

Abstract
Bone fragility is the product of defects in bone mass and bone quality, both of which show sex-specific differences. Despite this, the cellular and molecular mechanisms underpinning the sexually dimorphic control of bone quality remain unclear, limiting our ability to effectively prevent fractures, especially in postmenopausal osteoporosis. Recently, using male mice, we found that systemic or osteocyte-intrinsic inhibition of TGFβ signaling, achieved using the 9.6-kb DMP1 promoter-driven Cre recombinase (TβRIIocy−/− mice), suppresses osteocyte perilacunar/canalicular remodeling (PLR) and compromises bone quality. Since systemic TGFβ inhibition more robustly increases bone mass in female than male mice, we postulated that sex-specific differences in bone quality could likewise result, in part, from dimorphic regulation of PLR by TGFβ. Moreover, since lactation induces PLR, we examined the effect of TGFβ inhibition on the female skeleton during lactation. In contrast to males, female mice that possess an osteocyte-intrinsic defect in TGFβ signaling were protected from TGFβ-dependent defects in PLR and bone quality. The expression of requisite PLR enzymes, the lacuno- canalicular network, and the flexural strength of female TβRIIocy−/− bone was intact. With lactation, however, bone loss, and induction in PLR and osteocytic parathyroid hormone type I receptor (PTHR1) expression, were suppressed in TβRIIocy−/− bone, relative to the control littermates. Indeed, differential control of PTHR1 expression, by TGFβ and other factors, may contribute to dimorphism in PLR regulation in male and female TβRIIocy−/− mice. These findings provide key insights into the sex-based differences in osteocyte PLR that underlie bone quality and highlight TGFβ signaling as a crucial regulator of lactation-induced PLR.
2. Introduction
Evolution selects for skeletal adaptations that improve survival and reproductive fitness. A prime example is the ability of female mammals to release stored calcium from bone to support lactation. In spite of a sharp drop in bone mass during lactation, the female skeleton mechanically adapts to accommodate these metabolic demands(1–5). Changes in bone quality, including trabecular microarchitecture and cortical bone geometry, compensate for the loss of bone mass. Other aspects of bone quality, including the material properties of bone extracellular matrix (ECM), show sexual dimorphism(6,7). Through mechanisms that remain unclear, the protective advantage of such skeletal adaptations for reproductive females is lost later in life. Women over 50 have a 4-fold higher rate of osteoporosis relative to men of the same age(8,9). Furthermore, about half of osteoporotic fractures cannot be explained by clinically low bone mass(10,11). Therefore, understanding the sexually dimorphic regulation of bone quality will elucidate mechanisms of bone fragility in post-menopausal osteoporosis, which affects over 200 million people worldwide(12,13).
Skeletal variation in men and women arises in part from endocrine differences in hormones such as androgens, estrogens, and inhibin(14). These hormones act in concert with paracrine growth factors to control bone cell function(15,16). Osteoblasts and osteoclasts also exhibit sexual dimorphism at the cell-intrinsic level(17–20). Osteocytes respond to high levels of PTH/PTHrP during lactation by inducing perilacunar/canalicular remodeling (PLR)(21,22). In PLR, osteocytes resorb, and later replace, bone matrix surrounding their intricate lacunocanalicular network(21,23–25). Originally termed osteocyte osteolysis, PLR supports the metabolic demand for calcium during lactation through coordinated induction of several key genes, including matrix metalloproteinases (Mmps; namely Mmp2, Mmp13, and Mmp14), cathepsin K (Ctsk), carbonic anhydrase 2, and tartrate-resistant acid phosphatase (Acp5/TRAP) to mediate resorption(23,24,26–28). After weaning, osteocytes replace the bone lost during lactation(29), although the underlying molecular mechanism remains elusive. It is possible that genes such as soluble cytosolic phosphatase (Phospho1) and dentin matrix protein (Dmp1)(30–32), both of which are implicated in PLR, participate in replenishing the mineralized bone. Until recently, PLR was primarily considered a response to metabolic stress or disease. Work from our group and others reveals PLR to be crucial in homeostasis as well, where it regulates bone quality through TGFβ signaling(33). However, the extent to which the sexually dimorphic control of PLR extends dimorphism in bone quality remains unknown.
TGFβ is a key driver of bone mass and quality that dictates activities of all of the cells of the bone remodeling unit. Indeed, dysregulated TGFβ signaling plays a causal role in several skeletal diseases, including Camurati Engelman disease and osteogenesis imperfecta(34,35). This key cytokine, which is largely produced by osteoblasts and osteocytes, is embedded within the bone ECM in a latent form(36–38). Upon osteoclastic resorption, TGFβ is released and activated to promote recruitment and commitment of osteoblast progenitors to the resorption site, while inhibiting their terminal differentiation. TGFβ also promotes osteoclast formation and survival and couples bone resorption with formation(39). In osteocytes, we found that TGFβ signaling is indispensable for PLR(33,40). Using pharmacologic TGFβ antagonists and a novel mouse model, we demonstrated that osteocyte-intrinsic ablation of TGFβ signaling is sufficient to suppress PLR and cause bone fragility due to severely compromised bone quality(33). Although female mice accrue more bone mass in response to pharmacologic TGFβ antagonists than males(37), the sexually dimorphic regulation of PLR or bone quality by TGFβ has not been evaluated.
Therefore, we used mice with an osteocyte-specific deletion of TGFβ receptor II (TβRII) to investigate the sexually dimorphic regulation of PLR in mice at homeostasis and during the metabolic stress of lactation. Our findings reveal a dual role for TGFβ in the control of PLR in female bone. While TGFβ is required for lactation-induced PLR, at homeostasis, female bone is insensitive to loss of osteocytic TGFβ signaling. Here we elucidate a cellular mechanism that contributes to the dimorphic control of PLR in males and females, a mechanism that protects bone quality in reproductive age females.
3. Material and Methods
3.1. Mice.
The generation of viable osteocyte specific TβRIIocy−/− mice was described previously(33). In brief, homozygous TGFβ type II receptor (TβRII)-floxed mice were bred with the hemizygous 9.6-kb DMP1-Cre mice(41,42). Half of the mice resulting from the cross were DMP1-Cre Tg+; TβRIIflox/flox (labeled TβRIIocy−/− mice) and half were littermates lacking the DMP1-Cre transgene (TβRIIflox/flox) served as controls (labeled control mice). All the transgenic mouse lines were backcrossed for 8 generations into a congenic C57BL/6 background. In all the experiments 15-week old male and female TβRIIocy−/− and control mice were used. For lactation studies, TβRIIocy−/− mice and control mice were time-mated at 10 weeks of age. Females that did not become pregnant after 1 week were removed from the study to minimize age-related differences in the bone phenotype. Following delivery, for each lactating mother the litter size was adjusted to 8 pups in order to normalize for breast milk requirements. Mice were sacrificed after 7 days of lactation. For all experiments, we used control mice that were true littermates for the mutant mice. All studies were conducted with the approval of the Institutional Animal Care and Use Committee of the University of California San Francisco. Additional details on mice are provided in the Supplemental data.
3.2. Micro-computed tomography (micro-CT).
For skeletal phenotyping, 15-week-old male and female mouse bones were harvested by cleaning off the surrounding soft tissue. Cleaned bones were subsequently fixed in 10% neutral buffered formalin and stored in 70% ethanol. A 2 mm region of the femoral metaphysis and a 1 mm region of the femoral mid-diaphysis were scanned using a Scanco µCT50 specimen scanner to assess trabecular and cortical parameters, respectively. Scanning was performed with an X-ray potential of 55 kVp and current of 109 µA and at a resolution of 10 µm. Table 1 shows standard parameters for n≥8 mice per group(43). Additional details of micro-CT procedure are provided in the Supplemental data.
Table 1:
μCT of male and female (virgin and lactating) control and TβRIIocy−/− mouse bones.
| Male | Female | Lactation | ||||
|---|---|---|---|---|---|---|
| Control | TβRII ocy−/− | Control | TβRII ocy−/− | Control | TβRII ocy−/− | |
| Distal Femur | ||||||
| Tb.BV/TV(%) | 0.18 ± 0.04 | 0.30 ± 0.07* | 0.08 ± 0.04 | 0.1 ± 0.05 | 0.04 ± 0.01a | 0.08 ± 0.02b |
| Tb.N (1/mm) | 5.30 ± 1.08 | 6.11 ± 0.87 | 2.44 ± 0.72 | 2.68 ± 1.13 | 1.76 ± 0.45 | 2.89 ± 0.84 |
| Tb. Th (mm) | 0.03 ± 0.003 | 0.05 ± 0.01* | 0.033 ± 0.01 | 0.035 ± 0.01 | 0.022 ± 0.002 a | 0.027 ± 0.00a,b,c |
| Tb. Sp (mm) | 0.16 ± 0.05 | 0.12 ± 0.03* | 0.40 ± 0.12 | 0.42 ± 0.22 | 0.59 ± 0.2 a | 0.35 ± 0.15 b,c |
| SMI | 1.71 ± 0.36 | 0.52 ± 0.62* | 2.83 ± 0.43 | 2.74 ± 0.66 | 3.01 ± 0.27 | 2.48 ± 0.38 |
| Tb. Min (mg HA/cm3) | 1065.5 ± 18.9 | 1098.3 ± 16.3* | 1103.85 ± 30.2 | 1096.5 ± 23.6 | 1010.5 ± 27.0a | 1035.3 ± 40.9c |
| Midshaft Femur | ||||||
| Ct. BA/TA(%) | 0.28 ± 0.03 | 0.30 ± 0.03 | 0.28 ± 0.02 | 0.29 ± 0.02 | 0.21 ± 0.03a | 0.24 ± 0.02a,b,c |
| Ct. Th(mm) | 0.20 ± 0.01 | 0.22 ± 0.02* | 0.19 ± 0.01 | 0.20 ± 0.01 | 0.14 ± 0.006a | 0.16 ± 0.007c |
| Ct. SMI | 0.16 ± 0.41 | 2.82 ± 1.42* | 0.28 ± 0.14 | 0.12 ± 0.23 | 0.22 ± 0.03 | 0.10 ± 0.18 |
| Ct. Min (mg HA/cm3) | 1385.8 ± 14.4 | 1337.8 ± 18.7* | 1362.60 ± 46.6 | 1332.5 ± 66.4 | 1330.3 ± 29.1 | 1319.2 ± 36.5 |
p<0.05 significantly different from the male control virgin (Student’s t-test),
p<0.05 significantly different from the control virgin,
p<0.05 significantly different from the control lactation, and
p<0.05 significantly different from the virgin TβRIIocy−/− mice.
