

Abstract
One of the central aims in Alzheimer’s disease (AD) research is the identification of clinically relevant drug targets. A plethora of potential molecular targets work very well in preclinical model systems both in vitro and in vivo in AD mouse models. However, the lack of translation into clinical settings in the AD field is a challenging endeavor. Although it is long known that N-terminally truncated and pyroglutamate-modified Abeta (AβpE3) peptides are abundantly present in the brain of AD patients, form stable and soluble low-molecular weight oligomers, and induce neurodegeneration in AD mouse models, their potential as drug target has not been generally accepted in the past. This situation has dramatically changed with the report that passive immunization with donanemab, an AβpE3-specific antibody, cleared aymloid plaques and stabilized cognitive deficits in a group of patients with mild AD in a phase II trial. This review summarizes the current knowledge on the molecular mechanisms of generation of AβpE, its biochemical properties, and the intervention points as a drug target in AD.
Subject terms: Drug discovery, Neuroscience
Introduction
For more than three decades, the amyloid cascade hypothesis [1] claiming that amyloid (Aβ) peptides as cleavage products of the precursor amyloid precursor protein (APP) [2] and accumulation in plaques triggers neuron loss and neurofibrillary tangle formation. Since the first report on the peptide sequence of Aβ1-24 of the N-terminus isolated from cerebrovascular amyloid preparations from Alzheimer’s disease (AD) and Down syndrome brain [3] followed by peptide sequencing of amyloid-β isolated from plaques it became evident that amyloid in plaque cores must be predominantly N-truncated [4]. Later, Mori et al. [5] described an N-truncated Aβ variant starting with pyroglutamate at position 3 (AβpE3-X). Saido et al. [6] was the first to compare the staining pattern of Aβ1-X with AβpE3-X specific antibodies and showed that the N-truncated Aβ variant is a dominant fraction in plaques in AD brain. Russo et al. [7] reported that both Aβ1-X and AβpE-X can form stable water-soluble aggregates. Using matrix-assisted laser-desorption-time-of-flight mass spectrometry a variety of N-truncated Aβ peptides, including AβpE3-X in amyloid plaques very identified [8]. Wildburger et al. [9] used high-resolution mass spectrometry and identified a wide range of N- and C-truncated amyloid-β peptides from post-mortem brain of AD patients demonstrating no correlation with post-mortem interval. Portelius et al. [10] revealed the relative abundance of full-length and N-truncated variants using immunoprecipitation in combination with mass spectrometric analysis in different brain areas of sporadic and familial AD cases. The major variants were Aβ1-42, AβpE3-42, Aβ4-42 followed by Aβ1-40 in both sporadic and familial AD cases. Moreover, Upadhaya et al. [11] demonstrated that AβpE3-X western blotting can be used as an informative biomarker for biochemical amyloid-β staging in post-mortem brain tissue from symptomatic AD and preclinical AD cases. Besides AβpE-X and a phosphorylated Aβ variant found in plaques, they were also observed as soluble aggregates in a disease-specific manner. The authors concluded that the level of different Aβ variants occur in a hierarchical sequence allowing the distinction of three biochemical amyloid-β stages, with stage 1 and its characteristic marker Aβ1-X, followed by stage 2 with AβpE3-x and stage 3 with a phosphorylated Aβ [11]. In good agreement, Moro et al. [12] observed that deposition of AβpE-X is closely related to AD, but not with normal ageing and is found in plaques and neurons. In AD mouse models, similar observations were observed. In APP23 mouse brain, AβpE-X deposits appear during ageing as a maturation process of amyloid plaques starting with Aβ1-X first followed by AβpE3-X deposits [13]. In APP/PS1KI mice, AβpE3-X positive plaques increased with age, on the expense of Aβ1-X [14]. Transgenic mice expressing mutant AβQ3-42 elicit partial conversion of N-terminal Gln-3 into pyroglutamate Glu-3 in a cell-type dependent manner depending on the different mouse line likely due to specific genomic integration of the transgene. Besides abundant loss of Purkinje cells and ataxia [15], degeneration of hippocampus CA1 neurons and cognitive decline [16], or loss of striatal neurons associated with basal locomotor activity and sensorimotor gating (when coexpressed with human QC) [17] was reported in different transgenic lines.
