

Abstract
Erlotinib was covalently linked to 3-(1’-hexyloxy)ethyl-3-devinylpyropheophorbide a (HPPH) and structurally related chlorins and bacteriochlorins at different positions of the tetrapyrrole ring. The functional consequence of each modification was determined by quantifying the uptake and subcellular deposition of the Erlotinib conjugates, cellular response to therapeutic light treatment in tissue cultures, and in eliminating of corresponding tumors grown as xenograft in SCID mice. The experimental human cancer models included the established cell lines UMUC3 (bladder), FaDu (hypopharynx) and primary cultures of head & neck tumor cells. The effectiveness of the compounds was compared to that of HPPH. Furthermore, specific functional contribution of the carboxylic acid side group at position-172 and the chiral methyl group at 3-(1’) to the overall activity of the chimeric compounds was assessed. Among the conjugates investigated, the PS 10 was identified as the most effective candidate than HPPH for achieving tumor cell-specific accumulation and yielding improved long-term tumor control.
Graphical Abstract
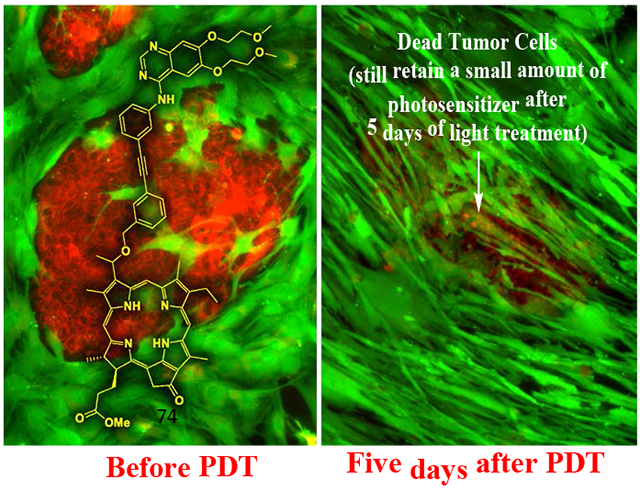
Among a series of Erlotinib-Photosensitizer conjugates, PS 10 in which the Erlotinib moiety was introduced at position 3(1’) of methyl pyropheophorbide a (a chlorophyll a analog) shows excellent tumor cell-specificity, e.g., HN 114: tumor cells derived from a head & neck cancer patient.
Red = Tumor cells, Green = Stromal cells.
INTRODUCTION
Photodynamic therapy (PDT) is a non-invasive cancer treatment modality and is an alternative to surgery, chemotherapy and radiotherapy.1,2 Since the approval of Photofrin, a hematoporphyrin-based photosensitizer (PS) by various health organizations, different laboratories have investigated new avenues to develop new PDT-agents with photophysical properties at longer wavelength, increased tumor cell specificity, and more favorable pharmacokinetic profile.2-5 During the last 30 years, a large number of PDT agents with excitation light wavelength ranging from 650-800 nm have been synthesized and evaluated.6,7 Tumor cells in general have a non-specific affinity to porphyrins, and there has not been much success in improving the tumor selectivity and specificity of these agents.8-10
Attempts have been made to direct PSs to cellular targets by conjugating with known tumor-targeting molecules.11,12 The targeting molecules include cholesterol,13 chemotherapy agents,14 monoclonal antibodies,15 carbohydrates16-20 and linear or cyclic peptide ligands.21-23 However, there is still a major challenge to gain a PS that is selectively taken up and is retained by malignant cells.
One of the potential tumor cell targets appears to be epidermal growth factor receptor (EGFR).24-27 Mutations that lead to EGFR overexpression28 or to dysregulation of its kinase activity29 have been associated with a number of cancers, including squamous-cell carcinoma of the bladder, head and neck (80-100%), lung (80%), anal cancers, and glioblastoma (50%).
The separate approach to identify optimal PS structures is by structure-activity relationship (SAR) and quantitative structure-activity relationship (QSAR).30-32 These studies indicated that in asymmetrical PSs, such as chlorophyll-a derivatives, the presence of the substituents at variable positions of the macrocycle and the overall lipophilicity, make a significant impact on tumor-uptake and pharmacokinetic profile. Such studies helped us in developing effective analogs, such as 3-(1’-hexyloxy)ethyl-3-devinylpyropheophorbide-a (HPPH),32 which is currently in Phase I/II human clinical trials of head & Neck (H & N),33 esophageal34 and lung cancer.35 The 3-[1’ (m-iodobenzyloxy)ethyl-3-devinylpyropheophorbide-a in radioactive (124I-, PET-ONCO) and non-radioactive (127I-) forms provide an opportunity to use a single agent for positron emission tomography (PET) imaging, fluorescence imaging and photodynamic therapy of a variety of cancers, including head & neck, lung and renal carcinoma.27,28 Among the near infrared (NIR) PSs derived from bacteriochlorophyll-a, 3-(1’-(butyloxy)ethyl-3-deacetyl-bacteriopurpurin-18-N-butylimide methyl ester (Photobac) showed potential for treating glioblastoma in SCID mice bearing U87 tumors.36,37
We have recently shown that the iodinated PS discussed above on conjugating with an Erlotinib moiety has applications in imaging (PET, fluorescence) and phototherapy (PDT) of bladder cancer.38 Interestingly, the in vivo results indicated that position of the erlotinib moiety at the peripheral position of basic skeleton made a significant difference in long-term control.38 Therefore, we sought to:(i) to extend the SAR study in a variety of tetrapyrrolic systems derived from chlorophyll-a and bacteriochlorophyll-a and compare their in vitro and in vivo photosensitizing ability in head & neck and bladder tumor cells and mice tumor models. and (ii) to compare the biological efficacy of the best candidates with HPPH, a leading agent undergoing human clinical trials33
Co-joining a porphyrin with Erlotinib demands a re-assessment of whether the distinct functionalities of each component are retained in the final product. Expected is that the lipophilic porphyrin contributes to the mode and specificity of cellular uptake and Erlotinib the interaction with the cytoplasmic domain of the ATP binding site of the plasma membrane-resident EGFR. However, it is not yet known whether the close proximity of the porphyrin with its preference for membrane binding will negate Erlotinib interaction with the EGFR domain. To address part of these questions, this study presents the synthesis of various porphyrin-Erlotinib variants and tests these for: (i) uptake, cellular deposition and retention by cells with prominent EGFR expression, (ii) photoreaction in vitro and (iii) in vivo localization and action in xenograft-bearing mice. A separate study will address the Erlotinib-specific function of the chimeric compounds.
RESULTS AND DISCUSSION
Chemistry:
For our study, a series of Erlotinib-PS conjugates related to (a) pyropheophorbide-a λmax: 660 nm, (b) purpurinimide, λmax: 700 nm, (c) bacteriopurpurinimide, λmax: 782 nm and (d) chlorin e6, λmax: 660 nm (Figure 1) were synthesized.31,39 These PSs were selected not only to determine the impact of the position of the Erlotinib moiety, but also the effects of inherent structural differences of these compounds in the photodynamic treatment of cancer.
Figure 1:

Structures of the starting chlorins and bacteriochlorin used in present study
1. Erlotinib moiety introduced at the lower half (position-172-) of the pyropheophorbide-a with different linkers:
For the synthesis of the conjugate 5 containing a triazole alkyl linker, Erlotinib 1 was first reacted with 1–azido-4-butyl amine 2, and the intermediate 3 was reacted with HPPH 4 in presence of EDC/DMAP and the desired conjugate 5 was obtained in 52% yield. To investigate the impact of the linker joining the PS-Erlotinib moieties, HPPH 4 was first reacted with 3-iodobenzylamine 6, and gave the corresponding iodobenzylamide analog 7 in 89% yield, which on reacting with Erlotinib under mild reaction conditions afforded the desired HPPH-Erlotinib conjugate 8 in 75% yield (Scheme 1).
Scheme 1:

Erlotinib moiety conjugated at position-17 of the hexyl ether analog of pyropheophorbide-a
2. Erlotinb moiety introduced at the upper half (position 3(1’) of the pyropheophorbide-a and bacteriopyropheophorbide-a:
Methyl 3-(1’-m-iodo-benzyloxy)ethyl-3-devinylpyropheophorbide 9 obtained by reacting pyropheophorbide-a with HBr/AcOH and m-iodobenzyl alcohol was reacted with Erlotinib in presence of palladium catalyst and the respective conjugate 10 as a methyl ester analog was isolated in 72% yield. The methyl ester functionality was then converted into the corresponding carboxylic acid 11 by reacting with aqueous lithium hydroxide under mild conditions in 95% yield. Following a similar approach, the bacteriopyropheophorbide 12 in which rings “B & D” are reduced was also converted into the corresponding methyl ester and carboxylic acid analogs 13 and 14 respectively (Scheme 2).
Scheme 2:

Erlotinib moiety conjugated at position-3 of pyropheophorbide-a and bacteriopyro-pheophorbide-a
3. Erlotinb conjugated at p-position of 3-(1’-p-iodo- benzyloxy)ethyl- and 3 (-m-iodobenzyloxy methyl) pyropheophorbide-a:
To investigate the impact of the position of the Erlotinib moiety (m- vs p- of the phenyl group) the methyl ester and carboxylic acid analogs 16 and 17 were synthesized in 72% and 95% yield respectively. For determining the necessity of the chiral center at position 3(1’-) in biological efficacy of 11, PS 18 (with no chiral center at position 3(1’-) was synthesized by following our own methodology, which on conjugation with Erlotinib gave the conjugate 19 as methyl ester in excellent yield.
4. Erlotinib moiety conjugated at position-20 of the methyl mesopyropheophorbide-a:
For introducing Erlotinib at position-20 of the PS, compound 20 containing a benzoic acid functionality 20 was prepared from 20-bromo-pyropheophorbide-a following the methodology developed in our laboratory.40 It was reacted with p-iodobenzyl amine and the intermediate 22 was obtained in 66% yield. It was then reacted with Erlotinib by following the methodology discussed for other compounds, and the desired erlotinib-PS conjugate 23 was obtained in 40% yield (Scheme 4).
Scheme 4:

Erlotinib moiety conjugated at position 20 of methyl meso-pyropheophorbide a
5. HPPH conjugated with multiple Erlotinib moieties at the lower half (position 17-) of the PS:
HPPH 4 was first reacted with triethyl ethyl amine 24, and the known HPPH tricarboxylic analog 25 was obtained in 80% yield. It was then reacted with erlotinib analog 26 (BOP. Et3N) and the tri-Erlotinib-PS conjugate 27 was obtained in 84% yield (Scheme 5).
Scheme 5:

The hexyl ether analog of pyropheophorbide-a (HPPH) conjugated with three Erlotinib moieties.
6. Erlotinib-N-imide substituted purpurinimide and bacteriopurpurinimides:
To investigate the impact of Erlotinib moiety in biological efficacy, the purpurinimide 28 and bacteriopurpurinimide 30 containing N-3-iodobenzyl group at position132- were reacted with Erlotinib 1 by following the standard methodology as discussed for other conjugates and the corresponding conjugates 29 and 31 were obtained in 88% and 75% yield respectively (Scheme 6).
Scheme 6:

Erlotinib moiety conjugated to the fused imide ring system of purpurinimide and Bacteriopurpurinimide
7. Erlotinib conjugated at position 13 and 15 of chlorin e6:
Recently, Smith et. al prepared a series of peptide analogs of chlorin e6 in which the peptides were attached either at position- 13,15 or 17 of chlorin e6 moieties41. In preliminary in vitro screening, the PS conjugated at position-13 and 15 were more effective than those conjugated at position-17. Therefore, to investigate the effect of the presence of Erlotinib moiety at these position on biological efficacy, methyl-3 devinyl-pheophorbide-a 32 and the corresponding 3-hexyloxy ethyl analog (methyl 3-(1’-hexyloxy)ethyl-pheophorbide-a 33 were individually reacted with 3-iodobenzyl amine 34, and the respective iodobenzyl amide analogs 35 and 36 were isolated in 90% yield. Reaction of these intermediates with Erlotinib moieties under similar reaction conditions as discussed for other compounds afforded the desired 13-substituted erlotinib analogs 37 and 38 in 79% yield (Scheme 7).
Scheme 7:

Erlotinib moiety conjugated at position-13 of chlorin e6 with an amide linker
To determine the impact of ethylene glycol with variable carbon units and also the nature of substituent at position-3 of chlorin e6, conjugates 43 and 50 with variable glycol linker joining the PS at position-13 with Erlotinib were synthesized in excellent yields (Scheme 8 & 9).
Scheme 8:

Erlotinib moiety conjugated at position-13 of chlorin e6 with an ethylene glycol linker
Scheme 9:

Erlotinib moiety conjugated at position-13 of chlorin by a longer ethylene glycol linker
For the synthesis of the conjugate in which Erlotinib was introduced at position-15 of the PS, chlorin e6 tricarboxylic acid 51 was reacted with 3-iodo benzyl amine 6 gave the 15-substituted iodobenzyl analog 52 in 51% yield, which on reacting with Erlotinib 1 afforded the 15-Erlotinib chlorin e6 dimethyl ester 53 in 45% yield (Scheme 10).
Scheme 10:

Erlotinib moiety conjugated at position-15 of chlorin e6
The structures of all intermediate and final products were confirmed by 1H and 13C NMR (1D and 2D). The NMR data are consistent with the proposed structures. In some cases, these data exhibited interesting features suggesting significant differences in molecular conformation, which depend on the location of the Erlotinib linkage point in the molecule.
Linkers joining the PS and the Erlotinib moiety through the 17-position of the macrocycle resulted in chemical shifts that differed substantially from those observed in other conjugates. Conjugates 5 and 8 exhibited shielding (relative to unconjugated compound 4) of protons in the vicinity of the macrocycle’s C-ring. In conjugate 8, the 10-H, 132-CHH, and 12-CH3 protons were observed at 8.97/8.95, 4.53, and 2.29/2.26 ppm, respectively. In unconjugated photosensitizer 4,42 however, these protons appear at 9.45, 5.11, and 3.63 ppm, respectively. So, conjugation to Erlotinib through the 17-position resulted in a significant shielding of each of these protons, which ranged from ~0.5 to ~1.4 ppm. In addition, the NMR spectra of 8 exhibited a marked deshielding of the quinazoline 5-H of Erlotinib, which was observed at 7.853/7.850 ppm. This contrasts with the chemical shifts observed for the corresponding protons of conjugates linked through other positions, as well as that of an unconjugated Erlotinib analog (similar to 26) observed in non-polar solvent. Under these conditions, the quinazoline 5-H typically resonates at ~7.2 ppm. A deshielding of ~0.7 ppm, therefore, resulted from conjugation to the photosensitizer’s 17-position.
Similar 1H NMR chemical shifts were observed for 5, which differs from 8 in linker structure only. Here, the 10-H, 132-CHH, and 12-CH3 protons were observed at 8.80/8.78, 4.98, and 2.77 ppm, respectively. Compared with the shieldings observed for 8, shieldings observed for 5 were larger for 10-H, but smaller for 12-CH3 and 132-CHH. The difference in linker structure is likely responsible for this. The quinazoline 5-H of 5 (7.77 ppm) was only slightly less deshielded than that observed in 8.
The chemical shifts observed for these specific protons of the PS and Erlotinib moieties of 5 and 8 indicate significant differences in the electronic environment, relative to the other conjugates and precursors examined here. Conjugates 5 and 8 are both 17-linked. Furthermore, no other linkage point resulted in such pronounced shielding/deshielding effects for quinazoline 5-H or for protons near the macrocycle’s C-ring (relative to unconjugated PS of similar structure or the free Erlotinib derivative).
In 5 and 8 the linkage point is reasonably close to the C-ring, and each linker consists of a flexible chain of several atoms that connect to the more rigid, aromatic portion of the Erlotinib-containing substituent. This arrangement may allow the Erlotinib moiety to interact with the atoms surrounding the C-ring. Stacking of the aromatic ring systems of the macrocycle and substituent might result in ring current-induced shielding/deshielding, accounting for the observed chemical shifts, both for the protons (near the C-ring) of the macrocycle and the quinazoline 5-H. Pi-pi stacking interactions could help stabilize such a conformation. In the 3- and 20-linked conjugates, the Erlotinib-containing substituent is much more distant from the C-ring. Moreover, the linkers are shorter and presumably much less flexible than those of 5 and 8. So, it is unlikely that these species can adopt conformations allowing a close approach of the C-ring and quinazoline moiety. This is consistent with the NMR observations. In conjugates with linkages at the 13-, 15-, or imide nitrogen position, the linkage point is certainly close enough to permit interactions between the C-ring and the quinazoline moiety, but linker length and flexibility likely prevent this. In several of these conjugates (e.g. 23, 29, 31, 37 and 53), the linker is too short and insufficiently flexible. In others (e. g. 43 and 47), the linker is much too long, likely permitting too much motional freedom. Again, the NMR spectra are consistent with these hypotheses.
Unlike 5 and 8, conjugate 27 showed no shielding of the protons near the C-ring, despite being linked through the 17-position. However, the substituent is much bulkier in 27, containing three Erlotinib moieties. Steric hindrance may prevent close approach to the C-ring by this very bulky substituent, accounting for the observed (unshielded) delta values of protons near the C-ring. The quinazoline 5-H signal was actually moderately deshielded in 27, but this likely resulted from the polar solvent used during observation. Similar to 27, 37, 17 also exhibited a moderate, solvent-induced (CDCl3/CD3OD (90:10)) deshielding of the quinazoline 5-H signal, but no shielding in the vicinity of the C-ring. Similar deshielding (δ5-H = 7.65 ppm) was observed in 26, which was examined in CDCl3/CD3OD (80:20) solvent36. The polar solvent may induce a conformational change responsible for the deshielding. In less polar solvent (i. e. CDCl3), in which most compounds reported here were observed, an Erlotinib analog similar to 26 showed no deshielding of the quinazoline 5-H signal.
The quinazoline 5-H protons of 23, 31, 43, and 50 were each deshielded, but to a much lesser extent than observed in 5 or 8. These conjugates, however, did not exhibit the wider effects demonstrated by 5 and 8: the PS protons near the C-ring were not shielded. This suggests that the deshielding of 5-H in each of these four conjugates was not due to interaction with the C-ring pi system. Some other affect was likely responsible, perhaps a change in conformation within the Erlotinib moiety itself. Although 10-H of 7 was shielded (δ = 9.07/9.05 ppm), neither its 12-CH3, nor its 132-CHH proton was significantly affected. This may be due to an interaction involving the aromatic ring of the iodobenzyl group, resulting from a conformation affecting only the proton at position-10.
COMPARATIVE IN VITRO AND IN VIVO EFFICACY:
Uptake and retention of the PS as function of the tumor cell phenotype:
Charge and position of hydrophobic residues critically influence the cell biological properties of porphyrins. Cultures of primary human head/neck tumor cells have been used to evaluate the structures that determines cell type-specific uptake and PDT of the synthesized Erlotinib-porphyrin compounds (Schemes 1-10). For facilitating comparison of activities, HPPH was included in the analyses as reference. A representative example is shown in Figure 2 in which tongue cancer derived HNT1 cells were used to compare the uptake of HPPH with that of the Erlotinib-conjugates 10 and 11. The analysis determined the effect of the substitution of the hexyl group at position 3 by Erlotinib (10) and the presence of a carboxyl group at position-172 (11). The results indicated a marked difference in the kinetics of accumulation and efflux. Retention of 10 after 24h chase period exceeded that of HPPH and 11, even though the latter two compounds were initially taken up at much higher rate. An equivalent difference due to the presence of a carboxyl residue at position 17-2 was observed for conjugates 16 and 17. The quantitative comparison of all Erlotinib-porphyrin compounds in separate preparations of head & neck cancer cell preparations carried out over the past 2 years indicated a consistent pattern of uptake and retention. Elevated retention over 48 hours by cancer cells were identified for 5, 8, 10, 16, 17, 43. In contrast, substantially lower uptake and retention level were determined for 13, 14, 19, 22, 23, 27, 29, 36, 38, 49 and 50 (data not shown, see also Figures 3 and 4 below)).
Figure 2.

Uptake and retention by HNT1 cells (passage 116). Confluent cell cultures were incubated for 30 min in serum-free medium containing 3.2 μM of the indicated PS. The cellular level and subcellular distribution of PS fluorescence indicates the binding to and mode of uptake by the cells. Subsequent treatment of the cells for 5 h with serum and PS-containing medium determines the competing effect of serum proteins on uptake and steady-state accumulation of the PSs. Follow-up incubation for 15 h in serum-containing medium but without PS defines the rate of egress of PS from cells. PS fluorescence is recorded at 100X magnification by different-length exposure of the camera. Images allow digital quantification of PS fluorescence level.
Figure 3.

Cell type specific retention of Erlotinib-conjugates. Five-day old co-culture of FaDu-49 and CFSE-stained HN-121 T-Fb were incubated with medium containing 10% FBS and 1.6 μM PS followed by a 48-h chase in medium without PS. Cell-associated fluorescence signals were recorded.
Figure 4.

