

SUMMARY
Rationally sequencing and combining PD-1/L1- and MAPK-targeted therapies may overcome innate and acquired resistance. Since increased clinical benefit of MAPK inhibitors (MAPKi) is associated with prior immune checkpoint therapy, we compare the efficacies of sequential and/or combinatorial regimens in subcutaneous murine models of melanoma driven by BrafV600, Nras, or Nf1 mutations as well as colorectal and pancreatic carcinoma driven by KrasG12C. Anti-PD-1/L1 lead-in preceding MAPKi combination optimizes response durability by promoting pro-inflammatory polarization of macrophages and clonal expansion of IFNγhi, CD8+ cytotoxic and proliferative (versus CD4+ regulatory) T cells that highly express activation genes. Since therapeutic resistance of melanoma brain metastasis (MBM) limits patient survival, we demonstrate that sequencing anti-PD-1/L1 therapy before MAPKi combination suppresses MBM and improves mouse survival with robust T-cell clonal expansion in both intracranial and extracranial metastatic sites. We propose clinically testing brief anti-PD-1/L1 (± anti-CTLA-4) dosing prior to MAPKi co-treatment to suppress therapeutic resistance.
eTOC blurb
Wang et al. couple in vivo preclinical therapeutic testing with temporal single-immune cell and T-cell clonotype analysis to identify a sequential-combinatorial regimen and cellular effectors associated with the most durable control of tumor growth and brain metastasis. Initiating immune checkpoint therapy briefly before adding MAPK-targeted therapy may improve patient survival.
Graphical Abstract
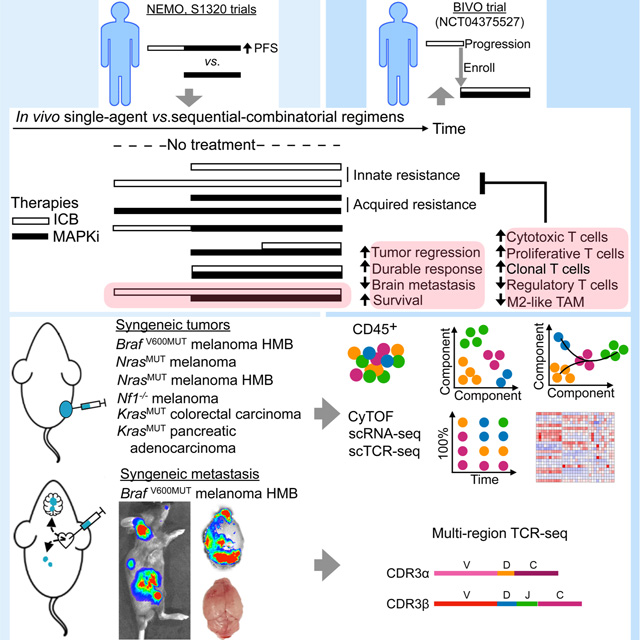
INTRODUCTION
Programmed cell death protein-1/programmed death-ligand 1 (PD-1/PD-L1 or PD-1/L1)- and mitogen-activated protein kinase (MAPK)-targeted therapies have revolutionized the treatment of BRAFV600MUT melanoma and beyond. For MAPK inhibitors (MAPKi) consisting of BRAF inhibitor (BRAFi) + MEK inhibitor (MEKi), 5-year survival is under 30%, and acquired resistance occurs within one year in the majority of patients with BRAFV600MUT melanoma. In contrast, for patients with BRAFV600WT melanoma, BRAFi is contraindicated, and MEKi monotherapy provides limited benefits due to innate or rapid development of resistance. Immune checkpoint therapy (ICT) with anti-PD-1 agents results in 30–40% response rates in patients with either BRAFV600MUT or BRAFV600WT melanoma. Combination ICT with anti-PD-1 + anti-CTLA-4 agents reduces the rate of innate resistance from 60–70% to 40–50%. Simultaneous initiation of treatments with anti-PD-1/L1 and BRAFi + MEKi (aka triplet therapy) in BRAFV600MUT has been tested in clinical trials (Ascierto et al., 2019; Gutzmer et al., 2020; Ribas et al., 2019) and has been hypothesized to reduce both innate anti-PD-1/L1 resistance and acquired MAPKi resistance.
Retrospective analyses of clinical data suggest that progression on MAPKi is associated with inferior responses to subsequent anti-PD-1 therapy and that any second-line therapy results in inferior outcomes versus the same therapy in the first-line setting (Ackerman et al., 2014; Johnson et al., 2017; Mason et al., 2020; Reijers et al., 2020; Simeone et al., 2017; Tetu et al., 2018). Whether treatment first with ICT until progression alters subsequent MAPKi responsiveness is still unclear. Prospectively, the optimal sequencing of MAPKi vs. ICT is being tested in multiple clinical trials. These trials test one therapy modality until disease progression before switching to the alternative. However, treatment until progression may induce cross-resistance. To date, the impact of shorter exposures to one therapy (to generate a priming effect) before switching to or combining with another therapy has not yet been evaluated. This sequential-combinatorial fusion may avoid the development of cross-resistance, raise the threshold for resistance evolution by stacking multiple therapeutic mechanisms of action, and permit one mode of therapy to prime responsiveness to the other, thereby creating synergy. Importantly, prior studies have implicated a role of antitumor immunity in prolonging, clinically and preclinically, the durability of MAPKi responses (Hong et al., 2021; Hugo et al., 2015).
Since prior ICT exposure seems to prime subsequent MEKi responsiveness in patients with NRASMUT melanoma (Dummer et al., 2017), we test the hypothesis that the same association exists in patients with BRAFV600MUT melanoma. We also test the hypothesis that brief anti-PD-1/L1 dosing or lead-in before MAPKi combination maximizes antitumor efficacy and identify intratumoral immune cell phenotypes that associate with superior efficacy. Since MAPKi or ICT appears to be less durably active against intracranial (vs. extracranial) melanoma metastases (Flaherty et al., 2012; Ribas et al., 2016), we evaluate whether the optimal sequential-combinatorial regimen suppresses resistance in an organ-specific context. In particular, BRAFi + MEKi elicit lower response rates against clinical melanoma brain metastasis (MBM) (Davies et al., 2017), and clinically acquired MAPKi resistance emerges preferentially in the brain (Seifert et al., 2016). Thus, we develop a murine model of experimental melanoma metastases where MBM limits survival and compare the relative efficacies of anti-PD-1/L1 (± anti-CTLA-4) and MAPKi sequential-combinatorial regimens and their impacts on T-cell clonality.
RESULTS
Prior ICT enhances MAPKi responses in melanoma patients
In a trial of the MEKi binimetinib vs. dacarbazine for patients with NRASMUT melanoma, greater clinical benefit (median progression-free survival or PFS, confirmed overall response, and median duration of objective response) of binimetinib was associated with prior ICT (Dummer et al., 2017). S1320 was a phase 2 randomized clinical trial comparing intermittent (n = 101) versus continuous (n = 105) dosing of the BRAFi dabrafenib and MEKi trametinib for BRAFV600E/K metastatic melanoma. The trial randomized patients between 2013 and 2019. After an eight-week lead-in period of continuous treatment, patients who did not progress were randomized to either continuous or intermittent dosing of both drugs on a 3-week-off, 5-week-on schedule. As reported (Algazi et al., 2020), PFS was longer with continuous vs. intermittent dosing. Prior exposure to ICT was a randomization stratification factor in the trial, with 61 patients (30%) with prior exposure (anti-CTLA-4 alone, n = 21; anti-PD-1 alone, n = 22; anti-CTLA-4 and anti-PD-1 separately, n = 6; anti-PD-1 alone and anti-PD-1 + anti-CTLA-4 separately, n = 1; anti-PD-1, anti-CTLA-4, anti-PD-1 + anti-CTLA-4 separately, n = 1; unknown, n = 4). Patient characteristics were similar between patients who did and did not have prior ICT (Table S1). PFS was longer among patients with prior ICT on univariate (hazard ratio (HR) = 0.64, 95% confidence interval (CI) 0.44–0.92, p = 0.017) and multivariable analysis (HR = 0.60, 95% CI 0.47–0.77, p = 0.009; Figure 1; Table S2A). There were no significant differences in overall survival (OS) by prior ICT on univariate (HR = 0.86, 95% CI 0.54–1.35, p = 0.51) or multivariable analysis (HR = 0.85, 95% CI 0.63–1.17, p = 0.52; Figure S1, Table S2B). There was no evidence of heterogeneity in these results by treatment arm (interaction p-value = 0.55 for PFS, p = 0.67 for OS).
Figure 1. Progression-free survival of patients with BRAFV600MUT melanoma treated with dabrafenib plus trametinib and stratified by prior exposure to ICT.

Results from the S1320 SWOG trial and analyzed by the univariate Cox regression model. ICT, immune checkpoint therapy.
Anti-PD-1/L1 lead-in preceding MAPKi combination optimizes antitumor efficacy
Using syngeneic subcutaneous tumor models, we tested whether brief anti-PD-L1 (or anti-PD-1) pretreatments (two doses over one week) can improve subsequent responses to MEKi, with or without continuing anti-PD-L1 dosing with MEKi. We defined time at which the average tumor volumes reach 120–140 mm3 as day 0 (d0) and d0 to d7 as the anti-PD-1/L1 lead-in period (Figure 2A). We treated tumor-bearing mice in the following control (no active treatment, single-agent treatment, or simultaneous combination treatment) groups: (i) vehicle (starting on d7), (ii) anti-PD-L1 (starting on d0, d7 or from d0 to d7 only), (iii) MEKi (starting on d0 or d7), and (iv) MEKi + anti-PD-L1 (simultaneously starting on d0 or d7). We expected superior antitumor activity with anti-PD-L1 lead-in before MEKi dosing in two experimental groups: (i) MEKi d7, anti-PD-L1 d0 to d7 (anti-PD-L1 started on d0 and stopped on d7 followed by MEKi started on d7), and (ii) MEKi d7, anti-PD-L1 d0 (anti-PD-L1 started on d0 followed by MEKi started on d7, while continuing anti-PD-L1) (Figure 2A). Trametinib dose level for each tumor model was chosen based on the minimal dose required to elicit tumor stabilization or regression early on-treatment and near complete p-ERK suppression on day 3 (Figure S2A). We used six murine syngeneic tumor models: (i) BrafV600E melanoma with high mutational burden (YUMM1.7ER; Figure 2B) (Wang et al., 2017), (ii) NrasQ61R melanoma (NIL; Figure 2C) (Hong et al., 2018), (iii) NrasQ61R melanoma with high mutational burden (NILER1–4; Figure 2D) (Hong et al., 2021), (iv) Nf1−/− melanoma (mSK-Mel254; Figure 2E), (v) KrasG12C colorectal carcinoma (CT26; Figure 2F), and (vi) KrasG12C pancreatic adenocarcinoma (KPC; Figure 2G).
Figure 2. Two doses of anti-PD-1/L1 before MAPKi combination forestall therapy resistance and induce pro-inflammatory TAM polarization.

(A) Schematic of timelines for subcutaneous tumor progression and for dosing anti-PD-L1 and/or MEKi therapies in (B to I) and for tumor sampling in (J and K). Anti-PD-L1 (200 μg/mouse) twice per week IP; MEKi (dosage variable depending on the tumor model) daily PO via chow. Gray circles indicate regimen and time points for CyTOF analysis in Figure 2 and scRNA-seq + scTCR-seq analysis in Figure 3.
