

Abstract
Rationale:
There is an intimate relationship between the endothelium and monocytes, and activated endothelial cells promote monocyte transformation to macrophages and dendritic cells (DCs). Recently, a subset of human DCs expressing the receptor tyrosine kinase, Axl and the lectin Siglec-6 has been described and termed AS DCs.
Objective:
We sought to determine if circulating AS DCs are increased in human hypertension and to examine how Axl signaling contributes to this disease.
Methods And Results:
We demonstrated that circulating AS DCs are increased in hypertensive humans compared to normotensive controls. Pulse pressure in humans also correlated with plasma levels of the Axl agonist growth arrest specific 6 (GAS6). Exposure of human endothelial cells to 10% cyclical stretch increased release of the GAS6, promoted Axl signaling and caused AS DC formation; events that were inhibited by blockade of Axl with R428 or by siRNA knockdown of either endothelial GAS6 or Axl. GAS6/Axl signaling in human monocytes potentiated interleukin 1-beta production through NLRP3/caspase-1 and caused accumulation of immunogenic isolevuglandin (isoLG)-protein adducts. In mice, the Axl inhibitor R428 or global deletion of Axl attenuated hypertension and renal inflammation caused by angiotensin II (Ang II) infusion. Bone marrow transplant studies demonstrated a role of both stromal and immunological Axl in Ang II-induced hypertension. Lastly, in freshly harvested human endothelial cells, a striking correlation was observed between the degree of endothelial cell activation as reflected by intracellular adhesion molecule 1 (ICAM-1), isoLG-adduct accumulation and intracellular GAS6 levels.
Conclusions:
We define a previously unrecognized interaction of human endothelial cells and monocytes that promote formation of AS DCs in hypertension and show a critical role of GAS6 and Axl signaling in both immune cells and endothelial cells. This pathway is potentially a novel therapeutic target to reduce inflammation and end organ damage in hypertension.
Keywords: Animal Models of Human Disease, Basic Science, Inflammation, Pathophysiology
Graphical Abstract
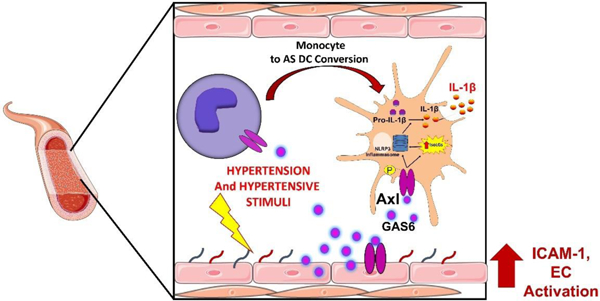
INTRODUCTION
Over the last decade, it has become increasingly evident that inflammation and immune activation contribute to the genesis of systemic hypertension. In particular immune cells including lymphocytes, monocytes and monocyte-derived dendritic cells (DCs) infiltrate the vasculature, kidney and other organs and promote end organ damage during hypertension.1 We and others have defined an important role of monocyte-derived cells in murine hypertension. Deletion of monocytes in mice prevents the hypertension caused by Ang II and abrogates the vascular dysfunction that accompanies this disease.2 We have shown that DCs of hypertensive mice potently drive T cell proliferation and produce increased amounts of IL-1β, IL-6 and IL-23. Adoptive transfer of DCs from hypertensive mice can prime hypertension in recipient mice.3 We have also shown that DCs accumulate isolevuglandin (isoLG)-modified proteins that seem to act as antigens to drive T cell proliferation, and that the kidney is an important site of DC activation in hypertension.4
In humans, DCs have been classified into three major subsets: myeloid/classical DC1 (cDC1), myeloid/classical DC2 (cDC2), and plasmacytoid DCs (pDCs).5 cDC1 exhibit surface expression of CD141 while cDC2 express CD1c. Both of these cDCs subsets are efficient professional antigen-presenting cells that stimulate CD4+ and CD8+ T cells, while pDCs, which express CD123, produce type I interferons in response to viral infection.5 Recently, Villani et al used unbiased single-cell RNA sequencing to identify a new subset of human DCs with surface expression of Axl, a tyrosine kinase receptor, and sialic acid-binding immunoglobulin-type lectin 6 (Siglec-6).6 The Axl+ Siglec-6+ DCs (AS DCs) exhibit varying surface expression profiles of CD11c, CD123 and CD1c. AS DCs were described to be on a continuum of DC phenotypes with the potential for transition to cDC2. Furthermore, Villani and colleagues demonstrated that AS DCs produce large amounts of IL-1β, IL-6, and TNFα and promote both CD4+ and CD8+ T cell proliferation in response to toll-like receptor (TLR) stimulation.6
A major question relates to the factors encountered in hypertension that can activate monocytes and monocyte-derived cells. Prior studies have implicated signals derived from the vascular endothelium in the transformation of monocytes to DCs and macrophages.7 Monocytes encounter the endothelium in the microcirculation, and endothelial cells that are activated by bacterial products, cytokines and mechanical factors increase expression of adhesion molecules and chemokines that promote adherence and transmigration of monocytes7. During this, monocytes receive information from the endothelium that influences their phenotype and function. In a recent study, we showed that increased mechanical stretch of human endothelial cells led to release of factors including hydrogen peroxide and IL-6, which in turn promoted transformation of classical (CD14++/CD16low) monocytes to an intermediate (CD14++/CD16+) phenotype with evidence of DC transformation.8 These cells were found to produce copious quantities of IL-6, IL-1β, TNFα and IL-23 and to potently drive autologous T cell proliferation, characteristics shared by the AS DC subset described by Villani et al.
Axl is a member of the TAM tyrosine kinase receptor family and is potently activated by growth arrest specific-6 (GAS6). Elevated levels of plasma GAS6 are found in numerous pathological conditions including sepsis, obesity, chronic renal disease, cardiac hypertrophy and systemic lupus erythematosus.9–13 Moreover, elevated plasma GAS6 levels are associated with end organ damage and renal failure.11 Taken together, these studies led us to test the hypothesis that the pro-inflammatory AS cells, endothelial cell activation, and GAS6/Axl signaling contribute to the development of hypertension and end-organ inflammation.
In this study we investigated the circulating levels of AS cells in human hypertension and performed studies both in cell culture and in vivo in experimental murine hypertension to examine mechanisms of GAS6/Axl in the formation and activation of AS cells, and the subsequent development of hypertension and end-organ inflammation. Using bone marrow transplant studies and siRNA mediated knockdown in cells in culture, we demonstrate that Axl/GAS6 signaling has profound effects on dendritic cells function and autocrine effects on the endothelium to modulate inflammation and hypertension.
METHODS
Data Availability.
The data that support the findings will be available from DGH upon reasonable request. An expanded methods section is available in the Online Supplemental Material.
Human subjects.
In experiments to determine the presence of AS DCs in vivo, we studied normotensive (n=23) and hypertensive (n=11) subjects between the ages of 23–67 (Online Table I). Patients were considered hypertensive if they had a systolic blood pressure higher than 140 mmHg, a diastolic blood pressure higher than 90 mmHg or had a diagnosis of hypertension and were currently treated with anti-hypertensive agents. Blood pressure was obtained as the average of 3 determinations using an automatic device after 15 minutes rest with the subject sitting upright with their back supported. Hypertensive subjects were defined as those with systolic pressures greater than 135 mmHg while normotensive participants had blood pressures between 120/80 – 135/80 mmHg.
In experiments to determine AS DC function in vitro, 65 additional subjects were recruited between the ages of 23–67. Clinical and demographic characteristics are shown in Online Table II. In experiments to determine plasma GAS6 concentrations, 28 additional subjects were recruited between the ages of 23–67. Clinical and demographic characteristic are shown in Online Table III. In additional experiments to determine isoLG-adduct accumulation, ICAM-1 expression, and GAS6 production from human endothelial cells, 23 additional subjects were recruited between the ages of 18–50. Clinical and demographic characteristics are shown in Online Table IV.
Exclusion criteria included the following: 1) Autoimmune disease or history of inflammatory diseases; 2) Vaccinations within the prior 3 months 3) Confirmed or suspected causes of secondary hypertension; 4) Severe psychiatric disorder; 5) HIV/AIDS; 6) Participants currently taking steroids or antihistamines; and 7) participants with malignancies; 7) Morbid obesity (body mass index > 35). The protocol was approved by the Vanderbilt Institutional Review Board and conformed to standards of the US Federal Policy for the Protection of Human Subjects.
Animals and blood pressure measurements.
C57BL/6J, Axl+/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). Axl+/− were bred to produce wild type (Axl/+/+) and homozygotic Axl deficient (Axl−/−) mice. C57BL/6J, Axl+/+, and Axl−/− male mice at 10 to 12 weeks of age were used for the study. Mice were randomly selected in cages to the normotensive or the hypertensive study group. Hypertension was induced by 14-day Ang II infusion (490 ng/min) by subcutaneous osmotic minipump. One group of mice was treated with daily gavage of the Axl inhibitor R428 (25 mg/kg in DMSO) while another group as treated with the DMSO vehicle during Ang II infusion. In a separate study, 6-week-old male Axl+/+ and Axl−/− mice were transferred to sterile cages and fed sterile chow and acidified water (pH 2.0) containing 1 mg/ml of sulfamethazine Na+ and 0.2 mg/ml of trimethoprim for prophylaxis 1 week prior to bone marrow transplant (BMT). On the day of BMT, mice were irradiated with a dose of 10 Gy using a 137Cs irradiator (J.L. Shepheard and Associates). Four hours later, bone marrow cells from pooled femurs and tibias collected from 1 mouse were used to reconstitute the marrows of 2 mice (20 × 106 cells for each mouse). Mice were allowed to recover for 8 weeks before implantation of radiotelemeters and 66 days before implanting osmotic minipumps. Mice were euthanized at the end of all experiments by CO2 inhalation. All experimental procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee and were conducted in an AAALAC-accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals and the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Renal and aortic single cell suspensions were prepared for flow cytometry as previously described.14
Statistics.
For all flow cytometric data, files were exported from the cytometer and were given a random identifier so that the analysis was performed blinded in Flowjo. For all blood pressure data, mice were given a blinded identifier until each experiment was completed during analysis of the data. Graphpad Prism version 9 was used to perfrom all statistical analysis. To assure values were not outliers, a Grubbs test was employed with an alpha set to 0.05. All data are expressed as mean ± SEM. In this study no experiment-wide comparions were made and only within experiment comarpisons were applied. One tailed-paired and unpaired Student’s t-tests were used to compare two groups. Normality and non-normality distribution were determined by the Shapiro-Wilk test. In case of the non-normality, the nonparametric test one-tailed Mann-Whitney U test or the Friedman test was used. For studies comparing the effect of inhibition of caspase-1 and NLRP3 inflammasome, for studies of the effect of GAS6 or Axl siRNA (1 µM) in HAECs on the formation AS DCs, or the effect of GAS6 and cyclical stretch stimulation on HAECs co-treated with Axl inhibitor, PEG-SOD, or 2-HOBA, one-way ANOVAs were employed, followed by a Bonferroni post hoc test when significance indicated. For tail cuff and telemetry blood pressure measurements over time, 2-way ANOVA with repeated measures was employed, followed with a Bonferroni post hoc test when significance was indicated. Multiple linear regression was used to analyze the association between hypertension and circulating AS DCs with adjustment of other factors. Linear regression was used to analyze associated between pulse pressure and GAS6, systolic blood pressure and GAS6, diastolic blood pressure and GAS6, endothelial cell GAS6 and isoLGs, endothelial cell GAS6 and ICAM-1, and endothelial cell ICAM-1 and isoLGs. Precise p values are reported in the figures and were considered significant when less than 0.05.
