

Abstract
Yes‐associated protein (YAP), a central effector in the Hippo pathway, is involved in the regulation of organ size, stem cell self‐renewal, and tissue regeneration. In this study, we observed YAP activation in patients with alcoholic steatosis, hepatitis, and cirrhosis. Accumulation of this protein in the nucleus was also observed in murine livers that were damaged after chronic‐plus‐single binge or moderate ethanol ingestion combined with carbon tetrachloride intoxication (ethanol/CCl4). To understand the role of this transcriptional coactivator in alcohol‐related liver injury, we knocked out the Yap1 gene in hepatocytes of floxed homozygotes through adeno‐associated virus (AAV8)‐mediated deletion utilizing Cre recombinase. Yap1 hepatocyte‐specific knockouts (KO) exhibited hemorrhage, massive hepatic necrosis, enhanced oxidative stress, elevated hypoxia, and extensive infiltration of CD11b+ inflammatory cells into hepatic microenvironments rich for connective tissue growth factor (Ctgf) during ethanol/CCl4‐induced liver damage. Analysis of whole‐genome transcriptomics indicated upregulation of genes involved in hypoxia and extracellular matrix (ECM) remodeling, whereas genes related to hepatocyte proliferation, progenitor cell activation, and ethanol detoxification were downregulated in the damaged livers of Yap1 KO. Acetaldehyde dehydrogenase (Aldh)1a1, a gene that encodes a detoxification enzyme for aldehyde substrates, was identified as a potential YAP target because this gene could be transcriptionally activated by a hyperactive YAP mutant. The ectopic expression of the human ALDH1A1 gene caused increase in hepatocyte proliferation and decrease in hepatic necrosis, oxidative stress, ECM remodeling, and inflammation during ethanol/CCl4‐induced liver damage. Taken together, these observations indicated that YAP was crucial for liver repair during alcohol‐associated injury. Its regulation of ALDH1A1 represents a new link in liver regeneration and detoxification.
Keywords: Aldh1a1, carbon tetrachloride (CCl4), ethanol, liver regeneration, Yes‐associated protein
Abbreviations
- AC
alcoholic cirrhosis
- ADH
alcohol dehydrogenase
- ALD
alcohol‐associated liver disease
- ALDH
aldehyde dehydrogenases
- AS
alcoholic steatosis
- ASH
alcoholic steatohepatitis
- Axin2
Axin‐related protein 2
- CBA
chicken β‐actin
- CCl4
carbon tetrachloride
- ctgf
connective tissue growth factor
- CYP2E1
cytochrome P450
- Dhrs3
dehydrogenase/reductase 3
- ECM
extracellular matrix
- Epcam
epithelial cell adhesion molecule
- GEO
Gene Expression Omnibus
- GFP
green fluorescence protein
- Hif1α
hypoxia‐inducible factor 1α
- HNE
4‐hydroxynonenal
- Hnf4ɑ
hepatocyte nuclear factor 4ɑ
- HSC
hepatic stellate cell
- iCre
codon‐improved Cre recombinase
- Itgam
Integrin subunit alpha M
- KO
Knockout
- KOMP
Knockout Mouse Project
- Krt19
cytokeratin 19
- Lgr5
leucine rich repeat containing G protein‐coupled receptor 5
- MDA
malondialdehyde
- pfu
plaque forming unit
- p‐YAP
phosphorylated Yap at serine 127 residue
- ROS
reactive oxygen species
- TAZ
transcriptional co‐activator with PDZ‐binding motif
- TBG
thyroxine‐binding globulin
- Tbx3
T‐Box 3
- Vegf‐a
vascular endothelial growth factor‐a
- vWF
von Willebrand factor
- WWTR1
the WW domain‐containing transcription regulator protein 1
- Yap
Yes‐associated protein
- αSMA
alpha smooth muscle actin
1. INTRODUCTION
Drinking too much alcohol can have many adverse effects on the body, particularly to the liver that is the main site for ethanol metabolism. Alcohol dehydrogenase (ADH), catalase, and cytochrome P450 (CYP)2E1 are responsible for the first step of ethanol oxidation leading to acetaldehyde production in the liver. This toxic intermediate is eventually converted into acetic acid by cytosolic and mitochondrial aldehyde dehydrogenases (ALDH). Alcohol abuse can activate the microsomal ethanol oxidizing system to induce oxidative stress and lipid peroxidation leading to release of byproducts that form adducts, cause protein dysfunction, and promote DNA mutagenesis. 1 Reactive oxygen species (ROS) attack hepatic cells and their signaling mediators leading to loss of hepatocyte function, inflammation, and abnormal liver repair. Consequently, a broad spectrum of liver pathologies starting from steatosis, hepatitis, fibrosis, to cirrhosis may occur in alcohol‐associated liver disease (ALD). Ethanol impairs liver regeneration, accumulating with chronic inflammation and liver scarring. Understanding the molecular mechanisms underlying ethanol‐induced hepatotoxicity and associated impairment of liver regeneration is essential for therapeutic intervention in ALD.
ALDH family1 member A1 (ALDH1A1) belongs to a group of nicotinamide‐adenine dinucleotide phosphate‐positive [NAD(P)+]‐dependent enzymes that convert toxic aldehyde substrates to their corresponding carboxylic acids. 2 It is a cytosolic enzyme and plays an important role in acetaldehyde removal. 3 It has been considered as a metabolic marker for stem cells and provides drug protection and chemo‐radiation resistance to cancer stem cells. 4 , 5 In the liver, upregulation of this enzyme is associated with a 57‐month recurrence‐free survival in patients with HBV‐related hepatocellular carcinoma (HCC). 6 Aldh1a1 has been shown to detoxify lipid peroxidation products such as 4‐hydroxynonenal (HNE) and malondialdehyde (MDA), whereas hepatic inflammation downregulates this enzyme in rodents. 7 , 8 In addition, polymorphic variants of ALDH1A1 resulting in low enzyme activity have been reported to be associated with alcohol sensitivity. 9 So far, the involvement of this enzyme during alcohol‐induced liver injury is not fully understood.
Yes‐associated protein (YAP) and its paralog—the WW domain‐containing transcription regulator protein 1 (WWTR1), also termed TAZ (transcriptional co‐activator with PDZ‐binding motif), are central effectors in the Hippo pathway critical for regulation of liver development, repair, cell fate determination and tumorigenesis. Their activity is inhibited through phosphorylation by Hippo kinases including MST1/2 and LATS1/2. 10 Phosphorylated YAP is inactive and undergoes proteasome degradation in the cytoplasm. YAP becomes accumulated upon liver injury. Active YAP is in an unphosphorylated form and can enter the nucleus where it binds to transcriptional factors, including members of the TEAD family, and activates target genes including connective tissue growth factor (Ctgf). 11 Upon transcriptional activation, the YAP targets can influence the cell cycle and enhance cell survival thereby promoting cell proliferation. High YAP activity has also been shown to reprogram hepatocytes into a progenitor‐like state and control the fate of injured mouse hepatocytes in vivo. 12 , 13 Activation of YAP can attenuate hepatic damage and fibrosis in liver ischemia‐reperfusion injury. 14 Moreover, YAP can integrate metabolic and nutrient‐sensing pathways leading to coordination of nutrient availability with the genetic program that sustains cell proliferation. 15 , 16 Emerging evidence has shown YAP dysregulation in alcoholic hepatitis. 17 Hence, we examined expression patterns of this transcriptional regulator in human livers from patients with alcoholism, and in murine livers that were damaged after chronic‐plus‐single binge or moderate ethanol ingestion combined with acute intoxication by carbon tetrachloride (ethanol/CCl4). Hepatocyte‐specific KO were utilized to determine the function of Yap1 gene in liver regeneration following ethanol/CCl4. Aldh1a1 was identified as one of the downregulated genes in the KO livers by RNA Sequencing analysis. YAP regulation of this detoxification gene and its function in alcohol‐related liver injury were investigated.
2. MATERIALS AND METHODS
2.1. Animal experiments
Binge ethanol drinking in combination with chronic ethanol exposure was performed to induce hepatocyte damage using the Lieber‐DeCarli liquid diet (BioServ) according to Bertola et al. 18 In brief, female C57Bl6 mice (8–10 weeks old) were fed ad libitum with the Lieber‐DeCarli ethanol diet (ethanol‐fed group) or an isocaloric maltose diet (pair‐fed group) (Bio‐Serv, Flemington, NJ) for 10 days. On day 11, an ethanol binge (single dose at 5 g/kg of body weight) was delivered through oral gavage. The animals were sacrificed at 0 or 9 h after the binge. Parallel experiments were performed in controls that were fed isocaloric maltose and received gavage of equal volume of water (pair‐fed/water).
Hepatocyte damage and liver regeneration were induced through combined treatment with ethanol and CCl4 according to our previous report. 19 Animals were allowed free access to diets with increasing concentrations of ethanol to 2% vol/vol (11% of Kcal) for 4 days. A single acute dose of CCl4 (1 μl/g body weight) prediluted 1:3 in olive oil was given through intraperitoneal injection.
Hepatocyte‐specific Yap1 KO was generated through tail‐vein injection into Yap1flox / flox mice (8‐week‐old age) with AAV8 virus expressing codon‐improved Cre recombinase (iCre) under the control of the human thyroxine‐binding globulin (TBG) promoter (AAV8‐TBG‐iCre). The virus was purchased from Vector Biolabs (Malvern, PA) and was given at 5 × 1012 plaque forming unit (pfu) per mouse. The same amount of AAV8‐green fluorescence protein (GFP) was injected into animals as control. The Yap1flox / flox mice were in C57BL6 background and were derived from FLPase‐mediated removal of the lacZ and neomycin reporters in Yap1 tm1a(KOMP)Mbp animals that were purchased from the Knockout Mouse Project (KOMP) Repository. Three weeks after the viral administration, the AAV8‐iCre infected Yap1flox / flox mice (hepatocyte‐specific Yap1 KO) and AAV8‐GFP‐infected controls were exposed to ethanol (1%–2% vol/vol) followed by single acute dose of CCl4 according to our previous report. 19 The livers were harvested at 0, 7, 24, or 48 h post the chemical induced liver injury.
