

Abstract
Juvenile neuronal ceroid lipofuscinosis (JNCL, aka. juvenile Batten disease or CLN3 disease) is a lysosomal storage disease characterized by progressive blindness, seizures, cognitive and motor failures, and premature death. JNCL is caused by mutations in the Ceroid Lipofuscinosis, Neuronal 3 (CLN3) gene, whose function is unclear. Although traditionally considered a neurodegenerative disease, CLN3 disease displays eye-specific effects: JNCL often first presents as vision loss; and vision loss has also been reported in non-syndromic CLN3 disease. Here we described the roles of CLN3 protein in maintaining healthy retinal pigment epithelium (RPE) and normal vision. Using electroretinogram, fundoscopy and microscopy, we showed impaired visual function, retinal autofluorescent lesions, and RPE disintegration and metaplasia/hyperplasia in a Cln3 ~1 kb-deletion mouse model [1] on C57BL/6J backgroun. Utilizing a combination of biochemical analyses, RNA-Seq, Seahorse XF bioenergetic analysis, and Stable Isotope Resolved Metabolomics (SIRM), we further demonstrated that loss of CLN3 increased autophagic flux, suppressed mTORC1 and Akt activities, enhanced AMPK activity, and up-regulated gene expression of the autophagy-lysosomal system in RPE-1 cells, suggesting autophagy induction. This CLN3 deficiency induced autophagy induction coincided with decreased mitochondrial oxygen consumption, glycolysis, the tricarboxylic acid (TCA) cycle, and ATP production. We also report for the first time that loss of CLN3 led to glycogen accumulation despite of impaired glycogen synthesis. Our comprehensive analyses shed light on how loss of CLN3 affect autophagy and metabolism. This work suggests possible links among metabolic impairment, autophagy induction and lysosomal storage, as well as between RPE atrophy/degeneration and vision loss in JNCL.
Graphical Abstract
Introduction
Neuronal ceroid lipofuscinoses (NCLs) are the most common pediatric neurodegenerative disorders [2]. Juvenile Neuronal Ceroid Lipofuscinosis (JNCL, aka. juvenile Batten disease or CLN3 disease), a form of NCLs with juvenile onset, is the most common form of NCLs [3]. Children with JNCL suffer from progressive vision loss, seizures, cognitive and motor failure, and premature death. Long considered a disease of the CNS, emerging evidence suggests disease pathology in the heart and the immune system [4–8], with non-syndromic CLN3 disease restricted to the eye [9–11]. JNCL is caused by mutations in the Ceroid Lipofuscinosis, Neuronal 3 (CLN3) gene, with the predominant mutation being a homozygous deletion of 966 bp in the region spanning exons 7 and 8 (aka. the ~1 kb-deletion mutation) [12]. Despite the monogenetic nature and severity of the disease, the mechanisms of its pathology are still ill-defined almost 200 years after its discovery [13].
Vision loss is often the earliest symptom of syndromic JNCL disease and the only symptom of non-syndromic disease, making ophthalmology the first point of care for patients (reviewed in [3, 14]). JNCL patients display a wide range of functional vision impairment, e.g., nystagmus, photophobia, nyctalopia, loss of visual acuity, and loss of peripheral and color vision [15]. JNCL ophthalmoscopic features include diffuse retinal pigment epithelium (RPE) atrophy of the macula, Bull’s eye macular dystrophy, optic disc pallor, attenuated vessels, and accumulation of autofluorescent cytoplasmic ceroid granules in photoreceptors and retinal ganglion cells (RGCs) (reviewed in [3]). JNCL patients also show profound electroretinogram (ERG) abnormalities, ranging from detectable reduction in rod- and coneresponse amplitudes early in disease to un-recordable rod-mediated activity and greatly reduced cone-mediated activity late in disease (reviewed in [3]). Recent utilization of optical coherence tomography (OCT) and confocal scanning laser ophthalmoscope (cSLO) reveals abnormalities in various retinal layers including RPE and papillomacular bundles of JNCL patients [16, 17].
Despite extensive ocular pathologies, the etiology of JNCL-related vision loss is poorly understood. Often occurred only at advanced disease stages, postmortem examination of the retina from JNCL patients reveals a substantial degeneration of photoreceptors and loss of the outer nuclear layer (ONL), accumulation of ceroids within RGCs, and a paradoxical reduction in lipofuscin granules within the RPE (Goebel et al., 1974; Bensaoula et al., 2000). Therefore, the primary or early defects due to JNCL in the vision system is uncertain: They could be either in the photoreceptors and the inner retinal neurons, as evidenced by the subnormal scotopic a-wave and the electronegative b-wave ERG [18], or external to the retina, as shown by the reported optic atrophy and retrograde degeneration of the optic nerve (i.e., the RGC axon bundle) and its downstream dorsal lateral geniculate nucleus (LGNd) thalamic neurons in the visual cortex [19]. Thus, it is important to identify the retinal cell type(s) that are crucial in JNCL-related vision loss initiation and progression.
The RPE is a monolayer of polarized pigmented cells that play critical roles in retinal physiology, including light absorption, phagocytosis of shed photoreceptor outer segments (POS), recycling retinoids in the visual cycle, synthesis and release of growth factors, as well as serving as the outer blood-retina barrier and transporting nutrients and metabolites into and out of the neural retina [20, 21]. Due to the importance of the RPE in retinal physiology, its dysfunction contributes significantly to retinal degenerative diseases, including age-related macular degeneration (AMD) [22, 23], vitelliform macular degeneration [24] and Sorsby fundus dystrophy [25]. Thus, it stands to reason that RPE dysfunction may contribute to vision loss in JNCL. The RPE is particularly important for energy metabolism in the retina: It supplies ~60% of the glucose consumed by the neural retina [26], one of the most metabolically active tissues [27]; and the RPE itself is highly metabolically active. Metabolic impairment, which includes suppression of mitochondrial metabolism [28], chronic HIF2αmediated metabolic stress [29], or deficient autophagy [30–32] in RPE cells, has been shown to cause RPE dysfunction, photoreceptor and retinal degeneration, and vision impairment. Despite observations of RPE abnormalities in both JNCL patients [3, 16, 17] and a mouse model [33], the contributions of RPE as well as RPE metabolism to JNCL-related vision loss remain elusive. Here we focus on RPE cell metabolism in JNCL-related vision loss.
CLN3, the sole gene mutated in JNCL, encodes a 438 amino acid-long protein named CLN3 or Battenin, whose structure, localization and functions remain elusive (reviewed in [34–37]). Although CLN3 has been postulated to have myriad of potential roles1, it is unknown which of these cellular functions, when impaired, lead to disease pathologies or in which order cellular functions are impaired upon losing CLN3 protein; and cell type-specific roles of CLN3 in the retina have rarely been studied beyond neurodegeneration. In this work, we focus on how CLN3 deficiency impacts RPE cell metabolism in JNCL-related vision loss. Using a homozygous CLN3 ~1 kb-deletion mutant mouse model [1] with a late retinal degeneration phenotype as compared to JNCL patients, we showed that loss of CLN3 protein results in RPE dystrophy and degeneration. Utilizing an integrated approach involving biochemical analyses, RNA-Seq, Seahorse XF bioenergetic analysis, and SIRM, we further demonstrate that CLN3 deficiency in RPE-1 cells results in autophagy induction and concomitant disturbances in glycolysis, the TCA cycle, oxidative phosphorylation (OXPHOS), de novo nucleotide synthesis, glycogen metabolism, and lipid metabolism. Our work suggests that CLN3 protein plays a critical role in maintaining RPE cellular metabolic homeostasis and normal vision. We also provide a working model for how RPE metabolic impairment and degeneration may be linked to each other upon CLN3 deficiency, leading to JNCL-related vision loss.
Results
1. The common ~1 kb-deletion CLN3 disease mutation caused RPE atrophy and degeneration, and vision impairment in mice.
To investigate JNCL-related vision loss, we characterized a mouse model (i.e., Cln3Δex7/8) which was generated by replacing the mouse Cln3 gene with the most common ~1 kb-deletion JNCL mutation of the CLN3 gene [1]. This “knock-in” eliminated exons 7 and 8 as well as surrounding non-coding DNA, causing a frameshift and premature termination [1]. Similar to other JNCL mouse models [33, 76–78], Cln3Δex7/8 mice recapitulated JNCL morphologies, including autofluorescent storage materials, electron-dense deposits with fingerprint profiles, and mitochondrial ATP synthase F0 sub-complex subunit C (abbreviated as subunit C hereafter) deposits, and exhibited neurodegeneration in the brain and retina [1, 79–81]. However, Cln3Δex7/8 mice used in these studies were all on genetic backgrounds that carried CLN3-independent retinal degenerative genetic loci [1, 79–81].
To determine retinal phenotype of Cln3Δex7/8 mice without other confounding retinal degenerative genetic loci, we investigated Cln3Δex7/8 mice on a C57B6/J background. We first confirmed that these mice and their wild-type (WT) controls did not carry either Crb1 (i.e., Rd8) or Pde6b (i.e., Rd1) mutation [82], both of which are spontaneous, retinal degenerative mutations [83, 84]. We found that the scotopic ERG response from dark adapted WT mice showed normal wave form with a trough (a-wave) followed by a peak (b-wave) (Supplemental Figure 1A). We observed a trend of reduction in both the scotopic a- and bwave amplitudes in homozygous Cln3Δex7/8 mice as compared to those in WT mice as early as 8-month old (Figure 1A, Supplemental Figure 1A). With aging, the scotopic a- and bwave amplitudes decrease in both WT and Cln3Δex7/8 mice (Figure 1A). Importantly, scotopic b-wave amplitudes in 14-month old Cln3Δex7/8 mice were significantly diminished compared to those in WT mice (Figure 1A). The ratio of b-wave amplitude to a-wave amplitude in 14month old Cln3Δex7/8 mice had a decreasing trend as compared to that in WT mice, even though it did not reach statistical significance (Supplemental Figure 1B). As a- and b-waves are derived from photoreceptors and inner retinal neurons, respectively [85], our data suggest that the ~1 kb-deletion mutation in the CLN3 gene led to decreased functions of inner retinal neurons and that photoreceptor responses might also be mildly diminished in this model.
Figure 1. Homozygous Cln3Δex7/8 mice show vision impairment as well as RPE atrophy and degeneration.
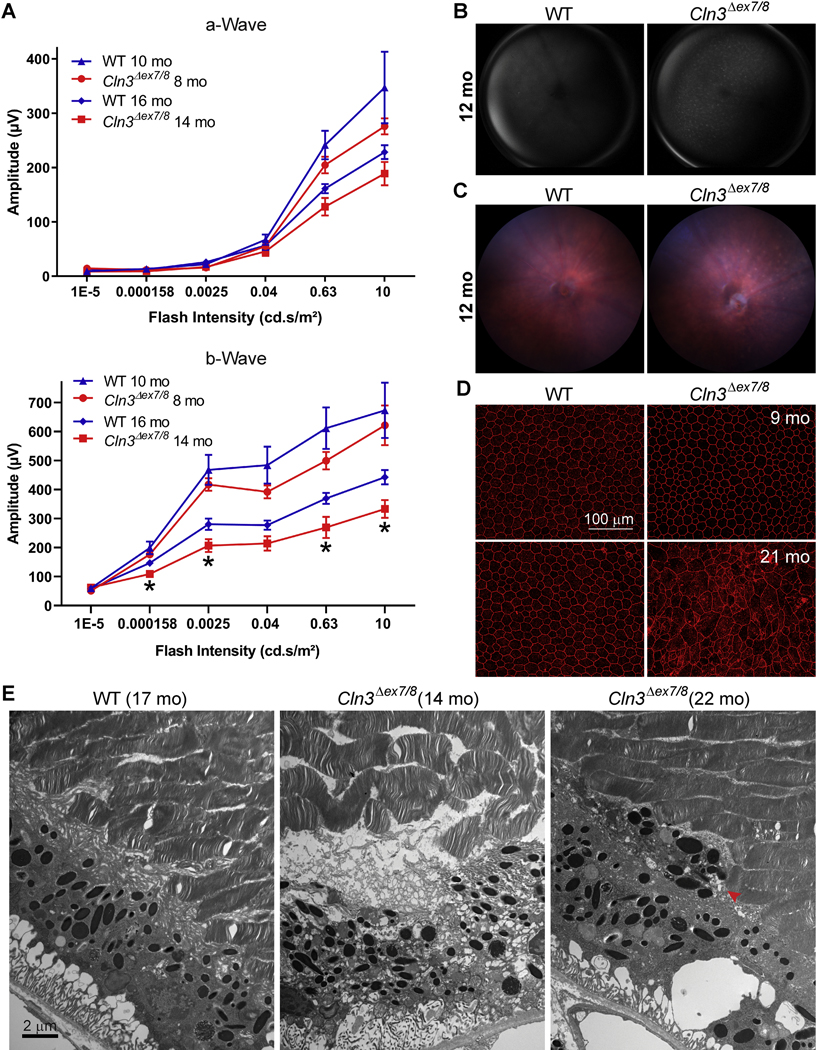
(A) Electroretinograms of 8- and 14-month old homozygous Cln3Δex7/8 mice showed vision impairment. As compared to WT, both a-wave and b-wave of homozygous Cln3Δex7/8 mice showed trends of reduction at both 8- and 14-month old of ages (e.g., p values were 0.23 and 0.17 for 0.63 cd.s/m2 at 8- and 14-month old of age, respectively), even though only the reduction in b-wave of 14-month old homozygous Cln3Δex7/8 mice was statistically significant (using unpaired, nonparametric Mann-Whitney test, with two-tailed p values <0.05; labeled by asterisks). Note that the light intensities were measured at the target area rather than at the light source. (B) Fundus autofluorescence imaging showed autofluorescent lesions in Cln3Δex7/8 but not WT mouse retinas at 12-month old. These autofluorescent lesions were visible in Cln3Δex7/8 mouse retinas at as early as 8-month old (data not shown). (C) Micron IV color fundus imaging showed diffuse hypo-pigmented punctate deposits consistent with subretinal drusen in Cln3Δex7/8 but not WT mouse retina at 12-month old. (D) ZO-1 staining of the fresh flat-mount RPEs showed RPE degeneration in 21-month (but not 9-month old) homozygous Cln3Δex7/8 (but not WT) mouse eyes. Scale bar: 100μm. (E) Electron micrographs showed RPE atrophies, including disintegrated RPE (middle panel) and RPE metaplasia/hyperplasia (right panel) in the 14- and 22-month old homozygous Cln3Δex7/8 mouse retina. The retina of a 17-month old WT mouse appeared normal (left panel). Scale bar: 2μm.
To address whether neurodegeneration was the only cause of vision loss in JNCL, we employed Fundus autofluorescence (FAF) imaging, which provides a density map of ocular fluorophores (predominantly lipofuscin) in the RPE [86, 87]. We observed diffuse autofluorescent lesions in the retina of Cln3Δex7/8 mice as young as 8-month old (data not shown); these retinal autofluorescent lesions were extensive in 12-month old Cln3Δex7/8 mice (Figure 1B). In addition, using Micron IV color fundus imaging, which can display subtler details of retinal degeneration as a result of its higher resolution, we observed diffuse hypo-pigmented lesions (Figure 1C). To determine if the retinal autofluorescent lesions seen by FAF imaging and the hypo-pigmented lesions seen by color fundus imaging were originated from ceroids/lipofuscins in the RPE, we examined autofluorescence in cryo cross sections of the Cln3Δex7/8 mouse retina using confocal fluorescence microscopy. As we reported in the companion Data in Brief article [82], detection of RPE autofluorescence was hindered by the extensive light absorption by the pigments in the melanosomes in the RPE, consistent with the literature [87]; rather, autofluorescent lesions were present mostly at the outer plexiform layer (OPL) and inner nuclear layer (INL) of Cln3Δex7/8 mouse retina, and positive for subunit C immunoreactivity, consistent with neural retinal dysfunction above. To characterize potential JNCL phenotype in RPE besides neural retina, we examined fresh flat-mount RPE isolated from both WT and Cln3Δex7/8 mice. Zonula occludens-1 (ZO-1) was used as a marker for RPE tight junctions. Immunostaining of 21-month old (but not 9-month old) Cln3Δex7/8 mouse retina with a ZO-1 antibody showed increased cell sizes, irregular cell shape, and discontinuous tight junctions, consistent with RPE atrophy and degeneration; in contrast, immunostaining of WT mouse retina with the same ZO-1 antibody showed regular hexagonal RPE monolayer regardless of age (Figure 1D).
We further performed electron microscopy to examine the ultrastructure of the Cln3Δex7/8 mouse retina. Toluidine blue (TB) stain of thick sections revealed drusen-like deposits in Cln3Δex7/8 retina, despite relatively normal photoreceptor and inner retinal layers (Supplemental Figure 2A). Ultrastructural images of thin sections showed a single RPE layer with organized POS in WT retina (Figure 1E, left). In contrast, disintegrated RPE (Figure 1E, middle; Supplemental Figure 2B, left) and RPE metaplasia/hyperplasia (Figure 1E, right; Supplemental Figure 2B, right) were present in Cln3Δex7/8 retina as early as 14-month old, even though POS were largely normal. In addition, double-membraned autophagosome-like structures filled with materials, including mitochondria and other vesicles, were present in the drusen-like deposits in the Cln3Δex7/8 retina (Supplemental Figure 2C). These data demonstrated RPE atrophy and degeneration in the Cln3Δex7/8 JNCL mouse model, consistent with frequent observations of RPE atrophy and degeneration in JNCL patients. Therefore, we focused the rest of this study on the roles of the JNCL disease gene CLN3 in the RPE.
2. CLN3 deficiency enhanced autophagy in RPE-1 cells
Our observations of double-membraned autophagosome-like structures in the drusenlike deposits in Cln3Δex7/8 retina (Supplemental Figure 2C) prompted us to investigate the roles of CLN3 in autophagy. Autophagy is an essential lysosomal degradation pathway that supplies cells with metabolites for energy production and anabolism, as well as clears unwanted or harmful intracellular materials, e.g., damaged mitochondria, protein aggregates, and oxidized lipids. CLN3 was found to be a component of the human autophagy proteinprotein interaction network [88] and a mutation in CLN3 (p.Gly165Glu) was associated with autophagic vacuolar myopathy [89]. We examined how loss of CLN3 alters autophagy in a human RPE cell line, RPE-1. To knock down CLN3 expression from RPE-1 cells, we tested three CLN3 siRNAs, and found that the one targeting exon 8 of CLN3 (labeled CLN3-Exon8, sequence 5′-GCC GUG AUC UCC UGG UGG U-UU-3’) was the most effective in decreasing the CLN3 protein levels in RPE-1 cells (Figures 2A–B). Of note, CLN3 protein migrated as a smeared band around 70 kDa (Figures 2A, Supplemental Figure 3B), which is upshifted compared with its theoretical molecular weight of 48 kDa. The CLN3 transcript levels were greatly reduced by the CLN3-Exon8 siRNA, as shown by both quantitative real-time polymerase chain reaction (qRT-PCR) (Supplemental Figure 4A) and RNA-Seq (Figure 4B and Supplemental Table 1). Autophagic flux was measured by two assays: 1) the protein levels of the cargo adaptor p62/SQSTM1 in the absence of lysosomal inhibitors and 2) the differences in the autophagosome membrane marker LC3II levels in the presence and absence of lysosomal inhibitors. Loss of CLN3 caused a reduction in p62/SQSTM1 protein levels in the absence of lysosome inhibitors (Figures 2A–C) without decreasing p62/SQSTM1 mRNA levels (Figure 4A and Supplemental Figure 4B), suggesting enhanced degradation of p62/SQSTM1. Treatment with the lysosomal inhibitor bafilomycin A1 (Baf A1), which inhibits autophagosome-lysosome fusion, drastically increased LC3II levels (Figure 2B, short exposure; quantified in Figure 2D), suggesting enhanced autophagosome biogenesis upon loss of CLN3. Together, these results indicate that loss of CLN3 caused enhanced autophagic flux in RPE-1 cells.
