

Abstract
Purpose:
Cell-free DNA (cfDNA) offers a noninvasive approach to monitor cancer. Here we develop a method using whole-exome sequencing (WES) of cfDNA for simultaneously monitoring the full spectrum of cancer treatment outcomes, including minimal residual disease (MRD), recurrence, evolution, and second primary cancers.
Experimental Design:
Three simulation datasets were generated from 26 patients with cancer to benchmark the detection performance of MRD/recurrence and second primary cancers. For further validation, cfDNA samples (n = 76) from patients with cancer (n = 35) with six different cancer types were used for performance validation during various treatments.
Results:
We present a cfDNA-based cancer monitoring method, named cfTrack. Taking advantage of the broad genome coverage of WES data, cfTrack can sensitively detect MRD and cancer recurrence by integrating signals across known clonal tumor mutations of a patient. In addition, cfTrack detects tumor evolution and second primary cancers by de novo identifying emerging tumor mutations. A series of machine learning and statistical denoising techniques are applied to enhance the detection power. On the simulation data, cfTrack achieved an average AUC of 99% on the validation dataset and 100% on the independent dataset in detecting recurrence in samples with tumor fractions ≥0.05%. In addition, cfTrack yielded an average AUC of 88% in detecting second primary cancers in samples with tumor fractions ≥0.2%. On real data, cfTrack accurately monitors tumor evolution during treatment, which cannot be accomplished by previous methods.
Conclusions:
Our results demonstrated that cfTrack can sensitively and specifically monitor the full spectrum of cancer treatment outcomes using exome-wide mutation analysis of cfDNA.
Translational Relevance.
Continuous cancer monitoring is clinically necessary for patients with cancer to detect minimal residual disease (MRD), recurrence, and progression, allowing for early intervention and therapy adjustment. Cell-free DNA (cfDNA) in blood has become an appealing option due to its noninvasiveness. Until now, cfDNA-based cancer monitoring methods have been focused on deep sequencing at a few known mutations, which is insufficient when tumors evolve or new tumors emerge. We present the method, cfTrack, which uses whole-exome sequencing of cfDNA to track the full range of cancer treatment outcomes for the first time, including MRD, recurrence, evolution, and second primary cancer. We demonstrate that, even with very low tumor fractions, cfTrack achieves sensitive and specific monitoring of tumor MRD/recurrence/evolution based on both simulation data and a cohort of patients with cancer. These findings demonstrate the clinical utility of cfTrack.
Introduction
Despite the rapid development of cancer treatments, a large fraction of patients experience recurrence, resistance, or progression of cancer during or after treatment (1). Even after the surgical removal of tumors, a patient can still have minimal residual disease (MRD), which is associated with an increased likelihood of recurrence (2). Thus, patients with cancer need continuous monitoring to detect MRD, recurrence, and progression, thereby facilitating early intervention and therapy adjustment (2, 3). Although cancer monitoring is clinically important, the sequential sampling of tumor tissue from the patient poses a significant challenge. In this context, liquid biopsy is an attractive option, especially the usage of cell-free DNA (cfDNA) in blood. Blood samples can be obtained noninvasively for continuous monitoring, and the tumor-derived DNA fragments in cfDNA can provide comprehensive genetic profiling even of heterogeneous tumors (4).
However, a major challenge associated with cfDNA-based cancer monitoring is low tumor content. In patients with cancer receiving treatment or with MRD, the fraction of tumor DNA in a cfDNA sample can be as low as 0.1% (5). Previous studies on cancer monitoring in plasma have used deep sequencing on a small mutation panel to discover the weak tumor signal (2, 5, 6, 7, 8). However, these methods have several crucial limitations: (i) the high cost of deep sequencing restricts the panels to a small number of known mutations (either common cancer mutations or mutations selected from the pretreatment tumor sample of a specific patient); (ii) personalized panels usually require a labor-intensive experimental design; (iii) panel-based monitoring cannot detect emerging tumors with a different mutation profile, for example, a second primary cancer, yet approximately 30% patients develop a second primary cancer (9), driven by de novo tumor mutations; and (iv) panels usually set detection thresholds by studying a cohort of noncancer individuals, which exposes the test to systemic bias from interindividual variations and interexperimental differences. Recently, two studies (10, 11) presented cancer monitoring methods using whole-genome sequencing (WGS), but they do not yet address limitations (iii) and (iv) mentioned above, and focus on mutations seen in pretreatment tumor samples. In addition, the high cost of WGS limits the clinical applications of those two methods.
In this study, we describe a new cancer monitoring approach, named cfTrack, based on the cfDNA whole-exome sequencing (WES). cfTrack addresses all the aforementioned limitations of existing methods. Specifically, not only can it monitor the preexisting cancer (i.e., the original, primary cancer) to detect recurrence or MRD, but it can also monitor tumor evolution by detecting cancer progression or the emergence of a second primary cancer. To monitor the preexisting cancer, cfTrack (i) uses exome-wide somatic mutations collected in pretreatment samples (solid tumor or blood samples) to provide a robust statistical index, then (ii) models sample-specific background noise in the cfDNA sequencing data to provide an unbiased detection threshold for each patient. To monitor tumor evolution, cfTrack performs detection of exome-wide de novo tumor mutations in the posttreatment plasma samples, using our recently developed cfSNV method (12). With exome-wide sequencing and comprehensive analysis of mutations, cfTrack can sensitively identify these previously undiagnosed patients with second primary cancers, comprehensively describe their tumor status, and enable early intervention and personalization of treatment. Using both simulation data and a cohort of patients with cancer (n = 35, 18 prostate cancer, 8 lung cancer, 4 ovarian cancer, 3 glioma, 1 bladder cancer, and 1 germ cell cancer), we show that cfTrack achieves sensitive and specific monitoring of tumor MRD/recurrence and evolution from cfDNA, even with very low tumor fractions. These results demonstrate that cfTrack enables full-spectrum monitoring of cancer treatment outcomes.
Materials and Methods
Data collection
We collected WES data from four public datasets. We collected data of 18 patients with metastatic cancer from Adalsteinsson and colleagues (13) under dbGaP accession code phs001417.v1.p1. Each patient's data include a white blood cell (WBC) sample, a tumor biopsy sample, and two plasma samples. We also collected WES data of 3 patients with cancer (1 bladder cancer, 1 prostate cancer, and 1 germ cell cancer) from Tsui and colleagues (14) under NCBI BioProject accession code PRJNA679359 and WES data of 3 patients with glioma under NCBI BioProject accession code PRJNA641696. Each patient has a WBC sample, a solid tumor sample, and a plasma sample. We collected WES data of 17 patients with prostate cancer from Ramesh and colleagues (15) under NCBI BioProject accession code PRJNA554329. All patients have one WBC sample; 8 of the 17 patients have a solid tumor sample (metastatic site); 5 (7, 2, 2, and 1) patients have one (2, 3, 4, and 5, respectively) plasma sample collected at different timepoints. We also collected samples from 8 patients with non–small cell lung cancer (NSCLC) and 4 patients with ovarian cancer and generated our own WES data as described below. For all 8 patients with NSCLC, a tumor biopsy sample, a WBC sample, and three plasma samples were collected. For all 4 patients with ovarian cancer, a WBC sample and two serum samples were collected. In addition, for the patient with ovarian cancer OV4, who underwent surgical resection at the first blood collection, we collected the patient's tumor tissue sample. For all sources, only one WBC sample (or its WES data) was collected for each patient with cancer.
