters and inflammatory reactions, disrupting cell configuration and altering their genoms. Therefore, it is rational to consider the possible role of the intestinal microbiota in gastric oncogenesis[76]. Furthermore, under the consideration that H. pylori plays a dominant role in Correa’s cascade (i.e., from NAG to atrophic gastritis and further to IM, dysplasia and GC), the inflammatory process of gastritis could be considered to be started and continued by Hp-I, which can colonize epithelium decades before neoplastic transformation. Ultimately, this transformation could develop owing to augmented pH of the stomach because of the loss of parietal cells and the multiplication of microbes other than H. pylori[18]. Certainly, the microbiota differs between patients with chronic gastritis, IM and GC. The later indicates the significant role of gut microbiota in H. pylori-related tumorigenic effect. In contrast, progressive alterations in gastric pH could also be anticipated through H. pylori-derived histological alterations, facilitating the gastric colonization from other bacteria[18]. Other investigators showed that the GC microbiota mainly included Citrobacter, Achromobacter, Clostridium, Lactobacillus, Phyllobacterium and Rhodococcs. Nevertheless, additional research is warranted to clarify the fingerprint of bacterial populations associated with gastric disorders in connection with the Correa’s cascade sequence.
Currently, the comprehension of dysbiosis-related genotoxicity and inflammation needs to move from descriptive studies to functionally based studies which investigate the effects of specific taxa and bacteria-derived metabolites on the gastric mucosa. In this regard, the potential introduction of probiotics should be studied thoroughly in order to delineate its effectiveness in the rebalance of human microbiota synthesis[77].
INTERACTION BETWEEN HP-I, GASTRIC MICROBIOTA AND GASTRIC CANCER
The perpetuation of Hp-I reduces microbiota diversity and is connected with atrophy, IM and GC[78]. Although it represents the main genus in chronic gastritis with a mean relative abundance of 42% (varying from 0.01%-95%), H. pylori presents a dramatic decrease in GC tissues with a relative abundance of 6%. In this regard, recent data based on RNA sequencing analyses revealed that H. pylori entirely dominated the microbiota not only in infected patients but also in the majority of individuals categorized as H. pylori-uninfected using conventional approaches, thus implying an active role in all cases of GC development[78].
The vast majority of information regarding the role of GM in carcinogenesis derives from preclinical studies in INS-GAS transgenic mouse models. Complex microbiota has been associated with intensive gastric inflammation, epithelial damage, oxyntic gland atrophy, hyperplasia, metaplasia and dysplasia[71]. Moreover, co-infection with H. pylori in INS-GAS rodents predisposed to more severe gastric lesions and earlier development of early GC in comparison to H. pylori-infected germ-free INS-GAS mice[71]. Concerning the co-infective bacteria, complex microbiota and restricted microbiota consisting of only three species of commensal murine bacteria (Clostridium sp., Lactobacillus murinus and Bacteroides sp.) predisposed similarly to neoplasia generation in H. pylori positive models[73]. Further in vivo studies with Hp-I revealed that the co-infection with commensal microbiota accelerated the progression to gastric intraepithelial neoplasia and the progression to cancer, whereas the treatment with antibiotics delayed the gastric tumorigenesis in H. pylori-free and specific pathogen-free INS-GAS mice[73,79,80]. Moreover, the environment of gastric atrophy reduces the density of H. pylori aggregates to give rise to bacteria from other locations of the GIT, thus perpetuating the inflammatory process and genotoxicity, to induce malignant transformation. The overgrowth of such microbiome could partially contribute to the “point of no return” of carcinogenesis prevention after H. pylori eradication[81]. As already known, eradication of H. pylori is associated with a reduced risk of GC, although ambiguity exists over whether this is an isolated result from the eradication of the H. pylori or the modification of the whole GM, as bacterial diversity increases probably beneficially[80].
Interestingly, Eun et al[82] reported variations in the composition and diversity of GM among patients with chronic gastritis, IM and GC. More specifically, in the early stages of carcinogenesis, H. pylori may trigger the development of CAG, rather than direct induction of GC[82]. Subsequently, the resulting increased pH provokes changes in the constitution of GM thus facilitating the progression from CAG to IM and finally to GC[83]. On the other hand, subjects with GC showed a significant increase in the Bacilli class and Streptococacceae family whereas the Epsilonproteobacteria class and Helicobacteriaceae family were decreased[82]. As suggested by Correa et al[84], chronic Hp-I triggers a CAG with the mentioned defective acid secretion, thus facilitating the excessive colonization of gastric micro-flora with bacteria capable of reducing nitrate to nitrite, to form N-nitroso compounds that are carcinogenic[84,85]. In this regard, the GC microbiome is different from atrophic gastritis and possesses increased representation of nitrate reductases, with Citrobacter, Achromobacter, Clostridium, Campylobacter, Deinococcus, Sulfurospirillum and Phyllobacterium representing ascendant species[79], thus accelerating the development of GC following Hp-I in INS-GAS mice when compared to germ-free mice that were monocolonized by H. pylori[71]. Relatively, chronic treatment with the mentioned PPIs increases the potential of atrophy among H. pylori positive subjects[86] in contrast to H. pylori negative individuals or patients receiving eradication treatment thus implying that the non-H. pylori microbiota could only promote gastric atrophy when co-existing with H. pylori[35,87].