(B to H) Tumor volumes of YUMM1.7ER (B), NIL (C), NILER1–4 (D), mSK-Mel254 (E, H), CT26 (F), and KPC (G) treated with indicated regimens in (A). Trametinib at 1 (B, C, D), 2 (G), 3 (E) or 5 (F) mg/kg/d PO via chow. Anti-PD-L1 (200 μg/mouse) and anti-PD-1 (300 μg/mouse first week only and then 200 μg/mouse) twice per week IP. N = 8 tumors/group. Data are means ± SEMs (P-values, Student’s t test) and representative of two independent experiments.
(I) Overall survival (cutoff tumor volume ≥ 1000 mm3) of mice bearing YUMM1.7ER tumors treated as indicated. PLX4032, 50 mg/kg/d PO; trametinib, 0.3 mg/kg/d PO. CTRL, historical control. All p-values (logrank test) are for pairwise comparisons relative to mice treated with anti-PD-L1 (d0 to d7) followed by the triplet (anti-PD-L1 + PLX4032 + trametinib) combination.
(J) t-SNE maps (left) of tumor-infiltrating CD45+ cells analyzed by CyTOF in three indicated syngeneic subcutaneous tumor models at time points and in treatment regimens indicated in (A). Heatmaps (right) showing the expression values of immune phenotypic protein markers normalized to the maximum mean value across subsets.
(K) Frequencies of iNOS+ TAMs in the CD45+ population of three syngeneic tumor models at time points and treatment regimens indicated in (A). Mean ± SEMs. Pairwise comparisons were performed in (i) vehicle vs. two doses of anti-PD-L1, (ii) MEKi d7 vs. MEKi d7, anti-PD-L1 d0 to d7, (iii) MEKi d7, anti-PD-L1 d7 vs. MEKi d7, anti-PD-L1 d0. P-value, Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001. See (A).
See also Figure S2.
In all models, anti-PD-L1 treatment alone (d0, d0 to d7, or d7) had minimal (YUMM1.7ER, NILER1–4, mSK-Mel254) to no (NIL, CT26, KPC) tumor growth-inhibitory effect (Figures 2B to 2G). In all models (Figures 2B to 2G), at the trametinib doses chosen, treatment with MEKi d7 vs. MEKi d7, anti-PD-L1 d7 elicited only transient tumor regression and small differences, if any, in the average tumor volumes over time. In general (Figures 2B and Figures 2D to 2G), MEKi d0 or MEKi d0, anti-PD-L1 d0 (i.e., treatments started on smaller tumors) still did not improve the durability of antitumor activities, except in CT26 (MEKi d0, anti-PD-L1 d0 vs. MEKi d7, anti-PD-L1 d7; p < 0.008). In one tumor model (mSK-Mel254) (Figure S2B), using MEKi started on d0 against smaller tumors, we observed that the efficacies of regimens MEKi d0, anti-CTLA-4 d0 and MEKi d0, anti-PD-L1 d7 were lower than that of the regimen MEKi d0, anti-PD-L1 d0. Thus, anti-CTLA-4 and delayed dosing of anti-PD-L1 (vs. MEKi) were not studied further in subcutaneous tumor models. Importantly, between the two regimens we hypothesized to elicit the most robust antitumor activity, anti-PD-L1 lead-in followed by MEKi combination consistently led to the most extensive and durable tumor regression (Figures 2B to 2G). This regimen was either not associated with body weight loss or associated with a weight loss that was < 10% compared to any other concurrent regimen. In contrast, the efficacy of the regimen of MEKi d7, anti-PD-L1 d0 to d7 was not superior to the efficacies of the regimens MEKi d7 or MEKi d7, anti-PD-L1 d7. Thus, brief dosing with anti-PD-L1 prior to its combination with MEKi in MAPK-addicted tumor models overcame anti-PD-L1 innate resistance and delayed acquired MEKi resistance.
Using mSK-Mel254, we tested whether anti-PD-1 would produce a similar priming effect (Figure 2H). As with anti-PD-L1 d0 (Figure 2E), anti-PD-1 d0 yielded little to no tumor growth inhibition. Clearly, the regimen of MEKi d7, anti-PD-1 d0 was superior to the regimen of MEKi d7, anti-PD-1 d7 (Figure 2H). Using YUMM1.7ER, we also tested regimens containing the combination of BRAFi + MEKi. We started BRAFi + MEKi ± anti-PD-L1 treatments either on d0 or d7. For the latter group (starting on d7), we tested the impact of anti-PD-L1 lead-in (2 doses). Consistent with previous data using MEKi alone (Figure 2B), anti-PD-L1 lead-in followed by BRAFi + MEKi combination consistently led to the most durable antitumor activity (Figure 2I), which was associated with mice weight loss that was < 10% compared to any other concurrent regimen.
Pro-inflammatory TAM polarization distinguishes the tumor microenvironment after sequential-combinatorial therapy
To identify immune cell alterations specifically elicited by the regimen of anti-PD-L1 lead-in prior to MEKi combination, we sampled tumors (n = 3–4/group) with vehicle or anti-PD-L1 (priming) treatments on d7 and tumors (n = 3–4/group) on d10 and d14 in four regimens: (i) MEKi d7, (ii) MEKi d7, anti-PD-L1 d7, (iii) MEKi d7, anti-PD-L1 d0 to d7, and (iv) MEKi d7, anti-PD-L1 d0 (Figure 2A). Dissociated cells from three tumor models (YUMM1.7ER, mSK-Mel254, CT26) were subjected to analysis by an immune cytometry by time-of-flight (CyTOF) panel published previously (Hong et al., 2021). We found that the three tumor models were highly distinct in the levels of infiltration by CD45+ cells, which ranged from 20–80% without treatment or after two doses of anti-PD-L1 treatments (Figure S2C). These percentages increased (YUMM1.7ER), decreased (mSK-Mel254), or decreased and then increased (CT26) over time after MEKi-containing treatments (Figure S2C). Among CD45+ cells, two doses of anti-PD-L1 increased the abundance of CD8+ T cell (vs. vehicle) only in YUMM1.7ER tumors (Figure S2D). After MEKi-containing treatments (vs. vehicle treatment), the percentages of CD8+ T cells among CD45+ cells increased only in YUMM1.7ER and CT26, but these increases did not associate with treatment efficacy (Figure S2D).
Among CD45+ cells in all treatment groups, tumor-associated macrophages (TAMs) were the most abundant subpopulation across the three tumor models (Figures 2J and S2D). We identified 6–9 TAM subpopulations across the three tumor models (Figure 2J). iNOS-high or iNOS+ M1-like TAMs were identified in 6 of 9 and 6 of 8 TAM subpopulations in YUMM1.7ER and mSK-Mel254 but only in 1 of 6 TAM subpopulations in CT26 (Figure 2J). Despite these differences, we observed specific induction of iNOS+ M1-like TAMs only in the regimen of MEKi d7, anti-PD-L1 d0 (Figure 2K). Moreover, after sub-clustering CD4+ T-cells, we found that Th1-like (T-bet high) CD4+ T cells were also specifically induced only in the regimen of MEKi d7, anti-PD-L1 d0 (Figure S2E). After sub-clustering CD8+ T-cells, we found that two doses of anti-PD-L1 elevated the levels of granzyme B-high CD8+ cytotoxic T-cells (TC) (Figures S2F and S2G). However, this elevation was not maintained after initiating any MEKi-containing regimen (YUMM1.7ER) or not specifically maintained in the most efficacious regimen (mSK-Mel254, CT26) (Figure S2G).
We sought to corroborate TAM findings above (Figure 2K) at the transcriptomic level. Sorted CD45+ cells from four mSK-Mel254 tumors per regimen, per time point (Figure 2A) were admixed and then subjected to coupled 5’ single-cell RNA-sequencing (scRNA-seq) and T-cell receptor-sequencing (scTCR-seq). In total, data from 53,841 CD45+ cells passed quality control. Based on expression profiles of known lineage marker genes, we annotated seven major immune cell types, including monocyte/macrophages (Mo/MΦ), T cells, B cells, natural killer (NK) cells, tumor-associated neutrophils (TANs), and two dendritic cell (DC) subsets (monocyte-derived DC and classical DC) (Figures 3A, S3A and S3B). Consistent with CyTOF analysis (Figures 2J and S2D), TAMs constituted the most abundant CD45+ subpopulation, followed by T cells (Figure S3C). Re-clustering the monocyte/macrophage population identified seven sub-populations (Figures 3B, 3C and S3D). Cells in MΦ1 and MΦ3 displayed significant upregulation of pro-inflammatory genes (e.g., Neat1, Malat1, Cxcl9, and Cxcl10). MΦ2, MΦ4, and MΦ5 subpopulations upregulated anti-inflammatory genes (e.g., Apoe, C1qa, Chil3, and Arg1). One subpopulation (Mo->MΦ) highly-expressed Ccrl2, Il1b, and Rgs1, suggesting monocytes transitioning into to macrophages. We then calculated the pro- vs. anti-inflammatory ratios and found that two lead-in doses of anti-PD-L1 (vs. vehicle treatment) enhanced this ratio from < 1 to > 1 (Figure 3D). However, this enhanced ratio was reversed subsequently with any MEKi-containing regimen, except the most efficacious regimen (MEKi d7, anti-PD-L1 d0), which further enhanced this ratio on day 10 (Figure 3D).
Figure 3. TAM and T-cell phenotypes associated with response to optimized therapy regimen.

(A) Uniform manifold approximation and projection (UMAP) of tumor-infiltrating CD45+ cells (n = 53,841) analyzed by scRNA-seq. Dissociated live, CD45+ cells were pooled from 4 tumors (mSK-Mel254) per regimen and per time point. Inferred cell types are indicated by clusters denoted by distinct colors.
(B) UMAP of tumor-infiltrating Mo/MΦ population (n = 28,857) analyzed by scRNA-seq. Clusters with differentially expressed genes are denoted by distinct colors.
(C) Heatmap showing differentially expressed genes (rows) among different Mo/MΦ subpopulations (columns). Representative genes of each cluster are highlighted (right).
(D) Ratios between the proportions of pro- and anti-inflammatory MΦ clusters across distinct regimens and time points. See Figure 2A.
(E) UMAP of tumor-infiltrating T cells (n = 13,803) analyzed by scRNA-seq. Clusters with differentially expressed genes are denoted by distinct colors.
(F) Heatmap showing differentially expressed genes (rows) among different T-cell clusters (columns) and highlighted genes of each cluster (right).
(G) UMAP in (E) colored by clonality based on scTCR-seq.
(H) Clonal expansion indices of T-cell subpopulations. P-value, Kruskal–Wallis test.
(I) Developmental transition indices of CD8+ TC cells with other CD8+ subpopulations. Pairwise comparisons were performed between Ki-67hi CD8+ T cells and each of the other CD8+ subpopulations with the Wilcoxon test. P-values, ***p < 0.001.
(J) Relative expansion ratios between TC vs. TREG (left) and Ki-67hi CD8+ vs. TREG (right) across different treatment regimens and time points. See Figure 2A.
(K) Developmental transition indices between CD8+ TC and Ki-67hi CD8+ T cells across distinct regimens and time points. See Figure 2A.
(L) Fold changes of top 5 (top) or 10 (bottom) clone sizes for CD8+ TC or Ki-67hi CD8+ T cells vs. CD4+ TREG cells. See Figure 2A.
