

Abstract
Premature termination codons (PTCs) in cystic fibrosis transmembrane conductance regulator (CFTR) produce nonfunctional protein. No approved therapies exist for PTC mutations, including W1282X. We hypothesized that ivacaftor, combined with readthrough therapy, may benefit W1282X patients. Two N-of-1 clinical trials were conducted with ataluren and ivacaftor in various combinations. No meaningful clinical benefit was observed in either patient with ivacaftor alone or ataluren/ivacaftor combination. However, isolated improvements of uncertain significance were noted by a nasal potential difference (NPD) and FEV1% with ivacaftor in Patient-1 and with ataluren/ivacaftor combination by NPD and body mass index in Patient-2. Drug regimen composed of readthrough agents and potentiators warrant further development for W1282X and other CFTR nonsense mutations.
Keywords: ataluren, G542X, ivacaftor, N-of-1 trial design, personalized medicine, PTC mutations, translational readthrough, W1282X
Approximately 11% of cystic fibrosis (CF) patients have premature termination codons (PTC), causing truncated cystic fibrosis transmembrane conductance regulator (CFTR) protein with minimal to no function.1 One emerging pharmacologic strategy is using readthrough agents to promote PTC suppression. Aminoglycosides induce PTC readthrough; however, toxicity limits the feasibility of long-term administration. The investigational readthrough agent ataluren did not show sufficient clinical efficacy.2 Improvements in CFTR-PTC mutations were achieved using CFTR modulators in vitro and this approach is specifically applicable to W1282X CFTR, since it exhibits partial function even in the truncated state.3 The combination of the CFTR potentiator ivacaftor to readthrough agents has shown to enhance W1282X CFTR activity compared with the readthrough agent alone.4 On the basis of this, we hypothesized that ivacaftor may increase efficacy of a readthrough agent, ataluren by activating the W1282X readthrough product. Here we present two open-label, single center, N-of-1 clinical studies in patients with nonsense mutations to evaluate the efficacy of ataluren/ivacaftor combination therapy. Of note, the two N-of-1 studies have different protocols due to the specific medications that the respective patients were already taking and the limited availability of those agents beyond our control posed to the study design.
Patient-1 is a 31-year-old Caucasian female (W1282X/G542X) with mild CF-lung disease. The N-of-1 trial took place over eight scheduled visits from March 2017 through July 2017 (Figure 1A). Patient-1 was already taking ataluren, so the purpose of their trial was to assess if ataluren/ivacaftor dual therapy or ivacaftor monotherapy provided clinical benefit than ataluren alone. From visit-1 through visit-6, Patient-1 remained on ataluren (10, 10, and 20 mg/kg three times daily) and received two 2-week on/off cycles of ivacaftor (150 mg twice daily), followed by ivacaftor monotherapy for 4 weeks; the off ataluren period was necessitated by discontinuation of the trial offering ataluren, which was terminated at 10 weeks. Clinical efficacy of ataluren monotherapy was assessed at visits 1, 2, 4, and 7 (N = 4; Figure 1A, black arrow), ataluren/ivacaftor combination therapy was measured at visits 3, 5(N = 2, red arrow) and ivacaftor monotherapy was evaluated at visit 8 (N = 1, blue arrow). No changes in sweat chloride (SC) levels were noted either during ataluren (93.9 mmol/L) and ivacaftor (98 mmol/L) monotherapy or ataluren/ivacaftor combination therapy (98.5 mmol/L) (Figure 1B,C). By contrast, CFTR-mediated chloride activity within airways was monitored using NPD measurements4 and revealed a modest benefit with ivacaftor. NPD (ΔCl-free plus isoproterenol) was −3.7 mV for ataluren monotherapy and −9.4 mV for ivacaftor monotherapy (Figure 1D–F). Mean FEV1 was 90.7% of predicted for ataluren monotherapy, 95% for ivacaftor monotherapy, and 95% for ataluren/ivacaftor combination therapy (Figure 1G,H). Of interest, Patient-1 had two pulmonary exacerbations during the N-of-1 trial, both occurred following cessation of ivacaftor. Body mass index (BMI) was not affected; 23.4 kg/m2 during ataluren monotherapy, 23.4 kg/m2 after ataluren/ivacaftor combination therapy, and 23.6 kg/m2 for ivacaftor monotherapy (Figure 1I,J). Respiratory symptoms by CF Questionnaire-Revised improved during ivacaftor treatment periods (Figure 1K). Overall, we did not observe a consistent effect that suggested clinically meaningful improvements given the inconsistency between FEV1% and CFTR function by NPD as compared with sweat chloride observed with ivacaftor.