The activity of gastritis is well known for its close relationship with Hp-I. A similar motif of diversity is suggested for further phyla, such as Bacteroidetes and increased abundances of Firmicutes or Proteobacteria, thus incriminating their dysbiosis for gastric carcinogenesis[87]. Nevertheless, despite the wide range of studies associating Hp-I with gastric dysbiosis, no data interpret the exact background of this interaction which seems to promote a sustained inflammation and genotoxicity[88]. A widely acceptable pattern suggests that chronic gastric inflammatory response to H. pylori may modify the gastric environment, paving the way to the growth of a dysbiotic gastric bacterial community; and H. pylori eradication reverses the gastric dysbiosis to a similar level to uninfected patients, and exerts beneficial effects on gut microbiota, achieving an increased probiotic and putative downregulation of drug-resistance[89]. More specifically, successful H. pylori eradication inhibited dysbiosis significantly (P < 0.001), although it remained higher than that of the H. pylori negative arm (P = 0.025). Nonetheless, treatment failure was associated with increased dysbiosis rate comparable to active Hp-I (P = 0.351)[89]. Intense dysbiosis was further found to be analogous to the progress from gastritis to atrophy, IM and GC (both P < 0.001)[89].
Pathophysiologically, the highly expressed VacA (vacuolating cytotoxin A), after Hp-I, binds to the receptor proteins tyrosine phosphatase α and β on gastric cells, thus generating pores to yield bacterial internalization[90]. Some data indicated that antibodies against VacA could be correlated with both peptic ulcer and gastric malignant disorders, thus it could be considered as a biomarker of both pathologies[91]. Additionally, H. pylori survival promoted by VacA is independent of CagA (cytotoxin-associated gene A) accumulation. VacA is connected with mucolopin 1 (transient receptor channel) which impedes the death of microbial cells through autophagic procedure and permits the formation of an intracellular niche in which H. pylori survives[91]. In this regard, infection of the AGS gastric adenocarcinoma cell line with H. pylori for 6 h, lead to autophagy that was dependent on VacA[92]. This implied that autophagy is activated by cells infected by H. pylori to evade the destructive effects of toxins thus promoting cell survival. In addition, others reported that 1 d exposure to VacA disturbs the antiphagocytic signaling and accumulates defective autophagosomes in cells[92]. Likewise, H. pylori controls the autophagocytic pathway as well as the expression of genes related to autophagy in both macrophages and gastric epithelial cells[93]. Therefore, it appears that during the initiation of carcinogenesis, the aforementioned pathway has a regulatory role and when suppressed, leads to premalignant disorders, induces oxidative stress, promotes cell growth, penetration and eventually metastases. Concerning GC, this could lead to precursor lesions extension[93]. Interestingly, there is a direct association between pathogens that induce dysbiosis and disturbed immune responses including apoptosis - autophagy and orodigestive cancers, including GC[93].
Besides, H. pylori releases a plethora of adhesins (BabA, BabB, SabA, AlpA and AlpB) which facilitate the opening of tight junctions (TJ) and adherent junctions (AJ)[94-96]. In this regard, in vivo CagA causes depolarization and disruption of the TJ barrier function in epithelial cells to the H. pylori attachment sites[7,94]. Additionally, after in vitro excessive administration, CagA binds to membrane e-cadherins, inhibits their interaction with β-catenin to disrupt the AJs’ integrity and tightness[97]. In vivo cagA with Lactobacillus enhances the effect of H. pylori to human monocyte-derived dendritic cells (DC) leading to DC maturation and induction, beyond H. pylori, additional inflammatory mediators[93]. This implies that the bacteria that produce lactic acid could increase H. pylori related inflammation promoting gastric oncogenesis. The latter are in concordance with human GM studies displaying a plethora of Lactobacillus in H. pylori-connected IM and GC (intestinal type) vs NAG[62] and the increased Lactobacillus in INS-GAS mouse model studies infected with H. pylori and reduced commensals (Clostridium, Lactobacillus, and Bacteroides) which develop gastric intraepithelial neoplasia[73]. Nevertheless, other findings indicate a probiotic Lactobacillus strain that inhibits H. pylori colonization in a Mongolian gerbil model[98]. More relevant to biofilm-associated H. pylori, Streptococcus mitis interacts with H. pylori in co-culture studies, converting it to coccoid cells, as proteomic analysis reveals, signifying an apparent impact on gastric oncogenesis linked with H. pylori[99,100]. Moreover, experimental data on INS-GAS mice co-colonized with H. pylori and Streptococcus Salivarius showed more severe gastritis when compared with solely Hp-I only at 5 mo post-infection. The latter data signify strong interactions among several bacteria and H. pylori that in turn may affect H. pylori-related tumorigenesis[101]. Of note, H. pylori-induced biofilms are associated with resistance to H. pylori antibiotic eradication regimens[102]; H. pylori biofilms appear to be one of the main barriers to H. pylori eradication, by inhibiting antibiotics penetration and augmenting the expression of efflux pumps and mutations, several therapeutic failures and chronic infections[103].