(M) Violin plots of Ifng expression in distinct T-cell subpopulations.
(N) Heatmap displaying the scaled mean expression levels of highlighted functional genes (rows) in TC cells across treatment regimens and time points (see Figure 2A). Gene expression levels were row-scaled for only TC cells.
Activated, proliferative, and cytolytic CD8+ T-cell gene expression tracks with optimized regimen
We next analyzed the T-cell population (n = 13,803 cells) using coupled scRNA- and scTCR-seq. By sub-clustering, we identified nine T-cells subpopulations (Figures 3E and 3F). These included three CD8+ subpopulations (naïve, TC and Ki-67hi); three CD4+ subpopulations (naïve, regulatory or TREG, T helper 1/2 or Th1/h2 that co-expressed Th1 and Th2 genes such as Cxcr3 and Gata3), NK T cells or NKTs, interferon (Ifn)- stimulated T cells, and gamma-delta T cells (Tγδ). As predicted, two lead-in doses of anti-PD-L1 elevated CD8+ TC levels (Figure S3E). Importantly, these elevated CD8+ TC levels were either maintained or surpassed with subsequently switching to or combining with MEKi (Figure S3E). By d14 in tumors treated with MEKi d7 or MEKi d7, anti-PD-L1 d7, CD8+ TC levels dropped clearly below that in the vehicle-treated tumors.
Among 7,017 TCR clonotypes identified with unique α and β chain pairs, 350 were represented by two or more cells, which resulted in 1,384 clonal T cells (Figure 3G). We calculated the expansion indices across all nine T-cell subpopulations and observed that CD8+ TC cells harbor the highest degree of clonal expansion, followed by the Ki-67hi CD8+ T cells (Figure 3H and Table S3). Consistently, clonal T cells were concentrated in the TC subpopulation (Figures 3E and 3G). Interestingly, transition index analysis associated Ki-67hi CD8+ T cells with TC subpopulations (Figure 3I), suggesting a differentiation trajectory from proliferative CD8+ T cells to the clonally expanded TC subpopulation. We then calculated the ratios of expansion indices of CD8+ TC or Ki-67hi CD8+ T cells to TREG to estimate the net antitumor status in each regimen and time point. Two lead-in doses of anti-PD-L1 increased both ratios, the elevation of which was either maintained or surpassed after MEKi combination (Figure 3J). In tumors treated with MEKi d7 or MEKi d7, anti-PD-L1 d7 (i.e., no prior priming doses of anti-PD-L1), these ratios persisted at low levels near that observed in the vehicle-treated group. When we examined the transition indices of CD8+ TC from Ki-67hi CD8+ T cells over time, we observed increased transition after anti-PD-L1 lead-in doses, and this transition was maintained and highest at both subsequent time points in and only in tumors on the regimen of MEKi d7, anti-PD-L1 d0 (Figure 3K).
To corroborate the specific association of CD8+ TC and Ki-67hi T cells with the most efficacious regimen, we analyzed the largest (i.e., most expanded) TCR clonotypes (Table S3), stemness, or expression of specific functional genes. The fold changes in the total clone size of top 5 or top 10 TCR clonotypes of either CD8+ TC or Ki-67hi T cells were calculated compared to that of CD4+ TREG cells. This revealed that the most efficacious regimen yielded the highest expansion of top TC clonotypes (vs. TREG) at both time points and of top Ki-67hi CD8+ T cell clonotypes (vs. TREG) at the last time point (Figure 3L). Tcf1+ stem-like CD8+ T cells are thought to be critical for tumor control in response to ICT (Siddiqui et al., 2019). Hence, we scored CD8+ T cell subpopulations for enrichment of this signature across regimens and time points (Figure S3F). For the Ki67hi CD8+ T cell subpopulation, this score trended higher (vs. vehicle treatment) after two anti-PD-L1 lead-in doses and stayed high on d10 only in the most efficacious regimen. By d14, this score dropped across all MEKi-containing regimens but remained highest in the tumors from the most efficacious regimen. Moreover, we visualized the expression levels of functional genes across T-cell subpopulations (Figure S3G). We noted that the critical gene, Ifnγ, was expressed (Figures 3M and S3G) by most CD8+ TC and Ki-67hi T cells, supportive of their antitumor functional importance. Critically, within the TC subpopulation, two anti-PD-L1 lead-in doses upregulated the expression of activation genes (Cd44), inhibitory genes (Lag3, Ctla4, Havcr2), cytolytic genes (Prf1, Gzmb), and effector genes (Ifnγ) (Figure 3N). On d10, treatment on the most efficacious regimen led to the highest levels of activation/exhaustion gene (Pdcd1) and Ifnγ. By d14, treatment on the most efficacious regimen led to the highest expression of both Pfr1 and Gzmb and the highest ratio of Pdcd1 to Tox, the master regulator of T-cell exhaustion. Interestingly, MEKi monotherapy induced the highest levels of Ctla-4 at both time points (Figure 3N), suggesting anti-CTLA-4 antibody as a combinatory agent. Foxp3 expression was down-regulated in TREG specifically in the most efficacious regimen (Figure S3G).
Functional contributions of TAM polarization and CD8+ T cells
We assessed whether targeting of M2-like TAMs by a peptide agonist of CD206 (Ghebremedhin et al., 2020; Jaynes et al., 2020) would augment the priming effects of anti-PD-L1 (Figure 4A). Using the mSK-Mel254 model, we observed that the CD206 agonist, RP832c, on its own (dosed from d0 to d14) elicited a small degree of growth inhibition, but, on top of MEKi d7, CD206 peptide d0 to d14, elicited no priming effect (Figure 4A). However, combining the CD206 agonist with anti-PD-L1 improved priming, as the regimen of MEKi d7, anti-PD-L1 d0, CD206 peptide d0 to d14 elicited deeper and more durable tumor regression when compared to the regimen of MEKi d7, anti-PD-L1 d0 (Figure 4A). We also tested using mSK-Mel254 whether neutralizing CD8+ T cells systemically would diminish or abolish the priming effect of two doses of anti-PD-L1. In the context of two regimens (MEKi d7; MEKi d7, anti-PD-L1 d7) without anti-PD-L1 lead-in (Figure 4B), CD8+ T-cell neutralization had no impact on tumor regression or resistance development. This lack of effect is consistent with the lack of additivity when MEKi is initiated with anti-PD-L1 on established (700–900 mm3) tumors (Figures 2B to 2G). Moreover, when we neutralized CD8+ T cells, we reversed the growth deceleration effect elicited by the two priming anti-PD-L1 doses from d0 to d7 (Figure 4C). Importantly, while CD8+ T-cell neutralization did not change the onset of acquired resistance in the regimen MEKi d7, anti-PD-L1 d0 to d7, it abolished the benefit (i.e., durability of tumor regression) of the regimen MEKi d7, anti-PD-L1 d0 (Figure 4C).
Figure 4. Functional roles of TAMs and CD8+ T cells in the development of acquired resistance.

(A to C) Tumor volumes of mSK-Mel254 without active treatment or treated with the indicated regimens. Trametinib at 3 mg/kg/d PO; RP-832c (CD206 peptide) at 10 mg/kg/d subcutaneously; anti-PD-L1 (200 μg/mouse IP twice per week); and anti-CD8 antibody at 200 μg/mouse IP initiated one day before any treatment regimen and then twice a week. N = 8 tumors/group. Data are means ± SEMs (P-values, Student’s t test).
PD-1/L1 plus CTLA-4 blockade before MAPKi combination prolongs MBM suppression and survival of mice
Since responses of MBM to therapies may be inferior, we tested sequencing-combinatorial regimens for their antitumor impacts across multiple organ sites. We engineered BrafV600E (YUMM1.7ER) melanoma cells to express luciferase and injected YUMM1.7ER-Luc cells into the left ventricle. By in vivo bioluminescence imaging (BLI), dissemination of BrafV600E melanoma cells resulted in tumor growth in 100% of mice in both intracranial and extracranial sites 8d after intracardiac (IC) injection (Figure 5A). In vivo BLI signals (ventral or dorsal extracranial and intracranial) grew from a nadir on d3 to d4 after IC injection and surpassed an average of 1e9 by ~d21 after IC injection (Figures 5B to 5E and S4A). Median survival of untreated mice (defined in Figure 5B) was 3.5 weeks after IC injection (n = 19) (Figure 5F). When the dorsal intracranial BLI signals averaged 1e6–1e7 on day 8 (when 100% of mice harbored MBM) after IC injection, we designated this time point of initiating MEKi-containing regimens as d0 (Figure 5A). At necropsy when untreated mice were moribund, ex vivo BLI revealed tumor burden in the lung, adrenal glands, ovaries, pancreas, and brain in 100% of mice (Figure 5B) as well as in the kidney (6/13 or 46%), heart (9/13 or 69%), and liver (6/13 or 46%) (Figure S4B).
Figure 5. Immune checkpoint blockade before MAPKi co-treatment prolongs MBM suppression and survival.

(A) Schematic of timelines for metastatic tumor progression of BrafV600MUT murine melanoma (YUMM1.7ER) and for dosing anti-PD-L1 and/or MEKi therapies in (B to F). Anti-PD-L1 (200 μg/mouse) IP on d-4 and d-2, when applicable, and, thereafter, twice per week; MEKi, 1 mg/kg/d PO. Gray circles indicate regimens and time points for TCR-seq analysis in Figure 6.
(B) Representative ex vivo BLI of organs from necropsies at experimental endpoints (total ventral or dorsal BLI signal in the range of 1e9–1e10, moribund state of health, or death). Scale for radiance, photons/sec.
(C, D) Temporal BLI quantification (radiance, photons/sec) based on the dosing timeline in (A) of dorsal extracranial (C) or intracranial (D) tumor burden. Data are mean ± SD based on the indicated numbers of mice in the untreated and treatment regimen groups. Pairwise comparisons (mixed model framework with Bonferronic correction for multiple testing) of the group MEKi d0, anti-PD-L1 d-4 vs. the group denoted by the specific color of symbol #. # p = 0.05–0.001, ## p < 0.001. *Indicates death of mouse or mice, resulting in drops in mean BLI values. True or confirmed complete responses or CRs defined in STAR Methods.
(E) In vivo BLI of representative mice from the untreated group vs. all mice treated with MEKi d0 monotherapy at indicated timepoints. All images were adjusted to the same radiance scale.
(F) Survival of untreated mice and mice on each treatment regimen. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant (logrank test). Survival criteria indicated in (B).
(G, H) Survival of untreated mice and mice on indicated treatment regimens. P values, logrank test. Day on which BRAFi + MEKi in (G) were started, defined as day 0, was the same as in (A) but pushed back by 2 days (day 10 after intracardiac injection) in (H). Low tumor burden model, MAPKi started on day 8 after IC injection; high tumor burden model, MAPKi started on day 10 after IC injection.
(I, J) As in (C, D).
(K) As in (F).
(L, M) As in (C, D), except anti-CTLA-4 at 200 μg/mouse twice a week IP and # p < 0.05, ## p < 0.01, ### p < 0.001, #### p < 0.0001. Pairwise comparisons (mixed model framework with Bonferronic correction for multiple testing) of the group BRAFi+MEKi d0, anti-PD-L1 d-4 vs. the group denoted by the specific color of symbol #.