Figure 2. CLN3 deficiency led to increased autophagic flux in RPE-1 cells.

(A) The siRNA against CLN3 exon 8 (as compared to the siRNA against CLN3 exon 6 or exon 10, or non-targeting siRNA) was the most effective in reducing both CLN3 and p62 protein levels and in increasing LC3II levels. Of note, CLN3 protein ran as a 64–76 kDa band (Supplemental Figure 3). (B) Representative immunoblotting data showed that CLN3 siRNA treatment resulted in decreased p62 protein levels in the absence of bafilomycin A1 (Baf A1) and increased LC3II levels in the presence Baf A1 (200 nM, 18 h). (C) Quantification of immunoblotting results for p62 in the absence of Baf A1 showed decreased p62 protein levels upon CLN3 exon 8 siRNA (versus non-targeting siRNA) treatments. (D) Quantification of immunoblotting results for LC3II in the presence of Baf A1 showed increased LC3II protein levels upon CLN3 exon 8 siRNA (versus non-targeting siRNA) treatments. For both (C) and (D), cells were cultured under non-starved (open bars) or 18 h FBS-starved (closed bars) conditions. Statistics were carried out using two-way ANOVA and p values for comparing CLN3 exon 8 siRNA versus non-targeting siRNA were displayed. For p62, experiments were performed 5 times under non-starved conditions and 3 times under FBS-starved conditions, respectively. For LC3II, experiments were performed 4 times under non-starved conditions and 3 times under FBS-starved conditions, respectively.
Figure 4. CLN3 deficiency led to transcriptional up-regulation of the autophagy-lysosomal system in RPE-1 cells.

(A) RNA-Seq analysis showed that CLN3 siRNA treatment resulted in significant up-regulation of autophagy-related gene transcripts. Fold changes are either >1 (i.e., up-regulation) or <−1 (i.e., down-regulation) in this presentation. Out of 38 significantly transcriptionally altered autophagy-related genes (FDR<0.05), only 4 were down-regulated: Nrbf2 (a negative autophagy regulator in RPE-1 cells), Atg10, Atg4C and TECPR2. Unchanged controls are GAPDH, FBXO11 and PPIA/CYPA. Transcriptional alterations for selected genes were confirmed by qRT-PCR (t-test; ** indicates p<0.05; *, 0.05<p<0.105; see Supplemental Figures 4), following the procedures described in the Supplemental Methods and our previous work [178]. Targets of FOXO3 are labeled by (F). (B) RNA-Seq analysis showed that CLN3 siRNA treatment resulted in significant up-regulation of lysosome-related gene transcripts. Fold changes are either >1 (i.e., up-regulation) or <−1 (i.e., down-regulation) in this presentation. Out of 71 significantly transcriptionally altered lysosome-related genes (FDR<0.05), only 6 were down-regulated: CLN3 (the target of siRNA), GLA, GALC, NAGA, HYAL3, ATP6V1E1 and LMBRD2. Of note, CLN6 and CLN8 are ER-resident proteins. Unchanged controls are GAPDH, FBXO11 and PPIA/CYPA (same as shown in A). Transcriptional alterations for selected genes were confirmed by qRT-PCR (t-test; ** indicates p<0.05; *, 0.05<p<0.16; see Supplemental Figures 5). Targets of FOXO3 and TFEB are labeled by (F) and (T), respectively. (C) Representative images showing increased LysoTracker staining in CLN3 (as compared to non-targeting) siRNA-treated RPE-1 cells. Magenta: LysoTracker Red DND-99; blue: Hoechst. Images were taken for live cells after 48 h of treatment with 40 nM non-targeting or CLN3 exon 8 siRNA. (D) Quantification of LysoTracker Red DND-99 total intensity per cell for RPE-1 cells treated respectively with 20 nM, 40 nM and 100 nM non-targeting (open bars) versus CLN3 (closed bars) siRNA for 48 or 96 h. T-test; *, p<0.05; **, p<0.01.
3. CLN3 deficiency triggered autophagy-inducing signaling cascades in RPE-1 cells
We next investigated possible mechanism(s) causing enhanced autophagic flux upon loss of CLN3. mTORC1, when activated by nutrients (e.g., amino acids), promotes protein synthesis and inhibits autophagy. Activated mTORC1 promotes protein synthesis through phosphorylating the ribosomal protein S6 kinase (S6K) at the amino acid T389. Phosphorylated S6K was not detectible in CLN3 siRNA-treated RPE-1 cells, suggesting inactive mTORC1 (Figures 3A–B). We further examined the kinases that regulate mTORC1. AMPK, when activated by low energy state (indicated by phosphorylation at T172), inhibits mTORC1. Akt activation, as indicated by phosphorylation at T308 and S473, stimulates mTORC1. Treating RPE-1 cells with CLN3 siRNA led to phosphorylation of AMPK at T172 (Figures 3A & 3C) and dephosphorylation of Akt at both T308 and S473 (Figures 3A, 3D, 3E), suggesting that loss of CLN3 increases AMPK activity and decreases Akt activity, both of which are consistent with inactive mTORC1. Together, these results show that loss of CLN3 triggers signaling cascades that induce autophagy in RPE-1 cells.
Figure 3. CLN3 deficiency led to autophagy-inducing mTORC1/Akt suppression and AMPK activation in RPE-1 cells.

(A) Representative immunoblotting data showed that CLN3 siRNA treatment of RPE-1 cells resulted in decreased S6K phosphorylation at T389, decreased Akt phosphorylation at T308 and S473, and increased AMPK phosphorylation at T172. Total S6K, Akt and AMPK protein levels as well as actin levels were largely unchanged. (B) Quantification of immunoblotting results showed decreased phospho-S6K (T389)/total S6K ratio that suggests mTORC1 suppression in response to CLN3 exon 8 siRNA (versus non-targeting siRNA) treatments under both non-starved (open bars) and 18 h FBS-starved (closed bars) conditions. (C) Quantification of immunoblotting results showed increased phospho-AMPK (T172)/total AMPK ratio that suggests AMPK activation in response to CLN3 exon 8 siRNA (versus non-targeting siRNA) treatments under both non-starved (open bars) and 18 h FBS-starved (closed bars) conditions. Quantification of immunoblotting results showed decreased (D) phospho-Akt (T308)/total Akt and (E) phospho-Akt (S473)/total Akt ratios that suggest Akt suppression in response to CLN3 exon 8 siRNA (versus non-targeting siRNA) treatments under non-starved conditions. Four-six independent experiments were averaged. Statistics were carried out using t-test.
4. CLN3 deficiency up-regulated the autophagy-lysosomal system through global transcriptional reprogramming
To define additional mechanism(s) leading to enhanced autophagic flux upon loss of CLN3, we compared transcription profiles of CLN3 siRNA- and non-targeting siRNA-treated RPE-1 cells by RNA-Seq analysis. The Gene Set Enrichment Analysis (GSEA) [90] of the RNA-Seq data show that autophagy and lysosome-related gene ontology (GO) terms were significantly enriched among the up-regulated genes in CLN3 siRNA-treated RPE-1 cells (Table 1 and Supplemental Table 2). Significantly up-regulated autophagy genes in response to CLN3 deficiency included those encoding the core autophagy machinery (e.g., ULK1, BECN1, ATG14, WIPI2, ATG9A, MAP1BLC3B, MAP1BLC3B2, GABARAP, GABARAPL1, GABARAPL2/GATE16, EPG5) and autophagy adaptors (e.g., p62/SQSTM1, OPTN, TAX1BP1, NBR1, TOLLIP) (Figure 4A and Supplemental Table 1). qRT-PCR analyses confirmed the down-regulation of CLN3 and the up-regulation of autophagy genes (e.g., ULK1, BECN1, ATG14, VPS34/PIK3C3, VPS15/PIK3R4, WIPI2, ATG7, MAP1BLC3B, and GABARAP; Supplemental Figure 4).
Table 1.
GSEA analysis on the C5: GO Gene Set database (total 3600 genes) shows that the GO terms closely associated with the autophagy-lysosomal system are highly enriched in the up-regulated RNA-Seq data of CLN3 siRNA-treated RPE-1 cells. Out of the 309 gene sets that are positively regulated with an FDR q-value less than 5% (41 are with an FDR q-value of 0), 21 are directly associated with the autophagy-lysosomal system. The whole list of our GSEA analysis output is provided in Supplemental Table 2.
| Rank | GO NAME | SIZE | NES | FDR q-val |
|---|---|---|---|---|
| 1 (total 41) | AUTOPHAGOSOME | 71 | 3.31 | 0 |
| 1 (total 41) | LYSOSOMAL_LUMEN | 62 | 3.65 | 0 |
| 1 (total 41) | LYTIC_V AGUOLE | 362 | 4.97 | 0 |
| 1 (total 41) | LYTIC_VACUOLE_MEMBRANE | 209 | 3.86 | 0 |
| 1 (total 41) | VACUOLAR_LUMEN | 76 | 3.61 | 0 |
| 1 (total 41) | VACUOLAR_MEMBRANE | 459 | 4.59 | 0 |
| 1 (total 41) | VACUOLAR_TRANSPORT | 216 | 3.20 | 0 |
| 1 (total 41) | VACUOLE_ORGANIZATION | 140 | 3.56 | 0 |
| 49 | AUTOPHAGOSOME_MEMBRANE | 22 | 3.03 | 2.40E-05 |
| 59 | PRE_AUTOP HAGO SOM AL_STRU CTU RE | 27 | 2.93 | 1.39E-04 |
| 65 | PROTEIN_LOCALIZATION_TO_LYSOSOME | 17 | 2.86 | 3.45E-04 |
| 68 | AUTOPHAGOSOMEJDRGANIZATION | 35 | 2.82 | 4.17E-04 |
| 82 | PROTEIN_L OCALIZATI ON_T 0_V AGUOLE | 42 | 2.69 | 1.07E-03 |
| 90 | AUTOPHAGY | 300 | 2.65 | 1.31E-03 |
| 101 | LYSOSOMAL_TRANSPORT | 60 | 2.57 | 2.24E-03 |
| 166 | MAGROAUTOPHAGY | 201 | 2.27 | 1.14E-02 |
| 189 | ESTABLISHMENT_OF_PROTEIN_LOCALIZATION_TO_VACUOLE | 30 | 2.21 | 1.50E-02 |
| 200 | PRE_AUTOPHAGOSOMAL_STRUCTURE_MEMBRANE | 15 | 2.18 | 1.80E-02 |
| 234 | REGULATION_OF_AUTOPHAGY | 217 | 2.10 | 2.54E-02 |
| 269 | POSITIVE_REGULATION_OF_AUT OPHAGY | 71 | 2.02 | 3.56E-02 |
| 275 | LYTIC_VACUOLE_ORGANIZATION | 43 | 2.01 | 3.75E-02 |
RNA-Seq and qRT-PCR analyses further indicated that loss of CLN3 significantly upregulated lysosomal genes, including some genes that are mutated in other forms of NCL − CLN1/PPT1, CLN2/TPP1, CLN5, CLN10/CTSD, CLN11/GRN and CLN13/CSTF; in contrast, CLN6 and CLN8, two NCL genes which encode ER membrane resident proteins [37], were down-regulated in the CLN3-deficient RPE-1 cells (Figure 4B, Supplemental Table 1, and Supplemental Figure 5). Significantly up-regulated lysosomal genes in CLN3 (vs. nontargeting) siRNA-treated RPE-1 cells also included lysosomal hydrolases (e.g., peptidases, proteases, glycosidases, lipases, sulfatases, nuclease), vacuolar ATP synthase, and other lysosomal membrane proteins (Figure 4B, Supplemental Table 1 and Supplemental Figure 5). These results suggest that CLN3 deficiency-ensued lysosomal gene up-regulation is a specific cellular response as opposed to a general decay/dying of cells. Similar conclusion was drawn when analyzing the RNA-Seq data using CategoryCompare2 [91].
We further performed Ingenuity Pathway Analysis (IPA) upstream analysis of the RNA-Seq data. FOXO3 is a forkhead box O family transcription factor that is sufficient and necessary for inducing autophagy in skeletal muscles [92, 93] and in haematopoietic stem cells [94]. We found that FOXO3 was significantly activated in CLN3-deficient RPE-1 cells, based on the expression pattern of its target genes (activation z-score 2.6, p value 3.1E-13); and the up-regulated FOXO3 target genes include autophagy genes ULK1, BECN1, SH3GLB1/Bif-1, MAP1BLC3B, GABARAPL1, TP53INP1, and BNIP3L/NIX as well as lysosome gene CTSB (Figure 4A–B and Supplemental Table 1). Moreover, TFEB, the master transcription factor of autophagy and lysosome biogenesis [95], was also significantly activated in CLN3-deficient RPE-1 cells (activation z-score 4.3, p value 1.6E-09); and the upregulated TFEB target genes include ARSA, ARSB, ATP6V0C, ATP6V1A, ATP6V1B2, CLCN7, CTSA, CTSB, CLN10/CTSD, CLN13/CTSF, GBA, GNS, HEXA, LAMP1, MCOLN1, NAGLU, PSAP, SGSH, TMEM55B and CLN2/TPP1 (Figure 4B and Supplemental Table 1).
We next asked if transcriptional up-regulation of the autophagy-lysosomal system led to an increase in degradative compartments in CLN3-deficient RPE-1 cells. To address this, we stained siRNA-treated cells with LysoTracker Red DND-99. LysoTracker staining was significantly increased on a per cell basis in CLN3 (vs. non-targeting) siRNA-treated RPE-1 cells (Figures 4C–D), suggesting more acidic and/or greater numbers of late endosomes and lysosomes. The observed transcriptional up-regulation of the autophagy-lysosomal system (in part due to FOXO3 and TFEB activation) and increase in acidity and/or numbers of degradative compartments in response to CLN3 deficiency are consistent with and likely contributed to increased autophagic flux.
5. CLN3 deficiency impaired energy metabolism in RPE-1 cells
Autophagy plays important roles in metabolism, e.g., providing fuels for energy metabolism, recycling building blocks for anabolism, and conducting mitochondrial quality control. Our observations of autophagy induction upon CLN3 deficiency (Fig. 2), concomitant AMPK activation (a response to low energy) and mTORC1 inhibition (a response to low energy and amino acid insufficiency) (Fig. 3), and transcriptional up-regulation of the autophagy-lysosomal system (Fig. 4) further support a link between autophagy and metabolism. Therefore, we investigated whether and how loss of CLN3 altered energy metabolism in RPE-1 cells.
5.1. CLN3 deficiency impaired glycolysis/lactic fermentation
We first investigated whether and how glycolysis/lactic fermentation (Fig. 5A) is affected by loss of CLN3. Seahorse XF bioenergetic analysis uses 1) oxygen consumption rate (OCR) as a real-time measurement of cellular oxygen level changes resulting primarily from mitochondrial respiration and OXPHOS, and 2) extracellular acidification rate (ECAR) as a real-time measurement of changes of medium acidification due to proton extrusion that correlates with lactic fermentation from glucose. We applied the Seahorse XF Glycolysis Stress Test to non-targeting and CLN3 siRNA-treated RPE-1 cells assayed in glucose/pyruvate-deprived medium. During the test, glucose (10μM), oligomycin A (1μM; a mitochondrial ATP synthase inhibitor), and 2-deoxy-D-glucose (2-DG, 100μM; a glucose analog whose metabolite blocks phosphoglucose isomerase) were sequentially injected (Fig. 5B). Our data show that loss of CLN3 reduced glucose-stimulated ECAR regardless of OXPHOS inhibition by oligomycin (i.e., glycolysis and glycolytic capacity, respectively) (Fig. 5B–C), suggesting that CLN3 impaired glycolysis/lactatic fermentation.
Figure 5. Loss of CLN3 led to decreased glycolysis from Seahorse and Stable-Isotope Resolved Metabolomics analyses.

(A) The glycolysis pathway with enzymes whose transcripts significantly changed (measured by RNA-Seq) upon CLN3 deficiency labeled. Red, up-regulation; blue, down-regulation; grey, unchanged. HK, hexokinase; PGI, phosphoglucose isomerase; PFK, phosphofructokinase; TPI, triosephosphate isomerase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PK, pyruvate kinase; LDH, lactate dehydrogenase. (B-C) Representative Seahorse glucose stress test showed that CLN3 siRNA treatment reduced extracellular acidification rate (ECAR) in RPE-1 cells under both non-starved and 18 h FBS-starved conditions. (C) Quantification of (B). Basal ECAR is the rate before glucose injection. Glycolysis is defined as the difference between the maximum rate upon glucose injection and the basal ECAR. Glycolytic capacity (Cap) is defined as the difference between the maximum rate after oligomycin injection and the basal ECAR. Glycolytic reserve (Res) is defined as the difference between Glycolytic capacity and Glycolysis. Two-way ANOVA test was performed and all non-targeting versus CLN3 comparisons were significant with p<0.0001. (D-F) 1H NMR spectra of growth media at 24 h after 13C6-glucose labeling, as compared to right before labeling, showed that CLN3-deficient RPE-1 cells had decreased (D) 13C6-glucose uptake, and decreased production and/or excretion of (E) 13C3-lactate and (F) 13C3-pyruvate. (G) 1H{13C}-HSQC NMR spectra of growth media at 24 h after 13C6-glucose labeling, as compared to right before labeling, showed that CLN3-deficient RPE-1 cells had decreased production and/or excretion of 13C3-alanine. (H) 1H NMR analysis of RPE-1 cell polar extracts showed increased levels of unlabeled and 13C-labeled glucose upon CLN3 deficiency. (I-O) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency decreased glycolysis/lactatic fermentation without reducing glucose 6-phosphorylation: (I) glucose 6-phosphate (G6P), (J) fructose 1,6-bisphosphate (F1,6BP), (K) 1,3-bisphospho-glycerate (1,3BPG), (L) 2-phophoglycerate/3-phosphoglycerate (2PG/3PG), (M) phosphoenolpyruvate (PEP), (N) pyruvate, and (O) lactate. m1-m5 indicate singly, doubly, triply, quadruply, and quintuply 13C-labeled isotopologues, respectively. (P) 1H NMR analysis of RPE-1 cell polar extracts showed decreased levels of unlabeled and 13C-labeled alanine upon CLN3 deficiency. Metabolites amounts were normalized by protein amounts of corresponding samples. Due to limit of resources, all SIRM experiments were performed with duplicated labeled samples but only single unlabeled sample for either non-targeting or CLN3 exon-8 siRNA treatment. Thus for all SIRM quantification results here (D-P) and hereafter, means and standard errors were only plotted for labeled samples.