Five-day old cocultures of HN-143 T-EC with HN-166 T-Fb were incubated with medium containing 10% FBS and 3 mM indicated PS for 5 h and then chased for 40 h with medium without PS. Phase contrast images at 100X magnification (upper panel in each section) and corresponding fluorescent images of all cultures (500 ms exposure time) were taken at the indicated time points (lower point in each section).
Microscopic images of the cells after short- and-long term treatment with Erlotinib conjugates indicated that each compound, like HPPH, was taken up by transmembrane diffusion and accumulated in mitochondria and ER compartment. The only exception was compound 27, which probably due to the presence of the triple Erlotinib side group, did not diffuse into the cells but was endocytosed and deposited in the lysosomal compartment.
One of the goals of this study was to achieve a preferential retention of an Erlotinib-conjugate in tumor epithelial cells. Previous studies have shown, that certain forms of pheophorbides, including HPPH, were several-fold less effectively retained by tumor stromal cells (fibroblasts) than by the tumor epithelial cells.43 To identify this property for he Erlotinib conjugates with appreciable uptake and retention activity, these were tested on reconstituted co-cultures of tumor and stromal cells from head and neck cancer tissue. This culture system allowed a simultaneous assay of the cell type-specific retention by comparing PS fluorescence in each cell type as a function of culture period post PS incubation period. A demonstration of this assay is shown by the example in Figure 3. In this particular experiment, the detection of the stromal cells separate from the tumor cell clusters were enhanced by tagging these with CFSE prior to the addition to FaDu cells (subclonal line 49). The comparison of HPPH, 10, and 19 demonstrated the difference in the magnitude of cellular retention. The PS level in the cell types served not only as measure for cell type specificity of the PS but also as marker for the expected response to treatment with therapeutic light.44
In order to rule out a modifying influence of the tagging, non-modified fibroblasts cultures combined with epithelial cells from different tumors were used to establish the level of cell type specificity and pinpoint the potential optimal Erlotinib conjugates. Despite quantitative variation in uptake and retention by separate tumor epithelial cell preparations, the most reproducible differential retention in epithelial cells was determined for 10. Although conjugates 5 and 43 were taken-up and retained by most tumor epithelial at higher level than 10, the similarly elevated retention of these PS, including HPPH-ME, by fibroblasts did reduce the tumor cell specificity (Figure 4). The comparative analyses also indicated that structural differences in the attachment of Erlotinib to the porphyrin have an impact on uptake properties. The rigid linker of 8 lowered uptake when compared to 5 with the flexible linker. The same feature was observed by comparing uptake of 38 with 43. The comparison of 10 and 16 highlighted the relevance of the position of Erlotinib attachment to the benzyl ring at position 3. Substitution in the meta- position 10 resulted in higher uptake than in the para position (16)
In vitro photosensitizing activity:
The proportional relationship of PS fluorescence with light-triggered photoreaction in cells have previously been demonstrated.45 The same biomarker for PDT, dose-dependent covalent crosslinking of latent STAT3 and degradation of EGFR 46 were detectable with Erlotinib conjugates in head & neck tumor cells (Figure 5).
Figure 5.

PS-dose dependent photoreaction in HN-85-1-3 T-EC. Level of PS taken up was quantified by fluorescence prior to 665 nm light treatment (3 J/cm2). A, Immunoblot analysis of STAT3 and EGFR immediately after light treatment. B, Duplicate cultures were incubated for 24 h after PDT and then the percentage of surviving cells determined.
To relate the PDT response mediated by the Erlotinib-conjugates in vitro with that of the same cells grown as xenograft in mice, two separate established cell systems, UMUC3 and FaDu, were applied. Compared to HPPH, several Erlotinib conjugates (e. g. 5, 8, 10, 13, 14, 16, 17, 35, 37, 43, 43 and 49) showed improved cytotoxicity in both UMUC3 and FaDu cell lines (Table 1).
Table 1:
Comparative in vitro photosensitizing efficacy (IC50) of PS-erlotinib conjugates with HPPH in UMUC3 (bladder cancer) and FaDu (head & neck cancer) cell lines.
| In Vitro PDT Efficacy (IC50) of photosensitizers (concentration: nM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound # |
UMUC3 | FaDu | Compound # |
UMUC3 | FaDu | Compound # |
UMUC3 | FaDu |
| 5 | 20.25 | 17.6 | 17 | 34.4 | 59.6 | 42 | 6.8 | 15.4 |
| 7 | 187.4 | 519.8 | 19 | 76.3 | 23.3 | 43 | 7.5 | 21.4 |
| 8 | 43.4 | - | 22 | 195.9 | 1,246 | 49 | 26.4 | 19.2 |
| 10 | 59.4 | 35.5 | 23 | - | >10 μM | 50 | 59.2 | 88.6 |
| 11 | 54.8 | 31.8 | 31 | - | 6,925 | 53 | 45.0 | 166.2 |
| 13 | 16.8 | - | 35 | 28.2* | 13.8 | HPPH | 68.4 | 43.8 |
| 14 | *8.9 | - | 37 | 39.6 | 33.42 | |||
| 16 | 20.9 | 59.6 | 38 | 3.502 | - | |||
Before selecting the best candidate for in vivo PDT efficacy, the PSs that showed higher tumor cell-specificity were investigated for their uptake in tumor, liver and skin at variable time points in SCID mice bearing UMUC3 and T24 tumors. PS 10 showed higher tumor uptake with a faster clearance from liver and skin with time than HPPH and other PS-Erlotinib conjugates. Previous reports from our laboratory have shown that among the 3-(1’) alkyl ether analogs of pyropheophorbide-a, HPPH containing a hexyl ether side chain at position-3 and carboxylic acid group at position 172- shows the best PDT efficacy. Replacing the carboxylic acid group at position-172 with methyl ester reduces its cell-selectivity and PDT efficacy in most of the tumor types. Besides, the presence and nature of chirality at position 3(1’) also seems to be important for higher efficacy. Therefore, to investigate the importance of these structural characteristics in PS-Erlotinib conjugate 10, the corresponding methyl ester analog 11, and 19 without chirality at position 3(1’) were investigated for comparative uptake and PDT efficacy (Figure 6; see also uptake and cell specificity in Figure 3 & 4).
Figure 6:

Compounds selected to determine the impact of structural requirements for tumor-uptake and in vivo PDT efficacy.
Uptake and PDT efficacy of HPPH with PS-erlotinib conjugates 10, 11 and 19 by UMUC3 bladder tumor:
All PSs were formulated in 1%Tween80 / 5% dextrose solution, and initially evaluated for tumor uptake at variable time points aa dose of 0.47 μmole selected HPPH dose for treating a variety of tumor types in animal models. Interestingly, compared to HPPH (Figure 7F), which showed highest uptake at 24 h postinjection. PS 10 had maximal tumor accumulation at 6h post-injection (Figure 7A) which was half as that of HPPH at the same time point. As expected, PS 10 uptake by all organs was progressively reduced at lower doses, 0.25 μmole/kg and 0.125 μmole/kg respectively (Figure 7B & C) . In contrast, PS 11, which is like HPPH bearing a carboxylic (-COOH) functionality at position-172 (Figure 7D) and PS 19 (achiral at position 3(1’) (Figure 7E) had significantly lower tumor uptake at all time points than 10. In addition, compared to HPPH, the liver and skin uptake of PS 19 was higher at initial timepoints, but cleared from the system significantly with time. Interestingly, the PS-Erlotinib conjugate 10 showed significantly improved long-term tumor control (74%, 17/23 mice were tumor free on day 60) than HPPH (30%, 3/10 mice were tumor free on day 30, Figure 7G) . At the reduced dose of 0.25 μmole/kg, PS 10 still yielded a long-term tumor cure.
Figure 7:

In vivo uptake in tumor, liverand skin (female SCID mice bearing UMUC3 tumor) of PS 10 at the dose of 0.47, 0.25 and 0.125 mmole/kg (A, B & C); PS 11 at a dose of 0. 47 mmole/kg (D); and PS 19 and HPPH at a dose of 0.47 mmol/kg (E & F).The comparative in vivo efficacy of PS 10 at variable drug dose (0.47, 0.25 and 0.125 mmol/kg), PS 19 and HPPH at a dose of 0.47 mole/kg is shown in Figure 7G. All tumors were exposed with the same light dose (665 nm, 135J/cm2,75 mW/cm2), at the time of maximal tumor uptake of the PS. For PS-uptake study (tumor, liver and skin), 3 female SCID mice bearing UMUC3 tumors were used (Figures 7A-7F). For determining in-vivo efficacy the number of female SCID mice used in each study are indicated in the Figure 7G). Statistical analysis; log rank (Mantel-Cox test), p-value:<0.0001.
cure (50%, 5/10 mice were tumor-free on day 60). HPPH at a dose of 0.47 μmole/kg. PS 19 at a dose of 0.47 μmole/kg and same light treatment parameters did not show any long-term tumor response (Figure 7G). The promising antitumor activity of PS 10 in vivo prompted us to compare its tumor uptake and PDT efficacy with HPPH , in FaDu head & neck cancer model.
Uptake PDT efficacy of HPPH with PS 10 and 19:
SCID mice bearing FaDu tumors (size: 4 to 5 mm diameter) were injected with HPPH or PS-Erlotinib conjugates 10 and 19 (PS dose: 0.47 μmol/kg) and the uptake of all the PSs was measured by fluorescence at various time points. HPPH and both Erlotinib conjugates yielded highest uptake at 24 h post injection. Unlike UMUC3 (Figure 7), FaDu tumors after 24 h indicated a higher level of PS 10 accumulation than of HPPH. Even at the reduced dose of 0.25 and 0.125 μmole/kg, PS 10 retained a similar biodistribution profile. In line with the results of the in vitro analysis (Figure 3), PS 19 showed significantly lower uptake than PS 10 and HPPH. The PDT efficacy of PS 10 was compared to HPPH using a larger group of FaDu tumor bearing mice (25 mice: 5 mice/group/experiment). The results depicted in Figure 8E shows that Erlotinib-PS conjugate 10 at a dose of 0.47 μmol/kg is more effective (92% cure, 23/25 mice were tumor free on day 60); than HPPH (30%, 3/10 mice were tumor-free on day 60).Even at a lower dose of 0.25 μmole/kg (10 mice: 5 mice/group) PS 10 mediated a more effective PDT response than HPPH.
Figure 8:

In vivo uptake of PS 10 at the dose of 0.47, 0.25 and 0.125 μmole/kg and HPPH in tumor, liver and skin of SCID mice bearing FaDu tumors (3 mice/group), is shown in A, B, C and D respectively. The in vivo PDT efficacy (long-term tumor response) of PS 10 at variable doses: 0.47, 0.25 and 0.125 μmole/kg and HPPH at a dose of 0.47 μmole/kg is shown in Figure 8E. Tumors were exposed to light (665 nm, 135 J/cm2, 75 mW/ cm2) at 24h post injection and tumor-growth was measured daily for 60 days. For PS-uptake study (tumor, liver and skin), 3 female SCID mice bearing FaDu tumors were used (Figures 8A-8D). For determining in-vivo efficacy, the number of female SCID mice used in each study are indicated in the Figure 8E). Statistical analysis; log rank (Mantel-Cox test), p-value:<0.0001.
Impact of positional isomers (10 vs. 16) in biological activity:
As already observed in tissue culture analyses (Figure 4) changing the Erlotinib moiety attachment from the meta to the para position of the 3-(1’-benzyloxy)ethyl side chain as shown for PS 10 and (Figure 9) reduces the uptake of the PS by tumor cells. This property attributed to PS was investigated my measuring PS 16 uptake by tumors and the PDT efficacy in SCID mice bearing either UMUC3 or FaDu tumors.
Figure 9:

Structure of the PS-Erlotinib conjugates in which erlotinib moiety is attached either at meta- ( PS 10) or para- position (PS 16) of the pyropheophorbide-a.
Mice were injected with PS 16 at a dose of 0.47μmol/kg and the uptake by tumor, liver, skin was compared (Figure 10). Maximum uptake of 16 was observed at 24 h postinjection, and the PS concentration was almost 2-fold higher in FaDu than UMUC3 tumors. The ratio of PS uptake between the tumor to liver and skin was higher in mice bearing FaDu tumors than in those with UMUC3 tumors. The in vivo PDT efficacy is illustrated in Figure 10C and D and indicates a 60% long-term cure of mice bearing bladder tumors and 66% mice with FaDu tumors. Although PS 16 was appreciably taken up By both the tumor types, compare to 10 ( Figures 7 and 8), it was still less effective. These results suggest that position of the Erlotinib moiety at 3-(1’)-benzyloxyethyl side chain ( meta- vs para-) makes a difference in directing uptake and intracellular retention resulting in a proportionally lower PDT efficacy.
Figure 10:

In vivo uptake of PS 16 at a dose of 0.47μmole/kg in tumor, liver and skin of SCID mice bearing (A) UMUC3 and (B) FaDu tumors. The in vivo PDT efficacy (long-term tumor response) of PS 16 at a dose of 0.47 μmole/kg and PS 16 for UMUC 3 and FaDu tumors are shown Figures C and D respectively. The tumors were exposed to light (665 nm, 135J/cm2, 75 mW/cm2) at 24 h post-injection of the PS. A direct correlation between the PS uptake and tumor response (cure) was observed./kg. Statistical analysis log rank (Mantel-Cox test), (C) UMUC3, p-value: <0.0001 and (D) FaDu tumors, p-value: 0.0019. For PS-uptake study (tumor, liver and skin), 3 female SCID mice bearing either UMUC3 or FaDu tumors were used (Figures 9A & 9B). For determining in-vivo efficacy the number of female SCID mice used in each study are indicated in the Figure 9C & 9D.
Tumor uptake and PDT efficacy of Erlotinib-PS conjugate 5:
Among all conjugates, PS 5 showed the highest initial cellular uptake, but on the expense of reduced tumor cell selectivity (Figure 4). Therefore, before selecting the optimal PS-Erlotinib compound for future studies, the tumor uptake and PDT efficacy of the PS 5 was also investigated as done for PS 16 in SCID mice bearing either UMUC3 or FaDu tumors (Figure 11). In UMUC3 tumor, the maximal uptake was observed at 24 h post injection, whereas the maximum uptake in FaDu tumor was measured at 6h post-injection. Moreover, the PS 5 accumulation in FaDu tumors exceeded 2 to 3-fold than that of the liver whereas the maximal PS 5 level in UMUC3 tumor relative to the liver was much lower and was 2 to 3-fold lower than that determined for FaDu tumors (Figures 11A & B). This difference in uptake was similarly manifested in the difference in PDT efficacy (Figures 11C & D).
Figure 11:

In vivo uptake of PS 5 at the dose of 0.47μmol/kg in tumor, liver and skin of SCID mice bearing (A) UMUC3 tumors and (B) FaDu tumors. The in vivo PDT efficacy (long-term tumor response) of PS 5 at a dose of 0.47 μmole/kg for UMUC3 and FADU tumors are shown in (C) and (D) respectively. The tumors were exposed to light (665 nm, 135J/cm2, 75 mW/cm2) at 24 h post-injection of the PS. A direct correlation between the PS uptake and tumor response (cure) was observed. For PS-uptake study (tumor, liver and skin), 3 female SCID mice bearing either UMUC3 or FaDu tumors were used (Figures 11A & 11B). For determining in-vivo efficacy the number of female SCID mice used in each study are indicated in the Figure 9C & 9D. Statistical analysis log rank (Mantel-Cox test), (C): p-value: <0.0046 and (D): p-value: 0.0153.
Statistical Analysis:
The standard log-rank test (Mantel-Cox)was used for statistical analysis. It is a hypothesis test and compares the survival based on Kaplan Meier survival curve. It is a test of significance to detect difference between groups to confirm if one group has risk of an event greater than the other. For analyzing the in vivo PDT/SDT efficacy (cure), the survival curves were plotted using the drug dose over tumor regrowth.
Ethical Approval for Using Animals:
The in vivo experiments discussed in this manuscript were performed in compliance with all state, local, federal laws and the PHS Policy on the Human Care and use of Laboratory Animals. This study was conducted in an AAALAC accredited facility. The animal study was approved by the ethics committee of the institute.
CONCLUSION:
A series of PSs derived from chlorophyll-a and bacteriochlorophyll-a were conjugated with Erlotinib moiety at different position of the macrocycle. The structural difference of the PS, the presence of Erlotinib at various position and the nature of linker joining the two moieties made a significant difference in tumor cell-specificity and in vitro PDT efficacy. The in vivo results suggest major tumor-specific differences in uptake and retention exist and these differences can only in part be detected in tissue culture systems. This in turn implies that the in vivo environment affects the availability and bioactivity of the porphyrin-Erlotinib compounds beyond what can be predicted from ex vivo SAR ad QSAR studies. The biology of the underlying processes responsible for in vivo fate of the compounds remain to be identified. Despite these unsolved questions, our study has allowed to identify in the example of PS 10 most promising agent for further development.
EXPERIMENTAL
All reactions were carried out in heat gun-dried glassware under an atmosphere of nitrogen and magnetic stirring. Thin layer chromatography (TLC) was done on pre-coated silica gel sheets (layer thickness 0.2 mm). Column chromatography was performed either using silica gel 60 (70-230 mesh) or neutral alumina grade III. In some cases preparative TLC was used for the purification of compounds. Purity of the compounds was ascertained by TLC and HPLC analysis. All compounds including the intermediates were >95% pure. UV-visible spectra were recorded on FT UV-visible spectrophotometer using methanol as a solvent. Mass spectrometry analyses were performed at the Mass Spectrometry Facility, University of Buffalo, NY.1H and 13C NMR spectra were recorded at 28 °C on a Bruker Avance III HD spectrometer equipped with a 9.4 T narrow-bore magnet, a 5-mm BBO Z-gradient probe, and Topspin 3.2 software. Observe frequencies were 400 MHz for 1H and 100 MHz for 13C. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane (TMS) and were calibrated to the residual solvent peak. Coupling constants (J) are reported in Hertz (Hz). 1H multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Compounds 10 and 11 are known compounds.
Synthesis of conjugate (5):
A mixture of erlotinib 1 (60 mg, 0.254 mmol), 4-azidobutylamine 2 (26.1 mg, 0.2286 mmol), CuSO4.5H2O (7.6 mg, 0.03 mmol), sodium ascorbate (9 mg, 0.046 mmol) was dissolved in 8 mL DMF, 4 mL H2O and 8 mL t-BuOH, and stirred at 40 °C under Ar for 8 h. The solvents were evaporated under reduced pressure and then the residue was diluted with H2O and extracted with DCM, dried over anhydrous sodium sulfate and the resulting reaction product was purified by alumina grade III column, using 10% methanol in dichloromethane as an eluent. After evaporating the solvents, the erlotinib analog 3 was obtained in 45% yield (77.3 mg). The erlotinib analog 3 ((47.82 mg, 0.0942 mmol) in 8 mL dichloromethane was reacted with HPPH 4 (30 mg, 0.0471 mmol) in presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 14.62 mg, 0.0942 mmol) and 4-(dimethylamino) pyridine (DMAP, 11.50 mg, 0.0942 mmol). The reaction mixture was stirred at room temperature under N2 atmosphere for overnight. It was then diluted with dichloromethane (40 mL), washed with water (3x50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by preparative plate using 20% methanol in dichloromethane to obtain pure final product 5 with 52% yield (25.85 mg). HRMS (ESI): calcd for C65H80N11O7 (M + H+) 1126.6197; found, 1126.6199. UV-vis (CH3OH, λmax, nm (abs)): 662 (4.65 x 104), 605 (8.77 x 103), 539 (8.77 x 103), 507 (8.77 x 103), 409 (8.84 x 103), 347 (4.78 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.76/9.73 (1H, s, meso 5-H), 8.80/8.78 (1H, br s, meso 10-H), 8.67 (1H, br s, amine H), 8.66 (1H, s, pyrimidine H), 8.471/8.466 (1H, s, meso 20-H), 8.10 (1H, d, J = 7.0, phenyl H), 7.98 (1H, d, J = 7.0 Hz, phenyl H), 7.77 (1H, s, quinazoline 5-H), 7.22-7.38 (4H, m, 3 x phenyl H & quinazoline 8-H), 6.32 (1H, m, amide H), 5.90/5.87 (1H, q, J = 6.7 Hz, 31-H), 5.28 (1H, d, J = 19.9 Hz, 132-CHH), 4.98 (1H, d, J = 19.9 Hz, 132-CHH), 4.47 (1H, dq, J = 7.3, 1.4 Hz, 18-H), 4.22-4.32 (5H, m, 17-H & 2 x -OCH2CH2OCH3), 3.75-3.86 (4H, m, -OCH2CH2OCH3 & >NCH2(CH2)3NH-), 3.71 (2H, m, -OCH2CH2OCH3), 3.57-3.70 (2H, m, -OCH2(CH2)4CH3), 3.35-3.57 (2H, m, 8-CH2CH3), 3.45 (3H, s, -OCH3), 3.35/3.34 (3H, s, ring CH3), 3.310/3.306 (3H, s, -OCH3), 3.25/3.24 (3H, s, ring CH3), 3.02 (1H, m, >N(CH2)3CHHNH-), 2.89 (1H, m, >N(CH2)3CHHNH-), 2.77 (3H, br s, ring CH3), 2.69 (1H, m, 17-CHHCH2-), 2.50 (1H, m, 17-CHHCH2-), 2.36 (1H, m, 17-CH2CHH-), 2.11/2.10 (3H, d, J = 6.4 Hz, 31-CH3), 2.06 (1H, m, 17-CH2CHH-), 1.67-1.81 (5H, m, -OCH2CH2(CH2)3CH3 & 18-CH3), 1.47-1.56 (5H, m, NCH2CH2CH2CH2N & 8-CH2CH3), 1.33-1.47 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.18-1.28 (4H, m, -O(CH2)3(CH2)2CH3), 1.12 (2H, m, NCH2CH2CH2CH2N), 0.80/0.77 (3H, distorted t, J = 6.9 Hz -O(CH2)5CH3), 0.41 (1H, br s, core NH), −1.59/−1.60 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.9, 173.03/173.01, 172.15/172.13, 160.4, 156.7, 155.5/155.4, 154.3, 153.5, 150.7, 149.0/148.9, 147.3, 147.12/147.08, 145.09/145.05, 141.6/141.5, 139.9, 139.82/139.80, 137.22/137.20, 136.09/136.06, 135.80/135.77, 132.5, 132.4, 131.1, 129.59/129.57, 129.3, 127.49/127.47, 121.6, 120.6, 119.58/119.56, 118.6, 109.6, 108.6, 105.9, 103.8/103.7, 102.9, 98.0/97.8, 92.6, 72.8, 70.8, 70.5, 69.7, 69.0, 68.4, 59.3, 59.2, 51.63/51.62, 50.0, 49.4, 48.1, 38.6, 33.04/32.98, 31.74/31.71, 30.6/30.5, 30.2, 27.2, 26.10/26.06, 25.78/25.75, 24.7/24.6, 22.94/22.93, 22.6/22.5, 19.2, 17.28/17.26, 14.0/13.9, 11.3, 11.2, 11.02/10.98. Note: Additional peaks, presumably due to grease impurity, were observed at 1.26 and 0.88 ppm in the 1H spectrum, and 29.7 ppm in the 13C spectrum.
Synthesis of 172-m-iodo-benzylamine-3-(1’-hexyloxy)ethyl-pyropheophorbide-a (7):
To a solution of 4 (50 mg, 0.0785 mmol) and 3-Iodobenzyl amine 6 (27.5 mg, 0.1178 mmol) in 10 mL of dry dichloromethane, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI, 30.0 mg, 0.1571 mmol) and 4-(dimethylamino(pyridine (DMAP, 19.2 mg, 0.158 mmol) were added. The reaction mixture was stirred at room temperature under N2 atmosphere for overnight. It was then diluted with dichloromethane (40 mL), washed with water (3x50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 2% methanol in dichloromethane to obtain pure product 7 in 89% (59.52 mg) yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.82/9.81 (1H, s, meso 5-H), 9.07/9.05 (1H, s, meso 10-H), 8.53 (1H, s, meso 20-H), 7.35-7.42 (2H, m, 2 x phenyl H), 6.89 (1H, m, phenyl H), 6.79 (1H, t, J ~ 7.7 Hz, phenyl H), 5.93/5.92 (1H, q, J = 6.6 Hz, 31-H), 5.82/5.80 (1H, t, J = 5.9 Hz, amide H), 5.181/5.177 (1H, d, J = 19.7 Hz, 132-CHH), 4.97/4.96 (1H, d, J = 19.7 Hz, 132-CHH), 4.51/4.50 (1H, dq, J = 7.4, 1.7 Hz, 18-H), 4.30 (1H, m, 17-H), 4.10/4.09 (1H, dd, J = 5.7, 15.1 Hz, -NHCHH-phenyl-), 3.98/3.96 (1H, dd, J = 5.8, 15.1 Hz, -NHCHH-phenyl-), 3.46-3.74 (4H, m, -OCH2(CH2)4CH3 & 8-CH2CH3), 3.393/3.387(3H, s, ring CH3), 3.30/3.29 (3H, s, ring CH3), 3.02/2.98 (3H, s, ring CH3), 2.63 (1H, m, 1H of 17-CH2CH2-), 2.45 (1H, m, 1H of 17-CH2CH2-), 2.25 (1H, ddd, J = 6.4, 9.8, 14.5 Hz, 1H of 17-CH2CH2-), 2.135/2.133 (3H, d, J = 6.7 Hz, 31-CH3), 1.92 (1H, ddd, J = 5.1, 9.8, 14.5 Hz, 1H of 17-CH2CH2-), 1.788/1.785 (3H, d, J = 7.3 Hz, 18-CH3), 1.77 (2H, m, -OCH2CH2(CH2)3CH3), 1.61/1.60 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.31-1.51 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.19-1.32 (4H, m, , -O(CH2)3(CH2)2CH3), 0.82/0.80 (3H, distorted t, J ~ 7.0 Hz, -O(CH2)5CH3), 0.44 (1H, br s, core NH), −1.63 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.2, 172.22/172.20, 171.9/171.8, 160.18/160.16, 155.30/155.29, 150.7, 148.9, 145.04/145.03, 141.5/141.4, 140.52/140.51, 139.79/139.76, 137.5/137.4, 136.45/136.43, 136.24/136.23, 136.17/136.16, 135.7/135.6, 132.4/132.3, 130.1, 130.00/129.98, 127.9/127.8, 126.78/126.77, 105.94/105.93, 103.91/103.90, 98.03/97.96, 94.3, 92.5, 72.83/72.81, 69.7, 51.59/51.57, 50.0, 48.0, 42.6/42.5, 32.7, 31.74/31.72, 30.4, 30.2, 26.09/26.06, 24.72/24.66, 23.0, 22.58/22.55, 19.3, 17.35/17.33, 13.98/13.95, 11.51/11.48, 11.3, 11.04/11.03. Note: 1H impurity peaks were observed at 0.91 ppm (probably grease CH3). Further 1H impurity peaks (probably grease CH2) interfere with the hexyl group methylene protons observed at ~1.25 ppm.
Synthesis of Conjugatev(8):
Triphenylarsine (8.62 mg, 0.0281 mmol) and Pd2dba3 (12.89 mg, 0.014 mmol) were added to a stirred solution of compound 7 (30 mg, 0.0352 mmol) and erlotinib 1 (20.78 mg, 0.0528 mmol) in dry THF (20 mL) and Et3N (4 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product 8. The remaining ¼ of 7 remain unreacted even by increasing the catalyst quantity, temperature and duration of the reaction. The desired conjugate 8 and the remaining starting material 7 were purified by preparative TLC plates. Based on the starting material 7 recovered, the desired erlotinib conjugate 8 was isolated in 75% yield (29.51 mg). MS (ESI) m/z: 1117.58 (M + H+). HRMS (ESI): calcd for C68H77N8O7 (M + H+) 1117.5871; found, 1117.5897. UV-vis (CH3OH, λmax, nm (abs)): 662 (4.58 x 104), 606 (8.84 x 103), 538 (9.02 x 103), 507 (8.66 x 103), 409 (8.52 x 104), 346 (5.06 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.79/9.76 (1H, s, meso 5-H), 8.97/8.95 (1H, s, meso 10-H), ~8.95 (1H, br s, amine NH), 8.68 (1H, s, pyrimidine H), 8.49 (1H, s, meso 20-H), 8.214/8.212 (1H, d, J = 8.1 Hz, phenyl H), 7.853/7.850 (1H, s, quinazoline 5-H), 7.64 (1H, m, phenyl H), 7.42 (1H, br s, phenyl H), 7.32 (1H, dd, J ~ 8.0, 8.0 Hz, phenyl H), 7.303/7.301 (1H, d, J ~ 7.5 Hz, phenyl H), 7.20-7.24 (2H, m, quinazoline 8-H & phenyl H), 7.17 (1H, br s, amide H), 7.02 (1H, ddd, J = 1.6, 7.6, 7.6 Hz, phenyl H), 6.91 (1H, d, J = 7.4 Hz, phenyl H), 5.91/5.90 (1H, q, J = 6.8 Hz, 31-H), 5.07 (1H, d, J = 19.8, 132-CHH), 4.53 (1H, d, J ~19.6 Hz, 132-CHH), 4.50 (1H, m, 18-H), 4.40 (1H, dd, J = 6.3, 14.8 Hz, -NHCHH-phenyl), 4.16-4.29 (5H, m, 17-H & 2 x -OCH2CH2OCH3), 3.99 (1H, m, -NHCHH-phenyl), 3.81 (2H, m, -OCH2CH2OCH3), 3.67 (2H, m, -OCH2(CH2)4CH3), 3.62 (2H, m, -OCH2CH2OCH3), 3.59 (m, 2H, 8-CH2CH3), 3.44 (3H, s, -OCH2CH2OCH3), 3.372/3.365 (3H, s, ring CH3), 3.29 (3H, s, ring CH3), 3.234/3.232 (3H, s, -OCH2CH2OCH3), 2.82 (1H, m, 1H of 17-CH2CH2-), 2.67 (1H, m, 1H of 17-CH2CH2-), 2.30 (1H, m, 1H of 17-CH2CH2-), 2.29/2.26 (3H, s, ring CH3), 2.13/2.12 (3H, d, J = 6.7 Hz, 31-CH3), 2.04 (1H, m, 1H of 17-CH2CH2-), 1.77 (2H, m, -OCH2CH2(CH2)3CH3), 1.75/1.74 (3H, d, J = 7.3 Hz, 18-CH3), 1.63/1.62 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.43 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.18-1.32 (4H, m, -O(CH2)3(CH2)2CH3), 0.82/0.79 (3H, distorted t, J ~ 7.1 Hz, -O(CH2)5CH3), 0.62 (1H, br s, core NH), −1.48/−1.49 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.8, 172.93/172.92, 172.20/172.19, 160.09/160.07, 156.7, 155.68/155.67, 154.4, 153.6, 150.9, 149.1, 148.9, 147.4, 145.09/145.08, 141.8/141.7, 139.94/139.92, 139.4, 138.5, 137.5, 136.28/136.27, 136.0/135.9, 132.6/132.5, 130.7, 130.6, 129.49/129.48, 129.0, 128.5, 127.78, 127.76, 126.9, 124.3, 123.7, 123.5, 122.1, 109.7, 108.6, 105.8, 104.0, 103.0, 98.05/97.97, 92.5, 89.6, 89.0, 72.83/72.80, 70.7, 70.5, 69.7, 69.0, 68.3, 59.3, 59.1, 51.5, 49.8, 47.8, 43.2, 32.71/32.69, 31.75/31.73, 30.9, 30.2, 26.09/26.07, 24.71/24.66, 22.92/22.91, 22.59/22.56, 19.4, 17.41/17.40, 13.98/13.95, 11.3, 11.2/11.1, 11.02/11.00. Note: Minor 1H impurity peaks appear at 7.65, 7.50, and 7.45 ppm. An additional 13C peak, presumably due to grease impurity, was observed at 29.7 ppm.
Synthesis of Conjugate (13) :
Triphenylarsine (18.72 mg, 0.0611 mmol) and Pd2dba3 (28.00 mg, 0.0305 mmol) were added to a stirred solution of compound 12 (60 mg, 0.07643 mmol) and erlotinib 1 (45.10 mg, 0.1146 mmol) in dry THF (30 mL) and Et3N (6 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product 13 The remaining ¼ unreacted 12 even by increasing the catalyst quantity, temperature and duration of the reaction. The product 13 and the remaining starting material 12 were purified by preparative TLC plates. On the basis of the compound 12 recovered, the title compound 13 was obtained in 72% yield (57.79 mg). MS (ESI) m/z: 1050.50 (M + H+). HRMS (ESI): calcd for C63H68N7O8 (M + H+) 1050.5051; found, 1050.5049. UV-vis (CH3OH, λmax, nm (abs)): 714 (4.15 x 104), 652 (1.74 x 104), 517 (3.1 x 104), 350 (1.4 x 105). 1H NMR (400 MHz, CDCl3, δ ppm): 8.69/8.68 (1H, s, pyrimidine H), 8.55/8.47 (1H, s, meso 5-H), 8.19 (1H, s, meso 10-H), 8.03/8.02 (1H, s, meso 20-H), 7.94 (1H, m, phenyl H), 7.70 (1H, m, phenyl H), 7.68/7.67 (1H, br s, erlotinib NH), 7.50 (1H, m, phenyl H), 7.42 (1H, m, phenyl H), 7.35/7.34 (1H, s, phenyl H), 7.313/7.309 (1H, dd, J = 8.0, 8.0 Hz, phenyl H), 7.23-7.27 (3H, m, quinazoline 5-H, quinazoline 8-H & phenyl H), 7.20/7.19 (1H, ddd, J ~ 7.7, 1.2, 1.2 Hz phenyl H), 5.72 (1H, q, J = 6.7 Hz, 31-H), 4.96 (1H, d, J = 19.8 Hz, 132-CHH), 4.79 (1H, d, J = 19.8 Hz, 132-CHH), 4.67/4.64 (1H, d, J ~ 12 Hz, -OCHH-phenyl-), 4.55/4.52 (1H, d, J = 12.2 Hz, -OCHH-phenyl-), 4.23-4.30 (4H, m, 2 x -OCH2CH2OCH3), 4.14 (1H, q, J = 7.2 Hz, 18-H), 4.08 (1H, dq, J = 7.2, 3.1 Hz, 7-H), 3.98 (1H, m, 17-H), 3.81-3.89 (3H, m, -OCH2CH2OCH3 & 8-H), 3.79 (2H, m, -OCH2CH2OCH3), 3.610/3.608 (3H, s, -COOCH3), 3.45 (3H, s, -OCH2CH2OCH3), 3.421/3.418 (3H, s, -OCH2CH2OCH3), 3.33 (3H, s, ring CH3), 3.15/3.14 (3H, s, ring CH3), 2.42-2.60 (2H, m, 17-CHHCHH-), 2.15-2.33 (3H, m, 8-CHHCH3 & 17-CHHCHH-), 2.05/2.03 (3H, d, J = 6.7 Hz, 31-CH3), ~2.02 (1H, m, 8-CHHCH3), 1.74/~1.67 (3H, d, J = 7.3 Hz, 7-CH3), ~1.68/1.66 (3H, d, J = 7.3 Hz, 18-CH3), 1.38/1.37 (1H, s, core NH), 1.09/1.07 (3H, t, J ~ 7.6 Hz, 8-CH2CH3), −0.13/−0.14 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 195.6, 173.60/173.57, 171.1/171.0, 170.34/170.32, 161.53/161.51, 156.3, 155.2, 154.9, 153.3, 149.1, 147.92/147.90, 141.0/140.9, 138.71/138.69, 138.66/138.65, 138.61/138.56, 137.38/137.37, 136.3/136.2, 133.75/133.72, 131.2/131.1, 131.0/130.9, 129.0, 128.84/128.82, 128.5/128.4, 128.0/127.9, 127.4, 124.4, 124.0, 123.4/123.3, 121.7, 118.11/118.07, 109.0, 108.7, 108.5, 102.7, 99.24/99.22, 95.2/94.9, 93.82/93.77, 89.5, 89.24/89.20, 71.8/71.2, 71.0, 70.7/70.46, 70.49, 69.5, 68.4, 59.4, 59.3, 54.33/54.27, 51.67/51.66, 50.45/50.43, 49.9, 49.11/49.07, 47.4, 30.83/30.82, 30.2/30.1, 29.91/29.90, 24.1/23.9, 22.7/22.53, 22.53/22.52, 11.3, 10.9/10.83, 10.81/10.7. Note: Minor 1H impurity peaks were observed at 7.77, 3.44, 3.43, 1.25 (likely grease CH2) and 0.88 (likely grease CH3) ppm. One 13C signal is missing, presumably due to aggregation-induced line broadening. 13C impurity peaks were observed at 131.7 and 128.3 ppm.