RESULTS
Hypertension is associated with increased circulating AS DCs.
In initial studies we examined the levels of circulating AS DCs in hypertensive and normotensive subjects. Demographic and clinical characteristics for these subjects are shown in Online Table I. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-gradient and flow cytometric analysis was performed on live single CD3−, CD19−, CD56−, and CD66b− cells to exclude T lymphocytes, B cells, granulocytes, and natural killer cells and gates were determined by fluorescence minus one experiments and isotype controls (Online Figure I A-B). Representative flow cytometric images from normotensive and hypertensive human subjects are shown in Figures 1A and B. Hypertensive subjects had a significantly greater CD1c+ AS DCs (Figure 1C) and CD123+ AS pDCs (Figure 1D). This difference remained highly significant following adjustment for BMI and age using multiple linear regression analysis (p=0.0005, Online Table I). No statistical differences were observed in HLA-DR+, CD11c+, CD1c+, CD141+, and CD123+ pDCs between normotensive and hypertensive subjects (Online Figure IC-G).
Figure 1: Hypertension is associated with increased circulating AS DCs.
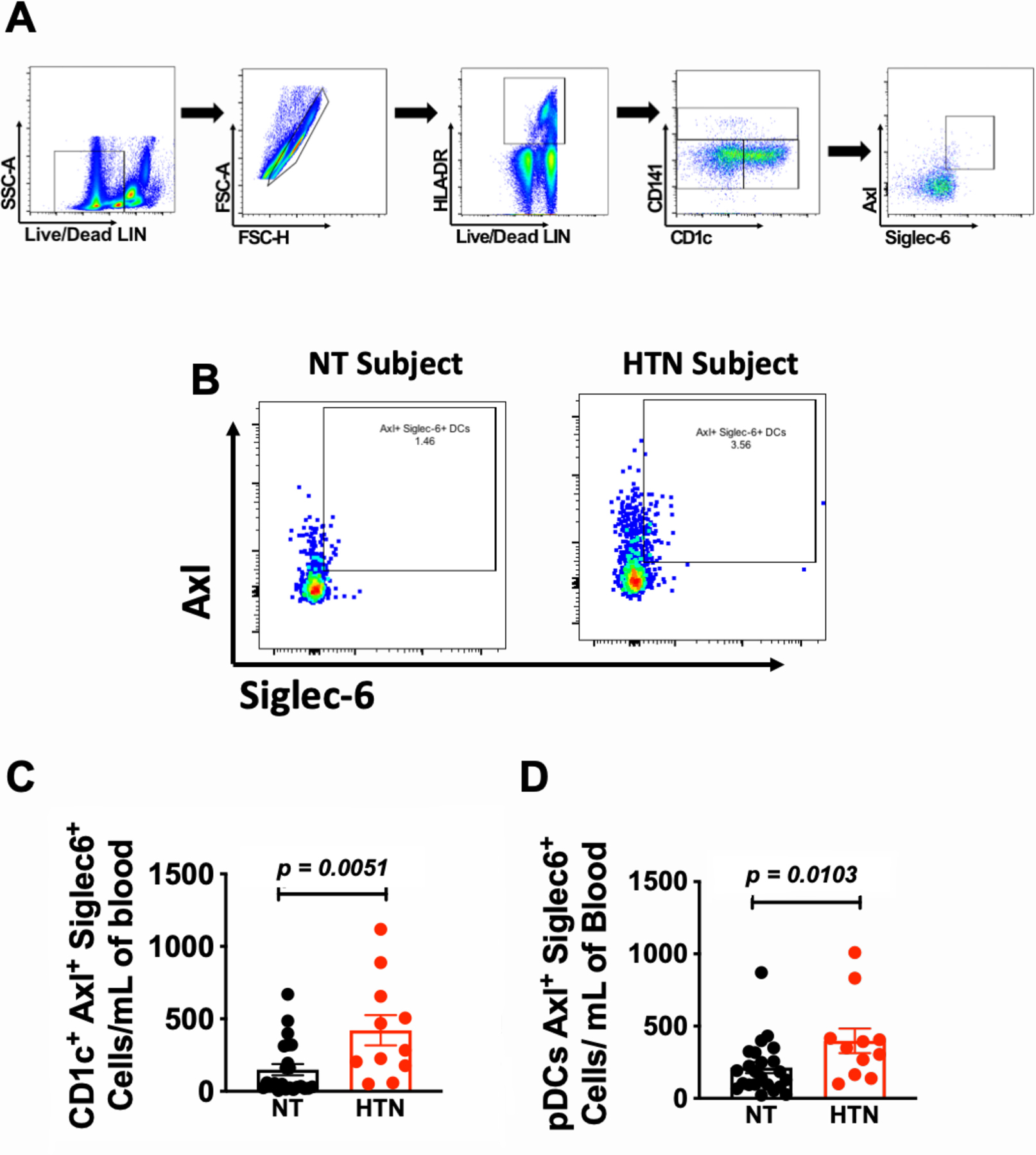
(A) Flow cytometry gating strategy to identify AS DCs from human PBMCs. (B) Representative flow cytometric images of CD1c+ AS DCs from normotensive and hypertensive human subjects. Summary data for (C) CD1c+ AS DCs and (D) AS pDCs from normotensive and hypertensive subjects. Data were analyzed using the Mann-Whitney non-parametric test; n=23 normotensive and 11 hypertensive subjects and expressed as a mean ± SEM. SSC-A indicates side scatter area. FSC-A indicates forward scatter area.
Hypertensive endothelial mechanical stretch promotes formation of CD1c+ AS DCs.
Endothelial cells activated by zymosan or lipopolysaccharide are capable of stimulating transformation of human monocytes to DCs.7 Recently, we demonstrated that endothelial cells activated by mechanical cyclic stretch can also promote conversion of human classical (CD14++/CD16−) monocytes an intermediate (CD14++/CD16+) phenotype and to CD14+ CD209+ (DC-SIGN) cells.15 In the present study, we isolated human CD14+ classical monocytes and exposed them to endothelial cells undergoing 5% or 10% cyclical stretch for 48 hours (Figure 2A) and then assessed the presence of AS DCs using the gating strategy shown in Figure 2B. We found that 10% endothelial cell stretch markedly enhanced the formation of AS DCs and that this could be completely prevented by inhibition of Axl with R428 (1 µM; DMSO; Figure 2D and Online Figure II). promoted Axl phosporylatinD and Online Figure II). Likewise, 10% cyclical stretch of endothelial cells released factors that promoted Axl phosphorylation as measured by flow cytometry and this was also prevented bye-treatment with R428 prevented Axl phosphorylation (Figure 2E). Notably, AS DCs formed from monocytes in response to 10% cyclical endothelial stretch predominately co-localized within the intermediate (CD14++/CD16+) monocyte population (Figure 2F; red cells and Online Figure III).
Figure 2: Hypertensive endothelial mechanical stretch promotes the formation of AS DCs.
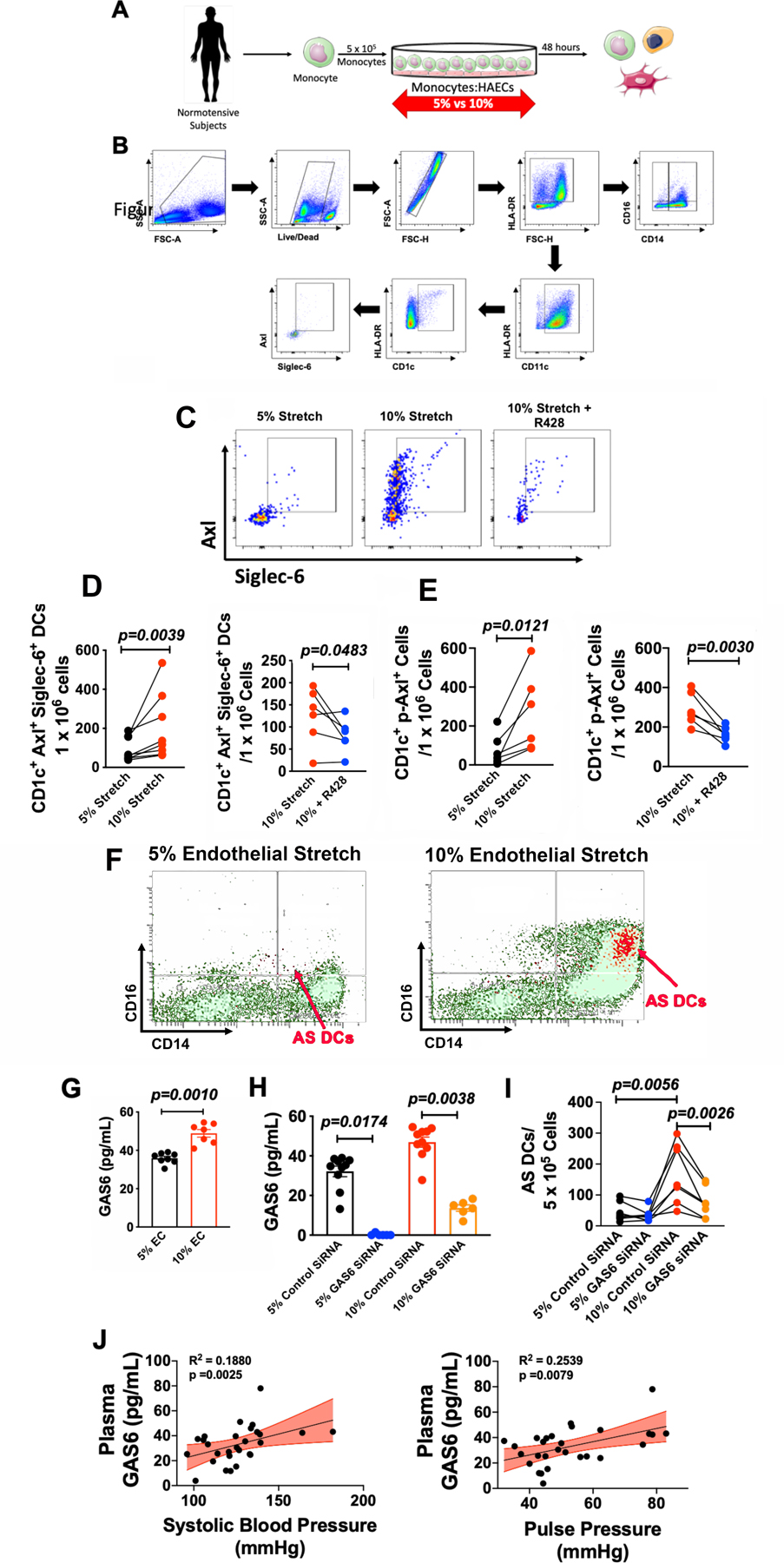
(A)Total PBMCs were isolated from normotensive human subjects and co-cultured with human aortic endothelial cells (HAECs) undergoing 5% and 10% cyclical stretch. (B) Flow cytometry gating strategy to identify Axl+ Siglec-6+ DCs. (C) Representative flow cytometry images for the relation of AS DCs to monocyte subpopulations. (D) Representative flow cytometry images for AS DC formation under co-culture conditions of 5% HAEC stretch, 10% HAEC stretch, and 10% stretch + R428 (1µM; Axl inhibitor; DSMO) for 48 hours. (E) Summary data of CD1c+ Axl+ Siglec-6+ cells and CD1c+ Axl+ Siglec-6+ cells undergoing 5% and 10% stretch compared using the Mann Whitney test (n=8). (F) Summary data of CD1c+ p-Axl+ cells undergoing 10% or 10% + R428 (1µM; DMSO) compared using the Student’s t test (n=6). (G) Summary data of GAS6 secretion from human aortic endothelial cells undergoing normotensive (5%) or hypertensive (10%) stretch. Student’s t-test (n=8 for 5% stretch and n=7 for 10% stretch). (H) Summary data of GAS6 secretion from human aortic endothelial cells from 5% + scrambled siRNA, 5% + GAS6 siRNA, 10% + scrambled siRNA, and 10% + GAS6 siRNA. Kruskal-Wallis non-parameteric test (n=10 for 5% control SiRNA, n=6 5% GAS6 SiRNA, n=10 10% control SiRNA, and n=6 10% GAS6 SiRNA). (I) Effect of GAS6 knockdown on the formation of AS DCs during co-culture of normotensive CD14+ monocytes and human aortic endothelial cells from 5% + scrambled siRNA (1 µM), 5% + GAS6 siRNA (1 µM), 10% + scrambled siRNA (1 µM, and 10% + GAS6 siRNA (1 µM). Friedman non-parameteric test (n=7). (J) Association between plasma GAS6 and systolic blood pressure and plasma GAS6 and pulse pressure. Linear regression and Spearman r correlation using Student’s t-test (n=16). SSC-A indicates side scatter area. FSC-A indicates forward scatter area. EC indicates endothelial cell. All data are represented as mean ± SEM.