SIRM utilizes stable-isotope tracing via a combination of high resolution NMR and MS to simultaneously track various metabolic pathways via isotopically enriched metabolites. We used 13C6-glucose as the tracer and SIRM to directly measure the altered metabolic network in CLN3 siRNA- and non-targeting siRNA-treated RPE-1 cells as well as the corresponding growth media. We first analyzed media samples. The 1H NMR spectra show that 13C6glucose levels in the media 24 h after labeling (red and black spectra) versus those before labeling (light red and grey spectra), were reduced for both CLN3-deficient and control cells (Supplemental Fig. 6A), indicating 13C6-glucose consumption from the media. Less 13C6-glucose was consumed from the 24 h media for the CLN3-deficient cells than that for the control cells (Fig. 5D and Supplemental Fig. 6A), suggesting reduced 13C6-glucose uptake upon CLN3 deficiency. This result was confirmed by 1H{13C}-HSQC NMR analysis (Supplemental Fig. 6C–D). Of note, 13C6-glucose uptake was reduced upon CLN3 deficiency despite transcriptional up-regulation of glucose transporter 3 (GLUT3/SLC2A3, the higher affinity and high capacity glucose transporter [96]) as shown by RNA-Seq (Supplemental Table 1). Moreover, both 1H (Fig. 5E and Supplemental Fig. 6B) and 1H{13C}-HSQC (Supplemental Fig. 6C,E) NMR analyses show lower levels of 13C3-lactate in the 24 h media of the CLN3-deficient cells versus the control cells, suggesting decreased lactatic fermentation and/or excretion upon CLN3 deficiency. NMR analyses of the media also showed decreased production and/or excretion of 13C3-pyruvate (Fig. 5F and Supplemental Fig. 6F). This was further confirmed by 1H{13C}-HSQC NMR analysis showing the decreased production and/or excretion of 13C-alanine (Fig. 5G), which is a direct transamination product of pyruvate. Together, these results from analyzing the media suggest overall decreased glycolysis in CLN3-deficient RPE-1 cells.
We then analyzed the polar fractions from the cell extracts. Both 1H (Fig. 5H) and 1H{13C}-HSQC (Supplemental Fig. 7A) NMR analyses of the polar cell extracts show surprisingly higher intracellular 13C6-glucose in the CLN3-deficient RPE-1 cells than in control cells, despite reduced 13C6-glucose consumption upon CLN3 deficiency (Fig. 5D and Supplemental Fig. 6A,C,D). Consistent with this result, analyses of these polar cell extracts using Ion Chromatography Mass Spectrometry (ICMS) show that loss of CLN3 increased intracellular incorporation of 13C into glucose 6-phosphate (G6P, Fig. 5I). As 13C6-glucose uptake was reduced upon CLN3 deficiency, CLN3-deficiency-ensued increase in intracellular 13C6-glucose and 13C6-G6P must result from decreased utilization of G6P. ICMS analyses further show that loss of CLN3 decreased intracellular incorporation of 13C into fructose 1,6-bisphosphate (Fig. 5J), 1,3-bisphosphoglycerate (Fig. 5K), 3- and/or 2-phosphoglycerate (Fig. 5L), phosphoenolpyruvate (PEP; Fig. 5M), pyruvate (Fig. 5N), and lactate (Fig. 5O). Also both 1H (Fig. 5P) and 1H{13C}-HSQC (Supplemental Fig. 7B) NMR analyses show decreased intracellular incorporation of 13C into alanine (thus pyruvate) upon loss of CLN3. In addition, RNA-Seq data show transcriptional down-regulation of hexokinase 1 (HK1), phosphofructokinase, platelet (PFKP), triosephosphate isomerase 1 (TPI1), phosphoglycerate kinase (PGK), and enolase 2 (ENO2) (Fig. 5A and Supplemental Table 1), consistent with reduced labeling of glycolysis/lactatic fermentation metabolites. Together, our data show that CLN3 deficiency impairs glycolysis/lactatic fermentation, leading to concurrent intracellular accumulation of glucose and G6P, and reduction in other intracellular glycolysis intermediates and glucose consumption from media.
5.2. CLN3 deficiency impaired mitochondrial TCA cycle and OXPHOS
We next examined the mitochondrial function in CLN3-deficient RPE-1 cells by monitoring the TCA cycle and OXPHOS activity. 13C3-pyruvate, generated from 13C6-glucose through glycolysis, can enter into the TCA cycle via either 13C2-acetyl-CoA or 13C3-oxaloacetate (Fig. 6A). ICMS analyses of polar cell extracts reveal that levels of 13C2-labeled TCA cycle metabolites, including the m2 isotopologues of citrate (Fig. 6B), cis-aconitate (Fig. 6C), α-ketoglutarate (Fig. 6D), succinate (Fig. 6E), fumarate (Fig. 6F) and malate (Fig. 6G), decreased upon CLN3 deficiency. These results are consistent with decreased 13C2-acetylCoA production from 13C3-pyruvate via the action of pyruvate dehydrogenase (PDH) and reduced citrate synthase (CS) activity. ICMS analyses further reveal that CLN3 siRNA-treated RPE-1 cells had reduced levels of 13C5-citrate (Fig. 6B), 13C3-fumarate (Fig. 6F), 13C3-malate (Fig. 6G), and 13C3-aspartate (Fig. 6H), consistent with reduced 13C3-oxaloacetate production via an ATP-dependent anaplerotic reaction catalyzed by pyruvate carboxylase (PC). Consistent with ICMS results, NMR analyses also show decreased levels of fumarate (Supplemental Fig. 7C) and aspartate (Fig. 6I and Supplemental Fig. 7D) in both unlabeled and labeled CLN3-deficient cells. In addition, RNA-Seq data show transcriptional downregulation of the TCA cycle enzymes, including CS, aconitase 2 (ACO2), isocitrate dehydrogenase 2 (IDH2), oxoglutarate dehydrogenase (OGDH), succinyl-CoA ligase (SUCLA2, SUCLG1, SUCLG2), fumarate hydratase (FH), and PC (Fig. 6A and Supplemental Table 1), consistent with reduced 13C incorporation from 13C6-glucose into the TCA cycle metabolites. We also observed decreased PC protein levels in CLN3 siRNA-treated RPE-1 cells (Fig. 6J), further supporting reduced carbon flux through pyruvate carboxylase in CLN3-deficient RPE-1 cells. Together, these results show that regardless of the route for pyruvate entry, CLN3 deficiency impairs the TCA cycle.
Figure 6. Loss of CLN3 led to impaired TCA cycle from Seahorse and Stable-Isotope Resolved Metabolomics analyses.

(A) The TCA cycle with enzymes whose transcripts significantly changed (measured by RNA-Seq) upon CLN3 deficiency labeled. Red, up-regulation; blue, down-regulation; grey, unchanged. PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; CS, citrate synthase; ACO2, aconitase 2; IDH2, isocitrate dehydrogenase 2; OGDH, oxoglutarate dehydrogenase; SUCLA2, succinyl-CoA ligase ADP-forming β subunit; SUCLG1/2, succinyl-CoA ligase α and GDP-forming β subunits; SDH, succinate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase. (B-H) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency impaired TCA cycle: (B) citrate, (C) cis-aconitate, (D) α-ketoglutarate (αKG), (E) succinate, (F) fumarate, (G) malate, and (H) aspartate. m1-m6 indicate singly, doubly, triply, quadruply, quintuply and sextuply 13C-labeled isotopologues, respectively. (I) 1H{13C}-HSQC NMR spectra of the polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells had decreased intracellular levels of 13C3-aspartate. Metabolite amounts were normalized to protein amounts of corresponding samples. Due to limited resources, the SIRM experiment only included single unlabeled sample and duplicated labeled samples for either non-targeting or CLN3 exon-8 siRNA treatment. For all SIRM quantification results (B-I), means and standard errors were plotted for labeled samples. (J) An immunoblot showed reduction of PC protein levels upon CLN3 deficiency in RPE-1 cells. Due to the discontinuation of the only available working CLN3 antibody (see Figure 2) by the manufacturer, loss of Nrbf2 was used as an indicator of CLN3 deficiency. (K-L) Representative Seahorse mitochondrial stress test showed that CLN3-deficiency led to decreased mitochondrial basal respiration, ATP production and maximal respiration in RPE-1 cells under both non-starved and 18 h FBS-starved conditions. (L) Quantification of (K). ATP production is reflected by the decrease in OCR upon inhibiting mitochondrial ATP synthase by oligomycin A. Spare respiration capacity is reflected by the increase in OCR from basal levels upon uncoupling mitochondrial ATP synthase by FCCP. Non-mitochondrial respiration is the minimal OCR upon inhibiting Complex III cytochrome bc1 and Complex I NAD:ubiquinone oxidoreductase by antimycin A and rotenone, respectively. Basal respiration is the OCR before oligomycin injection, with non-mitochondrial OCR subtracted. Maximal respiration is the maximal OCR in the presence of the uncoupler FCCP, with nonmitochondrial OCR subtracted. The unpaired, two-tailed t-test was used to compare nontargeting with CLN3 siRNA treated samples: **, p<0.002; ****, p<0.0001. (M) An immunoblot showed reduction of ATP5B protein levels upon CLN3 deficiency in RPE-1 cells. The same samples as those in Figure 2B were used for this blot.
Utilizing FADH2 and NADH generated from the TCA cycle, mitochondrial OXPHOS produces ATP through a five-member electron transport chain. To monitor mitochondrial OXPHOS, we performed Seahorse XF Cell Mito Stress Test, using DMEM-based XF modified media containing 25 mM glucose and 1 mM pyruvate, with subsequent addition of oligomycin A (1μM), FCCP (0.5μM), antimycin A and rotenone (1μM each). We found that loss of CLN3 greatly reduced ATP-producing oxygen consumption without any significant change in spare respiration capacity (Fig. 6K–L). Therefore, the Mito Stress Test results indicate that CLN3 deficiency likely impaired mitochondrial ATP synthase. Consistently, ATP5B/ATP5F1B, the gene encoding the β subunit of the mitochondrial ATP synthase catalytic core F1 was transcriptionally down-regulated as shown by RNA-Seq (Supplemental Table 1), and the corresponding ATP5B protein levels were modestly decreased (Fig. 6M) in CLN3 siRNA-treated RPE-1 cells. Together, this set of results support impaired mitochondrial metabolism, including both the TCA cycle and OXPHOS, upon CLN3 deficiency.
5.3. CLN3 deficiency led to decreased ATP levels in RPE-1 cells
Due to impaired glycolysis (Fig. 5) and mitochondrial TCA cycle and OXPHOS (Fig. 6), we predicted reduced cellular ATP levels upon CLN3 deficiency. 1H{13C}-HSQC and 1H NMR analyses of the polar cell extracts showed decreased intracellular incorporation of 13C into AXP (i.e., AMP, ADP and/or ATP; Fig. 7A) and decreased unlabeled AXP (Fig. 7B) respectively upon loss of CLN3. ICMS analyses of these polar cell extracts further showed that the levels of 13C5-ATP, 13C5-ADP and 13C5-AMP were all reduced in CLN3-deficient RPE1 cells (Fig. 7C–E); in contrast, although total ADP and ATP levels decreased, total AMP levels increased in CLN3-deficient RPE-1 cells (Fig. 7F), consistent with increased AMP/ATP ratio and AMPK activation (Fig. 3). As the depletion of labeled AXP (primarily 13C5-AXP) in response to loss of CLN3 (Fig. 7C–E) concurred with reduced levels of 13C5-ribulose/ribose 5phosphate (Fig. 7G), this 13C5-AXP response was at least in part due to reduced 13C incorporation into the pentose phosphate pathway (PPP) intermediate 13C5-ribulose/ribose-5phosphate and subsequently 13C5-labeled AXP during de novo nucleotide synthesis. This was consistent with the depletion of 13C-ribose-AXP (AXP-1’ in Fig. 7A) evident in the 1H{13C}-HSQC NMR data of CLN3-deficient RPE-1 cells. Taken as a whole, our data clearly showed that loss of CLN3 in RPE-1 cells led to impaired energy and anabolic metabolism by attenuating glycolysis, the TCA cycle and OXPHOS, as well as PPP and de novo nucleotide synthesis.
Figure 7. Loss of CLN3 led to decreased intracellular ATP levels.

(A) 1H{13C}-HSQC NMR spectra of polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells had depleted intracellular levels of 13C-labeled AXP (i.e., AMP, ADP and/or ATP) with labeling at position 2, 8 and 1’. The inset shows the atom positions of AMP. (B) 1H NMR spectra of polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells had reduced intracellular levels of unlabeled AXP when detected at positions 2, 8 and 1’. (C-E) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency led to reduced intracellular (C) 13C5-ATP, (D) 13C5-ADP and (E) 13C5-AMP. (F) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency led to reduced intracellular total ATP and ADP levels but increased intracellular total AMP levels. (G) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency led to reduced intracellular 13C5-ribulose-5-phosphate and/or 13C5-ribose-5-phosphate. m1-m7 indicate singly, doubly, triply, quadruply, quintuply, sextuply, and septuplet 13C-labeled isotopologues, respectively. Metabolite amounts were normalized by protein amounts of corresponding samples. Owing to limited resources, the SIRM experiment only included single unlabeled sample and duplicated labeled samples for either non-targeting or CLN3 exon-8 siRNA treatment. For all SIRM quantification results (A-G), means and standard errors were plotted for labeled samples.
6. CLN3 deficiency led to glycogen accumulation in RPE-1 cells.
As JNCL is a lysosomal storage disease and impaired autolysosomal degradation was reported in Cln3Δex7/8 mouse livers [56] and Cln3Δex1−6 mouse RPE [33], we were also particularly interested in identifying metabolites that accumulated in response to CLN3 deficiency. In addition to primary lysosomal storage of autofluorescent subunit C [97–100], JNCL is also known to accumulate lipids, particularly sphingolipids including ceramides, sphingomyelins (SM), galactosylceramides (GalCer), glucosylceramides (GluCer), and globosides in fibroblasts [64, 101, 102]. We analyzed the lipid-containing nonpolar cell extracts by direct infusion Fourier-transform ion cyclotron resonance mass spectrometry (FTICR-MS). Our data showed less fractional enrichment of even-number 13C-labeled SM, triacylglycerols (TAG), and phospholipids (e.g., phosphatidylcholines (PC) and phosphatidylethanolamines (PE)) in the CLN3 siRNA-treated RPE-1 cells than in nontargeting siRNA-treated cells (Supplemental Fig. 8, “Even”). These data suggested 1) reduced 13C2-acetyl-CoA incorporation into fatty acids (i.e., impaired lipid synthesis), 2) enhanced usage of non-glucose (unlabeled) carbon source(s) for lipid synthesis, and/or 3) block in lipid (primarily unlabeled) hydrolysis and/or fatty acid β oxidation upon loss of CLN3. The latter two effects are more consistent with lipid accumulation in JNCL.
Glycogen accumulation has been observed in glycogen storage diseases (GSDs) (reviewed in [103]). Lafora bodies, aggregates of abnormal form(s) of glycogen, has been definitively demonstrated to be the cause of neurodegeneration and neurobehavioral phenotypes in Lafora disease [104, 105]. However, to the best of our knowledge, there is no report on whether or not glycogen is accumulated in JNCL. We used 1H and 1H{13C}-HSQC NMR to determine the levels of intact glycogen particles in the polar extracts of RPE-1 cells treated with CLN3 or non-targeting siRNAs. Glycogen is a polymer of glucose units joined by α−1,4-glycosidic linkages with evenly distributed branches via α−1,6-glycosidic linkages, forming a largely water-soluble spherical-like structure with newly-added glucose units constitute a mobile hydrophilic surface [104, 106]. Both 1H and 13C NMR have long been used to detect intact glycogen in vitro and in vivo [107–109]. We showed a large buildup of unlabeled intact glycogen (Fig. 8A–B; the 1H resonance at ~5.4 ppm) in the polar extracts of CLN3 siRNA-treated RPE-1 cells, consistent with impairment in glycogen mobilization upon loss of CLN3.
Figure 8. Loss of CLN3 led to glycogen accumulation.

(A) 1H NMR spectra of polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells (in red; solid and dash traces for the duplicated samples) had increased intracellular levels of un-labeled glycogen than non-targeting siRNA-treated control cells (in black; solid and dash traces for the duplicated samples). (B) 1H NMR spectra of polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells had increased intracellular levels of unlabeled glycogen. (C) The glycogen metabolism with enzymes whose transcripts significantly changed (measured by RNA-Seq) upon CLN3 deficiency labeled. Red, up-regulation; blue, down-regulation; grey, unchanged. (D) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency led to increased levels of intracellular 13C6-glucose-1-phosphate (G1P), the product of glycogen phosphorylase-catalyzed glycogen breakdown, the substrate of UDP-glucose pyro-phosphorylase-catalyzed synthesis of UDP-glucose from G1P and UTP, and a metabolic intermediate that interconverses with G6P. (E) 1H{13C}-HSQC NMR spectra of polar extracts of the cells at 24 h after 13C6-glucose labeling showed that CLN3-deficient RPE-1 cells had depleted intracellular 13C-labeled UDP-glucose (UDP-Glc). (F) ICMS analyses of RPE-1 cell polar extracts showed that CLN3 deficiency led to increased levels of intracellular 13C6-UDP-glucose/galactose (UDP-Glc/Gal; labeling likely at glucose/galactose), but reduced levels of intracellular 13C11-UDP-Glc/Gal (labeling likely at both glucose/galactose and UDP ribose ring) and 13C5-UDP-Glc/Gal (labeling likely at UDP ribose ring). Metabolites amounts were normalized by protein amounts of corresponding samples. Due to limit of resources, the SIRM experiment only included single unlabeled sample and duplicated labeled samples for either non-targeting or CLN3 exon-8 siRNA treatment. For all SIRM quantification results (B, D-F), means and standard errors were plotted for labeled samples.