(N) As in (F) except anti-CTLA-4 at 200 μg/mouse twice a week IP.
(G to N) BRAFi, PLX4032, 50 mg/kg/d PO; MEKi, trametinib, 0.3 mg/kg/d PO.
See also Figure S4.
To evaluate impacts on multi-organ metastatic growth, we compared the following regimens: (i) anti-PD-L1 d-4 or d0; (ii) MEKi d0; (iii) MEKi d0 plus anti-PD-L1 d-4, d0, or d4 (Figure 5A). Although anti-PD-L1 d0 elicited no discernable impact on extracranial or intracranial metastatic growth (Figures 5C, 5D, S4C and S4D), anti-PD-L1 d-4 suppressed MBM (Figures 5D and S4D) and extended survival (Figure 5F). MEKi d0 clearly reduced extracranial metastatic tumor burden in surviving mice past 4 weeks (Figures 5B, 5C, 5E, and S4C) but only delayed MBM by ~1.5 weeks (Figures 5B, 5D, 5E, S4C and S4D), which limited the survival benefit of MEKi monotherapy (Figure 5F) (Davies et al., 2017; Seifert et al., 2016). Comparing among the group (iii) regimens, we observed that the regimen of MEKi d0, anti-PD-L1 d-4 was superior in controlling both extracranial and intracranial tumor burdens (Figures 5C, 5D, S4C and S4D). Survival of mice on the regimen of MEKi d0, anti-PD-L1 d-4 was superior to that in every other group, except the regimen of MEKi d0, anti-PD-L1 d0 (Figure 5E). In contrast, survival of mice on the regimen of MEKi d0, anti-PD-L1 d0 was not significantly superior to that on any other group except for the no treatment group and the anti-PD-L1 monotherapy groups (Figure 5F). Thus, in mice with metastatic BrafV600E melanoma, two doses of anti-PD-L1 prior to its combination with MEKi overcame innate anti-PD-L1 resistance and delayed acquired MEKi resistance in MBM.
We further evaluated the two most efficacious group iii regimens, namely MEKi d0, anti-PD-L1 d-4 and MEKi d0, anti-PD-L1 d0, by substituting MEKi monotherapy with BRAFi + MEKi combo therapy. As expected, both of these regimens were superior in efficacy to BRAFi + MEKi (Figure 5G). Importantly, the regimen of BRAFi + MEKi d0, anti-PD-L1 d-4 (median survival, not reached at 10 weeks) trended (p = 0.0543) toward greater survival benefit compared to the regimen of BRAFi + MEKi d0, anti-PD-L1 d0 (median survival of 4 weeks) (Figure 5G). We devised a protocol to follow surviving mice longer-term, up to about ~34 weeks (Figure S4E). For mice treated on the regimen of BRAFi + MEKi d0, anti-PD-L1 d-4, the median survival was reached at ~13.5 weeks (Figure S4F), which tripled mice survival on the regimen of BRAFi + MEKi d0, anti-PD-L1 d0. Moreover, two long-term survivors displayed confirmed or true CRs. We then challenged the two most efficacious regimens with greater metastatic tumor burden (i.e., high tumor burden) by delaying the start of BRAFi + MEKi treatment by two days post-IC injection (Figure 5H). Although the survival benefits of both regimens were decreased, run-in with anti-PD-L1 prior to combination with BRAFi + MEKi yielded superior survival benefit compared with simultaneously initiating all three therapeutic agents (p = 0.0006) (Figure 5H). In the context of high tumor burden, we also evaluated the most efficacious group (BRAFi + MEKi d0, anti-PD-L1 d-4) by substituting anti-PD-L1 with anti-PD-1. Similar to the observations from a subcutaneous tumor model (Figure 2H), the antitumor (Figures 5I and 5J) and pro-survival (Figure 5K) activity of the regimen BRAFi + MEKi d0, anti-PD-1 d-4 was greater than that of anti-PD-1 d-4. Furthermore, since anti-PD-1 plus anti-CTLA-4 improve anti-melanoma and anti-MBM efficacy clinically (Tawbi et al., 2018; Wolchok et al., 2017), we tested whether anti-CTLA-4 (2 doses from d-4 to d0) in the high tumor burden model would further the priming action of anti-PD-L1 (Figures 5L to 5N). Indeed, priming with combined ICT improved the antitumor activity (Figures 5L and 5M) and survival benefit (Figure 5N) of the sequential-combinatorial regimen.
Anti-PD-1/L1 priming of MAPKi responses via T-cell clonal expansion
We tracked the intratumoral (ovarian and brain tumors) T-cell clonotypes by TCR-seq of five regimens at d0 and d3 (Figure 5A). We excluded from this analysis two regimens (anti-PD-L1 d0; MEKi d0, anti-PD-L1 d4), which respectively lacked superiority over the no treatment group and over the regimens of MEKi d0 or MEKi d0, anti-PD-L1 d0. We calculated the Gini or clonality and diversity indices and observed that T-cell clonality (based on α and β chains) was higher on d3 after treatment with MEKi d0, anti-PD-L1 d-4 than that in any other regimen in both tumor-involved brain and ovarian tissues (Figure S4G). Consistently, TCR diversity was lower on d3 after treatment with MEKi d0, anti-PD-L1 d-4 especially compared to the d3 tissues on-treatment with MEKi d0 or MEKi d0, anti-PD-L1 d0. We also analyzed the sizes of large (≥ 5%) TCR clones and found that the regimen of MEKi d0, anti-PD-L1 d-4 on d3 led to the highest accumulation of large TCR clones (on average 10–20% in tumor-involved brain or 15–35% in tumor-involved ovaries) (Figure 6A). The pattern was consistent when we analyzed the sizes of large clones defined as ≥ 8% or of the largest, top 5 or top 10 clones (Figures S4H and S4I). Moreover, we calculated the fractions of overlapping TCR clones between two distinct regions of tumor-involved brain tissues in each animal (Figure 6B). Increasing overlap may reflect greater tumor-specific T-cell expansion. In this regard, the fraction ranged from 4.56–5.71% on d0 in untreated mice; 1.89–8.58% on d0 in mice treated with anti-PD-L1 d-4; 6.17–9.77% on d3 in mice treated with MEKi d0; 7.09–11.90% on d3 in mice treated with MEKi d0, anti-PD-L1 d0; and, importantly, 13.50–19.50% on d3 in mice treated with MEKi d0, anti-PD-L1 d-4. The differences in this overlap between the most efficacious regimen (MEKi d0, anti-PD-L1 d-4) and MEKi d0 monotherapy were significant (p = 0.045 and 0.017 for 〈 and ® chain, respectively; Student’s t-test). Thus, the regimen of anti-PD-L1 lead-in followed by MEKi combination led to greater geographic convergence of TCR clones across distinct brain tumor lesions. Furthermore, within the overlapping TCR clones between two distinct tumor-involved lesions from brain or ovary tissues in each mouse, we calculated the sizes of common large (≥ 5%) TCR clones (Figure 6C). For both α or β TCR chains and for both brain and ovary tissues, the average size was greatest from tumor-bearing mice treated with anti-PD-L1 lead-in followed by MEKi combination. In further analysis, we calculated the fractions of overlapping TCR clones between all tumor-involved brain and ovary tissues (Figure S4J). The average of fractions for the α and β TCR chains from the regimen of MEKi d0, anti-PD-L1 d-4 trended higher versus any other group. Hence, the regimen consisting of two doses of anti-PD-L1 followed by MEKi combination elicited the most robust T-cell clonal expansion and clonotypic convergence between distinct tumor-involved regions of each organ and across intracranial and extracranial organs.
Figure 6. Anti-PD-L1 lead-in before MAPKi co-treatment augments clonal T-cell expansion.

(A) Tumor cell-involved brain and ovarian tissues were collected from mice at time points and in groups as indicated in Figure 5A (brain, n = 2 mice per group, except the no treatment group; ovary, n = 1 mouse per group; two geographically distinct regions of each organ site were sampled for TCR-seq analysis). The total sizes of large TCR clones (≥ 5%) for the α or β chain in tumor-involved brain or ovarian tissues (red dots, average values). Pairwise comparisons were performed between the group of MEKi d0, anti-PD-L1 d-4 vs. each of the other groups with Student’s t-test. P-values, *p < 0.05, **p < 0.01, ***p < 0.001.
(B) The fractions of overlapping TCR clones (α or β chain) between two distinct geographic regions of tumor-involved brain tissues from individual mice.
(C) Total sizes of large clones (≥ 5%) shared by two distinct geographic regions of tumor-involved brain or ovarian tissues in each mouse. Pairwise comparisons were performed as in (A).
See also Figure S4.
DISCUSSION
Combination therapies are typically initiated together. Whether sequencing therapies instead of or in addition to combining therapies would augment antitumor efficacy remains under explored. Here, we found that sequencing of just two doses of anti-PD-1/L1 (± two doses of anti-CTLA-4, without further dosing) prior to MAPKi combination maximizes antitumor immunity and efficacy.
Post hoc analyses of clinical data performed here and published previously (Dummer et al., 2017) in patients (with BRAFV600MUT or NRASMUT melanoma) treated with MAPKi found increased benefit from MAPKi in those who have been treated with ICT immediately prior to MAPKi. That antitumor immunity may be a critical element elicited by and modulating the efficacy of MAPK-targeted therapy was suggested by our prior studies of patients with BRAFV600MUT melanoma whose acquired MAPKi-resistant melanoma display signs of immune evasion (Hugo et al., 2015; Hugo et al., 2016; Song et al., 2017). Based on the current study, superior antitumor activity of the regimen consisting of anti-PD-1/L1 lead-in followed by MAPKi combination was positively linked to multiple cell types, including M1-like TAMs, CD4+ Th1, and CD8+ T cells. Thus, strategies to target M2-like TAMs and to improve clonal expansion and persistence of tumor-specific CD8+ cytotoxic T cells may further improve the proposed sequential-combinatorial strategy.
The finding here that anti-PD-1/L1 lead-in followed by MAPKi combination can improve anti-MBM activity (and thereby survival) in mice has immediate clinical implications. This sequential-combinatorial strategy may present an alternative strategy to combination ICT with anti-PD-1 + anti-CTLA-4, which improves anti-MBM responses elicited by anti-PD-1 monotherapy (Tawbi et al., 2018). Melanoma has a high propensity for CNS metastasis (40–80%), which is associated with poor overall survival (median 4–5 months) (Davies et al., 2011). CNS is often the initial site of acquired resistance (which often persists in isolation without extracranial disease progression) in patients on MAPKi (Frenard et al., 2016; Long et al., 2016). The experimental metastatic model established here recapitulated the propensity of MBM to escape from MAPKi therapy. Importantly, sequencing of anti-PD-1/L1 therapy prior to MAPKi addition resulted in the most durable anti-MBM activity. This benefit may be extended by priming with anti-PD-1/L1 plus anti-CTLA-4 (with a limited duration of anti-CTLA-4 dosing to avoid added toxicities).