Synthesis of conjugate (14, carboxylic analog of 13):
Aqueous LiOH (36.02 mg in 3 mL of H2O) was added to a solution of compound 13 (30 mg) in dry THF:MeOH (4.5:3 mL), and the reaction mixture was stirred under argon at room temperature for 3 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and washed with 2% AcOH in H2O (18 mL) and then with H2O (3 x 50 mL). The organic layer was dried over Na2SO4, concentrated, and purified over a preparative TLC plate using 8% MeOH in CH2Cl2 as eluent to yield 26.93 mg (93%) of the desired product 14. MS (ESI) m/z: 1034.48 (M + H+). HRMS (ESI): calcd for C62H64N7O8 (M + H+) 1034.4814; found, 1034.4821. UV-vis (CH3OH, λmax, nm (ε)): 662 (4.66 x 104), 604 (8.4 x 103), 536 (8.89 x 103), 505 (8.89 x 103), 407 (8.9 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 8.54/8.28 (1H, s, meso 5-H), 8.33/8.23 (1H, s, pyrimidine H), 8.10/8.08 (1H, s, meso 10-H), 8.00/7.96 (1H, s, meso 20-H), 7.79/7.73 (1H, s, phenyl H), 7.51 (1H, d, J ~ 7 Hz, phenyl H), 7.27-7.39 (3H, m, 2 x phenyl H & quinazoline 5-H), 7.05-7.25 (4H, m, 3 x phenyl H & quinazoline 8-H), 7.00 (1H, m, phenyl H), 5.72/5.58 (1H, q, J = 6.7 Hz, 31-H), 4.95 (1H, d, J = 19.7 Hz, 132-CHH), 4.75 (1H, d, J = 19.7 Hz, 132-CHH), 4.63/4.55 (1H, d, J = 12.2 Hz, -OCHH-phenyl-), 4.52/4.47 (1H, d, J = 12.2 Hz, -OCHH-phenyl-), 4.10-4.25 (5H, m, 2 x -OCH2CH2OCH3 & 18-H), 4.06 (1H, m, 7-H), 3.97 (1H, d, J ~ 7 Hz, 17-H), 3.72-3.83 (3H, m, -OCH2CH2OCH3 & 8-H), 3.70 (2H, br s, -OCH2CH2OCH3), 3.40 (3H, s, -OCH2CH2OCH3), 3.34 (3H, s, -OCH2CH2OCH3), 3.23/3.22 (3H, s, ring CH3), 3.12/2.97 (3H, s, ring CH3), 2.40-2.55 (2H, m, 17-CHHCHH-), 2.15-2.34 (3H, m, 8-CHHCH3 & 17-CHHCHH-), 2.01/1.99 (3H, d, J = 6.7 Hz, 31-CH3), 1.98 (1H, m, 8-CHHCH3), 1.71/1.66 (3H, d, J = 7.2 Hz, 7-CH3), 1.632/1.628 (3H, d, J = 7.2 Hz, 18-CH3), 1.43/1.35 (1H, s, core NH), 1.07/1.05 (3H, t, J = 7.2 Hz, 8-CH2CH3), −0.07/−0.13 (1H, br s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 195.88/195.87, 176.84/176.75, 171.1/170.9, 170.8/170.7, 161.5/161.4, 156.5/156.4, 155.6/155.4, 154.9, 152.2/152.0, 148.9, 148.04/148.03, 145.2, 141.1/141.0, 139.0/138.6, 138.61/138.60, 138.29/138.27, 137.31/137.28, 136.3/136.2, 133.9, 131.6/131.5, 130.76/130.73, 128.72/128.71, 128.62/128.59, 128.41/128.38, 128.1/128.0, 127.34/127.28, 124.8/124.7, 123.7, 123.1/123.0, 121.81/121.76, 117.9/117.7, 108.7, 108.6/108.5, 107.0/106.7, 103.11/103.10, 99.02/98.99, 95.4/94.2, 94.0/93.9, 89.29/89.26, 89.22/89.16, 71.43/70.73, 70.80/70.49, 70.77, 70.4, 69.1/69.0, 68.4, 59.19, 59.17, 54.3/54.2, 50.6, 49.92/49.87, 49.1, 47.4, 31.5, 30.12/30.07, 30.0, 23.9/23.7, 22.64/22.50, 22.46/22.40, 11.2/11.1, 11.0/10.6, 10.88/10.86. Note: Two protons (one acid and one NH of erlotinib) are missing, presumably due to chemical exchange. Additional peaks, presumably due to grease impurity, were observed at 1.26 ppm in the 1H spectrum, and 29.7 ppm in the 13C spectrum.
Synthesis of Conjugate (16):
Triphenylarsine (25 mg, 0.082 mmol) and Pd2dba3 (37.5 mg, 0.041 mmol) were added to a stirred solution of 15 (80 mg, 0.103) and erlotinib 1 (60.3 mg, 0.153) in dry THF (20 mL) and Et3N (4 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product. The product 16 and the starting material 15 were purified by preparative TLC plates. On the basis of the starting material recovered the title compound 15 was obtained in 75% yield (80.35 mg). MS (ESI) m/z: 1048.49 (M + H+). HRMS (ESI): calcd for C63H66N7O8 (M + H+) 1048.4928; found, 1048.4957. UV-vis (CH3OH, λmax, nm (ε)): 661 (4.66 x 104), 606 (8.44 x 103), 537 (9.2 x 103), 506 (8.89 x 103), 407 (8.97 x 104), 334 (1.11 x 105). 1H NMR (400 MHz, CDCl3, δ ppm): 9.75 (1H, s, meso 5-H), 9.52 (1H, s, meso 10-H), 8.70 (1H, s, pyrimidine H), 8.55 (1H, s, meso 20-H), 7.93 (1H, m, phenyl H), 7.74 (1H, d, J = 8.1 Hz phenyl H), ~7.46 (1H, br s, erlotinib NH), 7.46 (2H, m, 2 x phenyl H), 7.37 (1H, dd, J ~ 7.9, 7.9 Hz, phenyl H), 7.27-7.34 (4H, m, 3 x phenyl H & 1 x quinazoline 5-H), 7.25 (1H, s, quinazoline 8-H), 5.99 (1H, q, J = 6.7 Hz, 31-H), 5.267/5.265 (1H, d, J = 19.9, 132-CHH), 5.117/5.116 (1H, d, J ~19.8 Hz, 132-CHH), 4.75 (1H, d, J = 12.2 Hz, -OCHH-phenyl-), 4.61/4.60 (1H, d, J = 12.2 Hz, -OCHH-phenyl-), 4.49 (1H, qd, J = 7.3, 1.8 Hz, 18-H), 4.25-4.33 (5H, m, 2 x -OCH2CH2OCH3 & 17-H), 3.84 (4H, m, 2x -OCH2CH2OCH3), 3.70 (2H, q, J = 7.7 Hz, 8-CH2CH3), 3.67 (3H, s, ring CH3), 3.610/3.607 (3H, s, -COOCH3), 3.47 (3H, s, -OCH2CH2OCH3), 3.46 (3H, s, -OCH2CH2OCH3), 3.363/3.359 (3H, s, ring CH3), 3.20/3.19 (3H, s, ring CH3), 2.69 (1H, m, 1H of 17-CH2CH2-), 2.56 (1H, m, 1H of 17-CH2CH2-), 2.21-2.40 (2H, m, 2H of 17-CH2CH2-), 2.17/2.16 (3H, d, J = 6.7 Hz, 31-CH3), 1.83/1.82 (3H, d, J = 7.3 Hz, 18-CH3), 1.71 (3H, t, J = 7.7 Hz, 8-CH2CH3), 0.43 (1H, br s, core NH), −1.71 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.3, 173.5, 171.4, 160.33/160.31, 156.3, 155.2, 154.7, 153.6, 151.0, 149.03, 148.99, 147.6, 145.0, 141.24/141.21, 138.9, 138.67/138.66, 138.61/138.59, 137.9, 136.4, 135.5/135.4, 132.73/132.67, 131.6 (2C), 130.5, 129.1, 128.4, 127.9 (2C), 127.3, 124.4, 124.0, 122.4, 121.7, 109.2, 108.9, 106.0, 104.2, 102.6, 97.9, 92.7, 89.5, 89.2, 71.8, 71.0, 70.7, 70.5, 69.4, 68.4, 59.34, 59.28, 51.7 (2C), 50.0, 48.1, 30.9, 29.9, 24.54/24.51, 23.2/23.1, 19.5, 17.4, 12.1, 11.2, 11.11/11.10. Note: The 13C peak at 51.7 ppm is due to two overlapped signals (C17 and ester CH3). 1H impurity peaks were observed at 1.27 (likely grease CH2), 0.89 (likely grease CH3), and 0.09 ppm (minor). Small 13C impurity peaks were observed at 29.4, 22.7, 14.1 (probably grease CH3) and 1.0 ppm. A somewhat larger 13C impurity peak – presumably CH2 of grease – was observed at 29.7 ppm
Synthesis of Conjugate (17 (carboxylic acid analog of 16):
Aqueous LiOH (36.02 mg in 3 mL of H2O) was added to a solution of compound 8 (30 mg) in dry THF: MeOH (4.5:3 mL), and the reaction mixture was stirred under argon at room temperature for 3 h. It was then diluted with CH2Cl2 (50 mL) and washed with 2% AcOH in H2O (18 mL) and with H2O (3 x 50 mL), and the organic layer was dried over Na2SO4, concentrated, and purified over a preparative TLC plate using 8% MeOH in CH2Cl2 as eluent to yield 26.93 mg (93%) of product 9. MS (ESI) m/z: 1034.48 (M + H+). HRMS (ESI): calcd for C62H64N7O8 (M + H+) 1034.4814; found, 1034.4821. UV-vis (CH3OH, λmax, nm (ε)): 662 (4.66 x 104), 604 (8.4 x 103), 536 (8.89 x 103), 505 (8.89 x 103), 407 (8.9 x 104). 1H NMR (400 MHz, 90:10 CDCl3/CD3OD, δ ppm): 9.54 (1H, s, meso 5-H), 9.36 (1H, s, meso 10-H), 8.403/8.402 (1H, s, meso 20-H), 8.36 (1H, s, pyrimidine H), 7.76 (1H, m, phenyl H), 7.532/7.527 (1H, dd, J = 7.9, 1.3 Hz, phenyl H), 7.51 (1H, s, quinazoline 5-H), 7.34 (2H, m, 2 x phenyl H), 7.24 (1H, dd, J ~ 7.9, 7.9 Hz, phenyl H), 7.15-7.21 (3H, m, 3 x phenyl H), 7.04 (1H, s, quinazoline 8-H), 5.851/5.849 (1H, q, J = 6.7 Hz, 31-H), 5.13/5.12 (1H, d, J = 20.0, 132-CHH), 4.96 (1H, d, J = 20.0 Hz, 132-CHH), 4.62 (1H, d, J = 12.1 Hz, -OCHH-phenyl-), 4.50/4.49 (1H, d, J = 12.1 Hz, -OCHH-phenyl-), 4.35 (1H, qd, J = 7.3, 1.8 Hz, 18-H), 4.12-4.19 (5H, m, 2 x -OCH2CH2OCH3 & 17-H), 3.73 (4H, m, 2x -OCH2CH2OCH3), 3.54 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.50 (3H, s, ring CH3), 3.36 (3H, s, -OCH2CH2OCH3), 3.34 (3H, s, -OCH2CH2OCH3), 3.21/3.20 (3H, s, ring CH3), 3.024/3.018 (3H, s, ring CH3), 2.54 (1H, m, 1H of 17-CH2CH2-), 2.44 (1H, m, 1H of 17-CH2CH2-), 2.04-2.20 (2H, m, 2H of 17-CH2CH2-), 2.02/2.01 (3H, d, J = 6.7 Hz, 31-CH3), 1.67/1.66 (3H, d, J = 7.3 Hz, 18-CH3), 1.55 (3H, t, J = 7.6 Hz, 8-CH2CH3); 13C NMR (100 MHz, 90:10 CDCl3/CD3OD, δ ppm): 197.2, 175.5, 171.9, 160.6, 157.0, 155.4, 154.5, 152.7, 150.8, 148.9, 148.7, 146.2, 145.0, 141.2, 138.7, 138.4/138.3, 138.31/138.29, 137.3, 136.2, 135.4/135.3, 132.93/132.87, 131.4 (2C), 129.6, 128.6, 128.1, 127.8 (2C), 127.3, 125.3, 123.5, 122.5, 122.3, 109.3, 107.2, 105.5, 104.0, 103.4, 97.5/97.4, 92.7, 89.1, 89.0, 71.6, 70.6, 70.5, 70.3, 68.6, 68.1, 58.9, 58.8, 51.4, 49.9, 47.7, 30.8, 29.8, 24.03/23.99, 22.71/22.68, 19.1, 17.0, 11.5, 10.8, 10.67/10.65. Note: One acid and three NH protons were not observed, presumably due to chemical exchange. 1H impurity peaks at 1.10 (likely grease CH2), 0.75 (likely grease CH3), and −0.08 ppm were observed. 13C peaks at 131.4 and 127.8 ppm are each comprised of two aromatic CH signals of the benzyl group. Low intensity 13C impurity peaks were observed at 31.6, 22.3, and 0.6 ppm. A somewhat larger 13C impurity peak – presumably CH2 of grease – was observed at 29.4 ppm).
Synthesis of Conjugate (19):
Tryphenylarsine (6.37 mg, 0.0208 mmol) and Pd2dba3 (28.06 mg, 0.0104 mmol) were added to a stirred solution of compound 18 (20 mg, 0.0260 mmol) and Erlotinib 1 (15.35 mg, 0.0390 mmol) in dry THF (15 mL) and Et3N (2 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product. The remaining ¼ unreacted even by increasing the catalyst quantity, temperature and duration of the reaction. The desired product 19 was purified by preparative TLC plates with 73% yield (19.64 mg). HRMS (ESI): Calcd. for C62H64N7O8 (M + H+) 1034.4772; found, 1034.4795. UV-vis (CH3OH, λmax, nm (abs)): 664 (4.6 x 104), 606 (8.7 x 103), 537 (9.1 x 103), 507 (9.1 x 103), 410 (8.8 x 104), 347 (5.8 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.45 (1H, s, meso10-H), 9.41 (1H, s, meso 5-H), 8.64 (1H, s, pyrimidine H), 8.55 (1H, s, meso 20-H), 7.75 (1H, m, phenyl H), 7.73 (1H, m, phenyl H), 7.66 (1H, s, phenyl H), 7.51 (1H, ddd, J = 7.5, ~1.5, ~1.5 Hz, phenyl H), 7.44 (1H, ddd, J = 7.5, ~1.5, ~1.5 Hz, phenyl H), 7.38 (1H, dd, J ~7.5, ~7.5 Hz, phenyl H), 7.33 (1H, dd, J ~7.9, ~7.9 Hz, phenyl H), 7.24 (1H, s, quinazoline 8-H), 7.21 (1H, ddd, J = 7.6, ~1.3, ~1.3 Hz, phenyl H), ~7.20 (1H, br s, erlotinib NH), 7.05 (1H, s, quinazoline 5-H), 5.73 (2H, s, 3-CH2O-), 5.26 (1H, d, J = 19.8 Hz, 132-CHH), 5.11 (1H, d, J = 19.8 Hz, 132-CHH), 4.83 (2H, s, -OCH2-phenyl-), 4.48 (1H, qd, J = 7.3, 2.0 Hz, 18-H), 4.26-4.32 (3H, m, 17-H & -OCH2CH2OCH3), 4.12 (2H, m, -OCH2CH2OCH3), 3.86 (2H, m, -OCH2CH2OCH3), 3.72 (2H, t, J = 4.7 Hz -OCH2CH2OCH3), 3.61 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.610 (3H, s, ring CH3), 3.608 (3H, s, -COOCH3), 3.48 (3H, s, -OCH2CH2OCH3), 3.41 (3H, s, -OCH2CH2OCH3), 3.36 (3H, s, ring CH3), 3.20 (3H, s, ring CH3), 2.70 (1H, m, 1H of 17-CH2CH2-), 2.56 (1H, m, 1H of 17-CH2CH2-), 2.24-2.36 (2H, m, 2H of 17-CH2CH2-), 1.81 (3H, d, J = 7.3 Hz, 18-CH3), 1.63 (3H, t, J = 7.6 Hz, 8-CH2CH3), 0.35 (1H, br s, core NH), −1.76 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.2, 173.5, 171.3, 160.3, 156.1, 155.2, 154.8, 153.4, 151.0, 149.0, 148.9, 145.0, 141.1, 138.7, 138.6, 137.9, 136.6, 136.3, 134.7, 133.9, 131.2, 131.0, 130.6, 129.0, 128.6, 128.4, 127.9, 127.2, 124.3, 123.9, 123.5, 121.5, 109.0, 108.7, 106.2, 104.0, 102.4, 97.2, 93.1, 89.5, 89.4, 71.8, 70.9, 70.5, 69.3, 68.4, 63.2, 59.3 (2C), 51.74, 51.68, 50.0, 48.1, 31.0, 29.9, 23.1, 19.4, 17.3, 12.0, 11.3 (2C). Note: 1H impurity peaks at 1.27 and 0.88 ppm are likely due to grease CH2 and CH3, respectively. One 13C peak is missing, presumably due to aggregation-induced line broadening. The 13C peak at 59.3 ppm is comprised of two erlotinib –OCH3 carbon signals. The 13C peak at 11.3 ppm is comprised of two photosensitizer ring CH3 carbon signals. The 13C impurity peak at 29.7 ppm is likely due to grease CH2.
Synthesis of methyl-20-phenyl-p-iodobenzulamide-pyropheophorbide-a (22):
The 20- benzoic acid analog of methyl mesopyropheophorbide-a [44.6 mg (0.067 mmol)] 20, synthesized using a previously established method, was dissolved in 10 ml of dichloromethane (DCM) dried over molecular sieves. To this 73.5 mg (0.166 mmol) of (Benzotriazol-1-yloxy)tris (dimethylamino) phosphonium hexafluorophosphate (BOP) and 250 μL (0.1806 g; 1.785 mmol) of triethylamine was added and the reaction mixture was allowed to stir for 30 minutes under Ar. To this reaction mixture, 35.6 mg (0.153 mmol) of 4-Iodobenzylamine HCl 21 was added. The reaction was allowed to stir overnight, protected from light and under Ar atmosphere. TLC in 3% methanol (MeOH) in DCM showed a disappearance of the starting material 20. The reaction mixture was diluted with DCM and washed once with 1M HCl, twice with DI H2O and once with brine (saturated aqueous sodium chloride). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated using a rotorary evaporator and dried under vacuum. The crude reaction mixture was purified using preparation TLC (3% MeOH in DCM). The desired product was collected with an Rf value of about 0.61 was then filtered over the silica gel using 50% MeOH in DCM. The organics were removed and concentrated using a rotary evaporator and dried to yield 22 in 84% (49.5 mg) yield. HRMS (ESI) for C48H48IN5O4 [MH+] calculated: 886.28237; found: 886.28239. UV–Vis (MeOH, λmax, nm): 270, 410, 510, 545 610, 665. 1H NMR (400 MHz, CD2Cl2, δ ppm): 9.57 (1H, s, meso 10-H), 9.43 (1H, s, ,meso 5-H), 8.17-8.23 (2H, m, 2 x phenyl H), 8.04 (1H, m, phenyl H), 7.75-7.79 (3H, m, 3 x phenyl H), 7.27 (2H, m, 2 x phenyl H), 6.84 (1H, t, J = 5.8 Hz, amide H), 5.18 (1H, d, J = 19.8 Hz, 132-CHH), 5.15 (1H, d, J = 19.8 Hz, 132-CHH), 4.72 (1H, dd, J = 5.8, 15.2 Hz, -NHCHH-phenyl-), 4.71 (1H, dd, J = 5.8, 15.2 Hz, -NHCHH-phenyl-), 4.24 (1H, q, J = 7.1 Hz, 18-H), 4.10 (1H, dd, J = 8.7, 3.4 Hz, 17-H), 3.81 (2H, q, J = 7.7 Hz, -CH2CH3), 3.73 (2H, q, J = 7.6 Hz, -CH2CH3), 3.66 (3H, s, ring CH3), 3.54 (3H, s, -COOCH3), 3.30 (3H, s, ring CH3), ~2.55 (1H, m, 1H of 17-CH2CH2-), ~2.44 (1H, m, 1H of 17-CH2CH2-), 2.31 (3H, s, 2-CH3), 2.13-2.28 (2H, m, 2H of 17-CH2CH2-), 1.73 (3H, t, J = 7.7 Hz, -CH2CH3), 1.65 (3H, t, J = 7.6 Hz, -CH2CH3), 1.04 (3H, d, J = 7.1 Hz, 18-CH3), −1.53 (1H, br s, core NH),; 13C NMR (100 MHz, CD2Cl2, δ ppm): 196.1, 173.7, 172.2, 167.4, 160.8, 153.7, 150.9, 148.8, 145.17, 145.16, 144.6, 140.8, 139.1, 139.0, 138.2 (2C), 136.7, 135.4, 135.1, 134.4, 133.0, 132.2, 131.6, 130.3 (2C), 129.1, 127.4, 126.8, 111.5, 107.0, 104.2, 97.3, 93.1, 52.6, 51.8, 49.0, 48.8, 44.0, 31.6, 30.2, 21.2, 19.83, 19.80, 17.6, 17.0, 14.3, 12.2, 11.4. Note: One core NH proton was not observed, presumably due to chemical exchange. 1H impurity peaks were observed at: 5.33 (DCM), 1.27 (grease CH2), and 0.89 (grease CH3) ppm. 13C peaks at 138.2 and 130.3 ppm are each comprised of two aromatic CH carbon signals of the iodobenzyl group.
The13C peak at 30.1 ppm is likely due to the CH2 carbon of grease impurity.
Synthesis of Conjugate (23):