Finally, the interplay between H. pylori and GM in the pathogenesis of GC can be dependent on Toll-like receptors through a perpetual stimulation by H. pylori and potentially by other microorganisms[104]. In this regard, Hp-I seems to create a premalignant environment of atrophy and IM and the subsequent alterations in GM in later stages play a more relevant role in carcinogenesis itself[105].
CONCLUSION
It is more than clear that Hp-I, GM and GC constitute a challenging tangle due to the strong interaction between them making it difficult to unroll it.
The stomach harbors a large and diverse bacterial community with H. pylori, a member of Proteobacteria phylum, being the most dominant and abundant genus. The main phyla colonizing the stomach are Proteobacteria, Bacteroidetes, Firmicutes, Fusobacteria and Actinobacteria. Most studies show that H. pylori has inhibitory effects on the colonization of other bacteria, harboring a lower diversity of them in the stomach. Other factors that influence GM are dietary habits, age, ethnicity, medication use (PPIs, antibiotics), gastric mucosa inflammation and GC. It is worthwhile to mention that GM differs in patients with chronic gastritis, IM, dysplasia or GC, but its role in GC has not yet been fully elucidated. Data shows that from a specific point and beyond, apart from H. pylori-related gastritis, the GC progress seems not to be related with H. pylori presence, since the gastric adenocarcinoma microbiota mainly consists of intestinal and oral bacterial genera, considering that this progression can happen even after successful H. pylori eradication. The above has been verified to an accountable level by well-designed animal model experiments. In accordance, beyond H. pylori’s role in gastric oncogenesis, other bacteria, H. pylori-stimulated or not, in GM also seem to be responsible for transformation of gastric epithelial cells.
To conclude, the aforementioned studies amongst others have begun to shed light into the maze of GC complex pathogenesis where abundant data show that beyond H. pylori related gastritis, additional pathogens might contribute to this type of cancer development. Nevertheless, large-scale experiments are needed to discern the exact role of different kinds of pathogens which reside in the stomach and their contribution to neoplasia emergence, aiding in the prediction of adverse prognosis of a specific microbiota diversity. Only then would the manipulation of GM be feasible, modifying the number and the types of the necessary commensals.
Footnotes
Conflict-of-interest statement: The authors declare having no conflict of interests for this article.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: March 19, 2021
First decision: May 3, 2021
Article in press: April 9, 2022
Specialty type: Gastroenterology and Hepatology
Country/Territory of origin: Greece
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Dong QJ, China S-Editor: Chang KL L-Editor: Filipodia P-Editor: Chang KL
Contributor Information
Christos Liatsos, Department of Gastroenterology, 401 General Military Hospital of Athens, Athens 11525, Greece. cliatsos@yahoo.com.
Apostolis Papaefthymiou, Department of Gastroenterology, 401 General Military Hospital of Athens, Athens 11525, Greece; Gastroenterology, University Hospital of Larissa, Larissa 41336, Greece.
Nikolaos Kyriakos, Department of Gastroenterology, 401 General Military Hospital of Athens, Athens 11525, Greece.
Michail Galanopoulos, Department of Gastroenterology, 401 General Military Hospital of Athens, Athens 11525, Greece.
Michael Doulberis, Division of Gastroenterology and Hepatology, Medical University Department, Kantonsspital Aarau, Aarau 1234, Switzerland.
Marios Giakoumis, Department of Gastroenterology, 401 General Military Hospital of Athens, Athens 11525, Greece.
Evangelia Petridou, Department of Microbiology, “Agia Sofia” Paediatric Hospital, Goudi, Athens 11527, Greece.
Christos Mavrogiannis, Gastrointestinal and Liver Unit, Faculty of Nursing, Kifissia General and Oncology Hospital, Kaliftaki, N.Kifisia 14564, Greece.
Theodore Rokkas, Gastroenterological Clinic, Henry Dunant Hospital, Athens 11525, Greece.
