

Abstract
Spreading depolarizations are highly prevalent and spatiotemporally punctuated events worsening the outcome of brain injury. Trigger factors are poorly understood but may be linked to sudden worsening in supply–demand mismatch in compromised tissue. Sustained or transient elevations in intracranial pressure are also prevalent in the injured brain.
Here, using a mouse model of large hemispheric ischaemic stroke, we show that mild and brief intracranial pressure elevations (20 or 30 mmHg for just 3 min) potently trigger spreading depolarizations in ischaemic penumbra (4-fold increase in spreading depolarization occurrence). We also show that 30 mmHg intracranial pressure spikes as brief as 30 s are equally effective. In contrast, sustained intracranial pressure elevations to the same level for 30 min do not significantly increase the spreading depolarization rate, suggesting that an abrupt disturbance in the steady state equilibrium is required to trigger a spreading depolarization. Laser speckle flowmetry consistently showed a reduction in tissue perfusion, and two-photon pO2 microscopy revealed a drop in venous pO2 during the intracranial pressure spikes suggesting increased oxygen extraction fraction, and therefore, worsening supply–demand mismatch. These haemodynamic changes during intracranial pressure spikes were associated with highly reproducible increases in extracellular potassium levels in penumbra. Consistent with the experimental data, a higher rate of intracranial pressure spikes was associated with spreading depolarization clusters in a retrospective series of patients with aneurysmal subarachnoid haemorrhage with strong temporal correspondence.
Altogether, our data show that intracranial pressure spikes, even when mild and brief, are capable of triggering spreading depolarizations. Aggressive prevention of intracranial pressure spikes may help reduce spreading depolarization occurrence and improve outcomes after brain injury.
Keywords: cerebral infarction, peri-infarct depolarization, intracranial pressure, clinical, experimental
Spreading depolarizations (SDs) have been linked to poor neurological outcomes after brain injury. Oka et al. show that intracranial pressure (ICP) spikes, even when mild and brief, are capable of triggering SDs. Aggressive prevention of ICP spikes may help reduce SD occurrence and improve outcomes after brain injury.
Introduction
Spreading depolarizations (SDs) are recurrent intense pandepolarization waves that emanate from the metabolically disturbed brain in an ostensibly spontaneous fashion and propagate far into the healthy tissue.1,2 Regardless of the mode of injury (e.g. ischaemic, haemorrhagic, traumatic), they worsen the supply–demand mismatch by increasing the metabolic demand and reducing the blood supply in viable tissue at risk.3 As a result, SDs have been linked to poor neurological outcomes after brain injury.4-6 Despite our wealth of knowledge on the characteristics and consequences of SDs,5,7 relatively little is known about how and why SDs originate. A sudden worsening in supply–demand mismatch in ischaemic penumbra triggers peri-infarct SDs, such as during hypoxic or hypotensive transients that are highly common in patients with brain injury.8 Intracranial pressure (ICP) elevations are also common in brain-injured patients, and ICP spikes occur during pain, agitation, respiratory distress and ventilator dyssynchrony. Whether brief or sustained, high ICP can diminish cerebral perfusion pressure (CPP) and worsen the supply–demand mismatch. Therefore, we hypothesized that ICP elevations can trigger SDs, and tested this in a hemispheric ischaemic stroke model in mice. We then corroborated our findings in a clinical series of aneurysmal subarachnoid haemorrhage (SAH) patients with subdural SD recordings coupled to ICP monitoring.
Materials and methods
Experimental study
All experimental procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication no. 85–23, 1996), and were approved by the institutional review board (MGH Subcommittee on Research Animal Care, SRAC). We used a total of 58 adult male mice (C57BL/6, 2–3 months old, 22–30 g: Charles River Laboratories).
Filament middle cerebral artery occlusion
Mice were anaesthetized with isoflurane (2.5% induction, 1.5% maintenance, in 70% N2O and 30% O2). The femoral artery was catheterized with a polyethylene catheter (PE10) for the measurement of blood pressure (ADInstruments) and arterial pH, PO2, and PCO2 (Supplementary Table 1; Rapidlab 248 blood gas/pH analyser; Siemens HealthCare). Rectal temperature was kept at 37°C using a thermostatically controlled heating pad (FHC). A nylon monofilament was inserted into the internal carotid artery via the external carotid artery and occluded the middle cerebral artery (MCA). Occlusion was confirmed using laser Doppler flowmetry (Perimed) over the ischaemic core. After filament MCA occlusion (fMCAO), mice were placed in a prone position and each head was fixed in a stereotaxic frame.
Measurement and manipulation of intracranial pressure
We measured and adjusted the ICP through a cisterna magna catheter. After midline scalp incision, the atlantooccipital membrane was exposed and punctured with a needle connected to a pressure transducer (ADInstruments) via a polyethylene catheter (PE10 tubing, filled with physiological saline). To manipulate the ICP, we adjusted the catheter height.
Laser speckle imaging
We used laser speckle flowmetry (LSF) through the intact skull to detect SDs and image the spatiotemporal characteristics of cerebral blood flow (CBF) changes during ICP spikes (n = 32), as described in detail previously.9 LSF imaging was initiated ∼15 min after the occlusion of MCA. Using a near infrared laser diode (Thorlabs; 785 nm, 75 mW) and a CCD camera (Cohu 4600; 640 × 480 pixels), raw speckle frames were continuously acquired at 2.5 Hz. The imaging field was positioned over both hemispheres. Data were recorded and analysed using custom-designed software written in LabWindows CVI (National Instruments, Austin, TX) and MATLAB (MathWorks, Natick, MA).
Infarct volume
In a separate group, neurological outcome was assessed, and infarction volume was measured at 24 h after reperfusion (n = 10). In this cohort, femoral artery cannulation was avoided, since this procedure confounds the neurological assessment. Fifteen minutes after the occlusion of MCA in the ICP elevation group, ICP was increased to 30 mmHg for 30 s every 9 min, for five times. At 60 min after occlusion, MCA was reperfused by removing the occluding monofilament. In the control group, the duration of ischaemia was the same and ICP was measured in the same way but not manipulated. Neurological deficits were assessed using 5-point grading score: 0, normal; 1, forepaw paresis; 2, circling to one side; 3, falling to one side; 4, no spontaneous walking and a depressed level of consciousness; and 5, death. Infarct volumes were calculated by integrating the areas of ipsilateral infarcted and non-infarcted tissue and the contralateral hemisphere on 10 1-mm thick 2,3,5-triphenyltetrazolium chloride (TTC) stained coronal sections, and subtracting the volume of ipsilateral non-infarcted tissue from the contralateral hemisphere volume.
Multispectral imaging
In a separate group (n = 4), LSF imaging of CBF was coupled to multispectral reflectance imaging of microvascular oxygen saturation, as described in detail previously.10,11 Illumination for multispectral reflectance imaging was provided by a tungsten halogen lamp attached to a fibre-optic bundle (Techniquip R150; Capra Optical). Before illuminating the tissue, light was first directed through a filter wheel containing six 10-nm wide bandpass filters (ranging from 560 to 610 nm). Diffuse reflectance images of cortical microvasculature were collected by a variable magnification objective (×0.75 to ×3; Edmund Optics), and reflected by a dichroic mirror onto a second CCD camera (Coolsnap fx; Roper Scientific; 1300 × 1030 pixels with 3 × 3 binning, resulting in a 434 × 343 image size for multispectral reflectance). The final multispectral imaging field was positioned over the left hemisphere (7 × 6 mm2). Imaging was performed continuously with 1.6 Hz frame rate for each wavelength. The data were subsequently interpolated to the common time base using the recorded filter wheel angular positions and exposure times of both cameras. Data were recorded and analysed using custom-designed software written in LabWindows CVI (National Instruments, Austin, TX) and MATLAB (MathWorks, Natick, MA).
Intravascular pO2 microscopy
In another group (n = 7), we measured oxygen partial pressure (pO2) in cerebral microvessels using confocal-based phosphorescence lifetime microscopy simultaneously with LSF, using our custom-designed multimodal imaging system.12–14 Following fMCAO, a 3 × 3 mm cranial window was implanted in the left parietal bone with intact dura and sealed with a 150-μm thick glass coverslip.15 The femoral artery was catheterized for arterial blood pressure, pH, pO2 and pCO2 measurements, and to administer dyes. Before imaging, Oxyphor R2, a phosphorescent O2 sensitizer (∼40 μM, Oxygen Enterprises) was injected via the retroorbital vein. Using custom-designed software, intravascular points of interest were selected for phosphorescence lifetime recordings. The fitted lifetimes were converted into absolute pO2 using a calibrated formula resembling the Stern–Vollmer relationship. Both pO2 and CBF were recorded continuously at a rate of 0.2 Hz for 10 min during ICP manipulations.
Measurement of tissue potassium concentration
Extracellular K+ concentration was measured using a K+-selective electrode (n = 5). After the induction of fMCAO, mice were placed in a stereotaxic frame (David Kopf Instruments) and the ICP catheter was inserted into the cisterna magna as above. A burr hole was drilled above the left hemisphere under saline cooling at 1.5 mm lateral and 1.0 mm lateral from the lambda. Dura was kept intact. The K+-selective glass micropipette was inserted 300 μm below pia. A laser Doppler flowmetry probe (Perimed) was placed next to the burr hole.
Clinical study
Inclusion criteria
Patients from a prospectively collected database approved by the Ethics Committee of Yamaguchi University16 were retrospectively included in this study according to the following criteria: age ≥20 years, SAH caused by ruptured anterior circulation aneurysm, aneurysm clipping within 72 h from onset, availability of simultaneous DC/AC and ICP recordings of sufficient quality from Days 3 to 14. Thirty-five patients enrolled between September 2013 and June 2018 met these criteria (Table 1). Written informed consent was obtained from legally authorized patient representatives.
Table 1.
Clinical characteristics of cases and controls
| Cluster (+) (n = 10) | Cluster (−) (n = 25) | P | |
|---|---|---|---|
| Age, years, mean [95% CI] | 63 [54–72] | 59 [53–66] | 0.504 |
| Sex, F: M | 10:0 | 18:7 | 0.084 |
| WFNS grade, n(%) | 0.528 | ||
| 1 | 4 (40) | 10 (40) | |
| 2 | 2 (20) | 9 (36) | |
| 3 | 0 (0) | 1 (4) | |
| 4 | 2 (20) | 4 (16) | |
| 5 | 2 (20) | 1 (4) | |
| Fisher grade, n (%) | >0.999 | ||
| 1 | 0 (0) | 0 (0) | |
| 2 | 1 (10) | 3 (12) | |
| 3 | 9 (90) | 22 (88) | |
| ICH, n (%) | 5 (50) | 6 (24) | 0.227 |
| Aneurysm location, n (%) | 0.481 | ||
| AComA | 2 (20) | 10 (40) | |
| PComA | 2 (20) | 7 (28) | |
| MCA | 5 (50) | 7 (28) | |
| ACA | 1 (10) | 1 (4) | |
| Cilostazol, n (%) | 5 (50) | 14 (56) | >0.999 |
| DCI, n (%) | 7 (70) | 1 (4) | <0.001 |
| Severe vasospasm n (%) | 9 (90) | 12 (48) | 0.028 |
| New infarction n (%) | 6 (60) | 0 (0) | 0.0001 |
| mRS at 3 months, median [IQR] | 3.50 [0.00–4.25] | 0.00 [0.00–2.00] | 0.021 |
| Recording time, h, median [IQR] | 312 [294–323] | 298 [272–330] | 0.477 |
| SD count, median [IQR] | 32.0 [23.5–54.8] | 0 [0.0–0.5] | <0.001 |
| SD/h, median [IQR] | 0.11 [0.08–0.17] | 0.00 [0.00–0.00] | <0.001 |
| ICP spike count, median [IQR] | 32.50 [2.00–49.50] | 2.00 [0.00–9.00] | 0.012 |
| ICP spike/h, median [IQR] | 0.10 [0.01–0.16] | 0.01 [0.00–0.03] | 0.011 |
ACA = anterior cerebral artery; AComA = anterior communicating artery; DCI = delayed cerebral ischaemia; IQR = interquartile range; MCA = middle cerebral artery; mRS = modified Rankin Scale; PComA = posterior communicating artery; WFNS = World Federation of Neurosurgical Society. Statistical testing was performed using t-test (age), Fisher’s exact test (sex, Fisher grade), Chi-square test (WFNS grade, aneurysm location) and Mann-Whitney U-test (recording time, SD count, SD/h, ICP spike count, ICP spike/h).
Neuroimaging
On admission, CT angiography and/or digital subtraction angiography (DSA) was performed for all patients to assess the aneurysm. To evaluate the degree of vasospasm, CT angiography or DSA was conducted on Day 9 ± 2. If patients were diagnosed with delayed cerebral ischaemia at any time, DSA was performed. A CT scan was performed on admission and 6 to 12 h after aneurysm treatment. Follow-up CT was performed 7 ± 2 and 14 ± 2 days and 1 and 3 months after onset, and whenever neurological symptoms worsened. New infarct considered to be due to delayed cerebral ischaemia was evaluated using follow-up CT or MRI. To assess the degree of vasospasm, diameters of distal internal carotid arteries (C1), M1 and M2 segments of middle cerebral arteries, and A1 segments of anterior cerebral arteries were compared to baseline diameters using the same imaging modality. Severity of vasospasm was categorized as none or mild (<33% decrease in arterial diameter on follow-up images), moderate (33–66% decrease) and severe (>66% decrease).17
Neuromonitoring
At the end of the surgery, a single, linear subdural electrode (SD-6P; Ad-Tech Medical Instrument Corporation) and parenchymal ICP probe (Codman & Shurtleff, Inc) were placed frontally and tunnelled through the craniotomy. The subdural strip (six platinum contacts, 10 mm intercontact distance) was connected in sequential unipolar fashion to a NicoletOne system (0.016-45 Hz; CareFusion) or a DC-coupled amplifier (KS217-023, UNIQUE MEDICAL) using an ipsilateral subgaleal reference electrode (Disposable Subdermal Needle Electrode; Nihon Koden) and recorded using a PowerLab (ADInstruments) and LabChart Software (v.8.0; ADInstruments; sampling rate: 2000 Hz). SDs were identified by slow potential changes (<0.05 Hz) consecutively appearing in neighbouring electrocorticogram (ECoG) channels. A cluster of SDs was defined as at least three SDs within three consecutive recording hours.18 ICP was also recorded using PowerLab (ADInstruments) and LabChart Software (Version 8.0; ADInstruments; sampling rate: 200 Hz), and lowpass filtered (0.05 Hz cutoff frequency). An ICP spike was defined as ICP rise above 30 mmHg regardless of its duration.
Assessment of delayed cerebral ischaemia and postoperative standard of care
Delayed cerebral ischaemia was defined as focal neurological deficits or a decrease of at least two points on the Glasgow Coma Scale that were not attributed to other causes by means of clinical assessment, CT or MRI, and appropriate laboratory studies.19 New infarcts on CT or MRI after exclusion of procedure-related infarction was also defined as delayed cerebral ischaemia. Prophylactic hypertension or hypervolaemia was not used. All patients were treated with a Rho-kinase inhibitor fasudil hydrochloride (Asahi Kasei) for 30 mg twice a day. A selective phosphodiesterase-3 inhibitor cilostazol (100 mg, twice a day) was administrated for 5 of 10 and 14 of 25 subjects in cluster (+) and (−) cohorts, respectively (Table 1).
Statistical analysis
Data are presented as box and whisker plots, mean ± standard error, proportions, or individual data-points, and analysed using paired or unpaired t-test, one- or two-way ANOVA for single or repeated measures, χ2, Mann-Whitney test, Wilcoxon matched-pairs signed rank test, Spearman or Pearson correlation, and mixed effects model (Prism 8; GraphPad Software, San Diego, CA), specified for each dataset in figure legends, tables or the text. Normality was checked using Shapiro-Wilk test. All P-values are two-tailed. In the absence of previous experience, initial sample sizes were selected empirically and adjusted based on power calculations using the initial datasets. Blinding was done only for infarct outcomes study; other experiments did not involve interventions or group comparisons. We did not exclude any mouse in this study.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
Experimental study
On endovascular fMCAO, a large portion of the ipsilesional hemisphere developed a perfusion defect, monitored in real time using LSF with high spatial and temporal resolution (Fig. 1A). Spontaneous SDs, reliably detected by their characteristic blood flow signature, occurred frequently but irregularly at an average rate of 3.4 ± 0.9/h during the 3-h monitoring in the control fMCAO group (Fig. 1B). During 3-h fMCAO, ICP, monitored through a cisterna magna catheter (Fig. 1C), remained below 15 mmHg (8.1 ± 3.0 mmHg), and did not significantly differ from naïve mice without cerebral ischaemia (8.4 ± 3.6 mmHg). ICP did not show a consistent change during each spontaneous SD (Fig. 1D).
Figure 1.

Experimental design and protocols. (A) Full-field LSF was used to image CBF changes over the dorsal cortex during fMCAO. Imaging field (green rectangle) was positioned to encompass both hemispheres. A representative speckle contrast image is also shown superimposed with the perfusion defect (blue shaded area) with ≤30% residual CBF, calculated using the average correlation time values from the contralateral hemisphere to serve as baseline. Perfusion changes (shown in artificial units as a marker of SD) in a typical penumbral region of interest (ROI) detected the characteristic hypoperfusion transients associated with peri-infarct depolarizations (green symbols). (B) Spontaneous SDs occurring in random fashion throughout 3 h of imaging in control mice (n = 5). Each horizontal line is one animal. Green symbols indicate the timing of SD occurrence. (C) ICP was monitored and modulated via a cisterna magna catheter connected to a fluid column. (D) A typical ICP tracing is shown recorded simultaneously with blood pressure (BP) to calculate the CPP (CPP = BP − ICP). ICP remained <10 mmHg in control animals with fMCAO for the duration of recordings (∼3 h after stroke onset), and did not show a consistent relationship to SD occurrence (green circles). Vertical scale bars indicate 5 mmHg for ICP, 50 mmHg for BP and CPP. The ICP, BP and CPP values at the onset of the tracings are also shown in mmHg. (E) Experimental protocol and timeline. After fMCAO (time 0), cisterna magna was quickly accessed and laser speckle imaging started within 15 min to obtain a baseline. Then six ICP spikes were induced at one every 12 min for 3 min or 30 s, at an ICP level of 20, 25 or 30 mmHg. In a separate group ICP was persistently kept high at 20 or 30 mmHg; slow decline was periodically corrected (asterisk).
Transient ICP elevations trigger spreading depolarizations and expand the infarct
To test the hypothesis that ICP elevations can trigger SDs in hemispheric ischaemic stroke, we manipulated the global ICP after fMCAO by connecting the cisterna magna catheter to a fluid column (Fig. 1C) and adjusting its height to 20, 25 or 30 mmHg for 3 min, 30 s or persistently (Fig. 1E). We repeated the ICP spikes every 12 min in each animal, in random order. If an SD emerged into the imaging field within 3 min after the ICP spike onset, we classified this as an SD induced by the ICP spike. All other SDs were classified as spontaneous SDs. We then calculated the occurrence rate (i.e. incidence) of ‘induced SDs’ and compared this to the ‘spontaneous SD’ occurrence rate. A representative experiment is shown in Fig. 2A, where 20, 25 and 30 mmHg ICP spikes are denoted by blue, orange and red bars, and induced or spontaneous SDs are marked by yellow or green circles, respectively. We found that 3-min ICP spikes triggered an SD in two out of every three attempts (67%) regardless of the ICP level (Fig. 2B, top). In contrast, the average spontaneous SD occurrence rate during any 3-min period outside of the ICP spikes (i.e. ICP < 10 mmHg) was only 16%, which did not differ from naïve animals without ICP spikes (3-min spontaneous SD incidence of 17%; Fig. 1B). Remarkably, when ICP was raised for only 30 s, 30 mmHg ICP spikes were still capable of inducing SDs at the same rate, whereas 20 and 25 mmHg ICP spikes were no longer effective (Fig. 2B, bottom). Most induced SDs emerged with a latency of only 1–2 min after the onset of ICP spike (Fig. 2B, right). These data showed that brief and mild ICP spikes can potently trigger SDs and suggested a dose–response relationship.
Figure 2.

Transient ICP elevations trigger SDs. (A) A representative experiment with six 3-min ICP spikes at 20 (blue), 25 (orange) and 30 (red) mmHg level, as indicated on each transient. Yellow circles indicate SDs triggered during the 3-min ICP spike (horizontal coloured bars), green circles indicate spontaneous SDs occurring outside of the ICP spikes. Lower tracing shows CBF in artificial units used to identify SD occurrence marked by sharp hypoperfusion transients. (B) Summary of SD occurrence rate linked to 3-min (top) or 30-s (bottom) ICP spikes. Timeline on the left shows the temporal relationship between ICP spikes (thick horizontal coloured bars) and SDs either occurring within 3 min after spike onset (yellow symbols) or outside of ICP spikes (i.e. spontaneous SD; green symbols). Each horizontal line is one animal (n = 6/group). ICP-induced SD was defined as an SD occurring within 3 min elevating the ICP (i.e. spike onset). In the bottom timeline, thinner bars show this 3-min period from the 30-s spike onset. Bar graphs in the middle show the 3-min SD incidence during normal ICP (<10 mmHg), and at ICP levels of 20, 25 or 30 mmHg. P < 0.0001 versus normal ICP (χ2). Whisker-bar plots on the right show the latency of ICP-induced SD from the onset of ICP rise.
In contrast, persistently elevated ICP at either 20 or 30 mmHg for 60 min caused only a small increase in SD occurrence rate compared with normal ICP (Supplementary Fig. 1). Indeed, the small number of SDs occurring during persistent ICP elevation were temporally linked to the periodic ICP corrections we had to do to maintain an ICP plateau for 60 min (Fig. 1E, asterisk), once again suggesting that an abrupt disturbance in metastable penumbra is required to trigger an SD. The SD occurrence rate within 3 min of ICP correction was 35%, whereas the spontaneous SD rate in the absence of an ICP correction was only 8% (P < 0.0001; χ2). The SD occurrence rate with these ICP corrections was lower than that during the ICP spikes presumably due to the smaller ICP steps (typically <5 mmHg) required to sustain the plateau. These data strongly suggested that ICP transients rather than steady elevation were critical for SD induction, reminiscent of our previous findings with supply–demand mismatch transients tipping the metastable penumbral hot zones over to anoxic depolarization thereby triggering an SD.8 Of note, although we did not specifically test whether a sudden ICP drop could also trigger an SD, the rather quick ICP normalization at the end of each spike (Fig. 1E) was not associated with a higher rate of SDs. Of a total of 36 3-min ICP spikes, only two were followed by an SD within the 3 min after the end of the ICP spike (<6%), which was even lower than the spontaneous 3-min SD incidence in the absence of an ICP spike.
Importantly, increased SD rate by five 30-s, 30 mmHg ICP spikes during 1-h transient fMCAO was associated with enlarged infarct volumes when measured in a separate cohort 24 h later, suggesting a detrimental effect on stroke outcome (Supplementary Fig. 2).
Origins of spreading depolarizations induced by ICP spikes
Our next aim was to determine the impact of elevated ICP in foci at which SDs originated, to better understand the trigger mechanisms. For this we needed to examine the SD origins in more detail. About 43% of all SDs originated from within the field of view, 34% propagated into the field from the anterior and the rest from the posterior perimeter of the infarct (Fig. 3A). This distribution did not significantly vary between the persistent ICP and ICP spike groups, or among different ICP levels (Fig. 3A, bottom; P = 0.77; χ2). An example of SD originating from within the field of view is shown in Fig. 3B. The latency between the ICP spike onset and the SD onset was shorter when SD emerged within the field of view than when SD propagated into the field of view anteriorly and to a lesser extent posteriorly (Fig. 3C), probably reflecting the time it takes for an SD to propagate into the field of view when its origin was outside the field.
Figure 3.

Origins of SDs. (A) Full-field laser speckle imaging of CBF over the dorsal cortex shows the origins of SDs in relation to the perfusion defect (blue overlay). About 1/3 of SDs propagated into the imaging field from the anterior (Ant, green) and a third from the posterior (Post, yellow), while the remainder originated from the peri-infarct regions within the imaging field of view (blue). Horizontal bars below show the distribution of SD origins in persistent ICP and ICP spike groups at different ICP levels. The distribution did not statistically differ among ICP levels or between persistent or transient ICP elevation groups (χ2). (B) Time lapse laser speckle images of relative CBF changes (blue indicates a decrease, red indicates an increase in CBF from baseline image, B) show the origin of a typical SD within the field of view (red circle) and its propagation throughout the peri-infarct cortex in a representative experiment during a 30-s, 30 mmHg ICP spike. The top left image shows the position of the imaging field as in Fig. 5A (a = anterior; p = posterior; i = ipsilateral to fMCAO; c = contralateral; ss = sagittal sinus; red circle, SD origin). Time (in seconds) after ICP spike onset is indicated on the lower left of each frame (B, baseline). SD onset is visible at 75 s marked by a focal hypoperfusion developing near lambda (*), as marked on the upper left frame by a red circle. (C) The latency between ICP spike onset and SD emergence in the field of view differed among SDs originating within the field of view and propagating into the field anteriorly or posteriorly (P = 0.0078; one-way ANOVA). (D) Spatial distribution of the origins of SDs that started within the field of view during steady state ICP (left) and during 30-s, 30 mmHg ICP spikes (right), regardless of ICP level or spike duration. Each yellow circle indicates an SD. The centre of the red cross indicates the average mediolateral and anteroposterior coordinates of SDs in each group with standard errors in mediolateral (vertical error bar) and anteroposterior (horizontal error bar). All SDs originated from the peri-infarct tissue. The mediolateral coordinates of SD origins did not differ between persistent and transient ICP groups (P = 0.49). In contrast, SDs originating during an ICP spike were significantly more posterior compared with persistent ICP group (P = 0.006; Student’s t-test). Indeed, most spike-induced SDs originated from the watershed region among anterior, middle and posterior cerebral arteries (red shaded area). ACA = anterior cerebral artery; PCA = posterior cerebral artery.
We next examined in more detail the point of origin of SDs that started within the imaging field. Under steady state, spontaneous SD origins within the field of view were evenly distributed along the medial border of the infarct, as expected (Fig. 3D, left). In contrast, the origins of SDs triggered during ICP spikes clustered more posteriorly (Fig. 3D, right; red crosses indicate the mean ± SEM of the coordinates of SD origins; P = 0.006 and 0.49 along the anteroposterior and mediolateral axes, respectively). This region represented the arterial watershed among the middle, anterior and posterior cerebral arteries, as well as the venous watershed between the transverse and sagittal sinuses, implicating local haemodynamics as a critical determinant.
Effects of ICP elevation on cerebral blood flow and oxygenation
We next quantified the impact of ICP spikes on cerebral haemodynamics. Both 3 min and 30 s ICP spikes reduced CPP in proportion to the magnitude of ICP rise (Supplementary Fig. 3A). In contrast, persistent ICP elevations caused an overall increase in CPP due to the hypertensive Cushing response that developed over minutes and persisted (Supplementary Fig. 3B). Despite reduced CPP, the impact of ICP spikes on CBF was surprisingly mild. CBF decreased during the ICP spikes by only 5–10% of prespike baseline in all cortical regions examined (Fig. 4A). When we measured the CBF within the tissue where an SD originated within the field of view during the ICP spike, we once again found only an 8% reduction in CBF compared to prespike baseline (Fig. 4B). Nevertheless, this CBF drop was highly reproducible regardless of the prespike perfusion level. Interestingly, blood flow in ipsilateral transverse and sagittal venous sinuses consistently showed a larger drop during the ICP spike (∼15% of prespike baseline; Fig. 4C), and while sagittal sinus flow recovered promptly after returning to normal ICP, ipsilateral transverse sinus flow remained significantly lower for almost 3 min. To test whether such small CBF drops can influence tissue oxygenation, we performed multispectral reflectance imaging of oxyhaemoglobin and deoxyhaemoglobin concentrations in ischaemic penumbra during 30 mmHg, 30-s ICP spikes. Consistent with the mild CBF reductions, we found only a very small and statistically insignificant decrease in oxyhaemoglobin and increase in deoxyhaemoglobin concentrations during such ICP spikes (Supplementary Fig. 4). Last, we also examined the impact of ICP elevation on the hypoperfusion response to SDs triggered by 3-min or 30-s ICP spikes. We found a slight but statistically insignificant worsening of hypoperfusion during SDs triggered by ICP spikes compared with that during spontaneous SDs that occurred under normal ICP (Supplementary Fig. 5).
Figure 4.

Effects of ICP elevations on CBF in relation to SDs. (A) CBF changes (relative to prespike baseline) in ischaemic core (C), penumbra (P), non-ischaemic cortex (N) and contralateral cortex (CL) regions of interest shown as a function of time after spike onset for 3 min (top row) and 30 s (bottom row) ICP spikes at different ICP levels (two-way ANOVA for repeated measures). (B) CBF changes at the origins of spike-induced SDs within the field of view (left) were calculated by measuring (middle panel) the grey shaded areas representing the prespike baseline, minimum CBF during the spike (30 s), and immediately before the SD (pre-SD), and expressed as a percentage of pre-ischaemic baseline (right). SDs originated from foci with a range of residual CBF levels that were usually <40% of pre-ischaemic baseline. There was a statistically significant drop in CBF during ICP spike, which persisted until an SD occurred (one-way ANOVA for repeated measures). (C) CBF changes in ipsilateral and contralateral transverse and sagittal venous sinuses during and after a 30-s, 30 mmHg ICP spike; 3-min ICP spikes caused similar changes (not shown). *P < 0.005 versus time 0 (one-way ANOVA for repeated measures).
Microvascular shunting is not involved in spreading depolarizations triggered by ICP spikes
One potential mechanism that might explain the ostensibly mild drop in CBF and oxygenation in penumbra was microvascular shunting and increased capillary transit time heterogeneity,20,21 which redistribute microvascular flow, causing ineffective tissue perfusion and oxygen delivery during high ICP states despite seemingly unchanged CBF and intravascular oxygen content. As a result, oxygen extraction fraction may decrease, leading to a paradoxical increase in venous pO2. To examine this possibility, we performed two-photon pO2 microscopy during ICP spikes after fMCAO (Fig. 5). We constructed a closed cranial window that permitted direct visual identification of pial arteries and veins to measure intravascular pO2 using phosphorescence lifetime.13 Baseline pial arterial and venous pO2 values were 60 ± 4 and 44 ± 2 mmHg, respectively. Arterial pO2 values did not show a significant change during ICP spikes (−1%; P = 0.2231; n = 25 spikes in seven mice; one-way ANOVA), whereas venous pO2 showed a small but reproducible reduction (−5%; P < 0.0001; n = 27 spikes in seven mice) that was in proportion to the mild CBF drop (Fig. 4). As expected, SDs triggered by ICP spikes induced a precipitous pO2 drop of 61 ± 5% often to levels below 20 mmHg, confirming the sensitivity of the pO2 imaging technique (Fig. 5). Altogether, reduced venous pO2 confirmed increased oxygen extraction fraction and worsening supply–demand mismatch in ischaemic penumbra during the ICP spikes, and did not suggest microvascular shunting or increased capillary transit time heterogeneity in this context.
Figure 5.

Mild, transient ICP elevations decrease venous pO2, detected by two-photon pO2 microscopy. (A) Brightfield and phosphorescence images of ischaemic penumbra from a representative experiment demonstrate the typical field of view with pial arteries and veins, and the points of interest used to measure intravascular pO2 in these compartments. Arteries and veins are distinguished by their colour, calibre and tracing the parent vessels. Points of interest 1–2 represent two separate veins, whereas points of interest 13–19 represent an artery. (B) Changes in venous pO2 during a 30 mmHg, 3 min ICP spike (red line) and the SD that was triggered during the spike are shown. Red circle shows the onset of ICP spike. Grey shaded areas indicate the averaged segments to obtain venous pO2 values at baseline and during the ICP spike, where a consistent decrease was found. Venous pO2 during SD was measured at the trough. (C) Cumulative data from all experiments (n = 7 mice, 27 ICP spikes) showing venous pO2 values at baseline, during the ICP spike and SD trough from individual ICP spikes. The wide range of baseline venous pO2 values is due to the spatiotemporal heterogeneity of venous outflow in ischaemic penumbra. With only one exception, all ICP spikes caused a reduction in venous pO2 (paired t-test) suggesting worsening oxygen supply–demand mismatch and increased oxygen extraction fraction, arguing against microvascular shunting or increased capillary transit time heterogeneity. SD occurred only in a subset of experiments as shown, and caused a major drop in venous pO2 (one-way ANOVA for repeated measures using only the ICP spikes that triggered an SD).
ICP spikes rapidly elevate extracellular K+ to trigger spreading depolarizations
Extracellular K+ concentration ([K+]e) may be a critical determinant of SD initiation. Sudden [K+]e elevations in ischaemic penumbra might therefore trigger SDs during the ICP spikes. To test this hypothesis, we performed K+-selective microelectrode recordings from the cortical watershed regions where SDs predominantly originated during the ICP spikes (n = 14 ICP spikes in five mice; Fig. 6A). We found a highly consistent rise in [K+]e during 3-min 30 mmHg ICP spikes (Fig. 6B). Nine of 14 ICP spikes were associated with an SD within 3 min (64%), and two of the remaining five ICP spikes were followed by an SD shortly thereafter (Fig. 6C). When an SD occurred during the spike, the massive [K+]e surge associated with the SD was superimposed on the background [K+]e elevation due to the ICP rise (Fig. 6B, arrowheads). When a spontaneous SD occurred during normal ICP between spikes, there was no such background [K+]e rise preceding the SD onset (Fig. 6B, sSD). When an SD did not occur during the ICP spike, [K+]e returned to prespike levels within a few minutes after resuming normal ICP (Fig. 6B, second ICP spike). Simultaneous laser Doppler flow recording from the same region once again confirmed the small CBF drop during the ICP spikes (19 ± 5% of prespike baseline, P = 0.0019; Fig. 6B, asterisk). Because K+-selective microelectrode was positioned in penumbra after the onset of fMCAO, it was not possible to ascertain the resting state (i.e. pre-ischaemic) baseline [K+]e values, precluding absolute [K+]e calculations during these recordings. Nevertheless, every single ICP spike was associated with a rise in [K+]e potential (∼2 mV) compared to prespike baseline regardless of the resting state [K+]e potential detected by the electrode (P = 0.0003; Fig. 6D), and the magnitude of [K+]e potential rise during the ICP spike did not differ between spikes that triggered an SD and spikes that did not (Fig. 6C, inset). Altogether, these data indicated a disturbance in [K+]e clearance caused by the ICP spikes in ischaemic penumbra.
Figure 6.
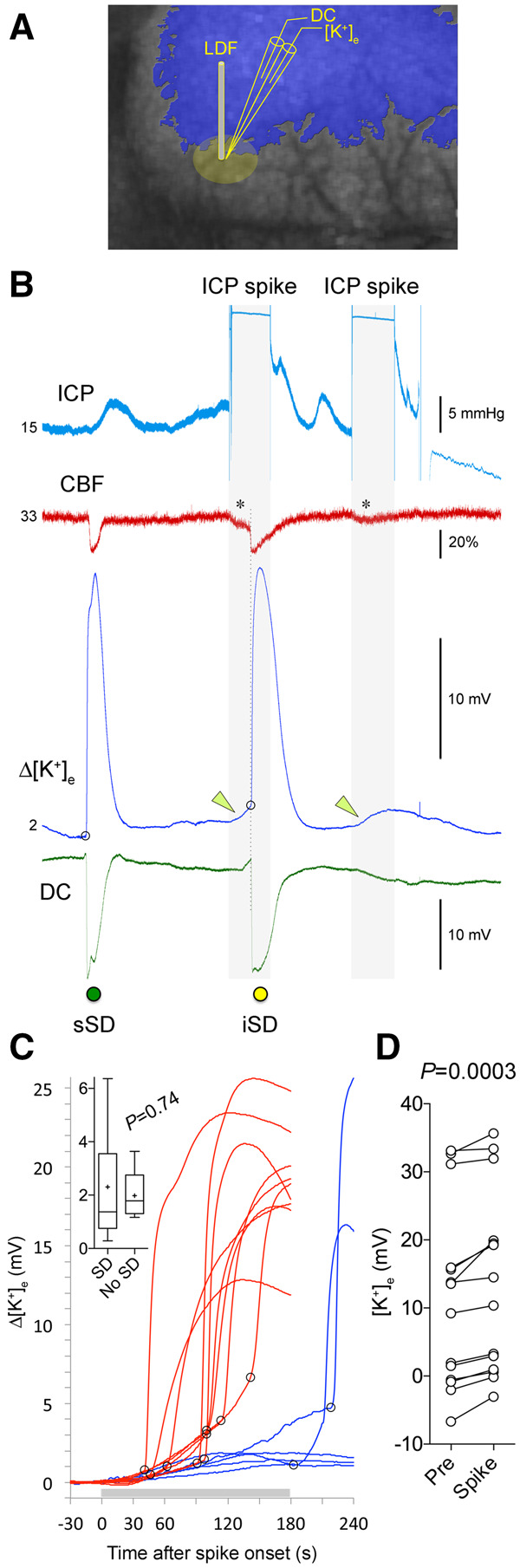
Mild, transient ICP elevations elevate resting extracellular K+ levels. (A) A double barrel K+-selective electrode was placed in the watershed region where most ICP-induced SDs originated. A laser Doppler flowmeter (LDF) was placed immediately adjacent to the electrode to record relative CBF changes. The laser speckle contrast image (greyscale) and the blue overlay are used only to demonstrate the electrode placement in relation to the typical perfusion defect (similar to that shown in Fig. 5C); no imaging was performed in these series of experiments. (B) A representative experiment testing two 3-min, 30 mmHg ICP spikes (blue tracing) on extracellular [K+] ([K+]e). During the initial portion of the recording a spontaneous SD (sSD, green symbol) occurred, associated with the characteristic CBF drop (red tracing), massive [K+]e elevation (blue tracing) and a negative slow potential shift (DC, green tracing). Both ICP spikes (grey shaded area) caused a slow but progressive rise in [K+]e (green arrowheads). About 1.5 min after its onset, the first ICP spike triggered an SD (iSD, yellow symbol), marked by the CBF drop, slow negative potential shift and a steep rise in [K+]e (onset of SD marked by the open circle). The second ICP spike did not trigger an SD despite a similar drop in CBF and slow rise in [K+]e. (C) Cumulative data from 14 ICP spikes in five mice showing the [K+]e tracings. Trials with an SD during the ICP spike (horizontal grey bar) are shown in red (9 of 14 spikes). Trials without an SD (3 of 14 spikes), or with an SD that developed after the end of the ICP spike (2 of 14 spikes), are shown in blue. Open circles mark SD onset. Inset shows the magnitude of [K+]e (mV) at the onset of SD (i.e. at open circles), or the maximum [K+]e if an SD did not occur (t-test). (D) The actual [K+]e electrode potentials before (Pre) and maximum during a spike (Spike) are shown in pairs (paired t-test). In all trials, [K+]e increased during the ICP spike. The wide range of [K+]e electrode potentials at baseline was due to the spatiotemporal heterogeneity of [K+]e in ischaemic penumbra where the electrode was placed in each experiment.
Clinical study
ICP spikes are associated with spreading depolarizations after subarachnoid haemorrhage
Based on these experimental findings, we sought clinical correlation between ICP elevations and SD clusters in patients with aneurysmal SAH. Among the 35 patients who met the inclusion criteria, 10 patients showed SD clusters. Imaging, ECoG and ICP from a representative cluster (+) patient are shown on Fig. 7. Patients with SD clusters did not significantly differ from those without clusters in terms of age, sex, aneurysm location and SAH severity (Table 1). As expected, cluster (+) patients had higher incidence of delayed cerebral ischaemia, severe vasospasm and infarction, and worse long-term outcomes, as well as higher SD counts and frequencies (Table 1 and Fig. 7C). More importantly, cluster (+) patients had significantly higher number and frequency of ICP spikes compared to cluster (−) patients (P = 0.011; Table 1 and Fig. 7C). In contrast, average ICP did not differ between the groups (Fig. 7C). The duration of ICP spikes exceeding 30 mmHg in the cluster (+) cohort was 32 ± 8 s. Because a subdural strip did not permit determination of the true time of onset and spatial origin of SDs the way full-field optical imaging did in our experimental study, we could not directly link each SD to an ICP spike. Nevertheless, in cluster (+) patients, occurrence of ICP spikes exceeding 30 mmHg and SDs showed a strong association when analysed daily (Pearson R = 0.79, P = 0.0025; Fig. 7D, left). The relationship was even stronger with the number of ICP spikes exceeding 30 mmHg and lasting >30 s (Pearson R = 0.82, P = 0.0010). A similar association emerged when analysed in 6-h epochs (Spearman R = 0.210, P = 0.015; Fig. 7D, right). In contrast, daily median ICP throughout the recordings did not correlate with daily number of SDs (Pearson R = −0.20, P = 0.5347). Of note, the number of ICP spikes did not differ between patients treated with cilostazol and those who were not [5.0 (1.0–20.0) versus 1.5 (0.0–24.5), median (25–75% range); P = 0.5447].
Figure 7.

Higher frequency of ICP spikes (≥30 mmHg) are associated with SD clusters in patients with aneurysmal SAH. (A) Representative CT and CT angiography images from the same patient. CT angiography on admission reveals right internal carotid-posterior communicating artery aneurysm (blue arrow). On Day 8, CT angiography shows vasospasm on the M2 portion of right MCA (black arrow). CT on Day 21 shows cerebral infarction in the right hemisphere. (B) Representative ECoG and ICP tracings in a patient who developed SD clusters. The patient was a 64-year-old female with SAH due to ruptured right internal carotid-posterior communicating artery aneurysm that was clipped on Day 0. Patient remained stable until Day 8 when level of consciousness declined. On Day 6, ICP was <20 mmHg and there was no SD (A, left). On Day 8, numerous ICP spikes were observed during a period of SD clusters (arrow) (A, right). (C) From left to right, the frequency of SDs and ICP spikes, and the average ICP values throughout the entire recordings (Mann-Whitney test), and the time course of median ICP values measured in 6-h epochs, are shown in cluster (−) and (+) patients (mixed effects model). (D) Left: The temporal association between the daily number of SDs and the number of ICP spikes in cluster (+) cohort is shown between 3 and 14 days after SAH. Number of ICP spikes and SDs were significantly correlated (Pearson R = 0.79, P = 0.0025; n = 10 patients). Right: Temporal association between the number of SDs and ICP spikes when analysed in 6-h bins in each patient shown as an xy plot (Spearman R = 0.21, P = 0.0150; n = 10 patients). Each symbol represents a 6-h period in one patient.
Discussion
Our data show that a mild and brief ICP spikes are capable of triggering SD in mice during focal cerebral ischaemia and that the mechanism probably involves worsening supply–demand mismatch and elevations of [K+]e in penumbra. We have previously shown that hypoxic and hypotensive transients, as well as functional activation of the peri-infarct tissue, trigger SDs and enlarge infarct volumes by similarly worsening the supply–demand mismatch in the metastable penumbra.8 Although the reduction in CBF during ICP spikes appeared to be small (5–10%), when superimposed on the already compromised perfusion it was sufficient to decrease venous pO2 (i.e. increased oxygen extraction fraction), suggesting supply–demand mismatch to promote SD. Elevated ICP might have additionally lead to microvascular shunting and increased capillary transit time heterogeneity, preventing effective tissue oxygen delivery even when CPP is maintained within normal limits.20–23
Nevertheless, the rise in [K+]e in penumbra was a consistent finding, resembling that observed during cerebral anoxia and ischaemia.24,25 Resting [K+]e is known to be already elevated in penumbra, and any further increase, no matter how small, can precipitate an SD. Although the drop in CBF and increase in supply–demand mismatch probably contribute to the rise in [K+]e, they were both mild, implicating additional mechanisms of impaired K+ clearance, such as reduced vascular washout or disruption in CSF flow.26–30 Indeed, elevated ICP increases cerebral venous pressure,31 diminishing the arteriovenous hydrostatic gradient driving the CSF flow. More work is needed to examine vascular and glymphatic K+ clearance mechanisms in acutely ischaemic brain.32
Although 3-min ICP spikes of 20, 25 or 30 mmHg triggered an SD to the same extent, 30-s ICP spikes triggered an SD only at the ICP level of 30 mmHg, suggesting a dose–response relationship. Regardless of the magnitude or duration of ICP spikes, however, SDs appeared to be more likely to originate from the watershed region between the middle, anterior and posterior cerebral arteries. This watershed region represents the most distal cortical vascular bed with potentially lower perfusion pressures. Moreover, both the arterial pulsatility and the hydrostatic pressure gradients between the arterial and venous perivascular spaces, which together drive the bulk CSF or interstitial fluid flow, are predicted to be lowest in the watershed region.33,34 These properties of the watershed may explain higher susceptibility to SDs induced by ICP spikes. Once triggered, SDs may then aggravate tissue swelling,35 creating a vicious cycle. Last, a sudden rise in ICP could have triggered an SD mechanically. However, we induced the ICP spikes globally via cisterna magna, cranial vault was sealed (i.e. no craniotomy), did not see any brain shifts on optical imaging, and SDs started with a latency of 1–2 min after spike onset, suggesting that mechanical SD induction by an ICP spike was unlikely.
Our clinical data corroborated the experimental findings by revealing an association between the number and frequency of ICP spikes and number and frequency of SDs. To examine the association, we needed a clinical cohort with abundant ICP spikes and SDs after subdural ECoG strips were placed. In most malignant hemispheric infarcts, SDs occur relatively early after stroke onset and diminish over time,36 whereas ischaemic oedema, swelling and thus craniectomies allowing invasive SD monitoring, usually happens only after 36–48 h.37 Moreover, presence of a wide craniectomy effectively diminishes ICP spikes. Therefore, we sought clinical correlation in patients with aneurysmal SAH because subdural electrophysiological recordings, considered as the gold standard for detection of SDs in human brain, are more readily available in this patient population at later time points when craniectomies are performed and closed. Nevertheless, we are cognisant of the pathophysiological differences between ischaemic stroke and aneurysmal SAH.5,38,39 It should be noted, however, that delayed cerebral ischaemia, often focal, is common after SAH and strongly associated with SD occurrence. Therefore, mechanisms underlying the clinical association between ICP spikes and SD occurrence after aneurysmal SAH probably overlap, at least in part, with mechanisms triggering SDs during ICP spikes in acute focal ischaemic brain as we showed experimentally. It was not possible to seek a one-to-one spatiotemporal correlation between ICP spikes and SDs in our clinical dataset for several reasons. First, subdural strips provide electrophysiological signals from a few focal points and, unlike the full-field optical imaging used in our experimental study, cannot resolve when and where an SD wave originated before propagating to one or more recording sites. Second, pressures recorded using a single intraparenchymal probe, as opposed to the global ICP recorded from cisterna magna in mice, may not reflect the true tissue pressures where SDs originated. Third, unlike the highly controlled experimental environment where ICP spikes were induced in an isolated manner, other concurrent triggers for SDs probably exist in patients with SAH and contribute to SD occurrence, confounding the association. Despite these caveats, our clinical series did reveal a clear link between ICP spikes and SD occurrence.
There have been several studies examining the association between ICP and SD occurrence in aneurysmal SAH and traumatic brain injury. Consistent with our findings, most studies in SAH did not reveal an association between the average ICP and SD occurrence38,40,41; one study noted significantly higher ICP levels during SD clusters than during isolated SDs albeit without showing data.42 In traumatic brain injury, higher ICP levels were associated with a higher risk of developing SDs, although a temporal relationship appeared only when hypotensive transients further decreased the CPP.43 Other studies in traumatic brain injury failed to show an association between average ICP and SDs.44,45 Notably, none of the previous studies examined the number or frequency of ICP transients, which, based on our experimental and clinical data, are more relevant than sustained ICP elevations to trigger SDs. Therefore, future studies will need to take a closer look at the ICP dynamics in addition to the average ICP levels.
In summary, we show that ICP spikes trigger SDs as a novel mechanism for a detrimental effect on the outcome of brain injury. Our data suggest that minimizing even mild and non-sustained ICP spikes (e.g. management of pain, agitation, ventilator dyssynchrony) or aggressive prevention and treatment of cerebral oedema may help improve the outcomes.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Supported by grants from the JSPS KAKENHI grant (19K09458), Japanese Heart Foundation/Bayer Yakuhin Research Grant Abroad, NIH (P01NS055104, R01NS102969 and R01NS091230), the Foundation Leducq, the Andrew David Heitman Foundation and the Ellison Foundation.
Competing interests
The authors declared no potential competing interests with respect to the research, authorship and/or publication of this article.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Abbreviations
- CBF
cerebral blood flow
- CPP
cerebral perfusion pressure
- fMCAO
filament middle cerebral artery occlusion
- ICP
intracranial pressure
- SAH
subarachnoid haemorrhage
- SD
spreading depolarization
References
- 1.Nedergaard M, Astrup J.. Infarct rim: Effect of hyperglycemia on direct current potential and [14C]2-deoxyglucose phosphorylation. J Cereb Blood Flow Metab. 1986;6(5):607–615. [DOI] [PubMed] [Google Scholar]
- 2.Hartings JA, Shuttleworth CW, Kirov SA, et al. . The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao's legacy. J Cereb Blood Flow Metab. 2017;37(5):1571–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayata C, Lauritzen M.. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015;95(3):953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strong AJ, Hartings JA, Dreier JP.. Cortical spreading depression: An adverse but treatable factor in intensive care? Curr Opin Crit Care. 2007;13(2):126–133. [DOI] [PubMed] [Google Scholar]
- 5.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17(4):439–447. [DOI] [PubMed] [Google Scholar]
- 6.Hartings JA, Bullock MR, Okonkwo DO, et al. ; Co-Operative Study on Brain Injury Depolarisations. Spreading depolarisations and outcome after traumatic brain injury: A prospective observational study. Lancet Neurol. 2011;10(12):1058–1064. [DOI] [PubMed] [Google Scholar]
- 7.Dreier JP, Isele T, Reiffurth C, et al. . Is spreading depolarization characterized by an abrupt, massive release of Gibbs free energy from the human brain cortex? Neuroscientist. 2013;19(1):25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Bornstadt D, Houben T, Seidel JL, et al. . Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron. 2015;85(5):1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA.. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24(7):744–755. [DOI] [PubMed] [Google Scholar]
- 10.Dunn AK, Devor A, Bolay H, et al. . Simultaneous imaging of total cerebral hemoglobin concentration, oxygenation, and blood flow during functional activation. Opt Lett. 2003;28(1):28–30. [DOI] [PubMed] [Google Scholar]
- 11.Dunn AK, Devor A, Dale AM, Boas DA.. Spatial extent of oxygen metabolism and hemodynamic changes during functional activation of the rat somatosensory cortex. Neuroimage. 2005;27(2):279–290. [DOI] [PubMed] [Google Scholar]
- 12.Yaseen MA, Srinivasan VJ, Sakadzic S, et al. . Optical monitoring of oxygen tension in cortical microvessels with confocal microscopy. Optics Express. 2009;17(25):22341–22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakadzic S, Roussakis E, Yaseen MA, et al. . Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nature Methods. 2010;7(9):755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yaseen MA, Srinivasan VJ, Gorczynska I, Fujimoto JG, Boas DA, Sakadžić S.. Multimodal optical imaging system for in vivo investigation of cerebral oxygen delivery and energy metabolism. Biomed Opt Express. 2015;6(12):4994–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D.. Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab. 2012;32(7):1277–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugimoto K, Nomura S, Shirao S, et al. . Cilostazol decreases duration of spreading depolarization and spreading ischemia after aneurysmal subarachnoid hemorrhage. Ann Neurol. 2018;84(6):873–885. [DOI] [PubMed] [Google Scholar]
- 17.Ayling OGS, Ibrahim GM, Alotaibi NM, Gooderham PA, Macdonald RL.. Anemia after aneurysmal subarachnoid hemorrhage is associated with poor outcome and death. Stroke. 2018;49(8):1859–1865. [DOI] [PubMed] [Google Scholar]
- 18.Dreier JP, Fabricius M, Ayata C, et al. . Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J Cereb Blood Flow Metab. 2017;37(5):1595–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergouwen MD, Vermeulen M, van Gijn J, et al. . Definition of delayed cerebral ischaemia after aneurysmal subarachnoid haemorrhage as an outcome event in clinical trials and observational studies: Proposal of a multidisciplinary research group. Stroke. 2010;41(10):2391–2395. [DOI] [PubMed] [Google Scholar]
- 20.Jespersen SN, Ostergaard L.. The roles of cerebral blood flow, capillary transit time heterogeneity, and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab. 2012;32(2):264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bragin DE, Bush RC, Nemoto EM.. Effect of cerebral perfusion pressure on cerebral cortical microvascular shunting at high intracranial pressure in rats. Stroke. 2013;44(1):177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bragin DE, Bush RC, Muller WS, Nemoto EM.. High intracranial pressure effects on cerebral cortical microvascular flow in rats. J Neurotrauma. 2011;28(5):775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemoto EM, Bragin D, Stippler M, et al. . Microvascular shunts in the pathogenesis of high intracranial pressure. Acta Neurochir Suppl. 2013;118:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen AJ. The extracellular potassium concentration in brain cortex following ischemia in hypo- and hyperglycemic rats. Acta Physiol Scand. 1978;102(3):324–329. [DOI] [PubMed] [Google Scholar]
- 25.Hansen AJ. Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiol Scand. 1977;99(4):412–420. [DOI] [PubMed] [Google Scholar]
- 26.Newman EA, Frambach DA, Odette LL.. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225(4667):1174–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kofuji P, Newman EA.. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iliff JJ, Wang M, Liao Y, et al. . A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iliff JJ, Wang M, Zeppenfeld DM, et al. . Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33(46):18190–18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaberel T, Gakuba C, Goulay R, et al. . Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: A new target for fibrinolysis? Stroke. 2014;45(10):3092–3096. [DOI] [PubMed] [Google Scholar]
- 31.Johnston IH, Rowan JO.. Raised intracranial pressure and cerebral blood flow. 3. Venous outflow tract pressures and vascular resistances in experimental intracranial hypertension. J Neurol Neurosurg Psychiatry. 1974;37(4):392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mestre H, Mori Y, Nedergaard M.. The Brain's glymphatic system: Current controversies. Trends Neurosci. 2020;43(7):458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. . Clearance systems in the brain–implications for Alzheimer disease. [published correction appears in Nat Rev Neurol. 2016;12(4):248]. Nat Rev Neurol. 2015;11(8):457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasmussen MK, Mestre H, Nedergaard M.. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mestre H, Du T, Sweeney AM, et al. . Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367(6483):eaax7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sueiras M, Thonon V, Santamarina E, et al. . Cortical spreading depression phenomena are frequent in ischemic and traumatic penumbra: A prospective study in patients with traumatic brain injury and large hemispheric ischemic stroke. J Clin Neurophysiol. 2021;38(1):47–55. [DOI] [PubMed] [Google Scholar]
- 37.Dohmen C, Sakowitz OW, Fabricius M, et al. ; Co-Operative Study of Brain Injury Depolarisations (COSBID). Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63(6):720–728. [DOI] [PubMed] [Google Scholar]
- 38.Dreier JP, Woitzik J, Fabricius M, et al. . Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129(Pt 12):3224–3237. [DOI] [PubMed] [Google Scholar]
- 39.Macdonald RL. Origins of the concept of vasospasm. Stroke. 2016;47(1):e11–e15. [DOI] [PubMed] [Google Scholar]
- 40.Winkler MK, Dengler N, Hecht N, et al. . Oxygen availability and spreading depolarizations provide complementary prognostic information in neuromonitoring of aneurysmal subarachnoid hemorrhage patients. J Cereb Blood Flow Metab. 2017;37(5):1841–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos E, Olivares-Rivera A, Major S, et al. . Lasting s-ketamine block of spreading depolarizations in subarachnoid hemorrhage: A retrospective cohort study. Critical Care. 2019;23(1):427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dreier JP, Major S, Manning A, et al. ; for the COSBID study group. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132(7):1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartings JA, Strong AJ, Fabricius M, et al. . Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma. 2009;26(11):1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartings JA, Wilson JA, Hinzman JM, et al. . Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol. 2014;76(5):681–694. [DOI] [PubMed] [Google Scholar]
- 45.Hartings JA, Andaluz N, Bullock MR, et al. . Prognostic value of spreading depolarizations in patients with severe traumatic brain injury. JAMA Neurol. 2020;77(4):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.