Human subjects
We collected blood samples, tumor samples, and WBC samples from 8 patients with NSCLC from KEYNOTE-001 (16) and KEYNOTE-010 (17), who all provided informed consent for research use. The blood and tissue collection protocols were described in the full protocol of KEYNOTE-001 and KEYNOTE-010. The project was approved by the Institutional Review Board (IRB) of University of California, Los Angeles, CA (IRB# 12-001891, IRB# 11-003066, and IRB# 13-00394) and was conducted in accordance with the Belmont Report. We also collected samples from 4 patients with ovarian cancer. Serum was harvested from whole blood by centrifugation (400 × g, 15′) and immediately flash frozen. Peripheral blood mononuclear cells were harvested from whole blood collected in a blue top phlebotomy tube with sodium citrate, centrifuged (400 × g, 15′), and aliquoted from the buffy coat before being immediately flash frozen. Portions of solid tumor from the operating room were brought back to the lab and flash frozen. Clinical information from consenting patients was obtained from medical records. Longitudinally collected clinical specimens from patients with ovarian cancer were obtained using IRB-approved protocols (IRB# 10-000727) and were studied in accordance with the Belmont Report. All patients provided written informed consent.
Genomic DNA WES library construction
For the 8 patients with NSCLC, WBC genomic DNA (gDNA) was isolated with QIAGEN DNeasy Blood & Tissue Kit. Library preparation and exon capture was performed with KAPA HyperPrep Kit and Nimblegen SeqCap EZ Human Exome Library v3.0 (Roche) and 2× 150 bp paired-end sequencing by Genewiz. For the 4 patients with ovarian cancer, WBC and tumor tissue gDNA were isolated with DNeasy Blood & Tissue Kit and sonicated by Covaris system. Ampure XP beads (Beckman-Coulter) size selection was performed to enrich 100–250 bp DNA fragments before library construction. In brief, 0.9 volume beads were added to the fragmented gDNA. After incubation, supernatant was transferred to a new tube and an additional 1.1 volume beads were added (“0.9×–1.1× bead size selection”). After wash, gDNA was eluted. gDNA WES library was constructed with SureSelect XT HS kit from Agilent Technologies according to manufacturer's protocol. In brief, 100 ng gDNA was used as input. The adaptor-ligated library was amplified with index primer in 8-cycle PCR. A total of 1,000 ng pre-capture library was hybridized to capture panel. The post-capture library was reamplified with 9-cycle PCR and 2× 150 bp paired-end sequenced by Genewiz.
Plasma cfDNA WES library construction
For each of the 8 patients with NSCLC, venipuncture was performed by trained phlebotomists. Plasma samples were isolated within 2 hours of blood collection and stored in −80°C until cfDNA extraction with QIAGEN QIAamp circulating nucleic acid kit. WES libraries were constructed with SureSelect XT HS kit. A total of 10 ng cfDNA was used, and 10-cycle PCR was performed for both pre- and post-capture libraries. A total of 700–1,000 ng pre-capture library was hybridized to the capture panel.
Serum cfDNA WES library construction
Serum cfDNA from the 4 patients with ovarian cancer was extracted by QIAamp circulating nucleic acid kit. To eliminate gDNA contamination, 0.5×–2.0× bead size selection was performed. WES libraries were constructed with SureSelect XT HS kit. Input was 5–20 ng cfDNA. Pre-capture libraries were amplified by PCR with 11 (10–20 ng input) or 12 (input less than 10 ng) cycles. Post-capture libraries were reamplified with 9-cycle PCR.
Data preprocessing
Both genomic DNA sequencing data and cfDNA sequencing data were preprocessed using the same procedure. Raw sequencing data (FASTQ files) were aligned to the hg19 reference genome by bwa mem (18) and sorted by samtools (19). Then, duplicated reads from PCR amplification were identified and removed by picard tools MarkDuplicates (20). After this step, read group information was added to the bam file using picard tools AddOrReplaceReadGroups, and reads were realigned around indels using GATK RealignerTargetCreator and IndelRealigner (21, 22). After realignment, base quality scores were recalibrated using GATK BaseRecalibrator and PrintReads. All tools in the data preprocessing pipeline were used with their default settings. After data preprocessing, the resulting bam files were used as inputs for mutation detection and MRD detection.
Comprehensive and personalized cancer monitoring using cfDNA
We present a new cancer monitoring method (Fig. 1A and B), cfTrack, that analyzes both preexisting tumor mutations and newly emerging mutations in posttreatment samples. Specifically, we collect a plasma or solid tumor sample and a matched WBC sample from a patient before the treatment to select markers that are specific to the preexisting tumor. In the posttreatment plasma samples, cfTrack both tracks preexisting tumor markers and detects new somatic mutations.
Figure 1.

Cancer monitoring in plasma samples by tracking preexisting tumor mutations and newly emerging tumor mutations. A, Illustration of the sample collection for cfDNA-based cancer monitoring. Prior to surgery or therapy, a plasma or tumor sample and a WBC sample are collected to generate the preexisting tumor profile. Serial blood samples are collected to detect MRD/recurrence and monitor tumor evolution after treatment. B, Illustration of the method workflow. In the pretreatment samples, clonal tumor mutations are identified for tumor tracking in the posttreatment samples. Given a posttreatment plasma sample, the tumor fraction is calculated from the preexisting clonal tumor mutations and compared with a sample-specific background distribution. The empirical P value of the tumor fraction is used to predict MRD/recurrence. Furthermore, de novo somatic mutations are detected using cfSNV between the posttreatment plasma and WBC samples. A second primary cancer is predicted by a logistic regression model that accounts for both the amount of de novo mutations and the corresponding tumor fraction.
Because of the low tumor fraction in the plasma samples from patients with MRD, we integrate all clonal tumor mutations from the pretreatment samples [Fig. 1B (1)]. Tumor mutations evolve, so any given somatic mutation observed in pretreatment samples may disappear in posttreatment samples. We perform WES of the pretreatment samples (solid tumor or plasma samples) and select clonal somatic mutations that appear in all preexisting cancer cells and have high variant allele frequencies (VAF; ref. 4). Compared with a predefined, limited panel of known tumor mutations, these clonal mutations, observed in WES, are more likely to appear in posttreatment samples and are more informative for monitoring the preexisting tumor (5). However, when the tumor fraction in cfDNA is very low, WES at medium depth (100× or 200×) may contain few variant supporting reads at a specific locus. Therefore, to provide a robust mutation-based statistic index in cfDNA, we aggregate variant supporting reads across all clonal somatic mutations (see Identification of clonal mutations in pretreatment samples and Supplementary Fig. S1A and S1B). We quantify the tumor fraction using the integrated VAF (IVAF), which is the sum of variant supporting reads divided by the sum of all reads at the clonal somatic mutations. Note that a recent publication developed a similar integrative approach using the reads from WGS of cfDNA (11). Here we show that WES of cfDNA can also be used for ultrasensitive cancer detection, and given its cost-effectiveness compared with WGS it is more feasible for clinical use.