Generation of pyroglutamate Aβ
Figure 1 shows a schematic presentation of the different steps for generation of N-terminal pyroglutamate peptides involved in AD etiology as well as the cellular pathways involved. In the last years, considerable progress was achieved in elucidating the different molecular steps for generation of pyroglutamate-modified Aβ. β-site APP cleaving enzyme 1 (BACE1) is the first and rate-limiting step in the production of full-length Aβ [18] from its precursor the APP [19]. BACE1 cleaves between Met (position −1 of Aβ peptide) and Asp-1 (position +1 of Aβ peptide). This cleavage can also be carried out by meprin-β (reviewed in [20]). Another cleavage site of meprin-β is between Asp-1 and Ala-2 (liberating Aβ2-X). Besides meprin-β, aminopeptidase A (APA) can trigger the initial first amino acid (Asp-1) cleavage of Aβ [21]. Valverde et al. [22] have convincingly demonstrated that pharmacological reduction of APA activity or lowering of APA expression by shRNA leads to reduced levels of plaque-associated AβpE3-X and Aβ1-X peptides, together with normalized memory deficits in 3xTg-AD mice. Of note, AβpE3-X in plaques was correlated with APA activity and early Braak stages in brains of patients with sporadic AD. Therefore, APA represents a key enzyme as a potential drug target involved in N-terminal truncation of Aβ peptides independent from dipeptidyl peptidase 4 (DPP4) activity [22].
Fig. 1. Generation of pyroglutamated (pE) peptides involved in Alzheimer’s disease.
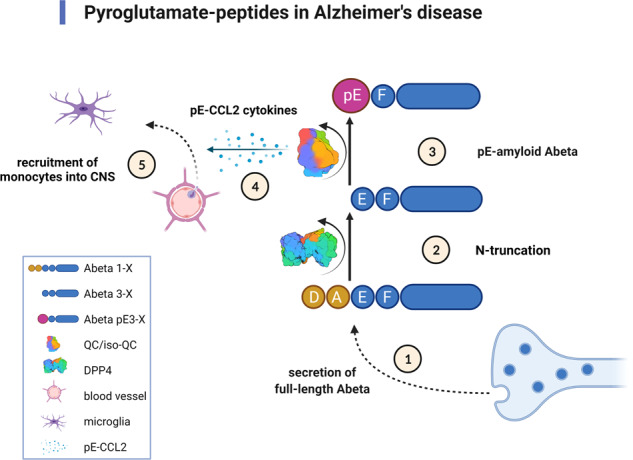
① The N-terminus of full-length Aβ is generated by BACE1 or meprin-β and secreted by neurons. ② Next, N-terminal amino acids are cleaved off by aminopeptidase A (APA), meprin-β or dipeptidyl peptidase 4 (DPP4). ③ Glutamate at position three of the N-terminus of Aβ is subsequently post-translationally modified into N-terminal pyroglutamate (pE) by dehydration catalyzed by glutaminyl cyclase (QC) activity. ④ The isoenzyme of QC, isoQC, predominantly converts the N-terminus of chemokine ligand 2 (CCL2) into pE-CCL2 ⑤ triggering monocyte recruitment into the central nervous system (CNS). The surface structure of DPP4 [84] and of QC [85] was taken from the Protein Data Bank (PDB). Created with BioRender.com.
The enzymatic cleavage between Ala-2 and Glu-3 (liberating Aβ3-X) is carried out by meprin-β or DPP4 [23, 24]. DPP4 is known to participate for example in cleaving neuropeptide Y [25]. Antonyan et al. [23] have demonstrated that purified DPP4 acts together with glutaminyl cyclase (QC) activity to generate AβpE3-40/42 in vitro by MALDI-TOF mass spectrometry. Valverde et al. [26] added convincing evidence that human recombinant DPP4 is the rate-limiting enzyme liberating Aβ3-X from its precursor Aβ1-X: (i) Pharmacological treatment with a DPP4-specific inhibitor elevated levels of full-length Aβ on the expense of AβpE3-40. (ii) Lentiviral expression of mutant APP cDNA in wild-type mouse brain and pharmacological blocking of DPP4 activity in brain in situ rescued dendritic spine morphology. (iii) This was further supported by studies using the 3xTg-AD mouse model. Amyloid-plaque load was reduced and memory deficits were normalized in this model by pharmacological and shRNA-driven inhibition of DPP4 activity. (iv) In addition, DPP4 activity seemed to be transiently elevated during AD pathogenesis. Hence, DPP4 may be a potential drug target against AD [27] and cognitive benefits in patients suffering from diabetes [28]. DPP4 is long known for treating diabetes as it metabolizes glucagon-like peptide-1 [29].