3.3. Quantitative RT-PCR analysis.
As previously described, RNA was extracted and purified from bones devoid of marrow using the miRNeasy mini kit (Qiagen, Valencia, CA)(33). Real-time quantitative PCR (qPCR) was performed using specific primers (iQ SYBR Green Supermix, BioRad) and Taqman probes (Applied Biosystems Taqman Assays, Thermo Fisher Scientific) following manufacturer’s protocol. Results are shown as relative gene expression, using expression of the 18s ribosomal RNA as an internal reference gene. Additional details on the RNA extraction and primers are provided in the Supplemental data (Table S1).
3.4. Histology.
For immunohistochemistry and immunofluorescence, paraffin embedded bone sections (7μm thickness) were incubated with rabbit polyclonal anti-MMP13 antibody (Abcam ab39012); rabbit polyclonal monoclonal anti-MMP14 antibody (Abcam ab53712); rabbit polyclonal anti-Cathepsin K (CTSK) antibody (Abcam ab19027), or rabbit monoclonal anti-TβRII antibody (Abcam ab186838) and mouse monoclonal anti-PTHR1 antibody (NBP1–40067: Novus Biologicals). Corresponding rabbit IgG negative control antibody was used to verify the specificity of primary antibodies. Ploton silver staining was performed to visualize the osteocyte lacuno-canalicular network in bones and the osteocyte network area was determined and normalized to total bone area, as previously described(33,44). Images were acquired using a Nikon Eclipse E800 bright-field microscope and analyzed with Image J. Sections were evaluated for one femur from each of n≥3 mice per group, as indicated in the figure legends. Quantitative averages represent data collected from 4–5 high-powered fields for each bone. Additional details on staining, image analysis, and quantification are provided in the Supplemental data.
3.5. Synchrotron radiation Micro-Tomography (SRµCT).
SRµCT studies were used to assess the degree of mineralization of the bone as well as the volume and degree of anisotropy of the lacunae. The mid-diaphysis of 15-week-old virgin and lactating female mouse femora were scanned with a 20 keV x-ray energy, a 300 ms exposure time and a 5x magnifying lens for a spatial resolution of 1.3 µm (n = 4 bones/group). Additional details of SRµCT procedure are described in the Supplemental data.
3.6. Flexural strength tests.
Whole bone flexural strength of femora was determined by three-point bending of intact, hydrated femurs isolated from TβRIIocy−/− and control mice. 15-week old males and female virgin and lactating mice of each genotype were used for macromechanical testing as previously described(33). Details on the flexural strength tests are described in Supplemental data.
3.7. Serum Analysis.
Blood was isolated from male and virgin and lactating female mice using a cardiac puncture protocol and processed for serum. Serum calcium and phosphorus were measured with colorimetric assays (Abcam). PTH in serum was analyzed by Enzyme-linked Immunosorbent Assay (ELISA) (Quidel Corporation, Catalog no. 60–2305).
3.8. Statistical Analysis.
Sample size was determined based on a power calculation that provides an 80% chance of detecting a significant difference (p<0.05). Technical replicates and biological replicates (n) used for all experiments are described in the figure legends. Where technical replicates are used, data are expressed as mean ± S.E.M. Otherwise, data are reported as mean ± S.D. Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA) was used for statistical analysis. Comparisons between two groups were evaluated by an unpaired two-tailed Student’s t test. For comparing between more than two groups, we used one-way ANOVA followed by Tukey’s test for multiple comparisons.
4. Results
4.1. Loss of osteocyte-intrinsic TGFβ signaling impacts bone mass and quality in male but not female mice.
Recent studies from our group implicate osteocyte-intrinsic TGFβ signaling to be key for controlling perilacunar/canalicular remodeling (PLR) and bone quality, however it is unclear if this regulatory function of TGFβ is subject to sex-specific differences. Indeed, we previously observed sexual dimorphism in the effect of TGFβ inhibition on bone mass and osteoblast and osteoclast numbers(37). To address this question, we used a previously developed mouse model for osteocyte-intrinsic inhibition of TGFβ signaling to assess the bone phenotype in both sexes. Using the 9.6-kb Dmp1-Cre transgene, the floxed TβRII allele was preferentially excised from odontoblasts and osteocytes of TβRIIocy−/− mice(33,41,42). The efficiency of this mouse model was confirmed by observing reduction in TβRII mRNA and protein levels in the long bone osteocytes of TβRIIocy−/− male and female mice (Figure 1A-D, Fig S1A-C and Fig 3 C-F).
Figure 1. Loss of osteocyte-intrinsic TGFβ signaling impacts bone mass and quality in male mice but does not affect female mice.
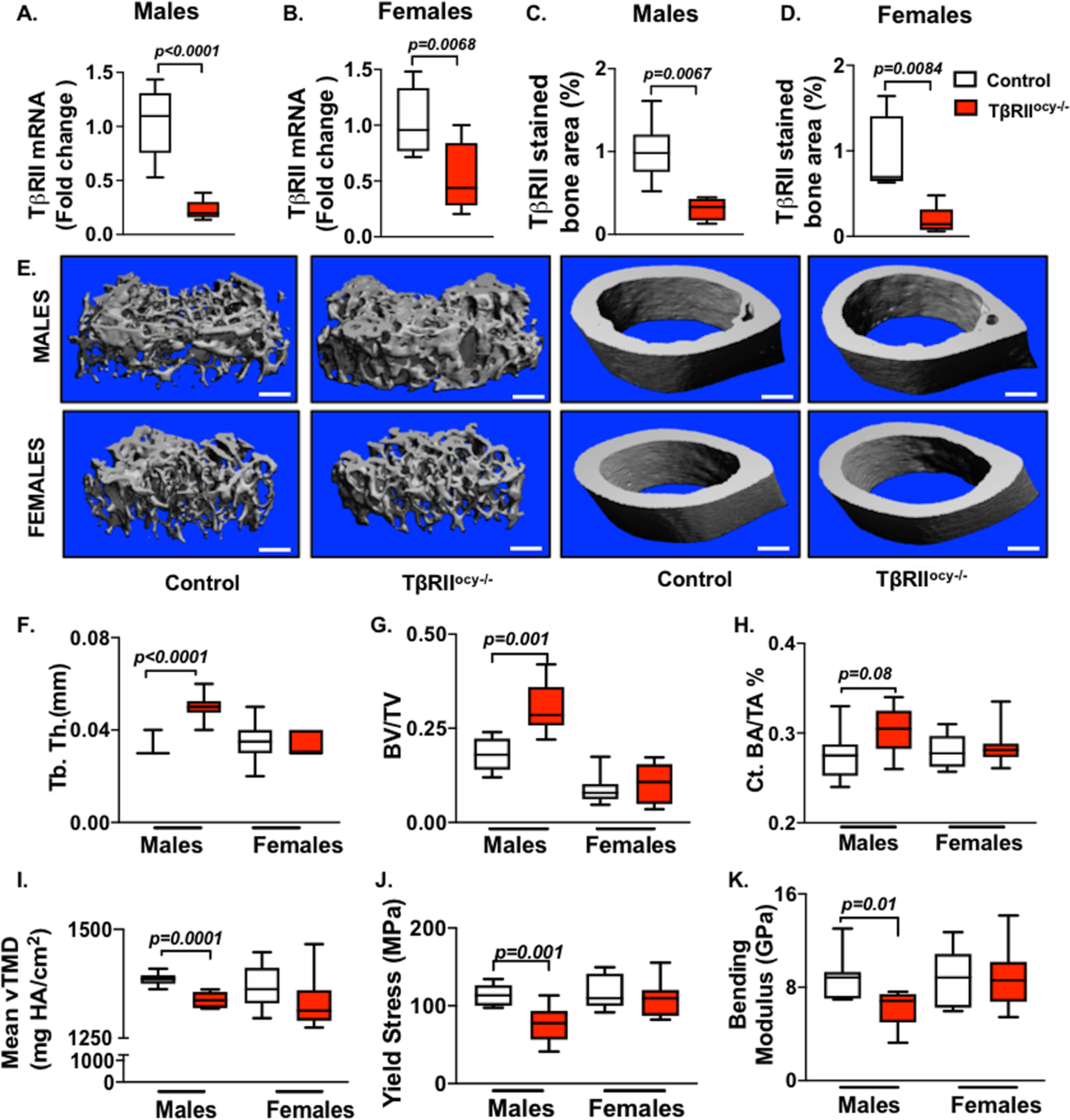
In male and female TβRIIocy−/− mice, knockdown of TβRII at mRNA (A, B) (n=6–9 mice/ group) and protein (C, D) (n=4–6 mice/ group) level was assessed. μCT analysis was conducted on femurs from control and TβRIIocy−/− male and female mice (15-week-old). Representative μCT reconstructions of trabecular (left) and cortical (right) bone (E) along with the trabecular bone thickness (Tb. Th.) (F) and volume (BV/TV) (G), and cortical bone area (BA/TA) (H) and mineralization (Ct. Min) (I) are shown (n=7–8 mice/group). Flexural testing of femurs from male and female control and TβRIIocy−/− mice shows bone quality parameters yield stress (J) and bending modulus (K) (n = 7 mice/group). Data for A-D are presented as mean ± SEM and as mean ± SD for E-K and statistically significant difference were determined with Student’s t test. Scale bar for E is 100μm (n = 7–8 mice/group).