To limit the accumulation of sequencing errors during integration, we suppress sequencing errors at the read level with a random forest model [Fig. 1B (2)]. When we integrate tumor reads across a large number of mutation sites to amplify the tumor signal, sequencing errors also accumulate. Therefore, we have developed a method to suppress individual sequencing errors and enhance the signal-to-noise ratio of cancer detection by differentiating the reads containing sequencing errors from those containing true variants. Specifically, this filter is based on a random forest model (see Machine learning model for suppressing sequencing errors). Previous work has shown that it is possible for machine learning to distinguish true cancer mutations from sequencing artifacts at the read level, and such filters have been used to predict mutations and detect cancer and MRD (11, 13). Unlike these previous works, our method is specifically designed for cfDNA WES data: it incorporates cfDNA fragmentation patterns and read sequence contexts (e.g., nucleotide substitution C>A). Both features are informative to distinguish tumor-derived true mutations and sequencing errors: tumor-derived cfDNA fragments are shorter than non–tumor-derived cfDNA fragments (23, 24); sequencing error rates are associated with nucleotide substitution types (25). By combining a wide variety of features (Supplementary Table S1), our model automatically discovers feature co-occurrence relationships that are associated with sequencing errors. The random forest model classifies all supporting reads at clonal somatic mutation loci as containing either a true variant or a sequencing error. Only those reads classified as “true variants” are counted as variant supporting reads.
We predict recurrence or MRD using sample-specific background noise distribution [Fig. 1B (3)]. To predict whether a patient has recurrence or MRD, we need to compare the estimated tumor fraction with a background noise distribution which represents the error allele fraction in samples from individuals without a tumor. Previous studies usually compared the posttreatment sample of a patient with a cohort of samples from healthy individuals, because the interindividual and interexperimental differences are difficult to model; however, this kind of comparison can introduce prediction bias, and the resulting detection thresholds are difficult to generalize to other experimental protocols. To avoid this limitation, we build the background noise distribution in the same sample. For a set of 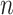 clonal tumor mutations, we calculate the IVAF repeatedly
clonal tumor mutations, we calculate the IVAF repeatedly 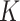 times (
times (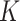 = 100) from
= 100) from 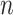 random genomic positions, excluding known mutations and positions associated with clonal hematopoiesis [see Identification of mutations and Clonal Hematopoiesis of Indeterminate Potential (CHIP) positions]. Ideally, all read pairs with nonreference alleles at random positions are from sequencing errors, so the observed frequency of these reads represents the background noise level. Therefore, we approximate the background noise distribution by the
random genomic positions, excluding known mutations and positions associated with clonal hematopoiesis [see Identification of mutations and Clonal Hematopoiesis of Indeterminate Potential (CHIP) positions]. Ideally, all read pairs with nonreference alleles at random positions are from sequencing errors, so the observed frequency of these reads represents the background noise level. Therefore, we approximate the background noise distribution by the 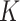 IVAFs from random positions, which represent the actual error rates observed in this specific sequencing experiment. Recurrence or MRD can then be detected using the empirical P value of the tumor fraction calculated from the preexisting clonal mutations with respect to the sample-specific background noise distribution. If the empirical P value is ≤ 0.05, the patient is regarded as having MRD/recurrence.
IVAFs from random positions, which represent the actual error rates observed in this specific sequencing experiment. Recurrence or MRD can then be detected using the empirical P value of the tumor fraction calculated from the preexisting clonal mutations with respect to the sample-specific background noise distribution. If the empirical P value is ≤ 0.05, the patient is regarded as having MRD/recurrence.
We monitor tumor evolution and newly emerging tumors by identifying newly emerging tumor mutations de novo [Fig. 1B (4)]. Previously described methods for cancer monitoring focus on a predefined mutation panel, which makes it difficult to detect tumor evolution or second primary cancers. Taking advantage of the WES data with broad genome coverage, cfTrack performs de novo mutation identification to accomplish both. For this, we utilize cfSNV (12), a method we recently developed for the sensitive and accurate calling of somatic mutations in plasma samples. cfSNV specifically accommodates key cfDNA-specific properties, including the low tumor fraction, short and nonrandomly fragmented DNA, and heterogeneous tumor content. It addresses the low tumor fraction and tumor heterogeneity in cfDNA by iterative and hierarchical mutation profiling and ensures a low false-positive rate by multilayer error suppression. On the basis of the mutation calling results from cfSNV, tumor evolution and newly emerging tumors are tracked by a logistic regression model, whose features are the tumor fraction and the number of detected mutations. The model is trained using a cohort of cancer and healthy plasma samples. A sample is predicted with evolved tumors or newly emerging tumors if its prediction score is larger than the 95th percentile of the prediction scores from the healthy samples in the training data.
Identification of clonal mutations in pretreatment samples
Tumor-derived somatic mutations are detected using cfSNV (12) from the pretreatment plasma sample; if only the pretreatment tumor sample is available, tumor-derived mutations are the common mutations detected by Strelka2 (26) somatic and MuTect (27) from the pretreatment tumor sample. The detected mutations are removed if there is at least one variant supporting read in the matched WBC sample. A mutation is considered clonal, and hence retained in the final marker list, if its VAF is > 25% of the average of the five highest VAFs in the sample (28). We require a minimum of 30 markers from the pretreatment plasma sample to obtain a robust prediction. If there are fewer than 30 clonal mutations, subclonal mutations with the highest VAFs will be included.
Identification of mutations and CHIP positions
We identify germline mutations in the pretreatment plasma sample and the matched WBC sample from the same patient using GATK HaplotypeCaller and Strelka2 Germline with the default settings. GATK HaplotypeCaller is applied to the plasma sample and the WBC sample individually; Strelka2 Germline is applied to the plasma-WBC sample pair. Somatic mutations are detected in the plasma sample and the matched WBC sample using cfSNV under default settings. The CHIP positions are identified from pileup files generated using samtools mpileup. If a nonmutated position has ≥ 3 variant supporting reads or a VAF > 1% in the matched WBC sample, it is regarded as a CHIP position. The selection of these parameters has little impact on the performance (Supplementary Fig. S2A and S2B). All the identified germline mutations, somatic mutations, and CHIP positions are excluded in the step of building the background noise distribution.
Machine learning model for suppressing sequencing errors
Although weak tumor signals in plasma samples can be amplified by integrating the variant supporting reads across a large genomic region, sequencing errors can also accumulate and possibly confound the tumor signal. Moreover, because of the low fraction of tumor DNA, the variant supporting reads at a single mutation are not sufficient to provide a robust and accurate estimation of site-level statistics (e.g., strand bias and average base quality) for error removal. Therefore, we developed a machine learning filter to eliminate reads with sequencing errors (Supplementary Fig. S3). Specifically, for a group of genomic positions (tumor mutations or random positions), we classify the variant supporting reads with a random forest model to distinguish sequencing errors from true variants. Because all data in this study were generated from paired-end sequencing, in the following section, we detail the model for paired-end reads, but the principle can also be applied to single-end reads. With paired-end sequencing data, there are two types of read pairs with regards to a specific mutation site: one (nonoverlapping read pair) covers the mutation site by one of its read mates, the other (overlapping read pair) covers the mutation site by both of its read mates (Supplementary Fig. S3A). The overlapping read pair can provide two readouts of the mutation site on the DNA fragment in the sequencing library, but the nonoverlapping read pair can only provide one readout. This means that the overlapping read pair naturally contains more information about the mutation site than the nonoverlapping read pair, and the two readouts can serve as validation for each other. Therefore, we trained two independent random forest models to fully utilize the information in the nonoverlapping read pair and the overlapping read pair. Please note that the random forest models in cfTrack classify sequencing errors and true variants in every read pair, that is, read-level error suppression. It is different from the empirical variant score model in Strelka2 and the variant quality score model in GATK, which rely on site-level statistics (such as averaged base quality in all reads) to classify sequencing errors and true variants.
