

Abstract
Agents that raise cyclic guanosine monophosphate (cGMP) by activating protein kinase G increase 26S proteasome activities, protein ubiquitination and degradation of misfolded proteins. Therefore, they may be useful in treating neurodegenerative and other diseases caused by an accumulation of misfolded proteins.
Mutations in myelin protein zero (MPZ) cause the peripheral neuropathy Charcot-Marie-Tooth type 1B (CMT1B). In peripheral nerves of a mouse model of CMT1B, where the mutant MPZS63del is expressed, proteasome activities are reduced, mutant MPZS63del and polyubiquitinated proteins accumulate and the unfolded protein response (p-eif2α) is induced. In HEK293 cells, raising cGMP stimulated ubiquitination and degradation of MPZS63del, but not of wild-type MPZ. Treating S63del mice with the phosphodiesterase 5 inhibitor, sildenafil—to raise cGMP—increased proteasome activity in sciatic nerves and reduced the levels of polyubiquitinated proteins, the proteasome reporter ubG76V-GFP and p-elF2α. Furthermore, sildenafil treatment reduced the number of amyelinated axons, and increased myelin thickness and nerve conduction velocity in sciatic nerves.
Thus, agents that raise cGMP, including those widely used in medicine, may be useful therapies for CMT1B and other proteotoxic diseases.
Keywords: cGMP, proteasome, protein degradation, Charcot Marie Tooth, proteostasis
VerPlank et al. report that raising cGMP levels with the PDE5 inhibitor sildenafil in a mouse model of Charcot Marie Tooth 1B neuropathy restores proteostasis, myelin thickness, and nerve conduction. Agents that increase cGMP may be a promising therapy for CMT1B neuropathy, which has no treatment, and other proteotoxic diseases.
Introduction
The ubiquitin proteasome system (UPS) degrades the great majority of proteins in mammalian cells.1 It also catalyses the selective destruction of misfolded, mutated and damaged proteins, and is therefore a critical line of defence against the accumulation of aggregation-prone proteins that cause many neurodegenerative diseases.2 In the UPS, proteins to be degraded are conjugated to chains of ubiquitin molecules, which mark them for rapid hydrolysis by the 26S proteasome. Most studies on the mechanisms regulating degradation rates have focused on ubiquitin conjugation to proteins.3 However, it is now well established that proteasome activity is also precisely regulated, especially by phosphorylation.4,5 The phosphorylation of proteasome components can increase or decrease proteasome activity and thus can influence the degradation rates of cell proteins.
A number of protein kinases have been reported to phosphorylate proteasomes and alter their activity. However, only three have been shown to affect intracellular protein degradation: dual specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2),6 cyclic guanosine monophosphate (cGMP)-dependent kinase (PKG)7 and cyclic adenosine monophosphate (cAMP)-dependent kinase (PKA).8,9 Pharmacological agents or hormones (e.g. epinephrine) that raise cAMP levels and activate PKA stimulate proteasomal activities and enhance the degradation of the short-lived fraction of cell proteins, which includes misfolded, damaged and regulatory proteins.8,9 A similar activation of proteasomes and protein degradation occurs in the skeletal muscles of humans and rats when epinephrine is released during intense exercise.8 Many neurodegenerative diseases are associated with the toxic accumulation of misfolded or mutated proteins.2 Therefore, activating their degradation by treatments that raise cAMP represents a promising new therapeutic strategy to slow the progression of these presently untreatable diseases. In fact, raising levels of cAMP can reduce the accumulation of mutant tau in cultured cells9 and in brains of a mouse model of frontotemporal dementia10 and in a zebrafish larval model of a tauopathy.7 However, raising cAMP levels in brains can have undesirable effects in humans (e.g. emesis).11 Consequently, we have investigated other signalling pathways that may also stimulate proteolysis.
cGMP is synthesized when nitric oxide (NO) binds to its receptor, the soluble guanylyl cyclase (sGC). The actions of cGMP are terminated by phosphodiesterases (PDEs), which hydrolyse it to guanosine monophosphate (GMP). cGMP influences many processes via activation of PKG, the most widely studied of which is to cause the relaxation of smooth muscles and vasodilation.12 Several pharmacological agents that raise cGMP have been approved by the US Food and Drug Administration and are widely used in medicine.11,13 Two classes of approved drugs raise cGMP via this NO pathway: phosphodiesterase 5 (PDE5) inhibitors (e.g. sildenafil, tadalafil), which are widely used to treat erectile dysfunction and pulmonary hypertension,11 and sGC stimulators (e.g. riociguat, vericiguat), which are used to treat pulmonary hypertension and cardiac failure.13
We showed that raising intracellular cGMP with PDE5 inhibitors or sGC stimulators activates 26S proteasomes within minutes via phosphorylation by PKG and stimulates the ubiquitination of cell proteins.7 These two actions led to enhanced degradation of both the short- and long-lived fractions of cell proteins without any change in autophagy. This faster degradation by the UPS of long-lived proteins, which comprise the bulk of cell proteins, differentiates cGMP’s effects on protein breakdown from those of cAMP.9 In zebrafish larval models of tauopathies, raising cGMP with a PDE5 inhibitor increased proteasome activity and the degradation of the mutant tau and reduced the associated neuronal death and morphological abnormalities. In addition, in a zebrafish larval model of Huntington’s disease, these agents reduced the amount of aggregates containing a mutant huntingtin with an expanded polyQ sequence.7 Similar beneficial effects in these disease models were seen when cAMP was raised with a PDE4 inhibitor.7 This shared ability of cAMP and cGMP to enhance proteasome activity and the degradation of short-lived proteins is likely to be responsible for their similar actions in these disease models.
Stimulation of proteasome activity is of therapeutic interest because there is growing evidence that 26S proteasome function and protein degradation by the UPS are impaired in neurodegenerative diseases caused by the expression of aggregation-prone proteins.2 A loss of 26S proteasomal activity has been observed in cellular and vertebrate models of hereditary peripheral neuropathies,14 amyotrophic lateral sclerosis,15 bovine spongiform encephalitis16,17 and tauopathies.10,18 Decreased protein degradation leads to further accumulation of the misfolded, toxic proteins and loss of protein homeostasis. Therefore, enhancing proteasomal activity by raising cGMP could be a new therapeutic approach to reduce the levels of the toxic proteins and to slow progression of these diseases.
Here we tested this possibility in the S63del transgenic mouse model of the hereditary peripheral neuropathy Charcot-Marie-Tooth disease type 1B (CMT1B),19 which is caused by a mutation in myelin protein zero (MPZ). The protein is synthesized by Schwann cells, the myelinating glia of the peripheral nervous system, and over 180 mutations in MPZ are known that cause hereditary peripheral neuropathies.20 The deletion of serine 63 (MPZS63del) causes CMT1B through gain of toxic function mechanisms.19 In S63del mice, the MPZS63del protein accumulates in the endoplasmic reticulum (ER) of Schwann cells, where it induces a canonical unfolded protein response (UPR).19,21,22 In the peripheral nerves of these mice, the ability of 26S proteasomes to degrade ubiquitinated proteins is impaired, and the rates of degradation of both short- and long-lived proteins are reduced.14 Thus, the processes that are impaired in S63del resemble ones that can be activated by cGMP.
This study was undertaken to test whether raising cGMP in S63del mice with the PDE5 inhibitor sildenafil can restore proteasome function and protein degradation by the UPS in the peripheral nerves. We also examined whether raising cGMP enhances the ubiquitination and degradation of MPZS63del and whether activating degradation with cGMP in these mice reduces the associated markers of proteotoxic stress in motor nerves. Most importantly, we tested whether sildenafil treatment not only has these beneficial biochemical actions, but can also reverse the defects in myelination and the slow nerve conduction associated with CMT1B.
Materials and methods
Mouse models
All experiments involving animals followed protocols approved by Institutional Animal Care and Use Committee at the University at Buffalo. S63del and ubG76V-GFP transgenic mice have been described previously.19,23 All mice were maintained on the congenic FVB background, housed on a 12-h light/dark cycle and both sexes were used equally. Genotyping of transgenic mice was performed by PCR on tail genomic DNA extracted with NaOH/EDTA. All experiments were performed on tissues from mice between postnatal Day 37 (P37) and P47.
Stock solutions of sildenafil (ApexBio) were prepared in dimethyl sulphoxide (DMSO) and then prior to injection, mixed with sterile PBS at a concentration of 0.3 mg/ml. For vehicle controls, DMSO was added to sterile PBS. Mice were injected intraperitoneally with 10 mg/kg sildenafil or vehicle. Injections were performed in the mouse home cages twice a day, one in the morning and one in the evening, ∼8 h apart.
Electrophysiology
Mice were anaesthetized with 20 mg/ml of tribromoethanol (Avertin), 0.02 ml/g of body weight. Nerve conduction velocity, F-wave latency and distal latency were measured as described previously.24
Morphology
Semithin section and electron microscopy were performed as described previously.24 Ultrastructural images were acquired on a FEI Tecnai G2 Spirit BioTWIN electron microscope. Quantification of images for g-ratio (axon diameter/fibre diameter) and axonal distribution was performed on electron microscopic images of thin sections of sciatic nerves. At least 15 images and 65 axons were quantified per mouse.
Quantification of amyelinated fibres was performed on three semithin images per sciatic nerve. Two hundred and fifty fibres were counted per image. Both the g-ratio and number of amyelinated fibres per nerve were measured using ImageJ software.
Lysis of sciatic nerves for immunoblotting
Flash-frozen sciatic nerves were ground with a mortar and pestle in liquid nitrogen and then suspended in RadioImmunoPrecipitation Assay (RIPA) lysis buffer: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM NaF, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 mM N-ethylmalmeimide. Samples were vortexed on high for 10 min at 4°C and then centrifuged at 10 000g for 10 min at 4°C. Relative protein concentration of each lysate was determined by bicinchoninic acid (BCA) assay (ThermoFisher Scientific). The volumes of lysates were adjusted with lysis buffer to ensure that all samples were of equal concentration. Lithium dodecyl sulphate (LDS) Bolt sample buffer (Invitrogen) was added to a final concentration of 1×, and then samples were boiled for 5 min at 95°C. Sodium dodecyl sulphate–polyacrylamide gel electrophoresis was performed with 4–12% Bis–Tris Plus polyacrylamide gels (ThermoFisher Scientific). Proteins were transferred to either nitrocellulose (Protran; VWR) or polyvinylidene fluoride (PVDF) (Immobilin FL; EMD Millipore) membranes and immunoblotting was performed using the following antibodies: Ubiquitin (VU1, Life Sensors), K48-linked polyubiquitin (D9D5, Cell Signaling), pVASP (3114, Cell Signaling), pPDE5 (36930, Genetex), PSMB5 (A303-847, Bethyl), Rpn6 (D1T1R, Cell Signaling), Protein Kinase G (C8A4, Cell Signaling), PSMA6 (A303-809, Bethyl), Rpt5 (BML-PW8770, Enzo), PSMA7 (15219, ProteinTech), PSMB1 (D-9, Santa Cruz), PSMD1 (C-7, Santa Cruz), GAPDH (G9545, Sigma), β-tubulin (AA2, EMD Millipore), GFP (C-2, Santa Cruz), HA (HA-7, Sigma), eIF2α (2103, Cell Signaling), p-eif2α (3597, Cell Signaling) and GRP78 (06274, Novus). Visualization was performed with the Odyssey CLx infrared imaging system (LiCor) and quantified with ImageStudio.
Measuring proteasome peptidase activity in sciatic nerve lysates
Flash-frozen sciatic nerves were ground with a mortar and pestle in liquid nitrogen and then suspended in a buffer containing: 25 mM HEPES-KOH pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM ATP, 1 mM DTT and the following inhibitors of proteases and phosphatases: 0.1 mM PMSF, 1 mM NaF, 10 mM β-glycerophosphate, 10 nM calyculin A and 50 nM okadaic acid. Samples were then sonicated and centrifuged at 10 000g for 10 min at 4°C. Relative protein concentration of each lysate was determined by the detergent-compatible Bradford Assay (ThermoFisher). Proteasome peptidase activity in the lysate was then measured as previously described.14
Purification of 26S proteasomes and measurements of proteasomal activity
The Ubl-method was used to purify 26S proteasomes from mouse brains, hearts, and skeletal muscles as described previously.25 Proteasomal hydrolysis of small peptides and ATP was measured as previously described.7 K48-linked superfolder-GFP was prepared as described26 and degradation by 26S proteasomes was measured by loss of green fluorescent protein (GFP) fluorescence (excitation: 475 nm; emission: 515 nm) on a SpectraMax M5 plate reader.
Reagents for cell culture
Sildenafil, riociguat and DETA NONOate were purchased from Cayman Chemical and cycloheximide was purchased from Sigma. Sildenafil and riociguat were suspended in DMSO and used at 1 μM. DETA NONOate was suspended in 0.01 M NaOH and used at 1 μM. Cycloheximide was added to media at a concentration of 150 μg/ml.
Degradation and ubiquitination of MPZS63del and MPZWT in HEK293 cells
HEK293 cells stably transfected with the Mpz or MpzS63del gene under a doxycycline-inducible promoter27 were treated with doxycycline (0.1 µg/ml) for 17 h prior to the beginning of the experiments. Cells were then exposed to DMSO, sildenafil (1 µM) or riociguat (1 µM) for 2, 4 or 6 h in the presence of cycloheximide (150 µg/ml) to block translation. The NO donor DETA NONOate (1 µM) was added with sildenafil or riociguat. Cells were washed twice in ice-cold PBS, lysed in RIPA buffer and centrifuged as described above. Protein levels of MPZWT or MPZS63del in each sample at each time point were measured by western blot against the haemagglutinin (HA)-tag.
To measure ubiquitinated MPZ proteins, expression was induced as above, and then cells were treated for 30 min in the presence of doxycycline with DMSO, DETA NONOate, DETA NONOate + sildenafil or DETA NONOate + riociguat. Following treatment, cells were lysed as above, and then incubated with anti-HA magnetic beads (ThermoFisher Scientific) rotating for 2 h at 4°C. Beads were then washed three times with lysis buffer and then HA-tagged proteins were eluted by boiling for 5 min in 2× non-reducing LDS sample buffer.
Data availability
All data in this report are available to all readers. The reagents used in this study, as well as further information, are available upon request from A.L.G.
Results
Sildenafil treatment of mice for 5 days increases 26S proteasome activity in peripheral nerves
To determine if raising cGMP can activate proteasomes in sciatic nerves of wild-type mice and those with CMT1B, S63del mice and wild-type littermates were treated with the PDE5 inhibitor sildenafil twice a day for 5 days starting at age P40, and then the proteasomes’ peptidase activity was measured in lysates of the sciatic nerves. The sildenafil treatment increased this activity by ∼50% in both wild-type and S63del mice (Fig. 1A). Because this increase in proteasome activity occurred without any increase in the levels of 26S proteasome subunits detected by western blot (Fig. 1B), it is most likely due to phosphorylation and activation of proteasomes by PKG, as occurs in many cultured cell lines and zebrafish larvae when PKG is activated with sildenafil.7
Figure 1.
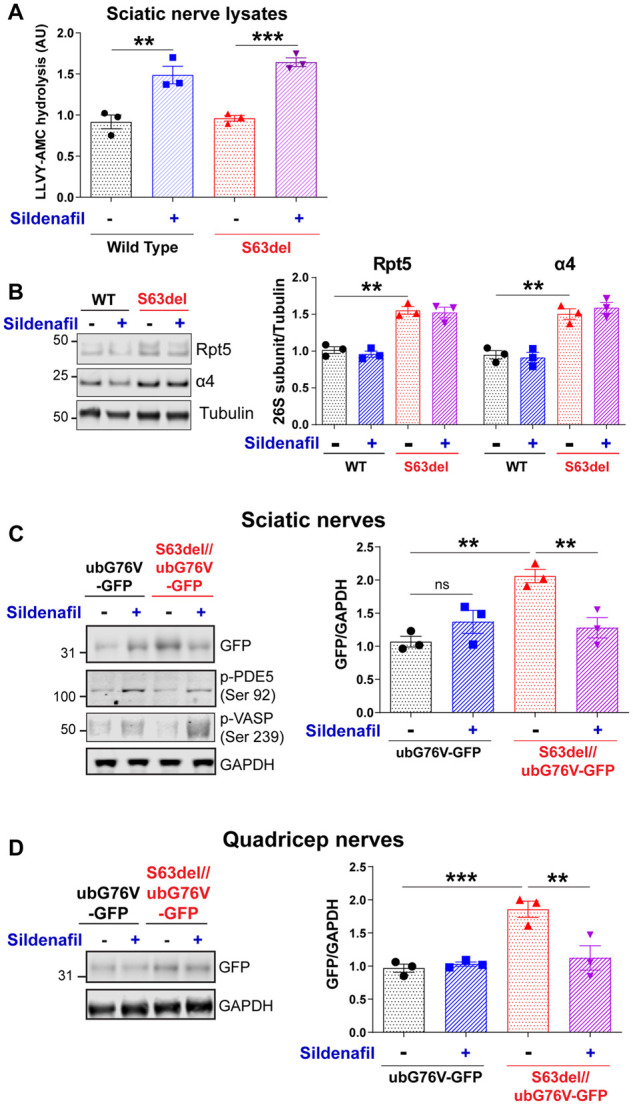
Sildenafil treatment for 5 days increases proteasome activity and protein degradation in the peripheral nerves of S63del mice. (A) Proteasomal chymotrypsin-like activity was increased in the lysates of sciatic nerves from wild-type (WT) and S63del mice following a 5-day treatment with the PDE5 inhibitor sildenafil to raise cGMP levels. n = 3 mice per genotype per condition. One-way ANOVA with Bonferroni post hoc analysis comparing wild-type vehicle and wild-type sildenafil, wild-type vehicle and S63del vehicle and S63del vehicle and S63del sildenafil. Here and throughout, error bars represent SEM and *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (B) Levels of proteasome subunits Rpt5 and α4 in sciatic nerve lysates were not changed by the sildenafil treatment of wild-type and S63del mice. The lysates from S63del sciatic nerves have higher levels of proteasome subunits than wild-type.14 Here and below all blots are cropped. Uncropped versions can be found in the Supplementary material. (C) In the sciatic nerves of S63del mice expressing the UPS reporter protein, ubG76V-GFP, the levels of ubG76V-GFP were higher than in wild-type and sildenafil treatment reduced ubG76V-GFP towards the levels seen in wild-type littermates. The phosphorylation of two well-characterized substrates of PKG, PDE5 and VASP, was increased in sciatic nerves from sildenafil-treated mice, indicating activation of PKG. (D) In the quadricep nerves, which are composed solely of motor fibres, the content of ubG76V-GFP in the S63del mice was twice that seen in wild-type. Sildenafil treatment reduced the amount of detectable ubG76V-GFP, as it did in the sensory–motor mixed sciatic nerves (C).
As reported previously, the lysates of sciatic nerve from untreated S63del mice contained higher levels of proteasome subunits than those from wild-type littermates (Fig. 1B).14 This increase in proteasome number in S63del is likely the reason why the total proteasomal activity was similar in sciatic nerve lysates from the wild-type and S63del mice, even though the specific activity of 26S proteasomes was reduced in the sciatic nerves of S63del mice.14 The Schwann cells in the S63del mice thus seem to compensate for their impaired proteasome activity by producing more 26S proteasomes, as occurs in other cells in response to pharmacological inhibition of proteasomes.28 However, the increased number of proteasomes in S63del does not fully compensate for their functional defects because polyubiquitinated proteins still accumulate in the affected sciatic nerves14 (see below).
Administering sildenafil to the wild-type mice for 5 days increased proteasome activity not only in the sciatic nerves, but also in heart, skeletal muscles and presumably other tissues without an increase in the levels of proteasome subunits (Supplementary Fig. 1A–D). Because these tissues are much larger than the sciatic nerves, they were used in further studies to confirm sildenafil’s actions. As expected, phosphorylation of the well-characterized PKG substrates, VASP or PDE5, increased in these tissues (Supplementary Fig. 1B and D), indicating an activation of PKG. To prove that sildenafil was activating 26S proteasomes, these particles were affinity-purified via the UBL-method25 from the hearts and skeletal muscles of the wild-type mice. The proteasomes of these tissues from sildenafil-treated mice exhibited faster hydrolysis of small peptides, ATP and a polyubiquitinated protein than of tissues of untreated controls (Supplementary Fig. 1E and F). Thus, raising cGMP levels and activating PKG in these tissues activates 26S proteasomes. Sildenafil poorly crosses the mammalian blood–brain barrier.29 Accordingly, in the brain lysates from these treated animals, we did not detect faster peptide hydrolysis by proteasomes or an increase in the phosphorylation of either VASP or PDE5 (Supplementary Fig. 2A and B). In addition, the 26S proteasomes affinity-purified from the brain lysates did not hydrolyse peptides or a polyubiquitinated protein faster than those from untreated controls (Supplementary Fig. 2C). Thus, the activation of proteasomes by sildenafil in tissues correlated closely with the activation of PKG and resembles our findings in cell cultures.7 It is also noteworthy that sildenafil is effective in peripheral nerve, although not in the brain, presumably because the blood nerve barrier is more permeable than the blood–brain barrier.30
Sildenafil treatment increases protein degradation in the sciatic nerves of S63del mice
To test if sildenafil treatment also increases protein degradation by the UPS in the sciatic nerves of S63del mice in vivo, we bred these mice with mice constitutively expressing the model UPS substrate ubG76V-GFP23 in all tissues. In cells, this fusion protein is rapidly ubiquitinated after synthesis and hydrolysed by 26S proteasomes via the ubiquitin fusion degradation pathway.31 Consequently, this protein is detected at low levels in normal conditions, but when the UPS is impaired, the protein is more stable and accumulates. As expected, in lysates of sciatic and quadriceps nerves from S63del mice, the levels of GFP detected by western blot were ∼2-fold greater than in wild-type lysates (Fig. 1C and D), because the 26S proteasomes are less active in the Schwann cells due to the expression of MpzS63del. 14 However, after sildenafil treatment for 5 days, the levels of GFP in S63del sciatic and quadricep nerves returned towards the levels in the controls (Fig. 1C and D). These findings are strong evidence for faster degradation of this substrate by the UPS, presumably a consequence of the proteasome activation (Fig. 1A).
cGMP enhances degradation and ubiquitination of MPZS63del protein in HEK293 cells
The MPZS63del protein accumulates in the ER of Schwann cells and causes CMT1B through gain of toxic function mechanisms.19,21 Therefore, the pathological consequences of MPZS63del expression may be lessened by stimulating its degradation and reducing the amount that accumulates in cells. To test whether raising cGMP can increase the degradation of MPZS63del, we used HEK293 cells that express HA-tagged MPZS63del or wild-type MPZ (MPZWT) under the control of a tetracycline-inducible promoter.27 After induction for 17 h, a time at which MPZS63del protein had accumulated in the ER,27 translation was blocked with cycloheximide and degradation of the mutant and wild-type protein was monitored by western blot. Raising cGMP levels in the presence of the NO donor DETA NONOate with either the PDE5 inhibitor sildenafil or the soluble guanylyl cyclase stimulator riociguat increased the rate of degradation of MPZS63del (Fig. 2A). In untreated HEK293 cells, MPZS63del showed an approximate half-life of 5 h, but about 3 h when cGMP was elevated. By contrast, the degradation rate of MPZWT was not affected by the rise in cGMP (Fig. 2B). Thus, raising cGMP levels accelerates the breakdown only of the misfolded, mutant MPZ.
Figure 2.

Raising intracellular cGMP increases the degradation and ubiquitination of P0S63del protein in HEK293 cells. (A) The rate of degradation of MPZS63del-HA in HEK293 cells is increased by incubation with sildenafil or riociguat, which stimulates cGMP synthesis by sGC. Experiments were performed in the presence of the NO donor DETA NONOate and cycloheximide was added at time 0. n = 3. (B) The degradation of MPZWT-HA was not changed when cGMP levels were raised as in A. (C) After immunoprecipitation of MPZS63del-HA, the amounts of ubiquitinated proteins in the precipitate was increased after a 30-min treatment with sildenafil or riociguat. Representative western blots from one of the three independent immunoprecipitations are shown. (D) After immunoprecipitation of MPZWT-HA, the ubiquitinated proteins in the precipitate were very low and did not increase with sildenafil or riociguat treatment for 30 min as in C.
In addition to causing proteasome activation, cGMP stimulates the ubiquitination of cell proteins and increases the total cellular content of ubiquitinated proteins.7 Presumably, those proteins that are ubiquitinated faster with cGMP are also degraded more rapidly. To test if cGMP enhances MPZS63del degradation by increasing its ubiquitination, we expressed HA-tagged MPZS63del in HEK293 cells for 17 h and then raised cGMP in the cells for 30 min with the NO donor and the PDE5 inhibitor or sGC stimulator. MPZS63del was then immunoprecipitated, and the amount of K48-linked ubiquitin in the precipitate was measured by western blot. The NO donor by itself increased the amounts of ubiquitinated MPZS63del, and when combined with sildenafil or riociguat to further raise cGMP caused an even greater increase in ubiquitinated MPZS63del (Fig. 2C). The immunoprecipitated MPZWT also contained some K48-linked polyubiquitin chains, but much less than the mutant protein (Fig. 2D), as expected because the wild-type protein was more stable than MPZS63del (Fig. 2A and B). Moreover, the ubiquitination of MPZWT did not increase with treatments that activate PKG (Fig. 2D). Thus, rates of ubiquitination correlated with degradation rates and raising cGMP increased selectively the ubiquitination and degradation of the mutant protein.
Sildenafil treatment for 14 days increases proteasome activity and decreases proteotoxic stress in S63del mice
Because sildenafil increased selectively the degradation of MPZS63del in HEK293 cells and activated proteasome activities in S63del mice, we extended the sildenafil treatment to 14 days (P25–P39) to evaluate its effects on proteostasis and myelination. Like the 5-day treatments, 14 days of sildenafil enhanced proteasome activity by ∼50% in the sciatic nerve lysates from both wild-type and S63del mice (Fig. 3A). Interestingly, unlike the 5-day treatment, sildenafil for 14 days caused a small but reproducible decrease in the levels of 26S proteasome subunits in S63del, but not in the wild-type sciatic nerves (Fig. 3B). Therefore, the cGMP-mediated increase in total proteasome activity measured in the S63del sciatic nerve lysates (Fig. 3A) must underestimate the actual increase in the proteasomes’ specific activity. When proteasomes are impaired, either by pharmacological inhibitors32,33 or by accumulation of MPZS63del in Schwann cells,14 the transcription factor NRF1 activates the expression of genes encoding 26S proteasome subunits. Because levels of proteasome subunits decreased only in S63del, it is likely that the sildenafil treatment reduced the need for this response that compensates for inadequate proteasome function.
Figure 3.
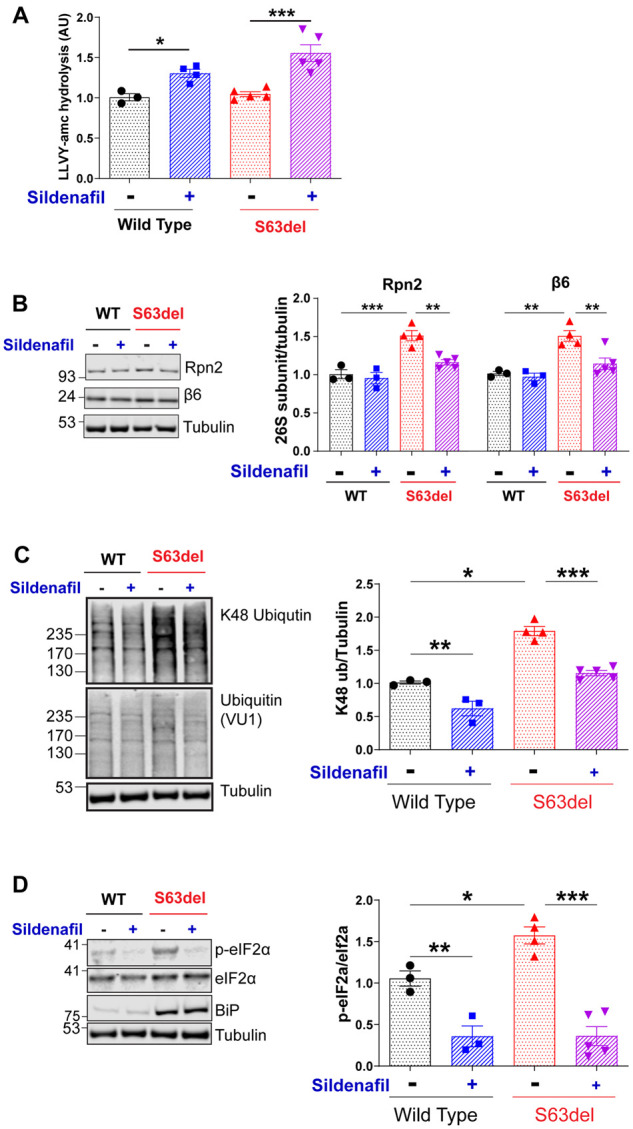
Sildenafil treatment for 14-day increases proteasome activity and reduces markers of proteotoxic stress in sciatic nerves of S63del mice. (A) Total proteasomal peptidase activity was increased in the lysates of sciatic nerves from wild-type (WT) and S63del mice following sildenafil treatment for 14 days. Here and below, n = 3 mice for wild-type vehicle, n = 4 for wild-type sildenafil, n = 5 for S63del vehicle and n = 5 for S63del sildenafil. One-way ANOVA with Bonferroni post hoc analysis comparing wild-type vehicle and wild-type sildenafil, wild-type vehicle and S63del vehicle and S63del vehicle and S63del sildenafil. (B) In S63del, sildenafil treatment for 14 days reduced the levels of 26S proteasome subunits Rpn2 and β6. (C) Total polyubiquitinated proteins, including K48-linked polyubiquitinated proteins, were higher in the sciatic nerves of S63del mice than in wild-type nerves. Sildenafil treatment reduced the levels of these ubiquitin conjugates. (D) In sciatic nerves, phosphorylation of elF2α on serine 51 was higher in S63del than in wild-type, but sildenafil treatment for 14 days reduced the phosphorylation of eIF2α in nerves from both wild-type and S63del mice.
The impairment of 26S proteasome function in Schwann cells of untreated S63del mice caused about a 50% increase in total content of polyubiquitinated proteins, specifically of K48-linked polyubiquitinated proteins, above levels in the wild-type littermates14 (Fig. 3C). Sildenafil treatment of the S63del mice for 14 days reduced the levels of these ubiquitin conjugates in sciatic nerves towards the levels seen in the wild-type mice (Fig. 3C), presumably due to their faster degradation by 26S proteasomes. The ubiquitinated proteins also decreased in wild-type mice with sildenafil treatment (Fig. 3C).
Schwann cells expressing MPZS63del manifest a canonical UPR,21 and levels of p-eIF2α are higher in S63del sciatic nerves than in wild-type littermates (Fig. 3D).21 Treatment with sildenafil for 14 days reduced the levels of p-eIF2α in sciatic nerve lysates from S63del and wild-type mice to almost undetectable levels without changing the total amount of eIF2α protein (Fig. 3D). The decreases in S63del in the levels of phosphorylated eIF2α, of polyubiquitinated proteins and of proteasome subunits strongly suggest that the 14-day sildenafil treatment abrogated the proteotoxic stress in the Schwann cells caused by the expression of MpzS63del.
Sildenafil treatment for 14 days increases myelination in S63del mice
A prominent feature of human patients with CMT1B is the demyelination of peripheral nerves, which leads to the loss of motor functions.34 The axons in the peripheral nerves of S63del mice also have thinner myelin sheaths than their wild-type littermates (Fig. 4A and B).19 To test whether the activation of 26S proteasomes and the stimulation of the degradation of MPZS63del and other proteins by cGMP can ameliorate this deficiency in myelination, S63del mice and wild-type littermates were treated for 14 days with sildenafil or vehicle, and the amount of myelin per axon was then quantified by electron microscopy of thin sections of the sciatic nerve. Myelin thickness is quantified as a g-ratio, which is defined as the axon diameter divided by the fibre diameter. Therefore, the higher the g-ratio, the thinner the myelin sheath relative to the axon diameter. In S63del, the sildenafil treatment reduced the average g-ratio, which indicates that it increased the thickness of the myelin sheath (Fig. 4A and B). This increase in thickness was most prominent around the small-calibre axons, those with diameters ≤3 µm, in which the g-ratio returned to wild-type levels after treatment (Fig. 4C and D). Sildenafil treatment of S63del mice also increased the myelin thickness surrounding the medium-calibre axons (3–5 µm) and the large-calibre axons (5–6 µm), but to a lesser extent (Fig. 4C and D). The percentage of myelinated axons of each diameter was unchanged after the sildenafil treatment in both S63del mice and the wild-type littermates (Fig. 4E).
Figure 4.

Sildenafil treatment for 14 days prevents the myelin defects seen in S63del sciatic nerves. (A) Representative electron microscopic images of thin sections of sciatic nerves showing that myelin thickness was increased in the sciatic nerves of S63del mice following sildenafil treatment for 14 days. Red arrows indicate amyelinated fibres. (B) The myelin thickness was lower in S63del sciatic nerves and was increased by sildenafil treatment. Quantification of average myelin thickness in sciatic nerves. g-ratio = axon diameter / fibre diameter. Therefore, the higher the g-ratio, the thinner the myelin sheath. (C) In S63del, the increase in myelin thickness caused by sildenafil treatment is more pronounced in small diameter axons (1–3 µm) than in the medium and large diameter axons (3–6 µm). (D) Scatterplot of g-ratio distribution of sciatic nerve thin sections from mice in C. S63del mice, compared to wild-type (WT) littermates, have less myelin (higher g-ratio) on axons of all diameters, indicated by the red line (S63del) always being above the black line (wild-type). Sildenafil treatment of S63del mice increased myelin thickness (lower g-ratio) around axons of all diameters, indicated by the purple line (S63del silden) always being lower than the red line (S63del). This increase was most pronounced in the small diameter axons (1–3 µm), indicated by the purple line migrating further away from the red line (S63del) and overlapping the black line (wild-type). (E) Sildenafil treatment did not change the percentage of myelinated axons of any diameter. (F) The amount of pathological amyelinated fibres (arrow in A) in S63del sciatic nerves was reduced by sildenafil treatment.
Another pathological hallmark of hereditary peripheral neuropathies is the presence of amyelinated axons. These axons are present in a 1:1 relationship with Schwann cells and have diameters greater than 1 µm. Therefore, these axons should be myelinated, but are not. Amyelinated fibres were rare in the sciatic nerves of wild-type mice (∼1% of myelinated fibres; Fig. 4F), but were much more prevalent in S63del (∼10% of myelinated fibres; Fig. 4E).24 The sildenafil treatment of S63del mice reduced the frequency of amyelinated fibres to ∼3% (Fig. 4F).
The 14-day treatment with sildenafil also increases nerve conduction in S63del mice
These improvements in nerve morphology with the sildenafil treatment prompted us to investigate whether nerve function also improved. S63del mice, like CMT1B patients, have impaired motor capacities associated with decreased nerve conduction velocities, increased distal latencies, and increased F-wave latencies due to uniform hypomyelination (Fig. 5A–C).22,24,35 F-wave latency measures the time it takes for an electrical signal to travel from the site of stimulation at the distal portion of a motor nerve to the spinal cord and then back down the length of that nerve to the muscle. In S63del, the sildenafil treatment restored nerve conduction velocities and distal latencies to wild-type levels (Fig. 5A and B) and partially restored F-wave latencies (Fig. 5C). Together, these data demonstrate that elevating cGMP levels with sildenafil not only corrected the defects in protein homeostasis in the nerves, but also prevented the myelin abnormalities and the associated functional deficits in nerves of the S63del mouse model of CMT1B.
Figure 5.
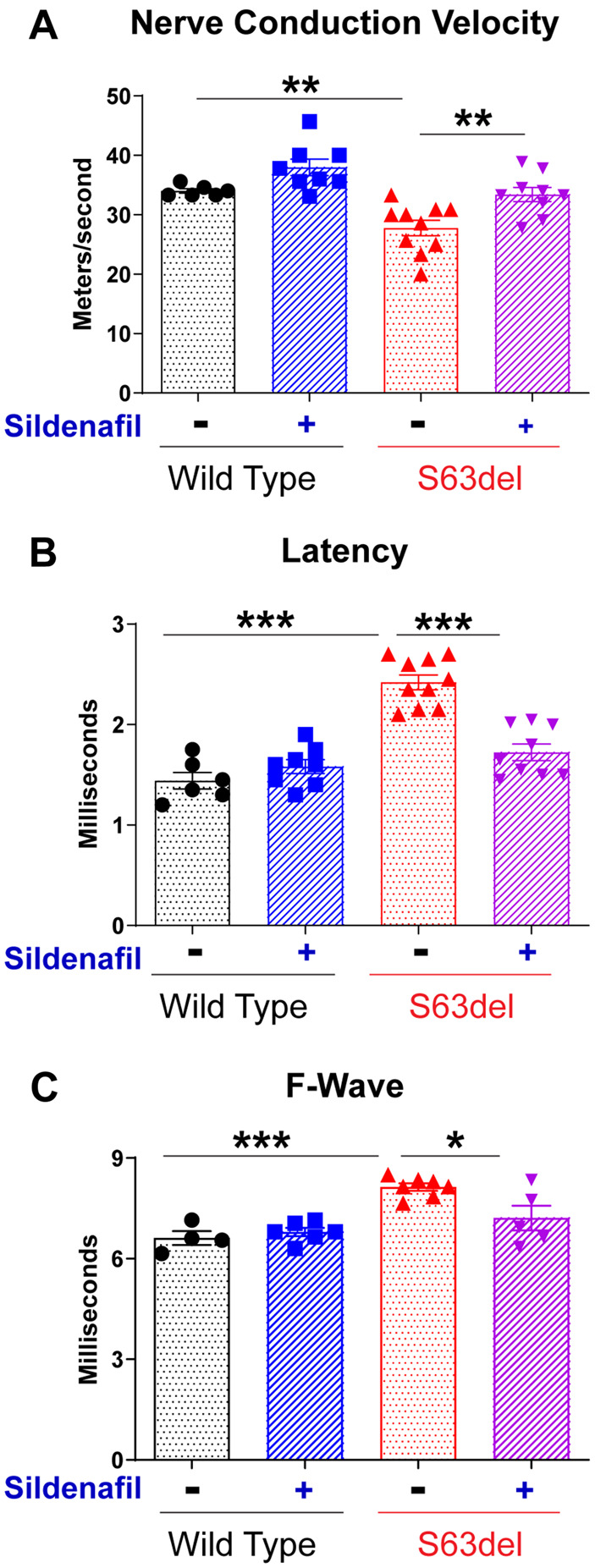
Sildenafil treatment increases nerve conduction in S63del mice. (A) Sildenafil treatment of S63del mice for 14 days increased conduction velocity in the peripheral nerves. In control mice, nerve conduction velocity in S63del mice is lower than in wild-type mice, but sildenafil treatment caused it to resemble that of wild-type littermates. n = 6 wild-type vehicle, n = 8 wild-type sildenafil, n = 10 S63del vehicle and n = 9 S63del sildenafil. (B) Sildenafil treatment restored the distal latency in peripheral nerves of S63del mice to that of wild-type littermates. (C) Sildenafil treatment reduced the longer F wave latencies of S63del mice towards that seen in wild-type littermates. n = 4 wild-type vehicle, n = 6 wild-type sildenafil, n = 7 S63del vehicle and n = 5 S63del sildenafil.
Discussion
Raising cGMP restores protein homeostasis in S63del mice
Although less studied than tauopathies, synucleinopathies or amyotrophic lateral sclerosis, CMT1B offers many advantages for investigating the mechanisms and potential treatments of proteotoxic diseases. Our knowledge of the molecular alterations, their cellular consequences and the resulting quantifiable defects in myelination and conduction are quite advanced largely through studies of the S63del mouse model. Building on these insights, we have shown here that raising levels of cGMP not only improves protein homeostasis in the Schwann cells, but also reverses the morphological and functional defects in the S63del mouse, and therefore appears to be a promising approach to combat CMT1B in humans.
In Schwann cells in S63del mice, the accumulation of the mutant MPZ in the ER leads to multiple signs of proteotoxic stress. One is the activation of the UPR, leading to the transcription of molecular chaperones and enzymes that increase the capacity of the ER for protein folding and also for degradation of misfolded proteins by the ER-associated degradation (ERAD) pathway.36 In addition, the UPR causes phosphorylation of eiF2α by PERK and consequently leads to a general reduction in overall protein synthesis, which decreases the continued production of proteins and lessens the substrate load on the cell’s proteostasis network.36 However, prolonged activation of the UPR can be deleterious, and in the S63del mice, signalling through C/EBP homologous protein (CHOP)21 and the activation of its downstream effector GADD3422,37 causes demyelination of the motor nerves. Treating S63del mice with sildenafil was found to decrease this activation of the UPR, presumably by PKG-mediated activation of the UPS and accelerated clearance of MPZS63del and perhaps other misfolded proteins.
Unfortunately, the levels of MPZS63del protein in Schwann cells cannot be detected reliably by western blot in the peripheral nerves due to the massive amount MPZ present in the myelin sheath. However, our finding that sildenafil treatment for 2 weeks abrogated the accumulation of polyubiquitinated proteins and the phosphorylation of eIF2α (Fig. 3) indicates that the build-up of misfolded proteins was reduced. Furthermore, in cultured HEK293 cells, the PDE5 inhibitor accelerated both the ubiquitination and degradation of MPZS63del (Fig. 2). These findings thus extend the earlier observations that raising cGMP in zebrafish larvae increases the degradation of mutant proteins that cause tauopathies and Huntington’s disease,7 and in cells, increases the degradation of a mutant protein that causes a hereditary cardiomyopathy.38
Another promising pharmacological approach to treat S63del neuropathy is with agents that further reduce protein synthesis. Increased myelin thickness and nerve conduction were demonstrated upon prolonged treatment (3 or 5 months) with Sephin137 or salubrinal,22 which promote the phosphorylation of eIF2α by blocking its dephosphorylation by GADD34. These prior studies were performed in S63del mice that were 2–4 months older than those used in the present study. We studied here 1-month-old S63del mice, because we had previously shown in them clear defects in proteasome function and protein degradation.14 To determine whether sildenafil treatment also improves motor performance, which is expected with an improvement in myelination and was seen with our previous Sephin1 treatments of older animals, it will be important in future studies to examine proteasome function in aged S63del mice. It will also be interesting to examine if administering one of these GADD34 inhibitors together with a PDE5 inhibitor, to enhance protein degradation while slowing translation, has additive or synergistic effects in improving this disease.
Defective proteasome function and its restoration by cGMP
The 26S proteasomes purified from the sciatic nerves of S63del mice have a decreased ability to hydrolyse small peptides and ubiquitinated proteins.14 These defects presumably account for the previously observed decrease in overall protein degradation in the sciatic nerve14 as well as the accumulation of polyubiquitinated proteins (Fig. 3)14 and the UbG76V-GFP reporter protein (Fig. 1). Although the specific activity of 26S proteasomes was reduced in S63del, the total proteasome activity in the lysates of sciatic nerve was similar to that seen in wild-type, due to a greater content of 26S proteasomes (Fig. 1).14 S63del sciatic nerves also have increased levels of the active form of the transcription factor, NRF1,14 which regulates the expression of all proteasome genes.32,33 These findings are strong evidence of an attempt by Schwann cells to compensate for the decreased degradative capacity by increasing proteasome production, as is also seen in various cell types upon treatment with proteasome inhibitors.32,33 However, the increased number of proteasomes was not sufficient to prevent the reduction in protein degradation and the accumulation of polyubiquitinated proteins observed in Schwann cells in S63del mice.14 Therefore, factors other than proteasome abundance must influence the proteasomes’ degradative capacity and prevent the greater number of proteasomes from restoring proteolysis in this disease model. Among these unknown factors may be subcellular localization of the 26S proteasomes, or changes in post-translational modifications, or association of regulatory factors.
Impairment of 26S function has also been demonstrated in several experimental models of other neurodegenerative diseases, including tauopathies,10,18 prion disease16,17 and amyotrophic lateral sclerosis caused by expansions in C9orf72,39 probably as a consequence of protein aggregates blocking proteasome function. Thibaudeau et al.40 demonstrated that soluble oligomers formed by the amyloid-β fragment (1–42) of APP, α-synuclein, or polyQ-extended huntingtin, which share an amyloid-like conformation, can bind to the proteasome and inhibit substrate entry. Presumably, MPZS63del acts in a similar way to impair proteasome function. However, in the S63del mouse and these other neurodegenerative disease models, it is unclear if the decreased 26S activity represents a partial inhibition of most cell proteasomes or a selective inactivation of a subset of the particles. In cells overproducing expanded C9orf 72 polyGA, the loss of proteasome activity seems to result from the sequestration of a large fraction of proteasomes in large insoluble aggregates of polyGA.15 By contrast, in the S63del mouse14 and tauopathy models,10 decreased activity was evident after purification of soluble 26S proteasomes.
Whatever the basis for the reduced 26S activity in the sciatic nerve, the sildenafil treatment increased proteasomal peptidase activity and lowered the content of UbG76V-GFP towards levels seen in wild-type littermates (Fig. 1). Recent studies involving cryo-electron tomography of hippocampal neurons41 and fungi42,43 have shown that the great majority of intracellular proteasomes are in an inactive conformation, as was also found after rapid purification of these particles from rabbit skeletal muscle,44 although these particles become activated upon binding proteins that contain UBL-domains.44 It is unclear in the sildenafil-treated S63del mice if cGMP and PKG improved protein homeostasis in the nerves by restoring the activity of impaired 26S proteasomes or by activating some of the cell’s previously latent particles.
The present findings and our related studies do not support the claim that increases in proteasome activity are necessarily associated with greater formation of the 26S complex.45,46 In human neuroblastoma cells (SH-SY5Y) and other cultured cell lines, PDE5 inhibitors and soluble sGC stimulators (separately or together) increased 26S proteasome activity within minutes, but these agents did not alter the levels of proteasome subunits or the relative amounts of singly capped or doubly capped proteasomes.7 In the present studies, sildenafil treatment of wild-type mice for 5 days did not increase the levels of proteasome subunits in sciatic nerves (Fig. 1) or in skeletal and cardiac muscles (Supplementary Fig. 1), nor did treatment for 14 days alter their levels in the sciatic nerves (Fig. 3B). Thus, the increased activity induced by PKG occurs through post-synthetic phosphorylation of pre-existent 26S particles. Even though this activation by PKG can be reversed by protein phosphatase treatment, and pure PKG can induce phosphorylation and activation of pure 26S proteasomes,7 we have thus far been unable, for reasons that are unclear, to identify the specific 26S component phosphorylated by PKG, despite repeated attempts using several mass spectrometry approaches.
Identifying the site(s) on the proteasome phosphorylated by PKG would not only aid our understanding of proteasome function but would also enable site-directed mutagenesis of the phosphosite to determine whether this modification is necessary and sufficient for the accelerated protein degradation, as well as the therapeutic effects of sildenafil in S63del mice. The observed enhancement of the cell’s degradative capacity by sildenafil may also require or may result only from the increased ubiquitin conjugation to cell proteins. In HEK293 cells, sildenafil enhanced ubiquitination and degradation of MPZS63del but not of the wild-type protein (Fig. 2). In cell extracts, cGMP stimulates ubiquitin conjugation to cell proteins, and in human neuroblastoma cells, raising cGMP with sildenafil leads to an increased content of ubiquitinated proteins within 15 min, despite their faster degradation by the activated proteasomes.7 Most likely, the improvement in protein homeostasis by cGMP and PKG results in part from the increased ubiquitination and in part from proteasome activation, both of which may be necessary to increase intracellular protein breakdown.
Raising cGMP may be a useful therapy for CMT1B and other neurodegenerative diseases
Hypomyelination, leading to defects in saltatory conduction, is a hallmark of human CMT1B and of the S63del mice. Not only did treatment with the PDE5 inhibitor correct the indicators of proteotoxic stress in the sciatic nerves, but it also increased the thickness of the myelin sheath (Fig. 4) and reversed the defects in nerve conduction (Fig. 5). The 14-day treatment with sildenafil decreased the number of amyelinated axons and increased the myelin thickness of most axons, especially the smaller-calibre axons. It is not clear why the magnitude of the effect seemed to vary with axon diameter, but manipulation of other pathways in Schwann cells, such as those induced by neuregulin, also affects small-calibre axons more prominently.47 The apparent refractoriness of the larger diameter neurons to these treatments may also reflect some inherent differences in their sensitivity to PKG’s activation. Alternatively, increased myelination of the large-calibre axons may simply require a longer treatment with sildenafil, or a larger sample size, to become evident. These trends in the larger axons may not have reached significance due to their low representation in the thin sections used to measure g-ratio in this study. Most importantly, the restoration of myelination in the sciatic nerve led to an increased conduction velocity and decreased latency, which are also functional characteristic features of the human disease. The ability to precisely quantitate these parameters in patients, the rapidity of this response and the lack of apparent toxicity of these widely used drugs all make this approach particularly promising to test for efficacy in human CMT1B patients.
Although raising cGMP stimulates the degradation of the bulk of the cell constituents, it did not cause significant weight loss or other deleterious effects, even after twice daily administration to the mice for 14 days (Supplementary Fig. 3). Similarly, in the developing zebrafish larvae, no harmful effects of sildenafil treatment were evident; on the contrary, sildenafil administration reduced neuronal death and decreased the morphological abnormalities caused by mutant tau.7 By itself, such a general enhancement of protein degradation should cause a marked loss of cell mass, as occurs for example in mouse skeletal muscles in 2–5 days upon fasting or denervation.48 This maintenance of body size, despite the large increase in proteolysis, is surprising and an intriguing subject for future research, but it further argues that a prolonged increase in cGMP levels is not likely to have harmful consequences. Unlike many novel pharmacological agents, which may trigger unanticipated cellular responses, raising cGMP to enhance proteolysis is well-tolerated probably because it activates a physiological mechanism that organisms have evolved to enhance protein turnover under specific conditions.
In addition to CMT1B, raising cGMP may also be beneficial in combatting other hereditary peripheral neuropathies. Proteasome activities have also been reported to decrease in two mouse models of CMT1A, which is the most common form of CMT and is caused by overexpression of peripheral myelin protein 22 (PMP22) in Schwann cells.49,50 Therefore, PDE5 inhibitors or sGC stimulators also merit study as potential treatments of this proteotoxic disease. Because decreased proteasome activity has been observed in other neurodegenerative disease models and appears to arise by a common mechanism,2,40 raising cGMP seems like an attractive new approach to combat various neurodegenerative diseases, as well as a number of myopathies affecting skeletal and cardiac muscle,51,52 which also result from the accumulation of aggregation-prone proteins. It is noteworthy that sildenafil had these effects in peripheral nerves, but did not affect PKG or proteasomes in brain. Thus, it is able to promote proteostasis in Schwann cells, but not in the CNS, as suspected from its known failure to permeate the blood–brain barrier. However, brain-penetrant PDE5 inhibitors and sGC stimulators are known13,29 and are probably more appropriate for testing against diseases of the brain and spinal cord. Moreover, because raising cGMP levels also causes vasodilation and increased blood flow to tissues,13 these treatments may have additional therapeutic benefits in vivo, in addition to enhancing the clearance of the toxic proteins in the affected cells.
Supplementary Material
Acknowledgements
We thank Dr Maurizio D’Antonio (San Raffaele Scientific Institute, Milan, Italy) for the HEK293 cells that express MPZWT or MPZS63del, Edward Hurley for his excellent technical assistance and Amelia Gould for assistance in preparing this manuscript.
Funding
We are grateful for funding from: NIH National Institute of General Medical Sciences (R01 GM051923-20), Cure Alzheimer’s Fund, Muscular Dystrophy Association Grant, and Project ALS (to A.L.G.); from the NIH National Institute of General Medical Sciences (F32 GM128322) (to J.J.S.V.); and from the NIH National Institute of Neurological Disorder and Stroke, 5R01-NS55256 and R56NS096104 (to L.W.).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- CMT1B
Charcot-Marie-Tooth type 1B
- GFP
green fluorescent protein
- PDE
phosphodiesterase
- PKG
cGMP-dependent kinase
- sGC
soluble guanylyl cyclase
- UPR
unfolded protein response
- UPS
ubiquitin proteasome system
References
- 1.Zhao J, Zhai B, Gygi SP, Goldberg AL.. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A. 2015;112(52):15790–15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith DM. Could a common mechanism of protein degradation impairment underlie many neurodegenerative diseases? J Exp Neurosci. 2018;12:1179069518794675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deol KK, Lorenz S, Strieter ER.. Enzymatic logic of ubiquitin chain assembly. Front Physiol. 2019;10:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.VerPlank JJS, Goldberg AL.. Regulating protein breakdown through proteasome phosphorylation. Biochem J. 2017;474(19):3355–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo X, Huang X, Chen MJ.. Reversible phosphorylation of the 26S proteasome. Protein Cell. 2017;8(4):255–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo X, Wang X, Wang Z, et al. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat Cell Biol. 2016;18(2):202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.VerPlank JJS, Tyrkalska SD, Fleming A, Rubinsztein DC, Goldberg AL.. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc Natl Acad Sci U S A. 2020;117(25):14220–14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.VerPlank JJS, Lokireddy S, Zhao J, Goldberg AL.. 26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation. Proc Natl Acad Sci U S A. 2019;116(10):4228–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lokireddy S, Kukushkin NV, Goldberg AL.. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015;112(52):E7176–E7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myeku N, Clelland CL, Emrani S, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2016;22(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baillie GS, Tejeda GS, Kelly MP.. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat Rev Drug Discov. 2019;18(10):770–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis SH, Busch JL, Corbin JD, Sibley D.. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandner P, Zimmer DP, Milne GT, Follmann M, Hobbs A, Stasch J-P.. Soluble guanylate cyclase stimulators and activators. Handb Exp Pharmacol. 2021;264:355–394. [DOI] [PubMed] [Google Scholar]
- 14.VerPlank JJS, Lokireddy S, Feltri ML, Goldberg AL, Wrabetz L.. Impairment of protein degradation and proteasome function in hereditary neuropathies. Glia. 2018;66(2):379–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Q, Lehmer C, Martínez-Sánchez A, et al. In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell. 2018;172(4):696–705.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kristiansen M, Deriziotis P, Dimcheff DE, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26(2):175–188. [DOI] [PubMed] [Google Scholar]
- 17.Deriziotis P, André R, Smith DM, et al. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 2011;30(15):3065–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez A, Lee SE, Wojta K, et al. ; Tauopathy Genetics Consortium. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain. 2017;140(4):1128–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wrabetz L, D'Antonio M, Pennuto M, et al. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci. 2006;26(8):2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jerath NU, Shy ME.. Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochim Biophys Acta. 2015;1852(4):667–678. [DOI] [PubMed] [Google Scholar]
- 21.Pennuto M, Tinelli E, Malaguti M, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Antonio M, Musner N, Scapin C, et al. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210(4):821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindsten K, Menéndez-Benito V, Masucci MG, Dantuma NP.. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003;21(8):897–902. [DOI] [PubMed] [Google Scholar]
- 24.Sidoli M, Musner N, Silvestri N, et al. Ablation of perk in Schwann cells improves myelination in the S63del Charcot-Marie-Tooth 1B Mouse. J Neurosci. 2016;36(44):11350–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo C-L, Collins GA, Goldberg AL.. Methods to rapidly prepare mammalian 26S proteasomes for biochemical analysis. Methods Mol Biol. 2018;1844:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodnar NO, Rapoport TA.. Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell. 2017;169(4):722–735.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volpi VG, Ferri C, Fregno I, et al. Schwann cells ER-associated degradation contributes to myelin maintenance in adult nerves and limits demyelination in CMT1B mice. PLoS Genet. 2019;15(4):e1008069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motosugi R, Murata S.. Dynamic regulation of proteasome expression. Front Mol Biosci. 2019;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuccarello E, Acquarone E, Calcagno E, et al. Development of novel phosphodiesterase 5 inhibitors for the therapy of Alzheimer’s disease. Biochem Pharmacol. 2020;176:113818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malong L, Napoli I, White IJ, Stierli S, Bossio A, Lloyd AC.. Macrophages enforce the blood nerve barrier. bioRxiv. [Preprint] doi: 10.1101/493494. [DOI] [Google Scholar]
- 31.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG.. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18(5):538–543. [DOI] [PubMed] [Google Scholar]
- 32.Sha Z, Goldberg AL.. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol. 2014;24(14):1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ.. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38(1):17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanmaneechai O, Feely S, Scherer SS, et al. ; Inherited Neuropathies Consortium—Rare Disease Clinical Research Consortium (INC-RDCRC). Genotype–phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain. 2015;138(Pt 11):3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musner N, Sidoli M, Zambroni D, et al. Perk ablation ameliorates myelination in S63del-Charcot-Marie-Tooth 1B neuropathy. ASN Neuro. 2016;8(2):175909141664235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costa-Mattioli M, Walter P.. The integrated stress response: From mechanism to disease. Science. 2020;368(6489):eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das I, Krzyzosiak A, Schneider K, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348(6231):239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranek MJ, Terpstra EJM, Li J, Kass DA, Wang X.. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128(4):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khosravi B, LaClair KD, Riemenschneider H, et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020;39(8):e102811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thibaudeau TA, Anderson RT, Smith DM.. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun. 2018;9(1):1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asano S, Fukuda Y, Beck F, et al. Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science. 2015;347(6220):439–442. [DOI] [PubMed] [Google Scholar]
- 42.Albert S, Schaffer M, Beck F, et al. Proteasomes tether to two distinct sites at the nuclear pore complex. Proc Natl Acad Sci U S A. 2017;114(52):13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albert S, Wietrzynski W, Lee C-W, et al. Direct visualization of degradation microcompartments at the ER membrane. Proc Natl Acad Sci U S A. 2020;117(2):1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins GA, Goldberg AL.. Proteins containing ubiquitin-like (Ubl) domains not only bind to 26S proteasomes but also induce their activation. Proc Natl Acad Sci U S A. 2020;117(9):4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rousseau A, Bertolotti A.. An evolutionarily conserved pathway controls proteasome homeostasis. Nature. 2016;536(7615):184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rousseau A, Bertolotti A.. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol. 2018;19(11):697–712. [DOI] [PubMed] [Google Scholar]
- 47.Taveggia C, Zanazzi G, Petrylak A, et al. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47(5):681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cohen S, Nathan JA, Goldberg AL.. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14(1):58–74. [DOI] [PubMed] [Google Scholar]
- 49.Fortun J, Go JC, Li J, Amici SA, Dunn WAJ, Notterpek L.. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22(1):153–164. [DOI] [PubMed] [Google Scholar]
- 50.Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L.. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem. 2005;92(6):1531–1541. [DOI] [PubMed] [Google Scholar]
- 51.Gilda JE, Gomes AV.. Proteasome dysfunction in cardiomyopathies. J Physiol. 2017;595(12):4051–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Wang H.. Priming the proteasome to protect against proteotoxicity. Trends Mol Med. 2020;26(7):639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in this report are available to all readers. The reagents used in this study, as well as further information, are available upon request from A.L.G.