20.0 mg (0.065 mmol) of triphenylarsine (AsPh3) and 22.0 mg (0.024 mmol) of tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba)3] were added to a stirred solution of 49.5 mg (0.056 mmol) of 22, 34.1 mg (0.087 mmol) of erlotinib 1 and 4 mL (2.89 g; 28.56 mmol) of triethylamine in 30 mL of freshly distilled THF under an atmosphere of Ar. The reaction was allowed to stir overnight, protected from light and under Ar atmosphere. TLC in 5% methanol (MeOH) in DCM showed a disappearance of the starting material 1. The reaction mixture was diluted with DCM and washed once with 1M HCl, twice with DI H2O and once with brine (saturated sodium chloride). The organic layer was dried over anhydrous Na2SO4. The organic solution was filtered and concentrated using a rotorary evaporator and dried using a high vacuum pump. The crude reaction mixture was purified using preparation TLC (4% MeOH in DCM). The desired product 10 was collected with an Rf value of about 0.24. The desired product was filtered off the silica gel using 50% MeOH in DCM. The organics were removed and concentrated using a rotary evaporator and dried using a high vacuum pump. The title compound was obtained in 40% yield (25.7). HRMS (ESI) for C70H70N8O8 [MH+] calculated: 1151.53894; found: 1151.54308. UV–Vis (MeOH, λmax, nm): 255, 335, 410, 510, 545 610, 665. 1H NMR (400 MHz, CDCl3, δ ppm) 9.48 (1H, s, meso 10-H), 9.34 (1H, s, meso 5-H), 8.54 (1H, s, pyrimidine H), ~8.4 (1H, br s, erlotinib NH), 8.26 (1H, dd, J = 1.6, 7.9 Hz, phenyl H), 8.17 (1H, dd, J = 1.6, 7.9 Hz, phenyl H), 8.11 (1H, dd, J = 1.6, 7.9 Hz, phenyl H), 7.95 (1H, s, phenyl H), 7.76 (1H, d, J = 8.0 Hz, phenyl H), 7.72 (1H, dd, J = 1.6, 7.9 Hz, phenyl H), 7.56 (1H, br s, quinazoline 5-H), 7.46 (2H, d, J = 7.9 Hz, 2 x phenyl H), 7.38 (2H, d, J = 8.1 Hz, 2 x phenyl H), 7.34 (1H, t, J ~ 7.8 Hz, phenyl H), 7.28 (1H, d, J = 7.7 Hz, phenyl H), 7.25 (1H, s, quinazoline 8-H), 7.16 (1H, br s, amide H), 5.15 (1H, d, J = 20.0 Hz, 132-CHH), 5.12 (1H, d, J = 20.0 Hz, 132-CHH), 4.77 (1H, dd, J ~ 5.5, 15.2 Hz, -NHCHH-phenyl-), 4.76 (1H, dd, J ~ 6.0, 15.2 Hz, -NHCHH-phenyl-), 4.36 (2H, m, -OCH2CH2OCH3), 4.25 (2H, m, -OCH2CH2OCH3), 4.17 (1H, q, J = 7.1 Hz, 18-H), 4.02 (1H, dd, J = 3.5, 8.2 Hz, 17-H), 3.83 (4H, m, 2 x -OCH2CH2OCH3), 3.74 (2H, q, J = 7.7 Hz, -CH2CH3), 3.69 (2H, q, J = 7.7 Hz, -CH2CH3), 3.65 (3H, s, ring CH3), 3.52 (3H, s, -COOCH3), 3.459 (3H, s, -OCH2CH2OCH3), 3.456 (3H, s, -OCH2CH2OCH3), 3.26 (3H, s, ring CH3), ~2.48 (1H, m, 1H of 17-CH2CH2-), ~2.38 (1H, m, 1H of 17-CH2CH2), 2.27 (3H, s, 2-CH3), 2.09-2.21 (2H, m, 2H of 17-CH2CH2), 1.70 (3H, t, J = 7.7 Hz, -CH2CH3), 1.61 (3H, t, J = 7.6 Hz, -CH2CH3), 1.01 (3H, d, J = 7.0 Hz, 18-CH3), −1.47 (1H, s, core NH); 13C NMR (100 MHz, 90:10 CDCl3/CD3OD, δ ppm): 196.8, 173.6, 171.7, 167.6, 159.6, 157.2, 155.2, 154.2, 151.4, 151.2, 149.2, 148.3, 144.8, 144.7, 144.1, 140.2, 138.7, 138.4, 138.3, 136.1, 135.0, 134.4, 133.6, 132.3, 131.8 (2C), 131.5, 130.4, 128.8, 128.4, 128.0, 127.8 (2C), 127.0, 126.5, 125.6, 123.7, 122.8, 122.2, 110.4, 108.7, 105.8, 105.7, 103.9, 103.8, 96.7, 89.24, 89.21, 70.8, 70.3, 69.0, 68.5, 59.2, 59.1, 51.9, 51.5, 48.4, 48.3, 43.7, 31.1, 29.7, 20.8, 19.3, 19.2, 17.2, 16.6, 13.9, 11.8, 11.1. Note: One of the core NH protons was not observed, presumably due to chemical exchange. 1H impurity peaks were observed at: 5.30 (DCM), 3.35, 2.02, 1.26 (grease CH2), and 0.88 (grease CH3) ppm. One carbon peak is missing, presumably due to line broadening caused by aggregation. 13C peaks at 131.8 and 127.8 ppm are each comprised of two aromatic CH signals of the benzyl group. Very low intensity 13C impurity peaks were observed at the following chemical shifts: 113.8, 55.8, 31.8, 31.7, 29.2, and 22.5 ppm. The 13C impurity peak at 29.6 ppm (likely due to grease CH2) is larger.
Synthesis of Conjugate (27):
The compounds 25 and 26 were synthesized by using our published synthetic procedures. To a stirred solution of compound 25 (30 mg, 0.035 mmol) and erlotinib analog 26 (26 mg, 0.052 mmol) in dry THF (10 mL), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimideHCl (EDCI, 40 mg, 0.21 mmol) and 4-dimethylaminopyridine (DMAP, 25 mg, 0.21 mmol) was added. The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvents were evaporated and diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated to yield the crude product, which was purified by preparative TLC plates to yield 27 in 45% yield (36 mg). MS (ESI) m/z: 2307.24 (M + H+). HRMS (ESI): calcd for C136H148N17O18 (M + H+) 2307.2413; found, 2307.2419. λmax, nm (ε)): 663 (4.5 x 104), 607 (8.8 x 103), 542 (8.8 x 103), 509 (8.4 x 103), 413 (8.4 x 104), 335 (9.2 x 104).. 1H NMR (400 MHz, 90:10 CDCl3/CD3OD, δ ppm): 9.66/9.62 (1H, s, meso 5-H), 9.37/9.36 (1H, s, meso 10-H), 8.45 (3H, s, 3 x pyrimidine H), 8.371/8.365 (1H, s, meso 20-H), 7.67 (3H, m, 3 x phenyl H), 7.64 (3H, m, 3 x phenyl H), 7.514/7.512 (3H, s, 3 x quinazoline 5-H), 7.21 (3H, t, J ~ 8 Hz, 3 x phenyl H), 7.18 (3H, d, J = 7.0 Hz, 3 x phenyl H), 7.09 (3H, m, 3 x phenyl H), 7.08 (3H, m, 3x phenyl H), 7.063/7.059 (3H, s, 3 x quinazoline 8-H), 6.98/6.97 (3H, t, J = 7.6 Hz, 3 x phenyl H), 6.94 (3H, m, 3 x phenyl H), 5.83/5.80 (1H, q, J = 6.7 Hz, 31-H), 5.14 (1H, d, J = 19.9 Hz, 132-CHH), 4.91 (1H, d, J = 19.9 Hz, 132-CHH), 4.38 (1H, dq, J = 7.4, 1.7 Hz,18-H), 4.19 (6H, m, 3 x -OCH2CH2OCH3), 4.11 (13H, m, 3 x -OCH2CH2OCH3 & 3 x -NHCH2-phenyl & 17-H), 3.77 (6H, m, 3 x -OCH2CH2OCH3), 3.69 (6H, m, 3 x -OCH2CH2OCH3), 3.50-3.64 (2H, m, -OCH2(CH2)4CH3), ~3.62 (2H, m, 8-CH2CH3), 3.50 (3H, s, 12-CH3), 3.40 (9H, s, 3 x -OCH2CH2OCH3), 3.364/3.361 (9H, s, 3 x -OCH2CH2OCH3), 3.25/3.24 (3H, s, 2-CH3), 3.184/3.179 (3H, s, 7-CH3), 2.56 (1H, m, 1H of 17-CH2CH2-), 2.30 (1H, m, 1H of 17-CH2CH2-), 2.24 (1H, m, 1H of 17-CH2CH2-), 2.12 (6H, m, 3 x CH2 of -CH2CH2C(=O)NH-), 2.05/2.04 (3H, d, J = 6.6 Hz, 31-CH3), 2.02 (1H, m, 1H of 17-CH2CH2-), 1.94 (6H, m, 3 x CH2 of -CH2CH2C(=O)NH-), 1.69/1.68 (3H, d, J = 7.1 Hz, 18-CH3), 1.68 (2H, m, -OCH2CH2(CH2)3CH3), 1.64 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.23-1.40 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.16 (4H, m, -O(CH2)3(CH2)2CH3), 0.71/0.70 (3H, distorted t, J ~ 6.9 Hz, -O(CH2)5CH3); 13C NMR (100 MHz, 90:10 CDCl3/CD3OD, δ ppm): 197.33/197.32, 174.08/174.06, 173.63/173.62, 172.2, 160.5, 157.0, 155.7, 154.6, 153.3, 150.9, 149.0, 148.8/148.7, 146.9, 145.22/145.20, 141.5/141.4, 139.6, 139.1, 138.39/138.37, 137.4, 136.35/136.32, 135.8/135.7, 132.7/132.6, 130.36/130.34, 130.26, 129.7, 128.8, 128.4, 128.1, 127.4, 127.2, 125.0, 123.5, 123.28/123.27, 122.5, 109.6, 107.7, 105.7, 104.2, 103.69/103.68, 97.9/97.7, 92.8, 89.37/89.36, 89.1, 72.9/72.8, 70.9, 70.6, 69.7, 68.9, 68.3, 59.19, 59.16, 57.9/57.8, 51.74/51.72, 50.1, 47.9, 42.9, 33.9, 31.7, 31.3, 31.0, 30.6, 30.2, 26.07/26.05, 24.6/24.5, 22.9, 22.57/22.55, 19.4, 17.4, 13.88/13.86, 11.8, 11.2, 10.90/10.86. Note: Two core NH, four amide NH, and three amine NH protons are missing, presumably due to chemical exchange. 1H impurity peaks at 1.21 (likely grease CH2), 0.83 ppm (likely grease CH3) were observed. The 13C impurity peak at 29.7 ppm is likely due to grease CH2.
Synthesis of Conjugate (29):
Triphenylarsine (9.25 mg, 0.0302 mmol) and Pd2dba3 (13.84 mg, 0.0151 mmol) were added to a stirred solution of compound 28 (30 mg, 0.0377 mmol) and erlotinib 1 (22.30 mg, 0.0569 mmol) in dry THF (10 mL) and Et3N (2.5 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC, only 50% amount of the starting material reacted to produce the desired product. The desired conjugate 29 was purified by preparative TLC plates and was obtained 68% yield (27.22 mg). MS (ESI) m/z: 1159.47 (M + H+). HRMS (ESI): calcd for C63H63N8O8 (M + H+) 1159.4724; found, 1159.4745. UV-vis (CH3OH, λmax, nm (abs)): 707 (4.4 x 104), 652 (8.5 x 103), 550 (2.3 x 104), 513 (6.3 x 103), 482 (4.8 x 103), 416 (1.16 x 105), 348 (6.7 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.46 (1H, s, meso H), 9.24 (1H, s, meso H), 8.60 (1H, s, pyrimidine H), 8.54 (1H, s, meso 20-H), 7.89 (1H, br s, phenyl H), ~7.85 (1H, br d, J ~ 8.0 Hz, phenyl H), 7.82 (1H, dd, J = 17.9, 11.5 Hz, -CH=CH2), 7.72 (1H, br d, J = 7.7 Hz, phenyl H), 7.66 (1H, br s, amine NH), 7.59 (1H, br s, phenyl H), 7.38 (1H, br d, J = 7.7 Hz, phenyl H), 7.32 (1H, dd, J ~ 7.7, 7.7 Hz, phenyl H), ~7.26 (1H, m, phenyl H), 7.24 (1H, br s, quinazoline 5-H), 7.20 (1H, ddd, J = 7.7, ~1.2, ~1.2 Hz, phenyl H), 7.18 (1H, s, quinazoline 8-H), 6.24 (1H, dd, J = 17.9, 1.2 Hz, -CH=CHH), 6.11 (1H, dd, J = 11.6, 1.2 Hz, -CH=CHH), 5.72 (1H, d, J = 14.4 Hz, >NCHH-phenyl-), 5.61 (1H, d, J = 14.4 Hz, >NCHH-phenyl-), 5.40 (1H, m, 17-H), 4.34 (1H, q, J = 7.3 Hz, 18-H), 4.23 (2H, m, -OCH2CH2OCH3), 4.16 (2H, t, J = 4.7 Hz, -OCH2CH2OCH3), 3.82 (2H, m, -OCH2CH2OCH3), 3.75 (3H, s, ring CH3), 3.70 (2H, m, -OCH2CH2OCH3), 3.53 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.52 (3H, s, -COOCH3), 3.45 (3H, s, -OCH2CH2OCH3), 3.36 (3H, s, -OCH2CH2OCH3), 3.31 (3H, s, ring CH3), 3.07 (3H, s, ring CH3), ~2.75 (1H, m, 1H of 17-CH2CH2-), 2.35-2.52 (2H, m, 2H of 17-CH2CH2-), 2.01 (1H, m, 1H of 17-CH2CH2-), 1.77 (3H, d, J = 7.3 Hz, 18-CH3), 1.60 (3H, t, J = 7.6 Hz, 8-CH2CH3), −0.06 (1H, s, core NH), −0.17 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 176.8, 175.0, 174.2, 167.5, 163.3, 156.2, 155.4, 154.4, 153.5, 149.8, 148.8, 147.3, 145.4, 143.0, 139.6, 139.0, 138.9, 137.6, 137.0, 136.3, 136.0, 131.8, 131.7, 131.1, 130.3, 128.89, 128.87, 128.6, 128.5, 126.9, 124.2, 123.9, 123.2, 123.0, 121.4, 115.6, 109.2, 108.7, 106.9, 102.6, 102.4, 97.3, 94.7, 89.8, 89.0, 70.8, 70.5, 69.1, 68.3, 59.3, 59.2, 54.7, 51.6, 49.1, 43.1, 32.5, 31.5, 24.0, 19.3, 17.4, 12.5, 11.9, 11.0. Note: 1H impurity peaks at 1.27 and 0.89 ppm are likely due to grease CH2 and CH3, respectively.
Synthesis of Conjugate (31):
Triphenylarsine (8.44 mg, 0.0275 mmol) and Pd2dba3 (12.63 mg, 0.0137 mmol) were added to a stirred solution of compound 30 (30 mg, 0.0344 mmol) and erlotinib 1 (20.34 mg, 0.0517 mmol) in dry THF (10 mL) and Et3N (2.5 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The product 11 was purified by preparative TLC plates and obtained in 75% yield (29.36 mg). MS (ESI) m/z: 1135.56 (M + H+). HRMS (ESI): calcd for C67H75N8O9 (M + H+) 1135.5612; found, 1135.5635. UV-vis (CH3OH, λmax, nm (abs)): 783 (4.15 x 104), 538 (3.98 x 104), 415 (4.37 x 104), 366 (1.07 x 105), 342 (7.5 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.46 (1H, s, meso H), 9.24 (1H, s, meso H), 8.60 (1H, s, pyrimidine H), 8.54 (1H, s, meso 20-H), 7.89 (1H, br s, phenyl H), ~7.85 (1H, br d, J ~ 8.0 Hz, phenyl H), 7.82 (1H, dd, J = 17.9, 11.5 Hz, -CH=CH2), 7.72 (1H, br d, J = 7.7 Hz, phenyl H), 7.66 (1H, br s, amine NH), 7.59 (1H, br s, phenyl H), 7.38 (1H, br d, J = 7.7 Hz, phenyl H), 7.32 (1H, dd, J ~ 7.7, 7.7 Hz, phenyl H), ~7.26 (1H, m, phenyl H), 7.24 (1H, br s, quinazoline 5-H), 7.20 (1H, ddd, J = 7.7, ~1.2, ~1.2 Hz, phenyl H), 7.18 (1H, s, quinazoline 8-H), 6.24 (1H, dd, J = 17.9, 1.2 Hz, -CH=CHH), 6.11 (1H, dd, J = 11.6, 1.2 Hz, -CH=CHH), 5.72 (1H, d, J = 14.4 Hz, >NCHH-phenyl-), 5.61 (1H, d, J = 14.4 Hz, >NCHH-phenyl-), 5.40 (1H, m, 17-H), 4.34 (1H, q, J = 7.3 Hz, 18-H), 4.23 (2H, m, -OCH2CH2OCH3), 4.16 (2H, t, J = 4.7 Hz, -OCH2CH2OCH3), 3.82 (2H, m, -OCH2CH2OCH3), 3.75 (3H, s, ring CH3), 3.70 (2H, m, -OCH2CH2OCH3), 3.53 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.52 (3H, s, -COOCH3), 3.45 (3H, s, -OCH2CH2OCH3), 3.36 (3H, s, -OCH2CH2OCH3), 3.31 (3H, s, ring CH3), 3.07 (3H, s, ring CH3), ~2.75 (1H, m, 1H of 17-CH2CH2-), 2.35-2.52 (2H, m, 2H of 17-CH2CH2-), 2.01 (1H, m, 1H of 17-CH2CH2-), 1.77 (3H, d, J = 7.3 Hz, 18-CH3), 1.60 (3H, t, J = 7.6 Hz, 8-CH2CH3), −0.06 (1H, s, core NH), −0.17 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 176.8, 175.0, 174.2, 167.5, 163.3, 156.2, 155.4, 154.4, 153.5, 149.8, 148.8, 147.3, 145.4, 143.0, 139.6, 139.0, 138.9, 137.6, 137.0, 136.3, 136.0, 131.8, 131.7, 131.1, 130.3, 128.89, 128.87, 128.6, 128.5, 126.9, 124.2, 123.9, 123.2, 123.0, 121.4, 115.6, 109.2, 108.7, 106.9, 102.6, 102.4, 97.3, 94.7, 89.8, 89.0, 70.8, 70.5, 69.1, 68.3, 59.3, 59.2, 54.7, 51.6, 49.1, 43.1, 32.5, 31.5, 24.0, 19.3, 17.4, 12.5, 11.9, 11.0. Note: 1H impurity peaks at 1.27 and 0.89 ppm are likely due to grease CH2 and CH3, respectively.
Synthesis of Conjugate (37):
Methyl pheophorbide-a 32 (50 mg, 0.0825 mmol), and 3-iodonenzlamine 34 (96 mg, 0.413 mmol) were dissolved in 10 mL dry THF. The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent was evaporated and the crude product was purified by silica column by using 3% methanol in dichloromethane to obtain pure product 35 with 91% (69.2 mg) yield. MS (ESI) m/z: 840.2617 (M +H+). HRMS (ESI): calcd for C43H47N5O5I (M + H+) 840.2617; found, 840.2616. 1H NMR (400 MHz, CDCl3, δ ppm): 9.66 (1H, s, meso H), 9.62 (1H, s, meso H), 8.80 (1H, s, meso 20-H), 8.07 (1H, dd, J = 17.9, 11.5 Hz, -CH=CH2), 7.91 (1H, t, J ~ 1.6 Hz, phenyl H), 7.67 (1H, ddd, J = 7.9, ~1.4, ~1.2 Hz, phenyl H), 7.49 (1H, br d, J = 7.7 Hz, phenyl H), 7.12 (1H, t, J = 7.8 Hz, phenyl H), 6.70 (1H, br t, J ~ 5.8 Hz, amide NH), 6.34 (1H, dd, J = 17.9, 1.5 Hz, -CH=CHH), 6.13 (1H, dd, J = 11.5, 1.5 Hz, -CH=CHH), 5.50 (1H, d, J = 18.8 Hz, 15-CHH-), 5.24 (1H, d, J = 18.8 Hz, 15-CHH-), 4.96 (1H, dd, J = 14.6, 6.4 Hz, -NHCHH-phenyl-), 4.64 (1H, dd, J = 14.6, 5.3 Hz, -NHCHH-phenyl-), 4.47 (1H, q, J = 7.2 Hz, 18-H), 4.38 (1H, m, 17-H), 3.77 (2H, q, J = 7.7 Hz, 8-CH2CH3), 3.72 (3H, s, -COOCH3), 3.61 (3H, s, -COOCH3), 3.51 (3H, s, ring CH3), 3.48 (3H, s, ring CH3), 3.31 (3H, s, ring CH3), ~2.55 (1H, m, 1H of 17-CH2CH2-), 2.12-2.27 (2H, m, 2H of 17-CH2CH2-), ~1.82 (1H, m, 1H of 17-CH2CH2-), 1.72 (3H, d, J = 7.2 Hz, 18-CH3), 1.71 (3H, t, J = 7.7 Hz, 8-CH2CH3), −1.56 (1H, br s, core NH), −1.76 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.9, 173.5, 169.3, 169.0, 166.7, 154.3, 149.0, 144.8, 140.4, 139.0, 137.1, 136.8, 136.1, 135.0, 134.93, 134.91, 134.6, 130.6, 130.3, 129.8, 129.4, 127.6, 127.4, 121.7, 102.1, 101.5, 98.8, 94.6, 93.7, 53.1, 52.2, 51.6, 49.3, 43.9, 38.0, 31.1, 29.7, 23.0, 19.7, 17.7, 12.1, 12.0, 11.3. Note: 1H impurity peaks at 1.27 and 0.89 ppm are likely due to grease.
Triphenylarsine (11.67 mg, 0.038 mmol) and Pd2dba3 (17.45 mg, 0.02 mmol) were added to a stirred solution of compound 35 (40 mg, 0.048 mmol) and erlotinib 1 (28 mg, 0.0715 mmol) in dry THF (20 mL) and Et3N (3 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. After completion of the reaction the reaction mixture was evaporated. The crude was diluted with DCM and washed with water. The organic layer was removed and concentrated using a rotary evaporator and dried using a high vacuum pump. The desired conjugate 37 was purified by preparative TLC plates with 79% yield (41.52 mg). MS (ESI) m/z: 1103.54 (M + H+). HRMS (ESI): calcd for C67H71N6O9 (M + H+) 1103.5441; found, 1103.5446. UV-vis (CH3OH, λmax, nm (abs)): 660 (5.1 x 104), 501 (1.4 x 104), 397 (1.6 x 105), 349 (5.5 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.59 (2H, s, meso 5-H & meso 10-H), 8.78 (1H, s, meso 20-H), 8.57 (1H, br s, pyrimidine H), 8.04 (1H, dd, J = 17.4, 11.6 Hz, -CH=CH2), 7.80 (1H, s, phenyl H), 7.66-7.74 (2H, m, 2 x phenyl H), ~7.50 (1H, br s, amine NH), 7.43-7.52 (2H, m, 2 x phenyl H), 7.37 (1H, t, J = 7.6 Hz, phenyl H), 7.22-7.32 (3H, m, 2 x phenyl H & quinazoline 5-H), 7.16 (1H, s, quinazoline 8-H), 6.83 (1H, br s, amide NH), 6.31 (1H, d, J = 17.7 Hz, -CH=CHH), 6.10 (1H, d, J = 11.4 Hz, -CH=CHH), 5.49 (1H, d, J = 18.8 Hz, 15-CHH-), 5.25 (1H, d, J = 18.8 Hz, 15-CHH-), 4.95 (1H, dd, J ~ 14, 5 Hz, -NHCHH-phenyl-), 4.72 (1H, dd, J = 14.3, 4.3 Hz, -NHCHH-phenyl-), 4.45 (1H, q, J = 7.2 Hz, 18-H), 4.37 (1H, m, 17-H), 4.18 (4H, m, 2 x -OCH2CH2OCH3), 3.76 (2H, m, -OCH2CH2OCH3), ~3.72 (2H, m, 8-CH2CH3), 3.71 (3H, s, -COOCH3), 3.66 (2H, m, -OCH2CH2OCH3), 3.59 (3H, s, -COOCH3), 3.45 (6H, br s, 2 x ring CH3), 3.41 (3H, s, -OCH2CH2OCH3), 3.34 (3H, s, -OCH2CH2OCH3), 3.27 (3H, s, ring CH3), ~2.54 (1H, m, 1H of 17-CH2CH2-), 2.10-2.27 (2H, m, 2H of 17-CH2CH2-), 1.81 (1H, m, 1H of 17-CH2CH2-), 1.69 (3H, d, J = 7.1 Hz, 18-CH3), 1.67 (3H, t, J = 7.5 Hz, 8-CH2CH3), −1.59 (1H, br s, core NH), −1.82 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.9, 173.6, 169.5, 168.9, 166.6, 156.3, 154.7, 154.3, 153.3, 149.05, 149.01, 144.8, 139.0, 138.7, 138.2, 136.1, 134.91, 134.90, 134.6, 131.4, 130.9, 130.2, 129.8, 129.4, 128.95, 128.94, 128.2, 127.6, 127.3, 124.6, 123.8, 123.7, 122.0, 121.6, 109.0, 108.6, 102.7, 102.1, 101.4, 98.8, 93.7, 89.6, 89.2, 70.8, 70.4, 69.3, 68.3, 59.24, 59.20, 53.1, 52.2, 51.6, 49.2, 44.4, 38.0, 31.2, 29.7, 23.0, 19.6, 17.7, 12.1, 12.0, 11.3. Note: 1H impurity peaks at 1.27 (likely grease CH2), 0.89 (likely grease CH3), and 0.08 ppm were observed. Two carbon peaks are missing, presumably due to line broadening caused by aggregation.
Synthesis of Conjugate (38):