The potential role of GAS6 in hypertension.
Axl is a member of the TAM tyrosine kinase receptor family and is uniquely activated by GAS6. We therefore sought to determine if GAS6 was increased by hypertensive stretch. We found that 10% stretch caused a 50% increase in the release of GAS6 from human aortic endothelial cells compared to 5% stretch as determined by ELISA (Figure 2G). We next used small interfering RNA (siRNA) against GAS6 (1 µM; Dharmacon) in HAECS to achieve knockdown GAS6 in HAECs exposed to both 5% and 10% cyclical stretch (Figure 2H). When monocytes were co-cultured with HAECs exposed to either scrambled or GAS6 siRNA, we found that knockdown of GAS6 in HAECs led to a significant reduction in the formation of AS DCs (Figure 2I).
We next sought to determine if plasma GAS6 levels in humans correlate with blood pressure. In a separate cohort of human subjects, we found positive associations between plasma GAS6 and systolic blood pressure (Figure 2J; R2=0.19, p=0.0025) and pulse pressure (Figure 2J; R2=0.25, p=0.0079). These relationship remained significant when corrected for age and sex using multivariate analysis (Online Table III). No statistical association was observed between diastolic blood pressure and plasma GAS6 (Online Figure IV; R2=0.005, p=0.7167.
Based on these in vitro and in vivo findings, we sought to determine if GAS6 potentiates formation of AS DCs. We treated CD14+ monocytes with or without GAS6 (80 nM; R&D Systems) for 48 hours (Figure 3A). We found that the concentration of GAS6 achieved when endothelial cells are exposed to 10% stretch caused a portion of CD14+ monocytes to acquire the conventional DC marker CD1c (Figure 3B and Online Figure V). GAS6 exposure also enhanced surface expression of Axl and Siglec-6, indicating that the formation of AS DCs is promoted by GAS6 (Figure 3B). Flow cytometric analysis demonstrated that GAS6 markedly increased formation of AS DCs, Axl+ DCs, and Siglec-6+ DCs (Figure 3C). Moreover, GAS6 treatment significantly increased the activation markers CD83 and CD86 and these effects of GAS6 were prevented by inhibition of Axl with R428 (Figure 3D). We also found that a portion of CD14+ monocytes exposed to GAS6 acquire dendritic morphology (Figure 3E). To determine AS DCs formed in this fashion can present antigen, we exposed CD14+ monocytes from 3 additional volunteers and exposed these to GAS6 for 24 hours. AS DCs were isolated from these by flow sorting and mixed with T cells from the same subject at a ratio of one AS DC to 10 T cells and then exposed to varicella zoster virus. As controls, T cells without AS DCs were exposed to VZV antigen. As evident in Online Figure VI A-B, VZV induced T cell activation as reflected by ELISPOT quantification of IFNg production only when AS DCs were present. Thus GAS6/Axl signaling promotes formation of monocyte-derived AS DCs even in the absence of other hypertensive stimuli. We considered the hypothesis that GAS6 might signal through other TAM receptors, however found that MerTK and Tyro3 receptors were virtually absent in human monocytes and did not change with GAS6/Axl signaling (Online Figure VII A-C).
Figure 3: GAS6 exposure promotes the formation of monocyte derived AS DCs.
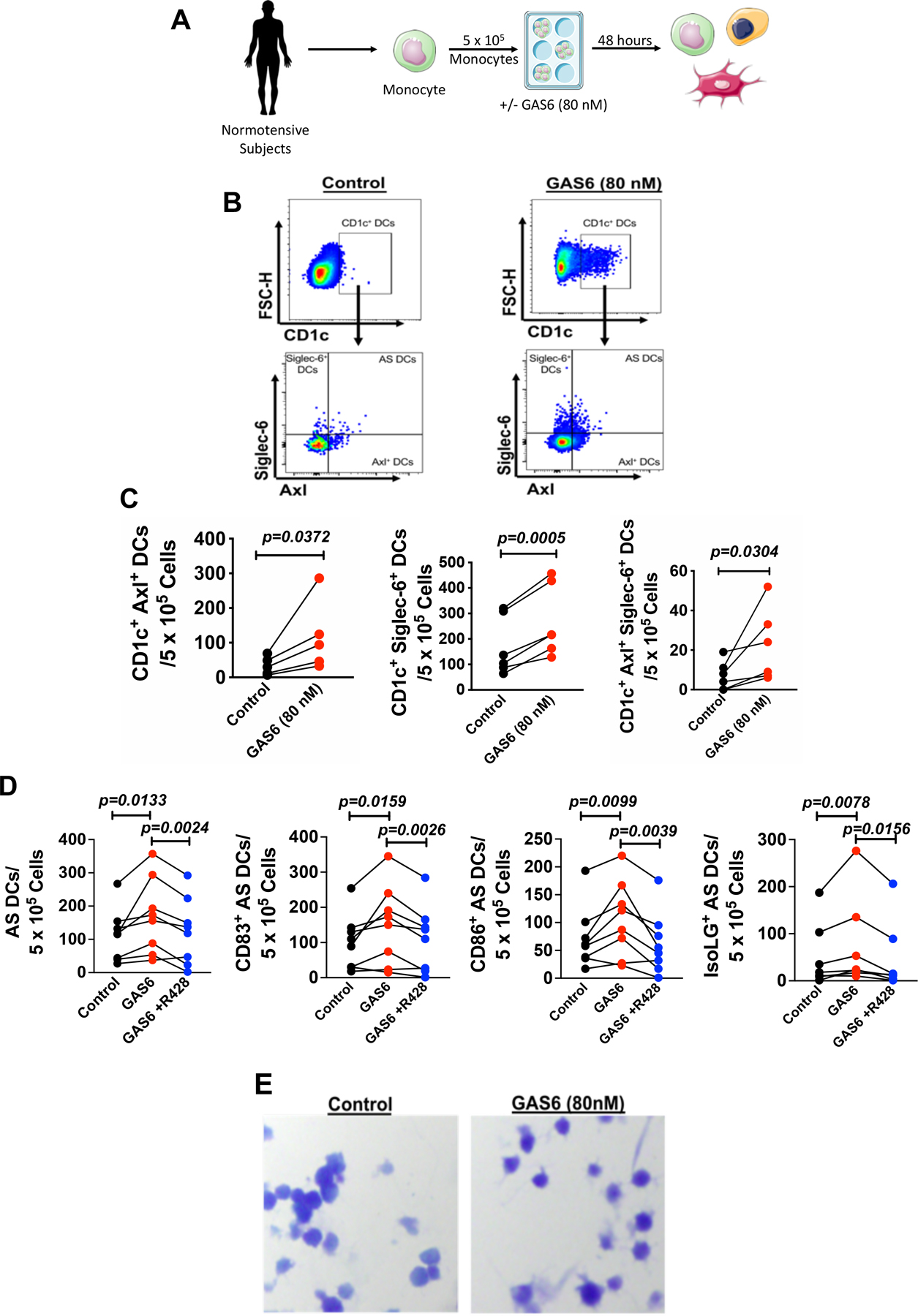
(A) Human CD14+ monocytes from normotensive human subjects were cultured in control media or control media + GAS6 (80nM) for 48 hours. (B) Representative flow cytometry data for surface expression of CD1c and AS DCs under control and GAS6 stimulation (80 nM). (C) Summary data for CD1c+ Axl+ cells, CD1c+ Siglec-6+ Cells, and CD1c+ Axl+ Siglec-6+ cells compared using a Paired Student’s t-test (n=6). (D) Summary data for the formation of AS DCs, CD83+ AS DCs, and CD86+ AS DCs upon GAS6 stimulation (80 nM) with or without Axl inhibition with R428 (1 µM; DMSO) analyzed using the Wilcoxon non-parametric test (n=8). (E) Representative Wright-Stained images for dendritic cell morphology in control monocytes and GAS6 treated monocytes from the same normotensive human subject. All data are represented as mean ± SEM.
GAS6 stimulates IL-1β production through NLRP3 inflammasome and caspase-1 activation in AS DCs.
We previously observed that DCs of hypertensive mice produce large amounts of IL-1β. 3, 4, 16 Likewise, Villani et al demonstrated that AS DCs are capable of producing this cytokine.6 Because inflammasome activation is required for mature IL-1β production we sought to determine if GAS6 stimulates this is human monocytes. We found that exposure to endothelial cells undergoing 10% hypertensive stretch markedly increased the transformation of human monocytes to IL-1β+ Axl+ DCs, and this could be prevented by the Axl inhibitor R428 (Figure 4A and B). Moreover, exposure of human CD14+ monocytes to GAS6 for 48 hours, using concentrations we observed to be released from endothelial cells, markedly enhanced IL-1β production (Figure 4C and D). The production of IL-1β in response to GAS6 was prevented by either inhibition of the NLRP3 inflammasome (MCC950; 1 µM) or caspase-1(Ac-YVAD-cmk; 1 µM) (Figure 4D). IL-1β production requires transcriptional activation of pro-IL-1β and subsequent cleavage to mature IL-1β. We found no significant difference in the mRNA levels of pro-IL-18, pro-IL-1β, and casp-1 in GAS6 stimulated monocytes (Figure 4E-G), indicating that the major action of GAS6 is likely mediated via NLRP3 activation of caspase-1 cleavage of pro-IL-1b.
Figure 4: GAS6 stimulates IL-1β production through NLRP3 inflammasome and caspase-1 activation in AS DCs.