FIGURE 1.
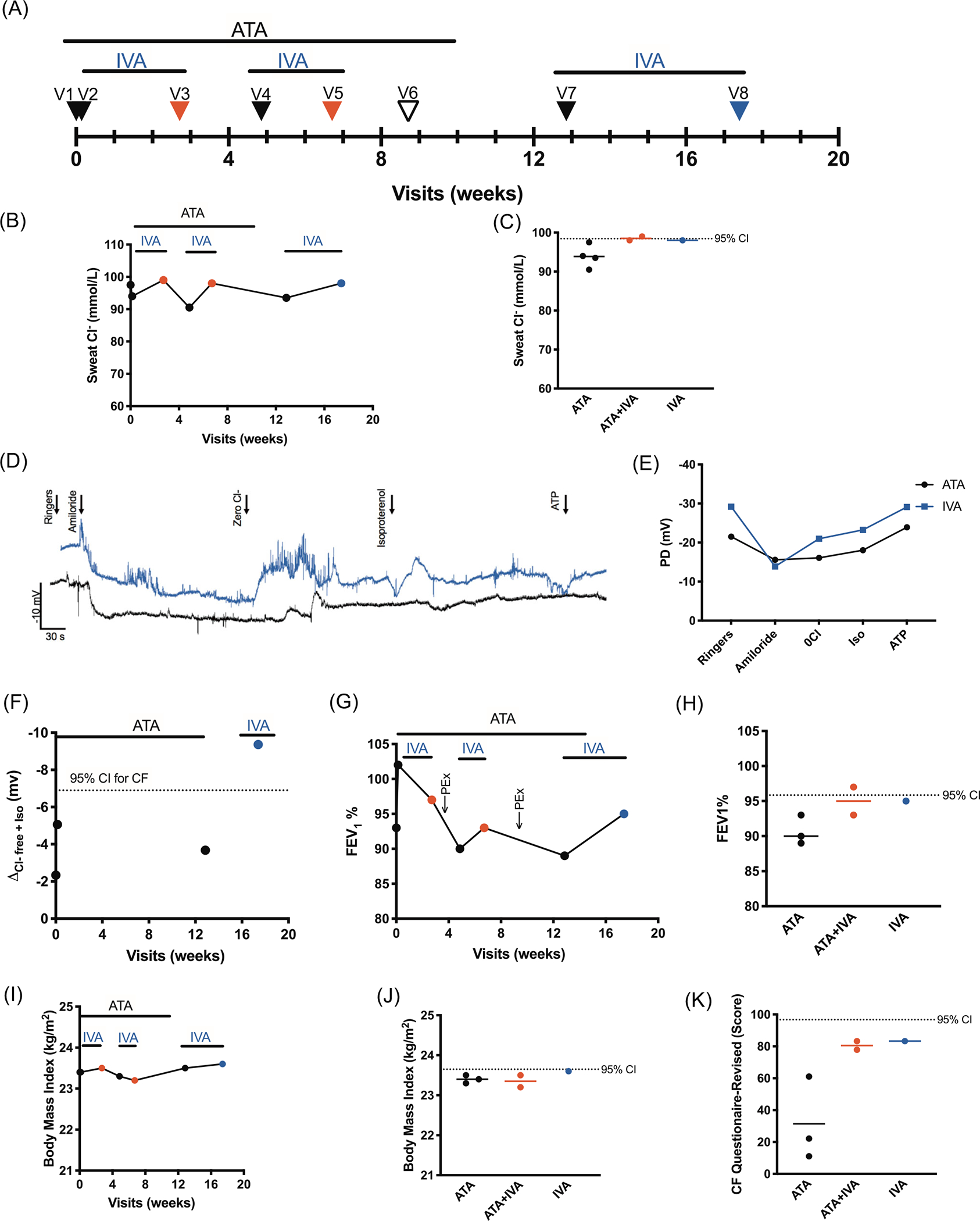
Effect of ataluren/ivacaftor combination therapy in cystic fibrosis (CF) patient with W1282X/G542X mutation (Patient-1). Combination therapy is denoted with red, ataluren (ATA) monotherapy denoted by black, and ivacaftor (IVA) monotherapy denoted by blue. A, Timeline of visits and treatment with ATA and IVA for Patient-1. B, Sweat chloride longitudinally. C, Sweat chloride by therapy. D, Representative tracings of nasal potential difference (NPD) for ATA and IVA monotherapies. E, Averaged NPD values for ATA and IVA. F, Mean chloride conductance PD (ΔCl-free plus isoproterenol) longitudinally. G, FEV1% longitudinally. Note the pulmonary exacerbations (PEx) following cessation of combination therapy. H, FEV1% by therapy. I, Body mass index longitudinally. J, Body mass index by therapy. K, CF Questionnaire-Revised Score by therapy
Patient-2 is a 32-year-old non-Hispanic, Caucasian female CF patient (W1282X/W1282X) with severe CF lung disease and in whom ivacaftor monotherapy was found to be potentially effective in a prior study.5 Since Patient-2 was already taking ivacaftor, the purpose of their trial was to assess if ataluren/ivacaftor dual therapy provided clinical benefit than ivacaftor alone. The N-of-1 occurred over seven scheduled visits from May 2016 through May 2017 (Figure 2A). For the first month, Patient-2 received ivacaftor monotherapy (150 mg twice daily), and for the remainder of the trial, she took ataluren/ivacaftor combination therapy (10,10,20 mg/kg three times daily). Clinical efficacy of ivacaftor monotherapy was assessed at visits 1 and 2 (N = 2; Figure 2A, blue arrow); ataluren/ivacaftor combination therapy was measured at visits 3 to 7 (N = 3–5; Figure 1A, red arrow).During ivacaftor treatment, mean SC was 102.3 mmol/L. Following ataluren/ivacaftor combination therapy, there was no improvement in SC (105.8 mmol/L) (Figure 2B,C) compared with ivacaftor monotherapy. NPD measurements demonstrated that ataluren/ivacaftor combination therapy improved total chloride conductance than