ALDH1A1 was fused with FLAG tagged epitope in its C terminus (ALDH1A1:FLAG) and was expressed under the control of the chicken β‐actin (CBA) promoter in AAV8 virus. AAV8‐ALDH1A1:FLAG and AAV8‐GFP (1 × 1012 pfu/mouse) were transduced into 8‐week‐old animals. Three weeks after the viral administration, the infected mice were exposed to ethanol followed by acute CCl4 injection as mentioned above. The livers were harvested at 48 h post the chemical induced liver injury.
All animal protocols were approved by the Animal Care and Usage Committee at Tulane University and were conducted in compliance with their guidelines.
2.2. Human ALD livers and histological analysis
Liver tissues from normal livers and ALD patients were purchased from Sekisui XenoTech (Kansas City, KS). Frozen tissues and fixed sections were used to examine expression patterns of YAP protein. Histological analyses for human or animal livers were performed with standard protocols using the following primary antibodies. Rabbit anti‐YAP (Cell Signaling, Danvers, MA), rabbit anti‐4‐hydroxynonenal (HNE) (Millipore, Burlington, MA), rabbit anti‐Ctgf (Abcam, Waltham, MA), rabbit anti‐hepatocyte nuclear factor (Hnf)4ɑ (Santa Cruz Technologies, Paso Robles, CA), rabbit anti‐Ki67 (Abcam), rabbit anti‐CD11b (Abcam), mouse anti‐α smooth muscle actin (SMA) (Sigma, St. Louis, MO), rabbit anti‐von Willebrand factor (vWF) (Dako, Santa Clara, CA), mouse anti‐pimonidazole adduct (Hypoxyprobe, Burlington, MA), rat anti‐CD163 (Thermo Fisher Scientific, Waltham, MA), and rabbit anti‐collagen IV (Thermo Fisher Scientific). Detection was carried out according to the manufacturer's instructions using the ABC‐Elite kit with ImmPACT DAB substrate (Vector Laboratories, Burlingame, CA). In addition, IHC for either Ctgf and CD11b in liver sections was detected using a VECTASTAIN ABC‐AP kit and Vector Alkaline Phosphatase Red substrate (Vector Laboratories). Alexa Fluor 488 or 594 conjugated donkey anti‐rabbit secondary antibody (Invitrogen, Carlsbad, CA) were used for the immunofluorescence staining. Necrosis was revealed by H&E staining. Collagen deposition was measured by Sirius Red staining according to previous publication. 20 For estimation of YAP‐stained positive areas, hepatic necrosis, Hnf4ɑ+ hepatocytes, and Ki67+ proliferating hepatocytes, images were captured with CellSens software using an Olympus BX 51 upright fluorescence microscope outfitted with an Olympus DP80 camera, Plan Fluorite objectives and an LED transmitted light source (Olympus, Waltham, MA). DAB stained areas were quantified from 10 random fields of images (200× magnification) using Image J software (http://rsb.info.nih.gov/ij/) and IHC profiler according to published methods. 21
2.3. RNA isolation, reverse transcriptase‐polymerase chain reaction (RT‐PCR), and RNA‐sequencing (RNA‐Seq) analysis
Total RNA isolation, RT‐PCR and Quantitative RT‐PCR (Q‐RT‐PCR) analyses, and RNA‐Seq were performed in the same conditions according to our previous publication. 19 The raw sequencing data and the processed data have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE123309 for Yap1 hepatocyte‐specific knockouts that received the ethanol/CCl4‐induced liver injury. The related raw and processed data for the chronic ethanol feeding‐plus‐single binge model are also in GEO database (under review). Primers used in RT‐PCR analysis were reported, 20 except for the following genes. Dehydrogenase/Reductase 3 (Dhrs3) primers were 5′ GCGCTGGTAGTGTTCCCTC 3′ and 5′ GGTGTTGACATGCTGGGACTT 3′; Aldh1a1 were 5′ AACACAGGTTGGCAAGTTAATCA 3′ and 5′ TGCGACACAACATTGGCCTT 3′; Yap1 were 5′ TTCAATGCCGTCATGAACC 3′ and 5′ ATCCTGAGTCATGGCTTGCT 3′; iCre were 5′ GTGCAAGCTGAACAACAGGA 3′ and 5′ CCAGCATCCACATTCTCCTT 3′; Adh1 were 5′ CCATCGAGGACATAGAAGTCGC 3′ and 5′ TGGTTTCACACAAGTCACCCC 3′; Hypoxia‐inducible factor 1α (Hif1α) were 5′ GTCCCAGCTACGAAGTTACAGC 3′ and 5′ CAGTGCAGGATACACAAGGTTT 3′; Vascular endothelial growth factor (Vegf)‐a were 5′ GCACATAGAGAGAATGAGCTTCC 3′ and 5′ CTCCGCTCTGAACAAGGCT 3′; Integrin subunit alpha M (Itgam) were 5′ ATGGACGCTGATGGCAATACC 3′ and 5′ TCCCCATTCACGTCTCCCA 3′; Epithelial cell adhesion molecule (Epcam) were 5′ CTGGCGTCTAAATGCTTGGC 3′ and 5′ CCTTGTCGGTTCTTCGGACTC 3′; Axin‐related protein 2 (Axin2) were 5′ ATGAGTAGCGCCGTGTTAGTG 3′ and 5′ GGGCATAGGTTTGGTGGACT 3′; Leucine rich repeat containing G protein‐coupled receptor 5 (Lgr5) were 5′ GGACCAGATGCGATACCGC 3′ and 5′ CAGAGGCGATGTAGGAGACTG 3′; T‐Box 3 (Tbx3) were 5′ AGATCCGGTTATCCCTGGGAC 3′ and 5′ CAGCAGCCCCCACTAACTG 3′; Cytokeratine 19 (Krt19) were 5′ GTTCAGTACGCATTGGGTCAG 3′ and 5′ GAGGACGAGGTCACGAAGC 3′; GFP were 5′ CAAATGGGCGGTAGGC 3′ and 5′ AGCGTGGATGGCGTCT 3′.
2.4. Hepatotoxicity measurement
Hepatic MDA in liver tissues were extracted and quantified based on the fluorescence intensity with excitation at 532 nm and emission at 553 nm according to manufacturer's instruction in a Lipid Peroxidation Assay Kit (Sigma).
ROS generation was determined in mouse livers by using 2′,7′‐dichlorofluorescin diacetate (DCFHDA) that reacts with ROS to produce 2′,7′‐dichlorofluorescein (DCF), a highly fluorescent compound. In brief, mouse livers were perfused to remove blood. The fluorescent product DCF was measured with excitation at 484 nm and emission at 530 nm after homogenization of the liver extracts.
For measurement of hepatic Aldh activity, cytosolic fractions were isolated by centrifugation at 108 000g from homogenized mouse livers. The Aldh activity was measured using fluorometric detection kit (Sigma) with excitation wavelength at 535 nm and emission wavelength at 587 nm.
For acetaldehyde determination, the liver was perfused and homogenized in PBS. After centrifugation, the liver supernatants were mixed with AldeLight™ blue reaction mixture in Amplite™ Fluorimetric Aldehyde Quantitation kit (AAT Bioquest, Inc., Sunnyvale, CA) and fluorescence was measured at 365 nm for excitation wavelength and 435 nm for emission wavelength.
For assessment of liver hypoxia, animals that received ethanol/CCl4 cotreatment were intraperitoneally injected with pimonidazole (120 mg/kg) 1 h before euthanasia. The harvested livers were paraffin embedded. Pimonidazole‐adducts were stained in IHC using a kit from Hypoxyprobe.
Alanine aminotransferase (ALT) in sera was measured using a kit from Sigma according to manufacturer's instruction.
2.5. Western blotting
Total proteins were extracted from mouse livers or cultured cells in RIPA buffer containing proteinase inhibitors (Sigma). Nuclear fractions were isolated using NE‐PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific, Waltham, MA). Nuclear fractions (10 μg) or total protein lysates (50 μg) were boiled in 1× Laemmli buffer containing 5% β‐mercaptoethanol, separated on 4%–12% Bis‐Tris protein gels (Novex, Carlsbad, CA), and electro‐transferred onto polyvinylidene difluoride membrane for immunoblotting. Primary antibodies used were mouse anti‐Aldh1a1 (Santa Cruz Technologies), rabbit anti‐Yap and anti‐phosphorylated Yap at serine 127 residue (p‐YAP) (Cell Signaling), rabbit anti‐Adh1 (Santa Cruz Technologies), mouse anti‐FLAG antibody (sigma), mouse anti‐Cyclin D1 (Santa Cruz Technologies), rabbit anti‐Hif1α (ThermoFisher Scientific), rabbit anti‐fibronectin (Fn1) (Abcam), rabbit anti‐Vegfa, rabbit anti‐CD11b (Abcam), mouse amti‐Aldh1a1 (Cell Signaling), and Dhrs3 (Proteintech, Rosemont, IL), mouse anti‐Actin (Abcam), and rabbit anti‐GAPDH (Abcam). Detection was carried out using horseradish peroxidase‐conjugated secondary antibodies (Santa Cruz biotechnologies) and the ECL Plus kit (Amersham Biosciences, Piscataway, NJ).