There is considerable evidence that QC is the rate-limiting enzyme in the final step the conversion of N-truncated Aβ3-X into pyroglutamate AβpE3-X [23, 30–35]. Besides, the normal function of QC to stabilize hormones, peptides, and proteins it may also contribute to neurodegenerative disorders, systemic inflammatory diseases and certain types of oncological conditions (reviewed recently [36]). In the 5XFAD mouse model for AD, overexpression of human QC and knock-out of murine QC clearly demonstrated that memory function correlated well with the formation of AβpE3-X [32]. Although complete loss of QC activity in homozygous QC knock-out mice did not eliminate AβpE3-X levels completely suggesting that there is a resting QC-like activity by other enzyme(s). Such an activity has been added by Cynis et al. [37] demonstrating that the isoenzyme of QC (isoQC) predominantly modulated chemokine ligand 2 (CCl2; synonyms: monocyte chemotactic protein 1 and small inducible cytokine A2) thereby fostering pE-CCL2 formation and monocyte infiltration. Moreover, pharmacological inhibition of QC/isoQC-activity reduced the pathology in a mouse model for atherosclerosis. The function of QC/isoQC activity in the etiology of AD addresses directly the generation of toxic AbpE3-X peptides, but equally important may modulate microglia function via recruitment of monocytes from the periphery. Of note, the dual function of microglia cells in AD pathology should be taken into account. They participate in both beneficial amyloid-β clearance, but also in destructive inflammation in AD brain (reviewed in detail [38]).
Properties of pyroglutamate Aβ
Saido et al. [39] hypothesized that the lactam ring of AβpE3-x peptides and loss of two negative and one positive charges leads to higher hydrophobicity, higher peptide stability, increased aggregation propensity and may escape enzymatic degradation. Another twist in the complex story was published by Nussbaum et al. [40], who pointed out that AβpE3-42 peptides can form soluble oligomers that potentially seed full-length Aβ1-42 and are toxic in a tau-dependent manner. Cross-seeding activities of AβpE3-X peptides with wild-type Aβ1-X were also reported by Hu et al. [41, 42]. In addition, AβpE3-42 and Aβ4-42 when expressed together elicited neurological deficits, which were significantly stronger as compared to expression of AβpE3-42 or Aβ4-42 alone [43]. Neuronal expression of AβpE3-42 triggered amyloid-plaque load and memory deficits when crossed to amyloid-plaque AD mouse models [44, 45].
The secondary structure of AβpE3-42, Aβ4-42 and Aβ1-42 peptides was analyzed by far-UV circular dichroism (CD) spectroscopy demonstrating that the CD spectra of monomers were characteristic of a disordered conformation [46]. The aggregation propensities of the Aβ variants analyzed by liquid-state nuclear magnetic resonance (NMR) spectroscopy suggested that the temporal loss of signal intensities was due to conversion of NMR-visible monomers and small aggregates into larger aggregates. The highest loss in signal intensity was found for N-truncated Aβ. Hence, it was reported that AβpE3-42 and Aβ4-42 rapidly formed aggregates with a high aggregation propensity in terms of monomer consumption and oligomer formation [46]. Acute treatment of primary cortical neurons indicated that AβpE3-42 and Aβ4-42 are equally toxic as Aβ1-42. This was further corroborated by induction of working memory deficits after intraventricular injection of AβpE3-42, Aβ4-42 or Aβ1-42 in wild-type mice [46, 47].
In aqueous solution and in 10% sodium dodecyl sulfate micelles AβpE3-40 peptides showed increased β-sheet formation using CD spectroscopy and aggregation behavior by sedimentation analysis when compared with Aβ1-40 [48]. The relative toxicity and abundance of N-truncated and full-length Aβ variants both in vitro and in vivo in AD mouse models was reviewed and discussed previously [21, 49–51].