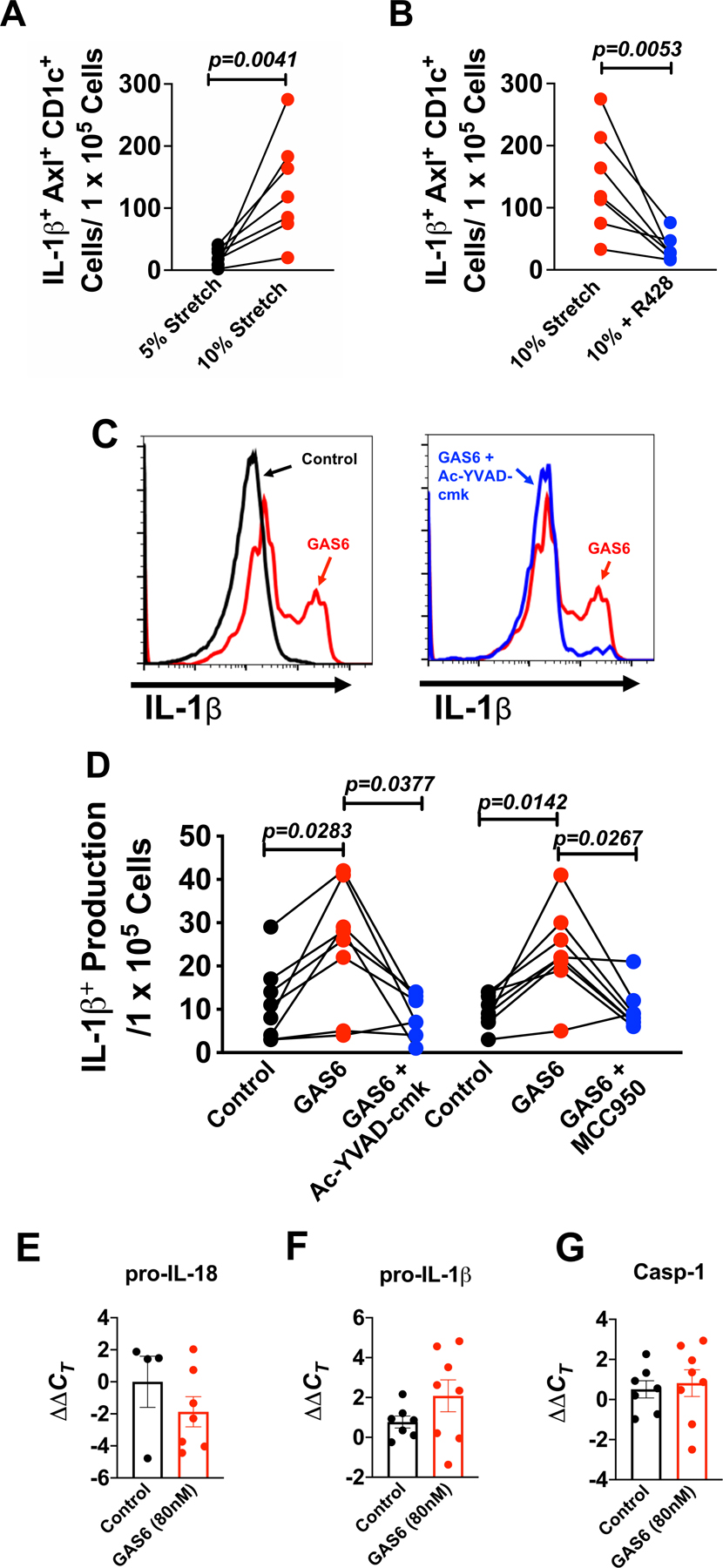
Human CD14+ monocytes were isolated by magnetic sorting from the buffy coat of normotensive human subjects and were co-cultured with HAECs undergoing either 5% or 10% stretch for 48 hours or human monocytes were cultured in control media, GAS6 media (80 nM), GAS6 + caspase-1 inhibitor (Ac-YVAD-cmk; 1µM) or GAS6 + NLRP3 inflammasome inhibitor (MCC950; 1 µM) for 48 hours. Summary data for (A) IL-1β+ Axl+ CD1c+ cells undergoing 5% and 10% endothelial cell stretch and (B) Summary data for IL-1β+ Axl+ CD1c+ cells undergoing 10% or 10% + R428 (1µM; DMSO). Significance was analyzed using the Paired Student’s t-test (n=7 per group). (C) Representative flow cytometry plots showing intracellular IL-1β staining in monocytes treated with control, GAS6, or GAS6 + Ac-YVAD-cmk. (D) Summary data for showing the effect of NLRP3 inflammasome (MCC950) and capase-1 (Ac-YVAD-cmk) inhibition in AS DCs (n=8 per group). Summary data showing the effect of GAS6 exposure on mRNA levels for (E) pro-IL-18 (n=4 control and n=7 GAS6), (F) pro-IL-1β (n=7 control and n=8 GAS6), and (G) caspase-1 (casp-1). All data are represented as mean ± SEM. Panels C and D were analyzed using the Freidman non-parametric test and panels E-G were analyzed using the Mann Whitney test.
GAS6 promotes isoLG-protein adduct formation in AS DCs.
Oxidative events are well established to contribute to hypertension and affect the vasculature, the kidney and central neural control. Our group has also demonstrated reactive oxygen species (ROS) formation is increased in myeloid cells in hypertension, and that this leads to formation of isoLGs that rapidly bind to self-proteins and seem to act as neoantigens that stimulate T cell activation and progression of hypertension.3, 4 We sought to determine if GAS6/Axl signaling in AS DCs leads to the formation of isoLG-protein adducts. We found that GAS6 stimulated isoLG-adduct formation predominantly in Siglec-6+ cells (Figure 5A and B). Given that isoLG formation is usually due to non-enzymatic oxidation of fatty acids,17 this suggests that ROS are involved formation of AS cells. To examine the role of ROS, and isoLG-adducts in AS cell formation we treated human monocytes with polyethylene glycol (PEG)-catalase (500 U/mL) or the isoLG-adduct scavenger 2-HOBA (10 µM) prior to GAS6 (80 nM) exposure. PEG-catalase prevented formation of isoLGs, suggesting a critical role of hydrogen peroxide in formation of these oxidation products. In addition, PEG-catalase exposure prevented surface expression of both Axl and Siglec-6 while scavenging of isoLGs with 2-HOBA had minimal effect on Axl+ cells, but completely suppressed Siglec-6 positive cells, and thus the formation of AS DCs (Figure 5C-E). In subsequent experiments, we tested if inhibition of Axl would prevent the formation of isoLGs in AS DCs under GAS6 stimulation. We found that co-treatment with R428 (1 µM) completely prevented the accumulation of isoLGs (Figure 5F). These findings suggest a pathway whereby oxidants like hydrogen peroxide stimulate formation of Axl and isoLGs and that isoLGs are upstream of Siglec-6 expression.
Figure 5: GAS6 promotes isoLG-protein adduct formation in AS DCs.
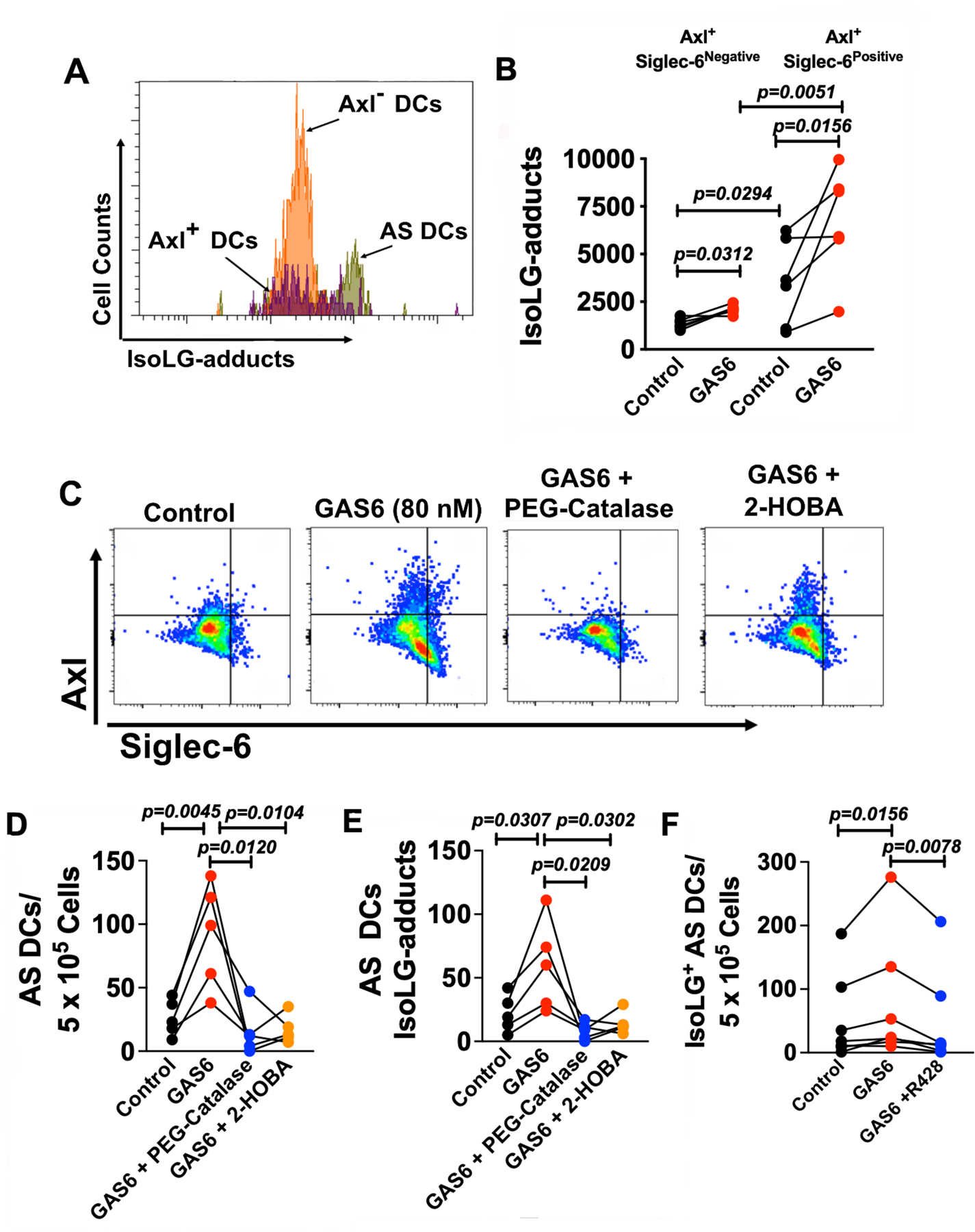
Human monocytes were isolated from total PBMCs of normotensive subjects by magnetic sorting and cultured in control media or media containing GAS6 (80nM) for 48 hours. (A) Representative flow cytometry plots illustrating isoLG-protein adduct formation in AS DCs. (B) Summary data demonstrating the effect of GAS6 exposure on IsoLG-protein adduct accumulation in Axl+ Siglec-6− DCs and Axl+ Siglec-6+ DCs by intracellular D-11 antibody staining (n=6 per group). (C) Representative flow cytometry plots illustrating the effects of GAS6, PEG-catalase (500 U/mL), and 2-HOBA (10 µM) on AS DC formation. Summary data demonstrating the effect of PEG-catalase and 2-HOBA during GAS6 exposure on (D) AS DC formation and (E) isoLG-protein adduct accumulation in AS DCs (n=5 per group). (F) Summary data for the accumulation of isoLG-adducts in AS DCs under GAS6 (80 nM) stimulation with and without R428 (n=8 per group). All data are represented as mean ± SEM. Data in panels B and E were analyzed using a Paired Student’s t test, and panel F by a Wilcoxon non-parametric test.