2.6. Electrophoretic mobility shift assay and luciferase reporter assay
Electrophoretic Mobility Shift Assay (EMSA) was carried out using Pierce™ Biotin 3′ End DNA labeling kit (ThermoFisher Scientific) to generate probes. A wild type Aldh1a1 probe was generated using primers 5′ AGCAGGAAAAGGGAATGGAAAAAAAA 3′ (sense) and 5′ TTTTTTTTCCATTCCCTTTTCCTGCT 3′ (antisense). 5′ AGCAGGAAAAGactagGAAAAAAAA 3′ (sense) and 5′ TTTTTTTTCctagtCTTTTCCTGCT 3′ (antisense) were for a mutant probe. A mobility shift reaction mixture was set up using LightShift™ Chemiluminescent EMSA kit (ThermoFisher Scientific). It contained crude nuclear extracts overexpressing murine Yap protein that was tagged with Myc epitope at its C terminal region (Yap:Myc) (2 mg), poly(dIdC) (1mg), 0.1 mg of sonicated denatured salmon sperm DNA, biotin‐labeled wild type or mutant probe (4 pmol). A cold wild type probe (20 fmol) was added in some experiments to determine binding specificities of tested probes. Complexes in reaction mixtures (20 μl) were separated in polyacrylamide gel and electro‐transferred onto a nylon membrane for immunoblotting. The biotin end‐labeled DNA probe was detected using streptavidin conjugated to horseradish peroxidase and chemiluminescent substrate according to previous publication. 19
The reporter plasmid carrying Gaussia luciferase (Gluc) under the control of the mouse Aldh1a1 promoter (Aldh1a1p‐Gluc) (NM_027406) was from Genecopoedia (Rockville, MD). This plasmid also expressed secreted alkaline phosphatase (SEAP) that enabled transfection normalization. The Aldh1a1p‐Gluc plasmid was co‐transfected in DMEM medium carrying 10% FBS together with constructs that express a human YAP2 protein or the YAP2S127A mutant in which the phosphorylation site at serine 127 residue was mutated to alanine (Addgene, Watertown, MA). GLuc activity was measured in proliferating cells at 24 h post transfection using Secrete‐Pair™ Dual Luminescence Assay Kit (Genecopoeia).
2.7. In vivo bioluminescence imaging
The mouse Aldh1a1 promoter was cloned to the 5′ region of a firefly luciferase gene at KpnI and BamHI restriction sites in AAV vector. The resulting clone was transfected together with pACG2c8 and phelper plasmids into AAV293 cells (Cell Biolabs, San Diego, CA). AAV8 virus expressing Aldh1a1 promoter‐driven luciferase (AAV8‐Aldh1a1p‐luc) was generated and purified. AAV8‐Aldh1a1p‐luc (1 × 1012 pfu/mouse) was administrated into mouse livers through tail vein injection. Three weeks later, animals were subjected to chronic‐ethanol‐plus single binge according to Bertola et al. 18 Thirty‐six hours before the binge, the plasmid carrying FLAG:YAP2S127A (10 µg/mouse) was delivered into mice through hydrodynamic tail vein injection. AAV8‐Aldh1a1p‐luc activities were measured at 0 or 9 h after the binge by IVIS in vivo imaging system (Perkin Elmer, Waltham, MA) after intraperitoneal injection of D‐luciferin (150 mg/kg body weight) (ThermoFisher Scientific).
2.8. Statistical analysis
GraphPad Prism 6.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Statistical significance (p < .05) was evaluated using the unpaired t‐test and one‐way analysis of variance (ANOVA).
3. RESULTS
3.1. YAP activation during ALD progression or in experimental models of ethanol‐associated liver injury
Alcohol abuse can cause a broad spectrum of liver pathology starting from fat accumulation, alcoholic steatohepatitis (ASH), to alcoholic cirrhosis (AC). We examined expression pattern of YAP protein in total liver homogenates from six ALD patients who suffered from alcoholic steatosis (AS), ASH, and AC. Elevated levels of total YAP protein were found in all tested ALD livers in comparison to two normal controls (Figure 1A). Given the fact that YAP is inactivated through phosphorylation, we also determined the levels of inactive YAP protein using a specific antibody that recognized the phosphorylation site at serine 127. The inactive phosphorylated YAP was fluctuated in the ALD livers compared to normal controls, but percentage of the active nonphosphorylated form were significantly increased in total liver homogenates of patients with AS, ASH, and AC after quantitative analysis using densitometry (Figure 1B). Further IHC in ASH livers showed appearance of YAP in parenchyma of ASH livers (Figure 1C), although there was constitutive expression of this protein in normal portal tracts (Figure S1). These results demonstrated YAP activation during ALD progression.
FIGURE 1.
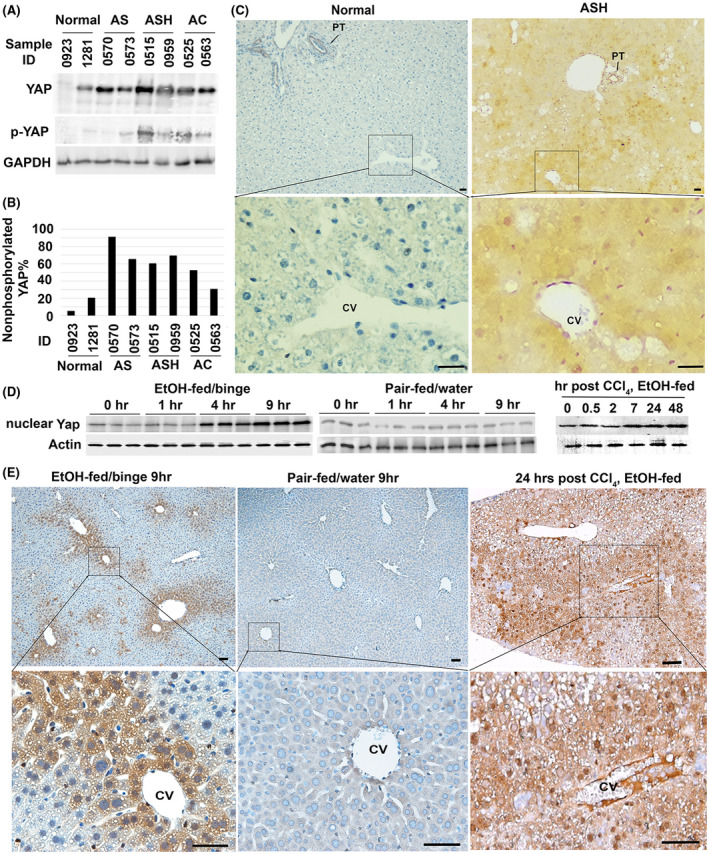
YAP activation during progression of alcohol‐related liver disease and in experimental models of alcohol‐associated liver injury. (A) Total YAP and its phosphorylated form at serine 127 residue in six patients with alcoholic liver disease were examined with specific rabbit antibodies in Western analysis. (B) Densitometrical analysis was performed to calculate percentage of the active nonphosphorylated YAP based on band intensity of phosphorylated (inactive) and total forms of this protein in (A). AS: alcoholic steatosis; ASH: alcoholic steatohepatitis; and AC: alcoholic cirrhosis. (C) IHC detected YAP accumulation in ASH livers. Scale bar: 50 μM. (D) Immunoblotting and densitometry analyses showed Yap accumulation in nuclear fraction of 5% ethanol‐fed animals that received ethanol binge (EtOH‐fed/binge), but not in pair‐fed livers that received water gavage (pair‐fed/water). Accumulation of nuclear Yap was also found in livers that were exposed to moderate ethanol and one acute dose of CCl4 intoxication. Quantification was performed based on three independent experiments from 5 animals per group. *p < .05. (E) IHC detected abundant Yap protein in pericentral hepatocytes of ethanol‐fed livers at 9 h after binge or 24 h post CCl4 intoxication. Scale bar: 100 μM
The NIAAA mouse model of chronic‐plus‐single‐binge ethanol feeding mimics hepatocyte damage, inflammation and steatosis in patients with acute‐on‐chronic alcoholic liver injury. 18 We utilized this model and fed C57BL6 mice 5% ethanol‐containing liquid diet followed by ethanol binge. Then RNA‐Seq was performed to identify genes or pathways that were differentially expressed at 9 h after binge in comparison to the livers without binge in ethanol‐fed mice. Functional enrichment analysis detected upregulation of gene signatures for triglyceride catabolic process, smooth muscle cell proliferation, inflammatory response, and Hippo signaling pathway, whereas genes in oxidation‐reduction process and retinol metabolism were downregulated in the ethanol/binge‐treated livers (Tables 1 and S1). To monitor active Yap content, we isolated nuclear fractions from total liver homogenates of mice that were exposed to ethanol‐fed/binge or pair‐fed/water. As shown in Figure 1D, accumulation of nuclear Yap was observed at 4 and 9 h post‐binge in ethanol‐fed livers but not in controls. IHC demonstrated Yap accumulation in pericentral hepatocytes where ethanol was presumably metabolized in these ethanol‐fed/binge livers (Figure 1E).
TABLE 1.