Figure 3. Lactation induced PLR is blocked by osteocyte-specific disruption of TβRII gene.

Bones from 15- week-old virgin and lactating control and TβRIIocy−/− female mice were harvested and processed for gene expression (A-B) and immunohistochemistry (C-D). qPCR analysis of TβRII and PLR genes Mmp2, Mmp13, Mmp14, Acp5, Ctsk, AtpD2 and AtpG1 (A-B) in virgin and lactating control and TβRIIocy−/− mouse bones was compared (n=6–9 mice/ group). Immunohistochemistry (IHC) for MMP13, MMP14, and CTSK was conducted on femoral cortical bones of virgin and lactating control and TβRIIocy−/− mice. Arrows in the representative images indicate positively stained area (D) that were quantified and normalized to total bone area (C) (n =3–4 mice/group and 4 ROI/mouse, scale bar is 50μm). Representative images of immunofluorescence of TβRII (top) and MMP13 (bottom) protein and (E) quantification of cells positive for TβRII and DAPI, or MMP13 and DAPIIn murine cortical bone (F and G) (scale bar is 40μm) is shown. Error bars indicate mean ± SEM, *p<0.05 compared to control virgins, #p<0.05 compared to control lactation group, and $p<0.05 compared to TβRIIocy−/− virgin, as calculated from one-way ANOVA with Tukey post-hoc test.
To assess the skeletal phenotype of TβRIIocy−/− male and female mice, micro-computed tomography (μCT) was used. These analyses revealed an increase in trabecular thickness and trabecular bone volume in male TβRIIocy−/− mice compared to the control littermates (Figure 1E-G, table 1). Although cortical bone mass was unchanged, the cortical bone mineralization of TβRIIocy−/− male mice was reduced by 3.5% relative to littermate controls (Figure 1E, H-I). Flexural strength tests showed that both yield stress and bending modulus of TβRIIocy−/− male bones was decreased by 20%, thereby indicating a reduction in the bone material quality of TβRIIocy−/− males compared to control mice (Figure 1J-K). As expected, based on our prior work on 8-week old TβRIIocy−/− males, the current findings confirm the critical role of osteocyte-intrinsic TGFβ signaling in maintaining bone quality in 15-week old male mice.
None of these bone quality defects in TβRIIocy−/− males were present in female TβRIIocy−/− mice. In particular, the trabecular and cortical bone mass and geometry of TβRIIocy−/− females remained intact, and the flexural strength of TβRIIocy−/− female bones was identical to that of the control group (Figure 1E-I, J-K, Table 2). These observations suggest that female mouse bones do not depend on osteocytic TGFβ signaling for maintaining bone mass and quality, and that osteocyte-intrinsic TGFβ impacts bone in a sexually dimorphic manner.
Table 2:
Flexural strength test derived macro-mechanical properties of 15-week-old male and virgin and lactating female control and TβRIIocy−/− mice.
| Flexural Strength parameters | Male | Female | Lactation | |||
|---|---|---|---|---|---|---|
| Control | TβRII ocy−/− | Control | TβRII ocy−/− | Control | TβRII ocy−/− | |
| Stiffness (N-mm2) | 189.8 ± 24.0 | 169.7 ± 28.9 | 173.9 ± 20.3 | 177.1 ± 24.4 | 129.6 ± 22.5a | 161.2 ± 14.7 |
| Yield load (N) | 14.4 ± 1.9 | 12.5 ± 2.3 | 14.0 ± 1.1 | 13.7 ± 1.1 | 12.0 ± 1.7a | 13.8 ± 1.2 |
| Post-yield displacement (%) | 0.87 ± 0.71 | 0.55 ± 0.25 | 0.54 ± 0.19 | 0.72 ± 0.19 | 0.59 ± 0.18 | 1.27 ± 0.51 |
| Modulus (GN/m2) | 9.06 ± 1.99 | 6.2 ± 1.51* | 9.0 ± 2.45 | 8.8 ± 2.63 | 7.88 ± 1.38 | 9.41 ± 1.40 |
| Yield Stress (Mpa) | 115.6 ± 13.9 | 80.4 ± 23.98* | 118.4 ± 21.8 | 108.7 ± 22.9 | 135.3 ± 29.1 | 135.9 ± 15.2 |
| Ultimate Stress (Mpa) | 189.5 ± 22.5 | 189.30 ± 37.5 | 191.05 ± 30.2 | 227.1 ± 43.5 | 167.3 ± 20.0 | 169.7 ± 4.45 |
| MOI (mm4) | 0.117 ± 0.02 | 0.158 ± 0.05 | 0.113 ± 0.03 | 0.118 ± 0.03 | 0.077 ± 0.02a | 0.084 ± 0.01 |
p<0.05 significantly different from the male control virgin group (Student’s t-test),
p<0.05 significantly different from the female control virgin group.
4.2. TGFβ signaling regulates osteocyte-mediated PLR in sex-specific manner.
Bone quality is in part regulated by PLR, and our prior evidence from 8-week-old TβRIIocy−/− male mice ascribes their defects in bone quality to PLR suppression(33). Therefore, we hypothesized that the distinct bone quality phenotype of male and female TβRIIocy−/− mice is driven by differences in osteocytic PLR. Since an intact lacuno-canalicular network (LCN) is a hallmark of functional PLR, we first examined the LCN in femora from 15-week old male and female TβRIIocy−/− mice. Compared to control male mouse bones, the LCN of 15-week old TβRIIocy−/− bones was severely deteriorated, with a 50% reduction in LCN area and blunted canalicular length (Figure 2A-B, D). It is possible that the deregulation of TGFβ signaling may affect the integration of osteocytes into the bone matrix and an inducible model would be needed to conclusively address this question. Nonetheless, our previous observation of similar lacuno-canalicular defects in response to pharmacologic inhibition of TGFβ signaling (SD208) suggests that this is not solely a developmental defect(33). In addition, the male TβRIIocy−/− bones also displayed a strong coordinated decrease in the expression of genes encoding factors involved in bone resorption during PLR, namely, Mmp2, Mmp14 (matrix metalloproteinases), Ctsk (Cathepsin K), Acp5 (tartrate-resistant acid phosphatase), Atp6v0d2 (AtpD2), and Atp6v1g1 (AtpG1) (vacuolar ATPase pump encoding genes) (Figure 2F and H). Moreover, osteocyte-intrinsic protein expression of MMP13, MMP14 and CTSK was reduced in TβRIIocy−/− male mouse bones (Figure 2J and Figure S1).
Figure 2. Osteocyte-specific disruption of TβRII reduces PLR genes in male but not female mice.