We next measured glycogen metabolism intermediates to gain further understanding of the effects of CLN3 deficiency on glycogen metabolism (Fig. 8C). ICMS analyses showed that 13C6-glucose 1-phosphate (G1P) accumulated but unlabeled G1P depleted upon CLN3 deficiency (Fig. 8D). As G1P is the product of both glycogen synthesis and degradation, these data suggest suppression of both glycogen synthesis downstream of G1P and glycogen degradation via glycogenolysis, respectively. Attenuated glycogen synthesis is further supported by the depletion of 13C-labeled (Fig. 8E–F) and unlabeled (Fig. 8F) UDPglucose, the direct precursor of glycogen synthesis, based on both 1H{13C}-HSQC NMR (Fig. 8E) and ICMS (Fig. 8F) analyses. ICMS analyses also showed that CLN3 deficiency led to increased m6 isotopologue but decreased m5 and m11 isotopologues of UDP-glucose (Fig. 8F), which respectively represent changes in the 13C6-glucose, 13C5-ribose, and 13C6-glucose + 13C5-ribose moieties of UDP-glucose [110]. These changes reflected those in the precursors of UDP-glucose, i.e., 13C6-glucose (Fig. 5H and Supplemental Fig. 7A) and 13C5ribulose/ribose 5-phosphate (Fig. 7G). Moreover, RNA-Seq analysis showed transcriptional down-regulation of UGP2 (encoding UDPG pyrophosphorylase which catalyzes the formation of UDP-glucose from G1P and UTP during glycogenesis) and PYGL (encoding liver glycogen phosphorylase, which catalyzes dephosphorylation of glycogen during glycogenolysis) (Fig. 8C and Supplemental Table 1), consistent with reduced UDP-glucose synthesis (Fig. 8E–F) and glycogenolysis (Fig. 8D), respectively, which in turn may lead to the buildup of unlabeled glycogen (Fig. 8A–B) and opposite changes in labeled and unlabeled G1P levels (Fig. 8D).
Discussion
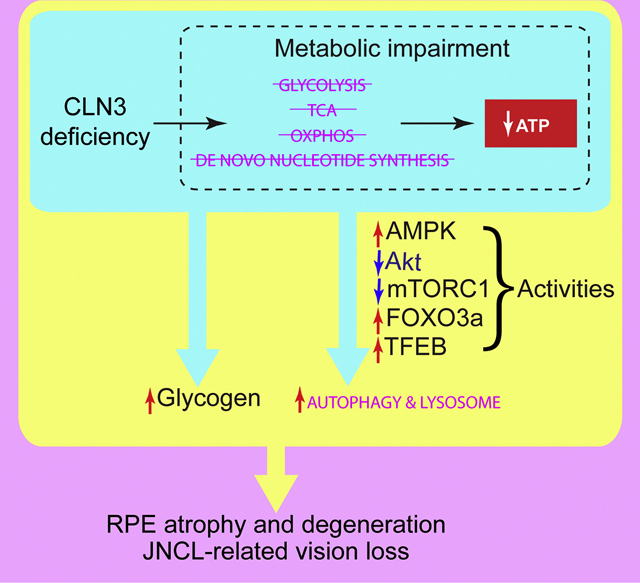
In this work, we characterized the effects of CLN3 deficiency on retinal function and morphology, as well as RPE cell biology, focusing on autophagy-lysosomal system and metabolism. JNCL mutant mice showed impaired retinal function, subunit C immunoreactive autofluorescent lesions in the OPL/INL, and RPE atrophy and degeneration. We further demonstrated in human RPE-1 cells that CLN3 deficiency led to enhanced autophagy and lysosomal system, and coincidently impaired energy metabolism and anabolism. In addition, CLN3 deficiency led to glycogen accumulation and possible lipid accumulation. Based on these results, we propose the following working model for JNCL-related vision loss (Fig. 9): CLN3 deficiency in RPE results in 1) an energy crisis and compensatory induction of autophagy, and 2) reduced utilization of glycogen and lipids as well as their accumulation; despite not knowing whether energy crisis or storage is the primary defect, these defects lead to RPE atrophy and degeneration, contributing to retinal degeneration and vison loss in JNCL.
Figure 9. Working models of how CLN3 deficiency in RPE may lead to metabolic impairment and autophagy induction.

(A) CLN3 deficiency-ensued energy deprivation may trigger signaling cascades and transcriptional activations that enhance the autophagy-lysosomal system (see Section 2 in the Discussion). Enhancement and reduction shown by our data are labeled by red and blue arrows, respectively. Our data are consistent with the literature [56, 93, 125–134]. (B) Is energy deprivation or glycogen/lipid storage the primary cause of JNCL phenotype? We observed impaired ATP synthesis and de novo synthesis (red arrows) as well as reduced flows of other metabolites (black arrows) in CLN3 siRNA-treated RPE-1 cells. We also observed glycogen accumulation. CLN3 deficiency-ensued energy deprivation may lead to JNCL phenotype including glycogen accumulation; alternatively, CLN3 deficiency may lead to glycogen accumulation which is toxic (like in the case of Lafora disease) and causes JNCL phenotype including metabolic impairment and energy deprivation.
1. Besides neurodegeneration, RPE atrophy and degeneration likely also contribute to JNCL-related vision loss
Although retinal phenotype in Cln3Δex7/8 mice has been examined previously, these earlier studies were confounded by genetic backgrounds, e.g., an outbred 129Sv/Ev/CD1 mixed background that carried unspecified CLN3-independent retinal degenerative genetic loci [1, 79], C57B6/N [79], a background that is known to carry the retinal degenerative Rd8 mutation [84], and an unspecified C57B6 background tested positive for the Rd8 mutation [80, 81]. We bred Cln3Δex7/8 mice on C57B6/J background which did not carry either Rd8 or Rd1 mutation [82]. Our study using these mice confirmed previously reported differential loss of scotopic ERG signals (with the b-wave amplitudes affected more than the a-wave amplitudes) in Cln3Δex7/8 mice on the above-mentioned confounding backgrounds. This ERG phenotype observed in Cln3Δex7/8 mice is consistent with the observations in Cln3Δex1−6 mice on C57B6/J background [78] and in JNCL patients [18]. The consensus in the ERG phenotype across different species, mouse models and genetic backgrounds suggests that differential loss of scotopic ERG signals is a strong phenotype of CLN3 deficiency. As the b-waves are generated by bipolar cells, prominent loss of the scotopic b-wave amplitudes suggest that bipolar cells are likely one of the retinal cell types that are affected early in the development of JNCL-related vision impairment. Unlike the consensus in the JNCL ERG phenotype, although extensive photoreceptor loss was reported in the original Cln3Δex7/8 mice on the outbred 129Sv/Ev/CD1 mixed background [1], we did not observe overt photoreceptor loss in Cln3Δex7/8 mice on C57B6/J background [82]. Our result is consistent with the reported photoreceptor phenotype in Cln3Δex1−6 mice also on C57B6/J background [78]. Therefore, extensive photoreceptor loss observed in the original Cln3Δex7/8 mice [1] likely reflected a mouse genetic background effect.
We observed diffuse autofluorescent (by FAF imaging) and hypo-pigmented (by color fundus imaging) lesions in the fundus of the CLN3Δex7/8 mice as early as 8-month old when visual function (e.g., as assessed by ERG) was barely affected. Our observations suggest that these fundoscopic features can serve as an early marker for JNCL-related vision impairment and that FAF and color fundus imaging can provide simple, sensitive, and noninvasive tools for monitoring JNCL eye pathologies. In accordance with this idea, autofluorescent lesions were previously reported in the fundus of 8–10 week old CLN3Δex7/8 mice (on C57B6/J background) carrying the light-sensitive RPE65 leucine variant [111] as well as the fundus of 16-month old Cln3Δex1−6 mice [77, 112] by the BluePeak Blue laser autofluorescence (BAF) method.
FAF provides a density map of ocular fluorophores, predominantly lipofuscin in the RPE [86, 87]. RPE autofluorescence was reported to either not decrease in Cln3Δex1−6 mice [78] or decrease in JNCL patients [113], the latter of which likely reflecting the degeneration of affected cells. Therefore, we postulated that autofluorescent lesions in the fundus of the CLN3Δex7/8 mice as seen by FAF imaging might originate from RPE. However, our cross-section images of CLN3Δex7/8 mouse retina showed predominant distribution of autofluorescent lesions in the OPL/INL; and these autofluorescent lesions were immunoreactive for subunit C, suggesting that they were JNCL storage materials [82]. These data are in accordance with bipolar cell dysfunction as indicated by the prominent loss of the scotopic b-wave amplitudes, as bipolar cells, together with horizontal cells, amacrine cells and photoreceptor pedicles, reside and form a dense network of synapses in the OPL/INL. Thus our study points to early JNCL defects at least in part in the bipolar cells and their communication with photoreceptors and other retinal neurons. Future studies are needed to identify the specific cell type(s) that harbor autofluorescent lesions in the OPL/INL and functional defects of these cells, so as to understand of the etiology of JNCL-related vision loss. To this end, our preliminary studies show close proximity of JNCL autofluorescent lesions to PNA-stained cone pedicles and CHX10 immunoreactive bipolar cell nuclei [82],
We did not observe high RPE autofluorescence in the cryo cross sections of mouse retinas, likely because detection of RPE autofluorescence was hindered by the extensive light absorption by the pigments in the melanosomes in the RPE [87]. However, we observed disorganized ZO-1 immunoreactivity in the flat mount RPE samples, as well as RPE metaplasia/hyperplasia and degeneration. These observations, together with others’ reports of RPE abnormalities in both JNCL patients [3, 16, 17] and Cln3Δex1−6 mice [33, 112], clearly demonstrated that besides neurons, RPE was also affected by CLN3 deficiency in vivo. Our in vitro studies in RPE-1 cells further revealed extensive alterations in the autophagy-lysosome system and metabolism in response to CLN3 siRNA treatment. Most recently, fingerprint lysosomal storage material that is positive for subunit C, and accumulation of autolysosomes and un-fused phagosome-lysosomes have been reported in the RPE of the CLN3Δex1−6 [112] JNCL mouse model [33]. As RPE plays critical roles in retinal physiology including providing growth factors and glucose to the neural retina (see Introduction), it is possible that impairment in RPE functions due to CLN3 deficiency may result in inadequate support of neural retina (even prior to pathologies of RPE itself becomes apparent), contributing to degeneration of retinal neurons including bipolar cells and ultimate vision loss in JNCL. Future studies such as those using RPE- and bipolar cell-specific CLN3-deficient mouse models are needed to further dissect the roles of individual retinal cell types in JNCL-related vision loss.
2. Enhancement of the autophagy-lysosome system as a compensatory stress response to impaired energy metabolism upon CLN3 deficiency
As discussed below, our results of enhanced autophagy-lysosomal system in the CLN3-deficient RPE-1 cells, despite consistent with numerous reported observations, are in discordance with JNCL being a lysosomal storage disease (see Section 2.1). Our results also show extensive metabolic impairment in the CLN3-deficient RPE-1 cells. As autophagy supplies cells with metabolites for energy production and anabolism, it is likely that CLN3 deficiency-ensued metabolic impairment boosts the autophagy-lysosome system as a compensatory stress response (see Section 2.2). Adopting the same strategy, it has recently been shown that overexpressing TFEB, the master transcription factor of the autophagy-lysosomal system, reversed the CLN3-deficient phenotypes in JNCL mouse models [114].
2.1. Autophagy-lysosomal system plays important roles in RPE; and JNCL leads to extensive autophagy-lysosomal dysregulation.
Autophagy plays important roles in virtually all cell types in the eye (reviewed in [115]), including the lens [116, 117], photoreceptors, and RPE [30, 32, 118]. Autophagy dysregulation is involved in numerous eye diseases (reviewed in [115]), including AMD – a major cause of vision loss among the elderly which initially affects RPE [119–121]. RPE cells are important phagocytic cells, engulfing 5% of POS on a daily basis. POS phagocytosis in RPE cells is a key step during the visual cycle. Autophagy plays critical roles in POS phagocytosis in RPE cells: RPE autophagy appears to follow a bimodal pattern of daily light-dark cycle in response to ingestion of POS [122]; a noncanonical autophagy process termed LC3-associated phagocytosis has been reported to be essential for POS phagocytosis [32, 123]. Deficient expression of autophagy genes in mouse RPE leads to not only impaired POS phagocytosis and visual cycle (e.g., in Atg5flox/flox;Best1-Cre/+ [32]), but also 1) RPE cell hypertrophy and vacuolization (e.g., in Atg7flox/flox;Tyr-Cre/+ [124]); 2) accumulation of p62/SQSTM1, ubiquitin-tagged debris, mitochondria, oxidatively-damaged proteins, and autofluorescent lesions; 3) RPE and secondary photoreceptor degeneration; and 4) impaired ERG (e.g., in Rb1cc1flox/flox;Best1-Cre/+ [30]).
CLN3, the protein mutated in JNCL, appears to be a component of the autophagy protein-protein interaction network [88]. A mutation in CLN3 (p.Gly165Glu) has been associated with autophagic vacuolar myopathy [89]. These studies suggest a role for CLN3 in autophagy and potential contribution of autophagy dysregulation to JNCL pathologies. Our data show that CLN3 siRNA-treated RPE-1 cells displayed increased autophagic flux, autophagy induction signaling (as indicated by Akt/mTORC1 suppression and AMPK activation), and transcriptional up-regulation of autophagy and lysosome genes including many targets of the FOXO3 and TFEB transcriptional factors. As discussed below, these results are not only in accordance with numerous reports but also self-consistent (Fig. 9A). For example, down-regulation of mTORC1 activity, which we observed in the CLN3 siRNA-treated RPE-1 cells, was previously shown in JNCL patient lymphoblastoid cells, homozygous Cln3Δex7/8 mouse brain, and CbCln3Δex7/8 cells, a cerebellar neuronal precursor cell model of JNCL [56]. AMPK is primarily activated by increased AMP:ATP ratio [125]. AMPK activation is known to induce autophagy, through 1) phosphorylating autophagy proteins ULK1 (S317 and S777) [126, 127] and Beclin 1 (S91/S94) [128], 2) phosphorylating TSC2 and Raptor thus suppressing mTORC1, a kinase complex that phosphorylates ULK1 (S757) and inactivates the ULK1 complex [127], and 3) phosphorylating FOXO3a in the nucleus thus resulting in transcriptional activation of TFEB via the transcriptional suppression of SKP2 and subsequently stabilization and increased levels of CARM1 protein, the transcriptional co-activator for TFEB [129, 130]. Moreover, mTORC1 phosphorylates TFEB, blocking it from entering the nucleus to activate transcription [131]. Therefore, our observations of ATP shortage and AMP accumulation, mTORC1 inhibition, AMPK activation, transcriptional up-regulation of FOXO3 and TFEB target genes, and autophagic flux increase in CLN3 siRNA-treated RPE-1 cells are consistent with the interplays among these molecules (Fig. 9A). FOXO3 [132] and mTORC1 are parallel pathways that are downstream of Akt to modulate the autophagy-lysosome system, with Akt suppressing autophagy-lysosomepromoting FOXO3 and activating autophagy-lysosome-inhibiting mTORC1 [93, 133]. Akt is phosphorylated (at T308 and S473 respectively) and activated by PDK1 and mTORC2 [134]. Therefore, our observations of enhanced FOXO3 activation and suppression of Akt, mTORC1 and mTORC2 in CLN3 siRNA-treated RPE-1 cells are also self-consistent (Fig. 9A). In addition, our observation of transcriptional up-regulation of autophagy and lysosome genes in CLN3 siRNA-treated RPE-1 cells is also consistent with increased levels of LAMP1 protein in cultured astrocytes from Cln3Δex7/8 mice [114], increased lysosomal proteins CLN1/PPT1, CLN2/TPP1, DPP7, GAA and ATP6AP1 in CbCln3Δex7/8 cells [55], and increased cathepsin D immunoreactivity in the RPE and OPL/INL of aged or light-exposed young, RPE65 leucine variant-carrying Cln3Δex7/8 mice [111]. Our results also agree with the notion that accumulation of lysosomal proteins and associated elevation in the activity of lysosomal enzymes is a common feature of lysosomal storage diseases (Burditt et al., 1980; Young et al., 1997). In addition, our observation of significantly increased total intensity of LysoTracker observed in CLN3 siRNA-treated RPE-1 cells is consistent with transcriptional up-regulation of the autophagy-lysosomal system and increased autophagic flux. This result is also in agreement with previously reported increase in LysoTracker staining with prolonged treatment of lysosomotropic compounds [135–137] and in other lysosomal storage diseases such as Mucolipidosis type IV [138] and Niemann-Pick disease type C1 [139, 140]. It is thought that such increase of LysoTracker staining likely reflects a compensatory attempt of cells to overcome lysosomal dysfunction [135].
Although CLN3-deficiency-elicited enhancement in the autophagy-lysosomal system was observed by us and others (see above), compromised autophagy-lysosomal system in the absence of CLN3 has also been reported. For example, lysosomal accumulation of storage materials (e.g., lipofuscin and subunit C [97–100]) occurs in JNCL. Treatment with inhibitors of lysosomal proteases recreates certain features of the pathology of Batten disease, resulting in the accumulation of lipofuscin-like dense bodies in neurons (Ivy et al., 1984). JNCL lysosomes displayed a 69.8% decrease in the activity of the v-ATPase compared with their matching controls [47]. LysoTracker staining [51] and levels of the mature cathepsin D heavy chain [39] in homozygous CbCln3Δex7/8 cells were reduced compared to those in wild-type controls. Impaired autolysosomal degradation has also been reported in the homozygous Cln3Δex7/8 knock-in mouse livers [56] and Cln3Δex1−6 mouse RPE [33]. Most recently, a lysosomal proteome analysis revealed decreased levels of numerous lysosomal proteins including lysosomal enzymes that catalyzes the degradation of lipids and glycans in CbCln3Δex7/8 cells [55]. These reports suggest that dysfunction of one or more aspects of lysosomal biology may contribute to JNCL pathogenesis [112].
There are several possible explanations for the discrepancies regarding whether the autophagy-lysosomal system is enhanced or compromised in the absence of CLN3. It is possible that: 1) dysfunction of one or more specific aspects concurs with overall enhancement of the autophagy-lysosomal system; 2) cell-type specific effects of CLN3 deficiency may occur for cerebellar granule neurons and RPE cells; 3) CbCln3Δex7/8 cell line may have acquired secondary mutation(s); and 4) different autophagy assays (e.g., EM [33, 56] versus the biochemical assays used in our study) may reveal different aspects of autophagy. Future studies are needed to resolve these discrepancies. In addition, the effects of CLN3 deficiency on LC3-associated phagocytosis in RPE is yet to be investigated.
2.2. RPE and retina are highly metabolically active; and JNCL leads to impaired energy metabolism, likely triggering compensatory autophagy.
Like brain or tumor tissue, the retina is one of the most metabolically active tissues in vertebrates [27]. Energy metabolism in the retina is highly important for visual functions, as shown by attenuated ERG signals in response to removal of glucose or O2 in both vascular [141–143] and avascular [144] retina. Retinal ischemia, i.e., shortage in nutrient (e.g., glucose) or O2 supply, is evident in most retinal diseases including glaucoma, artery or vein occlusions, diabetic retinopathy, and AMD [27].
The retina is highly glycolytic, converting 80–96% of glucose it consumes into lactate [145–147]. The majority of aerobic glycolysis occurs in the outer retina, i.e., the photoreceptors and RPE [148–150]. Pyruvate kinase (PK) catalyzes the last step in glycolysis to convert PEP to pyruvate. Deficiency in PKM2, the predominant isoform of PK expressed in photoreceptors, in mouse rods or cones leads to their degeneration [151, 152]. This suggests that glycolysis is essential for photoreceptor health and normal vision.