To train the random forest model, we used WES data from 18 patients: 12 with metastatic breast cancer (MBC) and 6 with metastatic prostate cancer [castrate-resistant prostate cancer (CRPC); ref. 13; Supplementary Fig. S3B]. Each patient had four samples sequenced: two plasma samples (collected at two different timepoints), a WBC sample, and a tumor biopsy sample. We use the supporting cfDNA read pairs at known mutation (error) sites as the training data. The known mutation sites include both germline and somatic mutation sites, where germline mutations are required to be detected in all four samples using Strelka2 germline, and somatic mutations are required to be detected from both the cfDNA-WBC pairs (cfDNA data vs. WBC data) and the tumor-WBC pair (tumor data vs. WBC data) using Strelka2 somatic and MuTect. Error sites are defined as sufficiently covered sites (>150×) with at most two high-quality nonreference read (base quality ≥ 20 and mapping quality ≥ 40) in only one of the four datasets. All high-quality labeled read pairs (base quality ≥ 30 and mapping quality ≥ 40) were extracted from raw cfDNA data using picard tools FilterSamReads. Multiple read pairs may be extracted covering the same mutation site, but these read pairs are similar and might cause redundancy in the training and testing data. Therefore, we solved the redundancy problem by retaining only one read pair per mutation/error site (Supplementary Table S2). Different features were extracted from the overlapping read pairs and the nonoverlapping read pairs (Supplementary Table S1). All categorical features were expanded using the one-hot encoding method. The hyperparameters of the random forest model were as follows: (i) the number of decision trees was 100, (ii) the maximum tree depth was 50, (iii) imbalanced classes were addressed by setting the class weights to “balanced,” and (iv) other parameters were left at their default values. Two separate random forest classifiers (one for overlapping read pairs and one for nonoverlapping read pairs) were trained on the extracted read pairs.
We validated the performance of the random forest model by cross-validation. For each patient, the labeled read pairs from the 17 other patients were used to train the model, while the patient's own data were used to test the model (results shown in Supplementary Fig. S4). The training data of the random forest model in all the simulation (MRD/recurrence and second primary cancers) also exclude the patient used for generating the simulation data to avoid data leakage. Therefore, the evaluation of cfTrack is independent of the training data. As an independent validation set, we used a group of patients with NSCLC (8 patients each with three samples) with sequential plasma cfDNA samples. The read pairs in these cfDNA samples were labeled in the same manner as described above. Then, these labeled read pairs were used as independent testing data for the random forest model trained by the data generated from the 12 patients with MBC and 6 patients with CRPC (results shown in Supplementary Fig. S5). On all cross-validation datasets, the random forest model can accurately distinguish sequencing errors from true variants (average AUC = 0.95, 95% confidence interval = 0.9496–0.9503).
Simulation of recurrence and MRD detection by tracking clonal somatic mutations in pretreatment samples
To evaluate the performance of our method, we generated simulation data to mimic patients with MRD/recurrence and patients with complete remission. The patients with MRD/recurrence have tumor content in the posttreatment plasma sample and will show the detection sensitivity; the patients with complete remission have no tumor content in the posttreatment plasma sample and will show the detection specificity. The simulation data were generated from two datasets independently: (i) validation dataset, 27 patients with MBC and 14 patients with CRPC and (ii) independent dataset, 8 patients with NSCLC.
In the validation dataset, only 12 patients with MBC and 6 patients with CRPC have two plasma cfDNA samples, so only these patients were used to generate the posttreatment cfDNA samples from the patients with MRD/recurrence. Note that these data were also used to generate the training data for the read-level error suppression model. Therefore, to avoid data leakage in the performance evaluation, the MRD/recurrence detection on the validation dataset was performed in a “leave-one-patient-out cross-validation” manner. In other words, for a simulated sample (generated from WES data from a specific patient) in the validation dataset, the random forest models used in the error suppression step were trained on the other 17 patients. In the independent dataset, the 8 patients with NSCLC have three plasma cfDNA samples. Only the first two timepoints of the plasma cfDNA samples were used in the simulation. These data were untouched and independent of the training of the read-level error suppression model, so the error suppression model used on the independent dataset was trained by all training data extracted from the 12 patients with MBC and 6 patients with CRPC.
To demonstrate the sensitivity of detection for preexisting cancer, we generated in silico dilution series to simulate patients with MRD/recurrence by mixing the plasma sample collected at the second timepoint and the matched WBC sample at varying concentrations of cfDNA reads (0.01%, 0.05%, 0.1%, 0.3%, 0.5%, 0.8%, 1%, 3%, 5%, and 8%) using samtools view and samtools merge. Five independent mixtures were generated at every concentration, at theoretical depths of 200×, 100×, or 50× on the WES targeted regions. Because read sampling is random, it is possible that there is no variant supporting read at a given marker, even across all markers. Thus, we removed samples with no variant supporting reads at all personalized markers (checked by samtools mpileup). In this simulation, the original matched WBC samples and the original plasma samples at the first timepoint were used as the WBC samples and the pretreatment plasma samples, respectively (Fig. 2). The in silico dilution series represents posttreatment plasma samples from patients with MRD/recurrence. For the validation dataset, we generated the data for each of the 12 patients with MBC and 6 patients with CRPC. The theoretical tumor fraction in each sample is calculated as the product of the original tumor fraction in the cfDNA sample and the dilution. The theoretical tumor fraction ranges from 0.001% to 6.114%, with a median of 0.270%. For the independent dataset, we generated the data for each of the 8 patients with NSCLC. The theoretical tumor fraction ranges from 0.001% to 1.867%, with a median of 0.103%. The different ranges of the theoretical tumor fractions in the two datasets are caused by differences in the tumor content levels in the original plasma samples. Note that the theoretical tumor fraction usually overestimates the true tumor fraction because of random sampling and the imperfect on-target rate.
Figure 2.

Settings to generate in silico spike-in simulation data. The simulation data are generated using WES data taken from (i) 12 patients with MBC and 6 patients with CRPC and (ii) 8 patients with NSCLC. Each patient has an early plasma sample (Blood T1), a WBC sample (WBC), and a late plasma sample (Blood T2). The three WES datasets from a patient are used directly or mixed to generate the simulation samples. To simulate the scenario of monitoring a patient for MRD or cancer recurrence, each case contains three simulation samples: a pretreatment plasma sample, a pretreatment WBC sample, and a posttreatment plasma sample. The raw data from Blood T1 are used directly as the pretreatment plasma sample for all cases. WBC and Blood T2 are mixed at specified dilutions to simulate the posttreatment plasma sample. To simulate remission cases, we generate two independent random samplings from the raw WBC data to use as the pretreatment WBC sample and the posttreatment plasma sample. To simulate the emergence of second primary cancers, each case contains two simulation samples: a pretreatment WBC sample and a posttreatment plasma sample. The generation of simulation samples for second primary cancer monitoring is the same as for MRD/recurrence monitoring, except that the pretreatment plasma sample (Blood T1) is not used.