Jannis Kountouras, Department of Internal Medicine, Second Medical Clinic, Ippokration Hospital, Aristotle University of Thessaloniki, Thessaloniki 41336, Macedonia, Greece.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, Malfertheiner P, Graham DY, Wong VWS, Wu JCY, Chan FKL, Sung JJY, Kaplan GG, Ng SC. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology. 2017;153:420–429. doi: 10.1053/j.gastro.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 3.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 4.Cheung KS, Leung WK. Long-term use of proton-pump inhibitors and risk of gastric cancer: a review of the current evidence. Therap Adv Gastroenterol. 2019;12:1756284819834511. doi: 10.1177/1756284819834511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liatsos C, Rokkas T. The Effect of Chronic Use of Proton Pump Inhibitors on Gastric Cancer: Should We Be Aware of It? Dig Dis. 2018;36:395–396. doi: 10.1159/000489629. [DOI] [PubMed] [Google Scholar]
- 6.Moss SF. The Clinical Evidence Linking Helicobacter pylori to Gastric Cancer. Cell Mol Gastroenterol Hepatol. 2017;3:183–191. doi: 10.1016/j.jcmgh.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kountouras J, Kapetanakis N, Zavos C, Polyzos SA, Romiopoulos I, Tsiaousi E, Anastasiadou K, Giorgakis N, Vardaka E, Nikolaidou C, Venizelos I, Katsinelos P. Helicobacter pylori might contribute to cancer and/or bone marrow-derived stem cell-related gastrointestinal oncogenesis. Oncogene. 2015;34:670. doi: 10.1038/onc.2013.602. [DOI] [PubMed] [Google Scholar]
- 9.Babu SD, Jayanthi V, Devaraj N, Reis CA, Devaraj H. Expression profile of mucins (MUC2, MUC5AC and MUC6) in Helicobacter pylori infected pre-neoplastic and neoplastic human gastric epithelium. Mol Cancer. 2006;5:10. doi: 10.1186/1476-4598-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kountouras J, Doulberis M, Papaefthymiou A, Polyzos SA, Vardaka E, Tzivras D, Dardiotis E, Deretzi G, Giartza-Taxidou E, Grigoriadis S, Katsinelos P. A perspective on risk factors for esophageal adenocarcinoma: emphasis on Helicobacter pylori infection. Ann N Y Acad Sci. 2019;1452:12–17. doi: 10.1111/nyas.14168. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Shi D, Li M, Li Y, Wang X, Li W. The relationship between gastric microbiota and gastric disease. Scand J Gastroenterol. 2019;54:391–396. doi: 10.1080/00365521.2019.1591499. [DOI] [PubMed] [Google Scholar]
- 12.Burkitt MD, Duckworth CA, Williams JM, Pritchard DM. Helicobacter pylori-induced gastric pathology: insights from in vivo and ex vivo models. Dis Model Mech. 2017;10:89–104. doi: 10.1242/dmm.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan G, Chen Y, He S. Family History of Gastric Cancer and Helicobacter pylori Treatment. N Engl J Med. 2020;382:2171. doi: 10.1056/NEJMc2003542. [DOI] [PubMed] [Google Scholar]
- 14.Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism. 2019;92:6–10. doi: 10.1016/j.metabol.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Choi IJ, Kim CG, Lee JY, Kim YI, Kook MC, Park B, Joo J. Family History of Gastric Cancer and Helicobacter pylori Treatment. N Engl J Med. 2020;382:427–436. doi: 10.1056/NEJMoa1909666. [DOI] [PubMed] [Google Scholar]
- 16.Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY, Kim CG, Park B, Nam BH. Helicobacter pylori Therapy for the Prevention of Metachronous Gastric Cancer. N Engl J Med. 2018;378:1085–1095. doi: 10.1056/NEJMoa1708423. [DOI] [PubMed] [Google Scholar]
- 17.Díaz P, Valenzuela Valderrama M, Bravo J, Quest AFG. Helicobacter pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front Microbiol. 2018;9:5. doi: 10.3389/fmicb.2018.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alarcón T, Llorca L, Perez-Perez G. Impact of the Microbiota and Gastric Disease Development by Helicobacter pylori. Curr Top Microbiol Immunol. 2017;400:253–275. doi: 10.1007/978-3-319-50520-6_11. [DOI] [PubMed] [Google Scholar]
- 19.Kountouras J, Boziki M, Polyzos SA, Katsinelos P, Gavalas E, Zeglinas C, Tzivras D, Romiopoulos I, Giorgakis N, Anastasiadou K, Vardaka E, Kountouras C, Kazakos E, Giartza-Taxidou E, Deretzi G, Dardiotis E, Kotronis G, Doulberis M. The Emerging Role of Helicobacter Pylori-Induced Metabolic Gastrointestinal Dysmotility and Neurodegeneration. Curr Mol Med. 2017;17:389–404. doi: 10.2174/1566524018666171219094837. [DOI] [PubMed] [Google Scholar]
- 20.Kountouras J, Polyzos SA, Doulberis M, Zeglinas C, Artemaki F, Vardaka E, Deretzi G, Giartza-Taxidou E, Tzivras D, Vlachaki E, Kazakos E, Katsinelos P, Mantzoros CS. Potential impact of Helicobacter pylori-related metabolic syndrome on upper and lower gastrointestinal tract oncogenesis. Metabolism. 2018;87:18–24. doi: 10.1016/j.metabol.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Saetang J, Sangkhathat S. Diets link metabolic syndrome and colorectal cancer development (Review) Oncol Rep. 2017;37:1312–1320. doi: 10.3892/or.2017.5385. [DOI] [PubMed] [Google Scholar]