Earlier reports also suggest that mitochondrial metabolism is essential for retinal health and normal vision. For example, conditional knocking-out of mitochondrial pyruvate carrier 1 (MPC1; using Six3-Cre) in the developing neural retina of mice led to degeneration of both photoreceptors and inner retinal neurons [153]. Genetic disruptions that caused loss of CS or PDH in Drosophila resulted in light-induced photoreceptor degeneration [154]. Loss-of-function mutation of IDH3B, the gene encoding the β subunit of the NAD-specific TCA cycle key enzyme isocitrate dehydrogenase, caused photoreceptor degeneration and retinitis pigmentosa [155]. Moreover, RPE utilizes the TCA cycle or reductive carboxylation for energy production via consuming lactate [148] and proline [156, 157]. Suppression of mitochondrial metabolism [28] or chronic HIF2α-mediated metabolic stress [29] in RPE cells caused RPE dysfunction and photoreceptor degeneration.
Despite growing knowledge about RPE and neural retinal metabolism, how they are involved in JNCL-related vision loss is unclear. As discussed above, a defect in PKM2, MPC1, CS, PDH, or IDH3B alone led to retinal degeneration [151–155], our observations of CLN3 deficiency-induced defects in glycolysis, and the mitochondrial TCA cycle and OXPHOS likely underlie RPE atrophy and degeneration in JNCL. The reduced ATP levels in CLN3-deficient RPE-1 cells is consistent with earlier findings in fibroblasts [158], CbCln3Δex7/8 cells [39], and Cln3Δex7/8 astrocytes [39, 159]. The reduced ATP levels without a decrease in spare respiratory capacity further suggests a defect in ATP synthesis. Indeed, we found transcriptional suppression of ATP5B and the resulting attenuation in the ATP5B protein levels in CLN3 siRNA-treated RPE-1 cells. As ATP5B is required for proper assembly of ATP synthase, our finding of CLN3 deficiency-ensuded attenuation in the ATP5B protein levels and ATP levels in RPE-1 cells are self-consistent and in accordance with previously reported JNCL mitochondrial abnormalities including 1) reduced mitochondrial ATP hydrolysis in JNCL skin fibroblasts [160]; 2) lysosomal accumulation of subunit C in JNCL brains [98, 100], Cln3Δex7/8 mouse brains [39, 51, 56], and Cln3Δex7/8 mouse RPE [33]; and 3) significant downregulation of genes associated with mitochondrial bioenergetics, including those encoding cytochrome b, cytochrome oxidase, and mitochondrial ATP synthase F0 sub-complex subunit B, in the whole eyes (but not the cerebellum) of the Cln3Δex1−6 mice prior to the appearance of lysosomal deposits [161]. Also, abnormal mitochondrial elongation and decreased cellular ATP levels were reported to precede subunit C accumulation in CbCln3Δex7/8 cells [39], suggesting that decreased ATP levels may be an earlier event leading to other events such as enhancement of the autophagy-lysosomal system and cell death during the course of JNCL (Fig. 9A–B). In addition, our observation of defective mitochondria, which may trigger mitophagy, is consistent with autophagy induction.
3. Storage as a cause or a consequence of CLN3 deficiency-ensued energy crisis
As stated above, JNCL is known to accumulate autofluorescent subunit C [97–100] and sphingolipids [55, 64, 101, 102]. However, our lipidomic analysis revealed reduced 13C fractional enrichment in fatty acyl chains of lipids (e.g., SM, TAG, PC and PE) in the CLN3 siRNA-treated cells than in control cells (Supplemental Fig. 8, “Even”). This result is consistent with the depletion of 13C2-citrate (Fig. 6B) and -cis-aconitate (Fig. 6C) in the CLN3 siRNA-treated cells, suggesting reduction in 13C2-acetyl-CoA incorporation into fatty acids thus impaired lipid synthesis upon loss of CLN3. In contrast, fractional enrichment of 13C-labeling in the glycerol backbone of TAG, PC and PE was unchanged upon loss of CLN3 (Supplemental Fig. 8, m3 isotopologues). Of note, we observed negligible m3 isotopologue in SM, consistent with lack of a glycerol backbone in SM (Supplemental Fig. 8 m3 isotopologue). Therefore, our SIRM analyses suggested impaired fatty acyl chain synthesis despite reported lipid accumulation [55, 64, 101, 102]. Although our RNA-Seq analysis showed global transcriptional up-regulation of the autophagy-lysosomal system, several key (glycol)sphingolipid metabolism genes were drastically down-regulated in CLN3-deficient RPE-1 cells (Supplemental Table 1). For example, the transcript of GALC, the gene encodes a lysosomal galactosylceramidase that hydrolyzes the galactose ester bonds of GalCer, psychosine, lactosylceramide (LacCer) and monogalactosyldiglyceride, was largely down-regulated. This result is consistent with decreased galactocerebrosidase protein levels reported in CbCln3Δex7/8 cells [55]. In addition, GALC mutations have been associated with Krabbe disease (aka. globoid cell leukodystrophy), with human pathologies similar to those of JNCL, including blindness, seizures, ataxia, and psychomotor decline [162]; and the dog model of GALC mutations also developed blindness [162]. Therefore, defects in lipid hydrolysis may contribute to lipid accumulation; likewise, impaired TCA cycle that we observed in CLN3-deficient RPE-1 cells may contribute to lipid accumulation as a result of defective fatty acid β oxidation by the TCA cycle (Fig. 9B). In addition, RPE cells not only phagocytose POS which is rich in lipids but also transport dietary lipids from blood vessels into the retina. Thus RPE cells require robust lipid handling capabilities and are likely to be sensitive to impaired lipid handling. Future investigation, including absolute lipid quantification, is needed to determine which aspect(s) of lipid handling are impaired upon CLN3 deficiency.
This work is the first to report glycogen accumulation upon CLN3 deficiency. A previous report using electron microscopy stated an increase in the storage of glycogen granules in the CLN3 siRNA-treated Hela cells without showing either quantification or micrographs of control cells [163]. Glycogen accumulation, which is characteristic of glycogen storage diseases (GSDs) (reviewed in [103]), may result from increased glycogenesis and/or defective glycogen utilization. Our results of reduced levels of both unlabeled and labeled UDP-glucose (Fig. 8E–F), and transcriptional down-regulation of UGP2 (Fig. 8C and Supplemental Table 1) argue against increased glycogenesis, despite the large accumulation of unlabeled glycogen (Fig. 8A–B). In contrast, decreased glycogenolysis is consistent with the transcriptional down-regulation of PYGL (Fig. 8C and Supplemental Table 1), leading to depletion of unlabeled G1P (Fig. 8D). Taken together, despite decreased glycogenesis, glycogen still accumulated in CLN3-deficient RPE-1 cells, likely due to impaired glycogenolysis. Future experiments are needed to further determine whether there is intracellular glycogen accumulation in JNCL patients and mouse models. As Lafora bodies have been demonstrated to be the cause of neurodegeneration and neurobehavioral phenotypes in Lafora disease [104, 105], in the future, it is of great importance to investigate whether glycogen accumulated upon loss of CLN3 is also toxic and contributes to neurodegeneration and vision loss in JNCL.
Although we observed impaired energy metabolism and glycogen accumulation in CLN3-deficient RPE-1 cells (Figs. 5–8), our results cannot distinguish whether impaired energy metabolism or storage is more upstream in the development of JNCL etiology (Fig. 9B). It is possible that defects in glycolysis, the TCA cycle, and/or OXPHOS may result in reduced production of ATP and other metabolites, leading to cell death, akin to retinal degradation resulting from a defect in PKM2, MPC1, CS, PDH, or IDH3B as we discussed above [151–155]. As such, lipids and glycogen storage is a consequence of ATP shortage that reduces consumption of lipids and glycogen. Conversely, it is possible that toxic storage of lipids and glycogens, rather than shortage of ATP or other essential metabolites, may lead to cell death in JNCL, as occurred in Lafora disease [104, 105]. Even if accumulated lipids and glycogen are not toxic, degrading these storage materials may over-consume ATP and result in a detrimental energy crisis. It is also possible that both ATP shortage and accumulation of toxic materials contribute to cell death in JNCL. Future investigations are needed to fully understand the causal relationship between storage and impaired production ATP and other metabolites, and their contributions to JNCL pathologies.
In summary, this work provides detailed characterization of the JNCL retinal phenotype, including retinal pathologies and visual function impairment in a mouse model as well as molecular and cellular deficits in a human RPE cell line. Our data suggest a possible contribution of RPE atrophy and degeneration to JNCL-related vision loss. Our work is the first study on the effect of CLN3 deficiency on the catastrophic disruption of RPE cell metabolism, establishing the importance of CLN3 for cell metabolism. Our work is also the first to report glycogen accumulation upon CLN3 deficiency. What we learned from RPE will likely facilitate our understanding of JNCL-related vision loss and neurodegeneration. Our study will shed light on developing novel therapeutics for treating JNCL, and potentially other NCL and lysosomal storage diseases, through targeting cell metabolism.
Experimental Procedures
Mouse strains and genotyping
All animal experiments were approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC) and were in accordance with the Association for Research in Vision and Ophthalmology Statement for the use of Animals in Ophthalmic and Visual Research. C57BL/6J wild-type (WT; 000664) and JNCL mutant B6.129(Cg)Cln3tm1.1Mem/J (Cln3Δex7/8; 017895, on the C57BL/6J background) mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and aged at the University of Kentucky under standard housing conditions with a 14:10 hour light-dark cycle. WT and Cln3Δex7/8 mutant mice were genotyped as described in the companion Data in Brief article [82] with WT primers (oIMR3012, 5’-CAC CTT CCT CTC ACT GAC TGC-3’; oIMR3013, 5’-ACC ACC ATG AGA TCA CAG CA-3’; ~145 bp band) and mutant primers (5’-GCC TTT ACT TGC TGC CTT ACA-3’; 5’-GGG TCT CGG TGC CTA TGA-3’; ~750 bp band).
Like the reported Cln3Δex1−6 mice [78], the Cln3Δex7/8 mice that we used in this study were on the C57B6/J background. We also genotyped the mice for the potential presence of the retinal degeneration Pde6b (Rd1) mutation and the Crb1 (Rd8) mutation [82]. Our genotyping results excluded either of these mutations in the Cln3Δex7/8 mice used in our study (data not shown). Therefore, retinal atrophy and degeneration that we observed in the Cln3Δex7/8 JNCL mouse model used in our study did not result from either Rd1 or Rd8 mutation. In contrast, the Cln3Δex7/8 mice previously reported by others were either on a C57B6/N background [79] that is known to have the retinal degenerative Rd8 mutation [84] or on an unspecified background that was tested positive for the Rd8 mutation [80, 81]. Moreover, none of the previous reports [78–81] examined the Rd1 mutation.
Electroretinography
Mice were dark adapted overnight before assessing the scotopic ERG response. Experiments were conducted with the aid of dim red lighting. Following anesthesia (i.p., ketamine hydrochloride (100 mg/kg body weight) combined with xylazine hydrochloride (10 mg/kg body weight)), the eyes were dilated using 1% tropicamide (Akorn Inc., IL) and 2.5% phenylephrine hydrochloride (Alcon Laboratories, TX). Mice were placed on a pre-warmed heating pad with both eyes at a constant relative position inside the Ganzfeld globe (Espion full field Ganzfeld; Diagnosys LLC, USA). Silver recording electrodes were placed over the corneas, a reference skin electrode at the base of the head, and a ground electrode at the base of the tail. Electrodes were impedance matched between both eyes. The eyes were kept moist using methylcellulose solution (Goniovisc; Sigma Pharmaceuticals, IA). The eyes were then exposed to flashes of light with increasing intensities (1E5, 0.000158, 0.0025, 0.04, 0.63 and 10 cd·s/m2; measured at the target area rather than at the light source), triggered using the Espion software (Espion v3) with a preset automatic protocol. Responses from multiple flashes (19, 13, 9, 6, 5 and 3 flashes for 1E-5, 0.000158, 0.0025, 0.04, 0.63 and 10 cd.s/m2, respectively) were averaged to obtain a single waveform at each light intensity.
Statistical analyses were performed using PRISM 7 (GraphPad, CA). All results were expressed as mean ± standard error of the mean (SEM). Differences between groups were compared using an unpaired, non-parametric Mann-Whitney test, with two-tailed p-values <0.05 considered statistically significant.
Fundus autofluorescence and photography
Fundus autofluorescence was imaged from unanesthetized mice using the TRC 50-IX imaging system with standard filters (excitation filter 536/40 nm and barrier filter 650/100 nm; Topcon, Tokyo, Japan) linked to a digital color camera (Sony, Tokyo, Japan). Mice were then anesthetized and their eyes were dilated as described above. Color images of the mouse fundus were taken using a Micron IV retinal imaging microscope (Phoenix Research Labs, CA, USA). Images were exported as TIFF files and processed identically using Adobe Photoshop.
RPE and retinal morphologies
Mouse eyes were enucleated after euthanization by CO2. RPE/choroid flat mounts were prepared and fixed with 4% paraformaldehyde for 30 min. Following washes with PBS for 3 times, the flat mounts were blocked for 1 h in 3% bovine serum albumin (BSA) and 4% goat serum at 4°C. Flat mounts were then incubated overnight with anti-ZO-1 antibody (Invitrogen 61–7300, rabbit pAb) followed by washing and 1 h incubation with fluorescent secondary antibody (Invitrogen A-11072, F(ab’)2-goat anti-rabbit IgG (H+L) cross-adsorbed, Alexa Fluor 594). For staining nuclei of RPE flat mounts, Hoechst 33342 (Invitrogen, H3570) was incubated for 5 min. RPE flat mounts were then imaged on an Acousto-Optical Beam Splitter SP5 confocal microscope (AOBS SP5, Leica GmbH, Germany). RPE and retinal morphologies were also assessed by Toluidine blue stain of cross sections of resinembedded retinas (see Supplemental Methods) and by differential interference contrast microscopy and confocal fluorescent microscopy of cross sections of cryo-preserved retinas (see the companion Data in Brief article [82]).
Transmission electron microscopy of retina
Mouse eyes were enucleated after euthanization, fixed with 4% paraformaldehyde/3.5% glutaraldehyde, post-fixed in 2% osmium tetroxide, dehydrated in ethanol, and embedded in resin. Ultrathin (80–90 nm) sections were cut and stained first with uranyl acetate (2% w/v in 70% ethanol) for 5 min and then lead citrate (0.4% w/v in boiled ddH2O cooled to room temperature and basified by adding 1/10 v/v of 1 M NaOH to avoid lead carbonate precipitation) for 2 min. Electron micrographs were collected on a transmission electron microscope (BioTwin 12, Phillips).
Cell culture
RPE-1 cells were authenticated by ATCC to be an exact match to the ATCC cell line CRL-4000 (hTERT RPE-1), using Short Tandem Repeat (STR) analysis [164, 165]. Cells were cultured in DMEM supplemented with 10% FBS in a cell culture incubator set at 37°C and with 5% CO2. Cells (6–8×105) were suspended to 3 ml of DMEM medium supplemented with 10% FBS without antibiotics before reverse transfected with 40 nM of non-targeting control siRNA, human CLN3 exon-6, exon-8, or exon-10 siRNA using Opti-MEM® I Reduced Serum Medium and Lipofectamine RNAiMAX (Invitrogen), following the manufacturer’s instructions. The siRNAs ON-TARGETplus non-targeting siRNA pool was purchased from Dharmacon (D-001810–10-20). The sequences of siRNAs against the human CLN3 gene were: 5′-GUG GGA UUU GUG CUG CUG GAA-UU-3’ (against exon-6), 5′-GCC GUG AUC UCC UGG UGG U-UU-3’ (against exon-8) and 5′-CCA GCC UCU CCC UUC GGG AAA-UU-3’ (against exon-10), with the corresponding target sequences underscored. The cell-siRNA mixtures were seeded onto 6-cm dishes. Cells were cultured for 3–4 days before harvesting. For Baf treatment, one day before harvesting, cells were washed with room temperature 1× PBS and cultured in freshly added DMEM either with or without 10% FBS in the presence or absence of 200 nM Baf for 18 h.
Antibodies and immunoblot analysis
Anti-CLN3 (ab87438, rabbit pAb; currently discontinued) was purchased from Abcam. Amnong many tested, this was the only commercial anti-CLN3 antibody that we verified to be able to recognize a 64–76 kDa band that was greatly suppressed in CLN3 siRNA-treated RPE-1 cells. Anti-p70 S6 kinase (2708, rabbit mAb), phospho-p70 S6 kinase (at T389; 9205, rabbit pAb), Akt (9272S, rabbit pAb), phospho-Akt (at S473; 4060S, rabbit mAb), phosphoAkt (at T308; 2965, rabbit mAb), AMPK (2532S, rabbit pAb) and phospho-AMPK (at T172; 2535S, rabbit mAb) were purchased from Cell Signaling. Anti-Nrbf2 (A301–852A, rabbit pAb) was purchased from Bethyl Laboratories (Montgomery, TX). Guinea pig anti-p62 antibody (03-GP62-C) was purchased from American Research Products (Waltham, MA). LC3–2G6 (0260–1000, mouse mAb) was purchased from nanoTools Antiköerpertechnik (Teningen, Germany). Anti-rabbit IgG-HRP (sc-2004) and anti-mouse IgG-HRP (sc-2005) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Peroxidase-rabbit anti-guinea pig IgG (H+L) was purchased from Invitrogen. Anti-β-actin (A5441, mouse mAb) was purchased from Sigma.
Cells were washed with ice-cold PBS and harvested using scrapers. Cell pellets were lysed with 1x RIPA buffer (Santa Cruz Biotechnology, Inc.) supplemented with Pierce halt protease and phosphatase inhibitor cocktail (78442, Thermo Fisher Scientific). Cell lysates were centrifuged at 13,000 x g for 5 minutes and supernatants were used for immunoblot analyses on an equal protein basis. For probing LC3, cell lysates were run on NuPAGE™ 12% BisTris gels (NP0343BOX, Invitrogen) using MOP (NP0001, Invitrogen) or MES (NP0002, Invitrogen) buffer. For probing all other proteins, cell lysates were run on NuPAGE™ 4–12% BisTris gels (NP0323BOX, Invitrogen) using MOP buffer. Proteins were transferred to methanol-pre-activated PVDF membranes using a Mini Trans-Blot apparatus (Bio-Rad) that was run at 4°C and 30 V overnight in NuPAGE™ transfer buffer (NP00061, Invitorgen), supplemented with NuPAGE™ antioxidant (NP0005, Invitrogen), 20% methanol, and 0.025% SDS. PVDF membranes were then incubated with primary antibodies at 4°C overnight, followed by secondary antibodies at the room temperature for 1 h. Chemiluminescent signals were detected by SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34096), except for β-actin, which was detected by SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34087). Films were exposed for various time periods and scanned using positive mode to ensure that the protein levels could be quantified within a linear range. The protein levels were quantified using ImageJ and normalized to either β-actin or, for phospho-proteins, corresponding total proteins.