Functional enrichment analyses identify genes and pathways that are upregulated or downregulated during indicated liver injury
| Injury type | Term | Fold enrichment | p value | Change |
|---|---|---|---|---|
| Chronic‐ethanol‐binge‐induced liver injury in wild type mice | GO:0055114~oxidation‐reduction process | −3.49554532 | 4.72E‐13 | Down |
| Mmu00830:Retinol metabolism | −5.194388162 | 4.33E‐05 | Down | |
| GO:0019433~triglyceride catabolic process | 11.02560976 | .00531723 | Up | |
| GO:0048661~positive regulation of smooth muscle cell proliferation | 5.088742964 | .00638916 | Up | |
| GO:0050729~positive regulation of inflammatory response | 5.25029036 | .005597808 | Up | |
| mmu04390:Hippo signaling pathway | 2.302091518 | .057852084 | Up | |
| Ethanol/CCl4 cotreatment in the hepatocyte‐specific Yap1 KO | GO:0055114~oxidation‐reduction process | −3.61466496 | 6.78E‐12 | Down |
| Mmu00830:Retinol metabolism | −7.63762278 | 2.47E‐08 | Down | |
| GO:0045766~positive regulation of angiogenesis | 8.160437763 | 1.08E‐09 | Up | |
| GO:0048661~positive regulation of smooth muscle cell proliferation | 7.714163823 | 5.61E‐06 | Up | |
| GO:0016477~cell migration | 3.5541697 | .001 | Up |
CCl4 and ethanol are metabolized in pericentral hepatocytes and induce hypoxia, oxidative stress, and hepatic damage. Moderate ethanol feeding in combination with CCl4 has been modeled in mice to investigate the effect of ethanol on hepatic apoptosis, liver regeneration, and fibrosis. 22 , 23 We have demonstrated that the ethanol/CCl4 cotreatment can induce Yap in damaged livers. 19 To further determine active Yap content, we extracted nuclear fractions from total liver homogenates and detected elevation of nuclear Yap protein within 2 days post CCl4 intoxication in ethanol‐fed livers (Figure 1D). Furthermore, IHC labeled plenty of nuclear Yap protein in pericentral hepatocytes within the first day post the chemical induced liver injury (Figure 1E). These observations indicated accumulation of active Yap in pericentral hepatocytes during ethanol‐associated liver injury.
3.2. Hepatocyte‐specific deletion of Yap1 reduces hepatocyte proliferation and cellular reprogramming to progenitor phenotypes during ethanol/CCl4‐induced liver injury
Hepatocytes are the main parenchymal cells to mediate liver regeneration. They harbor a heterogeneous capacity to proliferate. Subpopulations of pericentral hepatocytes express some early liver progenitor markers, such as Tbx3 and Axin2, and show self‐renewal abilities during homeostasis. 24 On the other hand, Hnf4α+ periportal hepatocytes can reconstitute the livers after chronic injury, such as CCl4 intoxication. 25 Hnf4α was deregulated in damaged hepatocytes after ethanol/CCl4‐induced damage. 19 To explore Yap function in regulation of hepatocyte proliferation and progenitor activation, we generated hepatocyte specific KO or controls through transduction of equal amounts of AAV8‐TBG‐iCre or AAV8‐GFP viruses into Yap1 flox/flox homozygotes prior to moderate ethanol feeding and acute CCl4 intoxication. Expression of iCre and GFP transgenes, and loss of Yap1 at mRNA and protein levels were detected in KO by RT‐PCR analysis, immunoblotting, and IHC (Figure 2A–C). Q‐RT‐PCR and immunoblotting analyses demonstrated significant reduction of Ccnd1 mRNA and Cyclin protein in the hepatocyte specific KO (Figure 2B,C). IHC revealed significantly enhanced loss of Hnf4α+ periportal populations and exacerbated necrosis in liver parenchyma of the damaged hepatocyte‐specific KO. There was concomitant reduction of hepatocyte proliferation as indicated by the decreased number of Ki67+ proliferating hepatocytes in the KO compared with controls (Figure 2C). RNA‐Seq and functional enrichment analysis demonstrated downregulation of progenitor‐related markers including Epcam, Lgr5, Axin, Tbx3, and the biliary marker cytokeratin 19 (Krt19) in the damaged Yap1 KO. Q‐RT‐PCR analysis verified 33% downregulation of Epcam, 87% decrease of Lgr5, 58% reduction of Axin2, 59% loss of Tbx3, and 82% decrease of Krt19 transcript in damaged Yap1 KO (Figure 2D). Collectively, these results indicated that Yap loss affected hepatocyte proliferation and cellular programming that regulated progenitor‐related genes during liver regeneration following ethanol/CCl4 cotreatment.
FIGURE 2.
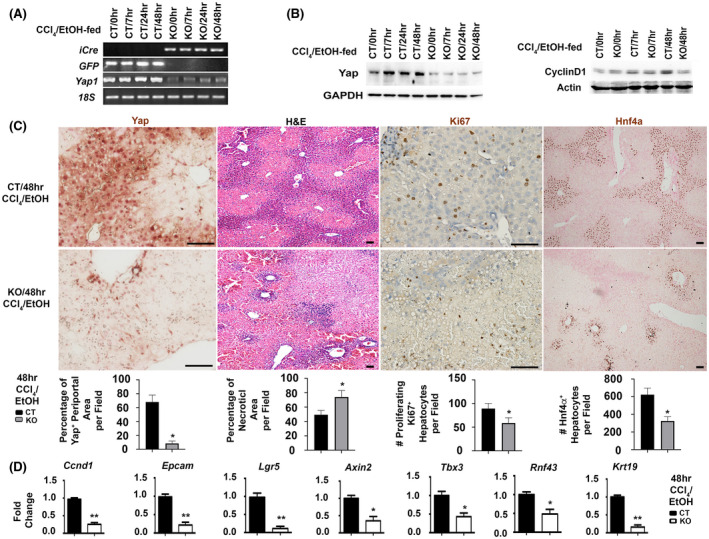
Yap1 deletion inhibits hepatocyte proliferation and downregulates progenitor related genes after ethanol/CCl4 treatment. Control (CT) animals or hepatocyte‐specific knockouts (KO) were generated by tail vein injection of AAV8‐GFP or AAV8‐iCre on floxed homozygotes followed by ethanol‐feeding and a single dose of CCl4 intoxication (n = 5/per group per time point). Yap1 deletion in hepatocytes was confirmed in RT‐PCR analysis (A), Western blotting (B), and IHC (C). Yap loss in periportal hepatocytes of KO livers at 48 h after ethanol/CCl4‐induced injury was associated with massive hepatic necrosis shown in H&E staining, reduced proliferating hepatocytes in Ki67 staining, and decreased number of Hnf4a+ periportal cells. Scale bar: 100 μm. Values were means ± SEM based on quantification of images from more than 10 fields per mouse (n = 5 mice per group). (D) Q‐RT‐PCR analysis detected downregulation of Ccnd1 and progenitor related transcripts in the ethanol‐fed Yap1 KO livers at 48 h post CCl4 intoxication. Values represent means ± SD in relation to controls from three independent experiments. *p < .05, **p < .01
3.3. Hepatocyte‐specific deletion of Yap1 enhances hemorrhages, hypoxia, aberrant extracellular matrix (ECM) remodeling, and recruitment of CD11b+ inflammatory cells during ethanol/CCl4‐induced liver injury
Hemorrhages can result from a breach in the hepatic parenchyma. Consistent with their impaired regeneration, the Yap1 KO developed morphologically distinguishable hemorrhage at 48 h post CCl4 intoxication after moderate ethanol pre‐exposure (Figure 3A). This hemorrhage was associated with abnormal ECM remodeling as detected by dual staining for the endothelial marker vWF and the activated hepatic stellate cell (HSC) marker αSMA. Spontaneous lesions positive for vWF and αSMA staining were found around pericentral zones only in the damaged Yap1 KO livers at 7 h and became more evident than controls at the 48‐h time point (Figure 3A). Since ethanol metabolism consumes oxygen and causes hypoxia leading to the stabilization of the oxygen‐sensitive transcription factor Hif1α, 26 we performed IHC for the hypoxia marker pimonidazole and found greater stained areas in ethanol‐fed Yap1 KO livers than controls at 7 and 48 h post CCl4 (Figure 3A). RNA‐Seq and functional enrichment analysis detected upregulation of gene signatures for hypoxia, angiogenesis, smooth muscle cell proliferation, and cell migration (Table 1). Q‐RT‐PCR analysis showed that Yap1 KO livers had about 2.31‐fold increase of Hif1α, 2.91‐fold upregulation of Vegf‐a, 4.83‐fold increase of Ctgf, 2.15‐fold elevation of Fn1, 2.41‐fold increase of Col4a1, 2.13‐fold upregulation of Col4a2, and 2.43‐fold induction of Itgam that encodes integrin αM subunit for CD11b (Figure 3B). Accordingly, Hif1α, Vegf‐a, Ctgf, Fn1, and Itgam proteins were elevated, and these elevations were confirmed after quantifications of band intensities based on Western analyses (Figure 3C). Further Sirius Red staining revealed larger areas of collagen deposition near perivenous areas in the ethanol‐fed Yap1 KO livers than CT controls at 48 h post CCl4 (Figure S2). Enhanced ECM remodeling in these damaged Yap1 KO livers was also verified after the immunofluorescent staining for collagen IV in Figure S3. We then monitored inflammatory cell infiltration based on IHC for CD11b since this cell surface protein is an adhesion receptor for Ctgf. 27 CD11b+ inflammatory cells were tightly associated with damaged pericentral hepatocytes, whereas the majority of CD11b+ inflammatory cells were in Ctgf‐enriched parenchyma of Yap1 KO livers where integrity of pericentral veins were disrupted (Figure 3D). M2‐type macrophages have been shown to control tissue repair by increasing the expression of growth factors such as Vegf‐a and transforming growth factor (Tgf)‐β. 28 , 29 Therefore, we examined the extent of M2 macrophage activation based on the immunofluorescent staining for cluster of differentiation 163 (CD163) protein. CD163 is a known marker for activated M2 macrophages and modulates inflammatory responses during physio‐pathological conditions in many organs including the livers. 30 , 31 , 32 More CD163+ activated M2 macrophages were found in the ethanol‐fed Yap1 KO livers than CT controls at 48 h post CCl4 (Figure S4). These observations indicated that Yap1 deficiency was associated with enhancement of hemorrhages, hypoxia, activation of stromal cells (vascular endothelial cells, HSC, and inflammatory cells), and aberrant ECM remodeling.