- 22.Schulz C, Koch N, Schütte K, Pieper DH, Malfertheiner P. H. pylori and its modulation of gastrointestinal microbiota. J Dig Dis. 2015;16:109–117. doi: 10.1111/1751-2980.12233. [DOI] [PubMed] [Google Scholar]
- 23.Sgambato D, Miranda A, Romano L, Romano M. Gut microbiota and gastric disease. Minerva Gastroenterol Dietol. 2017;63:345–354. doi: 10.23736/S1121-421X.17.02380-7. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Peng F, Peng C, Du JR. Gut Microbiota in Tumor Microenvironment: A Critical Regulator in Cancer Initiation and Development as Potential Targets for Chinese Medicine. Am J Chin Med. 2021;49:609–626. doi: 10.1142/S0192415X21500270. [DOI] [PubMed] [Google Scholar]
- 25.Sheh A, Fox JG. The role of the gastrointestinal microbiome in Helicobacter pylori pathogenesis. Gut Microbes. 2013;4:505–531. doi: 10.4161/gmic.26205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wroblewski LE, Peek RM Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanduleanu S, Jonkers D, De Bruine A, Hameeteman W, Stockbrügger RW. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: differential findings in gastric juice and gastric mucosa. Aliment Pharmacol Ther. 2001;15:379–388. doi: 10.1046/j.1365-2036.2001.00888.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu G, Torres J, Hu N, Medrano-Guzman R, Herrera-Goepfert R, Humphrys MS, Wang L, Wang C, Ding T, Ravel J, Taylor PR, Abnet CC, Goldstein AM. Molecular Characterization of the Human Stomach Microbiota in Gastric Cancer Patients. Front Cell Infect Microbiol. 2017;7:302. doi: 10.3389/fcimb.2017.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris PR, Smythies LE, Smith PD, Perez-Perez GI. Role of childhood infection in the sequelae of H. pylori disease. Gut Microbes. 2013;4:426–438. doi: 10.4161/gmic.26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 31.Adamsson I, Nord CE, Lundquist P, Sjöstedt S, Edlund C. Comparative effects of omeprazole, amoxycillin plus metronidazole vs omeprazole, clarithromycin plus metronidazole on the oral, gastric and intestinal microflora in Helicobacter pylori-infected patients. J Antimicrob Chemother. 1999;44:629–640. doi: 10.1093/jac/44.5.629. [DOI] [PubMed] [Google Scholar]
- 32.Schulz C, Schütte K, Malfertheiner P. Helicobacter pylori and Other Gastric Microbiota in Gastroduodenal Pathologies. Dig Dis. 2016;34:210–216. doi: 10.1159/000443353. [DOI] [PubMed] [Google Scholar]
- 33.Yang I, Woltemate S, Piazuelo MB, Bravo LE, Yepez MC, Romero-Gallo J, Delgado AG, Wilson KT, Peek RM, Correa P, Josenhans C, Fox JG, Suerbaum S. Different gastric microbiota compositions in two human populations with high and low gastric cancer risk in Colombia. Sci Rep. 2016;6:18594. doi: 10.1038/srep18594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, He LH, Xiao D, Liu GD, Gu YX, Tao XX, Zhang JZ. Bacterial flora concurrent with Helicobacter pylori in the stomach of patients with upper gastrointestinal diseases. World J Gastroenterol. 2012;18:1257–1261. doi: 10.3748/wjg.v18.i11.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engstrand L, Lindberg M. Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol. 2013;27:39–45. doi: 10.1016/j.bpg.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sung J, Kim N, Kim J, Jo HJ, Park JH, Nam RH, Seok YJ, Kim YR, Lee DH, Jung HC. Comparison of Gastric Microbiota Between Gastric Juice and Mucosa by Next Generation Sequencing Method. J Cancer Prev. 2016;21:60–65. doi: 10.15430/JCP.2016.21.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zilberstein B, Quintanilha AG, Santos MA, Pajecki D, Moura EG, Alves PR, Maluf Filho F, de Souza JA, Gama-Rodrigues J. Digestive tract microbiota in healthy volunteers. Clinics (Sao Paulo) 2007;62:47–54. doi: 10.1590/s1807-59322007000100008. [DOI] [PubMed] [Google Scholar]
- 39.Katsinelos T, Doulberis M, Polyzos SA, Papaefthymiou A, Katsinelos P, Kountouras J. Molecular Links Between Alzheimer's Disease and Gastrointestinal Microbiota: Emphasis on Helicobacter pylori Infection Involvement. Curr Mol Med. 2019;20:3–12. doi: 10.2174/1566524019666190917125917. [DOI] [PubMed] [Google Scholar]
- 40.Parsons BN, Ijaz UZ, D'Amore R, Burkitt MD, Eccles R, Lenzi L, Duckworth CA, Moore AR, Tiszlavicz L, Varro A, Hall N, Pritchard DM. Comparison of the human gastric microbiota in hypochlorhydric states arising as a result of Helicobacter pylori-induced atrophic gastritis, autoimmune atrophic gastritis and proton pump inhibitor use. PLoS Pathog. 2017;13:e1006653. doi: 10.1371/journal.ppat.1006653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett. 2018;414:147–152. doi: 10.1016/j.canlet.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llorca L, Pérez-Pérez G, Urruzuno P, Martinez MJ, Iizumi T, Gao Z, Sohn J, Chung J, Cox L, Simón-Soro A, Mira A, Alarcón T. Characterization of the Gastric Microbiota in a Pediatric Population According to Helicobacter pylori Status. Pediatr Infect Dis J. 2017;36:173–178. doi: 10.1097/INF.0000000000001383. [DOI] [PubMed] [Google Scholar]