Methylpheophorbide-a 32 (100.0 mg, 0.165 mmol) was dissolved in 12 mL DCM, HBr gas was bubbled for 5 minutes and the reaction mixture was stirred for 1 hour at room temperature. The solvent was evaporated under high vacuum, the resulting crude was dried, and the entire crude was dissolved in dry CH2Cl2 (10 mL). A 100 mg portion of dry K2CO3 was added to this mixture, followed by addition of 1-hexanol (0.62 mL, 4.95 mmol). The resulting reaction mixture was stirred for 1 h at room temperature and then worked up. The crude mixture was purified by flash column chromatography (silica gel, 40% ethyl acetate in hexane). Evaporation of the appropriate elutes gave the desired compound 33, 55% (64.26 mg). Compound 33 (50 mg, 0.053 mmol) was then reacted with 34 (61.9 mg, 0.266 mmol) dissolved in 10 mL dry THF. The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent was evaporated, and the crude product was purified by silica column by using 3% methanol in dichloromethane to obtain pure product 36 in 89% (59.13 mg) yield. MS (ESI) m/z: 942.36 (M + H+). 1H NMR (400 MHz, CDCl3, δ ppm): 9.93 (1H, s, meso 5-H), 9.71 (1H, s, meso 10-H), 8.77 (1H, s, meso 20-H), 7.94 (1H, s, phenyl H), 7.69 (1H, d, J = 7.8 Hz, phenyl H), 7.54 (1H, d, J = 7.7 Hz, phenyl H), 7.16 (1H, t, J = 7.8 Hz, phenyl H), 6.76 (1H, br t, J ~ 5.6 Hz, amide NH), 5.96 (1H, q, J = 6.7 Hz, 31-H), 5.51 (1H, d, J = 18.7 Hz, 15-CHH-), 5.26/5.25 (1H, d, J = 18.8 Hz, 15-CHH-), 5.02/5.00 (1H, dd, J = 14.5, 6.2 Hz, -NHCHH-phenyl-), 4.70/4.69 (1H, dd, J = 14.5, 4.6 Hz, -NHCHH-phenyl-), 4.46 (1H, q, J = 7.1 Hz, 18-H), 4.39 (1H, d, J = 9.5 Hz, 17-H), 3.81 (2H, q, J = 7.5 Hz, 8-CH2CH3), 3.74/3.73 (3H, s, -COOCH3), 3.59-3.70 (2H, m, -OCH2(CH2)4CH3), 3.60 (3H, s, -COOCH3), 3.56 (3H, s, ring CH3), 3.46/3.45 (3H, s, ring CH3), 3.34 (3H, s, ring CH3), 2.53 (1H, m, 1H of 17-CH2CH2-), ~2.21 (1H, m, 1H of 17-CH2CH2-), 2.133/2.125 (3H, d, J = 6.6 Hz, 31-CH3), ~2.12 (1H, m, 1H of 17-CH2CH2-), ~1.81 (1H, m, 1H of 17-CH2CH2-), ~1.75 (2H, m, -OCH2CH2(CH2)3CH3), 1.72 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.71 (3H, d, J = 7.1 Hz, 18-CH3), 1.29-1.47 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.23 (4H, m, -O(CH2)3(CH2)2CH3), 0.79/0.78 (3H, distorted t, J ~ 6.9 Hz, -O(CH2)5CH3), −1.59 (1H, br s, core NH), −1.78 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.9, 173.5, 169.4, 169.0, 166.4, 154.3, 149.0, 144.8, 140.4, 139.0, 137.20/137.19, 136.9, 136.2, 134.8, 134.5/134.3, 130.9, 130.6, 129.7, 127.52/127.51, 102.00/101.98, 101.5, 99.63/99.59, 94.7, 93.3/93.2, 72.9/72.8, 69.72/69.69, 53.0, 52.2, 51.6, 49.35/49.33, 44.0, 38.0, 31.8, 31.12/31.11, 30.24/30.22, 29.7, 26.1, 25.0/24.9, 23.02/23.00, 22.58/22.57, 19.7, 17.7, 14.0, 12.0, 11.40/11.39, 11.13/11.10. Note: 1H impurity peaks at 1.27 and 0.89 ppm are likely due to grease CH2 and CH3, respectively. An additional minor 1H impurity peak is observed at 0.09 ppm. Three carbon peaks are missing, presumably due to line broadening caused by aggregation. Very low intensity 13C impurity peaks were observed at the following chemical shifts: 31.9, 29.4, 22.7, and 14.1 (likely grease CH3) ppm.
Compound 36 (40 mg, 0.043 mmol) was reacted with erlotinib 1 (25 mg, 0.0637 mmol) in dry THF (20 mL) and triethylamine (3 mL) and Triphenylarsine (10.40 mg, 0.034 mmol) and Pd2dba3 (15.56 mg, 0.017 mmol) The reaction mixture was stirred at room temperature under an argon atmosphere overnight. After completion of the reaction the reaction mixture was evaporated. The crude was diluted with DCM and washed with water. The organic layer was removed and concentrated using a rotary evaporator and dried using a high vacuum pump. The product 15 was purified by preparative TLC plates with 79% yield (41.52 mg). MS (ESI) m/z: 1205.66 (M + H+). HRMS (ESI): calcd for C73H84N6O10 (M + H+) 1205.6634; found, 1205.6641. UV-vis (CH3OH, λmax, nm (abs)): 653 (5.7 x 104), 599 (5.4 x 103), 495 (1.4 x 104), 394 (1.7 x 105).. 1H NMR (400 MHz, CDCl3, δ ppm): 9.92/9.91 (1H, s, meso 5-H), 9.67 (1H, s, meso 10-H), 8.76/8.75 (1H, s, meso 20-H), 8.65 (1H, s, pyrimidine H), 7.85 (1H, br s, phenyl H), 7.71-7.76 (2H, m, 2 x phenyl H), 7.54 (1H, d, J = 7.6 Hz, phenyl H), 7.51 (1H, d, J = 7.7 Hz, phenyl H), ~7.50 (1H, br s, amine NH), 7.40 (1H, dd, J ~ 7.7, 7.7 Hz, phenyl H), 7.30 (1H, dd, J ~ 7.6 Hz, phenyl H), 7.28 (1H, d, J ~ 8 Hz, phenyl H), 7.26 (1H, s, quinazoline 5-H), 7.19 (1H, s, quinazoline 8-H), 6.89 (1H, br t, J ~ 5.6 Hz, amide NH), 5.95 (1H, q, J = 6.7 Hz, 31-H), 5.52 (1H, d, J = 18.7 Hz, 15-CHH), 5.27/5.26 (1H, d, J = 18.7 Hz, 15-CHH), 5.024/5.020 (1H, dd, J = 14.4, 5.9 Hz, -NHCHH-phenyl-), 4.79/4.78 (1H, dd, J = 14.4, ~4.6 Hz, -NHCHH-phenyl-), 4.44 (1H, q, J = 7.2 Hz, 18-H), 4.38/4.37 (1H, m, 17-H), 4.20 (4H, m, 2 x -OCH2CH2OCH3), 3.75-3.81 (4H, m, -OCH2CH2OCH3 & 8-CH2CH3), 3.723/3.721 (3H, s, -COOCH3), 3.69 (2H, m, -OCH2CH2OCH3), 3.57-3.69 (2H, m, -OCH2(CH2)4CH3), 3.58 (3H, s, -COOCH3), 3.53 (3H, s, ring CH3), 3.453/3.445 (3H, s, ring CH3), 3.42 (3H, s, -OCH2CH2OCH3), 3.36 (3H, s, -OCH2CH2OCH3), 3.32 (3H, s, ring CH3), 2.52 (1H, m, 1H of 17-CH2CH2-), 2.21 (1H, m, 1H of 17-CH2CH2-), 2.13/2.12 (3H, d, J = 6.6 Hz, 31-CH3), 2.11 (1H, m, 1H of 17-CH2CH2-), 1.82 (1H, m, 1H of 17-CH2CH2-), 1.75 (2H, m, -OCH2CH2(CH2)3CH3), 1.70 (3H, t, J = 7.7 Hz, 8-CH2CH3), 1.694/1.687 (3H, d, J = 7.2 Hz, 18-CH3), 1.28-1.47 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.23 (4H, m, -O(CH2)3(CH2)2CH3), 0.79/0.78 (3H, distorted t, J ~ 6.9 Hz, -O(CH2)5CH3), −1.62 (1H, br s, core NH), −1.80 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 174.0, 173.6, 169.5, 168.9, 166.39/166.37, 156.2, 154.7, 154.3, 153.6, 149.03/149.02, 148.98, 147.5, 144.80/144.79, 138.93/138.88, 138.87, 138.3, 136.2, 134.83, 134.78/134.77, 134.4/134.3, 131.49/131.48, 131.0, 130.9/130.8, 129.6, 129.01, 128.98, 128.3, 127.56/127.55, 127.3, 124.4, 123.9, 123.8, 121.8, 109.2, 108.9, 102.6, 102.02/102.01, 101.5, 99.62/99.58, 93.2, 89.6, 89.3, 72.9/72.8, 70.9, 70.4, 69.69/69.67, 69.3, 68.3, 59.24, 59.23, 53.0, 52.2, 51.6, 49.31/49.30, 44.5, 38.0, 31.7, 31.13/31.11, 30.23/30.21, 29.7, 26.08/26.07, 25.0/24.9, 23.01/23.00, 22.57/22.56, 19.7, 17.7, 13.96/13.95, 12.0, 11.38/11.37, 11.12/11.09. Note: 1H impurity peaks were observed at 1.27 (likely grease CH2), 0.89 (likely grease CH3), and 0.09 ppm (minor). One carbon peak is missing from this spectrum, presumably due to aggregation-induced line broadening. An additional 13C peak, presumably due to grease impurity, was observed at 29.7 ppm.
Synthesis of Conjugate (43):
Compound 32 (50 mg, 0.071 mmol) was dissolved in 10 mL dry THF and stirred under argon for 10 minutes. 39 (155.39 mg, 0.71 mmol) was added to the solution and the reaction mixture was stirred for 12 h. The reaction was monitored by spectrophotometry. The reaction mixture was evaporated and the residue was dissolved in 2.5% MeOH/CH2Cl2 and then chromatographed on a short silica gel column using 20% MeOH/CDM to obtain pure product 40 with 76% yield (49.81 mg). MS (ESI) m/z: 929.58 (M + H+). 1H NMR (400 MHz, 90:10 CDCl3/CD3OD, δ ppm): 9.94/9.93 (1H, s, meso 5-H), 9.72 (1H, s, meso 10-H), 8.803/8.801 (1H, s, meso 20-H), 5.95 (1H, q, J = 6.7 Hz, 31-H), 5.52 (1H, d, J = 19.1 Hz, 15-CHH), 5.273/5.265 (1H, d, J = 19.1 Hz, 15-CHH), 4.48 (1H, q, J = 7.1 Hz, 18-H), 4.35 (1H, m, 17-H), 3.86 (1H, m, -NHCHHCH2CH2O-), 3.80 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.75 (3H, s, -COOCH3), 3.74 (2H, t, J = 5.8 Hz, -NH(CH2)2CH2O-), ~3.69 (1H, m, -NHCHHCH2CH2O-), ~3.56-3.71 (2H, m, -OCH2(CH2)4CH3), 3.64 (2H, m, -OCH2CH2O-), 3.592/3.590 (3H, s, -COOCH3), ~3.56 (2H, m, -OCH2CH2O-), 3.55 (3H, s, 12-CH3), 3.443/3.437 (3H, s, 2-CH3), 3.31 (3H, s, 7-CH3), ~3.29 (2H, m, -OCH2CH2O-), 3.15/3.14 (2H, t, J = 5.8 Hz, -OCH2CH2CH2NH2), 3.09 (2H, m, -OCH2CH2O-), 2.78/2.77 (2H, t, J = 6.2 Hz, -OCH2CH2CH2NH2), 2.56 (1H, m, 17-CH2CHH-), ~2.20 (1H, m, 17-CHHCH2-), ~2.16 (1H, m, 17-CH2CHH-), 2.116/2.105 (3H, d, J = 6.6 Hz, 31-CH3), ~2.114 (2H, m, -NHCH2CH2CH2O-), 1.76 (1H, m, 17-CHHCH2-), 1.72 (2H, m, -OCH2CH2(CH2)3CH3), 1.71 (3H, t, J = 7.5 Hz, 8-CH2CH3), 1.69/1.68 (3H, d, J = 7.1 Hz, 18-CH3), 1.54 (2H, m, -OCH2CH2CH2NH2), 1.25-1.45 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.18 (4H, m, -O(CH2)3(CH2)2CH3), 0.74/0.72 (3H, t, J = 6.9 Hz, -O(CH2)5CH3); 13C NMR (100 MHz, 90:10 CDCl3/CD3OD, δ ppm): 174.6, 174.4, 170.7, 169.7, 167.09/167.07, 154.6, 149.4, 145.3, 139.23, 139.21/139.16, 136.8, 134.97/134.96, 134.8, 134.6/134.4, 131.6/131.5, 129.7, 128.21/128.20, 102.34/102.32, 101.7, 100.0/99.9, 93.8, 73.3/73.2, 70.7, 70.4, 70.07, 70.06/70.04, 69.82, 69.79/69.78, 69.68/69.67, 53.4, 52.5, 51.9, 49.60/49.58, 39.6, 38.8, 38.0, 32.01/32.00, 31.28/31.27, 30.43/30.41, 30.0, 29.6, 27.7, 26.34/26.32, 25.0/24.9, 23.2, 22.83/22.81, 19.9, 17.8, 14.05/14.03, 12.0, 11.50/11.48, 11.3/11.2. Note: Amide, amine and core NH protons were not observed, likely due to chemical exchange. Minor 1H impurity peaks were observed at various chemical shifts. The impurity at 1.23 ppm (likely grease CH2) is somewhat larger.
To a solution of compound 40 (40 mg, 0.0431 mmol) and 3-aminobenzyl acetic acid 41 (16.92 mg, 0.0646 mmol) in 10 mL of dry DMF, (benzotriazol-1-yloxy)tris(dimethylamino) phosphonium hexafluorophosphate (BOP, 38.1 mg, 0.0861 mmol) and 2 drops triethylamine were added. The reaction mixture was stirred at room temperature under Ar atmosphere for overnight. It was then diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 6% methanol in dichloromethane to obtain pure product 42 with 82% (41.42 mg) yield. MS (ESI) m/z: 1173.54 (M + H+). For the characterization details, including the 1H and 13C- NMR spectra please see the Supporting Material Information. 1H NMR (400 MHz, CDCl3, δ ppm): 9.95/9.93 (1H, s, meso 5-H), 9.72 (1H, s, meso 10-H), 8.78/8.77 (1H, s, meso 20-H), 7.33 (1H, dt, J = 7.8, ~1.3 Hz, phenyl H), 7.27 (1H, m, phenyl H), 7.19 (1H, t, J ~ 5 Hz, amide NH), 6.76 (1H, d, J = 7.6 Hz, phenyl H), 6.68 (1H, t, J = 7.7 Hz, phenyl H), 5.96/5.95 (1H, q, J = 6.6 Hz, 31-H), 5.62/5.60 (1H, d, J = 19.0 Hz, 15-CHH), 5.20-5.33 (2H, m, 15-CHH & amide NH), 4.46 (1H, q, J = 7.2 Hz, 18-H), 4.37 (1H, m, 17-H), 4.02 (1H, m, -NHCHHCH2CH2O-), 3.82 (2H, q, J = 7.7 Hz, 8-CH2CH3), 3.80/3.79 (3H, s, -COOCH3), ~3.77 (1H, m, -NHCHHCH2CH2O-), 3.74 (2H, m, -NH(CH2)2CH2O-), 3.57-3.70 (2H, m, -OCH2(CH2)4CH3), 3.622/3.621 (3H, s, -COOCH3), 3.58 (3H, s, 12-CH3), 3.56 (2H, m, -OCH2CH2O-), 3.46/3.45 (3H, s, 2-CH3), ~3.37 (2H, m, -OCH2CH2O-), 3.34 (3H, s, 7-CH3), 2.87/2.86 (1H, d, J = 15.1 Hz, -CHH-phenyl-), 2.82 (2H, m, -OCH2CH2O-), 2.81/2.80 (1H, d, J = 15.1 Hz, -CHH-phenyl-), 2.47-2.65 (3H, m, -NHCH2CH2CH2O- &17-CH2CHH-), 2.32-2.47 (4H, m, -NHCH2CH2CH2O- & -OCH2CH2O-), 2.05-2.27 (4H, m, 17-CHHCHH- & , -NHCH2CH2CH2O-), 2.13/2.11 (3H, d, J = 6.6 Hz, 31-CH3), 1.67-1.83 (3H, m, 17-CHHCH2- & -OCH2CH2(CH2)3CH3), 1.73 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.70/1.69 (3H, d, J = 7.2 Hz, 18-CH3), 1.29-1.47 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.23 (4H, m, -O(CH2)3(CH2)2CH3), 0.86 (2H, m, -NHCH2CH2CH2O-), 0.79/0.78 (3H, t, J = 6.9 Hz, -O(CH2)5CH3), −1.67 (1H, br s, core NH), −1.88 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.9, 173.5, 169.5, 169.3, 168.8, 166.7, 154.0, 149.0, 144.7, 138.90/138.89, 138.8/138.7, 137.8, 137.4, 136.3, 135.8, 134.99/134.97, 134.7, 134.3/134.1, 130.8/130.7, 130.0, 129.7, 128.63/128.62, 128.2, 102.35/102.33, 101.3, 99.7/99.6, 94.3, 93.2, 73.0/72.9, 70.4, 70.35, 70.33, 70.0, 69.72/69.69, 69.13, 69.13/69.11, 53.1, 52.1, 51.6, 49.29/49.26, 42.52/42.51, 39.1, 37.7, 37.50/37.48, 31.78/31.77, 31.1, 30.3/30.2, 29.7, 29.2, 28.0, 26.11/26.09, 25.0/24.9, 23.0, 22.59/22.57, 19.7, 17.8, 13.99/13.97, 12.0, 11.4, 11.2/11.1. Note: 1H impurity peaks were observed at 1.26 (likely grease CH2), and 0.87 ppm (likely grease CH3). Triphenylarsine (6.27 mg, 0.0205 mmol) and Pd2dba3 (9.37 mg, 0.0102 mmol) were added to a stirred solution of compound 42 (30 mg, 0.0256 mmol) and erlotinib 1 (15.09 mg, 0.0384 mmol) in dry THF (15 mL) and Et3N (2 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent evaporated and diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated to yield crude product 43. It was purified by preparative TLC plates and the pure compound was isolated in 65% yield (24 mg). MS (ESI) m/z: 1438.81 (M + H+). HRMS (ESI): calcd for C82H104N9O14 (M + H+) 1438.8131; found, 1438.8136. UV-vis (CH3OH, λmax, nm (abs)): 653 (5.3 x 104), 497 (1.4 x 104), 395 (1.6 x 105). 1H NMR (400 MHz, CDCl3, δ ppm): 9.95/9.93 (1H, s, meso 5-H), 9.67 (1H, s, meso 10-H), 8.768/8.765 (1H, s, meso 20-H), 8.63 (1H, s, pyrimidine H), 8.01 (1H, br s, amine NH), 7.83 (1H, m, phenyl H), 7.65 (1H, m, phenyl H), 7.40 (1H, s, quinazoline 5-H), 7.25-7.31 (3H, m, amide NH & 2 x phenyl H), 7.15-7.19 (2H, m, 2 x phenyl H), 7.07 (1H, s, quinazoline 8-H), 7.06 (1H, t, J = 7.6 Hz, phenyl H), 6.91 (1H, m, phenyl H), 5.962/5.958 (1H, q, J = 6.7 Hz, 31-H), 5.70/5.69 (1H, t, J ~ 5.6 Hz, amide NH), 5.58/5.57 (1H, d, J = 18.9 Hz, 15-CHH), 5.28/5.27 (1H, d, J = 18.9 Hz, 15-CHH), 4.45 (1H, q, J = 7.2 Hz, 18-H), 4.35 (1H, d, J ~ 10 Hz, 17-H), 4.05 (2H, m, -OCH2CH2OCH3), 4.02 (2H, m, -OCH2CH2OCH3), 3.94 (1H, m, -NHCHHCH2CH2O-), 3.80 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.753/3.750 (3H, s, -COOCH3), ~3.73 (1H, m, -NHCHHCH2CH2O-), ~3.67 (2H, m, -NH(CH2)2CH2O-), ~3.65 (2H, m, -OCH2CH2OCH3), ~3.64 (2H, m, -OCH2(CH2)4CH3), 3.606/3.603 (3H, s, -COOCH3), ~3.53 (2H, m, -OCH2CH2O-), 3.525 (3H, s, ring CH3), 3.46/3.45 (3H, s, ring CH3), 3.36-3.43 (4H, m, -OCH2CH2OCH3 & -OCH2CH2O-), 3.347/3.345 (3H, s, -OCH2CH2OCH3), 3.33 (3H, s, ring CH3), 3.17/3.16 (3H, s, -OCH2CH2OCH3), 3.10 (2H, m, -CH2-phenyl-), 2.89 (2H, m, -OCH2CH2O-), 2.67-2.85 (2H, m, -NHCH2CH2CH2O-), 2.42-2.61 (5H, m, 17-CH2CHH- & 2 x -OCH2CH2O-), 2.08-2.24 (2H, m, 17-CHHCHH-), 2.13/2.12 (3H, d, J = 6.6 Hz, 31-CH3), 2.07 (2H, m, -NHCH2CH2CH2O-), ~1.77 (1H, m, 17-CHHCH2-), ~1.75 (2H, m, -OCH2CH2(CH2)3CH3), 1.73 (3H, t, J = 7.7 Hz, 8-CH2CH3), 1.69/1.68 (3H, d, J = 7.2 Hz, 18-CH3), 1.29-1.47 (2H, m, -O(CH2)2CH2(CH2)2CH3), 1.23 (4H, m, -O(CH2)3(CH2)2CH3), 1.03 (2H, m, -NHCH2CH2CH2O-), 0.79/0.78 (3H, distorted t, J ~ 6.8 Hz, -O(CH2)5CH3), −1.66 (1H, s, core NH), −1.89 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.9, 173.53/173.52, 170.1, 169.5, 168.91/168.90, 166.61/166.60, 156.4, 154.4, 154.1, 153.5, 149.0, 148.8, 147.5, 144.7, 139.1, 138.89/138.88, 138.8/138.7, 136.2, 135.4, 134.88/134.86, 134.6, 134.3/134.1, 132.2, 130.9/130.7, 130.1, 129.6, 129.1, 128.8, 128.6, 128.4/128.3, 126.9, 124.7, 123.6, 123.5, 122.1, 109.3, 108.6, 102.7, 102.23/102.21, 101.3, 99.63/99.57, 93.2, 89.6, 89.2, 72.9/72.8, 70.6, 70.35, 70.31, 70.29, 70.2, 70.0, 69.69/69.67, 69.31/69.29, 69.22/69.21, 69.0, 68.1, 59.1, 59.0, 53.0, 52.1, 51.6, 49.24/49.21, 43.1, 39.1, 37.74/37.73, 37.71, 31.75/31.74, 31.10/31.08, 30.23/30.21, 29.7, 29.2, 28.1, 26.09/26.07, 25.0/24.9, 23.0, 22.57/22.55, 19.7, 17.7, 13.97/13.95, 12.0, 11.39/11.38, 11.12/11.08. Note: 1H impurity peaks were observed at 1.27 (likely grease CH2), and 0.89 ppm (likely grease CH3).
Synthesis of Conjugate (50):
Methyl pheophorbide-a 44 (200 mg, 0.330 mmol), and amine 45 (95.8 mg, 0.66 mmol) were dissolved in 12 mL dry THF. The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent was evaporated and the crude product was dissolved in 15 mL 70% TFA/DCM. The reaction mixture was stirred for 2 h and the solvent was evaporated and diluted with dichloromethane (20 mL), adjusted the pH to 6.6 by using saturated NaHCO3 solution. The crude was purified by silica column by using 5% methanol in dichloromethane to obtain pure product 46 with 71% (156 mg) over yield. MS (ESI) m/z: 667.36 (M + H+).
Compound 46 (120 mg, 0.18 mmol), was reacted with diglycolic anhydride (41.78 mg, 0.36 mmol) in 5 mL DMF. The reaction mixture was stirred for 5 hours to yield the intermediate carboxylic acid, which was activated twice with 0.5 M CDI (58. 36 mg, 0.36 mmol) for 1 hour, followed by agitating with 0.5 M HOBt (48.64 mg, 0.36 mmol) and di-(3-aminopropyl) digol (396.47 mg, 1.8 mmol) for 5 hours to afford PS-linker-NH2. The crude product was purified by silica column by using 20% methanol in dichloromethane to obtain pure product 47 with 59% (104.4 mg) yield. MS (ESI) m/z: 983.56 (M + H+).. 1H NMR (400 MHz, 80:20 CDCl3/CD3OD, δ ppm): 9.68 (1H, s, meso 10-H), 9.61 (1H, s, meso 5-H), 8.85 (1H, s, meso 20-H), 8.04 (1H, dd, J = 17.8, 11.5 Hz, 3-CH=CH2), 6.31 (1H, dd, J = 17.8, 1.3 Hz, 3-CH=CHH), 6.13 (1H, dd, J = 11.5, 1.3 Hz, 3-CH=CHH), 5.51 (1H, d, J = 19.0 Hz, 15-CHH), 5.28 (1H, d, J = 19.0 Hz, 15-CHH), 4.51 (1H, q, J = 7.2 Hz, 18-H), 4.36 (1H, m, 17-H), 4.14 (2H, s, -C(=O)CH2OCH2C(=O)-), 4.03 (2H, s, -C(=O)CH2OCH2C(=O)-), ~3.95 (1H, m, -NHCHHCH2NH-), ~3.87 (1H, m, -NHCHHCH2NH-), 3.78 (3H, s, -COOCH3), ~3.76 (2H, q, J = 7.5 Hz, 8-CH2CH3), ~3.73 (2H, m, -NHCH2CH2NH-), 3.64 (3H, s, -COOCH3), 3.49 (3H, s, ring CH3), 3.46 (3H, s, ring CH3), 3.28 (3H, s, ring CH3), 2.92 (2H, m, -NHCH2CH2CH2O-), 2.83 (2H, m, -OCH2CH2O-), 2.65 (1H, m, 17-CH2CHH-), 2.51 (2H, m, -NHCH2CH2CH2O-), 2.50 (2H, m, -OCH2CH2O-), 2.36 (2H, m, -NHCH2CH2CH2O-), ~2.27 (1H, m, 17-CH2CHH-), ~2.21 (1H, m, 17-CHHCH2-), ~2.15 (1H, m, -OCHHCH2O-), ~2.09 (1H, m, -OCHHCH2O-), ~2.08 (1H, m, -OCHHCH2O-), ~2.02 (1H, m, -NHCH2CH2CHHO-), ~1.97 (1H, m, -OCHHCH2O-), ~1.88 (1H, m, -NHCH2CH2CHHO-), ~1.73 (1H, m, 17-CHHCH2-), 1.703 (3H, t, J = 7.5 Hz, 8-CH2CH3), 1.695 (3H, d, J = 7.1 Hz, 18-CH3), ~1.25 (2H, m, -NHCH2CH2CH2O-), ~0.80 (1H, m, -NHCH2CHHCH2O-), ~0.68 (1H, m, -NHCH2CHHCH2O-); 13C NMR (100 MHz, 80:20 CDCl3/CD3OD, δ ppm): 174.5, 174.4, 172.4, 170.4, 170.0, 169.8, 167.6, 154.9, 149.5, 145.5, 139.5, 136.9, 135.4, 135.3, 135.1, 134.8, 131.1, 129.8, 129.5, 128.0, 122.5, 102.6, 101.8, 99.1, 94.3, 70.63, 70.57, 69.7, 69.3, 69.1, 68.9, 68.7, 67.8, 53.5, 52.6, 52.0, 49.6, 41.3, 40.1, 39.6, 37.9, 36.2, 31.4, 30.1, 28.7, 26.5, 23.3, 19.9, 17.9, 12.3, 11.9, 11.4. Note: Amide, amine and core NH protons were not observed, likely due to chemical exchange. 1H impurity peaks were observed at 1.23 (likely grease CH2), and 0.85 ppm (likely grease CH3). The 13C peak at 30.0 ppm is likely grease (CH2) impurity.