FIGURE 3.
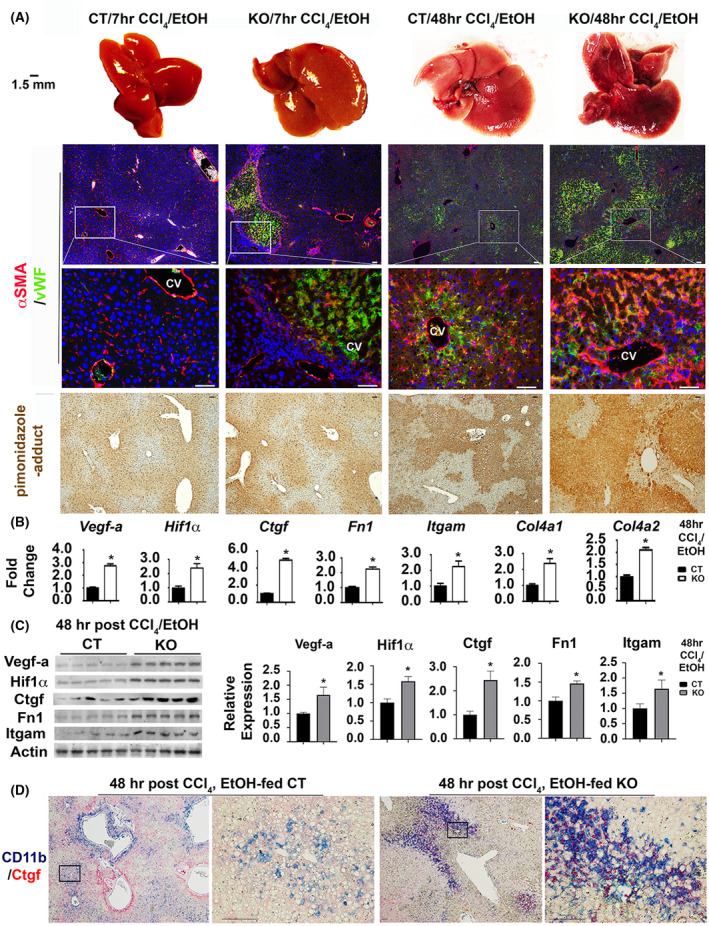
Yap1 deficiency in hepatocytes enhances hypoxia, vascular remodeling, and infiltration of CD11b+ inflammatory cells into Ctgf enriched microenvironments after ethanol/CCl4 treatment. (A) Macroscopic visualization shows hemorrhage in ethanol‐fed Yap1 KO livers at 48 h post CCl4 intoxication (first row). Vascular endothelial cells and activated hepatic stellate cells were stained with vWF and αSMA (second and third rows). Hypoxia was stained based on pimonidazole‐adducts (fourth row). Scale bar: 100 μm. Q‐RT‐PCR analysis (B) and Western blotting (C) detected upregulation of gene signatures for hypoxia and vascular remodeling. Values in (B) represent means ± SD from five animals. *p < .05. Relative levels of Vegf‐a, Hif1α, Ctgf, Fn1, and Itgam proteins in KO in comparison to CT groups (n = 5 per group) were quantified based on densitometrical analyses of band intensities in Western analyses and were expressed as means ± SD. *p < .05. (D) Dual staining showed extensive infiltration of CD11b+ macrophages into Ctgf enriched microenvironments in the damaged Yap1 KO livers. Scale bar: 100 μm
3.4. Hepatocyte‐specific deletion of Yap1 enhances susceptibility to hepatotoxicity induced by ethanol/CCl4 cotreatment
Transient accumulation of Yap was found in peri‐central hepatocytes at 7 h post the acute intoxication and in periportal hepatocytes at 48 h post ethanol/CCl4, whereas the hepatocyte‐derived Yap was lost in KO that received the same treatment during these time points (Figure 4A). IHC revealed 18% increase of areas that were positive for Yap staining in the damaged CT livers at 48 h compared with those at 7 h post ethanol/CCl4. KO livers lost this increase and stained negative for Yap in hepatocytes at both time points. The reactive aldehyde HNE is a major bioactive byproduct of lipid peroxidation and oxidative stress generated during metabolism of chemicals such as ethanol and CCl4. Increased intensity and areas positive for HNE staining were observed as indicated by in damaged livers of the ethanol‐fed Yap1 KO at 7 to 48 h post CCl4 (Figure 4A). Areas that were positive for HNE staining in the ethanol‐fed KO livers had 10 to 26% increases in comparison to CT at 7 to 48 h post CCl4. This enhancement of lipid peroxidation was verified based on quantification of hepatic MDA concentration (0.36 ± 0.02 in the ethanol pre‐exposed Yap1 KO versus 0.24 ± 0.04 nmol/mg protein in controls) at 48 h post the chemical induced injury (Figure 4B). Increased ROS production was observed (12.01 ± 1.50 pmol/min/mg protein in the KO livers versus 5.81 ± 0.51 pmol/min/mg protein in controls) (Figure 4C), indicating stronger oxidative stress in the Yap1 KO. Elevation of hepatic acetaldehyde was also found (89 ± 21 nmol/mg in the Yap1 KO livers versus 48 ± 14 nmol/mg in the control livers) (Figure 4D), indicating a slower removal of this toxic intermediate in absence of Yap during ethanol/CCl4 combined treatment. Adh1, Aldh1a1 and Dhrs3 encode cytosolic enzymes in ethanol and retinal metabolisms. There was a 1.81‐fold decrease of cytosolic Aldh activity in the damaged KO livers (Figure 4E). Additional qRT‐PCR and immunoblotting analyses demonstrated that the Yap1 KO livers had downregulation of Adh1, Aldh1a1, and Dhrs3 at mRNA and protein levels (Figure 4F,G). Further functional enrichment analysis detected 3.615‐fold decrease of the oxidation‐reduction pathway (p = 6.78E‐12) and 7.64‐fold reduction of retinal metabolism (p = 2.47E‐08) in the ethanol‐fed Yap1 KO livers at 48 h post CCl4 intoxication (Table 1). Related genes in the corresponding pathways identified in the functional enrichment analysis were listed in Table S1. Collectively, these results indicated that hepatocyte‐specific deletion of the Yap1 gene enhanced susceptibility to hepatotoxicity during ethanol/CCl4‐induced injury, likely due to alteration of detoxification pathways for these chemicals.
FIGURE 4.
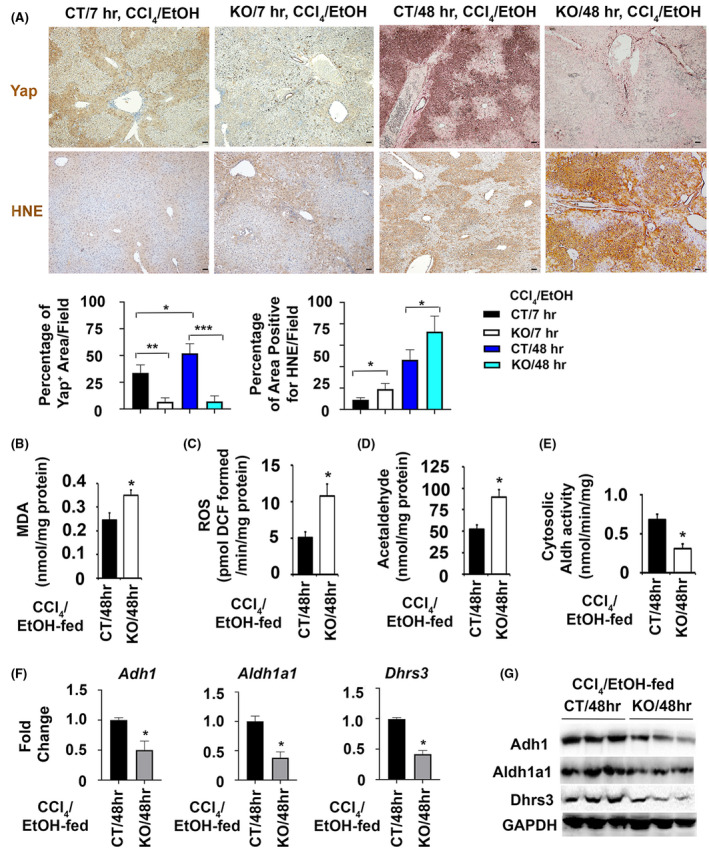
Yap1 loss in hepatocytes is associated with enhanced hepatotoxicity and downregulation of detoxification enzymes including Aldh1a1 after ethanol/CCl4 treatment. (A) The ethanol‐fed Yap1 null livers lost both pericentral and periportal induction of Yap protein post CCl4 intoxication, and exhibited elevated levels of lipid peroxidation as indicated by stronger staining of HNE‐adducts than controls. Scale bar: 100 μm. Areas immunoreactive to antibodies for Yap or HNE were quantified, and the percentages of positive areas were calculated as means ± SEM based on quantification of images from more than 10 fields (100× magnification) from 5 mice per group. *p < .05; **p < .01; ***p < .001. (B–E) Enhanced production of hepatic MDA (B), ROS (C), and acetaldehyde (D) were associated with decreased cytosolic Aldh activity (E) in the damaged Yap1 KO livers. (F and G) Downregulation of Adh1, Aldh1a1, and Drs3 were found at mRNA in Q‐RT‐PCR analysis (F) and protein levels in immunoblotting (G). Values in (B–F) represent means ± SD (n = 5 mice per group). *p < .05
3.5. The Aldh1a1 promoter contains a cis‐element for YAP and can be induced by a hyperactive mutant of this transcriptional coactivator
The human ALDH1A1 promoter contains a cis‐acting element for TEAD binding in a TAZ‐dependent manner. 33 Since YAP and TAZ are paralogous proteins with overlapping functions, we examined the mouse Aldh1a1 promoter (Aldh1a1p) and identified a putative YAP/TEAD binding site “GGAATG” located at −317 to −322 upstream of the transcription initiation site (Figure 5A). To test if Yap is able to be associated with this region, we performed EMSA assays using a biotinylated probe containing the consensus sequences and incubated it with nuclear extracts of HEK293 cells that overexpressed the murine Yap:Myc protein. This biotinylated Aldh1a1p probe bound to Yap:Myc protein and formed complexes, which were detected as a shift band in gel shift assays. The complexes could be disrupted with addition of excessive cold wild‐type probe (Figure 5B). Moreover, the interaction of Aldh1a1p with Yap:Myc was specific because a biotinylated mutant probe in which the putative consensus sequences were replaced to a SpeI site “actagt” was unable to form complexes (Figure 5A,B).