Drug target pyroglutamate Aβ
The different steps for therapeutic intervention against the pyroglutamate amyloid-β cascade in the etiology of AD are shown in Fig. 2. It is generated by a two-step process starting by a first cut of the APP between Met at postion-1 and Asp at position +1 by β-site APP cleaving enzyme 1 (BACE1), which generates the N-terminus of full-length Aβ1-X [33]. In addition, meprin-β can cut between Met-1 and Asp+2 independent from BACE1 [20]. DPP4 is responsible for the cleavage of the first two N-terminal amino acids of full-length Aβ1-X generating Aβ3-X [23, 26]. There is one clinical study published that investigated the influence of DPP4 inhibition in patients with dementia [52]. In this retrospective clinical study the effect of vildagliptin, a DPP4 inhibitor, was investigated on cognitive dysfunction in 60 elderly patients with diabetes with additional diagnosis of mild cognitive impairment (MCI). Fifty percent of the patients were treated with metformin as standard medication control; the other 50% received metformin plus vildagliptin. A significant beneficial effect on stabilizing cognitive scores was observed in the DPP4 inhibitor group [52]. This pilot study did not include however, the assessment of AD biomarkers commonly employed in clinical trials with MCI patients. In addition, it is not clear whether pharmacological inhibition of DPP4 activity is clinically meaningful in nondiabetic MCI and AD patients.
Fig. 2. Pyroglutamate Aβ as a potential drug target against Alzheimer’s disease.
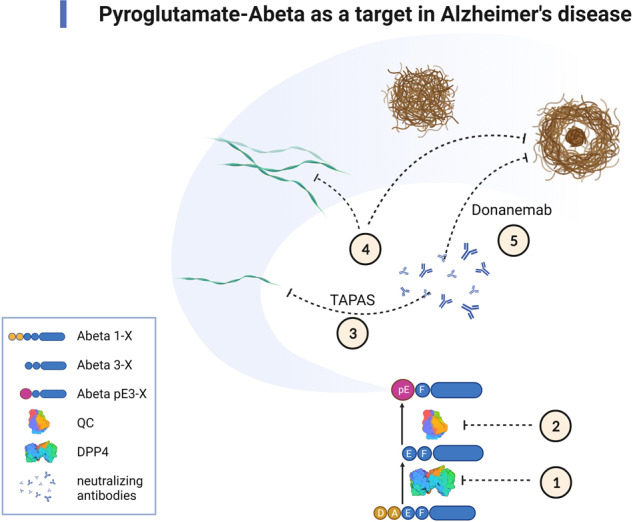
① Inhibition of DPP4 prevents N-terminal truncation of the first two amino acid residues of the full-length Aβ monomers (Aβ1-x). ② Inhibition of QC activity prevents the conversion of Aβ3-x into AβpE3-x monomers. ③–⑤ Neutralizing antibodies react with different conformational and structural variants of AβpE3-X. ③ Once AβpE3-x monomers are generated, they adopt a pseudo β-hairpin structure at the N-terminus, which is specifically recognized by the TAPAS family of antibodies. The pseudo β-hairpin epitope is neutralized by the TAPAS vaccine and by TAPAS monoclonal antibodies. ④ Pan-AβpE-X antibodies react with a range of conformations: soluble oligomers, protofibrils and fibrillar forms found in different plaque types. ⑤ Donanemab, a plaque-specific monoclonal antibody, reacts with AβpE3-X aggregates found in the amyloid-plaque cores. The surface structure of DPP4 [84] and of QC [85] was taken from the Protein Data Bank (PDB). Created with BioRender.com.
Alternatively, and independent from DPP4 activity, a sequential cleavage of full-length Aβ between Asp at position +1 and Ala at position +2 may also occur by aminopeptidase A activity thereby influencing formation of AβpE3-X [21, 22].
The next step in the cascade is the conversion of N-terminal Glu-3 of Aβ into AβpE3-X by QC. In preclinical experiments, pharmacological QC inhibition had significant beneficial effects on AβpE3 levels, plaque load, gliosis and memory function [33]. PQ912, a competitive inhibitor of QC, was analyzed in clinical trials in order to assess the safety, pharmacokinetics, and pharmacodynamics in healthy individuals [53], MCI and AD patients [54, 55] and was reported to be safe. QC-dependent blood biomarker levels were significantly altered by treatment with PQ912.