To a solution of compound 47 (80 mg, 0.0814 mmol) and 3-idobenzyl acetic acid 48 (32 mg, 0.122 mmol) in 12 mL of dry DMF, (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 72.1 mg, 0.163 mmol) and 3 drops triethylamine were added. The reaction mixture was stirred at room temperature under Ar atmosphere overnight. It was then diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 6% methanol in dichloromethane to obtain pure product 49 with 79% (78.9 mg) yield. MS (ESI) m/z: 1227.51 (M + H+). NMR (400 MHz, CDCl3, δ ppm): 9.57 (1H, s, meso 10-H), 9.46 (1H, s, meso 5-H), 8.79 (1H, s, meso 20-H), 8.21 (1H, t, J = 4.2 Hz, amide NH), 7.86 (1H, dd, J = 17.8, 11.5 Hz, 3-CH=CH2), 7.44 (1H, t, J = 5.7 Hz, amide NH), 7.373 (1H, m, phenyl H), 7.368 (1H, m, phenyl H), 7.00 (1H, bs, amide NH), 6.90 (1H, d, J = 7.8 Hz, phenyl H), 6.75 (1H, dd, J = 8.2, 7.7 Hz, phenyl H), 6.12 (1H, d, J = 18.0 Hz, 3-CH=CHH), 6.07 (1H, t, J ~ 5.0 Hz, amide NH), 5.96 (1H, d, J = 11.5 Hz, 3-CH=CHH), 5.32 (1H, d, J = 18.8 Hz, 15-CHH), 5.17 (1H, d, J = 18.8 Hz, 15-CHH), 4.49 (1H, q, J = 7.2 Hz, 18-H), 4.35 (1H, m, 17-H), 4.06, 4.04 (2H, ABq, JAB = 14.8 Hz, -C(=O)CH2OCH2C(=O)-), 3.98 (2H, s, -C(=O)CH2OCH2C(=O)-), 3.71 (2H, m, 8-CH2CH3), 3.70 (3H, s, -COOCH3), 3.67 (3H, s, -COOCH3), 3.59 (1H, br s, -NHCHHCH2NH-), 3.49 (1H, br s, -NHCHHCH2NH-), 3.358 (1H, br s, -NHCH2CHHNH-), 3.356 (3H, s, 2-CH3), 3.29 (1H, br s, -NHCH2CHHNH-), 3.24 (3H, s, 12-CH3), 3.12 (3H, s, 7-CH3), 3.06, 3.03 (2H, ABq, JAB = 15.1 Hz, -C(=O)CH2-phenyl-I), 2.99 (2H, t, J = 5.8 Hz, -NHCH2CH2CH2O-), 2.91 (2H, m, -NHCH2CH2CH2O-), 2.90 (2H, m, -OCH2CH2O-), 2.74 (2H, m, -OCH2CH2O-), ~2.65 (1H, m, 17-CH2CHH-), 2.59 (2H, q, J = 6.5 Hz, -NHCH2CH2CH2O-), 2.41 (2H, m, -OCH2CH2O-), ~2.30 (1H, m, 17-CH2CHH-), 2.25 (1H, m, 17-CHHCH2-), ~2.22 (2H, m, -OCH2CH2O-), ~2.23, ~2.14 (2H, 2 x m, -NHCH2CH2CH2O-), 1.81 (1H, m, 17-CHHCH2-), 1.70 (3H, d, J = 7.2 Hz, 18-CH3), 1.68 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.30 (2H, m, -NHCH2CH2CH2O-), 0.79 (2H, m, -NHCH2CH2CH2O-), −1.66 (1H, s, core NH), −1.85 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.6, 173.5, 171.5, 170.1, 169.2, 169.1, 168.2, 166.8, 154.4, 149.0, 144.8, 139.0, 137.9, 137.5, 136.3, 135.8, 134.9, 134.6, 134.5, 134.3, 130.4, 130.1, 129.4, 129.0, 128.3, 127.2, 121.7, 102.1, 101.4, 98.7, 94.3, 93.8, 70.7, 70.6, 69.5, 69.4, 69.30, 69.27, 68.8, 68.1, 53.1, 52.2, 51.7, 49.2, 42.7, 41.0, 39.6, 37.8, 37.4, 36.3, 31.1, 29.7, 28.32, 28.30, 23.0, 19.6, 17.7, 12.0, 11.5, 11.2. Note: 1H impurity peaks were observed at 1.27 (likely grease CH2), and 0.86 ppm (likely grease CH3). The 13C peak at 29.68 ppm is likely grease (CH2) impurity.
Triphenylarsine (9.97 mg, 0.0326 mmol) and Pd2dba3 (14.9 mg, 0.017 mmol) were added to a stirred solution of compound 49 (50 mg, 0.0407 mmol) and erlotinib 1 (24 mg, 0.0611 mmol) in dry THF (15 mL) and Et3N (2 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent evaporated and diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated to yield crude product 50. The crude 50 was purified by preparative TLC plates in 63% yield (38.32 mg). MS (ESI) m/z: 1492.7621 (M + H+). HRMS (ESI): Calcd for C83H102N11O5 (M + H+) 1492.7621; found, 1492.7626. UV-vis (CH3OH, λmax, nm (abs)): 663 (5.25 x 104), 610 (5.0 x 103), 498 (1.4 x 104), 400 (1.6 x 105), 350 (5.4 x 104). 1H NMR (400 MHz, CDCl3, δ ppm): 9.58 (1H, s, meso 10-H), 9.54 (1H, s, meso 5-H), 8.77 (1H, s, meso 20-H), 8.60 (1H, s, pyrimidine H), 8.31 (1H, br t, J = 4.6 Hz, -NHCH2CH2NH-), 8.17 (1H, s, amine NH), 7.98 (1H, dd, J = 17.8, 11.6 Hz, 3-CH=CH2), 7.88 (1H, m, phenyl H), 7.65 (1H, m, phenyl H), 7.50 (1H, br t, J = 5.7 Hz, -NHCH2CH2CH2O-), 7.45 (1H, s, quinazoline 5-H), 7.40 (1H, br t, J ~ 5.5 Hz, -NHCH2CH2NH-), 7.29 (1H, s, phenyl H), 7.22-7.28 (2H, m, 2 x phenyl H), 7.17 (1H, d, J = 7.7 Hz, phenyl H), 7.10 (1H, s, quinazoline 8-H), 7.06 (1H, t, J = 7.7 Hz, phenyl H), 6.97 (1H, d, J = 7.7 Hz, phenyl H), 6.33 (1H, t, J = 5.4 Hz, -NHCH2CH2CH2O-), 6.26 (1H, d, J = 17.9 Hz, 3-CH=CHH), 6.07 (1H, dd, J = 11.6, 1.0 Hz, 3-CH=CHH), 5.42 (1H, d, J = 18.8 Hz, 15-CHH), 5.21 (1H, d, J = 18.8 Hz, 15-CHH), 4.44 (1H, q, J = 7.2 Hz, 18-H), 4.32 (1H, dd, J = 10.1, ~1.4 Hz, 17-H), 4.11 (4H, m, 2 x -OCH2CH2OCH3), 4.09, 4.08 (2H, ABq, JAB = 15.1 Hz, -C(=O)CH2OCH2C(=O)-), 3.98 (2H, s, -C(=O)CH2OCH2C(=O)-), ~3.7-3.8 (6H, m, -NHCH2CH2NH- & 8-CH2CH3 & -OCH2CH2OCH3), 3.70 (3H, s, -COOCH3), 3.622 (3H, s, -COOCH3), 3.621 (2H, m, -OCH2CH2OCH3), 3.57 (2H, m, -NHCH2CH2NH-), 3.43 (3H, s, ring CH3), 3.39 (3H, s, -OCH2CH2OCH3), 3.36 (3H, s, ring CH3), 3.28 (3H, s, -OCH2CH2OCH3), 3.22 (3H, s, ring CH3), 3.20 (2H, s, -CH2-phenyl-I), 2.95-3.03 (4H, m, -NHCH2CH2CH2O- & -OCH2CH2O-), 2.93 (2H, m, -NHCH2CH2CH2O-), 2.88 (2H, m, -OCH2CH2O-), 2.70 (2H, q, J ~ 6.5 Hz, -NHCH2CH2CH2O-), 2.64 (2H, t, J ~ 4.6 Hz, -OCH2CH2O-), ~2.58 (1H, m, 17-CH2CHH-), 2.45 (2H, m, -OCH2CH2O-), ~2.37 (2H, m, -NHCH2CH2CH2O-), 2.13-2.27 (2H, m, 17-CHHCHH-), ~1.74 (1H, m, 17-CHHCH2-), 1.69 (3H, t, J = 7.7 Hz, 8-CH2CH3), 1.68 (3H, d, J = 7.1 Hz, 18-CH3), 1.29 (2H, m, -NHCH2CH2CH2O-), 0.93 (2H, m, -NHCH2CH2CH2O-), −1.63 (1H, s, core NH), -1.85 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.8, 173.5, 171.4, 170.6, 169.5, 169.1, 168.5, 166.8, 156.4, 154.5, 154.4, 153.6, 149.1, 148.7, 147.5, 144.9, 139.3, 139.1, 136.2, 135.6, 135.0, 134.7, 134.6, 134.4, 132.2, 130.4, 130.0, 129.5, 129.23, 129.18, 128.9, 128.6, 127.3, 126.7, 124.4, 123.4, 123.3, 122.0, 121.8, 109.4, 108.7, 103.3, 102.0, 101.4, 98.8, 93.8, 89.6, 89.2, 70.8, 70.75, 70.73, 70.4, 69.7, 69.50, 69.47, 69.4, 69.1, 69.0, 68.3, 68.2, 59.2, 59.1, 53.0, 52.2, 51.7, 49.2, 43.1, 40.8, 40.0, 37.8, 37.6, 36.5, 31.1, 29.7, 28.4, 28.3, 23.0, 19.6, 17.7, 12.1, 11.7, 11.3. Note: 1H impurity peaks were observed at 1.26 (likely grease CH2), and 0.86 ppm (likely grease CH3).
Synthesis of Conjugate (53):
Chlorin e6 51 (100 mg, 0.168 mmol) was dissolved in dry CH2Cl2. 35 mg DCC and 4.5 mg DMAP were added and allowed to stir until completely dissolved. After 3 h, 42 mg 3-idobenzyl amine 6 and 15 uL DIEA were mixed in CH2Cl2 and added to the reaction mixture. The reaction was allowed to stir overnight, and then diluted with CH2Cl2 and then washed with 5% aqueous citric acid, followed by washing with brine and water. It was dried over anhydrous Na2SO4 and then evaporated. The residue was dissolved in CH2Cl2 and treated with ethereal diazomethane. The residue was dissolved in 2% MeOH/CH2Cl2 and purified via silica gel column chromatography using the same mobile phase to yield compound 52 with 51% yield (71.8 mg). MS (ESI) m/z: 840.28 (M + H+). For the characterization details, including the 1H and 13C- NMR spectra please see the Supporting Material Information. 1H NMR (400 MHz, CDCl3, δ ppm): 9.73 (1H, s, meso 10-H), 9.57 (1H, s, meso 5-H), 8.76 (1H, s, meso 20-H), 8.04 (1H, dd, J = 17.9, 11.5 Hz, -CH=CH2), 7.42 (1H, br s, phenyl H), 7.38 (1H, dt, J = 7.8, ~1.3 Hz, phenyl H), 6.98 (1H, d, J = 7.7 Hz, phenyl H), 6.77 (1H, t, J = 7.8 Hz, phenyl H), 6.35 (1H, dd, J = 17.9, 1.4 Hz, -CH=CHH), 6.15 (1H, dd, J = 11.6, 1.4 Hz, -CH=CHH), 5.92 (1H, br s, amide NH), 5.30 (1H, d, J = 18.6 Hz, 15-CHH-), 5.19 (1H, br d, J = 18.6 Hz, 15-CHH-), 4.38-4.49 (3H, m, 18-H & 17-H & -NHCHH-phenyl-), 4.26 (3H, s, -COOCH3), 4.18 (1H, dd, J = 15.0, 5.7 Hz, -NHCHH-phenyl-), 3.78 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.62 (3H, s, ring CH3), 3.49 (3H, br s, -COOCH3), 3.47 (3H, s, ring CH3), 3.29 (3H, s, ring CH3), ~2.57 (1H, m, 17-CH2CHH-), ~2.26 (1H, m, 17-CHHCH2-), ~2.22 (1H, m, 17-CH2CHH-), ~1.80 (1H, m, 17-CHHCH2-), 1.73 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.60 (3H, d, J = 7.2 Hz, 18-CH3), −1.36 (1H, br s, core NH), −1.50 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.2, 172.2, 169.8, 169.4, 166.9, 155.1, 149.2, 145.2, 140.6, 139.7, 136.5, 136.3, 136.20, 136.15, 135.5, 135.0, 134.7, 130.8, 129.9, 129.5, 129.2, 127.0, 124.0, 122.0, 102.4, 101.5, 99.0, 94.2, 93.7, 53.4, 52.9, 51.7, 49.1, 42.9, 41.0, 31.2, 29.8, 23.1, 19.6, 17.7, 12.4, 12.1, 11.3. Note: 1H impurity peaks at 1.27 (likely grease CH2), 0.89 ppm (likely grease CH3) were observed.
Triphenylarsine (11.69 mg, 0.039 mmol) and Pd2dba3 (17.48 mg, 0.0191 mmol) were added to a stirred solution of compound 52 (40 mg, 0.048 mmol) and erlotinib 1 (28.32 mg, 0.072 mmol) in dry THF (12 mL) and Et3N (2 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. The solvent evaporated and diluted with dichloromethane (20 mL), washed with water (3 x 20 mL), dried over anhydrous sodium sulfate and concentrated to yield crude product 53, which was purified by preparative TLC plates with 45% yield (23.7 mg). MS (ESI) m/z: 1105.52 (M + H+). HRMS (ESI): calcd for C65H69N8O9 (M + H+) 1105.5242; found, 1105.5249. UV-vis (CH3OH, λmax, nm (ε)): 663 (3.6 x 104), 610 (4.3 x 103), 500 (1.0 x 104), 400 (1.2 x 105), 349 (4.3 x 104). For the characterization details, including the 1H and 13C- NMR spectra please see the Supporting Material Information. 1H NMR (400 MHz, CDCl3, δ ppm): 9.69 (1H, s, meso 10-H), 9.54 (1H, s, meso 5-H), 8.71 (1H, s, meso 20-H), 8.63 (1H, s, pyrimidine H), 8.01 (1H, dd, J = 17.8, 11.6 Hz, 3-CH=CH2), 7.86 (1H, dd, J = 8.1, ~1.3 Hz, phenyl H), 7.66 (1H, s, phenyl H), 7.43 (1H, br s, amine NH), 7.33 (1H, t, J ~ 7.9 Hz, phenyl H), 7.32 (1H, s, phenyl H), 7.23 (1H, d, J = 7.6 Hz, phenyl H), 7.20 (2H, s, quinazoline 5-H & quinazoline 8-H), 7.17 (1H, d, J = 7.7 Hz, phenyl H), 7.03 (1H, t, J = 7.6 Hz, phenyl H), 6.96 (1H, d, J = 7.7 Hz, phenyl H), 6.32 (1H, dd, J = 17.9, 1.4 Hz, 3-CH=CHH), 6.12 (1H, dd, J = 11.6, 1.4 Hz, 3-CH=CHH), 6.05 (1H, br t, J ~ 5.8 Hz, amide NH), 5.31 (1H, d, J = 18.6 Hz, 15-CHH), 5.17 (1H, br d, J = 18.6 Hz, 15-CHH), 4.59 (1H, dd, J = 14.7, 6.0 Hz, -CHH-phenyl-), 4.42 (1H, m, 17-H), 4.40 (1H, q, J = 7.3 Hz, 18-H), 4.26 (2H, m, -OCH2CH2OCH3), 4.24 (3H, s, -COOCH3), 4.18 (2H, m, -OCH2CH2OCH3), 4.14 (1H, dd, J ~ 15.1, 5.5 Hz, -CHH-phenyl-), 3.83 (2H, m, -OCH2CH2OCH3), 3.75 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.74 (2H, m, OCH2CHz 2OCH3), 3.57 (6H, br s, 12-CH3 & -COOCH3), 3.46 (3H, s, -OCH2CH2OCH3), 3.42 (3H, s, 2-CH3), 3.40 (3H, s, -OCH2CH2OCH3), 3.26 (3H, s, 7-CH3), 2.60 (1H, m, 1H of 17-CH2CH2-), 2.28 (1H, m, 1H of 17-CH2CH2-), 2.25 (1H, m, 1H of 17-CH2CH2-), 1.72 (1H, m, 1H of 17-CH2CH2-), 1.70 (3H, t, J = 7.6 Hz, 8-CH2CH3), 1.56 (3H, d, J = 7.2 Hz, 18-CH3), −1.41 (1H, br s, core NH), −1.55 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 173.5, 172.2, 169.8, 169.4, 167.0, 156.1, 155.1, 154.6, 153.6, 149.1, 148.9, 147.6, 145.2, 139.7, 139.0, 138.6, 136.4, 136.2, 135.6, 135.0, 134.8, 131.0, 130.8, 130.3, 129.5, 129.2, 129.0, 128.3, 127.7, 126.9, 124.0, 123.9, 123.8, 123.2, 122.0, 121.5, 109.2, 108.9, 102.5, 102.3, 101.5, 99.0, 93.8, 89.3, 89.2, 70.9, 70.5, 69.3, 68.3, 59.28, 59.27, 53.4, 52.9, 51.8, 49.1, 43.3, 41.1, 31.5, 29.7, 23.0, 19.6, 17.7, 12.3, 12.1, 11.3. Note: 1H impurity peaks were observed at 1.27 (likely grease CH2), and 0.89 ppm (likely grease CH3). Small 13C impurity peaks were observed at 31.9, 29.3, 22.7, and 14.1 ppm.
Cell biological analysis of PS uptake and cell type specificity
Human head & neck tumor cells were isolated from surgical specimens of primary tumor tissues provided by RPCCC Tissue Procurement Services under the terms of an IRB-approved protocol. Each sample received was labeled with a laboratory code (HN-number). Tissues were minced and mildly digested with trypsin and plated unto a collagen-1 matrix in tissue culture dishes. Proliferating tumor epithelial (T-EC) and stromal cells (T-Fb) were obtained by proliferation under selective media conditions as outlined prevously for human lung tumor cells47. Similarly, long-term proliferating T-EC (HNT1) were isolated from a PDX tongue cancer tissue. The established FaDu hypopharyngeal cell line had been purchased from ATCCC and subjected to single cell cloning (FaDu-49). To identify cell type-specific PS binding, reconstituted co-cultures T -EC and T-Fb (ratio 1:200) were established on collagen-1 matrix and grown to confluence in RPMI containing 10% FBS. In few cases, the experiments included CFSE-stained T-Fb for visualization of the stromal cell population in co-cultures. Monotypic and co-cultures were incubated with RPMI containing PS in the range of 0 to 3000 M. After various time points, cultures ware washed with PS-free medium containing 10% FBS and depending on the experimental setting, cultured for additional time periods (= chase). Cellular level of PS was recorded by imaging with an inverted fluorescence microscope (Zeiss or Nikon). The level of cell-associated fluorescent signal (pixel) was normalized to recording time (sec) and cell number (1 x 106) as described (Tracy et al). Photoreaction involved exposure of the culture dish at 37°C to 665-nm light of a diode laser at 6 mW/cm2 fluence rate to a total of 3J/cm2 fluence. Cells were either immediately extracted by lysis in RIPA buffer or cultured for additional 24 h in growth medium. Surviving cells were determined after tryspin release from culture support based on trypan blue exclusion. Proteins (STAT3, EGFR) in the cell lysates separated on SDS polyacrylamide gels and analyzed by immunoblotting as described previously48. The immune signals for STAT3 were quantified by scanning of the blots and expressed as percentage of conversion from monomeric to homodimeric forms.
In vitro photosensitizing efficacy of PS by MTT assay:
UMUC3 or FaDu cells were grown in T75 flasks in media supplemented with 10% FBS, 1% penicillin streptomycin. To begin the Cytotoxicity assay, cells were harvested from the T75 flasks, counted, and were plated in 96 well plates between 0.01x106 to 0.05x106 cells/ml and 100 μl per well. Photosensitizer was added after the cells have adhered to the plate (24 h after plating). The top photosensitizer dose on each plate was 1600 nM, with a 2-fold dilution for each subsequent dose. Plates were exposed to either no light (0 J/cm2) or 1 J/cm2 at 565 mW and a spot size of 15 cm. After 48 h, 20 μl of MTT was added to each well and 3 h later MTT was added to the cells. The metabolized MTT was solubilized in DMSO and read using a plate reader at 570 nm. Plates were analyzed using Graphpad Prism 8 to determine the IC50.
In vivo tumor uptake and PDT efficacy:
SCID mice bearing either UMUC3 or FaDu tumors at the flank/shoulder were subjected for whole body fluorescence imaging at various timepoints after PS injection intravenously. The tumor, liver and skin uptakes were measured by IVIS (Perkin Elmer) in vivo imaging system and pharmacokinetic profiles of the PSs was evaluated. Images were collected at various time points, and average radiant efficiency of PS in tumors was determined.
For evaluating in vivo PDT efficacy, the tumored mice restrained in acrylic holders after injecting the PS. The tumors were exposed to laser light at the longest wavelength absorption and maximal uptake time of the PS. The total light dose was 135 J/cm2 for a period of 30 min, with a fluence rate of 75mW/cm2. The tumor regrowth was measured daily, and as per approved protocol, the mice with tumor regrowth >400 mm3 were euthanized.
Supplementary Material
Scheme 3:

Erlotinib moiety conjugated either at p- position of the benzyloxy group having a chiral center at position 3(1’)- or at the m-position without a chiral center at position 3(1’) of the conjugate.
ACKNOWLEDFMENTS
The authors are highly thankful to Photolitec LLC, Buffalo, NY (RKP), Roswell Park Alliance Foundation (Pandey, Guru, Pawel, Koya), and NIH (PO1, CA055791, HB and RKP) for the financial support. A partial support from the shared resources of the RPCI support grant (P30CA16056) is also appreciated. The help rendered by Dr. Mykhaylo Dukh in providing the starting material for the synthesis of some of the final products discussed in this article is highly appreciated.
ABBREVIATIONS
- PS
photosensitizers
- PDT
photodynamic therapy
- EGFR
epidermal growth factor receptor
- NIR
near-infrared
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS publications website at DOI: NMR (1H and 13C-) details and spectra of new compounds.
Dr. Cheruku, Mr. Cacaccio, Dr. Durrani ,Dr. Siters, Mr. Missert, Dr. Guru, Dr. Baumann and Dr. Pandey listed as authors in this manuscript are also the inventors of a patent application submitted to the United States Patent Office.
REFERENCES
- (1).Juarranz A; Jaen P; Sanz-Rodriguez F; Cuevas J and Goncalez S Photodynamic therapy of cancer: Basic principles and applications, Clinical and Translational Oncology, 2008, 10, 148–154. [DOI] [PubMed] [Google Scholar]
- (2).Kessel D Photodynamic therapy: A brief history, J. Clin. Med, 2019, 8, 1581, and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hiramatsu R; Kawabata S; Tanaka H; Sakurai Y; Suzuki M; Ono K; Miyatake S-I; Kuroiwa T; Hao E and Vicente MGH, Tetrakis(p-carboranylthio-tetrafluorophenyl) chlorin (TPFC): Application for photodynamic therapy and boron neutron capture therapy. J. Pharmaceutical Sci, 2015, 104, 962–970 [DOI] [PubMed] [Google Scholar]
- (4).Lo P-C; Rodriguez-Morgade MS; Pandey RK; Ng DKP; Torres T and Dumoulin F The unique features and promises of phthalocyanines as advanced photosensitizers for photodynamic therapy of cancer. Chem. Soc. Rev, 2020, 49, 1041–1056. [DOI] [PubMed] [Google Scholar]
- (5).Shaker YM; Sweed AMK; Moylan C; Rogers L; Ryan AA; Pettidemange R and Senge MO, Current developments in using MESO-(TETRA) substituted porphyrins in PDT in HANDBOOK OF PHOTODYNAMIC THERAPY, Updates on Recent Applications of Porphyrin-Based Compound (Eds: Pandey RK; Kessel D and Dougherty TJ), World Scientific, New Jersey, 2016, pp 95–149. [Google Scholar]
- (6).Ethirajan M; Chen Y; Joshi P and Pandey RK The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev, 2011, 40, 340–362. [DOI] [PubMed] [Google Scholar]
- (7).Abrahamse H; Hamblin MR; New photosensitizers for photodynamic therapy, Biochem. J, 2016, 473, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Olivo M; Bhuvaneshwari R; Lucky SS; Dendukuri N and Thong PS-P , Targeted therapy of cancer using photodynamic therapy in combination with multi-faceted anti-tumor modalities. Pharmaceuticals (Basel), 2010, 3, 1507–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jeong H; Huh M; Lee SJ; Koo H; Kwon LC; Jeong SY and Kim K, Photosensitizers -conjugated with human serum albumin nanoparticles for effective photodynamic therapy. Theranostics, 2011, 1, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li C; Zhang W; Liu S; Hu X and Xie Z Mitochondria-rargeting organic nanoparticles for enhanced photodynamic/photothermal therapy. ACS Appl Mater. Interfaces, 2020, 12, 300077–30084. [DOI] [PubMed] [Google Scholar]
- (11).Zou Y; Li M; Xiong T; Zhao X; Du J; Fan J and Peng X A single molecule drug targeting photosensitizer for enhanced breast cancer photothermal therapy, Small, 2020, 16, 1907677. [DOI] [PubMed] [Google Scholar]
- (12).Mamblin MR; Miller JL; Rizvi I; Ortel B; Maytin EV and Hasan T, Pegylation of a chlorin e6 polymer conjugate increases tumor targeting of photosensitizer, Cancer Research, 2001, 61, 7155–7162. [PubMed] [Google Scholar]
- (13).Yoon Ii; Li JZ and Shim YK Advances in photosensitizers and light delivery for photodynamic therapy, Clin. Endosc 2013, 46, 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cai Y; Liang P; Tang Q; Si W; Chen P; Zhang Q; Song X Diketopyrrole-based photosensitizers conjugated with chemotherapeutic agents for multimodel tumor therapy, ACS Appl. Mater. Interfaces, 2017, 9, 30398–30405. [DOI] [PubMed] [Google Scholar]
- (15).Sandland J and Boyle RW, Photosensitizer antibody-drug conjugates: Past, present and future. Bioconjugate Chem, 2019, 30, 975–993, and references therein [DOI] [PubMed] [Google Scholar]
- (16).Chen X; Foster DA and Drain CM, Efficient synthesis and photodynamic activity of porphyrin saccharide conjugates: Targeting and incapacitating cancer cells. Biochemistry, 2004, 43, 10918–10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zheng G; Graham A; Shibata M; Missert JA; Oseroff AR; Dougherty TJ and Pandey RK Synthesis of b-galactose conjugated chlorins derived by enyne metathesis as galectin-specific photosensitizers for photodynamic therapy. J. Org. Chem, 2001, 66, 8709–8716. [DOI] [PubMed] [Google Scholar]
- (18).Zheng X; Morgan J; Pandey SK; Chen Y; Tracy E; Baumann H; Missert JR; Batt C; Jackson J; Bellnier D; Henderson BW and Pandey RK Conjugation of 2-(1’-hexyloxyethyl)-2-devinylpyropheophorbide-a (HPPH) to carbohydrates changes its subcellular distribution and enhances photodynamic activity in vivo. J. Med. Chem, 2009, 52, 4306–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Morgan J; Jackson JD; Zheng X; Pandey SK and Pandey RK Substrate affinity of photosensitizers derived from chlorophyll-a: The ABCG2 transporter affects the phototoxic response of the side population stem cell-like cancer cells to photodynamic therapy, Molecular Pharmaceutics, 2010, 7, 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pereira PMR; Silva S; Cavaleiro JAS; Rebeiro AAF; Tome JPC and Fernandes R, Galactodendritic phthalocyanine targets carbohydrate-binding proteins enhancing photodynamic therapy. PLoS One, 2014, 9, e95529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu Q; Pang M; Tan S; Wang J; Chen Q; Wang K; Wu W; Hong Z, Potent peptide-conjugated silicon phthalocyanines for tumor photodynamic therapy., J. Cancer, 2018, 19, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Srivastan A; Ethirajan E; Pandey SK; Dubey S; Zheng X; Liu T-H; Shibata M; Missert J; Morgan J and Pandey RK, Conjugation of cRGD peptide to chlorophyll-a based photosensitizer (HPPH) alters its pharmacokinetics with enhanced tumor-imaging and photosensitizing efficacy. Mol. Pharmaceutics, 2011, 8, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Miyoshi Y; Kadono M; Okazaki S; Nishimura A; Kitamatsu M; Watanabe K and Ohtsuki T, Endosomal escape of peptide-photosensitizer conjugates is affected by amino acid sequences near the photosensitizer, Bioconjugate Chem, 2020, 31, 916–922 [DOI] [PubMed] [Google Scholar]
- (24).Zhao N; Williams TM; Zhou Z; Fronczek FR; Vazquez M; Jois SD and Vicente MGH, Synthesis of BODIPY-peptide conjugates for fluorescence labeling of EGFR over-expressing cells. Bioconjugate. Chem, 2017, 28, 1566–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Siddiqui MR; Railkar R; Sanford T; Crooks DR; Eckhaus MA; Haines D; Choyke PL; Kobayashi H and Agarwal PK Targeting epidermal frowth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) expressing bladder cancer using combination photoimmunotherapy (PIT), Nature: SCIENTIFIC REPORTS, 2019, 9, 2084/ 10.1038/s41598-019-38575-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cadsson J; Wester K; Torre MDL; Malmstrom P-U; Gadmark T;EGFR-expression in primary urinary bladder cancer and corresponding metastasis and the relation to HER-2 expression. On the possibility to target these receptors with radionuclides, Radiol. Oncol, 2015, 49, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zimmermann M; Zouhair A; Azria D; Ozsahin M The epidermal growth factor receptor (EGFR) in head and neck cancer: its role and treatment implications. Radiation Oncology. 2006, 1, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nomura M; Shigemstsu H; Li L; Suzuki M; Takahashi, Estess P; Siegelman M; Feng Z; Kato H; Marchetij A; Shay JW; Spitz MR; Wistuba JI; Hohn DM and Cazdar AF polymorphisms, mutations, and amplification of the EGFR in non- small cell lung cancers. PLoS Med. 2007, 4, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gao SP; Mark KG; Leslie K; Pao W; Moloi N; Gerald WL; Travis WD; Mommann W; Veach D; Clarkson B; Bromberg JF, Mutations in the EGFR kinase domain mediate STAT3 activation vis IL-6 production in lung adenocarcinomas. J. Clin. Invest 2007, 117, 3846–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Henderson BW, Bellnier DA, Greco WR, Sharma A, Pandey RK, Vaughan LA, Weishaupt KR, Dougherty TJ An in vivo quantitative structure-activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy, Cancer Research, 1997, 57, 4000–4007. [PubMed] [Google Scholar]
- (31).Pandey RK; Goswami LN; Chen Y; Gryshuk A; Missert JR; Oseroff A and Dougherty TJ, Nature: A rich source for developing multifunctional agents: Tumor imaging and photodynamic therapy, Lasers Surg. Med, 2006, 28, 445–467. [DOI] [PubMed] [Google Scholar]
- (32).Miller JD; Baron ED; Scull H; Hsia A; Berlin JC; McCormick T; Colussi V; Kenney MF; Cooper KD and Olenick NL Photodynamic therapy with the phthalocyanine photosensitizer PC-4., Toxicol. Appl. Pharmacol, 2007, 224, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rigual N; Shafirstein G; Cooper MT; Baumann H; Bellnier DA; Sumar U; Tracy EC; Rohrbach DJ; Wilding G; Tan W; Sullivan M; Merzianu M and Henderson BW Photodynamic therapy with 3-(1’-hexyloxy)ethyl-pyropheophorbide-a for cancer of the oral cavity. Clinical Cancer Research, 2013, 19, 6605–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nava HR; Allamaneni SS; Dougherty TJ; Cooper MT; Tan W; Wilding G and Henderson BW, Photodynamic therapy (PDT) using HPPH for the treatment of precancerous lesions associated with Barett’s esophabus. Lasers Surg. Med 2011, 43, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Loewen GM; Pandey RK; Bellnier D; Henderson BW and Dougherty TJ, Endobronchial photodynamic therapy for lung cancer, Lasers Surg. Med 2006, 38, 364–370. [DOI] [PubMed] [Google Scholar]
- (36).Pandey SK; Gryshuk A; Sajjad M; Zheng X; Chen Y; Abouzeid MM; Morgan J; Charamisinau I; Nabi HN; Pserohh A; Pandey RK, Multimodality agents for tumor imaging (PET,Fluorescence) and photodynamic therapy. A possible “See and Treat” approach., J. Med. Chem, 2005, 48, 6286–6295. [DOI] [PubMed] [Google Scholar]
- (37).Srvatsan A; Pera P; Joshi P; Mrko AJ; Durrani F; Missert JR; Curtin L; Sexton M; Yao R; Sajjad M; Pandey RK Highlights on the imaging (nuclear/fluorescence) and phototherapeutic potential of a tri-functional chlorophyll-a analog with no significant toxicity in mice and rats. Journal of Photochem. Photobiol, B: Biology, 2020, 211998. [DOI] [PubMed] [Google Scholar]
- (38).Cheruku RR; Cacaccio J; Durrani F; Tabaczynski WA; Watson R; Marko A; Kumar R; Elkhouly ME; Fukuzumi S; Missert JR; Yao R; Sajjad M; Chandra D and Pandey RK Epidermal growth factor receptor-targeted multifunctional photosensitizers for bladder cancer imaging and photodynamic therapy. J. Med. Chem, 2019, 62, 2598–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Patel N; Pera P; Joshi P; Dukh M; Tabaczynski WA; Siters KE; Kryman M; Cheruku RR; Durrani F; Mossert JR; Watson R; Ohulchanskyy TY; Tracy EC; Baumann H and Pandey RK Highly effective dual-function near-infrared (NIR) photosensitizer for fluorescence imaging and photodynamic therapy (PDT) of cancer., J. Med. Chem, 2016, 59, 9774–9787. [DOI] [PubMed] [Google Scholar]
- (40).Ethirajan M, Joshi P, William WH, Ohkubo K, Fukuzumi S, Pandey RK. Remarkable Regioselective Position-10 Bromination of Bacteriopyropheophorbide-a and Ring-B Reduced Pyropheophorbide-a. Organic letters, 2011; 13, 1956–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jinadasa RGW; Hu X; Vicente MGH and Smith KM Syntheses and cellular investigations of 173-, 152-, and 131- amino acid derivatives of chlorin e6. J. Med. Chem, 2011, 54, 7464–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Pandey RK; Sumlin AB; Constantine S; Aoudia M; Potter WR; Bellnier DA; Henderson BW; Rodgers MA; Smith KM and Dougherty TJ Alkyl ether analogs of chlorophyll-a derivatives: Part 1. Synthesis , photophysical properties, and photodynamic efficacy. Photochem. Photobiol, 1996, 64,194–204. [DOI] [PubMed] [Google Scholar]
- (43).Tracy EC; Bowman MJ, Pandey RK, Henderson BW Baumann H Cell-type selective phototoxicity achieved with chlorophyll-a derived photosensitizers in a co-culture system of primary human tumor and normal lung cells. Photochem Photobiol. 2011, 87,1405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tracy EC; Bowman MJ, Henderson BW, Baumann H, Interleukin-1α is the major alarmin of lung epithelial cells released during photodynamic therapy to induce inflammatory mediators in fibroblasts. Br J Cancer. 2012,10, 1534–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tracy EC, Bowman MJ, Pandey RK, Baumann H Cell-specific Retention and Action of Pheophorbide-based Photosensitizers in Human Lung Cancer Cells. Photochem Photobiol. 2019; 95, 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liu W; Oseroff AR; Baumann H Photodynamic therapy causes cross-linking of signal transducer and activator of transcription proteins and attenuation of interleukin-6 cytokine responsiveness in epithelial cells. Cancer Res. 2004, 64, 6579–6587. [DOI] [PubMed] [Google Scholar]
- (47).Srivatsan A; Pera P: Joshi P; Wang Y; Missert JR; Tracy EC; Tabaczynski WA; Yao R; Sajjad M; Baumann H; Pandey RK Effect of chirality on cellular uptake, imaging and photodynamic therapy of photosensitizers derived from chlorophyll-a. Bioorg Med Chem. 2015. 13, 3603–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spernyak JA; White WH III; Ethirajan M; Patel NJ; Goswami L; Chen Y; Turowski S; Missert JR; Batt C; Mazurchuk R; Pandey RK Hexylether derivative of pyropheophorbide-a (HPPH) on conjugating with 3gadolinium(III) aminobenzyldiethylenetriaminepentaacetic acid shows potential for in vivo tumor imaging (MR, fluorescence) and photodynamic therapy. Bioconjugate Chem. 2010, 21, 828–835, DOI: 10.1021/bc9005317 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.