Axl inhibition prevents Ang II-induced hypertension and renal and aortic inflammation.
Using ELISA, we found that plasma GAS6 levels were increased in mice made hypertensive by a 2-week infusion of Ang II compared to sham infused mice (Figure 6A). We therefore sought to examine the role of Axl signaling in murine hypertension. During Ang II infusion, we found that Axl inhibition by gavage administration R428 (25 mg/kg daily; DMSO) reduced the increase in systolic blood pressure caused by Ang II by approximately 30 mmHg (Figure 6B). Furthermore, inhibition of Axl had no effect on blood pressure in mice not receiving Ang II or on body weight (Figure 6B and 6C).
Figure 6: Axl inhibition prevents Ang II-induced hypertension and renal inflammation.
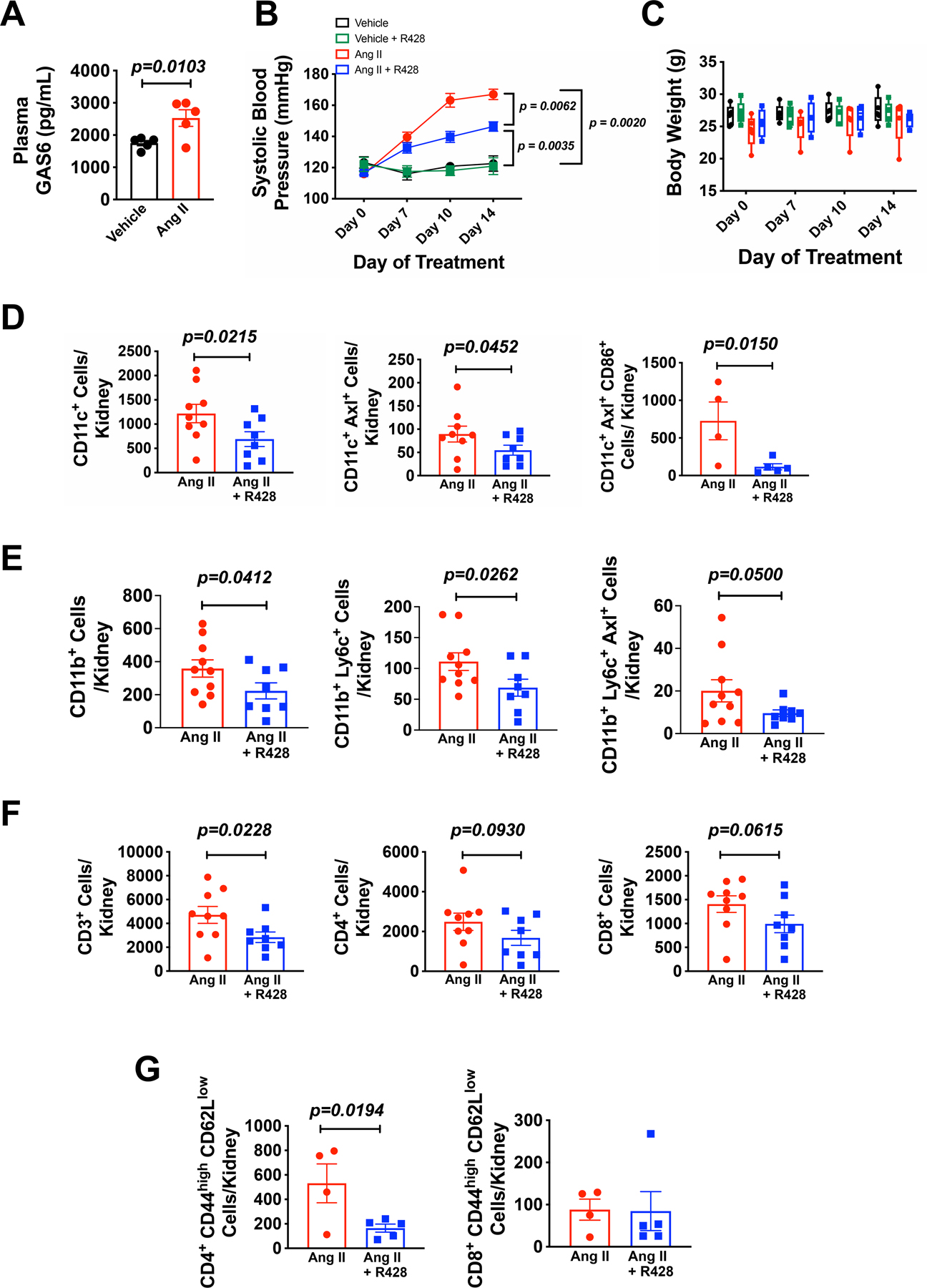
Male C57BL/6 mice received infusion of Ang II (490 ng/min; s.q.) or a sham infusion via osmotic minipumps for 14 days. One group of sham and Ang II mice were treated with the Axl inhibitor R428 (25 mg/kg; p.o.; DMSO) throughout the infusion. (A) Summary data of plasma GAS6 levels in mice subjected to Ang II-induced hypertension or control (n=5 per group). (B) Summary data for blood pressure measurements by tail cuff plethysmography of mice receiving vehicle (black circles; n=5), vehicle + R428 (green squares; n=9), Ang II (red circles; n=5) and Ang II + R428 (blue squares; n=9). (C) Summary data for body weight measurements of vehicle (black bars; n=5), vehicle + R428 (green bars; n=9), Ang II (red bars; n=5) and Ang II +R428 (blue bars; n=9). Flow cytometry summary data for (D) CD11c+ cells, CD11c+ Axl+ Cells (n=9 for Ang II and n=8 for Ang II + R428), and CD11c+ Axl+ CD86+ Cells (n=4 for Ang II and n=5 for Ang II + R428), (E) CD11b+ cells CD11b+ Ly6c+ cells, and CD11b+ Ly6c+ Axl+ cells (n=10 for Ang II and n=8 for Ang II + R428), (F) T lymphocytes (CD45+ CD3+ cells), CD4 T lymphocytes (CD3+ CD4+ cells), and CD8 T lymphocytes (CD3+ CD8+ cells; n=9 for Ang II and n=8 for Ang II + R428), and (G) effector memory CD4 lymphocytes (CD4+ CD44high CD62Llow) and effector memory CD8 lymphocytes (CD8+ CD44high CD62Llow; (n=4 for Ang II and n=5 for Ang II + R428) from single-cell suspensions from the kidney of Ang II and Ang II + R428 treated mice. All data are represented as mean ± SEM. Panels A, D-G were analyzed using a Student’s t-test, and panel B was analyzed by a two-way ANOVA with repeated measures with Bonferonni’s post hoc test.
Next we assessed renal and aortic inflammation by analzying single cell suspensions by flow cytometry using the gating strategy shown in Online Figure VIII A and B. R428 administration reduced renal infiltration of CD11c+, CD11c+ Axl+, and CD11c+ Axl+ CD86+ cells compared to Ang II alone (Figure 6D). Moreover, R428 reduced renal accumulation of CD11b+, CD11b+ Ly6c+, and CD11b+ Ly6c+ Axl+ cells compared to Ang II alone (Figure 6E). Axl inhibition also reduced renal infiltration of CD3+ T lymphocytes, accompanied by a trend in reduction in CD4+ and CD8+ T lymphocytes (Figure 6f), and suppressed renal accumulation of effector memory CD4+ T cells (CD4+ CD44high CD62Llow) T cells but had no significant effect on effector memory CD8+ cells during Ang II infusion (Figure 6G). We also found that inhibition of Axl markedly reduced the aortic infiltration of CD11c+, CD11c+ Axl+, CD11c+ Axl+ CD86+, and CD11b+ Ly6c+ Axl+ cells compared to Ang II alone (Online Figure VIII A-F). Inhibition of Axl had no significant effect on aortic infiltrating CD11b+, CD11b+ Ly6c+, CD3+, CD4+ or CD8+ T lymphocytes or CD4+, CD8+ effector memory T cells compared to Ang II alone (Online Figure IX D-K). Thus, Axl activation contributes to both renal and aortic immune cell infiltration and the genesis of Ang II-induced hypertension.
In accord with our pharmacological inhibition of Axl signaling in vivo, we found that Axl−/− mice exhibited markedly reduced systolic, diastolic, and mean blood pressure compared to Axl+/+ controls during Ang II infusion, as measured by radiotelemetry (Figure 7A). As Axl−/− mice exhibited a lower baseline blood compared to Axl+/+ mice, we compared the change in systolic blood pressure and found that Axl−/− mice had significantly less increase in systolic blood pressure in response to Ang II infusion as compared to Axl+/+ mice (Figure 7B). Cytokines produced by renal infiltrating immune cells can affect renal sodium transporters.1,2 To determine the role of Axl signaling in this process, we infused Ang II for 2 weeks in Axl+/+ and Axl−/− mice and then challenged these animals with an injection of normal saline equal to 10% body weight and monitored urine volume, sodium and chloride excretion over the ensuing 4 hours as previously described.18 As shown in Figure 7C, Axl−/− mice had a marked preservation in their ability to maintain sodium and volume excretion compared to Axl+/+ mice.
Figure 7: Genetic deletion of Axl prevents Ang II induced hypertension and end organ inflammation.