The conformation of the structure of amyloid-β fibrils is well established eliciting a cross-β structure identified by x-ray diffraction analysis [56]. The individual β-strands of amyloid fibrils are oriented perpendicular to the fibril axis [56]. NMR spectroscopy revealed that the N-terminus of full-length Aβ1-42 is unstructured [57, 58]. Ahmed et al. [59] reported that full-length Aβ oligomers are more toxic to mouse cortical than protofibrils and fibrils. Of note, these oligomers are composed of loosely aggregated strands and do not have the β-sheet structure characteristic of fibrillar amyloid-β.
Therefore, it is of upmost interest that the N-terminus of AβpE3-X monomers adopt a unique pseudo β-hairpin structure, which is recognized by the TAPAS family antibodies (murine and humanized variants) [60]. A stabilized cyclic form of Aβ1-14 (N-truncated amyloid peptide antibodies, the “TAPAS” vaccine) was engineered and shown that the vaccine adopted the same conformation as the native sequence when bound to TAPAS antibodies. Active immunization of two AD mouse models with the TAPAS vaccine led to a significant reduction in amyloid-plaque load, a rescue of brain glucose metabolism, rescue of neuron loss, and rescue of memory deficits. Both, active immunization with the TAPAS vaccine and passive immunization with the humanized TAPAS version of the murine precursor antibody [61, 62] had similar treatment effects. Of note the TAPAS binding motif is not found in amyloid plaques [60, 63].
The monoclonal antibody 9D5 reacted with low molecular weight oligomers (4–10 mers) of AβpE3-X and not with amyloid plaques in AD brain tissue [64]. Passive immunization of 5XFAD mice reduced amyloid plaques and rescued behavioral deficits.
Other antibodies preferentially react with amyloid plaques from AD patients, like the monoclonal antibody 3B8, which is specific for both conformational epitopes present on N-terminal truncated AβpE3-42 and its unmodified counterpart Aβ3-42 [65]. Another report supports the concept that pan-AβpE-X antibodies preferentially bind to amyloid plaques in brain tissue of sporadic and familial AD cases [14]. The AβpE3-X specific antibody 07/01 detected diffuse and senile plaques and cerebral amyloid angiopathy in the brain of human specimen and in nonhuman primates, as well as in a subset of focal and vascular deposits in transgenic mouse models for AD [66].
BAMB31 is another pan-AβpE monoclonal antibody designed to target plaques. Passive immunization elicited therapeutic effects in an acute application design using the APPPS1 mice [67]. Chronic cotreatment with a BACE1 inhibitor cleared preexisting plaques in APPLon and PDAPP mouse models of AD. The authors report that the plaque lowering effect was not accompanied by microhemorrhages.
Passive immunization with the monoclonal antibody 07/01 reduced plaque load and normalized behavioral deficits in the APPswe/PS1DeltaE9 model for AD [68, 69]. The 07/01 antibody was compared with other AβpE3-X antibodies using structural and functional assays. The authors reported that only 07/01 prevented in vitro toxicity of AβpE3-42 oligomers [70]. Furthermore, surface plasmon resonance revealed that all AβpE3-X antibodies preferentially reacted with amyloid fibrils. Enhanced phagocytosis of amyloid plaques could be achieved without inducing neuroinflammation using in vivo PET imaging in the APPSLxhQC model for AD [71]. The humanized antibody, PBD-C06, retained the binding properties, including mixed aggregates of full-length Aβ and AβpE3-X peptides [72].
Considerable attention in the AD field was raised by the publication of the results of the TRAILBLAZER-ALZ trial. The phase 2 trial evaluated safety, tolerability and efficacy of passive immunization with donanemab a plaque-specific humanized antibody against AβpE3-X [73]. The study design was unique, as the screening of early symptomatic AD patients was based on a tau threshold screening by brain imaging with flortaucipir PET scanning to assess tangle deposition in vivo. Only patients with intermediate levels of tangle formation were subsequently enrolled in the study. The primary outcome measures were tests to assess cognitive function. The secondary outcome measures assessed in addition amyloid-plaque load and tangle deposition by appropriate PET scans. Donanemab significantly slowed the disease process, slowed cognitive and functional decline on all secondary clinical endpoints, and reduced plaque load, and tau accumulation in a subgroup of patients. Of note, patients with the lowest tau accumulation demonstrated the highest benefit, while patients with the highest tau accumulation did not benefit at all. The safety profile was similar to findings of the phase 1 trial [74]. Although the major endpoints of the phase II donanemab trial were reached and are generally promising, the outcome on the neuropathological level is not entirely clear. For example, neurodegeneration and tau pathology progression were slowed down, but not reversed. Therefore, it is not entirely proven whether AβpE3-X directly triggers tau pathology. One needs to bear in mind, that many neuropathological studies have demonstrated that tau pathology precedes plaque pathology (for example [75, 76]). Of note, the sensitivity of biomarkers used in clinical studies is still lower in comparison with that of post-mortem neuropathological evaluations [77–82].