ivacaftor alone (PD −6.7 mV for ivacaftor and −22.5 mV for ataluren/ivacaftor combination; Figure 2D–F). Mean FEV1 was 35.0% predicted for ivacaftor monotherapy and 35.2% for ataluren/ivacaftor combination therapy (Figure 2G,H). Ataluren/ivacaftor combination therapy showed marginal effect on BMI (19.8 kg/m2) compared with ivacaftor monotherapy (19.3 kg/m2; Figure 2I,J). CFQ-R score was greater with ivacaftor monotherapy (88.9) compared with ataluren/ivacaftor combination therapy (78.9; Figure 2K). Overall, ataluren/ivacaftor combination therapy altered BMI and CFTR function by NPD in the patient homozygous for W1282X CFTR, but did not improve sweat chloride, FEV1 or respiratory symptoms. We interpret this as unlikely to be clinically meaningful, noting the possibility that as outcome measures become more sensitive, or that longer time domains, small effects can emerge.
FIGURE 2.

Effect of ataluren/ivacaftor combination therapy in cystic fibrosis (CF) patient with W1282X/W1282X mutation (Patient-2). Combination therapy is denoted with red and ivacaftor (IVA) monotherapy denoted by blue. A, Timeline of visits and treatment with ATA and IVA for Patient-1. B, Sweat chloride longitudinally. C, Sweat chloride by therapy. D, Representative tracings of nasal potential difference (NPD) for IVA monotherapy and ATA/IVA combination therapy. E, Averaged NPD values for ATA and IVA. F, Mean chloride conductance PD (ΔCl-free plus isoproterenol) longitudinally. G, FEV1% longitudinally. H, FEV1% by therapy. I, Body mass index longitudinally. J, Body mass index by therapy. K, CF Questionnaire-Revised Score by therapy
Messenger RNA (mRNA) isolated from human nasal epithelial (HNE) cells from each CF patient subjected to real-time polymerase chain reaction (RT-PCR) showed reduced CFTR mRNA expression compared to non-CF donors (3.6% and 10% of non-CF in Patient-1 and -2, respectively; data not shown) suggesting low levels of CFTR expression in both patients, likely due to nonsense-mediated decay. This may responsible for the lack of meaningful and consistent effect.