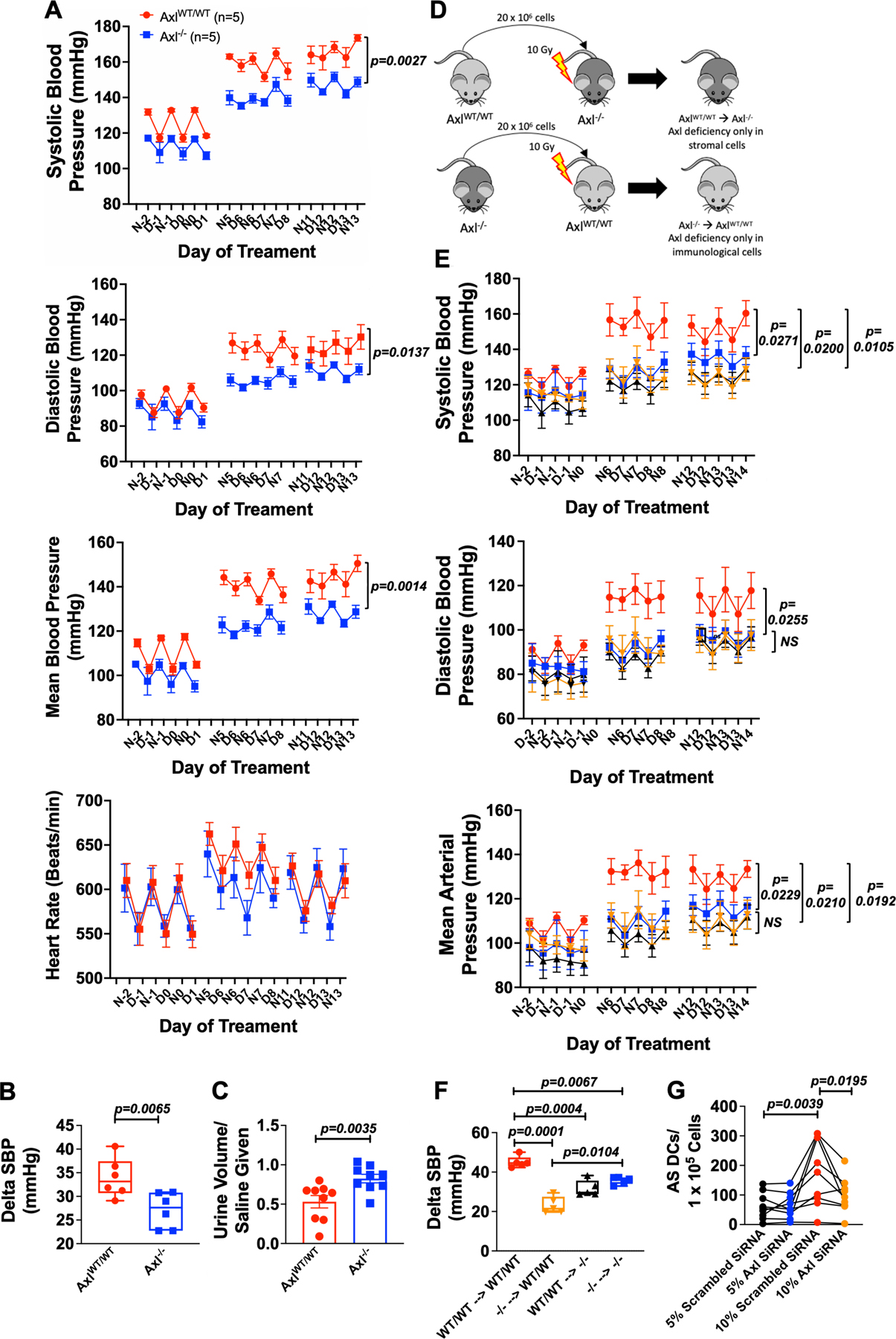
Male Axl WT and Axl−/− (Axl KO) mice were implanted with radiotelemeters and osmotic minipumps delivering Ang II (490 ng/min) for 14 days. Summary data of radiotelemetry for (A) systolic blood pressure, diastolic blood pressure, mean arterial pressure, and heart rate in Axl+/+ and Axl−/− mice (n=6 per group). (B) Summary data of the delta in systolic blood pressure from baseline to last week of Ang II infusion in Axl+/+ and Axl−/− mice. (C) Four-hour saline challenge in Axl WT and Axl KO mice (n=10 for Axl WT and n=9 for Axl KO). (D) Experimental design of BMT studies for the development of bone marrow chimeras. (E) Summary of radiotelemetry data for the BMT mouse experiment (n=5 per group). (F) Summary data of the delta in systolic blood pressure from baseline to last week of Ang II infusion in BMT mice (n=5 per group). (G) Summary data of the formation of AS DCs co-cultured with human aortic endothelial cells from 5% + scrambled siRNA (1 µM), 5% + Axl siRNA (1 µM), 10% + scrambled siRNA (1 µM), and 10% + Axl siRNA (1 µM). (n=9). All data are represented as mean ± SEM. Panels A, B and E-G were analyzed using two-way ANOVA with repeated measures with Bonferroni’s post hoc test and panel C by a Student’s t test.
Deficiency in both Stromal and Immunological Axl Prevent the Development of Hypertension.
Axl is present on hematopoietic cells, endothelial cells, and various stromal cells. To examine the cellular source of Axl that underlies the development of hypertension, we performed BMT experiments. BM (20 × 106 cells) from Axl+/+ or Axl−/− donor mice were transplanted into irradiated Axl+/+ or Axl−/− mice to create chimeric mice that have Axl deficiency in their stromal cells only (Axl+/+ Axl−/−) or hematopoietic cells only (Axl−/− Axl+/+; Figure 7D). Irradiated Axl+/+ transplanted with Axl+/+ BM and irradiated Axl−/− transplanted with Axl−/− BM served as controls. After 8 weeks of BM engraftment, radiotelemeters were implanted to measure blood pressures during infusion of Ang II. We found that baseline blood pressures were significantly lower in Axl+/+ Axl−/− and a trend for reduction in Axl−/− Axl+/+ and Axl−/− Axl−/− compared to Axl+/+ Axl+/+ controls (Online Figure X). Furthermore, we found that loss of hematopoietic cells only (Axl−/− Axl+/+) and stromal cells (Axl+/+ Axl−/−) completely prevented the augmentation of systolic blood pressure, diastolic blood pressure, and mean arterial pressure similar to Axl−/− Axl−/− mice (Figure 7E). Moreover, we assessed the change in systolic blood pressure and found that Axl−/− Axl−/−, Axl+/+ Axl−/−, and Axl−/− Axl+/+ had a significant reduction in the rise in systolic blood pressure compared to wild type mice transplanted with wild type bone marrow (Figure 7F). In keeping with the finding that stromal Axl plays a role in the development of hypertension, we treated HAECs with either scrambled or Axl siRNA and demonstrated effective knockdown of Axl gene expression (Online Figure XI A). We found that when monocytes were co-cultured with HAECs following Axl knockdown, there was a significant decrease in the formation of AS DCs compared to HAECs exposed to scrambled siRNA (Figure 7G). Taken together, these data demonstrated a critical interplay and role of both immunological and stromal Axl signaling in the development hypertension.
Given the above studies showing a role of endothelial Axl, we performed in vitro cell culture experiments where we exposed HAECs treated with vehicle (DMSO) or R428 (1 µM; DMSO) to either 5% or 10% cyclical stretch. We found that 10% cyclical stretch siginificantly increased ICAM-1. GAS6, isoLG-adduct accumulation, and class II MHC compared to 5% stretch. Moreover, inhibition of Axl with R428 significantly reduced ICAM-1, GAS6, accumulation of isoLG-adducts, and the expression of MHC II compared to vehicle, indicating that GAS6/Axl signaling leads to upregulation of activation and injury markers during hypertensive stimuli. (Online Figure XI B-E). To determine if GAS6/Axl signaling promotes oxidant signaling in endothelial cells, we exposed HAECs to either vehicle (control), GAS6 (80 nM), GAS6 + 2-HOBA (1 µM), or GAS6 + PEG-SOD (100 U/mL) for 48 hours. We found that GAS6 exposure significantly upregulated the adhesion molecule ICAM-1, the accumulation of isoLG-adducts, and MHC II. Treatment with the isoLG-adduct scavenger, 2-HOBA, prevented the increase in ICAM-1 and isoLG-adduct accumulation. Moreover, co-treatment with PEG-SOD prevented ICAM-1, GAS6, and isoLG-adduct accumulation in endothelial cells, but not MHC II (Online Figure XI F-I). Taken together with our BMT studies, these data demonstrate that GAS6/Axl signaling coordinates activation of both the endothelium and immune cells to promote an inflammatory environment and the development of hypertension.
GAS6 production is associated with vascular dysfunction and isoLG-adduct accumulation in human endothelial cells.
As shown in Figure 2G, we found that human endothelial cells undergoing hypertensive stretch markedly increase secretion of GAS6. Based on these findings, we sought to determine if GAS6 is associated with parameters of endothelial dysfunction and accumulation of isoLG-adducts in human volunteers. We recruited an additional 23 volunteers whose demographic and clinical characteristics are provided in Online Table IV. Endothelial cells were harvested by J-wire insertion into the brachial vein and subjected to flow cytometric analysis (Figure 8A). Using the gating strategy as shown in Figure 8B we found a positive significant association between GAS6 and ICAM-1, which is produced by activated endothelial cells (Figure 8C R2 = 0.39, p=0.0012). Moreover, we found a strong positive association between endothelial cell isoLG-adduct accumulation and ICAM-1 (Figure 8D; R2=.36, p=0.0026). Interestingly, we found a strikingly positive association between endothelial cell expression of GAS6 and the accumulation of isoLG-adducted proteins (Figure 8E; R2=0.87, p=0.0001), suggesting that activated and dysfunctional endothelial cells are a major source of GAS6 production in humans.
Figure 8: Association of GAS6 production with endothelial dysfunction and accumulation of isoLG-adducts in humans.
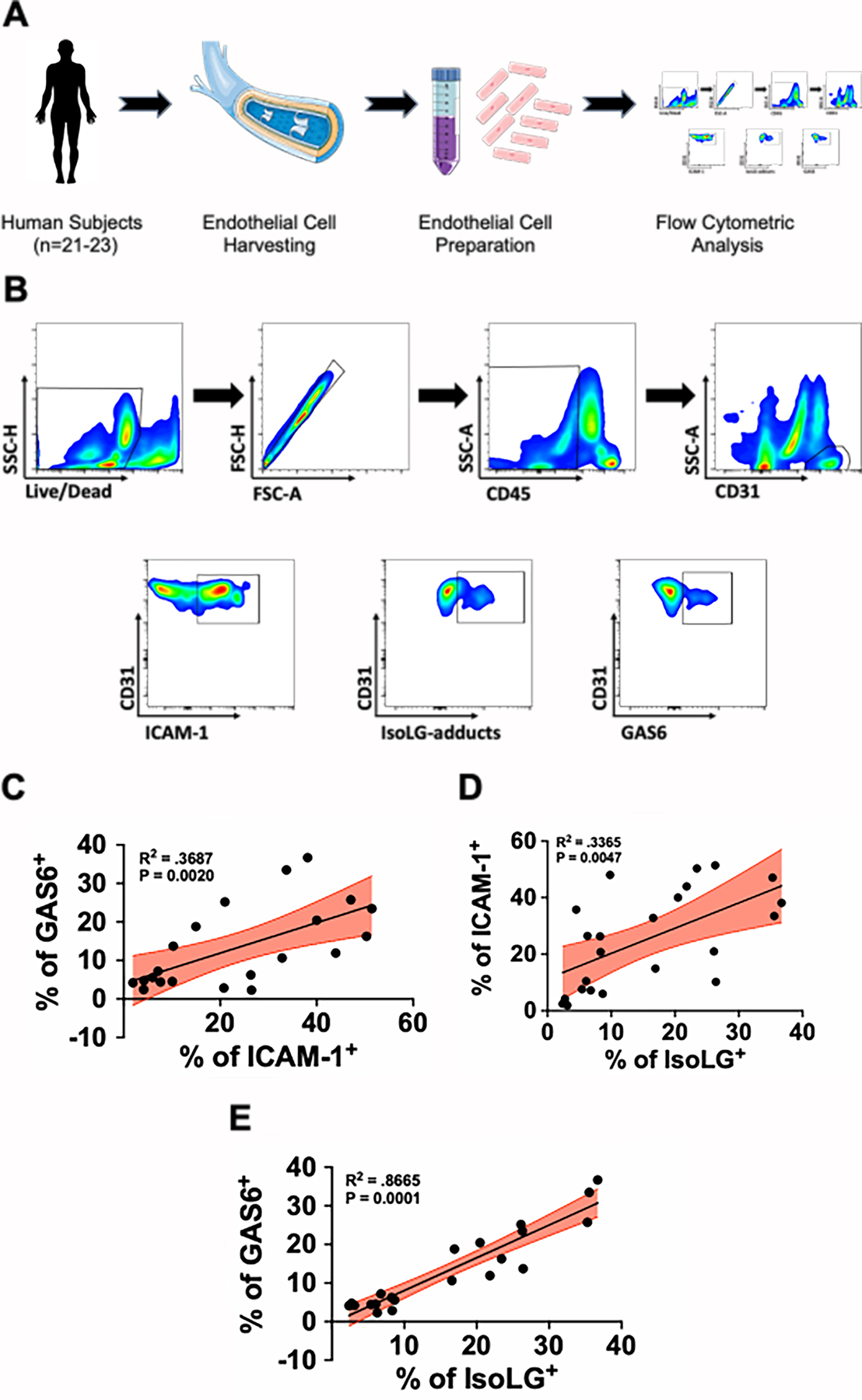
(A) Human venous endothelial cells were harvested by J-wire biopsy and prepared for flow cytometric analysis. (B) Flow cytometry gating strategy to identify human endothelial cells and their production of ICAM-1, isoLG-adducts, and GAS6. (C) Association between GAS6 and ICAM-1 (n=21), (D) ICAM-1 and isoLG-adducts (n=23), and (E) GAS6 and isoLG-adducts (n=21) in endothelial cells from human subjects. Data were analyzed using linear regression and significance computed using two tailed Spearman’s test.