Data presented here support the conclusions that anti-PD-1/L1 therapy primes a more durable MAPKi response and that MAPKi combination helps to overcome innate anti-PD-1/L1 resistance. These findings prompted the design and initiation of a single-site clinical trial (NCT04375527) testing the activity of binimetinib plus nivolumab in patients with BRAFV600WT melanoma that display innate resistance to ICT. Findings here should concentrate a larger effort to evaluate prospectively the activity of anti-PD-1/L1 agents combined with BRAFi + MEKi or MEKi (in BRAFV600MUT or BRAFV600WT melanoma, respectively) after distinct predefined periods of prior ICT exposure, with or without objective evidence of innate resistance to ICT. Moreover, the benefit of triplet therapy with anti-PD-1/L1 + BRAFi + MEKi may be clinically meaningful for anti-PD-1/L1-experienced (vs. naïve) patients despite objective evidence of innate ICT resistance and for patients with symptomatic or non-symptomatic MBM.
One limitation of this study is that the animal model studies did not address the impacts of prolonging anti-PD-1/L1 lead-in before MAPKi combination. However, these in vivo models collectively serve as an important platform to compare and mechanistically dissect alternative sequential-combinatorial regimens, including those inclusive of additional targeted agents. Future studies are needed to identify functional targets to prolong treatment benefits further and develop rational dosing strategies of higher-order combinations to minimize toxicities.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Roger S. Lo (Rlo@mednet.ucla.edu).
Materials availability
Mouse lines generated in this study are available upon request. There are restrictions to the availability of RP832c due to a MTA.
Data and code availability
Raw sequencing files of scRNA-seq, scTCR-seq, and bulk TCR-seq data have been deposited at GEO (GSE177902) and are publicly available as of the date of publication. Mass cytometry data of BrafV600E melanoma, Nf1−/− melanoma, and KrasG12C colorectal carcinoma syngeneic models have been deposited at FlowRepository (http://flowrepository.org/) under experiment IDs FR-FCM-Z43Z, FR-FCM-Z4YR, and FR-FCM-Z42V. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
There are no original codes generated in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 (Clone 30-F11) | Fluidigm | Cat#3089005B; RRID: AB_2651152 |
| Ki67 (Clone SolA15) | Invitrogen | Cat#14-5698-82; RRID: AB_10854564 |
| CD90.2/Thy-1.2 (Clone 30-H12) | Biolegend | Cat#105333; RRID: AB_2563765 |
| Ly-6G (Clone 1A8) | Biolegend | Cat#127602; RRID: AB_1089180 |
| CD69 (Clone H1.2F3) | Invitrogen | Cat#14-0691-82; RRID: AB_467325 |
| CD4 (Clone RM4-5) | Biolegend | Cat#116018; RRID: AB_2650936 |
| F4/80 (Clone BM8) | Biolegend | Cat#123102; RRID: AB_893506 |
| Eomes (Clone Dan11mag) | Invitrogen | Cat#14-4875-82, RRID:AB_11042577 |
| CD11b (Mac-1) (Clone m1/70) | Fluidigm | Cat#3148003B; RRID: AB_2814738 |
| CD62L (L-selectin) (Clone MEL-14) | Biolegend | Cat#104416; RRID: AB_313101 |
| Ly-6C (Clone HK1.4) | Biolegend | Cat#128002; RRID: AB_1134214 |
| CD25 (IL-2R) (Clone 3C7) | Biolegend | Cat#101913; RRID: AB_2562798 |
| CD3e (Clone 145-2C11) | Biolegend | Cat#100314; RRID: AB_312679 |
| TER119 (Clone TER119) | Biolegend | Cat#116214; RRID: AB_313715 |
| CD152 (CTLA-4) (Clone UC10-4B9) | Biolegend | Cat#106308; RRID: AB_2087654 |
| CD14 (Clone Sa14-2) | Biolegend | Cat#123302; RRID: AB_940592 |
| FoxP3 (Clone FJK-16s) | Invitrogen | Cat#14-5773-82; RRID: AB_467576 |
| CD279 (PD-1) (Clone 29F.1A12) | Biolegend | Cat#135202; RRID: AB_1877121 |
| iNOS (Clone CXNFT) | Fluidigm | Cat#3161011B |
| CD366 (Tim-3) (Clone RMT3-23) | Fluidigm | Cat#3162029; RRID: AB_2687841 |
| CD197 (CCR7) (Clone 4B12) | eBiosciences | Cat#16-1971-85; RRID: AB_494123 |
| CD182 (Clone SA044G4) | Biolegend | Cat#149302; RRID: AB_2565277 |
| CD19 (Clone 6D5) | Biolegend | Cat#115514; RRID: AB_313649 |
| CD335 (NKp46) (Clone 29A1.4) | Fluidigm | Cat#3167008B |
| CD8a (Clone 53-6.7) | Biolegend | Cat#100716; RRID: AB_312755 |
| T-bet (Clone 4B10) | Biolegend | Cat#644802; RRID: AB_1595503 |
| CD192 (Clone 475301R) | R&D | Cat#MAB55381R |
| Granzyme B (Clone GB11) | Fluidigm | Cat#3171002B; RRID: AB_2687652 |
| CD44 (Clone 1M7) | Biolegend | Cat#103014; RRID: AB_312965 |
| I-A/I-E (Clone M5/114.15.2) | Biolegend | Cat#107610; RRID: AB_313325 |
| CD278 (ICOS) (Clone 7E.17G9) | Invitrogen | Cat#14-9942-85; RRID: AB_468633 |
| CD11c (Clone N418) | Fluidigm | Cat#3209005B; RRID: AB_2811244 |
| CD16/CD32 (Clone 93, for blocking buffer) | eBioscience | Cat#14-0161-86; RRID: AB_467135 |
| CD8a (Clone 53-6.7) | Biolegend | Cat#100737; RRID: AB_10897101 |
| CD45 (Clone 30-F11) | Biolegend | Cat#103138; RRID: AB_2563061 |
| PerCP-anti-TER119 | Biolegend | Cat#116225; RRID: AB_893637 |
| inVivoMAb anti-mouse PD-L1 (B7-H1) | BioXcell | Cat#BE0101; RRID: AB_10949073 |
| inVivoMAb anti-mouse CD8α (Clone YTS169.4, in vivo antibody) | BioXcell | Cat#BE0117; RRID: AB_10950145 |
| Anti-Mouse CD279 (PD-1) (Clone RMP1-14) - Purified In vivo GOLD™ Functional Grade | Leinco Technologies, Inc. | Cat#P362; RRID: AB_2737557 |
| InVivoPlus anti-mouse CTLA-4 (CD152, Clone 9H10) | BioXcell | Cat#BE0131; RRID: AB_10950184 |
| phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat#4370; RRID: AB_2315112 |
| Goat Anti-Rabbit IgG (H+L) Highly Cross-adsorbed Antibody, Alexa Fluor555 | Life Technologies | Cat#A-21429; RRID: AB_2535850 |
| Bacterial and virus strains | ||
| NEB® Stable Competent E. coli | New England BioLabs | Cat#C3040I |
| Third generation lentivirus | Dr. Lo | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Trametinib | LC Laboratories | Cat#T-8123 |
| RP-832c | Riptide Bioscience | PMID: 32051227 |
| Vemurafenib | LC Laboratories | Cat#V-2800 |
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Cell-ID Intercalator Ir | Fluidigm | Cat#201192B |
| 7-AAD | BD Pharmigen | Cat#51-68981E |
| Blasticidin | Sigma-Aldrich | Cat#SBR00022 |
| Prolong Diamond Antifade Mountant with DAPI | ThermoFisher Scientific | Cat#P36962 |
| Critical commercial assays | ||
| 20-Plex Pd Barcoding Kit | Fluidigm | Cat#201060 |
| mirVana™ miRNA Isolation Kit, with phenol | Thermo Fisher Scientific | Cat#AM1560 |
| AllPrep DNA/RNA Mini Kit | Qiagen | Cat#80204 |
| MycoAlert™ Mycoplasma Detection Kit | Lonza | Cat#LT07-418 |
| MycoAlert™ Assay Control Set | Lonza | Cat#LT07-518 |
| Tumor Dissociation Kit | Miltenyi Biotec | Cat#130-095-929 |
| Single Cell 5′ Library and Gel Bead Kit v1.1 | 10X Genomics | Cat#1000167 |
| Enrichment Kit for Mouse T Cells | 10X Genomics | Cat#1000071 |
| QIAGEN QIAseq Immune Repertoire RNA Library Kit - T cell Receptor Panel | Qiagen | Cat#333705 |
| QIAseq Library Quant Array Kit | Qiagen | Cat#333304 |
| Foxp3 Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Deposited data | ||
| scRNA-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| scTCR-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| Bulk TCR-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| Mass cytometry data of mouse immune cells (BrafV600E melanoma model) | This Paper | FR-FCM-Z43Z |
| Mass cytometry data of mouse immune cells (Nf1−/− melanoma) | This Paper | FR-FCM-Z4YR |
| Mass cytometry data of mouse immune cells (KrasG12C colorectal carcinoma) | This Paper | FR-FCM-Z42V |
| Experimental models: Cell lines | ||
| Mouse cell line; NIL | Dr. Norman E. Sharpless and adapted by Dr. Roger S. Lo | PMID: 25252692 |
| Mouse cell line; NILER1-4 | Dr. Roger S. Lo | PMID: 33318037 |
| Mouse cell line; mSKMel-254 | Dr. David B. Solit and adapted by Dr. Roger S. Lo | This paper |
| Mouse cell line; YUMM1.7ER | Dr. Marcus W. Bosenberg | PMID: 27287723 |
| Mouse cell line; CT26 | ATCC | PMID: 33318037 |
| Mouse cell line; KPC (KP4662) | Dr. Robert Vonderheide | PMID: 27642636 |
| Mouse cell line; YUMM1.7ER-luciferase | Dr. Roger S. Lo | This paper |
| Experimental models: Organisms/strains | ||
| C57BL/6J; C57BL/6N mice | UCLA: Radiation-Oncology breeding colony | C57BL/6J/NROC |
| BALB/C mice | UCLA: Radiation-Oncology breeding colony | BALB/c ROC |
| C57BL/6J mice | Jackson Laboratory | Cat# 000664 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pLV-EF1a-IRES-Blast | Addgene | Cat#85133; RRID: Addgene_85133 |
| Software and algorithms | ||
| Cytofkit | Bioconductor | Version: 3.7 |
| R software | CRAN | Version: 3.5.1 |
| GraphPad Prism | https://swcstore.oit.ucla.edu/secure/browse_vendors.php | Version: 7 |
| Cytobank | https://www.cytobank.org/ | |
| Cell Ranger | 10x Genomics | Version: 2.1.0 |
| Seurat | CRAN | Version: 3.0.2 |
| STARTRAC | GitHub | Version: 0.1.0 |
| tcR | CRAN | Version: 2.2.4.1 |
| Living Image | PerkinElmer | Version: 4.7.2 Version |
| Other | ||
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human subjects
Patient characteristics and sample sizes are presented in Table S1. Patients were enrolled in the S1320 clinical trial with informed consents obtained from all patients and participation of sites approved by local institutional review boards.
Mice
C57BL/6 and Balb/c (for subcutaneous models) were obtained from the Radiation Oncology breeding colony at UCLA (Los Angeles, CA). C57BL/6 (for experimental metastasis model) were purchased from Jackson Laboratory. Female mice were used at 6–8 weeks of age. All animal experiments were conducted according to the guidelines approved by the UCLA Animal Research Committee.