FIGURE 5.
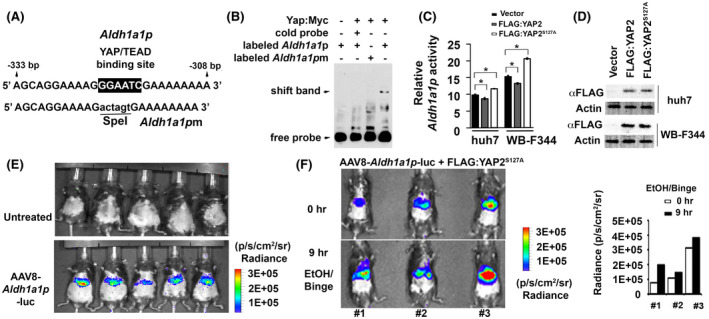
YAP can transcriptionally regulate Aldh1a1 promoter activity in vitro and in vivo. (A) Illustrates sequences flanking a putative YAP/TEAD binding site in wild type promoter (Aldh1a1p) while mutated fragments without this site (Aldh1a1pm) are used to determine binding specificity. (B) Complexes between a murine Yap protein that was fused with Myc epitope at its C terminal region (Yap:Myc) and biotinylated Aldh1a1p fragments were formed, whereas this binding was disrupted in presence of excessive cold or mutant probes in gel shift assays. (C) Aldh1a1 promoter activity as indicated by ratio between Gluc and SEAP could be induced by the FLAG:YAP2S127A mutant, but was inhibited by FLAG:YAP2 protein in Huh7 and WB‐F344 cells. Data were expressed as means ± SD in triplicate experiments. *p < .05. (D) Immunoblotting detected expression of FLAG:YAP2 and FLAG:YAP2S127A proteins in tested cells. (E) Bioluminescence showed Aldh1a1‐promoter driven luciferase activity in AAV8 virus that was transduced into mouse livers through tail vein injection, whereas no signal was seen in untreated control animals. (F) Bioluminescence detected induction of Aldh1a1p activity at 9 h compared to 0 h after binge in ethanol‐fed mouse livers that were transfected with the FLAG:YAP2S127A mutant. Graphs were quantification that determined the Aldh1a1p driven luciferase activity based on bioluminescent signals (Radiance, p/s/cm2/sr)
The regulation of Aldh1a1 promoter was further examined using YAP and mutant isoforms. Phosphorylation site at serine residue 127 has been known to control YAP cytoplasm retention. Abolishment of this phosphorylation site by alanine substitution gives rise to a hyperactive YAPS127A that escapes cytoplasmic sequestration and constitutively turns on signaling in nucleus. 34 , 35 YAP comprises two major splicing isoforms, YAP1 containing a single WW domain and YAP2 with two WW domains. 36 , 37 YAP2 was chosen for analysis in this study because it shares a similar protein structure with its mouse counterpart, which contains two WW domains. 38 We tested effects of YAP2 protein or the YAP2S127A mutant on Aldh1a1 promoter activity. The two proteins were tagged with FLAG epitope at their N terminal regions (FLAG:YAP2 and FLAG:YAP2S127A). SEAP was co‐expressed as internal control for normalization of transfection efficiency in the luciferase systems. Dual reporter analysis showed that the Aldh1a1 promoter activity as indicated by ratio between Gluc and SEAP could be induced by the FLAG:YAP2S127A mutant in the rat hepatic progenitor cell line WB‐F344 and the HCC cell line Huh7 (Figure 5C). Paradoxically, slight reduction of Aldh1a1 promoter mediated by FLAG:YAP2 was also observed (Figure 5C). Considering similar overexpression of FLAG:YAP2 and FLAG:YAPS127A proteins in the two types of cells (Figure 5D), these different patterns indicated that FLAG:YAP2 mediated negative regulation of Aldh1a1 promoter through unknown mechanisms, whereas the hyperactive FLAG:YAP2S127A protein transcriptionally activated this detoxification gene. To further verify the regulation of Aldh1a1 promoter by FLAG:YAP2S127A protein in vivo, we generated AAV8‐Aldh1a1p‐luc virus and transduced it into mouse livers. A strong luciferase activity driven by Aldh1a1 promoter was specifically found in transduced livers (Figure 5E). We then performed bioluminescence imaging and measured change of Aldh1a1p activity at 0 and 9 h after binge in ethanol‐fed mice that expressed FLAG:YAP2S127A. Quantitative analysis showed an increase of AAV8‐Aldh1a1p‐luc activity in all three tested mice according to elevated bioluminescent signals at 9 h after binge (Figure 5F). These data supported our observations that Aldh1a1 promoter could be transcriptionally activated by YAP2S127A.
3.6. Ectopic ALDH1A1 promotes hepatocyte proliferation, protects the livers from hepatotoxicity, reduces ECM remodeling, and attenuates recruitment of CD11b+ inflammatory cells during ethanol/CCl4‐induced liver damage
Previous reports show potential roles of ALDH1A1 to prevent oxidative stress‐induced pathology due to its remarkably low Km for HNE and MDA. 8 To study in vivo function of this enzyme, we ectopically expressed AAV8‐ALDH1A1:FLAG in ethanol‐fed livers that received one acute dose of CCl4. Elevation of ALDH1A1 mRNA and protein were confirmed at 48 h after the chemical‐induced liver damage compared to AAV‐GFP infected controls (Figure 6A,B). ALT level in sera was also significantly reduced (480.0 ± 86.86 in the AAV8‐ALDH1A1:FLAG infected mice versus 668.3 ± 96.43 Unit/L in AAV8‐GFP infected controls) (Figure 6C). ROS production in the AAV8‐ALDH1A1:FLAG‐infected livers had about 22% decrease (Figure 6D). Lower levels of hepatic MDA (0.075 ± 0.002 nmol/mg protein in AAV8‐ALDH1A1:FLAG infected livers versus 0.240 ± 0.030 nmol/mg protein in AAV8‐GFP infected controls) were also observed (Figure 6E), indicating ectopic ALDH1A1 ameliorated lipid peroxidation. Further IHC confirmed that ectopic ALDH1A1 was associated with decreased necrosis in H&E staining, reduced lipid peroxidation in HNE staining, and attenuated numbers of proliferating hepatocytes in Ki67 staining (Figure 6F). Consistent with the increased hepatocyte proliferation in AVV8‐ALDH1A1:FLAG‐transduced livers, we detected increased levels of Ccnd1 mRNA and Cyclin D1 protein (Figure 6A,B). Significant reduction of Ctgf, Fn1, and Itgam at mRNA and protein levels were observed after ectopic expression of ALDH1A1:FLAG protein (Figure 6A,B). Further immunofluorescent staining for αSMA, vWF, and CD11b demonstrated reduction in ECM remodeling and infiltration of inflammatory cells in the ethanol/CCl4‐damaged livers (Figure 6F). Additional Sirius Red staining verified less collagen deposition near perivenous areas in the AVV8‐ALDH1A1:FLAG‐transduced ethanol‐fed livers than controls at 48 h post CCl4 (Figure S5). Areas that were immunoreactive for collagen IV were also smaller in the livers that expressed ectopic ALDH1A1 than controls (Figure S6). Collectively, these data indicated that ectopic ALDH1A1 promoted liver regeneration, protected the transduced livers from hepatocyte death and oxidative stress, attenuated ECM remodeling, and reduced inflammation during ethanol/CCl4‐induced liver injury.
FIGURE 6.