To evaluate the specificity of the MRD detection pipeline, we generated the patients with complete remission by subsampling from the original WBC samples. Therefore, these subsamples are expected to have no tumor DNA. For a WBC sample from a patient with cancer, five subsamples were generated for each of 200×, 100×, and 50× theoretical depth of the targeted regions. These subsamples represent posttreatment plasma samples from patients without MRD. The original plasma samples at the first timepoint were used as the pretreatment plasma samples. Note that only one WBC sample was available for each patient. If the only original WBC sample was directly used as the pretreatment WBC sample, all data in the posttreatment plasma samples (i.e., subsamples) would have been observed in the pretreatment WBC sample (i.e., the full original sample), which is impossible in reality. Therefore, we used another subsample of the original WBC samples as the pretreatment data at a sampling rate of 95% (Fig. 2). In this simulation, we preserved some randomness between the WBC samples and the posttreatment plasma samples, which reflects real cases. For the validation dataset, we generated the remission samples for each of the 27 patients with MBC and 14 patients with CRPC. For the independent dataset, we generated the remission samples for each of the 8 patients with NSCLC.
To avoid potential bias from independently sampling replicates from the same patients, we randomly selected one replicate at every dilution (including 0% for remission samples) for every patient to calculate the performance (AUC, sensitivity, and specificity). After the selection, the performance metrics (AUC, sensitivity, and specificity) were evaluated on the MRD/recurrence samples grouped by the tumor fraction with a 0.01% step size and the remission samples (samples with WBC reads only). To provide a robust estimate, we randomly selected samples and calculated the performance 50 times. For the validation dataset, in each random selection, there are 41 simulated remission samples at each depth. At 200×, there are 143 simulated MRD/recurrence samples; at 100×, there are 142 simulated MRD/recurrence samples; at 50×, there are 128 simulated MRD/recurrence samples. For the independent dataset, in each random selection, there are eight simulated remission samples at each depth. At 200×, there are 68 simulated MRD/recurrence samples; at 100×, there are 65 simulated MRD/recurrence samples; at 50×, there are 56 simulated MRD/recurrence samples.
Simulation of second primary cancer detection
Similar to the simulation of recurrence and MRD detection, to evaluate the sensitivity of the method for second primary cancer detection, we generated an in silico dilution series by mixing the plasma samples at the second timepoint and the matched WBC samples from the 12 patients with MBC and 6 patients with CRPC at varying concentrations of cfDNA reads (from 1% to 10%: 1%, 3%, 5%, 8%, and 10%) using samtools view and samtools merge. Because no training and testing of new models is performed in the detection of second primary cancers, this is an independent testing dataset with respect to the detection method. Each spike-in sample contained a total number of randomly sampled reads theoretically equivalent to 200× depth of the targeted regions. Five independent mixtures were generated at every concentration. The tumor fraction in these spike-in samples was quantified by the variant supporting reads at the clonal somatic mutations identified in the original plasma sample. In this simulation, the original matched WBC samples were used as the WBC samples. To demonstrate the specificity of the method, we reused the complete remission samples at 200× generated in the simulation of recurrence and MRD detection. To avoid potential bias from independently sampling replicates from the same patients, we randomly selected one replicate at every dilution for every patient to calculate the performance (AUC, sensitivity, and specificity). To provide a robust estimate, we randomly selected samples and calculated the performance 10 times. In each random selection, there were 90 simulated samples from patients with second primary disease and 41 simulated samples from patients with complete remission. To evaluate the performance, after removing the replicates, the simulation data were randomly split into the training set (50%, n = 66) and the testing set (50%, n = 65) 10 times. A logistic regression model is trained on the training set and used to predict the presence of a second primary cancer in the testing set. The performance metrics (AUC, sensitivity, and specificity) are evaluated in the testing set on the second primary cancer samples grouped by tumor fraction with a 0.1% step size, but always using the complete set of remission samples.
Code availability statement
cfTrack is implemented in Python and is freely available for academic and research usage through https://zhoulab.dgsom.ucla.edu/pages/cfTrack.
Data availability statement
The data generated in this study are publicly available in the European Genome-Phenome Archive under the accession EGAD00001008454. The public data analyzed in this study were obtained from dbGaP under accession code phs001417.v1.p1 and from Sequence Read Archive under NCBI BioProject accession codes PRJNA679359, PRJNA641696, and PRJNA554329.
Results
Analytic performance of detecting cancer recurrence and MRD
To evaluate the performance of cfTrack on cancer MRD or recurrence, we use the in silico method of preparing spike-in simulation data. If a patient with cancer has cancer recurrence or MRD, the posttreatment plasma of the patient will contain DNA corresponding to the preexisting tumor. To simulate the posttreatment plasma samples from the patients with cancer recurrence or MRD, we computationally mix a plasma sample from a patient with cancer with a WBC sample from the same patient. The data, with known dilution ratios, can provide a sensitivity/specificity assessment of cfTrack.
We generated two sets of in silico spike-in simulation data: (i) validation dataset, using the WES data from 12 patients with MBC and 6 patients with CRPC (13) and (ii) independent dataset, using the WES data from 8 patients with NSCLC. For both datasets, each patient has sequencing data from two plasma samples (collected at two different timepoints T1 and T2, with 14–138 days in between for patients with MBC and CRPC, 42 days in between for patients with NSCLC), and the matched WBC sample. These patients underwent treatment between T1 and T2, so we consider the first plasma samples (at T1) the “pretreatment” samples, and the second plasma samples (at T2) the “posttreatment” samples. Tens to hundreds of clonal somatic mutations (for patients with MBC and CRPC, ranging from 49 to 674 with median 94; for patients with NSCLC, ranging from 30 to 1,239 with median 63) are found in the pretreatment samples when compared with their matched WBC samples. We then generated an in silico dilution series for each patient by mixing their posttreatment plasma sample with the matched WBC sample at varying fractions (the theoretical tumor fraction ranges from 0.001% to 6.114% with median 0.270% for the validation dataset, from 0.001% to 1.867% with median 0.103% for the independent dataset; see Material and Methods and Fig. 2). In addition, we simulated patients who achieved complete remission by subsampling the original WBC samples (the tumor fraction is 0%, see Material and Methods and Fig. 2). The simulation data were generated at three different depths, 50×, 100×, and 200×.
When applying cfTrack to the simulated datasets, we observed slightly increased detection performance with increasing sequencing depth (Fig. 3A–D; Supplementary Fig. S6A–S6D). This trend is expected because the higher the sequencing depth, the more tumor DNA fragments can be captured. Specifically, on the validation dataset, we achieved an average AUC of 99% (SD = 1%) when the tumor fraction was ≥ 0.05% at 200× depth (Fig. 3A; Supplementary Fig. S6A), with 100% average sensitivity (SD = 0%) and 96% average specificity (SD = 1%; Fig. 3B; Supplementary Fig. S6B). On the independent dataset, we achieved an average AUC of 100% (SD = 0%) when the tumor fraction was ≥ 0.05% at 200× depth (Fig. 3C; Supplementary Fig. S6C), with 89% average sensitivity (SD = 13%) and 100% average specificity (SD = 0%; Fig. 3D; Supplementary Fig. S6D). Considering the difference in the sample size and the higher specificity in the independent dataset, the performance on the two simulation datasets is comparable. This indicates that our method can achieve sensitive monitoring using only 200× WES data, offering a cost-effective solution for MRD detection. The detection limit can be further enhanced by increasing the sequencing depth.
Figure 3.