RNA-Seq
Three independent batches of RPE-1 cells were reversely transfected with 40 nM non-targeting and CLN3 exon-8 siRNA, respectively, and harvested 72 h post-transfection. Total RNA was extracted from freshly harvested cells using TRIzol™ reagent (Thermo Fisher Scientific, 15596018). Preliminary QC of total RNA samples was done using a NanoDrop microvolume spectrophotometer. Total RNA samples with A260/A280 or A260/A230 ratios less than 1.8 were cleaned by RNeasy Mini Kit (QIAGEN, 74104). Final QC of total RNA samples was run on an Agilent 2100 Bioanalyzer. All samples utilized for RNA-Seq were with A260/A280>2.03, A260/A230>1.98, 28s/18s>1.9, and RIN>7.4. RNA-Seq library preparations, next generation sequencing, and quality control steps were performed by the UT Southwestern Genomics and Microarray Core. Total RNAs were reverse transcribed and barcoded using a TruSeq™ RNA Sample Prep Kit v2 (RS-122–2001 or RS-122–2002 kit). Next-generation sequencing (paired-end, 100-bp read length) of the bar-coded cDNA libraries was carried out on an Illumina Hiseq1000/2000, with 10 randomized samples per lane.
For data analysis, low quality reads and reads containing adapters were removed from the raw sequencing data. FastQC [166] was used to investigate RNA sequencing quality. Reference transcripts were extracted using Ensembl GRCh38 v82 annotation gtf file. Transcriptome alignment was carried out using STAR [167]. Gene abundance was estimated using RSEM [168]. We performed differential expression analysis using edgeR [169], comparing CLN3 siRNA with non-targeting siRNA control.
Significantly up/down-regulated genes were determined using a FDR < 0.05. Functional analyses were performed using Ingenuity Pathway Analysis (IPA®, QIAGEN, Redwood City, www.qiagen.com/ingenuity). The gene set enrichment analysis (GSEA) was performed using the GSEA software (http://www.broad.mit.edu/gsea/) and the gene sets in the Molecular Signature Database (MSigDB) [90].
GO enrichment was also carried out using CategoryCompare2 [91] for transcripts with an adjusted p-value (FDR) <0.01. GO terms for each ENSEMBL transcript were extracted from the org.Hs.eg.db Bioconductor annotation resource [170], with ENSEMBL as the keytype, and returning all GO annotations for each (columns GOALL, EVIDENCEALL, and ONTOLOGYALL). For each of the up- and down-regulated gene sets, hypergeometric statistics were calculated. P-values were adjusted using a Benjamini-Hochberg correction [171]. GO terms with an FDR <0.001 and >2 genes annotated were considered significant and reported.
High content imaging
For LysoTracker® Red (LTR) imaging, 3,000 RPE-1 cells were seeded in a 96-well plate and treated with 20 nM, 40 nM or 100 nM non-targeting or CLN3 exon-8 siRNA for 48 h or 96 h, followed by LysoTracker® Red DND-99 staining (Thermo Fisher Scientific, Waltham, MA). The resultant images (>6 images or more per well) were automatically captured and quantified using ArrayScan XTI (Thermo Fisher Scientific, Waltham, MA). Quantification of fluorescence intensity was conducted using cell health profiling algorithm.
Seahorse XF bioenergetic analyses.
Seahorse analyses were done following the procedures described in [172] with minor modifications. In brief, RPE-1 cells were reverse transfected with non-targeting siRNA and CLN3 exon-8 siRNA, respectively, and seeded at ~5×105 (for 4-day growth) or ~1×106 (for 3-day growth) per 10-cm plate in DMEM media (Cellgro 10–013-cv) containing 10% FBS. The day before Seahorse analyses, cells were split into a 96-well plate at a seeding density of ~3×104 per well. The first and last columns of each 96-well plate were kept cell-free. These cell-free wells (16 in total) were filled with 175μL of corresponding assay media during OCR and ECAR measurements to do background correction and monitor any change in the baseline level of oxygen and pH.
For both glycolysis and mitochondrial stress tests, the Seahorse XFe96 Analyzer was programmed for 3 cycles of drug injection followed by 3 mixing and 3 measuring steps (3 min each). The glycolysis stress test was performed in Seahorse Biosciene XF assay media (102365–100) containing 2 mM GlutaMAX supplement but no glucose or pyruvate. D-glucose (10 mM), oligomycin A (1μM) and 2-DG (100 mM) were added sequentially. The mitochondrial stress test was carried out in DMEM-based XF modified media (10236–100) containing 25 mM glucose and 1 mM pyruvate (pH was adjusted to 7.40 ± 0.05 at 32°C, 37°C and 42°C). Oligomycin A (1μM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP; 0.5μM), and a combination of antimycin A and rotenone (1μM each) were added sequentially.
When data collection was completed, the assay medium from each well was removed and replaced with 25μl of cell lysis buffer containing 0.32 M sucrose, 2 mM EDTA, 2 mM EGTA, 20 mM HEPES, 0.2 mM PMSF, 4μg/mL leupeptin, 4μg/mL pepstatin and 5μg/mL aprotinin. Increasing volumes (1.25 to 12.5μL) of known protein standard (2μg/μl BSA) were added to the background wells to create a standard curve for the protein levels (ranging from 2.5 to 25μg). Thermo Scientific Pierce BCA Protein Assay Kit was used for determining protein concentrations.
The XFD raw data files, which were generated by Seahorse software (version 1.4.2.3), were processed using Seahorse Wave software (version 2.2.0), including detection of outliers and creation of base line phenograms. Automated data analysis for the OCR and ECAR signals were performed with XF Stress Test Report Generators V2. Figures and statistical analyses were generated with Prism 7 (GraphPad). For statistical analyses, two-way ANOVA and Welch’s t-test were used for glucose and mitochondrial stress tests, respectively.
Stable isotope-resolved metabolomics (SIRM).
SIRM experiment was carried out as previously described [173, 174] with minor modifications. In brief, non-targeting and CLN3 exon-8 siRNA transfected RPE-1 cells were grown in DMEM in 10 cm plates at 37°C with 5% CO2. Two days after transfection, when cells reached 70–80% confluence, cells were washed with glucose- and FBS-free DMEM, and traced in glucose-free DMEM supplemented with 10% dialyzed FBS and 4.5 g/L (25 mM) either unlabeled or 13C6-glucose (Cambridge Isotope). Due to limit of resources, only single unlabeled sample and duplicated labeled samples were set up for either non-targeting or CLN3 exon-8 siRNA treatment. Aliquots of 100μL of medium was collected at 0, 3, 6, 9, and 24 h after tracing began. Tracing was stopped at 24 h by quickly washing cells for 3 times with ice-cold PBS to remove medium components, and then lysing cells with 1 ml of cold acetonitrile (CH3CN; kept at −20°C) for 5 min. After addition of 0.55 ml ddH2O and 0.2 ml 0.2 mM Tris-HCl (pH 8, used as an internal standard for NMR), cell lysates in CH3CN:H2O (2:1.5 v/v) were collected. After addition of 1 ml of chloroform, cell lysates were rigorously mixed before phase separation by centrifugation at 4°C and 3500 x g for 20 min. A second extraction was carried out in 500μl of chloroform:methanol (2:1) with 1 mM BHT after the top and lower phases were transferred to a 5 ml Eppendorf tube and a glass vial, respectively. The pooled top aqueous phase (polar fraction) was aliquoted and lyophilized for downstream analyses using NMR and ICMS. The lower non-polar phase (lipid fraction) was dried in a Vacufuge at room temperature, reconstituted in chloroform:methanol (2:1) with 1 mM BHT, and analyzed by a Solarix FT-ICR MS. The protein-containing interphase was washed with 500μl of methanol, dried in Vacufuge at room temperature, solubilized in SDS buffer (62.5 mM Tris, 2% SDS at pH 6.8, with 1 mM final concentration of dithiothreitol), and denatured at 95°C for 10 min. Protein concentrations were determined by the Pierce Protein Assay Kit. Metabolites amounts were normalized by protein amounts of the corresponding samples.
For NMR experiments, lyophilized aliquots of polar fractions were reconstituted in D2O (>99.9%, Cambridge Isotope Laboratories, MA) containing 0.1 mM d16-EDTA (Ethylenediaminetetraacetic acid, Sigma Aldrich, St. Louis, MO) and 0.5 mM d6-2,2-dimethyl-2-silapentane-5-sulfonate (DSS) (Cambridge Isotope Laboratories, Tewksbury, MA) as the internal standard. The samples were centrifuged at 14,000 x g for 20 min to remove particulates. Supernatants were subsequently transferred into 1.7 mm NMR tubes for 1H-NMR and 1H{13C}-HSQC NMR data acquisition on a DD2 14.1 Tesla NMR spectrometer (Agilent Technologies, CA) equipped with a 3 mm inverse triple resonance HCN cold probe. Medium samples were extracted in −80% acetone, centrifuged to remove proteins, and aliquots lyophilized and reconstituted in phosphate buffer (pH 7) before NMR analyses. 1D 1H-NMR spectra were acquired with standard PRESAT pulse sequence at 15 °C. A total of 16384 data points were acquired with 2 s acquisition time, 512 transients, 12 ppm spectral width, and a 4 s relaxation delay time during which the residual HOD signal was suppressed with a weak transmitter pulse. The free induction decays were then linear predicted and zero filled to 128 k points and apodized with a 1 Hz line broadening exponential and a half Gaussian function. 1H{13C}-HSQC spectra were recorded with 13C adiabatic decoupling scheme for broad range decoupling during proton acquisition time of 0.25 s with a relaxation delay of 1.75 s. A total of 1024 transients (1796 data points per transient) were acquired with 12 ppm spectral width. The 1H{13C}-HSQC spectra were apodized with un-shifted Gaussian function and 4 Hz exponential line broadening and zero filling to 16k data points before Fourier transformation, baseline correcting and phasing. Of note, NMR detects intact glycogen. In particular, NMR detects the mobile glycogen which is on the outer shell of the glycogen particles and corresponds to newly synthesized glycogen. Although the relaxation delay times (in seconds) that we chose for acquiring the NMR spectra of the metabolites in the polar cell extracts and the media were much longer than the optimal (in ms) for detecting intact glycogen [109], we still saw large signals from intact glycogen in the 1H-NMR spectra. Metabolites were assigned by comparison with in-house [175] and public NMR databases, the Biological Magnetic Resonance Data Bank (BMRB) [176] and the Human Metabolome Database (HMDB) [177]. Metabolite and their 13C isotopomers were quantified using the MestReNova software (version 10, Mestrelab, Spain) by peak deconvolution. The peak intensities of metabolites obtained from the 1H-NMR spectra were converted into nmoles by calibration against the peak intensity of internal standard DSS at 0 ppm before normalized to total protein weight of each sample. The peak intensities of metabolites obtained from the 1H{13C}-HSQC spectra were calibrated against the peak intensity of lactate, whose concentrations were determined from the 1H-NMR spectra, before normalized to total protein weight of each sample.
For ICMS analyses of polar fractions, a lyophilized 55-compound mix was reconstituted in 1μM DSS, the internal standard, to obtain 1 mM concentration for each compound. Each lyophilized aliquot of polar fractions was reconstituted into 20μl ddH2O containing 1μM internal standard DSS. After 10 s centrifugation, the supernatant was transferred to a target PP polyspring insert (Thermo Fisher Scientific) that will be put in a 2 ml clear glass superstop vial (Thermo Fisher Scientific). Samples or the reconstituted 55-compound mix (10μl) will be injected for ICMS analysis. All ICMS analyses were performed on a ICS-5000+ ion chromatography (Dionex) coupled to a Thermo Fusion Orbitrap Tribrid mass spectrometer (Thermo Fisher Scientific) equipped with the heated electrospray ion source (HESI). For ion chromatography, analytes were separated on an IonPac AG11-HC-4μm guard (2×50mm) followed by an IonPac AS11-HC-4μm RFIC&HPIC column (2×250 mm) at 35˚C with the constant eluent flow rate at 0.38 ml/min. The following gradient program was employed: −8min-2min, kept at 1 mM KOH for equilibration; 2min-25min, KOH concentration increased from 1 mM to 40 mM; 25min-39.1min, KOH concentration increased from 40 mM to 100 mM; 39.1min-50min, kept at 100 mM KOH; 50min-50.1min, changed back to 1 mM KOH. KOH can be removed through the Dionex AERS 500 suppressor (2 mm) with regeneration flow rate at 0.75 ml/min. MS analyses were carried out in the negative ionization mode and 0.06 ml/min methanol was added post-column as the makeup solvent to facilitate vaporization. The following MS parameter settings were used: capillary voltage 2800 V, sheath gas 35 Arb, Aux gas 4 Arb, ion transfer tube temperature 300°C, vaporizer temperature 400˚C, mass scan range 80–700 m/z, automatic gain control (AGC) 2×105, and maximum injection time (MIT) 100 ms. The resolving power was set at 450,000 which can achieve resolution of 360,000 at m/z 400. Raw data were collected using Xcalibur and then imported to Thermo TraceFinder (version 3.3) software for peak area integration.
Supplementary Material
Highlights.
A juvenile neuronal ceroid lipofuscinosis (JNCL) mouse model on a C57BL/6J background displayed vision impairment and retinal abnormalities including retinal pigment epithelium (RPE) disintegration and metaplasia/hyperplasia.
Treating RPE-1 cells with CLN3 siRNA led to autophagy induction, as shown by increased autophagic flux that coincided with suppressed mTORC1 and Akt activities, enhanced AMPK activity, and transcriptional up-regulation of the autophagy-lysosomal system.
Treating RPE-1 cells with CLN3 siRNA also led to extensive metabolic abnormalities, including glycogen accumulation and ATP depletion, both of which are novel findings that provide critical clues to potential CLN3 disease etiology.
Acknowledgements
This work was supported by a New Scholar in Aging award from Ellison Medical Foundation (to Q.J.W.), National Institutes of Health (sub-awards to Q.J.W. from Center of Biomedical Research Excellence (COBRE) awards P20GM103486 and P20GM121327), National Institutes of Health EY028206 and BrightFocus Foundation (to M.E.K.), and National Science Foundation grant NSF 1419282 (to H.N.B.M.). This research was also supported by the Redox Metabolism Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558).
The authors thank Chaoying Liang at The UT Southwestern Genomics and Microarray Core for preparing the cDNA libraries and for running the HiSeq sequencing. The authors thank Dr. Kuey-Chu Chen and Donna Wall and in the Microarray Core Facility at the University Kentucky for discussing about RNA-Seq experimental design and for running the Bioanalyzer, respectively. The authors thank Jim Begley in the Imaging Facility at the University of Kentucky for EM sample processing. The authors also thank Dr. Andrew Lane and Dr. Sidney Whiteheart for critically reading the manuscript.
Footnotes
CLN3 has been postulated to have roles in maintenance of vacuolar/lysosomal sizes [38–40] and pH [40–45], transport of small molecules and amino acids (e.g., arginine) into vacuoles/lysosomes [46, 47], endosome-Golgi /TGN transport [48, 49], anterograde/retrograde transport of lysosomal enzymes [39], endocytic trafficking [39, 45, 50–55], autophagy and organelle fusion [45, 56–58], degradation of proteins [45], maintenance of normal mitochondrial morphology and cellular ATP levels [39, 51], lipid metabolism [53, 55, 58–64], RNA metabolism [65], the nitrogen compound biosynthesis process [62, 66], intracellular signaling [62, 65, 67, 68], osmoregulation [53, 69–71], and cell death, cell cycle and cell proliferation [58, 61, 63, 69, 72–75].
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cotman SL, Vrbanac V, Lebel LA, Lee RL, Johnson KA, Donahue LR, Teed AM, Antonellis K, Bronson RT, Lerner TJ, and MacDonald ME, Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet, 11 (2002) 2709–21. 10.1093/hmg/11.22.2709. [DOI] [PubMed] [Google Scholar]
- 2.NIH/NINDS. Batten Disease Fact Sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Batten-Disease-Fact-Sheet, 2019, (accessed 09/23/2019).