DISCUSSION
In this study, we defined a previously unknown role of GAS6/Axl signaling in human monocyte activation and show that the endothelium can serve as a source of GAS6. Human subjects with hypertension had a significant increase in the presence of circulating AS DCs compared to normotensive subjects and circulating levels of GAS6 correlate with both pulse pressure and systolic blood pressure. In vitro, we found that endothelial cell stretch markedly enhances release of GAS6 which in turn seems to stimulate the formation of human monocyte derived AS DCs. These events are completely abolished by inhibition of Axl and knockdown of endothelial-derived GAS6 and Axl. Stimulation of human monocytes by GAS6 promotes their transformation to AS DCs and markedly enhances IL-1β production through a NLRP3/caspase-1-dependent mechanism. In in vitro studies, we show that exposure of monocytes to GAS6 promotes the formation of immunogenic isoLG-protein adducts in both CD1c+ Axl+ DCs and CD1c+ AS DCs. Moreover, in vivo inhibition of Axl by pharmacological and genetic deletion (Axl−/− mice) markedly reduced systolic blood pressure and renal immune cell infiltration following Ang II-induced hypertension. Our BMT studies demonstrated that both stromal and immunological Axl are required for the development of hypertension following Ang II-infusion. In keeping with this, knockdown of Axl in endothelial cells prevented the ability of stretch to release GAS6 and to activate monocytes, suggesting a feed-forward autocrine role of GAS6 to promote its own release. Lastly, we found that in human endothelial cells there is a positive correlation between GAS6, ICAM-1, and isoLG-adduct formation.
In hypertension, the interaction of monocytes with the vascular endothelium likely leads to formation of monocyte-derived DCs and is at least one source of AS DCs. In keeping with this, Randolph et al showed that monocytes cultured with endothelial cells that had been stimulated with IL-1β, lipopolysaccharide or zymosan differentiate into either DCs or macrophages.7 Furthermore, Randolph et al demonstrated that a subpopulation of monocytes with upregulation of CD86 and HLA-DR potently drive allogenic T-cell proliferation.19 We recently showed that endothelial cells undergoing hypertensive cyclical stretch release factors that cause differentiation into CD14++ CD16+ intermediate monocytes that robustly express IL-1β, IL-6, IL-23 and TNF-α. We also observed that monocytes exposed to endothelial cells undergoing 10% stretch exhibit increased surface CD209, a surface marker of DCs and activated macrophages.8 An important finding in the current study is that endothelial cell hypertensive stretch, which promotes monocyte differentiation also promotes the formation of AS DCs.
The definition of monocyte-derived cells and related myeloid cells is evolving, and it is now clear that these cells exhibit division of duty, serving both pro-and anti-inflammatory roles.5, 20 Increasing evidence has demonstrated that myeloid cells play a critical role in the development and maintenance of hypertension.21, 22 Wenzel et al. showed that ablation of myeloid cells expressing lysozyme M in mice completely abrogated the increase in systolic blood pressure in response to Ang II-induced hypertension and that adoptive transfer of monocytes to these mice reestablished Ang II-induced hypertension.2 Kirabo et al. demonstrated that monocyte derived DCs present immunogenic isoLG-modified proteins that lead to T cell activation and the development of hypertension. Adoptive transfer of DCs from hypertensive mice primed hypertension in recipient mice. Mice lacking Macrophage Colony-Stimulating Factor (Op/Op mice), that lack macrophages and related myeloid cells, are protected from vascular injury and inflammation during both Ang II-induced and DOCA-salt induced hypertension.23, 24 Shah et al. showed that a subpopulation of inhibitory myeloid cells, myeloid-derived suppressor cells, limit the increase in blood pressure and inflammation.25 While our data suggest that AS DCs play a critical role in the development of human hypertension, it is likely that other myeloid cells, including monocytes and macrophages also contribute.
An important finding in the current study is that hypertensive stretch of human endothelial cells induces GAS6 secretion and that GAS6 levels are increased in experimental hypertension. Likewise, pulse pressure, which affects vascular stretch in vivo, correlated with GAS6 plasma levels in humans. GAS6 is the ligand for the tyrosine kinase receptor Axl.26 Elevated GAS6 plasma levels have been observed in various human disease states including sepsis, cardiac hypertrophy, chronic renal disease, and systemic lupus erythematosus.9–11, 13 Lee et al. demonstrated that plasma GAS6 levels increase with the severity of chronic kidney disease.11 Zhao and colleagues demonstrated that transgenic mice over-expressing GAS6 developed cardiac hypertrophy, and that genetic deletion of GAS6 prevented cardiac hypertrophy in response to transaortic constriction.13 Moreover, nephrotoxic nephritis is associated with increased GAS6 production by glomerular capillaries, further suggesting that endothelial cells produce this mediator. Adipocytes have also been shown to produce GAS6 in mice fed a high fat diet and seems to contribute to hypertrophy of these cells. Our data further suggest that endothelial production of GAS6 could promote monocyte transformation, as exposure to this mediator upregulates surface expression of the classical DC marker CD1c and promotes dendrite-like projections by these cells.
Recently the NLRP3 inflammasome has been shown to play an important role in the progression and development of salt-sensitive hypertension. Krishnan et al. demonstrated that in vivo inhibition of the NLRP3 inflammasome with MCC950 reduces blood pressure, renal inflammation, fibrosis, and dysfunction and this was associated with a reduction in pro-inflammatory and injury markers (IL-1beta, IL-17A, ICAM-1, VCAM-1).27 IL-1b is commonly increased in DCs of hypertensive animals, and inhibition of IL-1b signaling abrogates Ang II-induced hypertension.28 Likewise, inhibition of IL-1b reduced cardiovascular events most significantly in humans with the highest quartile of blood pressure in the recent CANTOS trial.29 An important finding of this study is that GAS6 released from endothelial cells undergoing hypertensive stretch promote IL-1β production through a NLRP3/capase-1-dependent mechanism in AS DCs. In keeping with this, macrophage Axl signaling has been demonstrated to promote NLRP3/caspase-1 dependent IL-1β secretion in a model of myocardial infraction.30 It is generally accepted that the synthesis of inflammasome products involves a priming step, in which pro-IL-1b and pro-IL-18 and components of the inflammasome are transcribed, often in response to NFkB activation, and a triggering phase requiring inflammasome assembly and subsequent cleavage of pro-cytokines to mature forms.31 We did not observe an increase in mRNA for caspase 1, pro-IL-1b or IL-18, indicating that GAS6/Axl signaling does not activate the priming step of inflammasome activation, but likely the triggering phase. In keeping with this, we found that caspase inhibition completely abrogated IL-1b production of human monocytes in response to GAS6. The precise mechanisms by which GAS6/Axl signaling leads to inflammasome triggering requires further investigation.
An important finding of the current study is that surface expression of both Axl and Siglec-6 increase in response to GAS6 stimulation while R428 decreased Axl and Siglec-6 surface levels in response to GAS6. These data suggest that Axl ligation by GAS6 signals formation of additional Axl receptors and likewise stimulates Siglec-6 formation. Thus, the interaction of GAS6 with Axl leads to a feed-forward formation of AS cells. We considered the possibility that GAS6 might activate other monocyte receptors including MerTK or Tyro3, however found these receptors to be very low in circulating human monocytes.
The formation of isoLG-adducts, and AS cells in response to GAS6 was also prevented by hydrogen peroxide scavenging, indicating that redox signaling is downstream of Axl activation. While the oxidation of arachidonic acid by hydrogen peroxide is energetically favorable,32 other oxidants that are formed from or act in concert with hydrogen peroxide could also contribute. Indeed both catalase and superoxide dismutase can prevent lipid peroxidation caused by xanthine oxidase, suggesting both superoxide and hydrogen peroxide are involved.33 Peroxidases like myeloperoxidase could also use hydrogen peroxide as a co-substrate to catalyze formation of isoLGs. The precise mechanism by which Axl stimulation induces ROS formation in monocytes requires further investigation however we have previously shown an indispensable role of the NADPH oxidase in formation of isoLGs in response to other stimuli.16
Axl activation has been associated with various cardiovascular disease states including atherosclerosis, myocardial infarction, hypertension, and chronic kidney disease.30, 34–37 Batchu and colleagues demonstrated that deletion of Axl in RAG-1−/− mice caused a profound reduction in systolic blood pressure, renal inflammation and renal dysfunction after the first week of DOCA-salt hypertension.35 Recently, DeBerge et al. demonstrated that Axl activation of macrophages promotes secretion of IL-1β and intramyocardial inflammation. Administration of the selective molecule Axl inhibitor, R428, improved cardiac healing and a reduction in intramyocardial inflammation.30 In accord with this, we found that treatment with R428 and or global Axl deletion reduced hypertension by approximately 20 mmHg. Moreover, R428 reduced Axl+ DCs in both the aorta and kidney with a decreased in renal T lymphocyte immune cell infiltration during Ang II infusion. An important mechanism of hypertension is impaired pressure natriuresis. Data from our group and others have implicated several inflammatory cytokines in modulation of renal sodium transporters.18, 38, 39 In keeping with this, we found that Axl-deficient mice exhibited a preserved ability to excrete a volume challenge during ang II infusion. This is compatible with the scenario that the decrease in renal immune cell infiltration preserved renal excretory capacity.
In keeping with the above, studies have demonstrated that both immunological and stromal Axl play a role in the development of cardiovascular disease. In this study, we found that deletion of either stromal or hematological Axl reduced hypertension in response to Ang II. These data demonstrate the critical and novel GAS6/Axl signaling mechanisms on both immunological and stromal Axl in the development of hypertension. As Axl is ubiquitously expressed in stromal and immunological cells, our data supports additional lines of investigation into this critical signaling mechanism. Moreover, these in vivo data are supported by the knockdown of either GAS6 or Axl in vitro in human aortic endothelial cells on the formation of pro-inflammatory AS DCs. Therefore, one could speculate that therapies targeting GAS6/Axl signaling could favorable impact cardiovascular disease development and progression.