![Figure 3. Performance of cancer recurrence and MRD detection using the simulation data. The area under the ROC curve (AUC) of the MRD/recurrence detection on the validation dataset (A) and the independent dataset (C) with different tumor fractions and sequencing depths. The sensitivity and specificity with different tumor fractions and sequencing depth on the validation dataset (B) and the independent dataset (D). Supplementary Figure S6A–S6D is the zoom-in of A–D at low tumor fraction ranging from 0% to 0.2%. E, AUCs of MRD/recurrence detection with and without error suppression (ES) on the validation dataset at 200× depth with different tumor fractions. F, The sensitivity and specificity of MRD/recurrence detection with and without error suppression on the validation dataset at 200× depth with different tumor fractions. In A, C, and E, the dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). In B, D, and F, the dots show the average sensitivity using a cut-off P value = 0.05 for the background noise distribution; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the legend in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with logit functions.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9365349_1841fig3.jpg)
Performance of cancer recurrence and MRD detection using the simulation data. The area under the ROC curve (AUC) of the MRD/recurrence detection on the validation dataset (A) and the independent dataset (C) with different tumor fractions and sequencing depths. The sensitivity and specificity with different tumor fractions and sequencing depth on the validation dataset (B) and the independent dataset (D). Supplementary Figure S6A–S6D is the zoom-in of A–D at low tumor fraction ranging from 0% to 0.2%. E, AUCs of MRD/recurrence detection with and without error suppression (ES) on the validation dataset at 200× depth with different tumor fractions. F, The sensitivity and specificity of MRD/recurrence detection with and without error suppression on the validation dataset at 200× depth with different tumor fractions. In A, C, and E, the dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). In B, D, and F, the dots show the average sensitivity using a cut-off P value = 0.05 for the background noise distribution; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the legend in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with logit functions.
Our method can achieve high detection power thanks to three key features: the exome-wide integration of tumor signals, the sample-specific decision threshold, and the read-level error suppression. Read-level error suppression greatly improves the detection power, especially in samples with a low tumor fraction. For example, based on our in silico samples with a 0.05% tumor fraction, employing read-level error suppression improved the AUC by 35% on the validation dataset (see Fig. 3E and F) and improved the AUC by 40% on the independent dataset (see Supplementary Figs. S6E and S2F).
Analytic performance of detecting second primary cancers
Sensitive monitoring of tumor evolution and newly emerging tumors requires the de novo detection of mutations from previously unobserved tumors. Pretreatment plasma samples and tumor biopsy samples cannot provide sufficient tumor markers for this purpose. In contrast with previous cancer monitoring methods, we can detect de novo tumor-derived single-nucleotide variants in the posttreatment plasma samples, which allow us to identify mutations that come from new tumors. In this section, we specifically evaluate cfTrack for the detection of second primary cancers, which depends solely on the detection of emerging tumors.
Detecting a second primary cancer is equivalent to detecting a new tumor without prior knowledge. To simulate this scenario, we generate an in silico dilution series from the 12 patients with MBC and 6 patients with CRPC by mixing their posttreatment plasma samples with the matched WBC samples (13). The mixed samples are prepared at varying fractions (the theoretical tumor fraction ranges from 0.111% to 7.680%, with a median of 2.984%; see Material and Methods and Fig. 2). For each dilution level, simulation data are generated with a depth of 200×. The samples simulating complete remission are the same as those used for MRD/recurrence detection (in the previous section). Because the detection of a second primary cancer involves no training or testing of new models, this simulation dataset is an independent dataset with respect to the detection method. In this simulation, we do not use the pretreatment plasma samples, representing the scenario where no preexisting tumor profile has been observed.
For each pair of simulated plasma and simulated WBC samples, we use cfSNV to identify somatic mutations. Then cfSNV estimates a tumor fraction from these mutations. We predict a second primary cancer by a logistic regression model using both the tumor fraction and the number of detected mutations as features. We randomly split the samples into a training set (50%) and a testing set (50%). A patient is predicted to have a second primary cancer if they have a large prediction score (≥95th percentile of prediction scores from the remission samples in the training set). The AUC is calculated on the basis of the prediction results in the testing sets for all complete remission samples and for the subset of simulation samples with a specific tumor fraction (see Material and Methods). We achieve an average AUC of 88% (SD = 10%) when tumor fraction ≥ 0.2% at 200× depth (Fig. 4A), with an average sensitivity of 76% (SD = 23%) and an average specificity of 93% (SD = 5%; Fig. 4B). The sensitivity of the methodology is lower for detecting second primary cancers than for detecting recurrence and MRD, because no preexisting tumor information is available and all novel somatic mutations need to be confirmed. The detection of a novel somatic mutation requires more variant supporting reads than just observing a weak signal at a known locus. Nevertheless, cfTrack still achieves high performance in detecting a new tumor. Therefore, cfTrack can be used for monitoring tumor evolution and detecting second primary cancers and cancer progression.
Figure 4.
![Figure 4. Performance of second primary cancer detection with the simulation data. A, AUC of the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). B, The sensitivity and specificity in the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots show the average sensitivity using a cutoff of the 95th percentile of prediction scores from the remission samples in the training data; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the text in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with a logit function.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9365349_1841fig4.jpg)
Performance of second primary cancer detection with the simulation data. A, AUC of the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). B, The sensitivity and specificity in the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots show the average sensitivity using a cutoff of the 95th percentile of prediction scores from the remission samples in the training data; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the text in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with a logit function.
Monitoring tumors in patients with cancer on treatments through cfDNA
Developments in immunotherapy and targeted therapy have improved the outcomes of patients with cancer in recent years (29, 30, 31). For example, immunotherapy, which activates a patient's own immune system to fight cancer, has remarkably improved clinical outcomes in a subset of patients with NSCLC (32). Despite these results, the majority of patients eventually develop resistance and fail to respond to treatment (33–35). Therefore, it is essential to closely monitor the response of patients and quickly recognize when the need for alternative treatment arises. However, because the development of resistance may be associated with tumor evolution (36), this type of monitoring cannot only rely on markers derived from the preexisting tumor, but requires constant reevaluation of the tumor profile during treatment. Our WES-based method, which detects mutations from both pretreatment and treated samples, can comprehensively track a patient's response.
To test our method in this clinical scenario, we not only collected samples from our cancer patients but also exhaustively surveyed available datasets from public databases. Specifically, we applied our cancer monitoring method to plasma/serum samples (n = 76, eight serum samples for 4 patients with ovarian cancer and 68 plasma samples for other patients) from a cohort of patients with cancer (n = 35) who received various treatments. This cohort contains 18 patients with prostate cancer (14, 15), 8 patients with lung cancer, 4 patients with ovarian cancer, 3 patients with glioma, 1 patient with bladder cancer (14), and 1 patient with germ cell cancer (14). All plasma/serum samples were collected when the patients did not have complete remission or had recurrence, so tumor content was expected in all samples. After applying our method, tumor-derived DNA was detected in all cfDNA samples except three plasma samples from patients with glioma (Supplementary Fig. S7). Because the detection of tumor-derived cfDNA is only possible in a very small fraction of patients with glioma due to the blood–brain barrier (37), our results were reasonable and consistent with the literature.