Subcutaneous syngeneic tumor models
For syngeneic subcutaneous tumor models, C57BL/6 (YUMM1.7ER, NIL, NILER1–4, mSK-Mel254 and KP4662) or Balb/c (CT26) mice were injected on both flanks with one million cells. Tumors were measured with a calliper every 2 days, and tumor volumes were calculated using the formula (length × width2)/2. Once tumors reached a size of 120–140 mm3, mice were assigned randomly into experimental groups. Special mouse diets (for C57BL/6 and Balb/c) were generated by incorporating trametinib at 1, 2, 3 or 5 mg/kg to facilitate daily drug dosing and to reduce animal stress (TestDiet). The combination of BRAFi+MEKi (PLX4032 50 mg/kg/day and trametinib 0.3 mg/kg/day; both resuspended in 10% DMSO with 0.1% methylcellulose) was administered to mice via oral gavage (subcutaneous model) or incorporated in chow (TestDiet). Anti-PD-L1 (200 μg/mouse, BioXcell), anti-PD-1 (300 μg/mouse for the first 2 doses, then 200 μg/mouse, Leinco Technologies) and anti-CTLA-4 (200 μg/mouse, BioXcell) were intraperitoneally administered twice per week. Anti-CD8 antibody was intraperitoneally administrated (200 μg/mouse, BioXcell) on day −1, day 0 or on day 6, day 7, and then twice a week. RP-832c was subcutaneously administrated (10mg/kg, Riptide Bioscience) from day 0 to day 14, daily. On indicated days, tumors were excised from mice, minced, and digested to single-cell suspensions using a tumor dissociation kit and gentleMACS™ Octo Dissociator (Miltenyi Biotech) and prepared for CyTOF staining or/and 10X Genomics single cell RNA sequencing followed by sorting for live CD45+ cells.
Experimental metastasis of syngeneic melanoma
YUMM1.7ER melanoma cells were engineered to express firefly luciferase (YUMM1.7ER-luc). Before every IC injection, cells were selected for stable luciferase expression by blasticidin treatment, yielding cells (1 million) that emitted luciferase light units of 5e8 to 5e9 in vitro. C57BL/6 (6–8 weeks old, female), after anesthesia with vaporized isoflurane (2.0–2.5%), were injected with 1×106 cells/mouse into the left ventricle for intracardiac inoculation. Progression of metastatic tumor burden and treatment effects in mice were monitored in vivo and ex vivo by bioluminescent imaging (BLI). We acquired BLI on both ventral and dorsal sides twice weekly until study endpoints (total ventral or dorsal BLI signal in the range of 1e9–1e10, moribund, or death). Mice were anesthetized with vaporized isoflurane; D-luciferin (150 mg luciferin/kg, Caliper Life Sciences) dosed by intraperitoneal injections; and, 10 minutes after injections, luciferase activities were measured with IVIS Lumina II Imaging System (PerkinElmer). Image analysis was performed with Living Image 4.7.2 Version (PerkinElmer). For ex vivo BLI, mice were euthanized 10 minutes after intraperitoneal injection of D-luciferin by cervical dislocation and organs were excised and imaged ex vivo by the IVIS system. Special mouse diets (for C57BL/6 mice) were generated by incorporating trametinib at 1 mg/kg/day or PLX4032 at 50 mg/kg/day plus trametinib at 0.3 mg/kg/day to facilitate daily drug dosing and to reduce animal stress (TestDiet). Anti-PD-L1 (200 μg/mouse, BioXcell) or anti-PD-1 (200 μg/mouse, Leinco Technologies) was intraperitoneally administered twice per week. Anti-CTLA-4 (200 μg/mouse, BioXcell) was intraperitoneally administered every four days starting at d-4 and ending d+4. We visualized BLI data on a logarithmic scale (means and corresponding standard deviations). Down error bars were not presented when they extended to zero or negative values. For mice in the experimental metastasis model treated with MEKi + anti-PD-L1 regimens, mice with no BLI signal beyond background for > ~60 days after starting treatment were deemed to have unconfirmed complete responses (CRs). For mice in the experimental metastasis model treated with BRAFi + MEKi + anti-PD-1/L1 regimens, mice with no BLI signal beyond background for ~75–100 days after starting treatment were deemed to have unconfirmed complete responses (CRs). To confirm CRs in these mice, treatment was stopped. Mice were confirmed to display complete CRs if only background radiance was detected twice weekly for two consecutive months after treatment cessation or until the experimental endpoint (death by euthanasia in situations of deteriorating health), whichever comes first. If tumor burden becomes detectable after treatment cessation, the protocol in the flowchart (Figure S4E) was followed.
Cell lines
All mouse cancer cell lines were routinely tested for mycoplasma and profiled and identified by RNA-seq and the GenePrint 10 system (Promega) at periodic intervals during the course of this study for banking and experimental studies. All cell lines were maintained in either DMEM (YUMM1.7ER, YUMMER1.7-luc, NIL, NILER1–4, mSK-Mel254 and KP4662) or RPMI (CT26) supplemented with high glucose with 10% heat-inactivated FBS (Omega Scientific) and 2 mM glutamine in humidified, 5% CO2 incubator. To establish YUMM1.7ER-luc, the pHIV-Luc-Zsgreen vector (Addgene) was subcloned into the lentiviral vector pLV-EF1a-IRES-Blast (Addgene). YUMM1.7ER cells transduced by luciferase-expressing lentiviruses were selected using blasticidin (Sigma-Aldrich).
METHOD DETAILS
Mass cytometry of murine tumors
2×106 or fewer cells were incubated with 2% of FBS in PBS with 25 μg/mL of 2.4G2 antibody at 4°C for 10 min prior to surface staining with an antibody cocktail at 4°C for 30 min in a 50 μL volume. Cells were incubated with 2.5 μM 194Pt monoisotopic cisplatin (Fluidigm) at 4°C for 1 min. Cells were then washed twice with FACS buffer and barcoded using palladium metal barcoding reagents according to manufacturer’s protocol (Fluidigm). Subsequently, fixation and permeabilization were performed using the Foxp3 fix and permeabilization kit according to the manufacturer’s protocol (Fluidigm). Cells were then stained with an intracellular stain antibody cocktail (Foxp3, Ki67, granzyme B, T-bet, iNos, Eomes) for 30 min at room temperature. Cells were then washed twice with Foxp3 permeabilization buffer, twice with FACS buffer, and incubated overnight in 1.6% PFA PBS with 100 nM iridium nucleic acid intercalator (Fluidigm). Cells were then washed twice with PBS with 0.5% BSA, filtered, and washed twice with water with 0.1% BSA prior to analysis. Samples were analyzed using a Helios mass cytometer based on the Helios 6.5.358 acquisition software (Fluidigm).
Single cell 5’ gene expression and V(D)J sequencing
Four different tumors per regimen and per time point were dissociated to single-cell suspensions using a tumor dissociation kit (Miltenyi Biotech) and gentleMACS™ Octo Dissociator (Miltenyi Biotech). 5×105 cells per tumor were pooled together (2×106 in total) as one sample for each regimen and each time point. Cells were incubated with 20% FBS in PBS with 25 μg/mL of anti-mouse CD16/CD32 antibody (eBioscience) at 4°C for 10 min to minimize background antibody binding. Then cells were stained with BV510-anti-CD45 (1 μg/mL, Biolegend) and PerCP-anti-TER119 (2 μg/mL, Biolegend) at room temperature for 20 minutes, followed by 7AAD (10 μL in 500 μL PBS per sample, BD Pharmigen) staining for 5 minutes on ice. Cells after staining were sorted by BD FACSAria II sorting system to harvest the BV510 (CD45) positive and PerCP (TER119, 7AAD) negative population as live CD45+ cells. Cells recovered were subjected to 10X Genomics standard protocol for coupled scRNA-seq and scTCR-seq library preparation using Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit v1.1 (10X Genomics) and V(D)J Enrichment Kit for Mouse T Cells (10X Genomics). Libraries were sequenced by Novaseq 6000 S2 flow cell with 2×50 reads targeting a minimum of 20,000 read pairs per cell for scRNA-seq library and 5,000 read pairs per cell for scTCR-seq library.
Generation of bulk tumor TCR-seq data
Total RNA was extracted from frozen tissue stored in RNALater using the QIAGEN All Prep DNA/RNA Mini Kit. RNA quality was measured using Bioanalyzer (Agilent). 710 ng-1,000 ng of RNA from ovary and brain tissues (RNA Integrity Number (RIN) score > 7) was used as input to construct libraries with the QIAGEN QIAseq Immune Repertoire RNA Library Kit – T cell Receptor Panel (Qiagen). Briefly, RNA was reverse transcribed using a pool of TCR gene-specific primers against the constant region for the T cell receptor alpha, beta, gamma, and delta genes. The resulting cDNA was then ligated to an oligo containing one side of sample index and unique molecular index (UMI). After reaction cleanup, a single primer extension was used to capture the T cell receptor using a pool of gene-specific primers. The resulting captured sequences were amplified and purified using QIAseq beads. Libraries were then sample-indexed on the other side by using a unique sample index primer and an universal primer to amplify the library and introduce platform-specific adapter sequences. The dual-indexed sample PCR fragment was purified and then quantified for absolute quantification of amplifiable libraries (DNA with adaptors at both ends) in triplicate by real-time qPCR using QIAGEN QIAseq Library Quant Array Kit. For sequencing, each library was diluted to 4 nM, pooled, and denatured. 12 pM of denatured library pool was run with QIAseq A Read1 Primer on Illumina NextSeq 500 Mid Output Kit using v2.5 chemistry for 300 cycles with an asymmetrical paired end 261/41 bp read for CDR3 region.
Tissue staining
For immunofluorescence (IF), tissues were fixed in formalin followed by embedding in paraffin. After deparaffinization and rehydration, tissue sections were subjected to heat for antigen retrieval. After tissue sections were permeabilized and blocked, primary antibodies (phospho-ERK1/2 (Cell Signaling Technology, #4370)) were added overnight. IF was performed with Alexa Fluor–conjugated secondary antibodies (Life Technologies, #A21429). Nuclei were counterstained by DAPI (ThermoFisher Scientific). Signals were captured with a Zeiss microscope (AXIO Imager A1) mounted with a charge-coupled device camera (Retiga EXi QImaging), and the images captured by Image-pro plus 6.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Clinical trial data analysis
Details of S1320 have been published with the primary outcome manuscript (Algazi et al., 2020). Association between prior ICT and categorical and quantitative variables were compared using Fisher’s exact and Wilcoxon tests, respectively. PFS and OS were estimated using the Kaplan-Meier method, and Cox regression models were used to evaluate associations.
CyTOF data analysis
Mass cytometry flow cytometry standard (FCS) data files were concatenated, bead-normalized and debarcoded using Helios software (Fluidigm), and then exported into individual files for each sample. Total CD45+ and CD8+ and CD4+ T-cell populations were manually identified and exported using negative and positive gating strategies of lineage markers in Cytobank (Kotecha et al., 2010). We applied Cytofkit (Chen et al., 2016) to perform the t-Distribution Stochastic Neighbor Embedding (t-SNE) analysis separately on the manually gated CD45+, CD4+ and CD8+ populations. We selected 5,000 (in sub-clustering CD45+) or 2,000 events (in sub-clustering CD4+/CD8+ T-cells) in each sample to ensure equal representation of cells across samples. All the cell lineage markers in the immune panel were used in CD45+ analysis. For T-cell analysis, all markers excluding the following were used: CD90, CD14, F480, Ly6G, CXCR2, CD19, and CD335. We chose the 3,000 iterations, perplexity of 30 and theta of 0.5, as the standard t-SNE parameters. Mean intensity values of markers in each cluster were calculated and visualized via heatmaps. Cells were assigned to different functional populations on the basis of the local gradient expression of known cell lineage markers. The percentages of different immune cell subsets were calculated for each sample.