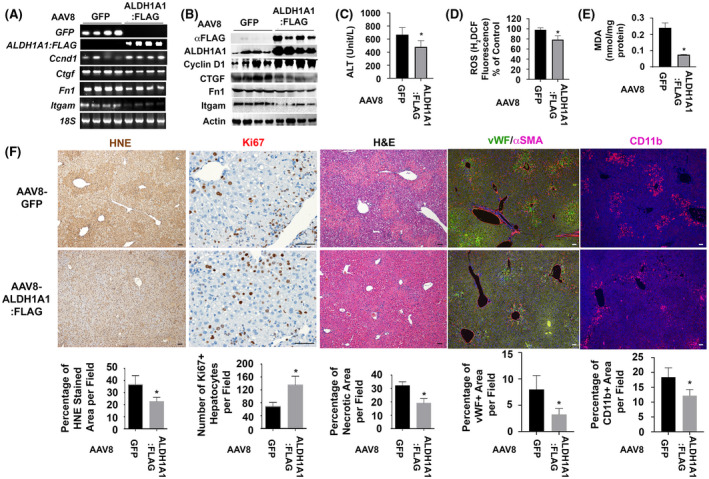
Ectopic expression of ALDH1A1 reduces oxidative stress, decreases hypoxia, attenuates vascular remodeling, ameliorates hepatic inflammation, and enhances hepatocyte proliferation during ethanol/CCl4–induced liver damage. (A and B) RT‐PCR analysis and Western blotting detected AAV8‐ALDH1A1:FLAG or AAV8‐GFP in transduced ethanol‐fed livers at 48 h post CCl4 intoxication. Ectopic expression of ALDH1A1 was associated with upregulation of Cyclin D1 and downregulation of Ctgf, Fn1, and Itgam at mRNA and protein levels. (C–E) Reduction of serum ALT (C), hepatic MDA (D), and ROS (E) were observed in the EtOH/CCl4‐damaged AAV8‐ALDH1A1:FLAG‐transduced livers. Values in (C–E) represent means ± SD (n = 5 mice per group). *p < .05. (F) IHC showed that ectopic expression of ALDH1A1 enhanced number of proliferating hepatocytes (Ki67 staining) and caused reduction in lipid peroxidation (HNE staining), hepatic necrosis (H&E staining), vascular remodeling (αSMA/vWF staining), and inflammation (CD11b staining) during ethanol/CCl4‐induced liver injury. Values were means ± SEM based on quantification of images from more than 10 fields (200× magnification) from 5 mice per group. *p < .05
4. DISCUSSION
YAP is an important regulator in liver regeneration following chemical and surgical‐induced damage. Consistent with our recent findings, 19 this study demonstrated Yap activation upon hepatocyte damage caused by insults such as ethanol and CCl4. Impaired regenerative responses were observed in hepatocyte‐specific Yap1 KO as summarized in Figure 7. Su et al have also characterized hepatocyte‐specific KO in which the murine Yap gene was deleted using albumin‐promoter‐driven Cre during liver damage after a single injection of CCl4. 39 They have found that Yap null hepatocytes proliferate significantly less than their WT counterparts throughout the repair phase at days 1–3 after the injury. In bile duct ligation‐induced injury, the liver‐specific Yap deletion suppresses hepatocyte proliferation, enhances hepatocyte necrosis, and compromises bile duct proliferation. 40 Active human YAP is also accumulated in duct cells and parallels fibrosis progression in non‐alcoholic fatty liver disease. 41 Similarly, deletion of Yap/Taz genes in murine livers has been known to reduce liver regeneration after partial hepatectomy owing to reduced ability of cell cycle entry and hepatocyte proliferation. 42 , 43 Moreover, YAP can regulate hepatocyte fate determination during injury. 12 Additionally, YAP has been considered as a stress sensor to maintain homeostasis in response to hepatic insults such as ethanol by eliminating injured cells. 13 In presence of second signal such as tissue damage or inflammation, YAP switches this homeostatic function from injured cell elimination to growth promotion leading to clonal expansion and hepatocyte reprogramming into progenitor phenotypes for liver regeneration. 41 , 44 Notably, YAP protein levels and phosphorylation seemed to correlate with alcoholic liver disease severity when we characterized its expression patterns in AS, ASH, and AC patients in this study. YAP’s roles from cell elimination to growth stimulation likely occur during the ALD progression and these diverse functions explain dynamic distributions of this protein during liver injury caused by CCl4 and ethanol in this study. We found Yap induction in pericentral hepatocytes at 7 h post CCl4 and at 9 h post chronic‐ethanol‐plus‐single binge, whereas this molecule was re‐distributed in periportal zones at 48 h post CCl4 in ethanol‐fed wild type livers. Considering that pericentral zones are damaged areas where CCl4 and ethanol are metabolized, it is conceivable that the pericentral‐derived Yap functions to selectively eliminate injured hepatocytes and the periportal‐derived Yap works to promote hepatocyte proliferation for liver regeneration in response to these chemical‐induced injuries.
FIGURE 7.
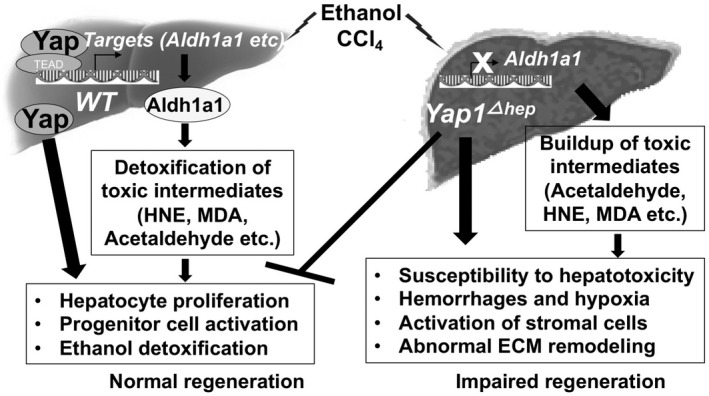
A diagram illustrating different regenerative responses in hepatocyte Yap1 specific KO and wild type mice during liver injury. Yap protein is activated upon hepatocyte damage caused by insults such as ethanol and carbon tetrachloride. Active Yap has been known to form complexes with TEAD transcriptional factors in nucleus and mediate transcriptional reprograming leading to hepatocyte proliferation and progenitor cell activation. Moreover, the active Yap can transcriptionally activate target genes, including Aldh1a1 that encodes an enzyme for removal of toxic aldehydes during hepatocyte damage. However, Yap1 hepatocyte specific KO loses normal regeneration ability. Without Yap, target genes such as Aldh1a1 can't be induced for effective removal of toxic aldehydes. The impaired regeneration eventually causes susceptibility to hepatotoxicity, hemorrhages, hypoxia, activation of stromal cells and abnormal ECM remodeling
Another important finding of this study is about potential roles of hepatocyte‐derived Yap against hypoxia, stromal cell activation and inflammatory cell infiltration. Abnormal ECM remodeling has been reported in mice deficient for Yap after CCl4 intoxication. 39 Consistent with the previous report, this study demonstrated enhancement of necrosis and severe hypoxia as well as abnormal ECM remodeling and inflammatory cell infiltration in hepatocyte‐specific Yap1 KO after ethanol/CCl4 treatment. Hepatotoxins such as ethanol and CCl4 cause hypoxia in the hepatic pericentral area due to competitive oxygen consumption by their metabolisms. HIF1α is normally degraded but becomes stabilized in response to low oxygen tension and pro‐inflammatory signals. Active HIF1α is translocated into the nucleus and induces expression of pro‐angiogenic genes including Vegf‐a, Ctgf, and fibronectin for ECM remodeling. Upregulation of Hif1α and its downstream targets in angiogenesis might result from increased hepatotoxicity due to downregulation of detoxification enzymes (Aldh1, Aldh1a1 and Dhrs3) in Yap null hepatocytes as shown in Figure 7. In addition, emerging evidence has shown a link of Yap regulation with oxidative stress. Weak oxidative stress defense can result from Yap decrease due to acetaminophen‐induced glutathione depletion. 45 Yap acts as a nuclear co‐factor and induces antioxidant genes leading to enhanced cardiomyocyte survival and reduced oxidative stress after ischemia/reperfusion. 46 Yap overexpression protects cardiomyocytes against H2O2‐induced cell death, whereas Yap1 deficiency exacerbates injury in response to chronic myocardial infarction. 47 Therefore, it is easy to postulate that transcriptional regulation of antioxidant and detoxification enzymes by Yap provides protective mechanisms against hypoxia and cellular toxicity caused by lipid peroxidation/ROS‐triggered signaling during tissue injury.
Ethanol abuse causes alcoholic liver disease through oxidative stress, acetaldehyde toxicity, and altered retinoid metabolism. This study demonstrated that forced expression of ALDH1A1 reduced hepatotoxicity by attenuating production of ROS, lipid peroxidation products (HNE, MDA etc), and acetaldehyde. ALDH1A1 can not only detoxify acetaldehyde in ethanol oxidation but also convert retinaldehyde into retinoic acid (RA). 48 RA is a potent inducer of hepatocyte proliferation. 49 , 50 This RA‐mediated action may contribute to the enhanced proliferation of hepatocytes observed in AAV8‐ALDH1A1:FLAG‐transduced livers after ethanol/CCl4 treatment. Furthermore, Dhrs3 is also involved in RA metabolism. This enzyme converts retinaldehyde into retinol and inhibits RA synthesis. 51 , 52 Interestingly, the promoter of Dhrs3 gene contains regulatory elements for YAP/TEAD binding. 52 Thus, YAP may regulate both Aldh1a1 and Dhrs3 promoters. Loss of this regulation may explain why Aldh1a1 and Dhrs3 as well as retinol metabolism (mmu00830) were downregulated in Yap1 KO livers during ethanol/CCl4‐induced liver injury.
Aldehydes may arise from metabolisms of amino acids, alcohols, lipids, vitamins, cytotoxic drugs, and environmental agents in the liver. These toxic intermediates easily engage in numerous secondary reactions that attack cells, resulting in protein or DNA adduct formation as well as lipid peroxidation, with varying degrees of toxicity at the site of generation or when transported to distant cells. The liver serves as the primary metabolism site and produces enzymes such as Aldh1a1 for hepatic protection after injury. High levels of Aldh isoforms are expressed in mouse livers, whereas Aldh1a1 deficiency in Hepa‐1c1c7 cells enhances susceptibility to aldehyde‐induced cell death, accumulation of acrolein‐protein adducts, caspase 3 activation and generating oxidative stress. 7 Moreover, regulation of ALDH1A1 by YAP and TAZ in some cancer cells has been reported. 33 , 53 Taking into considerations that ALDH activity is often altered in cirrhotic livers of AC patients, 54 understanding the regulation of ALDH1A1 by YAP has therapeutic implications against impaired liver regeneration and ethanol‐induced hepatotoxicity during development of ALD.
DISCLOSURES
The authors declare that there is no conflict of interest regarding publication of this manuscript.
AUTHOR CONTRIBUTIONS
Designed research: Liya Pi, Bryon Petersen, Qi Cao, Andrew Bryant, and Chenglong Li. Performed experiments and analyzed data: Junmei Zhou, Chunbao Sun, Lu Yang, Natacha Jn‐Simon, Chen Zhou, Jinhui Wang and Liya Pi. Wrote the article, and all authors read a draft of this article, suggested improvements, and approved the final version of this article: Liya Pi and Junmei Zhou.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
We want to thank Ms. Marda Jorgensen and Haven Thomas for technical assistance about immunohistochemistry. We also thank Drs. Chen Ling and Gai Ran in AAV production.
Zhou J, Sun C, Yang L, et al. Liver regeneration and ethanol detoxification: A new link in YAP regulation of ALDH1A1 during alcohol‐related hepatocyte damage. FASEB J. 2022;36:e22224. doi: 10.1096/fj.202101686R
Junmei Zhou and Chunbao Sun contributed equally to this study.
Funding information
This study is supported by National Institutes of Health NIAAA KO1AA024174 and RO1AA028035 grants as well as Children Miracle Research Foundation grant awarded to LP. This study is also partially supported by the National Natural Science Foundation of China (82060124), the Natural Science Foundation of Guangxi (2019JJA140718), and the Guangxi Distinguished Experts Special Fund (2019B12)
DATA AVAILABILITY STATEMENT
All data supporting the findings of this study are available within the paper and within its supplementary materials published online.
REFERENCES
- 1. Lieber CS, DeCarli LM. Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science. 1968;162:917‐918. [DOI] [PubMed] [Google Scholar]
- 2. Chen Y, Thompson DC, Koppaka V, Jester JV, Vasiliou V. Ocular aldehyde dehydrogenases: protection against ultraviolet damage and maintenance of transparency for vision. Prog Retin Eye Res. 2013;33:28‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine‐derived 3,4‐dihydroxyphenylacetaldehyde and 3,4‐dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125‐150. [DOI] [PubMed] [Google Scholar]
- 4. Ginestier C, Hur MH, Charafe‐Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steg AD, Bevis KS, Katre AA, et al. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin Cancer Res. 2012;18:869‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki E, Chiba T, Zen Y, et al. Aldehyde dehydrogenase 1 is associated with recurrence‐free survival but not stem cell‐like properties in hepatocellular carcinoma. Hepatol Res. 2012;42:1100‐1111. [DOI] [PubMed] [Google Scholar]
- 7. Makia NL, Bojang P, Falkner KC, Conklin DJ, Prough RA. Murine hepatic aldehyde dehydrogenase 1a1 is a major contributor to oxidation of aldehydes formed by lipid peroxidation. Chem Biol Interact. 2011;191:278‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rebollido‐Rios R, Venton G, Sánchez‐Redondo S, et al. Dual disruption of aldehyde dehydrogenases 1 and 3 promotes functional changes in the glutathione redox system and enhances chemosensitivity in nonsmall cell lung cancer. Oncogene. 2020;39:2756‐2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasiliou V, Pappa A. Polymorphisms of human aldehyde dehydrogenases. Consequences for drug metabolism and disease. Pharmacology. 2000;61:192‐198. [DOI] [PubMed] [Google Scholar]
- 10. Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao B, Ye X, Yu J, et al. TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev. 2008;22:1962‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miyamura N, Hata S, Itoh T, et al. YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat Commun. 2017;8:16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y, Lu T, Zhang C, et al. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia‐reperfusion injury. J Hepatol. 2019;71:719‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mo JS, Meng Z, Kim YC, et al. Cellular energy stress induces AMPK‐mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Enzo E, Santinon G, Pocaterra A, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015;34:1349‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bou Saleh M, Louvet A, Ntandja‐Wandji LC, et al. Loss of hepatocyte identity following aberrant YAP activation: a key mechanism in alcoholic hepatitis. J Hepatol. 2021;75:912‐923. [DOI] [PubMed] [Google Scholar]
- 18. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou J, Sun X, Yang L, et al. Hepatocyte nuclear factor 4alpha negatively regulates connective tissue growth factor during liver regeneration. FASEB J. 2020;34:4970‐4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pi L, Robinson PM, Jorgensen M, et al. Connective tissue growth factor and integrin alphavbeta6: a new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology. 2015;61:678‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9:e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiang DJ, Roychowdhury S, Bush K, et al. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol‐exacerbated liver fibrosis. PLoS One. 2013;8:e69114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deshpande KT, Liu S, McCracken JM, et al. Moderate (2%, v/v) ethanol feeding alters hepatic wound healing after acute carbon tetrachloride exposure in mice. Biomolecules. 2016;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self‐renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature. 2015;524:180‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Font‐Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell. 2015;162:766‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roychowdhury S, Chiang DJ, McMullen MR, Nagy LE. Moderate, chronic ethanol feeding exacerbates carbon‐tetrachloride‐induced hepatic fibrosis via hepatocyte‐specific hypoxia inducible factor 1alpha. Pharmacol Res Perspect. 2014;2:e00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schober JM, Chen N, Grzeszkiewicz TM, et al. Identification of integrin alpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate‐early gene products expressed in atherosclerotic lesions. Blood. 2002;99:4457‐4465. [DOI] [PubMed] [Google Scholar]
- 28. Lai YS, Wahyuningtyas R, Aui SP, Chang KT. Autocrine VEGF signalling on M2 macrophages regulates PD‐L1 expression for immunomodulation of T cells. J Cell Mol Med. 2019;23:1257‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034‐2042. [DOI] [PubMed] [Google Scholar]
- 30. Nielsen MC, Hvidbjerg Gantzel R, Claria J, Trebicka J, Moller HJ, Gronbaek H. Macrophage activation markers, CD163 and CD206, in acute‐on‐chronic liver failure. Cells. 2020;9:1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polfliet MM, Fabriek BO, Daniels WP, Dijkstra CD, van den Berg TK. The rat macrophage scavenger receptor CD163: expression, regulation and role in inflammatory mediator production. Immunobiology. 2006;211:419‐425. [DOI] [PubMed] [Google Scholar]
- 32. Svendsen P, Graversen JH, Etzerodt A, et al. Antibody‐directed glucocorticoid targeting to CD163 in M2‐type macrophages attenuates fructose‐induced liver inflammatory changes. Mol Ther Methods Clin Dev. 2017;4:50‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao AY, Dai YJ, Lian JF, et al. YAP regulates ALDH1A1 expression and stem cell property of bladder cancer cells. Onco Targets Ther. 2018;11:6657‐6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747‐2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β‐catenin signalling. EMBO J. 2012;31:1109‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Webb C, Upadhyay A, Giuntini F, et al. Structural features and ligand binding properties of tandem WW domains from YAP and TAZ, nuclear effectors of the Hippo pathway. Biochemistry. 2011;50:3300‐3309. [DOI] [PubMed] [Google Scholar]
- 37. Hoshino M, Qi ML, Yoshimura N, et al. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol. 2006;172:589‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sudol M, Bork P, Einbond A, et al. Characterization of the mammalian YAP (Yes‐associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem. 1995;270:14733‐14741. [DOI] [PubMed] [Google Scholar]
- 39. Su T, Bondar T, Zhou X, Zhang C, He H, Medzhitov R. Two‐signal requirement for growth‐promoting function of Yap in hepatocytes. eLife. 2015;4:e02948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bai H, Zhang N, Xu Y, et al. Yes‐associated protein regulates the hepatic response after bile duct ligation. Hepatology. 2012;56:1097‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Machado MV, Michelotti GA, Pereira TA, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non‐alcoholic fatty liver disease. J Hepatol. 2015;63:962‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moya IM, Halder G. Hippo‐YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211‐226. [DOI] [PubMed] [Google Scholar]
- 43. Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med. 2018;50:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swiderska‐Syn M, Xie G, Michelotti GA, et al. Hedgehog regulates yes‐associated protein 1 in regenerating mouse liver. Hepatology. 2016;64:232‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu H, Xiao Y, Zhang S, et al. The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep. 2013;3:1663‐1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shao D, Zhai P, Del Re DP, et al. A functional interaction between Hippo‐YAP signalling and FoxO1 mediates the oxidative stress response. Nat Commun. 2014;5:3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Del Re DP, Yang Y, Nakano N, et al. Yes‐associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288:3977‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fan X, Molotkov A, Manabe S, et al. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003;23:4637‐4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shabtai Y, Fainsod A. Competition between ethanol clearance and retinoic acid biosynthesis in the induction of fetal alcohol syndrome. Biochem Cell Biol. 2018;96(2):148‐160. [DOI] [PubMed] [Google Scholar]
- 50. Liu HX, Ly I, Hu Y, Wan YJ. Retinoic acid regulates cell cycle genes and accelerates normal mouse liver regeneration. Biochem Pharmacol. 2014;91:256‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adams MK, Belyaeva OV, Wu L, Kedishvili NY. The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J Biol Chem. 2014;289:14868‐14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiao Y, Hill MC, Zhang M, et al. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev Cell. 2018;45(2):153‐169.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu J, Alharbi A, Shan H, Hao Y, Snetsinger B, Rauh MJ, Yang X. TAZ induces lung cancer stem cell properties and tumorigenesis by up‐regulating ALDH1A1. Oncotarget. 2017;8(24):38426‐38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vidal F, Toda R, Gutierrez C, et al. Influence of chronic alcohol abuse and liver disease on hepatic aldehyde dehydrogenase activity. Alcohol. 1998;15:3‐8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
All data supporting the findings of this study are available within the paper and within its supplementary materials published online.