- 44.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shannon CE. A Mathematical Theory of Communication. Bell Syst Tech J. 1948;27:379–423. [Google Scholar]
- 46.Simpson EH. Measurement of Diversity. Nature. 1949;163:688–688. [Google Scholar]
- 47.Gantuya B, El-Serag HB, Matsumoto T, Ajami NJ, Oyuntsetseg K, Azzaya D, Uchida T, Yamaoka Y. Gastric Microbiota in Helicobacter pylori-Negative and -Positive Gastritis Among High Incidence of Gastric Cancer Area. Cancers (Basel) 2019:11. doi: 10.3390/cancers11040504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao R, Wan C, Wang Z. The relationship of gastric microbiota and Helicobacter pylori infection in pediatrics population. Helicobacter. 2020;25:e12676. doi: 10.1111/hel.12676. [DOI] [PubMed] [Google Scholar]
- 49.Wroblewski LE, Peek RM Jr. Helicobacter pylori, Cancer, and the Gastric Microbiota. Adv Exp Med Biol. 2016;908:393–408. doi: 10.1007/978-3-319-41388-4_19. [DOI] [PubMed] [Google Scholar]
- 50.Chan YK, Estaki M, Gibson DL. Clinical consequences of diet-induced dysbiosis. Ann Nutr Metab. 2013;63 Suppl 2:28–40. doi: 10.1159/000354902. [DOI] [PubMed] [Google Scholar]
- 51.Fan W, Huo G, Li X, Yang L, Duan C. Impact of diet in shaping gut microbiota revealed by a comparative study in infants during the six months of life. J Microbiol Biotechnol. 2014;24:133–143. doi: 10.4014/jmb.1309.09029. [DOI] [PubMed] [Google Scholar]
- 52.Goldsmith JR, Sartor RB. The role of diet on intestinal microbiota metabolism: downstream impacts on host immune function and health, and therapeutic implications. J Gastroenterol. 2014;49:785–798. doi: 10.1007/s00535-014-0953-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takagi T, Naito Y, Inoue R, Kashiwagi S, Uchiyama K, Mizushima K, Tsuchiya S, Okayama T, Dohi O, Yoshida N, Kamada K, Ishikawa T, Handa O, Konishi H, Okuda K, Tsujimoto Y, Ohnogi H, Itoh Y. The influence of long-term use of proton pump inhibitors on the gut microbiota: an age-sex-matched case-control study. J Clin Biochem Nutr. 2018;62:100–105. doi: 10.3164/jcbn.17-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo WK, Chan WW. Proton pump inhibitor use and the risk of small intestinal bacterial overgrowth: a meta-analysis. Clin Gastroenterol Hepatol. 2013;11:483–490. doi: 10.1016/j.cgh.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 56.Vesper BJ, Jawdi A, Altman KW, Haines GK 3rd, Tao L, Radosevich JA. The effect of proton pump inhibitors on the human microbiota. Curr Drug Metab. 2009;10:84–89. doi: 10.2174/138920009787048392. [DOI] [PubMed] [Google Scholar]
- 57.Paroni Sterbini F, Palladini A, Masucci L, Cannistraci CV, Pastorino R, Ianiro G, Bugli F, Martini C, Ricciardi W, Gasbarrini A, Sanguinetti M, Cammarota G, Posteraro B. Effects of Proton Pump Inhibitors on the Gastric Mucosa-Associated Microbiota in Dyspeptic Patients. Appl Environ Microbiol. 2016;82:6633–6644. doi: 10.1128/AEM.01437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malfertheiner P, Kandulski A, Venerito M. Proton-pump inhibitors: understanding the complications and risks. Nat Rev Gastroenterol Hepatol. 2017;14:697–710. doi: 10.1038/nrgastro.2017.117. [DOI] [PubMed] [Google Scholar]
- 59.Mason KL, Erb Downward JR, Falkowski NR, Young VB, Kao JY, Huffnagle GB. Interplay between the gastric bacterial microbiota and Candida albicans during postantibiotic recolonization and gastritis. Infect Immun. 2012;80:150–158. doi: 10.1128/IAI.05162-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aviles-Jimenez F, Vazquez-Jimenez F, Medrano-Guzman R, Mantilla A, Torres J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci Rep. 2014;4:4202. doi: 10.1038/srep04202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noto JM, Peek RM Jr. The gastric microbiome, its interaction with Helicobacter pylori, and its potential role in the progression to stomach cancer. PLoS Pathog. 2017;13:e1006573. doi: 10.1371/journal.ppat.1006573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, Wu WK, Wong SH, Chen Z, Sung JJY, Yu J. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67:1024–1032. doi: 10.1136/gutjnl-2017-314281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brusselaers N, Wahlin K, Engstrand L, Lagergren J. Maintenance therapy with proton pump inhibitors and risk of gastric cancer: a nationwide population-based cohort study in Sweden. BMJ Open. 2017;7:e017739. doi: 10.1136/bmjopen-2017-017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song H, Zhu J, Lu D. Long-term proton pump inhibitor (PPI) use and the development of gastric pre-malignant lesions. Cochrane Database Syst Rev. 2014:CD010623. doi: 10.1002/14651858.CD010623.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eslami L, Nasseri-Moghaddam S. Meta-analyses: does long-term PPI use increase the risk of gastric premalignant lesions? Arch Iran Med. 2013;16:449–458. [PubMed] [Google Scholar]