Analysis of scRNA-seq data
Alignment to GRCm38 reference genome, barcode and unique molecular identifier (UMI) counting were performed using Cell Ranger (10x Genomics, v2.1.0). Seurat package (Butler et al., 2018) was used for downstream analysis. Cells with fewer than 500 genes detected or greater than 10% mitochondrial RNA content were excluded from further analysis. Raw UMI counts were normalized to UMI count per million total counts and log-transformed. Variable genes were detected based on average expression and dispersion for each dataset independently. We then use CellCycleScoring function to calculate scores of S and G2/M cell cycle phases for each cell. Single cells from different conditions were integrated into a single assay based on variable genes identified from each sample. We then use the ScaleData function to calculate scaled z-scores of each variable gene in the integrated assay and regress out the effect of number of genes per cell, mitochondrial RNA content, and cell cycle scores (S phase score and G2/M phase score). This scaled data set was then used for principal component analysis (PCA) for cells. Clusters and UMAP projections were generated based on the top 30 PCA dimensions. Clusters were annotated based on expression of known marker genes, including Cd14 (myeloid), Igtam, Csf1r (monocyte/macrophage), Flt3 (dendritic cell), S100a8, S100a9 (neutrophil), Ncr1 (NK cell), Cd19, Cd79a (B-cell), Cd3d, Cd3e, Cd3g (T-cell). Cell clusters co-expressing markers of multiple cell types were defined as doublets and excluded from further analysis. We next isolated the monocyte/macrophage and T-cell populations identified from the broad clustering analysis and performed re-clustering analysis on them separately. Cells were re-clustered as described above and functional subpopulations were inferred and annotated by identifying differentially expressed marker genes with log-fold change higher than 0.4 using MAST in FindAllMarkers function. The pro/anti-inflammation ratio in the macrophage population was calculated as the fold changes of proportions between the inferred pro- vs. anti-inflammatory clusters. For the identified CD8+ T-cell subpopulations, we calculated the score of Tcf1+ stem-like signature for each cell by using Seurat’s AddModuleScore function based on the gene sets previously reported (Siddiqui et al., 2019).
Analysis of scTCR-seq data
Alignment to the GRCm38 reference genome and TCR contig annotation were performed by Cell Ranger vdj pipeline (10× Genomics, v2.1.0). For the TCR clonotype analysis, only cells assigned with both productive TRA and TRB sequences were kept for further analysis. If one cell had two or more TRA-TRB pairs identified, the pair with higher UMIs was considered as the dominant TRA-TRB pair in the corresponding cell and used in the analysis. We defined each unique TRA-TRB pair as a clonotype. The clonal status of TCR clones were characterized as non-clonal (n = 1) and clonal (n ≥ 2) based on their cell numbers. The TCR clonotype of each cell was further linked to inferred functional subsets based on the barcode information. We used STARTRAC package to calculate the expansion and transition indices of distinct T-cell subsets.
Bulk tumor TCR-seq data analysis
As we reported previously (10), raw reads were submitted to the QIAGEN GeneGlobe Data Analysis Center (https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/) to estimate the abundance of reads of unique CDR3 sequence and generate TCR clonotype calls. R package tcR (Nazarov et al., 2015) was used to perform all the statistical analysis for TCR repertoires, including: i) size of large clones with frequency not less than 5%; ii) diversity estimates using ecological diversity and Gini-Simpson indices, and iii) common TCR repertories (identified based on unique alpha or beta chains’ CDR3 sequences) shared by distinct tumor-involved regions within an organ tissue or by different organ tissues.
Statistical analysis of non-clinical data
No statistical methods were used to predetermine sample size. We used the paired t-test to determine the statistical significance of differences between two variables. Survival curves were compared via the logrank test. We applied the linear mixed effects model on the log10-transformed BLI intensity data. In this model, treatment group, days, and treatment*day interactions were treated as fixed effects, while a random intercept and time slope were assumed for individual mouse. Pairwise comparisons between treatment regimen groups were conducted within the mixed model framework with Bonferroni correction for multiple testing. All other statistical analyses were carried out using R and GraphPad Prism 7.
Supplementary Material
Highlights.
Prior ICT is associated with longer PFS of melanoma patients treated with MAPKi
Anti-PD-1/L1 before MAPKi combination prolongs durability of tumor regression
Targeting M2-TAMs augments and CD8+ T cells abolishes priming-associated benefit
Anti-PD1/L1 plus anti-CTLA-4 priming may further control melanoma brain metastasis
Acknowledgements
This research was supported by grants (to R. S. Lo) from the National Institutes of Health (NIH) (1R01CA176111A1; 1R21CA215910–01; R21CA255837–01), the Melanoma Research Alliance (MRA), and the V Foundation for Cancer Research. Additional funding was provided by the MRA (to Y. Wang, S. Liu, Z. Yang, G. Moriceau), Jonsson Comprehensive Cancer Center (JCCC) (to S. Liu, Z. Yang), NIH T32CA009120 Tumor Immunology Postdoctoral Fellowship (to A. Hong), NIGMS P20GM121293 (to A. J. Tackett), and Parker Institute for Cancer Immunotherapy and NIH R35 CA197633 (to A. Ribas). We also acknowledge the support of the NIH (1P01CA168585) (to R. S. Lo, A. Ribas and G. Moriceau), the Steven C. Gordon Family Foundation (to R. S. Lo), and Mary Tanner and Maurizio Grimaldi (to R. S. Lo and A. Ribas). We would like to thank S. Xu (Preclinical Imaging Technology Center) and X. Li (Technology Center for Genomics and Bioinformatics) at UCLA for excellent technical support. CyTOF was performed in the UCLA JCCC Flow Cytometry Core Facility (NIH award P30 CA016042). R. S. Lo and A. Ribas are grateful to the Ressler Family Foundation for its long-term support. S1320 is an intergroup trial led by the SWOG Cancer Research Network with funding and supervision from NCI-CTEP.
Footnotes
Declaration of Interests
R.S.L.—research support from Merck, Pfizer, BMS, and OncoSec. Consultant for Amgen, Novartis, Array BioPharma, Genentech, and Merck. A.R.—consultant for Amgen, BMS, Chugai, Genentech, Merck, Novartis, Roche, Sanofi, and Vedanta. Advisory board member for and stock shareholder of Advaxis, Apricity, Arcus, Compugen, CytomX, Five Prime, Highlight, ImaginAb, Isoplexis, Kalthera, Kite-Gilead, Merus, PACT Pharma, RAPT, Rgenix, and Tango. Research funding from Agilent and BMS through SU2C. A.P.A.—research support, advisory board member, consultant, shareholder and honorarium recipient, OncoSec. Advisory board member for and stock shareholder in Valitor Biosciences. Advisory board member and honorarium recipient for Regeneron and Array. Research support from Acerta, Amgen, AstraZeneca, BMS, Dynavax, Genentech, Idera, Incyte, ISA, LOXO, Merck, Novartis, Sensei and Tessa. M.O.—consultant for Merck, Biosight, and Daiichi Sankyo; independent data safety monitoring committee member for Celgene and Glycomimetics. C.C.Y.—consultant for QED Therapeutics and Riptide Biosciences and owner of stocks in Riptide Biosciences. H.L.—co-founder and Executive Vice President of Riptide Biosciences. M.W.B.— consultant for Eli Lilly and research support from AstraZeneca. D.B.S.—consultant for Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, Fore Therapeutics, and BridgeBio.
ADDITIONAL RESOURCES
ClinicalTrials.gov Identifier: NCT02196181
URL: https://clinicaltrials.gov/ct2/show/NCT02196181
SWOG Identifier: S1320
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP, Gunturi A, Flaherty KT, Hodi FS, Kefford R, et al. (2014). Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer 120, 1695–1701. [DOI] [PubMed] [Google Scholar]
- Algazi AP, Othus M, Daud AI, Lo RS, Mehnert JM, Truong TG, Conry R, Kendra K, Doolittle GC, Clark JI, et al. (2020). Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: a randomized phase 2 trial. Nat Med 26, 1564–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Di Giacomo AM, et al. (2019). Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 25, 941–946. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, and Chen J. (2016). Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput Biol 12, e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MA, Liu P, McIntyre S, Kim KB, Papadopoulos N, Hwu WJ, Hwu P, and Bedikian A. (2011). Prognostic factors for survival in melanoma patients with brain metastases. Cancer 117, 1687–1696. [DOI] [PubMed] [Google Scholar]
- Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, Chiarion-Sileni V, Thomas L, Lesimple T, Mortier L, et al. (2017). Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol 18, 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, Rutkowski P, Del Vecchio M, Gutzmer R, Mandala M, et al. (2017). Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 18, 435–445. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. (2012). Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenard C, Peuvrel L, Jean MS, Brocard A, Knol AC, Nguyen JM, Khammari A, Quereux G, and Dreno B. (2016). Development of brain metastases in patients with metastatic melanoma while receiving ipilimumab. J Neurooncol 126, 355–360. [DOI] [PubMed] [Google Scholar]
- Ghebremedhin A, Salam AB, Adu-Addai B, Noonan S, Stratton R, Ahmed MSU, Khantwal C, Martin GR, Lin H, Andrews C, et al. (2020). A Novel CD206 Targeting Peptide Inhibits Bleomycin Induced Pulmonary Fibrosis in Mice. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P, Demidov L, et al. (2020). Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 395, 1835–1844. [DOI] [PubMed] [Google Scholar]