Among the 35 patients, 8 patients with NSCLC, 4 patients with ovarian cancer, and 12 patients with prostate cancer have at least two plasma/serum samples collected at different timepoints, between which the patients received treatments. To monitor the tumor changes in these patients, two tumor fractions are calculated separately for the preexisting tumor mutations (preexisting tumor fraction) and for the de novo tumor mutations (de novo tumor fraction) from cfTrack. The two tumor fractions allow us to track possible tumor mutations during treatment.
The 8 patients with NSCLC received anti-PD-1 immunotherapy and their plasma samples were collected from each patient at 0 (baseline), 6, and 12 weeks, measured from the start of treatment. Among these patients, 4 are “durable responders” whose progression-free survival (PFS) is longer than 18 months; the other 4 patients are “early progressors” whose PFS is shorter than 6 months (see Supplementary Table S3). In general, we observed a decreasing or low tumor fraction in the durable responders and an elevated tumor fraction in the early progressors (Fig. 5A). An unusual example in the sample is early progressor LC-2, whose preexisting tumor fraction remained at a low level during immunotherapy treatment, while de novo tumor fraction increased. This implies a potential clonality change during treatment. In other words, the responding clone might have shrunk while the other clones grew. Existing cancer monitoring methods, which do not consider newly emerging mutations, could not have recognized this tumor growth and would have misled further treatments.
Figure 5.

Longitudinal cfDNA monitoring in patients with cancer who received treatments. The lines show the tumor fraction in cfDNA during treatment. A, Tumor fraction in plasma samples of 8 patients with NSCLC who received anti-PD-1 immunotherapy. B, Tumor fraction in serum samples of 4 patients with ovarian cancer. C, Tumor fraction in plasma samples of 12 patients with prostate cancer.
The 4 patients with ovarian cancer received chemotherapy (OV1, OV2, and OV3) or chemotherapy and surgery (OV4) between the collection of two serum samples (Supplementary Table S4). At the time of the second collection, patients OV1, OV2, and OV3 underwent surgery. Surgical and pathologic findings demonstrated a moderate treatment effect from chemotherapy. We observed a decrease in both tumor fractions using cfTrack (Fig. 5B), which indicated a decline in tumor burden. Patient OV4 had a recurrence after chemotherapy and surgery at the time of the second serum collection. Consistently, we observed an increase in both tumor fractions (Fig. 5B). Therefore, our results are consistent with the clinical outcomes of these patients.
We also tracked the tumor changes in the 12 patients with prostate cancer who received various treatment types during the time between the two plasma collections. During treatment, 9 patients (P8, P9, P10, P14, P15, P16, P18, P19, and P20) had clonal expansion and 3 patients (P6, P17, and P21) had persistent clones (15). The clonality change can be reflected by the discordance of the two estimated tumor fractions. In general, we observed discordance between the two tumor fractions in the majority of the patients with clonal expansion (Fig. 5C). There are no or only minor differences between the two tumor fractions in the patients with relatively stable clones (Fig. 5C). These observations are consistent with those from the patients with NSCLC.
From the analysis of this heterogeneous cohort of patients with cancer with different cancer types and various treatments, we showed that our method can not only closely track the change in tumor fraction, but also detect changes in mutation clonality. The latter is essential for the detection of resistance clones to promptly guide subsequent treatments, but it cannot be achieved by existing cancer monitoring methods.
Discussion
Cancer monitoring is essential to assess the effectiveness of treatment and improve the life quality of patients with cancer. Unlike traditional tumor biopsies, cfDNA can provide noninvasive and continuous monitoring of patients with cancer, but the very low tumor content of cfDNA remains a major challenge. Most current cfDNA-based methods rely on deeply sequencing a small gene panel to detect the weak tumor signal, but this approach cannot comprehensively cover the patient population or detect evolving tumors. Therefore, we have developed a new cfDNA-based cancer monitoring method that can effectively and sensitively track changes in tumors, detect cancer MRD/recurrence, and identify the presence of a second primary cancer. We present a new computational method for cancer monitoring using cfDNA WES data to overcome the limitations of previous methods. Taking advantage of the wide genome coverage of WES data, cfTrack (i) enhances the tumor signal by integrating a large number of clonal tumor mutations identified in pretreatment samples; (ii) suppresses sequencing errors at the read level with an accurate random forest model; (iii) builds sample-specific background noise distributions to predict MRD/recurrence, avoiding biases due to interindividual and interexperimental variations; and (iv) detects tumor evolution and second primary cancers by de novo identifying emerging tumor mutations.
Combining these techniques, cfTrack achieves sensitive and specific detection of recurrence, MRD, and second primary cancers. In detecting recurrence in samples with a 0.05% tumor fraction, cfTrack achieved an AUC of 99% (100% sensitivity and 96% specificity) on the validation dataset and an AUC of 100% (89% sensitivity and 100% specificity) on the independent dataset. In detecting second primary cancers in samples with a 0.2% tumor fraction, cfTrack yielded an AUC of 88% (76% sensitivity and 93% specificity). Because the performance of the method increases with the sequencing depth, these results can be further improved in practice. To evaluate cfTrack in clinical scenarios, we not only collected samples directly from our patients with cancer, but also exhaustively surveyed and utilized available datasets in public databases. On these data, we show that cfTrack achieved accurate and comprehensive monitoring of the changes in tumors for patients with different cancer types and undergoing various treatments, which cannot be accomplished by methods focusing only on a small panel of mutations from pretreatment tumor samples.
This study has its limitations. First, cfTrack has only been validated and evaluated using in silico spike-in simulation data and on a limited number of patients with cancer. To address this limitation, we generated simulation data that mimic real scenarios, including tumor evolution during treatment. For example, simulated plasma samples with varying tumor contents are generated by subsampling the original plasma sample from the second timepoint, which already contains a different tumor profile compared with the sample at baseline. Nevertheless, we acknowledge that real cases of MRD, recurrence, and second primary cancers could be more complicated. Applying cfTrack to larger datasets would enable a more comprehensive evaluation and possible optimization of parameters. Second, tumor fraction is calculated as an average across all reads for a predefined list of tumor markers. Tumor evolution and tumor heterogeneity could bias the selection of markers, resulting in the absence of important variant supporting reads in the posttreatment cfDNA samples and causing the model to infer a lower tumor fraction. Third, given the medium depth of WES data and the low tumor fraction in the cfDNA samples, cfTrack focuses on tracking the overall tumor changes rather than specific clones/subclones. For the same reason, cfTrack can detect de novo mutations to monitor newly emerging tumors, but it does not guarantee the detection of specific variants directly related to treatment targets.
In this study, for some patients, we use plasma samples to detect the preexisting tumor mutations, with no need for solid tumor biopsy samples. This is possible as long as the tumor content in plasma samples is sufficient for mutation detection. For patients who receive surgical tumor removal or for patients whose tumor biopsy samples are available, our method can also use a solid tumor sample to identify the preexisting tumor mutations. However, it is worth noting that a plasma sample may still offer a more comprehensive mutation profile than a biopsy sample (38). In practice, given a pretreatment blood sample or a solid tumor sample of a patient, we envision cfTrack being used on this patient's posttreatment blood sample to predict MRD/recurrence and the second primary cancers. To predict MRD/recurrence, cfTrack uses a within-sample error distribution, which does not rely on any baseline samples. To predict the second primary cancers, our classification model has only two variables, therefore only a limited number of training samples are needed to achieve a good performance.