- 66.Vogiatzi P, Cassone M, Luzzi I, Lucchetti C, Otvos L Jr, Giordano A. Helicobacter pylori as a class I carcinogen: physiopathology and management strategies. J Cell Biochem. 2007;102:264–273. doi: 10.1002/jcb.21375. [DOI] [PubMed] [Google Scholar]
- 67.Rajilic-Stojanovic M, Figueiredo C, Smet A, Hansen R, Kupcinskas J, Rokkas T, Andersen L, Machado JC, Ianiro G, Gasbarrini A, Leja M, Gisbert JP, Hold GL. Systematic review: gastric microbiota in health and disease. Aliment Pharmacol Ther. 2020;51:582–602. doi: 10.1111/apt.15650. [DOI] [PubMed] [Google Scholar]
- 68.Vogtmann E, Goedert JJ. Epidemiologic studies of the human microbiome and cancer. Br J Cancer. 2016;114:237–242. doi: 10.1038/bjc.2015.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dicksved J, Lindberg M, Rosenquist M, Enroth H, Jansson JK, Engstrand L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol. 2009;58:509–516. doi: 10.1099/jmm.0.007302-0. [DOI] [PubMed] [Google Scholar]
- 70.Robinson KM, Crabtree J, Mattick JS, Anderson KE, Dunning Hotopp JC. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome. 2017;5:9. doi: 10.1186/s40168-016-0224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC, Fox JG. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rolig AS, Cech C, Ahler E, Carter JE, Ottemann KM. The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect Immun. 2013;81:1382–1389. doi: 10.1128/IAI.00044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, Ge Z, Wang TC, Fox JG. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54–63. doi: 10.1136/gutjnl-2013-305178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu X, Shao L, Liu X, Ji F, Mei Y, Cheng Y, Liu F, Yan C, Li L, Ling Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine. 2019;40:336–348. doi: 10.1016/j.ebiom.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jo HJ, Kim J, Kim N, Park JH, Nam RH, Seok YJ, Kim YR, Kim JS, Kim JM, Lee DH, Jung HC. Analysis of Gastric Microbiota by Pyrosequencing: Minor Role of Bacteria Other Than Helicobacter pylori in the Gastric Carcinogenesis. Helicobacter. 2016;21:364–374. doi: 10.1111/hel.12293. [DOI] [PubMed] [Google Scholar]
- 76.Eyvazi S, Vostakolaei MA, Dilmaghani A, Borumandi O, Hejazi MS, Kahroba H, Tarhriz V. The oncogenic roles of bacterial infections in development of cancer. Microb Pathog. 2020;141:104019. doi: 10.1016/j.micpath.2020.104019. [DOI] [PubMed] [Google Scholar]
- 77.Homan M, Orel R. Are probiotics useful in Helicobacter pylori eradication? World J Gastroenterol. 2015;21:10644–10653. doi: 10.3748/wjg.v21.i37.10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li TH, Qin Y, Sham PC, Lau KS, Chu KM, Leung WK. Alterations in Gastric Microbiota After H. Pylori Eradication and in Different Histological Stages of Gastric Carcinogenesis. Sci Rep. 2017;7:44935. doi: 10.1038/srep44935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorell K, Bengtsson-Palme J, Liu OH, Palacios Gonzales RV, Nookaew I, Rabeneck L, Paszat L, Graham DY, Nielsen J, Lundin SB, Sjöling Å. In Vivo Analysis of the Viable Microbiota and Helicobacter pylori Transcriptome in Gastric Infection and Early Stages of Carcinogenesis. Infect Immun. 2017;85 doi: 10.1128/IAI.00031-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee CW, Rickman B, Rogers AB, Muthupalani S, Takaishi S, Yang P, Wang TC, Fox JG. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009;69:8166–8174. doi: 10.1158/0008-5472.CAN-08-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mera RM, Bravo LE, Camargo MC, Bravo JC, Delgado AG, Romero-Gallo J, Yepez MC, Realpe JL, Schneider BG, Morgan DR, Peek RM Jr, Correa P, Wilson KT, Piazuelo MB. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut. 2018;67:1239–1246. doi: 10.1136/gutjnl-2016-311685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, Song KS, Kim YS, Kim JF. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter. 2014;19:407–416. doi: 10.1111/hel.12145. [DOI] [PubMed] [Google Scholar]
- 83.Dias-Jácome E, Libânio D, Borges-Canha M, Galaghar A, Pimentel-Nunes P. Gastric microbiota and carcinogenesis: the role of non-Helicobacter pylori bacteria - A systematic review. Rev Esp Enferm Dig. 2016;108:530–540. doi: 10.17235/reed.2016.4261/2016. [DOI] [PubMed] [Google Scholar]
- 84.Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975;2:58–60. doi: 10.1016/s0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 85.Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10:324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Naylor G, Axon A. Role of bacterial overgrowth in the stomach as an additional risk factor for gastritis. Can J Gastroenterol. 2003;17 Suppl B:13B–17B. doi: 10.1155/2003/350347. [DOI] [PubMed] [Google Scholar]