- 3.Ouseph MM, Kleinman ME, and Wang QJ, Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Ann N Y Acad Sci, 1371 (2016) 55–67. 10.1111/nyas.12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofman IL, van der Wal AC, Dingemans KP, and Becker AE, Cardiac pathology in neuronal ceroid lipofuscinoses-a clinicopathologic correlation in three patients. Eur J Paediatr Neurol, 5 Suppl A (2001) 213–7. 10.1053/ejpn.2000.0465. [DOI] [PubMed] [Google Scholar]
- 5.Ostergaard JR, Rasmussen TB, and Molgaard H, Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology, 76 (2011) 1245–51. 10.1212/WNL.0b013e31821435bd. [DOI] [PubMed] [Google Scholar]
- 6.Lebrun AH, Moll-Khosrawi P, Pohl S, Makrypidi G, Storch S, Kilian D, Streichert T, Otto B, Mole SE, Ullrich K, Cotman S, Kohlschutter A, Braulke T, and Schulz A, Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol Med, 17 (2011) 1253–61. 10.2119/molmed.2010.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chattopadhyay S, Ito M, Cooper JD, Brooks AI, Curran TM, Powers JM, and Pearce DA, An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease. Hum Mol Genet, 11 (2002) 1421–31. 10.1093/hmg/11.12.1421. [DOI] [PubMed] [Google Scholar]
- 8.Castaneda JA and Pearce DA, Identification of alpha-fetoprotein as an autoantigen in juvenile Batten disease. Neurobiol Dis, 29 (2008) 92–102. 10.1016/j.nbd.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, Wang X, Zaneveld JE, Salvo JS, Siddiqui S, Mao L, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Wen C, Flagg K, Ferreyra H, Pei J, Khan A, Ren H, Wang K, Lopez I, Qamar R, Zenteno JC, Ayala-Ramirez R, Buentello-Volante B, Fu Q, Simpson DA, Li Y, Sui R, Silvestri G, Daiger SP, Koenekoop RK, Zhang K, and Chen R, Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotypephenotype correlation and clinical refinements. Hum Genet, 133 (2014) 331–45. 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ku CA, Hull S, Arno G, Vincent A, Carss K, Kayton R, Weeks D, Anderson GW, Geraets R, Parker C, Pearce DA, Michaelides M, MacLaren RE, Robson AG, Holder GE, Heon E, Raymond FL, Moore AT, Webster AR, and Pennesi ME, Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol, 135 (2017) 749–760. 10.1001/jamaophthalmol.2017.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen FK, Zhang X, Eintracht J, Zhang D, Arunachalam S, Thompson JA, Chelva E, Mallon D, Chen SC, McLaren T, Lamey T, De Roach J, and McLenachan S, Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc Ophthalmol, 138 (2019) 55–70. 10.1007/s10633-018-9665-7. [DOI] [PubMed] [Google Scholar]
- 12.The-International-Batten-Disease-Consortium, Isolation of a novel gene underlying Batten disease, CLN3. Cell, 82 (1995) 949–57. 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- 13.Stengel OC, Eyr, (1826) [Google Scholar]
- 14.Bozorg S, Ramirez-Montealegre D, Chung M, and Pearce DA, Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Surv Ophthalmol, 54 (2009) 463–71. 10.1016/j.survophthal.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins J, Holder GE, Herbert H, and Adams GG, Batten disease: features to facilitate early diagnosis. Br J Ophthalmol, 90 (2006) 1119–24. 10.1136/bjo.2006.091637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen MS, Hove MN, Jensen H, and Larsen M, Optical coherence tomography in juvenile neuronal ceroid lipofuscinosis. Retin Cases Brief Rep, 10 (2016) 137–9. 10.1097/ICB.0000000000000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dulz S, Wagenfeld L, Nickel M, Richard G, Schwartz R, Bartsch U, Kohlschutter A, and Schulz A, Novel morphological macular findings in juvenile CLN3 disease. Br J Ophthalmol, Online (2015) 1–5. 10.1136/bjophthalmol-2015-307320. [DOI] [PubMed] [Google Scholar]
- 18.Weleber RG, The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond), 12 ( Pt 3b) (1998) 580–90. 10.1038/eye.1998.148. [DOI] [PubMed] [Google Scholar]
- 19.Weimer JM, Custer AW, Benedict JW, Alexander NA, Kingsley E, Federoff HJ, Cooper JD, and Pearce DA, Visual deficits in a mouse model of Batten disease are the result of optic nerve degeneration and loss of dorsal lateral geniculate thalamic neurons. Neurobiol Dis, 22 (2006) 284–93. 10.1016/j.nbd.2005.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strauss O, The retinal pigment epithelium in visual function. Physiol Rev, 85 (2005) 845–81. 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 21.Sparrow JR, Hicks D, and Hamel CP, The retinal pigment epithelium in health and disease. Curr Mol Med, 10 (2010) 802–23. 10.2174/156652410793937813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ambati J. and Fowler BJ, Mechanisms of age-related macular degeneration. Neuron, 75 (2012) 26–39. 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ao J, Wood JP, Chidlow G, Gillies MC, and Casson RJ, Retinal pigment epithelium in the pathogenesis of age-related macular degeneration and photobiomodulation as a potential therapy? Clin Exp Ophthalmol, (2017) 10.1111/ceo.13121. [DOI] [PubMed] [Google Scholar]
- 24.Guziewicz KE, Sinha D, Gomez NM, Zorych K, Dutrow EV, Dhingra A, Mullins RF, Stone EM, Gamm DM, Boesze-Battaglia K, and Aguirre GD, Bestrophinopathy: An RPEphotoreceptor interface disease. Prog Retin Eye Res, 58 (2017) 70–88. 10.1016/j.preteyeres.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christensen DRG, Brown FE, Cree AJ, Ratnayaka JA, and Lotery AJ, Sorsby fundus dystrophy - A review of pathology and disease mechanisms. Exp Eye Res, 165 (2017) 35–46. 10.1016/j.exer.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Foulds WS, Retinal metabolism and the choroidal circulation. Eye (Lond), 4 ( Pt 2) (1990) ix–x. 10.1038/eye.1990.34. [DOI] [PubMed] [Google Scholar]
- 27.Country MW, Retinal metabolism: A comparative look at energetics in the retina. Brain Res, 1672 (2017) 50–57. 10.1016/j.brainres.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 28.Zhao C, Yasumura D, Li X, Matthes M, Lloyd M, Nielsen G, Ahern K, Snyder M, Bok D, Dunaief JL, LaVail MM, and Vollrath D, mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest, 121 (2011) 369–83. 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurihara T, Westenskow PD, Gantner ML, Usui Y, Schultz A, Bravo S, Aguilar E, Wittgrove C, Friedlander M, Paris LP, Chew E, Siuzdak G, and Friedlander M, Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. Elife, 5 (2016) 10.7554/eLife.14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao J, Jia L, Khan N, Lin C, Mitter SK, Boulton ME, Dunaief JL, Klionsky DJ, Guan JL, Thompson DA, and Zacks DN, Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy, 11 (2015) 939–53. 10.1080/15548627.2015.1041699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Cross SD, Stanton JB, Marmorstein AD, Le YZ, and Marmorstein LY, Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol Vis, 23 (2017) 228–241. [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, and Ferguson TA, Noncanonical autophagy promotes the visual cycle. Cell, 155 (2013) 725–6. 10.1016/j.cell.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wavre-Shapton ST, Calvi AA, Turmaine M, Seabra MC, Cutler DF, Futter CE, and Mitchison HM, Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Deltaex1–6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease). Hum Mol Genet, (2015) 10.1093/hmg/ddv406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce DA, Localization and processing of CLN3, the protein associated to Batten disease: where is it and what does it do? J Neurosci Res, 59 (2000) 19–23. [PubMed] [Google Scholar]
- 35.Phillips SN, Benedict JW, Weimer JM, and Pearce DA, CLN3, the protein associated with batten disease: structure, function and localization. J Neurosci Res, 79 (2005) 573–83. 10.1002/jnr.20367. [DOI] [PubMed] [Google Scholar]
- 36.Cotman SL and Staropoli JF, The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin Lipidol, 7 (2012) 79–91. 10.2217/clp.11.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carcel-Trullols J, Kovacs AD, and Pearce DA, Cell biology of the NCL proteins: What they do and don’t do. Biochim Biophys Acta, 1852 (2015) 2242–55. 10.1016/j.bbadis.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 38.Kitzmuller C, Haines RL, Codlin S, Cutler DF, and Mole SE, A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum Mol Genet, 17 (2008) 303–12. 10.1093/hmg/ddm306. [DOI] [PubMed] [Google Scholar]
- 39.Fossale E, Wolf P, Espinola JA, Lubicz-Nawrocka T, Teed AM, Gao H, Rigamonti D, Cattaneo E, MacDonald ME, and Cotman SL, Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci, 5 (2004) 57. 10.1186/1471-2202-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gachet Y, Codlin S, Hyams JS, and Mole SE, btn1, the Schizosaccharomyces pombe homologue of the human Batten disease gene CLN3, regulates vacuole homeostasis. J Cell Sci, 118 (2005) 5525–36. 10.1242/jcs.02656. [DOI] [PubMed] [Google Scholar]
- 41.Pearce DA, Ferea T, Nosel SA, Das B, and Sherman F, Action of BTN1, the yeast orthologue of the gene mutated in Batten disease. Nat Genet, 22 (1999) 55–8. 10.1038/8861. [DOI] [PubMed] [Google Scholar]
- 42.Pearce DA, Nosel SA, and Sherman F, Studies of pH regulation by Btn1p, the yeast homolog of human Cln3p. Mol Genet Metab, 66 (1999) 320–3. 10.1006/mgme.1999.2819. [DOI] [PubMed] [Google Scholar]
- 43.Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, and Wisniewski KE, CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Mol Genet Metab, 70 (2000) 203–13. 10.1006/mgme.2000.3006. [DOI] [PubMed] [Google Scholar]
- 44.Holopainen JM, Saarikoski J, Kinnunen PK, and Järvelä I, Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs). Eur J Biochem, 268 (2001) 5851–6. 10.1046/j.00142956.2001.02530.x. [DOI] [PubMed] [Google Scholar]
- 45.Vidal-Donet JM, Carcel-Trullols J, Casanova B, Aguado C, and Knecht E, Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One, 8 (2013) e55526. 10.1371/journal.pone.0055526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim Y, Ramirez-Montealegre D, and Pearce DA, A role in vacuolar arginine transport for yeast Btn1p and for human CLN3, the protein defective in Batten disease. Proc Natl Acad Sci U S A, 100 (2003) 15458–62. 10.1073/pnas.2136651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez-Montealegre D. and Pearce DA, Defective lysosomal arginine transport in juvenile Batten disease. Hum Mol Genet, 14 (2005) 3759–73. 10.1093/hmg/ddi406. [DOI] [PubMed] [Google Scholar]
- 48.Kama R, Kanneganti V, Ungermann C, and Gerst JE, The yeast Batten disease orthologue Btn1 controls endosome-Golgi retrograde transport via SNARE assembly. J Cell Biol, 195 (2011) 203–15. 10.1083/jcb.201102115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metcalf DJ, Calvi AA, Seaman M, Mitchison HM, and Cutler DF, Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic, 9 (2008) 1905–14. 10.1111/j.1600-0854.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- 50.Luiro K, Yliannala K, Ahtiainen L, Maunu H, Jarvela I, Kyttala A, and Jalanko A, Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum Mol Genet, 13 (2004) 3017–27. 10.1093/hmg/ddh321. [DOI] [PubMed] [Google Scholar]
- 51.Cao Y, Staropoli JF, Biswas S, Espinola JA, MacDonald ME, Lee JM, and Cotman SL, Distinct early molecular responses to mutations causing vLINCL and JNCL presage ATP synthase subunit C accumulation in cerebellar cells. PLoS One, 6 (2011) e17118. 10.1371/journal.pone.0017118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uusi-Rauva K, Kyttala A, van der Kant R, Vesa J, Tanhuanpaa K, Neefjes J, Olkkonen VM, and Jalanko A, Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell Mol Life Sci, 69 (2012) 2075–89. 10.1007/s00018-011-0913-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tecedor L, Stein CS, Schultz ML, Farwanah H, Sandhoff K, and Davidson BL, CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J Neurosci, 33 (2013) 18065–79. 10.1523/JNEUROSCI.0498-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uusi-Rauva K, Luiro K, Tanhuanpaa K, Kopra O, Martin-Vasallo P, Kyttala A, and Jalanko A, Novel interactions of CLN3 protein link Batten disease to dysregulation of fodrin-Na+, K+ ATPase complex. Exp Cell Res, 314 (2008) 2895–905. 10.1016/j.yexcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 55.Schmidtke C, Tiede S, Thelen M, Kakela R, Jabs S, Makrypidi G, Sylvester M, Schweizer M,Braren I, Brocke-Ahmadinejad N, Cotman SL, Schulz A, Gieselmann V, and Braulke T, Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J Biol Chem, 294 (2019) 95929604. 10.1074/jbc.RA119.008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, and Cotman SL, Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem, 281 (2006) 20483–20493. 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- 57.Lojewski X, Staropoli JF, Biswas-Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U, Sheridan SD, Lucente D, Sims KB, Gusella JF, Sondhi D, Crystal RG, Reinhardt P, Sterneckert J, Scholer H, Haggarty SJ, Storch A, Hermann A, and Cotman SL, Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet, 23 (2014) 2005–22. 10.1093/hmg/ddt596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang S, Heo TH, and Kim SJ, Altered levels of alpha-synuclein and sphingolipids in Batten disease lymphoblast cells. Gene, 539 (2014) 181–5. 10.1016/j.gene.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 59.Narayan SB, Rakheja D, Tan L, Pastor JV, and Bennett MJ, CLN3P, the Batten’s disease protein, is a novel palmitoyl-protein Delta-9 desaturase. Ann Neurol, 60 (2006) 570–7. 10.1002/ana.20975. [DOI] [PubMed] [Google Scholar]
- 60.Hobert JA and Dawson G, A novel role of the Batten disease gene CLN3: association with BMP synthesis. Biochem Biophys Res Commun, 358 (2007) 111–6. 10.1016/j.bbrc.2007.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rusyn E, Mousallem T, Persaud-Sawin DA, Miller S, and Boustany RM, CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatr Res, 63 (2008) 625–31. 10.1203/PDR.0b013e31816fdc17. [DOI] [PubMed] [Google Scholar]
- 62.Scifo E, Szwajda A, Debski J, Uusi-Rauva K, Kesti T, Dadlez M, Gingras AC, Tyynela J, Baumann MH, Jalanko A, and Lalowski M, Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: a label-free quantitative proteomics approach. J Proteome Res, 12 (2013) 2101–15. 10.1021/pr301125k. [DOI] [PubMed] [Google Scholar]
- 63.Kang S, Kim JB, Heo TH, and Kim SJ, Cell cycle arrest in Batten disease lymphoblast cells. Gene, 519 (2013) 245–50. 10.1016/j.gene.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 64.Persaud-Sawin DA, McNamara JO 2nd, Rylova S, Vandongen A, and Boustany RM, A galactosylceramide binding domain is involved in trafficking of CLN3 from Golgi to rafts via recycling endosomes. Pediatr Res, 56 (2004) 449–63. 10.1203/01.PDR.0000136152.54638.95. [DOI] [PubMed] [Google Scholar]
- 65.Tuxworth RI, Vivancos V, O’Hare MB, and Tear G, Interactions between the juvenile Batten disease gene, CLN3, and the Notch and JNK signalling pathways. Hum Mol Genet, 18 (2009) 667–78. 10.1093/hmg/ddn396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osorio NS, Carvalho A, Almeida AJ, Padilla-Lopez S, Leao C, Laranjinha J, Ludovico P, Pearce DA, and Rodrigues F, Nitric oxide signaling is disrupted in the yeast model for Batten disease. Mol Biol Cell, 18 (2007) 2755–67. 10.1091/mbc.E06-11-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang JW, Choi H, Cotman SL, and Jung YK, Lithium rescues the impaired autophagy process in CbCln3(Deltaex7/8/Deltaex7/8) cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition. J Neurochem, 116 (2011) 659–68. 10.1111/j.14714159.2010.07158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tuxworth RI, Chen H, Vivancos V, Carvajal N, Huang X, and Tear G, The Batten disease gene CLN3 is required for the response to oxidative stress. Hum Mol Genet, 20 (2011) 2037–47. 10.1093/hmg/ddr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huber RJ, Myre MA, and Cotman SL, Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS One, 9 (2014) e110544. 10.1371/journal.pone.0110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stein CS, Yancey PH, Martins I, Sigmund RD, Stokes JB, and Davidson BL, Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am J Physiol Cell Physiol, 298 (2010) C1388–400. 10.1152/ajpcell.00272.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Getty A, Kovacs AD, Lengyel-Nelson T, Cardillo A, Hof C, Chan CH, and Pearce DA, Osmotic stress changes the expression and subcellular localization of the Batten disease protein CLN3. PLoS One, 8 (2013) e66203. 10.1371/journal.pone.0066203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lane SC, Jolly RD, Schmechel DE, Alroy J, and Boustany RM, Apoptosis as the mechanism of neurodegeneration in Batten’s disease. J Neurochem, 67 (1996) 677–83. 10.1046/j.1471-4159.1996.67020677.x. [DOI] [PubMed] [Google Scholar]
- 73.Narayan SB, Rakheja D, Pastor JV, Rosenblatt K, Greene SR, Yang J, Wolf BA, and Bennett MJ, Over-expression of CLN3P, the Batten disease protein, inhibits PANDER-induced apoptosis in neuroblastoma cells: further evidence that CLN3P has anti-apoptotic properties. Mol Genet Metab, 88 (2006) 178–83. 10.1016/j.ymgme.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 74.Zhu X, Huang Z, Chen Y, Zhou J, Hu S, Zhi Q, Song S, Wang Y, Wan D, Gu W, Zhou H, Zhang B, Cao W, and He S, Effect of CLN3 silencing by RNA interference on the proliferation and apoptosis of human colorectal cancer cells. Biomed Pharmacother, 68 (2014) 253–8. 10.1016/j.biopha.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 75.Persaud-Sawin DA, VanDongen A, and Boustany RM, Motifs within the CLN3 protein: modulation of cell growth rates and apoptosis. Hum Mol Genet, 11 (2002) 2129–42. 10.1093/hmg/11.18.2129. [DOI] [PubMed] [Google Scholar]
- 76.Seigel GM, Lotery A, Kummer A, Bernard DJ, Greene ND, Turmaine M, Derksen T, Nussbaum RL, Davidson B, Wagner J, and Mitchison HM, Retinal pathology and function in a Cln3 knockout mouse model of juvenile Neuronal Ceroid Lipofuscinosis (batten disease). Mol Cell Neurosci, 19 (2002) 515–27. 10.1006/mcne.2001.1099. [DOI] [PubMed] [Google Scholar]
- 77.Groh J, Stadler D, Buttmann M, and Martini R, Non-invasive assessment of retinal alterations in mouse models of infantile and juvenile neuronal ceroid lipofuscinosis by spectral domain optical coherence tomography. Acta Neuropathol Commun, 2 (2014) 54. 10.1186/20515960-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Katz ML, Johnson GS, Tullis GE, and Lei B, Phenotypic characterization of a mouse model of juvenile neuronal ceroid lipofuscinosis. Neurobiol Dis, 29 (2008) 242–53. 10.1016/j.nbd.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Staropoli JF, Haliw L, Biswas S, Garrett L, Holter SM, Becker L, Skosyrski S, Da Silva-Buttkus P, Calzada-Wack J, Neff F, Rathkolb B, Rozman J, Schrewe A, Adler T, Puk O, Sun M, Favor J, Racz I, Bekeredjian R, Busch DH, Graw J, Klingenspor M, Klopstock T, Wolf E, Wurst W, Zimmer A, Lopez E, Harati H, Hill E, Krause DS, Guide J, Dragileva E, Gale E, Wheeler VC, Boustany RM, Brown DE, Breton S, Ruether K, Gailus-Durner V, Fuchs H, de Angelis MH, and Cotman SL, Large-scale phenotyping of an accurate genetic mouse model of JNCL identifies novel early pathology outside the central nervous system. PLoS One, 7 (2012) e38310. 10.1371/journal.pone.0038310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Volz C, Mirza M, Langmann T, and Jagle H, Retinal function in aging homozygous Cln3 (Deltaex7/8) knock-in mice. Adv Exp Med Biol, 801 (2014) 495–501. 10.1007/9781-4614-3209-8_63. [DOI] [PubMed] [Google Scholar]
- 81.Volz C, Mirza M, Langmann T, and Jagle H, Further Characterization of the Predominant Inner Retinal Degeneration of Aging Cln3 (Deltaex7/8) Knock-In Mice. Adv Exp Med Biol, 1074 (2018) 403–411. 10.1007/978-3-319-75402-4_50. [DOI] [PubMed] [Google Scholar]
- 82.Wang QJ, Jung KS, Mohan K, and Kleinman ME, Imaging data on characterizing the retinal autofluorescence puncta in a mouse model of juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Data In Brief, (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, and Heckenlively JR, Retinal degeneration mutants in the mouse. Vision Res, 42 (2002) 517–25. 10.1016/s00426989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- 84.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, and Caspi RR, The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci, 53 (2012) 2921–7. 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perlman I, The Electroretinogram: ERG, in: Kolb H, Fernandez E, and Nelson R, (Eds.), The Organization of the Retina and Visual System, University of Utah Health Sciences Center, Salt Lake City (UT), 2001. [Google Scholar]