- Hong A, Moriceau G, Sun L, Lomeli S, Piva M, Damoiseaux R, Holmen SL, Sharpless NE, Hugo W, and Lo RS (2018). Exploiting Drug Addiction Mechanisms to Select against MAPKi-Resistant Melanoma. Cancer Discov 8, 74–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong A, Piva M, Liu S, Hugo W, Lomeli SH, Zoete V, Randolph CE, Yang Z, Wang Y, Lee JJ, et al. (2021). Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity. Cancer Discov 11, 714–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. (2015). Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 162, 1271–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Homet-Moreno B, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Resistance and Sensitivity to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, Li D, Calvo R, Dashnyam M, Singh A, et al. (2020). Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Pectasides E, Feld E, Ye F, Zhao S, Johnpulle R, Merritt R, McDermott DF, Puzanov I, Lawrence D, et al. (2017). Sequencing Treatment in BRAFV600 Mutant Melanoma: Anti-PD-1 Before and After BRAF Inhibition. J Immunother 40, 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, and Irish JM (2010). Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom Chapter 10, Unit10 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Grob JJ, Nathan P, Ribas A, Robert C, Schadendorf D, Lane SR, Mak C, Legenne P, Flaherty KT, and Davies MA (2016). Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: a pooled analysis of individual patient data from randomised trials. Lancet Oncol 17, 1743–1754. [DOI] [PubMed] [Google Scholar]
- Mason R, Dearden HC, Nguyen B, Soon JA, Smith JL, Randhawa M, Mant A, Warburton L, Lo S, Meniawy T, et al. (2020). Combined ipilimumab and nivolumab first-line and after BRAF-targeted therapy in advanced melanoma. Pigment Cell Melanoma Res 33, 358–365. [DOI] [PubMed] [Google Scholar]
- Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, Chudakov DM, Lebedev YB, and Mamedov IZ (2015). tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics 16, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijers ILM, Rozeman EA, Wilgenhof S, van Thienen JV, Haanen J, and Blank CU (2020). Switch to checkpoint inhibition after targeted therapy at time of progression or during ongoing response: A retrospective single-centre experience in patients with BRAF-mutated melanoma. Pigment Cell Melanoma Res 33, 498–506. [DOI] [PubMed] [Google Scholar]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, et al. (2016). Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 315, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, Fisher R, Long GV, Hodi FS, Tsoi J, et al. (2019). Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 25, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert H, Hirata E, Gore M, Khabra K, Messiou C, Larkin J, and Sahai E. (2016). Extrinsic factors can mediate resistance to BRAF inhibition in central nervous system melanoma metastases. Pigment Cell Melanoma Res 29, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110. [DOI] [PubMed] [Google Scholar]
- Simeone E, Grimaldi AM, Festino L, Giannarelli D, Vanella V, Palla M, Curvietto M, Esposito A, Palmieri G, Mozzillo N, and Ascierto PA (2017). Correlation between previous treatment with BRAF inhibitors and clinical response to pembrolizumab in patients with advanced melanoma. Oncoimmunology 6, e1283462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X, Zhang H, Lomeli S, Qian J, Yu CC, et al. (2017). Recurrent Tumor Cell-Intrinsic and -Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation. Cancer Discov 7, 1248–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. (2018). Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 379, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetu P, Mangana J, Dummer R, Dutriaux C, Beneton N, Dalle S, Meyer N, Oriano B, Michielin O, and Lebbe C. (2018). Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer 93, 147–149. [DOI] [PubMed] [Google Scholar]
- Wang J, Perry CJ, Meeth K, Thakral D, Damsky W, Micevic G, Kaech S, Blenman K, and Bosenberg M. (2017). UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res 30, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. (2017). Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing files of scRNA-seq, scTCR-seq, and bulk TCR-seq data have been deposited at GEO (GSE177902) and are publicly available as of the date of publication. Mass cytometry data of BrafV600E melanoma, Nf1−/− melanoma, and KrasG12C colorectal carcinoma syngeneic models have been deposited at FlowRepository (http://flowrepository.org/) under experiment IDs FR-FCM-Z43Z, FR-FCM-Z4YR, and FR-FCM-Z42V. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
There are no original codes generated in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 (Clone 30-F11) | Fluidigm | Cat#3089005B; RRID: AB_2651152 |
| Ki67 (Clone SolA15) | Invitrogen | Cat#14-5698-82; RRID: AB_10854564 |
| CD90.2/Thy-1.2 (Clone 30-H12) | Biolegend | Cat#105333; RRID: AB_2563765 |
| Ly-6G (Clone 1A8) | Biolegend | Cat#127602; RRID: AB_1089180 |
| CD69 (Clone H1.2F3) | Invitrogen | Cat#14-0691-82; RRID: AB_467325 |
| CD4 (Clone RM4-5) | Biolegend | Cat#116018; RRID: AB_2650936 |
| F4/80 (Clone BM8) | Biolegend | Cat#123102; RRID: AB_893506 |
| Eomes (Clone Dan11mag) | Invitrogen | Cat#14-4875-82, RRID:AB_11042577 |
| CD11b (Mac-1) (Clone m1/70) | Fluidigm | Cat#3148003B; RRID: AB_2814738 |
| CD62L (L-selectin) (Clone MEL-14) | Biolegend | Cat#104416; RRID: AB_313101 |
| Ly-6C (Clone HK1.4) | Biolegend | Cat#128002; RRID: AB_1134214 |
| CD25 (IL-2R) (Clone 3C7) | Biolegend | Cat#101913; RRID: AB_2562798 |
| CD3e (Clone 145-2C11) | Biolegend | Cat#100314; RRID: AB_312679 |
| TER119 (Clone TER119) | Biolegend | Cat#116214; RRID: AB_313715 |
| CD152 (CTLA-4) (Clone UC10-4B9) | Biolegend | Cat#106308; RRID: AB_2087654 |
| CD14 (Clone Sa14-2) | Biolegend | Cat#123302; RRID: AB_940592 |
| FoxP3 (Clone FJK-16s) | Invitrogen | Cat#14-5773-82; RRID: AB_467576 |
| CD279 (PD-1) (Clone 29F.1A12) | Biolegend | Cat#135202; RRID: AB_1877121 |
| iNOS (Clone CXNFT) | Fluidigm | Cat#3161011B |
| CD366 (Tim-3) (Clone RMT3-23) | Fluidigm | Cat#3162029; RRID: AB_2687841 |
| CD197 (CCR7) (Clone 4B12) | eBiosciences | Cat#16-1971-85; RRID: AB_494123 |
| CD182 (Clone SA044G4) | Biolegend | Cat#149302; RRID: AB_2565277 |
| CD19 (Clone 6D5) | Biolegend | Cat#115514; RRID: AB_313649 |
| CD335 (NKp46) (Clone 29A1.4) | Fluidigm | Cat#3167008B |
| CD8a (Clone 53-6.7) | Biolegend | Cat#100716; RRID: AB_312755 |
| T-bet (Clone 4B10) | Biolegend | Cat#644802; RRID: AB_1595503 |
| CD192 (Clone 475301R) | R&D | Cat#MAB55381R |
| Granzyme B (Clone GB11) | Fluidigm | Cat#3171002B; RRID: AB_2687652 |
| CD44 (Clone 1M7) | Biolegend | Cat#103014; RRID: AB_312965 |
| I-A/I-E (Clone M5/114.15.2) | Biolegend | Cat#107610; RRID: AB_313325 |
| CD278 (ICOS) (Clone 7E.17G9) | Invitrogen | Cat#14-9942-85; RRID: AB_468633 |
| CD11c (Clone N418) | Fluidigm | Cat#3209005B; RRID: AB_2811244 |
| CD16/CD32 (Clone 93, for blocking buffer) | eBioscience | Cat#14-0161-86; RRID: AB_467135 |
| CD8a (Clone 53-6.7) | Biolegend | Cat#100737; RRID: AB_10897101 |
| CD45 (Clone 30-F11) | Biolegend | Cat#103138; RRID: AB_2563061 |
| PerCP-anti-TER119 | Biolegend | Cat#116225; RRID: AB_893637 |
| inVivoMAb anti-mouse PD-L1 (B7-H1) | BioXcell | Cat#BE0101; RRID: AB_10949073 |
| inVivoMAb anti-mouse CD8α (Clone YTS169.4, in vivo antibody) | BioXcell | Cat#BE0117; RRID: AB_10950145 |
| Anti-Mouse CD279 (PD-1) (Clone RMP1-14) - Purified In vivo GOLD™ Functional Grade | Leinco Technologies, Inc. | Cat#P362; RRID: AB_2737557 |
| InVivoPlus anti-mouse CTLA-4 (CD152, Clone 9H10) | BioXcell | Cat#BE0131; RRID: AB_10950184 |
| phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat#4370; RRID: AB_2315112 |
| Goat Anti-Rabbit IgG (H+L) Highly Cross-adsorbed Antibody, Alexa Fluor555 | Life Technologies | Cat#A-21429; RRID: AB_2535850 |
| Bacterial and virus strains | ||
| NEB® Stable Competent E. coli | New England BioLabs | Cat#C3040I |
| Third generation lentivirus | Dr. Lo | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Trametinib | LC Laboratories | Cat#T-8123 |
| RP-832c | Riptide Bioscience | PMID: 32051227 |
| Vemurafenib | LC Laboratories | Cat#V-2800 |
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Cell-ID Intercalator Ir | Fluidigm | Cat#201192B |
| 7-AAD | BD Pharmigen | Cat#51-68981E |
| Blasticidin | Sigma-Aldrich | Cat#SBR00022 |
| Prolong Diamond Antifade Mountant with DAPI | ThermoFisher Scientific | Cat#P36962 |
| Critical commercial assays | ||
| 20-Plex Pd Barcoding Kit | Fluidigm | Cat#201060 |
| mirVana™ miRNA Isolation Kit, with phenol | Thermo Fisher Scientific | Cat#AM1560 |
| AllPrep DNA/RNA Mini Kit | Qiagen | Cat#80204 |
| MycoAlert™ Mycoplasma Detection Kit | Lonza | Cat#LT07-418 |
| MycoAlert™ Assay Control Set | Lonza | Cat#LT07-518 |
| Tumor Dissociation Kit | Miltenyi Biotec | Cat#130-095-929 |
| Single Cell 5′ Library and Gel Bead Kit v1.1 | 10X Genomics | Cat#1000167 |
| Enrichment Kit for Mouse T Cells | 10X Genomics | Cat#1000071 |
| QIAGEN QIAseq Immune Repertoire RNA Library Kit - T cell Receptor Panel | Qiagen | Cat#333705 |
| QIAseq Library Quant Array Kit | Qiagen | Cat#333304 |
| Foxp3 Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Deposited data | ||
| scRNA-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| scTCR-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| Bulk TCR-sequencing data of melanoma mouse model | This Paper | GSE177902 |
| Mass cytometry data of mouse immune cells (BrafV600E melanoma model) | This Paper | FR-FCM-Z43Z |
| Mass cytometry data of mouse immune cells (Nf1−/− melanoma) | This Paper | FR-FCM-Z4YR |
| Mass cytometry data of mouse immune cells (KrasG12C colorectal carcinoma) | This Paper | FR-FCM-Z42V |
| Experimental models: Cell lines | ||
| Mouse cell line; NIL | Dr. Norman E. Sharpless and adapted by Dr. Roger S. Lo | PMID: 25252692 |
| Mouse cell line; NILER1-4 | Dr. Roger S. Lo | PMID: 33318037 |
| Mouse cell line; mSKMel-254 | Dr. David B. Solit and adapted by Dr. Roger S. Lo | This paper |
| Mouse cell line; YUMM1.7ER | Dr. Marcus W. Bosenberg | PMID: 27287723 |
| Mouse cell line; CT26 | ATCC | PMID: 33318037 |
| Mouse cell line; KPC (KP4662) | Dr. Robert Vonderheide | PMID: 27642636 |
| Mouse cell line; YUMM1.7ER-luciferase | Dr. Roger S. Lo | This paper |
| Experimental models: Organisms/strains | ||
| C57BL/6J; C57BL/6N mice | UCLA: Radiation-Oncology breeding colony | C57BL/6J/NROC |
| BALB/C mice | UCLA: Radiation-Oncology breeding colony | BALB/c ROC |
| C57BL/6J mice | Jackson Laboratory | Cat# 000664 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pLV-EF1a-IRES-Blast | Addgene | Cat#85133; RRID: Addgene_85133 |
| Software and algorithms | ||
| Cytofkit | Bioconductor | Version: 3.7 |
| R software | CRAN | Version: 3.5.1 |
| GraphPad Prism | https://swcstore.oit.ucla.edu/secure/browse_vendors.php | Version: 7 |
| Cytobank | https://www.cytobank.org/ | |
| Cell Ranger | 10x Genomics | Version: 2.1.0 |
| Seurat | CRAN | Version: 3.0.2 |
| STARTRAC | GitHub | Version: 0.1.0 |
| tcR | CRAN | Version: 2.2.4.1 |
| Living Image | PerkinElmer | Version: 4.7.2 Version |
| Other | ||