- 87.Gao JJ, Zhang Y, Gerhard M, Mejias-Luque R, Zhang L, Vieth M, Ma JL, Bajbouj M, Suchanek S, Liu WD, Ulm K, Quante M, Li ZX, Zhou T, Schmid R, Classen M, Li WQ, You WC, Pan KF. Association Between Gut Microbiota and Helicobacter pylori-Related Gastric Lesions in a High-Risk Population of Gastric Cancer. Front Cell Infect Microbiol. 2018;8:202. doi: 10.3389/fcimb.2018.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pereira-Marques J, Ferreira RM, Pinto-Ribeiro I, Figueiredo C. Helicobacter pylori Infection, the Gastric Microbiome and Gastric Cancer. Adv Exp Med Biol. 2019;1149:195–210. doi: 10.1007/5584_2019_366. [DOI] [PubMed] [Google Scholar]
- 89.Guo Y, Zhang Y, Gerhard M, Gao JJ, Mejias-Luque R, Zhang L, Vieth M, Ma JL, Bajbouj M, Suchanek S, Liu WD, Ulm K, Quante M, Li ZX, Zhou T, Schmid R, Classen M, Li WQ, You WC, Pan KF. Effect of Helicobacter pylori on gastrointestinal microbiota: a population-based study in Linqu, a high-risk area of gastric cancer. Gut. 2020;69:1598–1607. doi: 10.1136/gutjnl-2019-319696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rieder G, Fischer W, Haas R. Interaction of Helicobacter pylori with host cells: function of secreted and translocated molecules. Curr Opin Microbiol. 2005;8:67–73. doi: 10.1016/j.mib.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 91.Li Q, Liu J, Gong Y, Yuan Y. Serum VacA antibody is associated with risks of peptic ulcer and gastric cancer: A meta-analysis. Microb Pathog. 2016;99:220–228. doi: 10.1016/j.micpath.2016.08.030. [DOI] [PubMed] [Google Scholar]
- 92.Terebiznik MR, Raju D, Vázquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5:370–379. doi: 10.4161/auto.5.3.7663. [DOI] [PubMed] [Google Scholar]
- 93.Castaño-Rodríguez N, Kaakoush NO, Lee WS, Mitchell HM. Dual role of Helicobacter and Campylobacter species in IBD: a systematic review and meta-analysis. Gut. 2017;66:235–249. doi: 10.1136/gutjnl-2015-310545. [DOI] [PubMed] [Google Scholar]
- 94.Dubois A, Borén T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell Microbiol. 2007;9:1108–1116. doi: 10.1111/j.1462-5822.2007.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weydig C, Starzinski-Powitz A, Carra G, Löwer J, Wessler S. CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp Cell Res. 2007;313:3459–3471. doi: 10.1016/j.yexcr.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 96.Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, Ohno S, Hatakeyama M. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 97.Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM Jr, Azuma T, Hatakeyama M. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 98.Merino JS, García A, Pastene E, Salas A, Saez K, González CL. Lactobacillus fermentum UCO-979C strongly inhibited Helicobacter pylori SS1 in Meriones unguiculatus. Benef Microbes. 2018;9:625–627. doi: 10.3920/BM2017.0160. [DOI] [PubMed] [Google Scholar]
- 99.Krzyżek P. Commentary: Proteomics Analysis Revealed that Crosstalk between Helicobacter pylori and Streptococcus mitis May Enhance Bacterial Survival and Reduces Carcinogenesis. Front Microbiol. 2017;8:2381. doi: 10.3389/fmicb.2017.02381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khosravi Y, Dieye Y, Loke MF, Goh KL, Vadivelu J. Streptococcus mitis induces conversion of Helicobacter pylori to coccoid cells during co-culture in vitro. PLoS One. 2014;9:e112214. doi: 10.1371/journal.pone.0112214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shen Z, Dzink-Fox J, Wilson KT, Whary MT, Muthupalani S, Piazuelo MB, Bravo LE, Suerbaum S, Fox JG, Josenhans C. Tu1288 - Co-Colonization of Helicobacter Pylori with Staphylococcus Epidermidis or Streptococcus Salivarius Differ in the Progression of Gastritis in Ins-Gas Mice. Gastroenterology. 2018;154:s924–s925. [Google Scholar]
- 102.Kazakos EI, Dorrell N, Polyzos SA, Deretzi G, Kountouras J. Comment on "Effect of biofilm formation by clinical isolates of Helicobacter pylori on the efflux-mediated resistance to commonly used antibiotics". World J Gastroenterol. 2017;23:6194–6196. doi: 10.3748/wjg.v23.i33.6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moghadam MT, Chegini Z, Khoshbayan A, Farahani I, Shariati A. Helicobacter pylori Biofilm and New Strategies to Combat it. Curr Mol Med. 2021;21:549–561. doi: 10.2174/1566524020666201203165649. [DOI] [PubMed] [Google Scholar]
- 104.Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, Afonso L, Lopes P, Roncon-Albuquerque R Jr, Henrique R, Moreira-Dias L, Leite-Moreira AF, Dinis-Ribeiro M. Helicobacter pylori induces increased expression of Toll-like receptors and decreased Toll-interacting protein in gastric mucosa that persists throughout gastric carcinogenesis. Helicobacter. 2013;18:22–32. doi: 10.1111/hel.12008. [DOI] [PubMed] [Google Scholar]
- 105.González Torre JA, Cruz-Gómez ÁJ, Belenguer A, Sanchis-Segura C, Ávila C, Forn C. Hippocampal dysfunction is associated with memory impairment in multiple sclerosis: A volumetric and functional connectivity study. Mult Scler. 2017;23:1854–1863. doi: 10.1177/1352458516688349. [DOI] [PubMed] [Google Scholar]