Currently, cfTrack utilizes tumor somatic mutations to detect cancer. In a future version, more cancer-specific features in cfDNA can be incorporated. Recent studies have discovered that copy-number variations, fragment length, and jagged ends of cfDNA are all associated with tumor-derived cfDNA. In our random forest model, we incorporated the fragment length of the DNA fragments to discriminate true variants from sequencing errors. By integrating other features, we may further empower cancer monitoring to provide actionable information and treatment guidance for patients.
Authors' Disclosures
S. Li reports other support from EarlyDiagnostics Inc. outside the submitted work; in addition, S. Li has a patent for US20210125683A1 pending. W. Zeng reports personal fees from EarlyDiagnostics Inc. during the conduct of the study, as well as personal fees from EarlyDiagnostics Inc. outside the submitted work. X. Ni reports grants from NCI during the conduct of the study and is an employee at EarlyDiagnostics Inc. Y. Zhou reports grants from NIH and personal fees from EarlyDiagnostics Inc. outside the submitted work. E.B. Garon reports grants from NCI during the conduct of the study. E.B. Garon also reports grants and personal fees from ABL Bio, Bristol Myers Squibb, Eli Lilly, EMD Serono, Merck, and Novartis; grants from AstraZeneca, Dynavax Technologies, Genentech, Biotherapeutics, and Mirati; and personal fees from Boehringer Ingelheim, Dracen Pharmaceuticals, Eisai, Gilead, GSK, Natera, Neon Therapeutics, Personalis, Regeneron, Sanofi, Shionogi, and Xilio Therapeutics outside the submitted work. In addition, E.B. Garon has a patent for PCT/US2021/071754 pending to University of California. S.M. Dubinett reports other support from EarlyDiagnostics Inc. during the conduct of the study, as well as other support from LungLife AI outside the submitted work. W. Li reports grants from NIH and EarlyDiagnostics Inc. during the conduct of the study, as well as personal fees and other support from EarlyDiagnostics Inc. outside the submitted work; in addition, W. Li has a patent for US20210125683A1 pending to EarlyDiagnostics Inc. W. Li is a co-founder and a shareholder of EarlyDiagnostics Inc. X.J. Zhou reports grants from NIH and EarlyDiagnostics Inc. during the conduct of the study, as well as other support from EarlyDiagnostics Inc. outside the submitted work; in addition, X.J. Zhou has a patent for US20210125683A1 pending to EarlyDiagnostics Inc. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
This work was supported by the NCI U01 CA230705, to X.J. Zhou; NCI R01 CA246329, to X.J. Zhou, S.M. Dubinett, and W. Li; NCI U01 CA237711, to W. Li; NCI R43 CA246941, to X. Ni; NCI R01 CA208403, to E.B. Garon; the National Science Foundation Graduate Research Fellowship DGE-1418060, to M.L. Stackpole; and Department of Veterans Affairs Merit Award I01BX004651, to S. Memarzadeh and A. Neal.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
S. Li: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. W. Zeng: Data curation, formal analysis, investigation, writing–review and editing. X. Ni: Data curation, formal analysis, investigation, writing–review and editing. Y. Zhou: Data curation, formal analysis, investigation, writing–review and editing. M.L. Stackpole: Data curation, writing–review and editing. Z.S. Noor: Resources, data curation, writing–review and editing. Z. Yuan: Data curation, investigation, writing–review and editing. A. Neal: Resources, writing–review and editing. S. Memarzadeh: Conceptualization, resources, writing–review and editing. E.B. Garon: Conceptualization, resources, data curation, writing–review and editing. S.M. Dubinett: Writing–review and editing. W. Li: Conceptualization, formal analysis, validation, investigation, writing–original draft, writing–review and editing. X.J. Zhou: Conceptualization, supervision, writing–original draft, project administration, writing–review and editing.
References
- 1. Mahvi DA, Liu R, Grinstaff MW, Colson YL, Raut CP. Local cancer recurrence: the realities, challenges, and opportunities for new therapies. CA Cancer J Clin 2018;68:488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar SK, Rajkumar SV. The current status of minimal residual disease assessment in myeloma. Leukemia 2014;28:239–40. [DOI] [PubMed] [Google Scholar]
- 4. Murtaza M, Dawson S-J, Pogrebniak K, Rueda OM, Provenzano E, Grant J, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun 2015;6:8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133. [DOI] [PubMed] [Google Scholar]
- 7. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016;8:346ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McDonald BR, Contente-Cuomo T, Sammut S-J, Odenheimer-Bergman A, Ernst B, Perdigones N, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med 2019;11:eaax7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbosh C, Frankell A, Garnett A, Harrison T, Weichert M, Licon A, et al. Phylogenetic tracking and minimal residual disease detection using ctDNA in early-stage NSCLC: a lung TRACERx study [abstract]. In: Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; 2020 Apr 27–28 and Jun 22–24. Philadelphia (PA): AACR; Cancer Res 2020;80(16 Suppl):Abstract nr CT023. [Google Scholar]
- 10. Wan JCM, Heider K, Gale D, Murphy S, Fisher E, Mouliere F, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med 2020;12:eaaz8084. [DOI] [PubMed] [Google Scholar]
- 11. Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med 2020;26:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S, Noor ZS, Zeng W, Stackpole ML, Ni X, Zhou Y, et al. Sensitive detection of tumor mutations from blood and its application to immunotherapy prognosis. Nat Commun 2021;12:4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017;8:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsui DWY, Cheng ML, Shady M, Yang JL, Stephens D, Won H, et al. Tumor fraction-guided cell-free DNA profiling in metastatic solid tumor patients. Genome Med 2021;13:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramesh N, Sei E, Tsai PC, Bai S, Zhao Y, Troncoso P, et al. Decoding the evolutionary response to prostate cancer therapy by plasma genome sequencing. Genome Biol 2020;21:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med 2015;372:2018–28. [DOI] [PubMed] [Google Scholar]
- 17. Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet North Am Ed 2016;387:1540–50. [DOI] [PubMed] [Google Scholar]
- 18. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Broad Institute. Picard tools. 2019. Available from: https://broadinstitute.github.io/picard/.
- 21. Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017:201178. [Google Scholar]
- 22. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A 2015;112:E1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mouliere F, Rosenfeld N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. Proc Natl Acad Sci U S A 2015;112:3178–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma X, Shao Y, Tian L, Flasch DA, Mulder HL, Edmonson MN, et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol 2019;20:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 27. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strickler JH, Loree JM, Ahronian LG, Parikh AR, Niedzwiecki D, Pereira AAL, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov 2018;8:164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eisenhauer EA. Real-world evidence in the treatment of ovarian cancer. Ann Oncol 2017;28:viii61–5. [DOI] [PubMed] [Google Scholar]
- 30. Nevedomskaya E, Baumgart SJ, Haendler B. Recent advances in prostate cancer treatment and drug discovery. Int J Mol Sci 2018;19:1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med 2020;383:640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horvath L, Thienpont B, Zhao L, Wolf D, Pircher A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC)-novel approaches and future outlook. Mol Cancer 2020;19:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res 2009;15:7471–8. [DOI] [PubMed] [Google Scholar]
- 35. Raguz S, Yagüe E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer 2008;99:387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol Detect Quantif 2019;17:100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are publicly available in the European Genome-Phenome Archive under the accession EGAD00001008454. The public data analyzed in this study were obtained from dbGaP under accession code phs001417.v1.p1 and from Sequence Read Archive under NCBI BioProject accession codes PRJNA679359, PRJNA641696, and PRJNA554329.