- 86.Yung M, Klufas MA, and Sarraf D, Clinical applications of fundus autofluorescence in retinal disease. Int J Retina Vitreous, 2 (2016) 12. 10.1186/s40942-016-0035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rudolf M, Vogt SD, Curcio CA, Huisingh C, McGwin G Jr., Wagner A, Grisanti S, and Read RW, Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology, 120 (2013) 821–8. 10.1016/j.ophtha.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behrends C, Sowa ME, Gygi SP, and Harper JW, Network organization of the human autophagy system. Nature, 466 (2010) 68–76. 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cortese A, Tucci A, Piccolo G, Galimberti CA, Fratta P, Marchioni E, Grampa G, Cereda C, Grieco G, Ricca I, Pittman A, Ciscato P, Napoli L, Lucchini V, Ripolone M, Violano R, Fagiolari G, Mole SE, Hardy J, Moglia A, and Moggio M, Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology, 82 (2014) 2072–6. 10.1212/WNL.0000000000000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 102 (2005) 15545–50. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flight RM, Harrison BJ, Mohammad F, Bunge MB, Moon LD, Petruska JC, and Rouchka EC, categoryCompare, an analytical tool based on feature annotations. Front Genet, 5 (2014) 98. 10.3389/fgene.2014.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, and Sandri M, FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab, 6 (2007) 458–71. 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 93.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, and Goldberg AL, FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab, 6 (2007) 472–83. 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 94.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, and Passegue E, FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature, 494 (2013) 323–7. 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, and Ballabio A, TFEB links autophagy to lysosomal biogenesis. Science, 332 (2011) 1429–33. 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, and Vannucci SJ, The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab, 295 (2008) E242–53. 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, and Jolly RD, Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid-binding subunit of mitochondrial ATP synthase have the same NH2-terminal sequence. J Biol Chem, 264 (1989) 5736–40. [PubMed] [Google Scholar]
- 98.Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, and Jolly RD, Mitochondrial ATP synthase subunit c storage in the ceroidlipofuscinoses (Batten disease). Am J Med Genet, 42 (1992) 561–7. 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- 99.Palmer DN, Barry LA, Tyynela J, and Cooper JD, NCL disease mechanisms. Biochim Biophys Acta, 1832 (2013) 1882–93. 10.1016/j.bbadis.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 100.Hall NA, Lake BD, Dewji NN, and Patrick AD, Lysosomal storage of subunit c of mitochondrial ATP synthase in Batten’s disease (ceroid-lipofuscinosis). Biochem J, 275 ( Pt 1) (1991) 269–72. 10.1042/bj2750269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rylova SN, Amalfitano A, Persaud-Sawin DA, Guo WX, Chang J, Jansen PJ, Proia AD, and Boustany RM, The CLN3 gene is a novel molecular target for cancer drug discovery. Cancer Res, 62 (2002) 801–8. [PubMed] [Google Scholar]
- 102.El-Sitt S, Soueid J, Al Ali J, Makoukji J, Makhoul NJ, Harati H, and Boustany RM, Developmental Comparison of Ceramide in Wild-Type and Cln3 (Deltaex7/8) Mouse Brains and Sera. Front Neurol, 10 (2019) 128. 10.3389/fneur.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hicks J, Wartchow E, and Mierau G, Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol, 35 (2011) 183–96. 10.3109/01913123.2011.601404. [DOI] [PubMed] [Google Scholar]
- 104.Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, and Serratosa JM, Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem, 293 (2018) 7117–7125. 10.1074/jbc.R117.803064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brewer MK, Uittenbogaard A, Austin GL, Segvich DM, DePaoli-Roach A, Roach PJ, McCarthy JJ, Simmons ZR, Brandon JA, Zhou Z, Zeller J, Young LEA, Sun RC, Pauly JR, Aziz NM, Hodges BL, McKnight TR, Armstrong DD, and Gentry MS, Targeting Pathogenic Lafora Bodies in Lafora Disease Using an Antibody-Enzyme Fusion. Cell Metab, 30 (2019) 689705 e6. 10.1016/j.cmet.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prats C, Graham TE, and Shearer J, The dynamic life of the glycogen granule. J Biol Chem, 293 (2018) 7089–7098. 10.1074/jbc.R117.802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tkac I, Starcuk Z, Choi IY, and Gruetter R, In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med, 41 (1999) 649–56. . [DOI] [PubMed] [Google Scholar]
- 108.Laughlin MR, Petit WA Jr., Dizon JM, Shulman RG, and Barrett EJ, NMR measurements of in vivo myocardial glycogen metabolism. J Biol Chem, 263 (1988) 2285–91. [PubMed] [Google Scholar]
- 109.Soares AF, Gruetter R, and Lei H, Technical and experimental features of Magnetic Resonance Spectroscopy of brain glycogen metabolism. Anal Biochem, 529 (2017) 117–126. 10.1016/j.ab.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 110.Moseley HN, Lane AN, Belshoff AC, Higashi RM, and Fan TW, A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on (13)C mass isotopologue profiles under non-steady-state conditions. BMC Biol, 9 (2011) 37. 10.1186/1741-7007-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dannhausen K, Mohle C, and Langmann T, Immunomodulation with minocycline rescues retinal degeneration in juvenile neuronal ceroid lipofuscinosis mice highly susceptible to light damage. Dis Model Mech, 11 (2018) 10.1242/dmm.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mitchison HM, Bernard DJ, Greene ND, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, Gardiner RM, Lake BD, Taschner PE, and Nussbaum RL, Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. Neurobiol Dis, 6 (1999) 321–34. 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- 113.Bensaoula T, Shibuya H, Katz ML, Smith JE, Johnson GS, John SK, and Milam AH, Histopathologic and immunocytochemical analysis of the retina and ocular tissues in Batten disease. Ophthalmology, 107 (2000) 1746–53. 10.1016/s0161-6420(00)00264-5. [DOI] [PubMed] [Google Scholar]
- 114.Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, and Sardiello M, mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun, 8 (2017) 14338. 10.1038/ncomms14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frost LS, Mitchell CH, and Boesze-Battaglia K, Autophagy in the eye: implications for ocular cell health. Exp Eye Res, 124 (2014) 56–66. 10.1016/j.exer.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Costello MJ, Brennan LA, Basu S, Chauss D, Mohamed A, Gilliland KO, Johnsen S, Menko S, and Kantorow M, Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res, 116 (2013) 141–50. 10.1016/j.exer.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, and Mizushima N, Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem, 288 (2013) 11436–47. 10.1074/jbc.M112.437103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yao J, Jia L, Feathers K, Lin C, Khan NW, Klionsky DJ, Ferguson TA, and Zacks DN, Autophagy-mediated catabolism of visual transduction proteins prevents retinal degeneration. Autophagy, 12 (2016) 2439–2450. 10.1080/15548627.2016.1238553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, and Neufeld AH, Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One, 4 (2009) e4160. 10.1371/journal.pone.0004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, Boulton ME, and Petrovski G, Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy, 9 (2013) 973–84. 10.4161/auto.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kaarniranta K, Tokarz P, Koskela A, Paterno J, and Blasiak J, Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol Toxicol, 33 (2017) 113–128. 10.1007/s10565-016-9371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yao J, Jia L, Shelby SJ, Ganios AM, Feathers K, Thompson DA, and Zacks DN, Circadian and noncircadian modulation of autophagy in photoreceptors and retinal pigment epithelium. Invest Ophthalmol Vis Sci, 55 (2014) 3237–46. 10.1167/iovs.13-13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, and Green DR, Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol, 17 (2015) 893–906. 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Sukseree S, Chen YT, Laggner M, Gruber F, Petit V, Nagelreiter IM, Mlitz V, Rossiter H, Pollreisz A, Schmidt-Erfurth U, Larue L, Tschachler E, and Eckhart L, Tyrosinase-Cre-Mediated Deletion of the Autophagy Gene Atg7 Leads to Accumulation of the RPE65 Variant M450 in the Retinal Pigment Epithelium of C57BL/6 Mice. PLoS One, 11 (2016) e0161640. 10.1371/journal.pone.0161640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hardie DG, Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface, 15 (2018) 10.1098/rsif.2017.0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Egan D, Kim J, Shaw RJ, and Guan KL, The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy, 7 (2011) 643–4. 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim J, Kundu M, Viollet B, and Guan KL, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol, 13 (2011) 132–41. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, and Guan KL, Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell, 152 (2013) 290–303. 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, and Brunet A, The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem, 282 (2007) 30107–19. 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 130.Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, and Baek SH, AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature, 534 (2016) 553–7. 10.1038/nature18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, and Ballabio A, A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J, 31 (2012) 1095108. 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, and Greenberg ME, Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell, 96 (1999) 857–68. 10.1016/s00928674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 133.Mammucari C, Schiaffino S, and Sandri M, Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy, 4 (2008) 524–6. 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 134.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, and Sabatini DM, Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell, 11 (2006) 859–71. 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 135.Khoh-Reiter S, Sokolowski SA, Jessen B, Evans M, Dalvie D, and Lu S, Contribution of membrane trafficking perturbation to retinal toxicity. Toxicol Sci, 145 (2015) 383–95. 10.1093/toxsci/kfv059. [DOI] [PubMed] [Google Scholar]
- 136.Lu S, Jessen B, Strock C, and Will Y, The contribution of physicochemical properties to multiple in vitro cytotoxicity endpoints. Toxicol In Vitro, 26 (2012) 613–20. 10.1016/j.tiv.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 137.Logan R, Kong AC, Axcell E, and Krise JP, Amine-containing molecules and the induction of an expanded lysosomal volume phenotype: a structure-activity relationship study. J Pharm Sci, 103 (2014) 1572–80. 10.1002/jps.23949. [DOI] [PubMed] [Google Scholar]
- 138.Curcio-Morelli C, Charles FA, Micsenyi MC, Cao Y, Venugopal B, Browning MF, Dobrenis K, Cotman SL, Walkley SU, and Slaugenhaupt SA, Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol Dis, 40 (2010) 370–7. 10.1016/j.nbd.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xu M, Liu K, Swaroop M, Porter FD, Sidhu R, Firnkes S, Ory DS, Marugan JJ, Xiao J, Southall N, Pavan WJ, Davidson C, Walkley SU, Remaley AT, Baxa U, Sun W, McKew JC, Austin CP, and Zheng W, delta-Tocopherol reduces lipid accumulation in NiemannPick type C1 and Wolman cholesterol storage disorders. J Biol Chem, 287 (2012) 39349–60. 10.1074/jbc.M112.357707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, and Platt FM, Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med, 14 (2008) 1247–55. 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 141.Winkler BS, Glycolytic and oxidative metabolism in relation to retinal function. J Gen Physiol, 77 (1981) 667–92. 10.1085/jgp.77.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Winkler BS, The electroretinogram of the isolated rat retina. Vision Res, 12 (1972) 1183–98. 10.1016/0042-6989(72)90106-x. [DOI] [PubMed] [Google Scholar]
- 143.Peachey NS, Green DJ, and Ripps H, Ocular ischemia and the effects of allopurinol on functional recovery in the retina of the arterially perfused cat eye. Invest Ophthalmol Vis Sci, 34 (1993) 58–65. [PubMed] [Google Scholar]
- 144.Ames A 3rd and Li YY, Energy requirements of glutamatergic pathways in rabbit retina. J Neurosci, 12 (1992) 4234–42. 10.1523/JNEUROSCI.12-11-04234.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Winkler BS, A quantitative assessment of glucose metabolism in the isolated rat retina, in: Christen Y, Doly CY, and Droy-LeFaix M-T, (Eds.), Les Seminaires Ophthalmologiques d’IPSEN: Vision et Adaptation, Elsevier, Amsterdam, 1995, pp. 78–96. [Google Scholar]
- 146.Winkler BS, Pourcho RG, Starnes C, Slocum J, and Slocum N, Metabolic mapping in mammalian retina: a biochemical and 3H-2-deoxyglucose autoradiographic study. Exp Eye Res, 77 (2003) 327–37. 10.1016/s0014-4835(03)00147-7. [DOI] [PubMed] [Google Scholar]
- 147.Du J, Rountree A, Cleghorn WM, Contreras L, Lindsay KJ, Sadilek M, Gu H, Djukovic D, Raftery D, Satrustegui J, Kanow M, Chan L, Tsang SH, Sweet IR, and Hurley JB, Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. J Biol Chem, 291 (2016) 4698–710. 10.1074/jbc.M115.698985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hurley JB, Lindsay KJ, and Du J, Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J Neurosci Res, 93 (2015) 1079–92. 10.1002/jnr.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang L, Tornquist P, and Bill A, Glucose metabolism in pig outer retina in light and darkness. Acta Physiol Scand, 160 (1997) 75–81. 10.1046/j.1365-201X.1997.00031.x. [DOI] [PubMed] [Google Scholar]
- 150.Wang L, Tornquist P, and Bill A, Glucose metabolism of the inner retina in pigs in darkness and light. Acta Physiol Scand, 160 (1997) 71–4. 10.1046/j.1365-201X.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 151.Rajala A, Wang Y, Soni K, and Rajala RVS, Pyruvate kinase M2 isoform deletion in cone photoreceptors results in age-related cone degeneration. Cell Death Dis, 9 (2018) 737. 10.1038/s41419-018-0712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rajala A, Wang Y, Brush RS, Tsantilas K, Jankowski CSR, Lindsay KJ, Linton JD, Hurley JB, Anderson RE, and Rajala RVS, Pyruvate kinase M2 regulates photoreceptor structure, function, and viability. Cell Death Dis, 9 (2018) 240. 10.1038/s41419-018-0296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Grenell A, Wang Y, Yam M, Swarup A, Dilan TL, Hauer A, Linton JD, Philp NJ, Gregor E, Zhu S, Shi Q, Murphy J, Guan T, Lohner D, Kolandaivelu S, Ramamurthy V, Goldberg AFX, Hurley JB, and Du J, Loss of MPC1 reprograms retinal metabolism to impair visual function. Proc Natl Acad Sci U S A, 116 (2019) 3530–3535. 10.1073/pnas.1812941116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jaiswal M, Haelterman NA, Sandoval H, Xiong B, Donti T, Kalsotra A, Yamamoto S, Cooper TA, Graham BH, and Bellen HJ, Impaired mitochondrial energy production causes lightinduced photoreceptor degeneration independent of oxidative stress. PLoS Biol, 13 (2015) e1002197. 10.1371/journal.pbio.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, and Colman RF, Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet, 40 (2008) 1230–4. 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Du J, Yanagida A, Knight K, Engel AL, Vo AH, Jankowski C, Sadilek M, Tran VT, Manson MA, Ramakrishnan A, Hurley JB, and Chao JR, Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc Natl Acad Sci U S A, 113 (2016) 1471014715. 10.1073/pnas.1604572113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chao JR, Knight K, Engel AL, Jankowski C, Wang Y, Manson MA, Gu H, Djukovic D, Raftery D, Hurley JB, and Du J, Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J Biol Chem, 292 (2017) 12895–12905. 10.1074/jbc.M117.788422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Das AM, von Harlem R, Feist M, Lucke T, and Kohlschutter A, Altered levels of high-energy phosphate compounds in fibroblasts from different forms of neuronal ceroid lipofuscinoses: further evidence for mitochondrial involvement. Eur J Paediatr Neurol, 5 Suppl A (2001) 143–6. 10.1053/ejpn.2000.0451. [DOI] [PubMed] [Google Scholar]
- 159.Bosch ME and Kielian T, Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities. J Neurochem, 148 (2019) 612–624. 10.1111/jnc.14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Das AM and Kohlschutter A, Decreased activity of the mitochondrial ATP-synthase in fibroblasts from children with late-infantile and juvenile neuronal ceroid lipofuscinosis. J Inherit Metab Dis, 19 (1996) 130–2. 10.1007/BF01799411. [DOI] [PubMed] [Google Scholar]
- 161.Chattopadhyay S, Kingsley E, Serour A, Curran TM, Brooks AI, and Pearce DA, Altered gene expression in the eye of a mouse model for batten disease. Invest Ophthalmol Vis Sci, 45 (2004) 2893–905. 10.1167/iovs.04-0143. [DOI] [PubMed] [Google Scholar]
- 162.Sabourdy F, Kedjouar B, Sorli SC, Colie S, Milhas D, Salma Y, and Levade T, Functions of sphingolipid metabolism in mammals-lessons from genetic defects. Biochim Biophys Acta, 1781 (2008) 145–83. 10.1016/j.bbalip.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 163.Pears MR, Codlin S, Haines RL, White IJ, Mortishire-Smith RJ, Mole SE, and Griffin JL, Deletion of btn1, an orthologue of CLN3, increases glycolysis and perturbs amino acid metabolism in the fission yeast model of Batten disease. Mol Biosyst, 6 (2010) 1093–102. 10.1039/b915670d. [DOI] [PubMed] [Google Scholar]
- 164.S.D.O. ATCC, Authentication of Human Cell Lines: Standardization of STR Profiling. 2012, The ATCC® Standards Development Organization. [Google Scholar]
- 165.Capes-Davis A, Reid YA, Kline MC, Storts DR, Strauss E, Dirks WG, Drexler HG, MacLeod RA, Sykes G, Kohara A, Nakamura Y, Elmore E, Nims RW, Alston-Roberts C, Barallon R, Los GV, Nardone RM, Price PJ, Steuer A, Thomson J, Masters JR, and Kerrigan L, Match criteria for human cell line authentication: where do we draw the line? Int J Cancer, 132 (2013) 2510–9. 10.1002/ijc.27931. [DOI] [PubMed] [Google Scholar]
- 166.Andrews S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc, 2010, (accessed 9/23/19).
- 167.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29 (2013) 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li B. and Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12 (2011) 323. 10.1186/14712105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Robinson MD, McCarthy DJ, and Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26 (2010) 139–40. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carlson M, org.Hs.eg.db: Genome wide annotation for Human. R package version 3.6. 2019. [Google Scholar]
- 171.Benjamini Y. and Hochberg Y, Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 57 (1995) 289–300. [Google Scholar]
- 172.Mitov MI, Harris JW, Alstott MC, Zaytseva YY, Evers BM, and Butterfield DA, Temperature induces significant changes in both glycolytic reserve and mitochondrial spare respiratory capacity in colorectal cancer cell lines. Exp Cell Res, 354 (2017) 112–121. 10.1016/j.yexcr.2017.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fan TW, Lane AN, Higashi RM, and Yan J, Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics, 7 (2011) 257–269. 10.1007/s11306010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fan TW, Bandura LL, Higashi RM, and Lane AN, Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer. Metabolomics, 1 (2005) 325–339. [Google Scholar]
- 175.Fan TW-M and Lane AN, Assignment Strategies for Nuclear Magnetic Resonances in Metabolomic Research, in: Lutz NW, Sweedler JV, and Wevers RA, (Eds.), Methodologies for Metabolomics: Experimental Strategies and Techniques, Cambridge University Press, Cambridge, 2013, pp. 525–584. [Google Scholar]
- 176.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, and Markley JL, BioMagResBank. Nucleic Acids Res, 36 (2008) D402–8. 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, Sayeeda Z, Lo E, Assempour N, Berjanskii M, Singhal S, Arndt D, Liang Y, Badran H, Grant J, Serra-Cayuela A, Liu Y, Mandal R, Neveu V, Pon A, Knox C, Wilson M, Manach C, and Scalbert A, HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res, 46 (2018) D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhong Y, Morris DH, Jin L, Patel MS, Karunakaran SK, Fu YJ, Matuszak EA, Weiss HL, Chait BT, and Wang QJ, Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem, 289 (2014) 26021–37. 10.1074/jbc.M114.561134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.