The meaning of the data of the TRAILBLAZER-ALZ is a matter of ongoing scientific debates as aggregated fibrillar amyloid is generally thought to be pathologically inert as discussed above [59]. Ackley et al. [83] added further evidence to the discussion. The authors reported on a meta-analysis of randomized controlled clinical trials of drugs for the prevention or treatment of AD targeting amyloid mechanism. They concluded that amyloid-plaque reduction strategies did not substantially improve cognition.
In conclusion, there is ample evidence that pyroglutamate Aβ is involved in the etiology of AD. The pyroglutamate amyloid-β cascade provides several attractive therapeutic intervention points. DPP4 or QC inhibitors can modulate the stepwise formation of pyroglutamate Aβ enzymatically. Pyroglutamate Aβ induced toxicity can be neutralized by antibodies specific for the TAPAS epitope formed by the N-terminus of monomers or by antibodies against amyloid aggregates formed by oligomers, protofibrils and other high-molecular weight structures.
Author contributions
The author solely wrote the manuscript and designed the figures using software as indicated in the figures’ legends.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Competing interests
University Medicine Göttingen holds patents on parts of the discussed research.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem. 1995;65:1431–44. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer´s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 4.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem. 1992;267:17082–6. [PubMed] [Google Scholar]
- 6.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, Abeta N3(pE), in senile plaques. Neuron. 1995;14:457–66. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 7.Russo C, Saido TC, DeBusk LM, Tabaton M, Gambetti P, Teller JK. Heterogeneity of water-soluble amyloid beta-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 1997;409:411–6. doi: 10.1016/s0014-5793(97)00564-4. [DOI] [PubMed] [Google Scholar]
- 8.Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 9.Wildburger NC, Esparza TJ, LeDuc RD, Fellers RT, Thomas PM, Cairns NJ, et al. Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Sci Rep. 2017;7:9520. doi: 10.1038/s41598-017-10422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120:185–93. doi: 10.1007/s00401-010-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rijal Upadhaya A, Kosterin I, Kumar S, von Arnim CA, Yamaguchi H, Fandrich M, et al. Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain. 2014;137:887–903. doi: 10.1093/brain/awt362. [DOI] [PubMed] [Google Scholar]
- 12.Moro ML, Phillips AS, Gaimster K, Paul C, Mudher A, Nicoll JAR, et al. Pyroglutamate and isoaspartate modified amyloid-beta in ageing and Alzheimer’s disease. Acta Neuropathol Commun. 2018;6:3. doi: 10.1186/s40478-017-0505-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balakrishnan K, Rijal Upadhaya A, Steinmetz J, Reichwald J, Abramowski D, Fandrich M, et al. Impact of amyloid beta aggregate maturation on antibody treatment in APP23 mice. Acta Neuropathol Commun. 2015;3:41. doi: 10.1186/s40478-015-0217-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirths O, Bethge T, Marcello A, Harmeier A, Jawhar S, Lucassen PJ, et al. Pyroglutamate Abeta pathology in APP/PS1KI mice, sporadic and familial Alzheimer’s disease cases. J Neural Transm. 2010;117:85–96. doi: 10.1007/s00702-009-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009;118:487–96. doi: 10.1007/s00401-009-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexandru A, Jagla W, Graubner S, Becker A, Bäuscher C, Kohlmann S, et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate–Aβ formation. J Neurosci. 2011;31:12790–801. doi: 10.1523/JNEUROSCI.1794-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker A, Kohlmann S, Alexandru A, Jagla W, Canneva F, Bauscher C, et al. