

Abstract
Background
We explored combining the multiple molecular mechanisms of PRRA and docetaxel with an oncolytic prostate-restricted replication competent Ad (PRRA) as a first-line therapy for advanced prostate cancer (PCa) with the potential to overcome the therpautic limitations of poor virus distribution inside solid tumors.
Methods
We investigated docetaxel-mediated transgene expression and evaluated the antitumor efficacy and efficiency of virus transduction, transgene expression and virus distribution in a PSA/PSMA-positive tumor xenograft model. We also evaluated apoptosis induction, cell killing, and the efficiency of transgene expression and virus replication in vitro.
Results
Tumor growth inhibition was significantly enhanced when docetaxel was administrated before intratumor injection of PRRA. In vivo dual-photon microscopy and ex vivo fluorescence microscopy and immunohistochemistry showed that docetaxel increased transgene expression and expanded virus distribution. The combination of docetaxel and PRRA also increased cell apoptosis. In vitro, docetaxel significantly increased cell killing in PRRA-treated PCa cells. Docetaxel significantly increased Ad-mediated trangene expression independent of Ad binding receptors and replication capability. Docetaxel increased the activity of CMV promoter but not of PSES enhancer, resulting in higher transgene expression. The enhanced CMV promoter activity resulted from activation of p38 MAPK since inhibition of p38 MAPK blocked the docetaxel-induced increase in CMV promoter activity.
Conclusions
Combining docetaxel with an oncolytic PRRA improved therapeutic potential by expanding virus distribution and enhancing cell apoptosis and killing. These studies suggested a novel mechanism for enhancing the effect of therapeutic genes delivered by a PRRA.
Keywords: docetaxel, chemotherapy, oncolytic adenovirus, gene therapy, CMV promoter, p38 MAPK
Introduction
Prostate cancer (PCa) is the most commonly diagnosed noncutaneous malignancy and the second leading cause of cancer death in American men. Patients frequently have locally advanced disease and/or detectable distant bone metastases at initial presentation. Androgen ablation remains the main treatment modality recommended for patients with advanced disease, with an emerging role for chemotherapy. However, hormonal ablation is not curative, and PCa inevitably progresses to an androgen-independent (AI) lethal phenotype.
Docetaxel is standard first-line chemotherapy in advanced AI PCa, as single reagent or in combination regimens, because of the significant survival benefits [1]. Patients who received docetaxel chemotherapy had a significantly longer median survival of 18.9 months from the initiation of treatment compared with 16.5 months in patients who received mitoxantrone chemotherapy [2, 3]. However, docetaxel has been reported to produce adverse events such as anemia, alopecia, fatigue and nausea and grade 3 and 4 neutropenia and febrile neutropenia [4, 5]. Therefore, decreasing the dose of docetaxel by combining other modalities with docetaxel may enhance or retain the antitumor efficacy but reduce the adverse events.
Intratumoral injection of therapeutic vehicles, including gene therapy agents, is possible in the prostate gland due to easy transrectal or perineal access guided by ultrasonic probes. Many viral and non-viral vectors have been used widely for gene therapy. Adenoviruses have many advantages over other gene delivery vehicles [6]. Some clinical studies of oncolytic adenovirus have demonstrated promising results in cancer therapy mainly when administrated in combination with chemotherapy [7] or radiation therapy [8]. However, adenovirus vectors have a broad host range and can infect both normal and tumor cells [9]. It is important to develop tissue/tumor-restricted replication competent adenoviral vectors (TRRA). In theory, TRRA should exhibit superior antitumor efficacy and safety compared to replication-deficient adenovirus because each tumor cell can be an active virus producer and the adenovirus only propagates in and lyses targeted cancer cells and spares normal cells. Recently, we developed a novel prostate-restricted replication competent adenoviral vector (PRRA) by placing both adenoviral E1a and E4 genes under the control of PSES enhancer in order to direct viral replication in a tissue/tumor-specific manner. In addition, we also encoded an EGFP transgene under the control of CMV promoter [10]. PSES is a chimerical prostate specific enhancer sequence integrating the enhancer elements from the PSA and PSMA genes, the two best-studied prostate-specific biomarkers. PSES demonstrated high tumor specific activity in PSA/PSMA positive PCa cell lines [11]. PRRA showed tumor-restricted replication and killing activities in PSA/PSMA positive PCa cell lines in vitro and in vivo, but a single treatment of PRRA showed limited antitumor efficacy. It inhibited tumor growth only in the first two weeks, and virus infection and distribution inside the solid tumors was limited to the boundary between the tumor necrotic area and “healthy” tumor tissues [10]. Similar phenomena have been reported in other oncolytic adenovirus-mediated gene therapies, such as ONYX-015 [12] and herpes simplex virus (HSV) [13].
We are developing novel strategies to improve the antitumor efficacy of PRRA. One is to develop therapeutic gene “armed” PRRAs to deliver the therapeutic proteins and enhance antitumor efficacy [14–16]. Another strategy is to combine PRRA with conventional chemotherapy and/or radiotherapy to improve anti-tumor efficacy. In the present study, we evaluated the therapeutic efficacy of combining docetaxel with oncolytic PRRA in PSA/PSMA-positive PCa tumor models. We found that docetaxel significantly enhanced virus distribution and the transgene expression level of PRRA inside the tumors. In addition, the combination of PRRA and docetaxel enhanced apoptosis in cancer cells. The enhanced cell apoptosis or death resulted in increased gaps among cells, expanding the virus distribution inside the solid tumors. The enhanced transgene expression resulted from docetaxel up-regulation of CMV promoter activity by the activation of p38 mitogen-activated protein kinase (MAPK).
Materials and Methods
In vivo animal studies.
A CWR22rv subcutaneous tumor xenograft model was established as described previously [10]. Mice were inoculated with CWR22rv cells (1× 106), and when tumor size reached ~5 mm in diameter at 2–3 weeks after cells inoculation the mice were randomly assigned to 4 groups. In Group 1 the mice were treated with docetaxel, 12.5 mg/kg intravenously on day 1, 4 and 7 (n=6) [17]; in Group 2, PRRA 1.5 × 108 PFU via intratumoral injection on day 8 (n=10); in Group 3, combination docetaxel on days 1, 4 and 7, and PRRA on day 8 (n=10); in Group 4, PBS injections on days 1, 4, 7 and 8 as vehicle control (n=6). Four mice from groups 2 and 3 were used for tumor monitoring by dual-photon imaging. In the remaining 6 animals per group, tumor sizes were continuously measured with calipers on days 1, 4, 9, 14, 21, 28, 35 and 42, and the tumor volumes were calculated as length × width2 × 0.5236 [18]. Mice were sacrificed when tumor size exceeded 1500 mm3 or on day 42 if tumor size did not reach 1500 mm3. One-way ANOVA was used to compare the tumor sizes among all treatment groups, and the pair-wise comparisons between treatment groups were adjusted with the Bonferroni’s correlation.
Real-time intravital dual photon imaging of prostate tumor.
Forty-eight hours after PRRA injection, 4 mice each from groups 2 and 3 were anesthetized (ketamine/xylazine cocktail) for tumor examination. The tumors were exposed by a skin-flap window and covered by a glass bottom microwell dish (MatTek; Ashland, MA) bathed in isotonic saline [19]. The in vivo real-time intravital imaging of tumors was conducted under a Bio-Rad MRC1024 MP laser scanning confocal /multiphoton scanner (Hercules, CA) attached to a Nikon ×60 NA 1.2 water-immersion objective lens. Fluorescence excitation was provided by a tunable Titanium-Sapphire laser (using a 5w Millennia diode solid state pump laser). The mice were placed on a heating pad maintained at 37°C in all procedures until recovery. The virus-infected green fluorescent cells inside the tumor were observed. The images were collected as single focal planes, time series or Z-series for volume rendering. The images were subsequently analyzed and processed by the Metamorph program (Universal Imaging Corp., Downingtown, PA). The numbers of green fluorescent cells were measured in 10 randomly selected pictures in each animal. Statistical analysis for paired data was performed by Student’s t test.
Histology, immunohistochemistry and in situ Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
After observing the tumors by dual-photon microscopy, 4 mice each from groups 2 and 3 were sacrificed. Tumor samples were collected and cut into two pieces parallel to the direction of the needle tip. One piece was embedded in OCT for frozen sectioning. The frozen sections were fixed in 4% paraformaldehyde and the virus-infected green fluorescent cells were observed under a fluorescence microscope. The green fluorescence distribution area was determined using stereological principles in randomly selected 10 pictures in each animal [20]. The other piece was immediately fixed in buffered formalin, processed, embedded in paraffin and cut into histological sections. Sections were stained with hematoxylin and eosin according to a standard protocol. For the adenovirus infection and distribution analysis, mouse monoclonal (SPM 230) antibody (ready for use) to adenovirus type 5 E1a (Abcam, Cambridge, MA) and a super sensitive biotinylated second mouse antibody (BioGenex) were used. Determination of in situ apoptosis was performed with the TUNEL assay as per manufacturer’s instructions (Roche Dianostics, Indianapolis, IN) [10]. The nuclei-condensed, dark brown nuclear-staining cells were counted in 10 randomly selected vision fields (× 40) in each tumor, and the difference was compared among three groups with ANOVA. Results were expressed as means ± standard deviation of ten measurements. Statistical analyses were 2-sided and performed at the alpha = 0.05 statistical significance level.
Cell Culture.
PSA/PSMA positive PCa cell lines CWR22rv and C4-2 were all maintained in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin.
Cell clonogenic assay.
CWR22rv cells were seeded in 12-well plates (2.5 × 105/well) and treated with 10 nM docetaxel for 16 hours, followed by PRRA infection at a dose of 100 vp/cell, or by either docetaxel, PRRA or control alone at the same time. Two thousand cells were re-seeded in 100-mm culture dishes 48 hours after virus infection. The cells were cultured for an additional 2 weeks and cell clone formation was observed by crystal violet staining. The cell clones were counted and averaged from three 100-mm culture dishes with the same treatment.
Cell killing assay.
C4-2 and CWR22rv cells were seeded in 24-well plates (1 × 105/well) and treated with 5 nM docetaxel for 16 hours, followed by PRRA infection at serial virus doses, ranging from 12.5 v.p. per cell to 200 v.p. per cell. Cells were examined by light microscope daily. Five days after virus infection, crystal violet staining was performed to detect the living cells. The cells were lysed and optical density (OD) read at 590 nm [21]. We also tested the killing effect when the cells were treated with docetaxel and PRRA in different sequences. The cells were treated with 5 nM docetaxel or 100 vp/cell PRRA for 16 hours, followed by the administration of 100 vp/cell PRRA or 5 nM docetaxel respectively for 24 hours. Concurrent treatment with docetaxel and PRRA at time zero and 16 hours were used as the controls. Four wells were used for each treatment. All cells were stained with crystal violet dye 5 days after virus infection, and were lysed and the OD read at 590 nm. The data in various groups were compared using one-way ANOVA followed by Bonferroni’s correlation.
Flow cytometric analysis to detect cell apoptosis.
CWR22rv and C4-2 cells were seeded in 12-well plates (2.5 × 105/well) and treated with vehicle control (DMSO) or 10 nM docetaxel 16 hours before infection with PRRA at 100 vp/cell. Cells were harvested with 0.25% trypsin 48 hours post-virus infection and washed once with PBS. An Apo 2.7 antibody conjugated with PE (B.D. Biotechnology. San Diego, CA) was used to detect cell apoptosis by flow cytometric analysis.
Flow cytometric analysis to detect GFP transgene expression.
CWR22rv and C4-2 cells were seeded in 12-well plates (2.5 × 105 cells per well) and treated with 10 nM docetaxel 16 hours before infection with replication-deficient adenoviruses expressing EGFP gene with various types of fibers, such as Ad5EGFP, Ad5RGDEGFP, Ad5/35EGFP (gifts from Andre Liber in Washington University, Seattle, WA) and PRRA at 100 virus particles per cell. PBS-treated cells served as the control. Cells were harvested using 0.25% trypsin 24 hours post-infection, washed with FACS buffer (PBS with 5% FBS and 0.1% sodium azide) on ice, then fixed in 0.5 ml of cold 1% paraformaldehyde solution for flow cytometric analysis.
Western blotting analysis.
CWR22rv and C4-2 cells (4 × 106) were plated in 100-mm culture dishes and treated with 2.5 nM, 5 nM and 10 nM docetaxel for 24 hours. The same amounts of protein (40 μg) were subjected to SDS-PAGE separation and electroblotted to a nitrocellulose membrane. Antibodies against CAR and integrin αυ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and antibody against phosphorylated P38 MAPK was purchased from Cell Signaling (Danvers, MA). Primary antibodies were detected using horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (Cell Signaling). Supersignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL.) was used to detect the signal.
Flow cytometric analysis to detect integrin β1 and β3 expression on the cell surface.
CWR22rv and C4-2 cells were seeded in 12-well plates (2.5 × 105/well) and treated with vehicle control or docetaxel at 5 nM and 10 nM for 16 hours. Cells were harvested using 0.25% trypsin, washed with FACS buffer on ice, re-suspended in 50 μl of buffer and incubated on ice for 30 minutes with anti-integrin β1 or β3 (R&D, Minneapolis, MN) followed by washing three times. The cells were then incubated with a fluorescein-conjugated IgG secondary antibody (R&D) for 30 min followed by three additional washings. Finally, the cells were fixed in 0.5 ml of cold 1% paraformaldehyde solution for flow cytometric analysis.
Transient Reporter Assay.
CWR22rv and C4-2 cells (2.5 × 105/well) were seeded in 12-well plates. Cells were treated with 10 nM docetaxel for 16 hours followed by DNA transfection. The cells were transfected with 1.6 μg pGL3-CMV-luciferase (a plasmid encoding CMV promoter controlled luciferase gene expression cassette) together with 10 ng Renilla luciferase expressing vector pRL-SV40 (as an internal control) using Lipofectamin 2000 (Invitrogen, Carlsbad, CA). The cells without docetaxel treatment or with treatment with 10 μM of p38 MAPK inhibitor SB 203580, JNK inhibitor SP800125 and NF-κB inhibitor Bay-11-7082 (Sigma Aldrich, St. Louis, MO) were used as controls. Cells were collected 48 hours after transfection for dual luciferase assay (Promega, Madison, WI) according to the manufacturer’s protocol. Data are expressed as the mean of triplicate wells, and the statistical comparisons were analyzed by Two-tailed Student’s t tests.
Results
Docetaxel significantly increased the antitumor effect of oncolytic PRRA in vivo.
We evaluated the antitumor efficacy of docetaxel and oncolytic PRRA alone and in combination in PSA/PSMA positive, androgen-independent CWR22rv subcutaneous tumor xenografts in nude mice hosts. Compared to PBS-treated tumors, tumor growth was significantly delayed in mice treated with docetaxel. Consistent to the previous reports, PRRA inhibited the tumor growth but only in the first two weeks after virus injection and then the tumors grew exponentially [10]. Treatment with docetaxel followed by PRRA significantly delayed tumor growth compared to treatment with either docetaxel or PRRA alone (P<0.01, Fig. 1).
Fig.1.
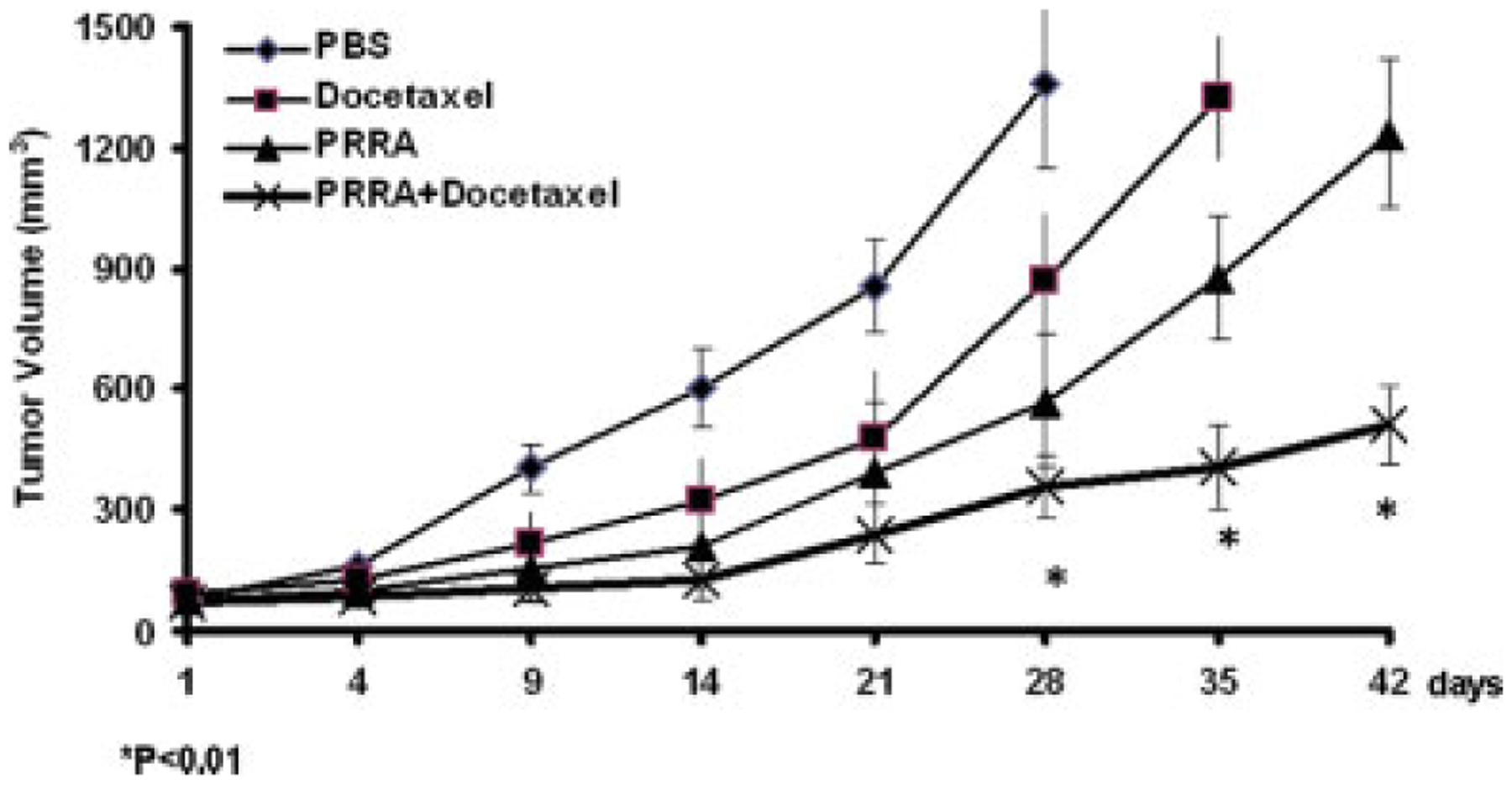
Docetaxel significantly increased the antitumor effects of a prostate-restricted replication competent adenovirus in vivo. CWR22rv subcutaneous tumors were established in the flanks of athymic nude mice. Mice were randomly assigned into four groups (n=6/group) and treated with PBS, docetaxel (12.5 mg/kg, i.v. at day 1, 4 and 7), PRRA (1.5 × 108 PFU, intratumoral injection at day 8), or docetaxel plus PRRA (docetaxel i.v. at day 1, 4 and 7 and PRRA at day 8). Mice were sacrificed when tumor size exceeded 1500 mm3 or at day 42 if tumor did not reach 1500 mm3. The mean (± standard deviation) tumor sizes of the 4 groups are shown. The tumor volume of 1500 mm3 was reached in the sacrificed mice at days 28, 35 and 42. *P< 0.01.
Docetaxel significantly increased in vivo adenovirus-mediated transgene expression in CWR22rv subcutaneous tumors.
PRRA encodes an EGFP gene, so we can continuously monitor the virus-infected green fluorescent cells inside tumors in living animals via a multiple-photon microscopy. Tumors in mice treated with PRRA (n=4) or docetaxel plus PRRA (n=4) were imaged 48 hours after virus injection. The number of virus-infected green fluorescent tumor cells was significantly higher in mice treated with docetaxel plus PRRA compared to PRRA alone (P<0.01; Fig. 2A). Consistent with this, significantly more and brighter green fluorescent cells were seen in the frozen tumors sections in mice treated with docetaxel plus PRRA compared to PRRA alone (10 ×, Fig. 2B). The results suggested that pre-treatment with docetaxel significantly increased in vivo PRRA-mediated EGFP transgene expression.
Fig.2.
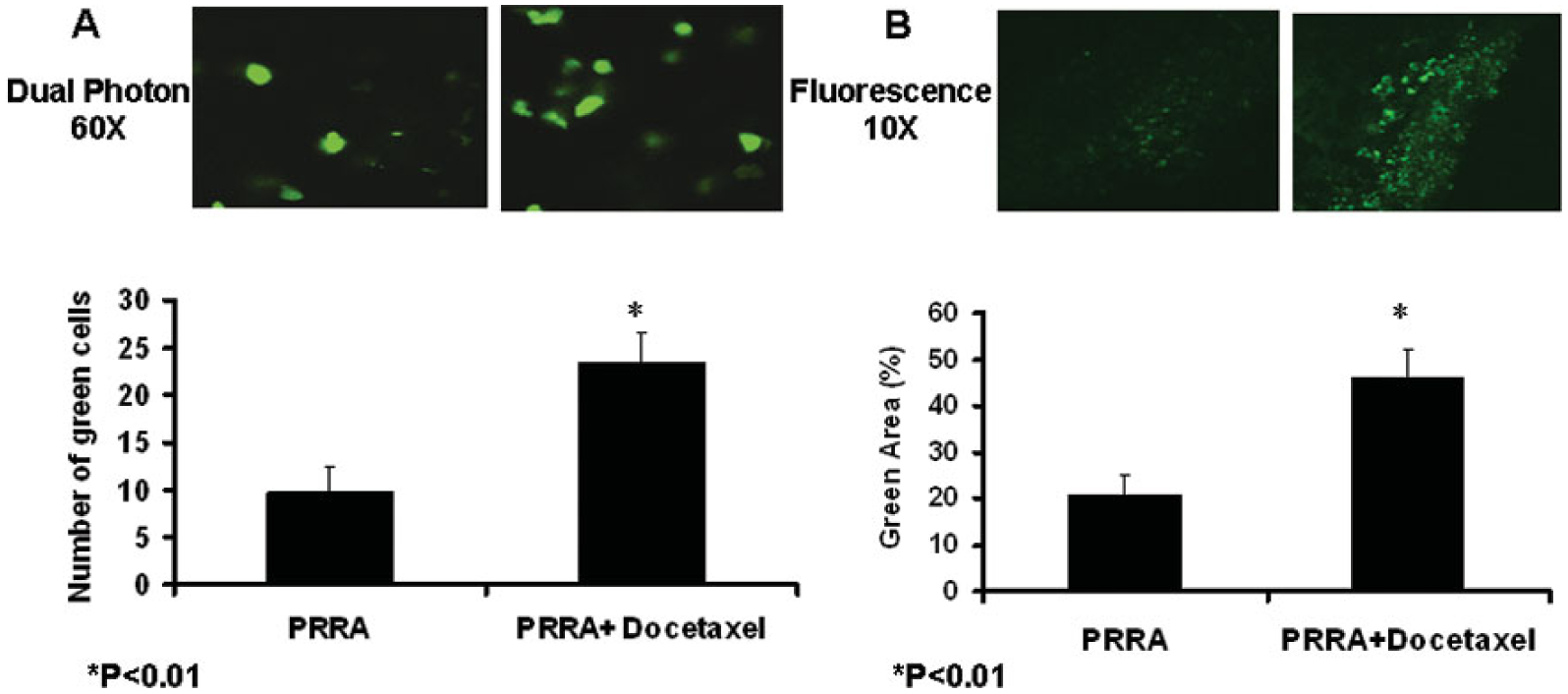
Docetaxel significantly enhanced in vivo adenovirus-mediated transgene expression inside CWR22rv subcutaneous tumors 48 hours after virus injection. A, The tumors treated by PRRA or PRRA plus docetaxel were observed in anesthetized mice using intravital biological dual-photon imaging microscopy (60 ×). The number of green fluorescent cells was counted in 10 randomly selected pictures in each animal. *P< 0.01. B, Tumors were harvested after imaging and embedded in OCT for frozen sections, and the tissues were observed with fluorescent microscopy. The positive area was measured using stereological principles in 10 randomly selected pictures in each mouse. The positive area is bigger and the fluorescence is significantly stronger in the docetaxel plus PRRA-treated tumors than the PRRA-treated tumors alone. *P< 0.01.
Docetaxel significantly increased virus distribution and cell apoptosis in CWR22rv subcutaneous tumors.
We observed the virus distribution in tumor sections using immunohistochemical staining with an antibody against adenovirus 5 E1a. Higher amplification (40 ×) showed that the virus-infected cells were limited to the boundary between necrotic areas and tumor areas in mice treated with PRRA alone. This suggests that there was limited virus distribution inside solid tumors. In comparison, there were significantly more virus-infected positive-cells in tumors in mice treated with docetaxel plus PRRA vs. those treated with PRRA alone, and the positive cells were located at both boundary areas and inside the tumor tissues (P< 0.01, Fig. 3A).
Fig.3.
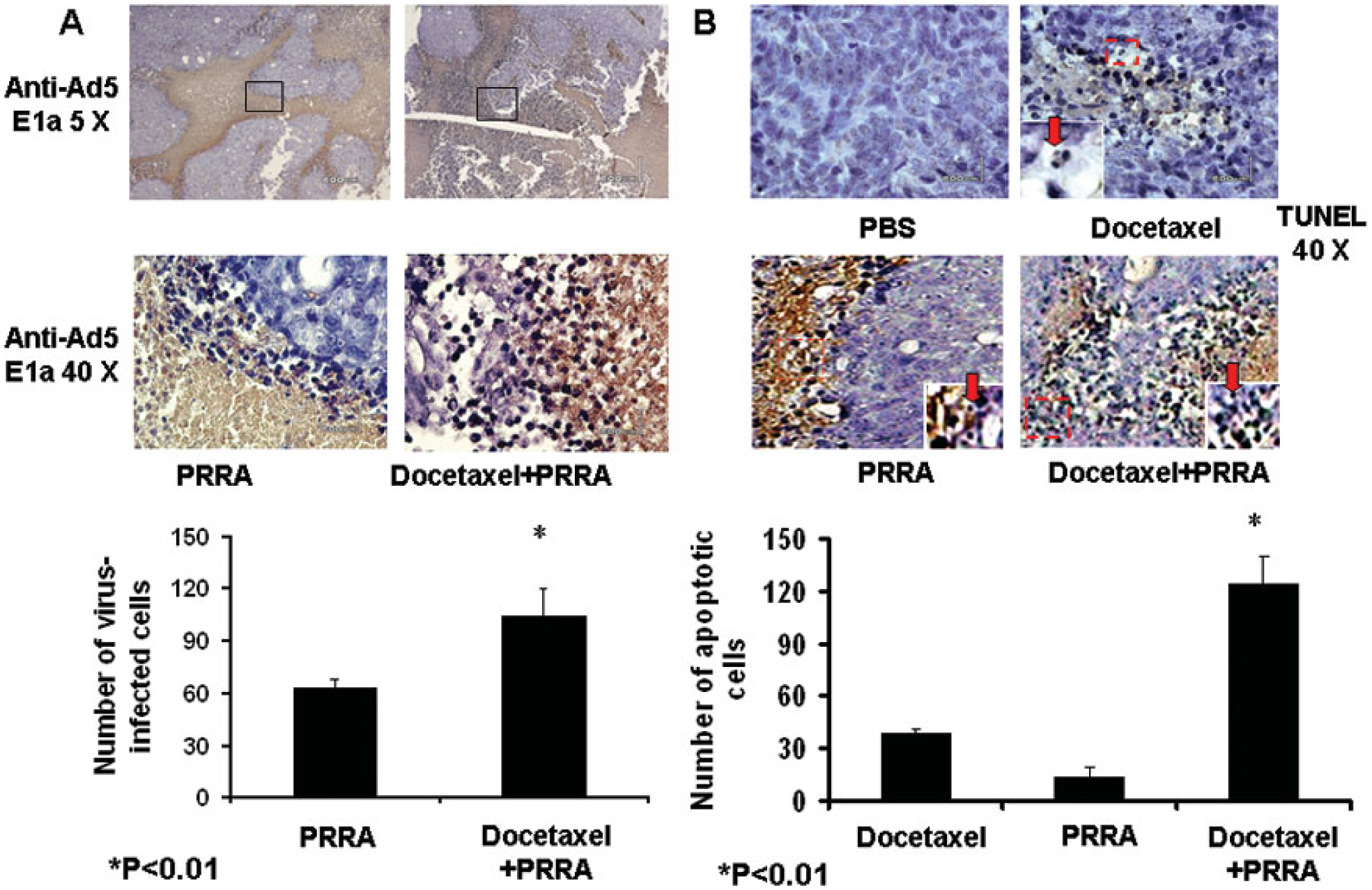
Docetaxel significantly enhanced virus distribution and cell apoptosis. Tumors in PRRA-treated or docetaxel plus PRRA-treated mice were harvested after dual-photon imaging. A, Adenovirus distribution was evaluated by immunohistochemical staining (anti-adenovirus 5 E1a). The number and distribution area of PRRA-infected tumor cells was significantly greater in mice treated with PRRA plus docetaxel compared to mice treated with PRRA alone. *P<0.01. B. Apoptotic cells in the tumor tissue were determined by an in situ TUNEL assay. The number of nuclei-condensed, dark brown nuclear staining apoptotic cells was counted in 10 randomly selected vision fields (40X). *P<0.01.
We also determined cell apoptosis with the in situ TUNEL assay. Some nuclei-condensed or broken, dark brown nuclear staining apoptotic cells could be determined inside the tumor masses in docetaxel-treated mice, and the tumor texture appeared looser than in PBS-treated tumors. There were few apoptotic cells in tumors in mice treated with PRRA alone. In contrast, tumors in mice treated with docetaxel plus PRRA contained significantly more apoptotic cells (P< 0.01, Fig. 3B).
Docetaxel plus PRRA decreased in vitro clonogenic capability and enhanced cell killing and apoptosis.
To confirm the in vivo findings that docetaxel plus PRRA significantly promoted antitumor efficacy compared to the individual treatments, we evaluated clonogenic capability, cell killing and apoptosis induction when CWR22rv cells were treated with docetaxel or PRRA alone and in combination in vitro. In a cell clonogenic assay 48 hours after treatment, two thousands cells were re-seeded in 100 mm culture dishes and studied 2 weeks later. The result showed that docetaxel plus PRRA significantly decreased the number of cell clones compared to docetaxel or PRRA alone (P<0.01, Fig. 4A).
Fig. 4.
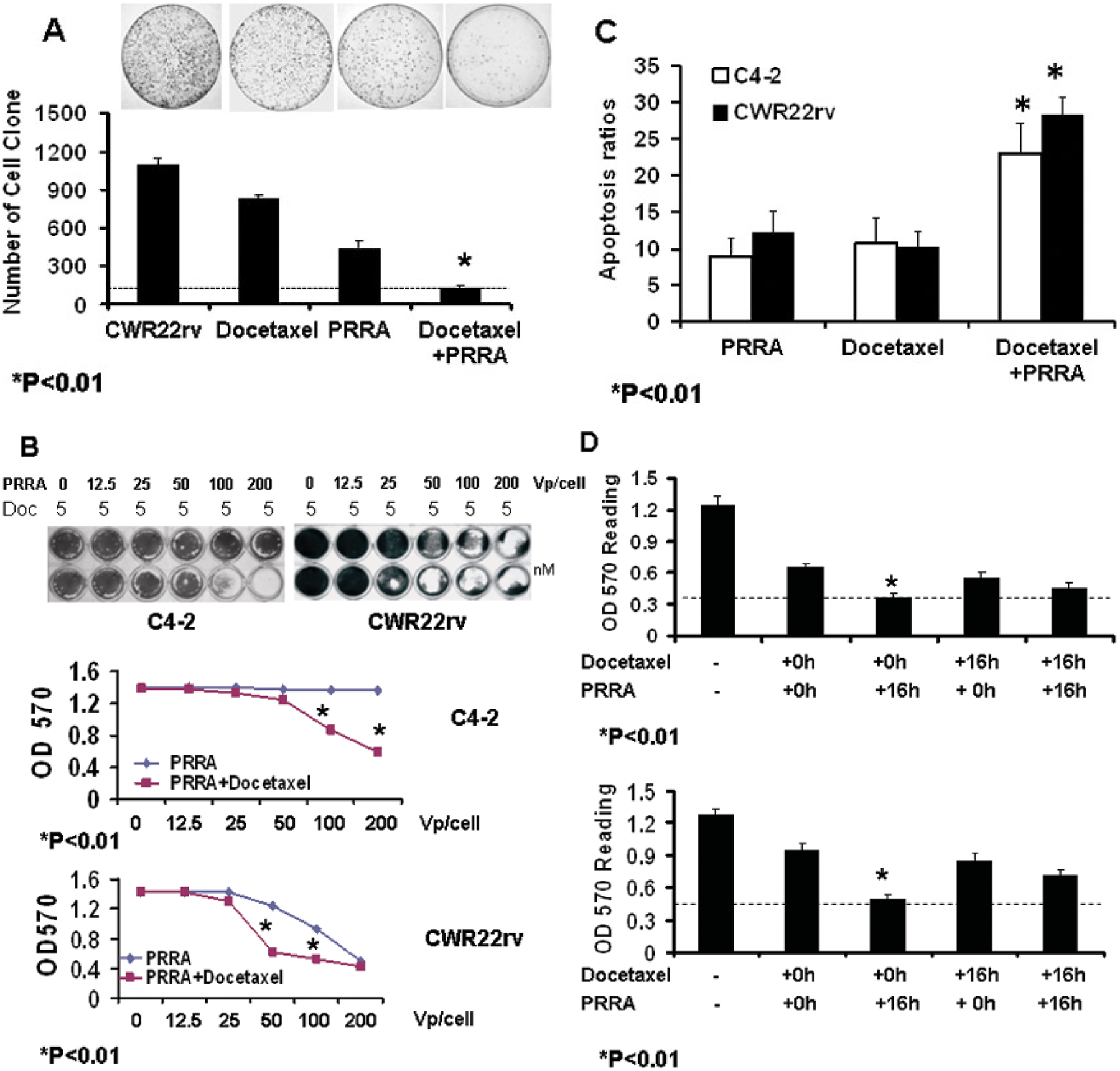
Docetaxel plus PRRA decreased clonogenesis and enhanced cell apoptosis and killing in prostate cancer cells. A, CWR22rv cells were treated with 10 nM of docetaxel for 16 hours, and then the cells were infected with PRRA (100 vp/cell). Cells treated with or without docetaxel or PRRA were used as controls. Two thousand cells were re-seeded in 100-mm culture dishes 48 hours after virus infection. The number of cell clones at 2 weeks after cell re-seeding was determined by crystal violet staining. B, CWR22rv and C4-2 cells were treated with 5 nM docetaxel for 16 hours, followed by infection with PRRA at stepwise-diluted doses. The cells were stained with crystal violet dye 5 days after virus infection, lysed and the OD read at 590 nm. *P<0.01.C, CWR22rv and C4-2 cells were treated with 10 nM docetaxel and PRRA (100 vp/cell), alone and in combination (docetaxel for 16 hours followed by infection with PRRA). Cells were harvested and labeled by Apo 2.7 antibody conjugated with PE for flow cytometric analysis 24 hours after treatment. *P<0.01. D, The sequence of treatments affected cell-killing capability. Cells were treated with 5 nM docetaxel or 100 vp/cell of PRRA for 16 hours, followed by treatment with 100 vp/cell of PRRA or 5 nM of docetaxel, respectively. Cells were treated concurrently with docetaxel and PRRA at time zero and 16 hours as controls. Four wells were used for each treatment. All cells were stained with crystal violet dye 5 days after virus infection, lysed and the OD read at 590 nm. *P<0.01.
To determine the cell killing effect of docetaxel plus oncolytic PRRA, C4-2 and CWR22rv, two PSA/PSMA-positive PCa cells, were treated with docetaxel followed by infection with PRRA at stepwise-diluted doses. In C4-2 cells, undetectable cell killing was observed at 200 vp/cell of PRRA or 5 nM of docetaxel alone; but when administered after 5 nM of docetaxel, PRRA induced cell killing at 100 or 200 vp/cell (P<0.01). In CWR22rv cells, undetectable killing was observed with up to 50 vp/cell of PRRA or 5 nM of docetaxel alone;, but whenPRRA was administered after 5 nM of docetaxel, significantly more killing at 50 vp/cell and significantly more killing at 100 vp/cell was shown (P<0.01, Fig. 4B). Thus, combining docetaxel with PRRA significantly enhanced the cell killing effect compared to docetaxel or PRRA alone.
To confirm whether the enhanced cell killing derived from enhanced cell apoptosis, CWR22rv and C4-2 cells were treated with docetaxel or PRRA alone and in combination. Cells were harvested 24 hours later and stained with Apo 2.7 antibody conjugated with PE to detect cell apoptosis. Docetaxel plus PRRA induced significantly more apoptosis than docetaxel or PRRA alone (P< 0.01, Fig. 4C).
We also tested whether the sequence of administration of docetaxel and PRRA altered the killing effect of PRRA. CWR22rv and C4-2 cells were treated with docetaxel followed 16 hours later with PRRA, or the cells were infected with PRRA followed 16 hours later with docetaxel. Cells treated with both agents together/concurrently at time zero and 16 hours served as controls. The treatment order of docetaxel followed 16 hours later by PRRA had a significantly greater killing effect compared to PRRA followed by docetaxel, and to both agents given together at time zero and 16 hours (P< 0.01, Fig. 4D).
Docetaxel enhanced adenovirus-mediated transgene expression independent of virus binding receptors and replication capability.
To determine whether docetaxel enhanced adenovirus transduction efficiency or replication activity, we used replication competent adenovirus vector PRRA and replication-deficient adenovirus vectors with various fiber types encoding the CMV-EGFP expression cassette. The replication deficient adenoviruses tested included Ad5CMVEGFP containing adenovirus 5 fiber binding to CAR, Ad5/35CMVEGFP containing a hybrid Ad5/35 fiber binding to CD46, and AdRGDCMVEGFP containing a modified Ad5 fiber shaft by RGD motif. CWR22rv and C4-2 cells were treated with docetaxel for 16 hours, virus infection alone, and in combination. Viral-infected cells were determined by flow cytometry of the green fluorescent cells. The percentage of GFP positive cells was significantly higher in cells treated with docetaxel plus viruses compared to the cells treated with viruses alone (Fig. 5A). This suggests that docetaxel enhanced adenovirus-mediated transgene expression independent of the levels of adenovirus-binding receptors and replication status.
Fig.5.
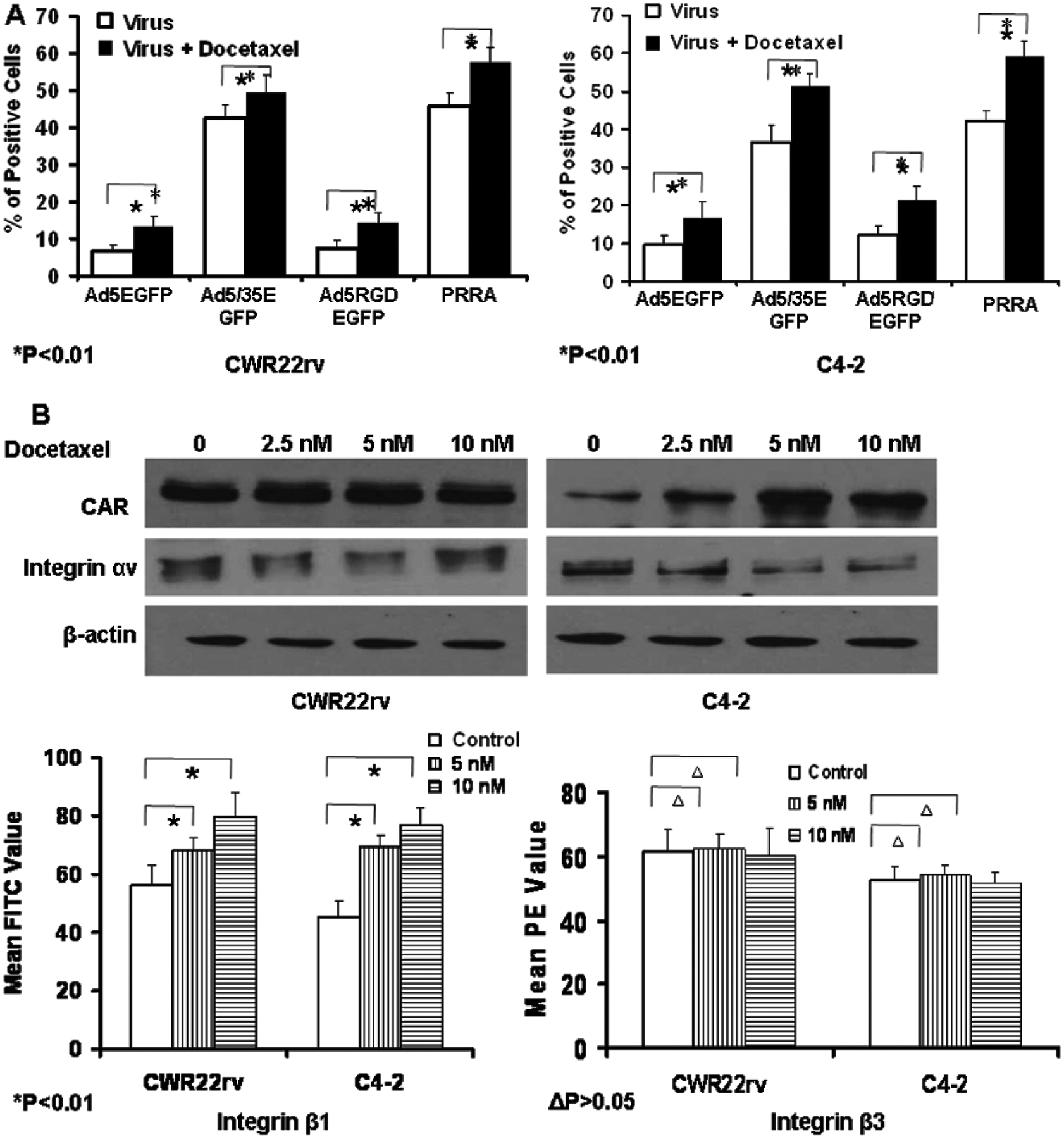
Docetaxel significantly enhanced Ad-mediated transgene expression independent of the levels of Ad binding receptors and virus replication. (A) CWR22rv and C4-2 cells were treated with vehicle or 10 nM docetaxel followed by infection with a panel of adenoviral vectors expressing EGFP, including replication-deficient Ads Ad5EGFP (fiber binding to CAR), Ad5/35EGFP (a hybrid Ad5/35 fiber binding to CD46) and Ad5RGDEGFP (a fiber-mutated by RGD), and replication-competent PRRA. The cells were harvested for flow cytometry 24 h after virus infection and the percentage of green fluorescent cells was determined. *p < 0.01. (B) The impact of docetaxel on the expression levels of CAR and integrins. CWR22rv and C4-2 cells were treated with vehicle or 2.5, 5 and 10 nM docetaxel for 24 h. The cells were lysed and the expression levels of CAR and integrin αν were determined by western blotting. The expression levels of integrin β1 and β3 were determined by flow cytometry. *p < 0.01, Δp > 0.05.
We also studied the impact of docetaxel on expression levels of CAR, and of integrins αv, β1 and β3 at the cell surface of C4-2 and CWR22rv cells by either Western blot or flow cytometry. In C4-2 cells, docetaxel significantly increased the expression of CAR and integrin β1 but had no impact on integrins αv and β3; in CWR22rv cells, docetaxel enhanced the expression of integrin β1 only (Fig. 5B). This suggests that the impact of docetaxel on adenovirus-binding receptors is cell-dependent. To test the influence of docetaxel on adenovirus uptake and replication, CWR22rv cells were treated with docetaxel at doses up to 10 nM, followed by infection with PRRA. The virus copy numbers was determined by RT-PCR for the evaluation of adenovirus uptake capability in the cells, and an adenovirus replication assay was performed. Docetaxel did not significantly increase adenovirus uptake and PRRA replication in CWR22rv cells (Supplement Fig. and Table).
Docetaxel significantly increased the activity of CMV promoter, but not PSES enhancer.
Docetaxel increased the transgene expression in replication-deficient Ad, which demonstrated that docetaxel increased CMV promoter activity. We determined whether docetaxel also increased PSES enhancer activity. C4-2 and CWR22rv cells were transfected with DNA plasmids encoding either a gene expression cassette of CMV promoter controlled EGFP/luciferase or PSES enhancer controlled EGFP/luciferase. This was followed by treatment with docetaxel. EGFP expression was evaluated by flow cytometry and the luciferase activity was evaluated with a luminometer. Docetaxel significantly enhanced EGFP or luciferase expression controlled by CMV promoter, but had no effect on EGFP or luciferase expression controlled by PSES promoter (Fig. 6A and 6B). The data suggest that docetaxel selectively increased CMV promoter activity, but had no impact on PSES enhancer activity.
Fig.6.
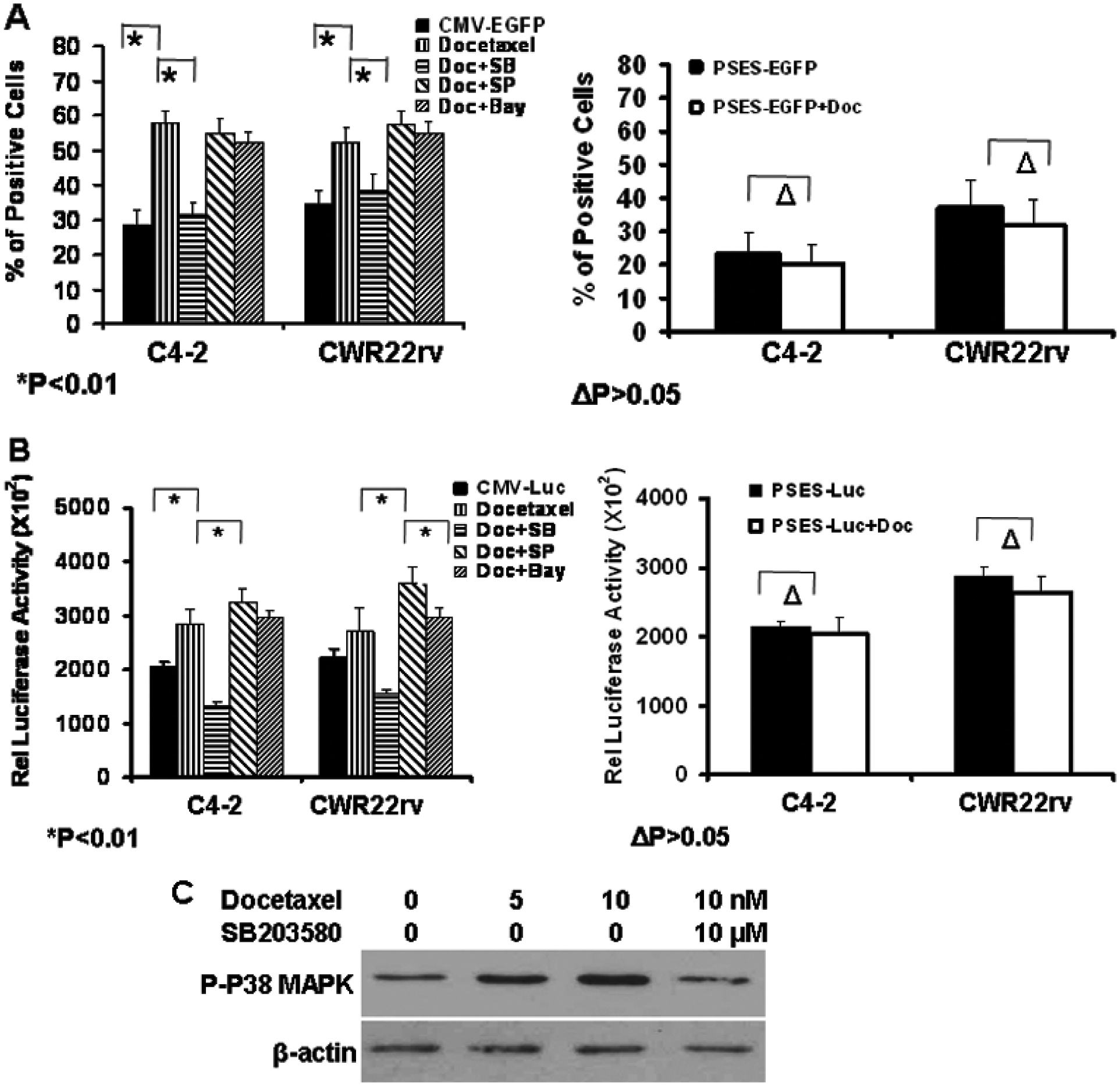
Docetaxel selectively enhanced the activity of CMV promoter but not of PSES enhancer. The enhancement was decreased by inhibition of p38 MAPK. DNA plasmids were transfected into CWR22rv and C4-2 cells by Lipofectamin 2000. 24 hours after transfection, the cells were treated with DMSO or 10 nM docetaxel together with 10 uM of p38 MAPK inhibitor SB205438, JNK inhibitor SP800125 or NF-κB inhibitor Bay-11-7082. Transgene expression was analyzed 24 hours later. A. Two DNA plasmids including CMV promoter-controlled EGFP or PSES enhancer-controlled EGFP expression cassettes were transfected into cells, and the green fluorescent cells were determined by flow cytometry. B, Two DNA plasmids including CMV promoter-controlled luciferase or PSES enhancer-controlled luciferase expression cassettes were transfected into cells. The luciferase activity was determined 24 hours later with a luminometer. *P<0.01, ΔP> 0.05. C. Docetaxel enhanced CMV promoter activity by activating p38 MAPK. CWR22rv cells were treated with 5nM and 10 nM docetaxel, or 10 nM docetaxel plus 10 uM SB203580. The phosphorylation of p38 MAPK was detected by Western blotting. The immunblotting for β-actin confirms equal protein loading.
Docetaxel increased CMV promoter activity by activation of p38 mitogen-activated protein kinase (MAPK).
Several previous reports have suggested that CMV promoter can be up-regulated in different cell lines under certain conditions, such as with increased cAMP in lymphoid cell lines [22], and radiation in pancreatic cancer cells [23]. Several cellular signaling pathways play roles in the activation of CMV promoter activity, such as NF-κB [24], c-Jun NH2-terminal kinases (JNK), and p38 stress-activated mitogen-activated protein kinases (MAPK) [23, 25, 26]. To determine if docetaxel enhanced transgene expression via one of these pathways, we tested the effects of kinase inhibitors on docetaxel enhancement of CMV promoter activity. CWR22rv and C4-2 cells were transfected with a plasmid DNA encoding CMV-EGFP or CMV-luciferase followed by treatment with p38 MAPK inhibitor SB203580, JNK inhibitor SP800125 or NF-κB inhibitor BAY-11-7082 for 1 hour before treatment with docetaxel. The p38 MAPK inhibitor impaired docetaxel-enhanced CMV promoter activity (Fig. 6A and 6B), suggesting the elevation of CMV promoter activity likely resulted from activation of p38 MAPK. This was supported by increased expression of phospho-p38 MAPK in CWR22rv cells when treated with 5 nM or 10 nM docetaxel. This increase was blocked by the p38 MAPK inhibitor SB203580 (Fig. 6C).
Discussion
Oncolytic adenovirus-mediated gene therapy holds great promise for cancer treatment, but some limitations need to be overcome before it can be considered as a first line therapy. For example, oncolytic adenovirus-mediated gene therapy (H101) has been used in clinical trials to treat several cancer types, but efficient spread of the virus within solid tumor masses was a problem [27]. This was also observed in a mouse model of prostate cancer where a prostate-restricted replication competent and oncolytic adenovirus vector inhibited tumor growth only in the first two weeks after virus injection, and virus distribution was limited to the boundary between “healthy” and necrotic areas inside the solid tumors [10].
Recent experimental and clinical studies showed that the therapeutic efficacy of adenovirus-mediated gene therapy can be enhanced when combined with chemotherapeutic agents [17, 28, 29] and radiation therapy [23, 30, 31]. For example, the antitumor effect of oncolytic replication competent adenoviruses ONYX-015 and CV787, and replication-deficient adenovirus-delivered p53, was enhanced when these agents were administered with microtubule-inhibiting taxane [17, 29, 32]. Therefore, in this study we tested the therapeutic efficacy of the combination of docetaxel and PRRA in a prostate cancer xenograft model. Docetaxel itself is used as a first-line chemotherapeutic agent for the treatment of androgen-refractory prostate cancer, but the clinical benefit is limited [2, 3], and at the dose level used it induces several adverse events. Thus a combination therapy may provide better efficacy than either agent alone, and if lower doses can be used then the safety profile of docetaxel may be improved also.
We found that the combination of docetaxel and PRRA had significantly better antitumor efficacy than either agent alone in a PSA/PSMA-positive prostate cancer xenograft model in immune-deficient mice. The enhanced cell killing and apoptosis were confirmed in PSA/PSMA positive prostate cancer cells in vitro. The greatest killing effect was produced when the cells were treated initially with docetaxel for 16 hr followed by infection with PRRA. There are several possible molecular mechanisms to explain this interaction. Docetaxel may enhance adenovirus E1a–induced p53 dependent apoptosis [33–35], or as a microtubule-interacting agent docetaxel may suppress adenovirus-induced S-phase arrest and instead cause G2/M arrest [36]. Also possible is that adenovirus augments the antitumor efficacy of docetaxel via E1a-induced cell sensitization [33, 35]. In this report, we defined several important effects on in vivo adenovirus distribution inside solid tumors, on in vivo and in vitro adenovirus transduction and on transgene expression.
When PRRA was given alone, the virus-infected cells were located at the boundary between necrotic area and “healthy” tumor tissue [10]. This suggests that the narrow gaps between cancer cells in the compact “healthy” tumor tissue are a major physical barrier to the movement of virus particles. The increased cell apoptosis and cell death with the combination therapy caused the tumor masses to become loose in texture with larger gaps between cancerous cells. These structural changes allowed virus particles to penetrate the boundary of the necrotic areas and “healthy” tumor tissue. Several strategies have been reported to promote oncolytic adenovirus distribution in solid tumors by altering the extracellular matrix [37, 38]. Paclitaxel showed the ability to induce cell apoptosis and death in collagen-rich breast tumor masses, which resulted in the enhanced spread and efficacy of oncolytic herpes simplex virus (HSV) [39]. In contrast to collagen-rich breast cancer, less extracellular matrix structure exists in CWR22rv prostate tumors. The tumor cell kill caused by docetaxel caused larger gaps between cells, which allowed the movement and spread of virus particles.
In addition to enhancing in vivo virus distribution and cell apoptosis, we also tested whether other mechanisms might contribute to the enhanced antitumor efficacy of docetaxel and PRRA, such as virus uptake and replication. In this study, docetaxel significantly increased adenovirus-mediated transgene expression in both CWR22rv and C4-2 cells. This effect was seen in both replication-competent adenovirus and replication-deficient adenoviruses with various types of hybrid fiber, which suggest that the effect was independent of adenovirus-binding receptors and replication capability. Although paclitaxel and docetaxel did not reduce virus replication [17, 36], and paclitaxel enhanced virus assembly and the release of adenovirus particles [36], we did not observe that docetaxel significantly increased the replication capability of PRRA. In addition, docetaxel increased one or more Ad-binding receptors including CAR and integrin β1 in C4-2 cells and integrin β1 in CWR22rv cells. The influence of docetaxel on the expression levels of cellular surface receptors depends on cell types and cell cycle phases [40]. The increase of integrin β1 in CWR22rv cells by docetaxel did not significantly increase adenovirus uptake efficiency. Therefore, docetaxel significantly increased adenovirus-mediated transgene expression possibly by increasing promoter activity.
Docetaxel increased CMV promoter activity but did not increase PSES enhancer activity. A variety of environmental stresses such as arsenic [25], chemotherapeutic agents including staurosporine [26], docetaxel [41], paclitaxel [26, 41] and doxorubicin [24, 26], and UV irradiation [23, 42] can all up-regulate CMV promoter activity. We speculate that docetaxel, as a microtubule-interacting agent, results in changes in several transcriptional factors leading to enhanced CMV promoter activity. CMV promoter has cis-regulatory elements recognized by transcription factors such as AP-1, CREB, retinoid acid nuclear receptors (RARs), NF-κB [24] and p38 MAPK. Phosphorylation and activation of these protein cascades are influenced by external or internal cellular stresses, but we believe these effects are cell-specific and/or dependent on the genetic status of the cell type [26]. In the present study, we found that SB203580, a kinase inhibitor of p38 MAPK cascade inhibited the docetaxel-enhanced CMV promoter activity, but SP800125 and BAY-11-7082, inhibitors of JNK and NF-κB, did not inhibit the docetaxel effect.
In summary, we tested the antitumor effects of docetaxel and a novel oncolytic and prostate-restricted replication competent adenovirus PRRA. Docetaxel significantly enhanced the antitumor efficacy of PRRA. This resulted from increased apoptosis and death in the tumor cells, and increased inter-cellular gaps allowing greater spread of virus particles within the tumor. Docetaxel increased virus-mediated transgene expression independent of virus binding receptors and replication capability. Docetaxel significantly enhanced CMV promoter activity by activation of p38 MAPK. These results provide a strong rationale for the clinical testing of a combination of therapeutic gene-armed PRRA with chemotherapy for the treatment of advanced PCa. This strategy could enhance the efficacy of therapeutic genes by increased transgene expression without increasing the safety risk when the therapeutic genes are delivered by PRRA vectors to specific tumor tissues/cells. Novel combination treatments with other chemotherapeutic agents and oncolytic viral delivery vectors might also provide advances in the treatment of other solid tumor types.
Supplementary Material
Acknowledgements
The authors acknowledge financial support from the Maine Institute for Human Genetics and Health Startup (Xiong Li.), NIH grant CA074042 (Chinghai Kao), and DOD grant W23RX-3270-N729 (Chinghai Kao). The work was supported in part by NIH National Research Service Award Number 1 T32 HL007910, Basic Science Studies on Gene Therapy of Blood Disease (Xiong Li).
References
- 1.Beardsley EK, Chi KN Systemic therapy after first-line docetaxel in metastatic castration-resistant prostate cancer. Curr Opin Support Palliat Care 2008; 2: 161–166. [DOI] [PubMed] [Google Scholar]
- 2.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–1520. [DOI] [PubMed] [Google Scholar]
- 3.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–1512. [DOI] [PubMed] [Google Scholar]
- 4.Friedland D, Cohen J, Miller R Jr. et al. A phase II trial of docetaxel (Taxotere) in hormone-refractory prostate cancer: correlation of antitumor effect to phosphorylation of Bcl-2. Semin Oncol 1999; 26: 19–23. [PubMed] [Google Scholar]
- 5.Picus J, Schultz M Docetaxel (Taxotere) as monotherapy in the treatment of hormone-refractory prostate cancer: preliminary results. Semin Oncol 1999; 26: 14–18. [PubMed] [Google Scholar]
- 6.Thomas CE, Ehrhardt A, Kay MA Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 2003; 4: 346–358. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Gao L, Yeagy B, et al. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther 2008; 10: 371–379. [PubMed] [Google Scholar]
- 8.Freytag SO, Kim JH, Brown SL, et al. Gene therapy strategies to improve the effectiveness of cancer radiotherapy. Expert Opin Biol Ther 2004; 4: 1757–1770. [DOI] [PubMed] [Google Scholar]
- 9.Green NK, Seymour LW Adenoviral vectors: systemic delivery and tumor targeting. Cancer Gene Ther 2002; 9: 1036–1042. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Zhang YP, Kim HS, et al. Gene therapy for prostate cancer by controlling adenovirus E1a and E4 gene expression with PSES enhancer. Cancer Res 2005; 65: 1941–1951. [DOI] [PubMed] [Google Scholar]
- 11.Lee SJ, Kim HS, Yu R, et al. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol Ther 2002; 6: 415–421. [DOI] [PubMed] [Google Scholar]
- 12.Kirn D Replication-selective oncolytic adenoviruses: virotherapy aimed at genetic targets in cancer. Oncogene 2000; 19: 6660–6669. [DOI] [PubMed] [Google Scholar]
- 13.Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res 2006; 12: 6737–6747. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Liu YH, Lee SJ, et al. Prostate-restricted replicative adenovirus expressing human endostatin-angiostatin fusion gene exhibiting dramatic antitumor efficacy. Clin Cancer Res 2008; 14: 291–299. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Liu YH, Zhang YP, et al. Fas ligand delivery by a prostate-restricted replicative adenovirus enhances safety and antitumor efficacy. Clin Cancer Res 2007; 13: 5463–5473. [DOI] [PubMed] [Google Scholar]
- 16.Ahn M, Lee SJ, Li X, et al. Enhanced combined tumor-specific oncolysis and suicide gene therapy for prostate cancer using M6 promoter. Cancer Gene Ther 2009; 16: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu DC, Chen Y, Dilley J, et al. Antitumor synergy of CV787, a prostate cancer-specific adenovirus, and paclitaxel and docetaxel. Cancer Res 2001; 61: 517–525. [PubMed] [Google Scholar]
- 18.Gleave M, Hsieh JT, Gao CA, et al. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res 1991; 51: 3753–3761. [PubMed] [Google Scholar]
- 19.Kelly KJ, Sandoval RM, Dunn KW, et al. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol 2003; 284: C1309–1318. [DOI] [PubMed] [Google Scholar]
- 20.ER W Stereological methods. Practical Methods for Biological Morphometry. Academic Press: New York, 1980; 40–160. [Google Scholar]
- 21.Hemminki A, Belousova N, Zinn KR, et al. An adenovirus with enhanced infectivity mediates molecular chemotherapy of ovarian cancer cells and allows imaging of gene expression. Mol Ther 2001; 4: 223–231. [DOI] [PubMed] [Google Scholar]
- 22.Stamminger T, Fickenscher H, Fleckenstein B Cell type-specific induction of the major immediate early enhancer of human cytomegalovirus by cyclic AMP. J Gen Virol 1990; 71 ( Pt 1): 105–113. [DOI] [PubMed] [Google Scholar]
- 23.Egami T, Ohuchida K, Mizumoto K, et al. Radiation enhances adenoviral gene therapy in pancreatic cancer via activation of cytomegalovirus promoter and increased adenovirus uptake. Clin Cancer Res 2008; 14: 1859–1867. [DOI] [PubMed] [Google Scholar]
- 24.Kim KI, Kang JH, Chung JK, et al. Doxorubicin enhances the expression of transgene under control of the CMV promoter in anaplastic thyroid carcinoma cells. J Nucl Med 2007; 48: 1553–1561. [DOI] [PubMed] [Google Scholar]
- 25.Bruening W, Giasson B, Mushynski W, et al. Activation of stress-activated MAP protein kinases up-regulates expression of transgenes driven by the cytomegalovirus immediate/early promoter. Nucleic Acids Res 1998; 26: 486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svensson RU, Barnes JM, Rokhlin OW, et al. Chemotherapeutic agents up-regulate the cytomegalovirus promoter: implications for bioluminescence imaging of tumor response to therapy. Cancer Res 2007; 67: 10445–10454. [DOI] [PubMed] [Google Scholar]
- 27.Yu W, Fang H Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets 2007; 7: 141–148. [DOI] [PubMed] [Google Scholar]
- 28.Lopez CA, Kimchi ET, Mauceri HJ, et al. Chemoinducible gene therapy: a strategy to enhance doxorubicin antitumor activity. Mol Cancer Ther 2004; 3: 1167–1175. [PubMed] [Google Scholar]
- 29.You L, Yang CT, Jablons DM ONYX-015 works synergistically with chemotherapy in lung cancer cell lines and primary cultures freshly made from lung cancer patients. Cancer Res 2000; 60: 1009–1013. [PubMed] [Google Scholar]
- 30.Freytag SO, Barton KN, Brown SL, et al. Replication-competent adenovirus-mediated suicide gene therapy with radiation in a preclinical model of pancreatic cancer. Mol Ther 2007; 15: 1600–1606. [DOI] [PubMed] [Google Scholar]
- 31.Freytag SO, Stricker H, Peabody J, et al. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol Ther 2007; 15: 636–642. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen LL, Lipari P, Dell J, et al. Adenovirus-mediated p53 gene therapy and paclitaxel have synergistic efficacy in models of human head and neck, ovarian, prostate, and breast cancer. Clin Cancer Res 1998; 4: 835–846. [PubMed] [Google Scholar]
- 33.Sanchez-Prieto R, Quintanilla M, Cano A, et al. Carcinoma cell lines become sensitive to DNA-damaging agents by the expression of the adenovirus E1A gene. Oncogene 1996; 13: 1083–1092. [PubMed] [Google Scholar]
- 34.Lowe SW, Ruley HE, Jacks T, et al. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993; 74: 957–967. [DOI] [PubMed] [Google Scholar]
- 35.Debbas M, White E Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 1993; 7: 546–554. [DOI] [PubMed] [Google Scholar]
- 36.AbouEl Hassan MA, Braam SR, Kruyt FA Paclitaxel and vincristine potentiate adenoviral oncolysis that is associated with cell cycle and apoptosis modulation, whereas they differentially affect the viral life cycle in non-small-cell lung cancer cells. Cancer Gene Ther 2006; 13: 1105–1114. [DOI] [PubMed] [Google Scholar]
- 37.Libertini S, Iacuzzo I, Perruolo G, et al. Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A-defective adenovirus dl922–947. Clin Cancer Res 2008; 14: 6505–6514. [DOI] [PubMed] [Google Scholar]
- 38.McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res 2006; 66: 2509–2513. [DOI] [PubMed] [Google Scholar]
- 39.Nagano S, Perentes JY, Jain RK, et al. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res 2008; 68: 3795–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seidman MA, Hogan SM, Wendland RL, et al. Variation in adenovirus receptor expression and adenovirus vector-mediated transgene expression at defined stages of the cell cycle. Mol Ther 2001; 4: 13–21. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Okegawa T, Lombardi DP, et al. Enhanced transgene expression in androgen independent prostate cancer gene therapy by taxane chemotherapeutic agents. J Urol 2002; 167: 339–346. [PubMed] [Google Scholar]
- 42.Hingorani M, White CL, Zaidi S, et al. Radiation-mediated up-regulation of gene expression from replication-defective adenoviral vectors: implications for sodium iodide symporter gene therapy. Clin Cancer Res 2008; 14: 4915–4924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.