Using two individualized N-of-1 trials impacted by limitation in drug availability, we demonstrate preliminary evidence that ivacaftor provides potential for augmentation of translational readthrough, noting findings were small and mixed. One possibility is that ivacaftor is needed to observe clinical benefit of ataluren or other weakly active readthrough agents. Patient-1 had isolated improvements in FEV1, NPD, and respiratory symptoms (CF questionnaire) with ataluren/ivacaftor combination therapy over ivacaftor monotherapy and the addition of ataluren to ivacaftor regimen slightly improved BMI and NPD in Patient-2. While we did not conduct inferential testing since measurements were conducted within single patients, and thus not independent, changes were sufficient in magnitude to raise the possibility that effects were related to drug. In contrast, SC levels did not substantially change in either treatment. Further research is needed to understand this discrepancy, but could be related to the sensitivity of sweat glands to nonsense mutation repair or the magnitude of CFTR benefit. In total, the N-of-1 trials presented here do not provide convincing evidence that ataluren/ivacaftor combination therapy is an efficacious therapy for patients with PTC mutations due to a lack of consistent effects in these two patients, partially driven by reduced mRNA levels; thus drug development for patients with nonsense mutations must continue.
W1282X is a mutation that exhibits function even in truncated state, making it potentially amenable to potentiator treatment. While results recapitulated beneficial effects of ivacaftor for W1282X,5 results with ataluren were mixed, potentially due to low activity of ataluren and diminished mRNA levels, a known negative covariate for readthrough efficacy.6 Novel corrector-potentiator combination regimens could benefit patients with the W1282X-CFTR mutation. Note that N-of-1 trial designs have limitations with potential carryover effects of short treatment blocks impacting measurements of treatment efficacy. Furthermore, appropriate statistical models for such trials remain controversial since multiple complementary outcome measures increase type-1 error, especially when findings are not consistent across all outcomes, as in this case. Future N-of-1 trials may require blinding to improve validity. Nevertheless, N-of-1 studies could accelerate the assessment of rationally selected multi-drug therapy regimens for rare mutation groups.
ACKNOWLEDGMENTS
The authors acknowledge the assistance of the two CF patients who volunteered for this study and the dedicated individuals on her care team. We also acknowledge inspiration from Drs. Kevin Foskett and Peter Haggie. Thank you to our funding sources and support systems for making this study possible. This study was funded by NIH F31HL146083 (to JEPL); Emily’s Entourage LLC (to VM); and NIH (P30DK072482) and the Cystic Fibrosis Foundation, each to SMR.
Funding information
National Institue of Health, Grant/Award Numbers: F31HL146083, P30DK072482; Emily’s Entourage, Grant/Award Number: Venkateshwar Mutyam; Cystic Fibrosis Foundation, Grant/Award Number: Steven Rowe
Footnotes
CONFLICT OF INTERESTS
Dr. Rowe reports grants from Bayer, grants from Forest Research Institute, grants from AstraZeneca, grants from N30/Nivalis, grants from Novartis, grants from Galapagos/AbbVie, grants from Proteostasis, grants from Eloxx, grants and personal fees from Celtaxsys, grants from PTC Therapeutics, grants, personal fees and nonfinancial support from Vertex Pharmaceuticals Incorporated, personal fees from Bayer, personal fees and nonfinancial support from Novartis, personal fees from Renovion, grants and personal fees from Synden/Synspira, personal fees from Genentech, personal fees and nonfinancial support from Boeringher Ingelheim, grants from Jannssen, Vivus, Actelion, Johnson and Johnson, and other related entities, outside the submitted work.
REFERENCES
- 1.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73(7):1251–1254. [DOI] [PubMed] [Google Scholar]
- 2.Zainal Abidin N, Haq IJ, Gardner AI, Brodlie M. Ataluren in cystic fibrosis: development, clinical studies and where are we now? Expert Opin Pharmacother. 2017;18(13):1363–1371. [DOI] [PubMed] [Google Scholar]
- 3.Rowe SM, Varga K, Rab A, et al. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007;37(3):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mutyam V, Du M, Xue X, et al. Discovery of clinically approved agents that promote suppression of cystic fibrosis transmembrane conductance regulator nonsense mutations. Am J Respir Crit Care Med. 2016;194(9):1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mutyam V, Libby EF, Peng N, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros. 2017;16(1): 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aksit MA, Bowling AD, Evans TA, et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J Cystic Fibros. 2019;18(5):606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]