With regard to the importance of GAS6/Axl signaling in vivo, we found a profound positive correlation with endothelial GAS6 production, isoLG-adduct accumulation, and endothelial cell dysfunction, as evidenced by ICAM-1 expression, in human subjects with cardiometabolic disease. A limitation of our study is that we harvested venous endothelial cells for this analysis and thus cannot make conclusions regarding the direct effect of hypertension on arterial cells. Indeed, only 4 of our subjects with cardiometabolic syndrome had overt hypertension and did not exhibit greater GAS6 expression than the non-hypertensive subjects in their venous endothelial cells. Nevertheless, our data indicate that human endothelial cells in vivo can exhibit a coordinated increase in an important adhesion molecule, oxidatively modified proteins and GAS6 production. Of interest, mice with global deletion of GAS6 fail to upregulate endothelial ICAM-1 and VCAM-1 when treated with TNF-α both in vitro and in vivo compared to wildtype controls.40 Moreover, GAS6 stimulation of human umbilical vein endothelial cells leads to upregulation of both ICAM-1 and VCAM-1 through a Mer-dependent mechanism.41 Taken together, out data are comparable with a scenario in which exposure to hypertensive pressures or increased cyclical stretch causes endothelial cells to simultaneously upregulate expression of adhesion molecules like ICAM-1 that enhance the interaction of monocytes with the endothelium and GAS6, which signals formation of the pro-inflammatory AS DCs.
In summary, our current study provides new insight how the endothelium can promote immune activation in hypertension and show a potential new role for AS DCs and Axl signaling via GAS6 activation in this disease. AS DCs may not only be a biomarker of inflammation in hypertension but can serve as a source of IL-1β and downstream cytokines such as IL-6. Importantly, production of such cytokines and presentation of isoLG-adducts can drive T cell polarization and the production of cytokines like IL-17A, which are pathogenic in hypertension. It is therefore likely that the activated endothelium, in addition to expressing chemokines and adhesion molecules, can promote formation monocyte-derived DCs leading to end organ dysfunction and worsening blood pressure elevation in hypertension.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Inflammation and immune activation have been observed in human and experimental hypertension for more than 50 years.
The interaction with the activated endothelium leads monocytes to transform to dendritic cells and macrophages by mechanisms that have been poorly defined.
Recently dendritic cells with the surface markers Axl and Siglec 6 (AS cells) have been described as similar to cells observed in hypertension.
What New Information Does This Article Contribute?
Circulating AS cells are increased in hypertensive humans compared to non-hypertensive subjects.
The ligand for Axl, GAS6 is also increased in hypertensive humans and correlates with the level of systolic pressure and pulse pressure.
Human endothelial cells undergoing hypertensive cyclical stretch release of GAS6 and promote the transformation of co-cultured monocytes to AS cells that produce IL-1β and present antigen to autologous T cells.
Axl signaling in both hematopoietic and stromal cells contributes to Ang II-induced hypertension in mice, and Axl blockade with R428 reduced blood pressure and renal inflammation.
Axl signaling in the endothelium also leads to endothelial activation in an autocrine fashion, promoting the expression of reactive oxygen species and the intracellular adhesion molecule ICAM-1.
This study shows that the activated endothelium in hypertension releases GAS6, which potently activates nearby monocytes to transform into proinflammatory antigen presenting cells. This is a novel example of vascular/immune cross-talk likely relevant to many cardiovascular disorders. Furthermore, we show that either pharmacological or genetic blockade of this pathway in vivo lowers blood pressure and reduces end-organ damage. Our findings improve understanding of immune activation in hypertension and identify a potential therapeutic target to treat this devastating chronic disease.
SOURCES OF FUNDING
This work was supported by the American Heart Association grants 18POST34020009 to NRB, 20PRE35080177 to CDS, 17SDG33670829 to LX and National Institutes of Health grants F32HL132937 to JPVB, F30HL151069 to CDS, K08HL153789, F32HL144050 and T32GM007569 to DMP 5R01DK111175–01 to CAS, 5R35HL140016–02 and 5P01HL129941–03 to DGH, Department of Veteran Affairs grants 1IK2BX005605–01 to JPVB and 1IK2BX005376 to DMP, the Vanderbilt Institute for Clinical and Translational Research, and National Center for Advancing Translational Sciences (NCATS) Clinical Translational Science Award (CTSA) program 5UL1TR002243–03 to CAS.
Nonstandard Abbreviations And Acronyms:
- Ang II
angiotensin II
- AS DC
Axl+ Siglec-6+ dendritic cell
- BM
Bone marrow
- BMT
Bone marrow transplant
- cDC1
myeloid/classical dendritic cell 1
- cDC2
myeloid/classical dendritic cell 2
- DC
dendritic cell
- FMO
fluorescence minus one
- GAS6
growth arrest specific 6
- HAEC
human aortic endothelial cell
- IL-1β
interleukin 1-beta
- ICAM-1
intracellular adhesion molecule 1
- IsoLG
isolevuglandin
- pDC
plasmacytoid dendritic cell
- PEG
polyethylene glycol
- PBMC
peripheral blood mononuclear cell
- ROS
reactive oxygen species
- Siglec-6
sialic acid-binding immunoglobulin-type lectin 6
- TLR
toll-like receptor
- VZV
varicella zoster virus
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
None.
REFERENCES
- 1.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme m-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381 [DOI] [PubMed] [Google Scholar]
- 3.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. J Clin Invest. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circ Res. 2015;117:547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collin M, Bigley V. Human dendritic cell subsets: An update. Immunology. 2018;154:3–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N. Single-cell rna-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483 [DOI] [PubMed] [Google Scholar]
- 8.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand CM, Guzik TJ, Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of stat3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114:1547–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekman C, Linder A, Akesson P, Dahlback B. Plasma concentrations of gas6 (growth arrest specific protein 6) and its soluble tyrosine kinase receptor saxl in sepsis and systemic inflammatory response syndromes. Crit Care. 2010;14:R158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekman C, Jonsen A, Sturfelt G, Bengtsson AA, Dahlback B. Plasma concentrations of gas6 and saxl correlate with disease activity in systemic lupus erythematosus. Rheumatology (Oxford). 2011;50:1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee IJ, Hilliard B, Swami A, Madara JC, Rao S, Patel T, Gaughan JP, Lee J, Gadegbeku CA, Choi ET, Cohen PL. Growth arrest-specific gene 6 (gas6) levels are elevated in patients with chronic renal failure. Nephrol Dial Transplant. 2012;27:4166–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu KS, Hung YJ, Lee CH, Hsiao FC, Hsieh PS. The involvement of gas6 signaling in the development of obesity and associated inflammation. Int J Endocrinol. 2015;2015:202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao YF, Xu DC, Zhu GF, Zhu MY, Tang K, Li WM, Xu YW. Growth arrest-specific 6 exacerbates pressure overload-induced cardiac hypertrophy. Hypertension. 2016;67:118–129 [DOI] [PubMed] [Google Scholar]
- 14.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, Itani HA, Himmel LE, Harrison DG, Kirabo A. High salt activates cd11c(+) antigen-presenting cells via sgk (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand C, Guzik TJ, Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of stat3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018 [DOI] [PMC free article] [PubMed]
- 16.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies SS, Amarnath V, Roberts LJ 2nd. Isoketals: Highly reactive gamma-ketoaldehydes formed from the h2-isoprostane pathway. Chem Phys Lipids. 2004;128:85–99 [DOI] [PubMed] [Google Scholar]
- 18.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Randolph GJ, Sanchez-Schmitz G, Liebman RM, Schakel K. The cd16(+) (fcgammariii(+)) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J Exp Med. 2002;196:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcantara-Hernandez M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, Davis MM, Nolan GP, Idoyaga J. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity. 2017;47:1037–1050 e1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu X, Crowley SD. Inflammation in salt-sensitive hypertension and renal damage. Curr Hypertens Rep. 2018;20:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. 2018;215:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin ii-infused macrophage colony-stimulating factor-deficient mice: Evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:2106–2113 [DOI] [PubMed] [Google Scholar]
- 24.Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, Schiffrin EL. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: Evidence from m-csf-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292:H1789–1795 [DOI] [PubMed] [Google Scholar]
- 25.Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y, Zhao T, Harrison DG, Bernstein KE, Shen XZ. Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res. 2015;117:858–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, Jones DS 2nd, Kapur S, Mathews II, Koong AC, Rankin EB, Cochran JR, Giaccia AJ. Inhibition of the gas6/axl pathway augments the efficacy of chemotherapies. J Clin Invest. 2017;127:183–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, Diep H, Kett MM, Samuel CS, Kemp-Harper BK, Robertson AAB, Cooper MA, Peter K, Latz E, Mansell AS, Sobey CG, Drummond GR, Vinh A. Pharmacological inhibition of the nlrp3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. 2019;115:776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the nkcc2 co-transporter in the nephron. Cell Metab. 2016;23:360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, Libby P, Glynn RJ, Ridker PM. Effects of interleukin-1beta inhibition on blood pressure, incident hypertension, and residual inflammatory risk: A secondary analysis of cantos. Hypertension. 2020;75:477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBerge M, Glinton K, Subramanian M, Wilsbacher LD, Rothlin CV, Tabas I, Thorp EB. Macrophage axl receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J Clin Invest. 2021;131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buettner GR. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543 [DOI] [PubMed] [Google Scholar]
- 33.Kellogg EW 3rd, Fridovich I. Superoxide, hydrogen peroxide, and singlet oxygen in lipid peroxidation by a xanthine oxidase system. J Biol Chem. 1975;250:8812–8817 [PubMed] [Google Scholar]
- 34.Batchu N, Hughson A, Wadosky KM, Morrell CN, Fowell DJ, Korshunov VA. Role of axl in t-lymphocyte survival in salt-dependent hypertension. Arterioscler Thromb Vasc Biol. 2016;36:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batchu SN, Dugbartey GJ, Wadosky KM, Mickelsen DM, Ko KA, Wood RW, Zhao Y, Yang X, Fowell DJ, Korshunov VA. Innate immune cells are regulated by axl in hypertensive kidney. Am J Pathol. 2018;188:1794–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerloff J, Korshunov VA. Immune modulation of vascular resident cells by axl orchestrates carotid intima-media thickening. Am J Pathol. 2012;180:2134–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu F, Zhuang S. Role of receptor tyrosine kinase signaling in renal fibrosis. Int J Mol Sci. 2016;17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17a regulates renal sodium transporters and renal injury in angiotensin ii-induced hypertension. Hypertension. 2016;68:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tjwa M, Bellido-Martin L, Lin Y, Lutgens E, Plaisance S, Bono F, Delesque-Touchard N, Hervé C, Moura R, Billiau AD, Aparicio C, Levi M, Daemen M, Dewerchin M, Lupu F, Arnout J, Herbert JM, Waer M, García de Frutos P, Dahlbäck B, Carmeliet P, Hoylaerts MF, Moons L. Gas6 promotes inflammation by enhancing interactions between endothelial cells, platelets, and leukocytes. Blood. 2008;111:4096–4105 [DOI] [PubMed] [Google Scholar]
- 41.Furukawa M, Wang X, Ohkawara H, Fukatsu M, Alkebsi L, Takahashi H, Harada-Shirado K, Shichishima-Nakamura A, Kimura S, Ogawa K, Ikezoe T. A critical role of the gas6-mer axis in endothelial dysfunction contributing to ta-tma associated with gvhd. Blood Adv. 2019;3:2128–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings will be available from DGH upon reasonable request. An expanded methods section is available in the Online Supplemental Material.