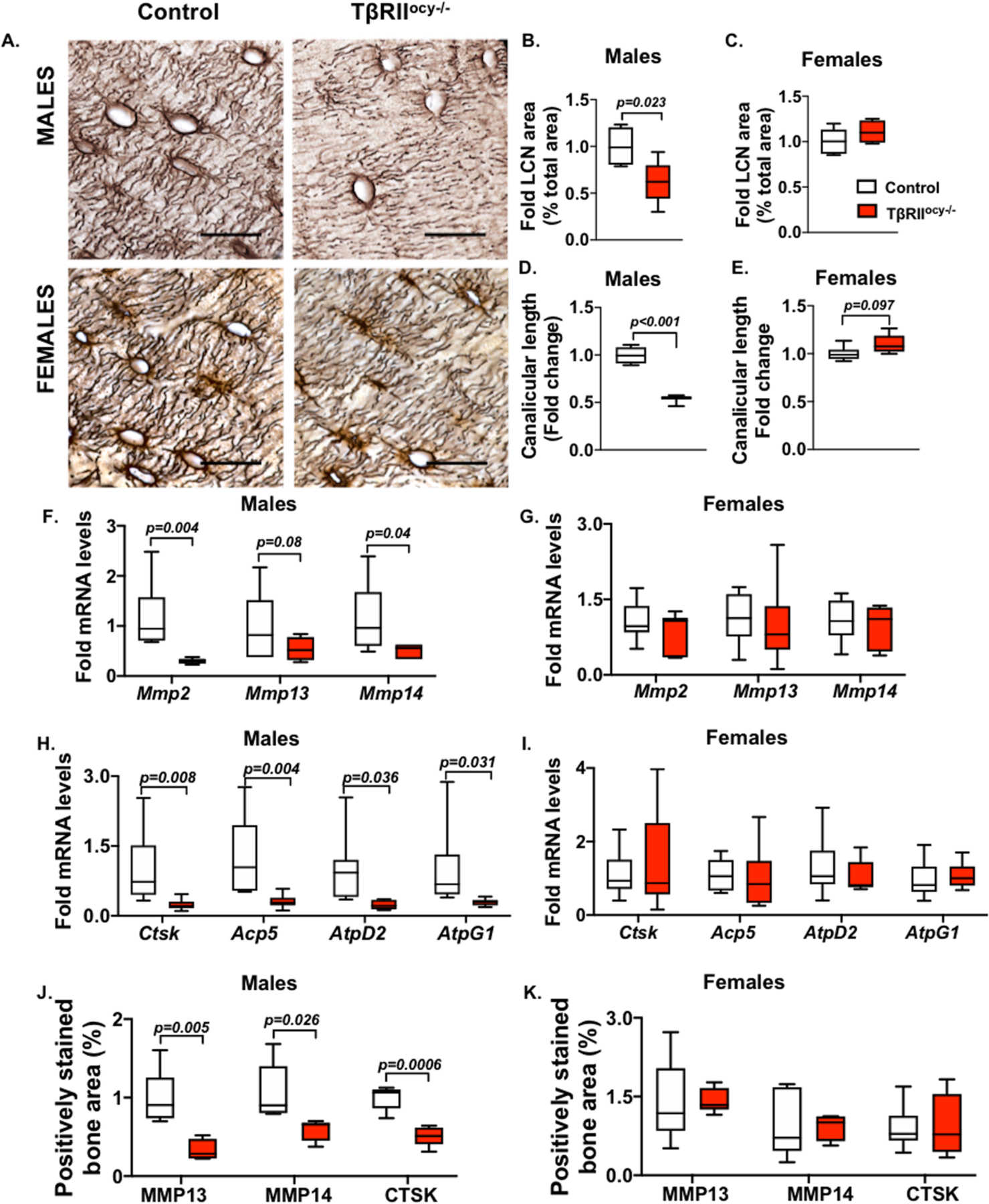
15-week-old male and female control and TβRIIocy−/− mouse bones were processed for mRNA and protein. Osteocyte lacuno-canalicular network (LCN) in the femoral cortical bones was assessed by Ploton stain. Representative images (A) and quantification of LCN area (B, C) and canalicular length (D, E) are shown. Scale bar, 20μm; n=4–6 mice/group and 4 ROI/mouse. Expression of PLR genes Mmp2, Mmp13 and Mmp14 (F, G) and Ctsk, Acp5, Atp6v0d2 (AtpD2) and Atp6v1g1 (AtpG1) are shown (H-I) (n=6–9 mice/ group). Immunohistochemistry (IHC) for MMP13, MMP14, and CTSK on femoral cortical bones of male and female control and TβRIIocy−/− mice shows percent positively stained bone area (J- K) (n =3–4 mice/group and 4 ROI/mouse). Scale bar, 30μm. Error bars indicate mean ± SEM, statistically significant differences were calculated with Student’s t test.
Female TβRIIocy−/− mice responded differently to osteocyte-intrinsic TGFβ ablation. In the female TβRIIocy−/− mice, the osteocyte LCN was intact with no changes observed in LCN area or canalicular length (Figure 2A, C and E). Consistent with these results, the expression of PLR genes in the bone of female TβRIIocy−/− mice did not differ from that in control mice (Figure 2G, I and K). Thus, the homeostatic regulation of PLR and bone quality in female mice occurs independently of TGFβ signaling. Since the absence of osteocytic TGFβ signaling in males adversely affects these outcomes, our data show a sexually dimorphic role for TGFβ in the control of PLR and bone quality.
4.3. Osteocytic deletion of TβRII prevents lactation induced PLR.
Although the homeostatic regulation of PLR in females is TGFβ-independent, we hypothesized that intact TGFβ signaling may be required to induce PLR in response to the metabolic stress of lactation. Using an established murine model of lactation, we assessed PLR outcomes in 15-week old lactating TβRIIocy−/− and lactating control mice. These results were compared to those in the 15-week old female TβRIIocy−/− and control virgin mice, described above. Upon validation of targeted TβRII deletion at the mRNA and protein levels in the lactating TβRIIocy−/− mice (Figure 3A, C-F), we monitored the effect of lactation on the expression of PLR genes. During lactation, control mice showed a 2.5 to 4-fold induction in the expression of the Mmp14, Ctsk, Acp5 and Atp6v0d2 genes (Figure 3A-B). The increased expression of PLR enzymes was also apparent at the protein level, with a 3 to 5-fold increase in the percentage of MMP13, MMP14 and CTSK-stained cortical bone area in the lactating control group (Figure 3C-D, E-G). Similar to resorptive genes, the lactation-induced expression of bone formation genes, Dmp1 and Runx2, was absent in TβRIIocy−/− mice (Figure S2A-B).
Consistent with these observations, lactating control mice undergo a 50% increase in the LCN area, evident from the Ploton silver staining (Figure 4A-B). Although no changes in the canalicular length were observed in either of the virgin or lactating TβRIIocy−/− mice compared to control group, changes in osteocyte lacunar features were detected with lactation. Using synchrotron X-ray tomography (SRμT), we quantified the lacunar volume of thousands of osteocyte lacunae, visualized in 3D. By conducting a tertile analysis of small, medium, and large- sized osteocytes, we observed that the lactating controls have significantly fewer small-sized lacunae, but more medium and large lacunae, compared to virgin controls. On the other hand, this lactation-induced shift from small to larger osteocyte lacunae is impaired in TβRIIocy−/− mice, where lactation-dependent differences are only apparent in the largest osteocytes. This suggests that osteocyte-intrinsic TGFβ signaling is required for maximal induction of PLR by lactation (Figure 4C).
Figure 4. Osteocyte-specific disruption of TβRII gene suppresses lactation induced increase in osteocyte lacunar volume.
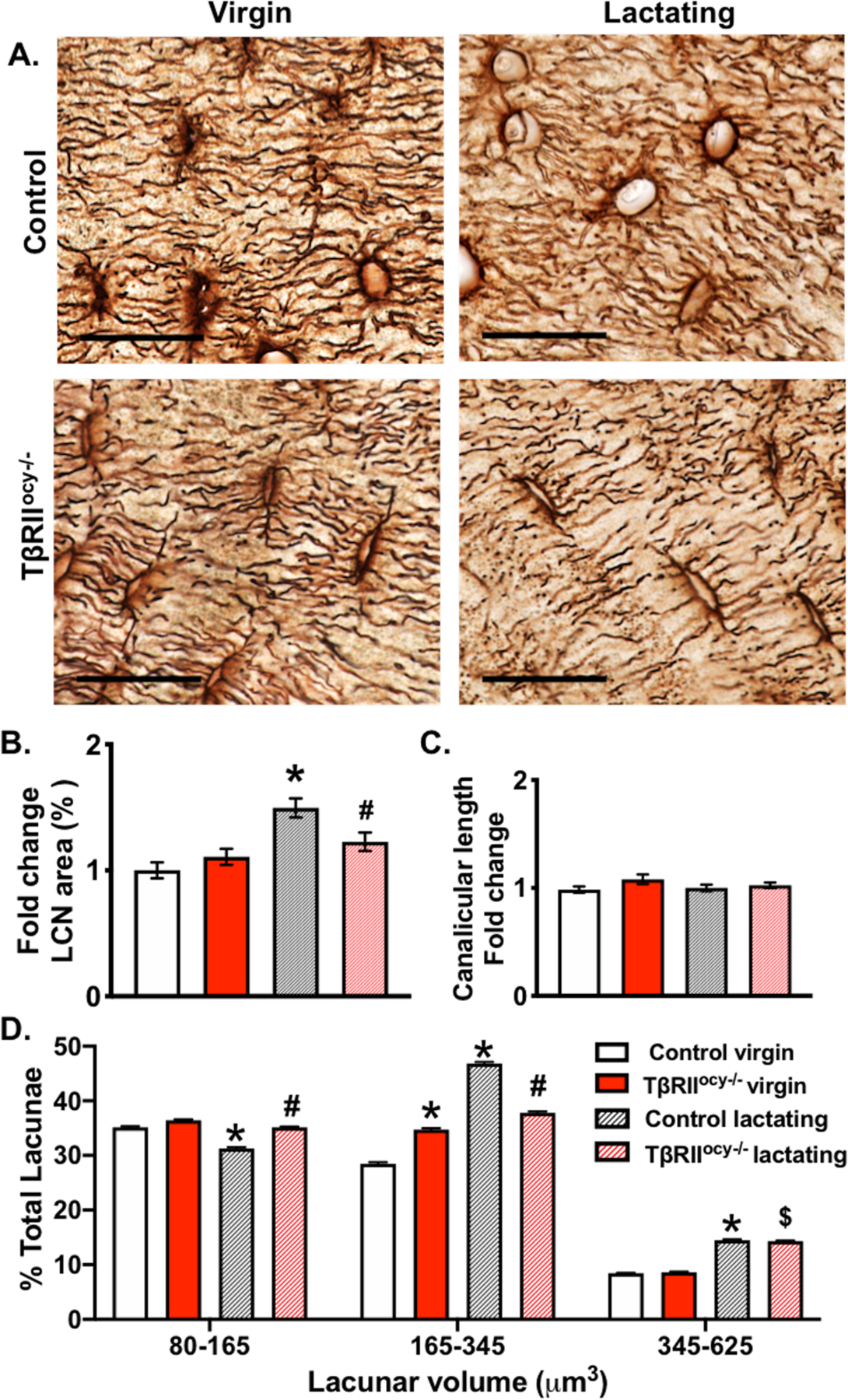
Representative Ploton stained histological sections of femoral cortical bones of 15-week-old virgin and lactating control and TβRIIocy−/− female mice (A) show the lacuno-canalicular network integrity (scale bar = 20μm). Quantification of average percent lacuno-canalicular area (normalized to total area) (B) and canalicular length (C) after 7 days of lactation is shown. XTM was used to determine osteocyte lacunar volume and the overall lacunar size distribution in cortical bone is shown in a graph, in control mice significant reduction in the overall percentage of smaller osteocytes and increase in larger osteocytes is observed in response to lactation. Scale bar for A is 20μm, and error bars indicate mean ± SEM, *p<0.05 compared to control virgin, #p<0.05 compared to control lactation, and $ p<0.05 compared to TβRIIocy−/− virgin, as calculated from one-way ANOVA with Tukey post-hoc test (n=4–5 mice/group).
Overall, lactation failed to induce PLR in TβRIIocy−/− mice. Lactating TβRIIocy−/− mice did not exhibit a significant induction in PLR genes, LCN area, lacunar volume and canalicular length (Figure 3 and 4). Therefore, distinct from the TGFβ-independent regulation of PLR in female bone at homeostasis, osteocyte-intrinsic TGFβ signaling is required for the induction of PLR during lactation.
4.4. Targeted deletion of osteocytic TβRII mitigates lactation-induced bone loss.
µCT analyses revealed that during lactation, control mice lose more than 52% of trabecular bone volume fraction (BV/TV, Table 1). This decrease in BV/TV is reflected by the corresponding reduction in trabecular thickness (32%) and increase in trabecular spacing (45%) during lactation in control mice (Table 1, Figure 5A-E). The trabecular bone mineralization is also reduced by 9% in the lactating control mice. A decrease in cortical bone thickness (27% reduction) and mass (25% reduction) was observed in lactating controls but the cortical mineralization remained intact (Table 1, Figure 5F-G). In contrast to control group, TβRIIocy−/− mice did not lose trabecular bone mass during lactation, and their trabecular number remained unaltered, while their trabecular thickness and spacing decreased only slightly with lactation. TβRIIocy−/− mice lost some cortical thickness and bone mass during lactation, however the magnitude of cortical bone loss was significantly lower in lactating TβRIIocy−/− mice than control mice (i.e. 14% vs. 25%, p<0.05) (Figure 5A-G).
Figure 5. Lactation induced bone loss in TβRIIocy−/− is mitigated.
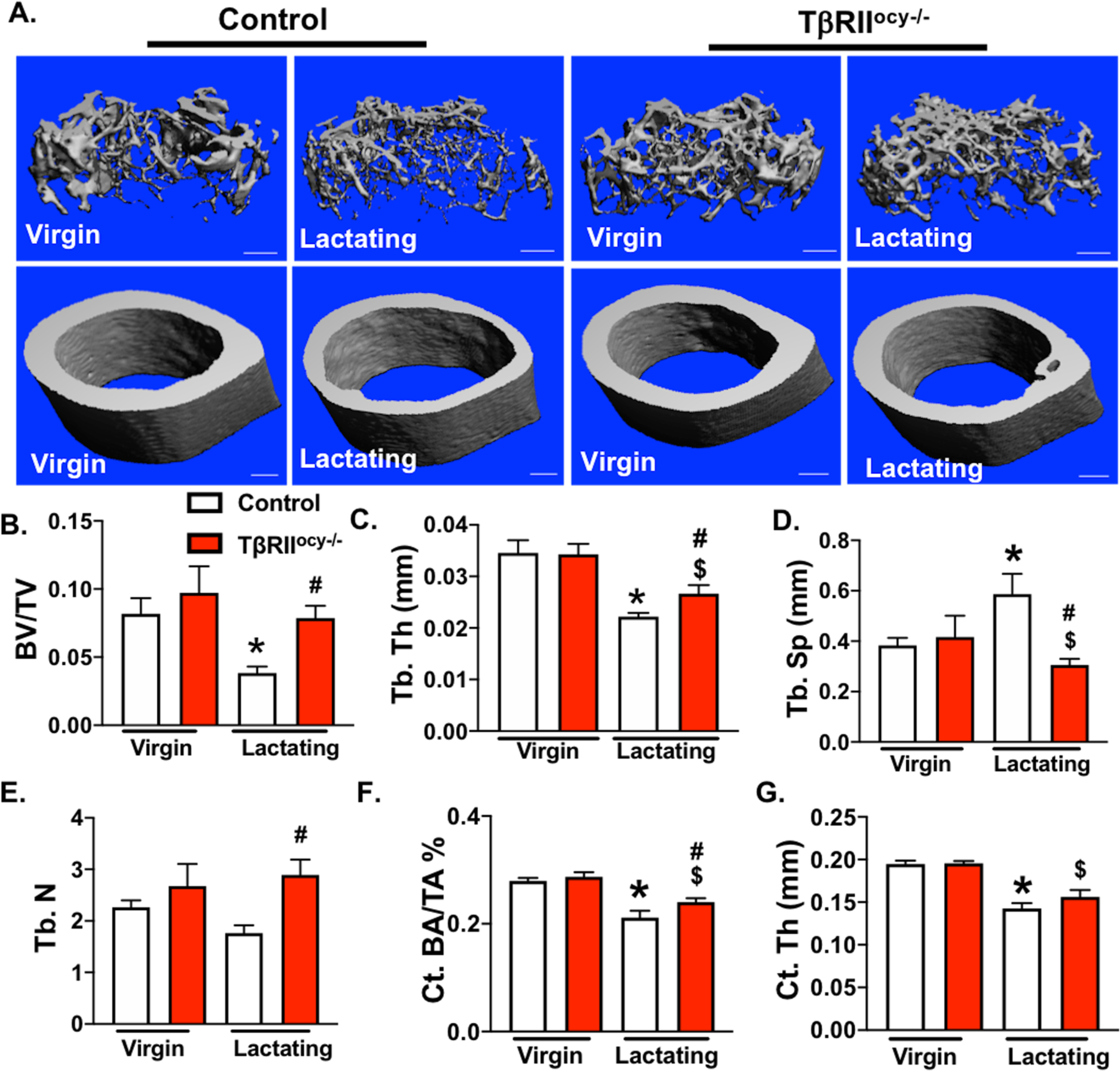
μCT analysis of femurs from 15-week old control and TβRIIocy−/− lactating female mice. Representative μCT reconstructions show trabecular (top) and cortical (bottom) bones (A). Decline in trabecular bone parameters: bone volume fraction (BV/TV), trabecular thickness (Tb.Th.), trabecular spacing (Tb.Sp.), and trabecular number (Tb.N) (B-E), and cortical bone parameters: cortical bone volume (Ct.BA/TA) and cortical thickness (Ct. Th), (F-G) in virgin and lactating mice of both genotypes is shown below. Scale bar, 100 μm (n = 6–10 mice/group). Error bars indicate mean ± SEM, *p<0.05 compared to control virgins, $p<0.05 compared to TβRIIocy−/− virgin group and #p<0.05 compared to control lactation group, as calculated from one-way ANOVA with Tukey post-hoc test.
The high-resolution quantitative imaging available through synchrotron SRµCT further revealed a comparable trend towards increased mineralization around osteocyte lacunae with lactation that was apparent in both genotypes (Figure S3). However, µCT data did not show significant differences in overall cortical bone mineralization during lactation in either genotype (Table 1). As expected, stiffness and yield load were significantly reduced in lactating control mice relative to their virgin counterparts (Table 2), consistent with the loss of cortical bone. However, these parameters were unaffected by lactation in the TβRIIocy−/− mice. When normalized for structural parameters, tissue properties such as bone bending modulus, yield stress, and ultimate stress were unaffected by lactation in either genotype (Table 2). Collectively, these data establish the role of osteocyte-intrinsic TGFβ signaling in lactation, such that absence of this mechanism alleviates lactation-induced bone remodeling by osteocytes and surface cells.
4.5. PTH involvement in the sexually dimorphic effects of osteocytic TGFβ.
As we previously reported, the elevated bone mass in 8-week old TβRIIocy−/− male mice is accompanied by reduced Rankl (Tnfsf11) expression and decreased osteoclast numbers(33). Therefore, we hypothesized that the sex-specific differences in bone mass in TβRIIocy−/− male and female mice were driven by dimorphic regulation of Rankl. Here we find that 15-week old male TβRIIocy−/− mice also express low levels of Rankl mRNA, with a stark reduction in the Rankl/Opg ratio, compared to control males. Female TβRIIocy−/− mice, on the other hand, showed intact Rankl and Rankl/Opg ratios during homeostasis (Figure 6A-B). During lactation, the high demand for calcium drives bone resorption, in part, through robust Rankl expression(45). Indeed, lactating control mice increase Rankl mRNA expression with a resulting 7-fold increase in the Rankl/Opg ratio (Figure 6C and S2C). Lactating TβRIIocy−/− mice, however, fail to stimulate Rankl mRNA expression. Instead Opg mRNA levels are suppressed in the lactating TβRIIocy−/− mice, through an apparent compensatory mechanism to increase the Rankl/Opg ratio 2-fold to release calcium for lactation (Figure 6C and S2C).
Figure 6. PTHR1 dependent signaling is reduced in TβRIIocy−/− males and lactating females.

qPCR analysis for Rankl and Opg in control and TβRIIocy−/− males (A) and virgin (B) and lactating (C) females is presented as a ratio of Rankl/Opg mRNA, with lactating groups shown relative to corresponding virgin groups. Pthr1 mRNA was also quantified by qPCR(D-F) and by immunohistochemistry (G-J). Percentage of osteocytes that stained positive for PTHR1 in bones of control and TβRIIocy−/− male and virgin and lactating female mice were quantified and normalized against total bone area as shown (H-J) (n=3–4 mice/group, 4 ROI/mouse). Error bars indicate mean ± SEM, statistically significant differences were calculated from Student’s t-test.
These differences in Rankl/Opg expression led us to focus on the role of PTH signaling in the sexually dimorphic effects of TGFβ in osteocytes. Several lines of evidence motivated our focus: First, PTH signaling directly regulates Rankl gene expression in osteocytes(46,47). Second, PTH and parathyroid hormone related peptide (PTHrP), key regulators of calcium and phosphate homeostasis, induce PLR during lactation(5,21,48–50). Third, PTH and PTHrP signal through a common parathyroid hormone receptor (PTHR1) in osteocytes; and PTHR1 ablation in osteocytes blocks lactation-inducible PLR(21). Fourth, as in PTHR1ocy−/− mice, TβRIIocy−/− mice are unable to induce PLR or bone loss during lactation(21). Fifth, TβRII and PTHR1 interact in osteoblasts to augment PTH signaling and increase bone formation(51). Based on these findings, we hypothesized that TGFβ and PTH signaling utilize a common molecular mechanism to regulate PLR.
To test this hypothesis, we first examined the effect of TβRII ablation on the expression of PTHR1 in the osteocytes. Both immunohistochemistry and quantitative PCR revealed a stark reduction (>70%) in the expression of PTHR1 in osteocytes of TβRIIocy−/− male mice compared to controls (Figure 6D, G, H). This was consistent with the low Rankl mRNA and Rankl/ Opg ratios seen in the TβRIIocy−/− males. In females during homeostasis, no changes were detected in the expression of PTHR1 at the mRNA or protein level between control and TβRIIocy−/− mice, which agrees with the lack of changes in Rankl/Opg in TβRIIocy−/− females (Figure 6E, G, I). During lactation, PTHR1 expression is robustly increased in control females, but loss of osteocytic TβRII mitigates this response in lactating TβRIIocy−/− mice (Figure 6F, G, J), consistent with the lower overall Rankl/Opg ratio in lactating TβRIIocy−/− mice compared to lactating controls.
Lastly, we evaluated serum levels of calcium, phosphate, and PTH in each group (Table S2). Phosphate levels did not differ in any condition. The two groups that had reduced PTHR1 expression, male and lactating female TβRIIocy−/− mice, had elevated levels of serum calcium relative to control mice. In male TβRIIocy−/− mice, the hypercalcemia may result from increased serum PTH levels. Importantly, in non-lactating female mice, none of these parameters differed between TβRIIocy−/− mice and controls, which is consistent with the insensitivity of these mice to osteocyte-intrinsic TGFβ ablation in all the other outcomes we analyzed. Taken together these findings suggest a key role of TβRIIIn regulating PTHR1 expression in osteocytes, and that this coordination between TGFβ and PTH signaling dictates the PLR response in osteocytes during homeostasis and metabolic stress.
5. Discussion
This study provides insight into sex-specific variation in bone fragility by revealing dimorphism in the regulation of osteocyte perilacunar/ canalicular remodeling (PLR). Using a genetic model of osteocyte-specific deletion of TGFβ receptor II (TβRII), we show that loss of osteocyte-intrinsic TGFβ signaling affects the skeleton of male, but not female, mice in homeostatic conditions. Male TβRIIocy−/− mice have defective bone quality, despite normal bone mass, resulting from impaired osteocytic PLR with deteriorated canalicular networks and reduced expression of requisite PLR enzymes. On the contrary, female TβRIIocy−/− mice are protected from these deficits in both PLR and bone quality. Osteocytic TGFβ signaling is necessary for PLR induction in female mice during lactation. Lactation-induced PLR is mitigated in TβRIIocy−/− females, as is lactation-induced bone loss. These findings highlight distinct roles for TGFβ signaling in regulating PLR in males and females to maintain bone homeostasis and implicate TGFβ as an essential regulator of PLR in females principally during the metabolic stress of lactation.
Gender disparity in fracture risk stems from differences in peak bone mass accrual, bone size, and geometry(52). Sex steroids that essentially drive skeletal variations in men and women integrate systemic cues such as that from vitamin D, IGF/GH, and PTH to dictate dimorphism at the cellular level(19,53–55). Indeed, sex differences in the cellular function of osteoblasts and osteoclasts have been attributed to differences in the bone phenotype of male and female mice(17–20). Differences in bone quality also contribute to the gender disparities in fracture risk.
Osteocyte PLR is one cellular mechanism that plays a pivotal role in maintaining bone quality. Although the contribution of PLR to sex differences in bone quality was unknown, osteocytes clearly have a dimorphic response to aging(56,57). The osteocyte lacuno-canalicular network, which is maintained by PLR, deteriorates more rapidly in aging females than in aging males(56,57). Here we describe sexually dimorphic regulation of osteocytes in the control of PLR and bone quality by TGFβ signaling. Whereas in males, loss of osteocytic TGFβ signaling suppresses PLR and increases bone fragility, female TβRIIocy−/− mice demonstrate a complete lack of this regulatory mechanism. Such skeletal adaptations in females could be one of the mechanisms by which the female skeleton compensates to meet the skeletal demands of pregnancy and lactation. The extent to which this ‘protective mechanism’ in reproductive age females is lost following menopause, or if it contributes to the increased risk of post-menopausal bone fragility remains unknown. Nevertheless, our study shows that sex specific differences in osteocyte sensitivity to TGFβ signaling contribute to the sexual dimorphism in PLR and bone quality.
TGFβ signaling has previously been implicated as one of the mechanisms recruited to confer sex specific differences in bone. An intricate crosstalk exists between TGFβ signaling and estrogen in bone. Estrogen promotes de novo synthesis and activation of TGFβ ligand in bone. Conversely, the loss of estrogen in ovariectomized rats leads to reduced TGFβ expression in bone(58–62). TGFβ, in turn, mediates bone protective effects of estrogen by increasing osteoclast apoptosis(59,63). With systemic inhibition of TGFβ signaling using a pharmacologic TGFβ receptor I kinase inhibitor (TβRI-I), we observed increased sensitivity of the female skeleton to TGFβ inhibition, with increased osteoblast proliferation and bone mass accrual in female mice than males(37). Here, in osteocytes of female mice with ablated TβRII, we observed that loss of osteocytic TGFβ signaling has no impact on PLR gene expression, the osteocyte lacuno-canalicular network, and bone quality. The extent to which these sex specific differences in osteocyte TGFβ signaling are related to estrogen levels remains to be determined. The remarkably distinct regulation of TGFβ signaling by sex-specific factors in osteoblasts, osteoclasts and osteocytes could afford independent, yet coordinated, regulation of bone quantity and quality to maintain mineral homeostasis and bone strength during hormonal and reproductive cycles in females.
Systemic calcium homeostasis is tightly regulated by the parathyroid gland that controls the interplay between the calciotropic hormones like, parathyroid hormone (PTH), parathyroid hormone related protein (PTHrP), calcitonin, and calciferol (1α,25-dihydroxycholecalciferol, vitamin D3). Together these hormones establish an equilibrium between dietary calcium absorption through the intestines, calcium reabsorption and excretion through kidneys, and its storage and release from bones(64). In bone, osteocytes show particularly high sensitivity to calcium fluctuations and are a target of PTH action(21,29,65). PTHR1, the receptor that binds both PTH and PTHrP, is highly expressed in bone and kidney and mediates the PTH-dependent regulation of calcium homeostasis. Although, little is known about the crosstalk of PTH and PTHrP with TGFβ signaling in bone, PTHR1 interacts directly with TβRII. In osteoblasts, PTHR1 and TβRII coordinately modulate bone formation(51). In osteocytes, however, we observed that ablation of TβRII causes a marked reduction in PTHR1 expression and Rankl/ Opg ratios only in male mice. Given that PTHR1 is a key regulator of PLR, the PLR and bone quality defects observed in the male TβRIIocy−/− may result from reduced PTHR1-dependent signaling. The reduced PTHR1-dependent signaling in TβRIIocy−/− male mice appears to be compensated by increased systemic PTH levels, which in turn increase systemic calcium levels, likely by targeting tissues other than bone.
In female mice, the role of TGFβ in osteocytes differs dramatically between homeostatic conditions and the metabolic stress of lactation. In homeostatic conditions, female TβRIIocy−/− mice differ from TβRIIocy−/− males, in that they show intact osteocytic PTHR1 expression and PLR. Other pathways that are active in females, but not in males, may converge on PTHR1 to sustain its expression in female mice at homeostasis. This capacity of female TβRIIocy−/− osteocytes to normalize PTHR1 levels at homeostasis may contribute to the intact bone quality of female TβRIIocy−/− mice. This capacity is challenged by the metabolic stress of lactation, which normally induces PLR to meet the high calcium demands for breastmilk production. PLR induction during lactation occurs in a hormonal milieu with low PTH and estradiol, but with high PTHrP levels. While we demonstrated reduction in circulating PTH with lactation in both genotypes, the circulating level of PTHrP was not significantly increased in either genotypes, most likely due to insufficient sensitivity of our assay. PLR induction during lactation also requires osteocytic PTHR1. Although PTHR1 mRNA is induced in lactation in both genotypes, the induction of PTHR1 protein in osteocytes is abrogated in lactating TβRIIocy−/− mice relative to control littermates. Accordingly, with reduced osteocytic PTHR1 protein levels, PLR induction in female TβRIIocy−/− mice during lactation is blocked. Our results point to the possibility that the level of PTHR1 is a pivotal determinant of the effect of TGFβ on PLR. Thus, the differential PLR response of osteocytes in the TβRIIocy−/− mice in males vs. females, and in homeostasis vs. metabolic stress, may result from differences in the ability to stimulate PTHR1-mediated bone resorption by osteocytes. It is likely that such molecular mechanisms are in place to compensate for reproductive bone loss and protect the female skeleton from higher fracture risk. Another such compensatory mechanism that was recently suggested includes accrual of higher trabecular bone mass than mechanically necessary by female rats compared to males(1,66).
In conclusion, our study highlights sexual dimorphism in TGFβ’s regulation of osteocyte perilacunar/canalicular remodeling activity. With implication of PLR in both physiological and pathological conditions, including renal osteodystrophy, glucocorticoid-induced osteonecrosis, osteoarthritis, and hyperparathyroidism(21,44,67–71), defining the sex differences in these cell-intrinsic cues and molecular networks in osteocytes will be critical to address sexual dimorphism in the prevalence and manifestation of bone fragility, as well as refining therapeutics for reducing fracture risk.
Supplementary Material
7. Acknowledgments.
The authors gratefully acknowledge J.J. Woo for expert technical assistance. This research was supported by NIH-NIDCR grant R01 DE019284 (T.A.), including a supplement from the Office of Research on Women’s Health, Department of Defense (DoD) grant PRORP OR130191 (T.A.), NSF grant 1636331, NIH-NIAMS grant R21 AR067439, NIH-NIAMS grant P30 AR066262–01 (T.A.), and the Read Research Foundation (T.A.). The authors acknowledge the use of the x-ray synchrotron beamlines 8.3.2 at the Advanced Light Source (ALS) at LBNL, which is supported by the Director of the U.S. Department of Energy under contract DE-AC02–05CH11231. We are grateful for helpful conversations with our colleagues in the San Francisco Veteran’s Administration Medical Center (SF-VAMC) Endocrine Unit throughout the development of this project.
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
References
- 1.de Bakker CMJ, Altman-Singles AR, Li Y, Tseng WJ, Li C, Liu XS. Adaptations in the Microarchitecture and Load Distribution of Maternal Cortical and Trabecular Bone in Response to Multiple Reproductive Cycles in Rats. J. Bone Miner. Res 2017;32(5):1014–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sowers M, Corton G, Shapiro B, Jannausch ML, Crutchfield M, Smith ML, et al. Changes in Bone Density With Lactation. JAMA 1993;269(24):3130–5. [PubMed] [Google Scholar]
- 3.Kent GN, Price RI, Gutteridge DH, Allen JR, Barnes MP, Hickling CJ, et al. Human lactation: Forearm trabecular bone loss, increased bone turnover, and renal conservation of calcium and inorganic phosphate with recovery of bone mass following weaning. J. Bone Miner. Res 1990;5(4):361–9. [DOI] [PubMed] [Google Scholar]
- 4.Liu XS, Ardeshirpour L, Vanhouten JN, Shane E, Wysolmerski JJ. Site-specific changes in bone microarchitecture, mineralization, and stiffness during lactation and after weaning in mice. J. Bone Miner. Res 2012;27(4):867–75. [DOI] [PubMed] [Google Scholar]
- 5.Vanhouten JN, Wysolmerski JJ. Low Estrogen and High Parathyroid Hormone-Related Peptide Levels Contribute to Accelerated Bone Resorption and Bone Loss in Lactating Mice. Endocrinology 2003;144(12):5521–9. [DOI] [PubMed] [Google Scholar]
- 6.Alswat KA. Gender Disparities in Osteoporosis. J. Clin. Med. Res 2017;9(5):382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieves JW, Formica C, Ruffing J, Zion M, Garrett P, Lindsay R, et al. Males have larger skeletal size and bone mass than females, despite comparable body size. J. Bone Miner. Res 2005;20(3):529–35. [DOI] [PubMed] [Google Scholar]
- 8.Daly RM, Rosengren BE, Alwis G, Ahlborg HG, Sernbo I, Karlsson MK. Gender specific age-related changes in bone density, muscle strength and functional performance in the elderly: A-10 year prospective population-based study. BMC Geriatr 2013;13(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao X, Carleton SM, Kettle AD, Melander J, Phillips CL, Wang Y. Gender-dependence of bone structure and properties in adult osteogenesis imperfecta murine model. Ann Biomed Eng 2013;41(6):1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright NC, Looker AC, Saag KG, Curtis JR, Delzell ES, Randall S, et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J. Bone Miner. Res 2014;29(11):2520–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wainwright SA, Marshall LM, Ensrud KE, Cauley JA, Black DM, Hillier TA, et al. Hip Fracture in Women without Osteoporosis. J. Clin. Endocrinol. Metab 2005;90(5):2787–93. [DOI] [PubMed] [Google Scholar]
- 12.Cooper C, Campion G, Melton L. International Original Article Hip Fractures in the Elderly : A World-Wide Projection. Osteoporos. Int 1992;2(6):285–9. [DOI] [PubMed] [Google Scholar]
- 13.Kanis JA, Oden A, Johnell O, Johansson H, De Laet C, Brown J, et al. The use of clinical risk factors enhances the performance of BMD in the prediction of hip and osteoporotic fractures in men and women. Osteoporos. Int 2007;18(8):1033–46. [DOI] [PubMed] [Google Scholar]
- 14.Nicks KM, Fowler TW, Gaddy D. Reproductive Hormones and Bone. Curr. Osteoporos. Rep 2010;8(2):60–7. [DOI] [PubMed] [Google Scholar]
- 15.Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol. Rev 2016;97(1):135–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karasik D, Kiel DP. Evidence for pleiotropic factors in genetics of the musculoskeletal system. Bone 2010;46(5):1226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jepsen KJ, Lane NE, Healey JH, Tosi LL. Editorial Comment: Symposium: Sex Differences in Musculoskeletal Disease and Science. Clin. Orthop. Relat. Res 2015;473(8):2474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jevon M, Sabokbar A, Fujikawa Y, Hirayama T, Neale SD, Wass J, et al. Gender- and age-related differences in osteoclast formation from circulating precursors. J. Endocrinol 2002;172(3):673–81. [DOI] [PubMed] [Google Scholar]
- 19.Babey M, Wang Y, Kubota T, Fong C, Menendez A, Elalieh HZ, et al. Gender-specific differences in the skeletal response to continuous PTH in mice lacking the IGF1 receptor in mature osteoblasts. J. Bone Miner. Res 2015;30(6):1064–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanotti S, Kalajzic I, Aguila HL, Canalis E. Sex and genetic factors determine osteoblastic differentiation potential of murine bone marrow stromal cells. PLoS One 2014;9(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qing H, Ardeshirpour L, Divieti Pajevic P, Dusevich V, Jähn K, Kato S, et al. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res 2012;27(5):1018–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jähn K, Kelkar S, Zhao H, Xie Y, Tiede-Lewis LM, Dusevich V, et al. Osteocytes Acidify Their Microenvironment in Response to PTHrP In Vitro and in Lactating Mice In Vivo. J. Bone Miner. Res 2017;32(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qing H, Bonewald LF. Osteocyte Remodeling of the Perilacunar and Pericanalicular Matrix. Int. J. Oral Sci 2009;1(2):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang SY, Herber R-P, Ho SP, Alliston T. Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J. Bone Miner. Res 2012;27(9):1936–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaya S, Basta-Pljakic J, Seref-Ferlengez Z, Majeska RJ, Cardoso L, Bromage T, et al. Lactation Induced Changes in the Volume of Osteocyte Lacunar-Canalicular Space Alter Mechanical Properties in Cortical Bone Tissue. J. Bone Miner. Res 2017;32(4):688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, et al. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J. Biol. Chem 2006;281(44):33814–24. [DOI] [PubMed] [Google Scholar]
- 27.Yamada S, Billinghurst RC, Chrysovergis K, Inoue S, Poole AR, Holmbeck K, Pidoux I, Birkedal-Hansen H, Bianco P, Wu W. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J. Cell Sci 2004;118(1):147–56. [DOI] [PubMed] [Google Scholar]
- 28.Arnett TR. Osteocytes: Regulating the Mineral Reserves? J. Bone Miner. Res 2013;28(12):2433–5. [DOI] [PubMed] [Google Scholar]
- 29.Wysolmerski JJ. Osteocytes remove and replace perilacunar mineral during reproductive cycles. Bone 2013;54(2):230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol 2011;209(1):21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nango N, Kubota S, Hasegawa T, Yashiro W, Momose A, Matsuo K. Osteocyte-directed bone demineralization along canaliculi. Bone 2016;84:279–88. [DOI] [PubMed] [Google Scholar]
- 32.Harris SE, Gluhak-Heinrich J, Harris MA, Yang W, Bonewald LF, Riha D, et al. DMP1 and MEPE expression are elevated in osteocytes after mechanical loading in vivo: theoretical role in controlling mineral quality in the perilacunar matrix. J. Musculoskelet. Neuronal Interact 2007;7(4):313–5. [PMC free article] [PubMed] [Google Scholar]
- 33.Dole NS, Mazur CM, Acevedo C, Lopez JP, Monteiro DA, Fowler TW, et al. Osteocyte-Intrinsic TGF-β Signaling Regulates Bone Quality through Perilacunar/Canalicular Remodeling. Cell Rep 2017;21(9):2585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, Ghadami M, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat. Genet 2000;26(1):19–20. [DOI] [PubMed] [Google Scholar]
- 35.Grafe I, Yang T, Alexander S, Homan E, Lietman C, Jiang MM, et al. Excessive TGFβ signaling is a common mechanism in Osteogenesis Imperfecta. Nat. Med 2014;20(6):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 2016;4. [DOI] [PMC free article] [PubMed]
- 37.Mohammad KS, Chen CG, Balooch G, Stebbins E, McKenna CR, Davis H, et al. Pharmacologic inhibition of the TGF-β type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One 2009;4(4):12–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filvaroff E, Erlebacher A, Ye J, Gitelman SE, Lotz J, Heillman M, et al. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 1999;126(19):4267–79. [DOI] [PubMed] [Google Scholar]
- 39.Weivoda MM, Ruan M, Pederson L, Hachfeld C, Davey RA, Zajac JD, et al. Osteoclast TGF-β Receptor Signaling Induces Wnt1 Secretion and Couples Bone Resorption to Bone Formation. J. Bone Miner. Res 2016;31(1):76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simpson AE, Stoddart MJ, Davies CM, Jähn K, Furlong PI, Gasser JA, et al. TGF β 3 and loading increases osteocyte survival in human cancellous bone cultured ex vivo. Cell Biochem. Funct 2009. Jan;27(1):23–9. [DOI] [PubMed] [Google Scholar]
- 41.Levéen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjöstrand LJ, et al. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood 2002;100(2):560–8. [DOI] [PubMed] [Google Scholar]
- 42.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted Cre Expression in Odontoblasts and Osteocytes. J. Dent. Res 2007;86(4):320–5. [DOI] [PubMed] [Google Scholar]
- 43.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 44.Fowler TW, Acevedo C, Mazur CM, Hall-Glenn F, Fields AJ, Bale HA, et al. Glucocorticoid suppression of osteocyte perilacunar remodeling is associated with subchondral bone degeneration in osteonecrosis. Sci. Rep 2017;7. [DOI] [PMC free article] [PubMed]
- 45.Ardeshirpour L, Dann P, Adams DJ, Nelson T, VanHouten J, Horowitz MC, et al. Weaning Triggers a Decrease in Receptor Activator of Nuclear Factor-κB Ligand Expression, Widespread Osteoclast Apoptosis, and Rapid Recovery of Bone Mass after Lactation in Mice. Endocrinology 2007;148(8):3875–86. [DOI] [PubMed] [Google Scholar]
- 46.Ben-awadh AN, Delgado-Calle J, Tu X, Kuhlenschmidt K, Allen MR, Plotkin LI, et al. Parathyroid hormone receptor signaling induces bone resorption in the adult skeleton by directly regulating the RANKL gene in osteocytes. Endocrinology 2014;155(8):2797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellido T, Saini V, Pajevic PD. Effects of PTH on osteocyte function. Bone 2013;54(2):250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishizawa Y, Yajima A, Ikeda K, Tominaga Y, Ito A, Inaba M. Increased osteocyte death and mineralization inside bone after parathyroidectomy in patients with secondary hyperparathyroidism. J. Bone Miner. Res 2010;25(11):2374–81. [DOI] [PubMed] [Google Scholar]
- 49.Yajima A, Tsuchiya K, Burr DB, Minner DE, Condon KW, Miller CA, et al. Osteocytic perilacunar/canalicular turnover in hemodialysis patients with high and low serum PTH levels. Bone 2018;113:68–76. [DOI] [PubMed] [Google Scholar]
- 50.Goltzman D Emerging roles for calcium-regulating hormones beyond osteolysis. Trends Endocrinol. Metab 2010;21(8):512–8. [DOI] [PubMed] [Google Scholar]
- 51.Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X. TGF-β type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol 2010;12:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seeman E The structural basis of bone fragility in men. Bone 1999;25(1):143–7. [DOI] [PubMed] [Google Scholar]
- 53.Riggs BL, Melton LJ, Robb RA, Camp JJ, Atkinson EJ, McDaniel L, et al. A population-based assessment of rates of bone loss at multiple skeletal sites: evidence for substantial trabecular bone loss in young adult women and men. J. Bone Miner. Res 2008;23(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan JW, Starczak Y, Tsangari H, Sawyer RK, Davey RA, Atkins GJ, et al. Sex-related differences in the skeletal phenotype of aged vitamin D receptor global knockout mice. J. Steroid Biochem. Mol. Biol 2016;164:361–8. [DOI] [PubMed] [Google Scholar]
- 55.Locatelli V, Bianchi VE. Effect of GH/IGF-1 on Bone Metabolism and Osteoporsosis. Int. J. Endocrinol 2014;2014:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vashishth D, Gibson GJ, Fyhrie DP. Sexual dimorphism and age dependence of osteocyte lacunar density for human vertebral cancellous bone. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 2005;282(2):157–62. [DOI] [PubMed] [Google Scholar]
- 57.Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, et al. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging 2017;9(10):2190–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao Y, Qian W-P, Dark K, Toraldo G, Lin ASP, Guldberg RE, et al. Estrogen prevents bone loss through transforming growth factor β signaling in T cells. Proc. Natl. Acad. Sci 2004;101(47):16618 LP–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF–β. Nat. Med 1996;2(10):1132–6. [DOI] [PubMed] [Google Scholar]
- 60.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming Growth Factor-β1 to the Bone. Endocr. Rev 2005;26(6):743–74. [DOI] [PubMed] [Google Scholar]
- 61.Thrailkill KM, Jo C-H, Cockrell GE, Moreau CS, Lumpkin CK Jr, Fowlkes JL. Determinants of undercarboxylated and carboxylated osteocalcin concentrations in type 1 diabetes. Osteoporos. Int 2012;23(6):1799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oursler MJ, Cortese C, Keeting P, Anderson MA, Bonde SK, Riggs BL, et al. Spelsberg TC. Modulation of Transforming Growth Factor-β Production in Normal Human Osteoblast-Like Cells by 17β-Estradiol and Parathyroid Hormone. Endocrinology 1991;129(6):3313–20. [DOI] [PubMed] [Google Scholar]
- 63.Hawse JR, Subramaniam M, Ingle JN, Oursler MJ, Rajamannan NM, Spelsberg TC. Estrogen-TGFβ cross-talk in bone and other cell types: Role of TIEG, Runx2, and other transcription factors. J. Cell. Biochem 2008;103(2):383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovacs CS. Bone Development and Mineral Homeostasis in the Fetus and Neonate: Roles of the Calciotropic and Phosphotropic Hormones. Physiol. Rev 2014;94(4):1143–218. [DOI] [PubMed] [Google Scholar]
- 65.Prideaux M, Dallas SL, Zhao N, Johnsrud ED, Veno PA, Guo D, et al. Parathyroid hormone induces bone cell motility and loss of mature osteocyte phenotype through L-calcium channel dependent and independent mechanisms. PLoS One 2015;10(5):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Bakker CMJ, Zhao H, Tseng W-J, Li Y, Altman-Singles AR, Liu Y, et al. Effects of reproduction on sexual dimorphisms in rat bone mechanics. J. Biomech 2018;77:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonucci E, Gherardi G. Osteocyte ultrastructure in renal osteodystrophy. Virchows Arch. A Pathol. Anat. Histol 1977;373(3):213–31. [DOI] [PubMed] [Google Scholar]
- 68.Alemi AS, Mazur CM, Fowler TW, Woo JJ, Knott PD, Alliston T. Glucocorticoids cause mandibular bone fragility and suppress osteocyte perilacunar-canalicular remodeling. Bone Reports 2018;9:145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ciani A, Toumi H, Pallu S, Tsai EHR, Diaz A, Guizar-Sicairos M, et al. Ptychographic X-ray CT characterization of the osteocyte lacuno-canalicular network in a male rat’s glucocorticoid induced osteoporosis model. Bone Reports 2018;9:122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nalla RK, Balooch M, Kinney JH, Bonewald LF, Lane NE, Habelitz S, et al. Glucocorticoid-Treated Mice Have Localized Changes in Trabecular Bone Material Properties and Osteocyte Lacunar Size That Are Not Observed in Placebo-Treated or Estrogen-Deficient Mice. J. Bone Miner. Res 2006;21(3):466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazur CM, Woo JJ, Yee CS, Fields AJ, Acevedo C, Bailey KN, et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. Bone Res 2019;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.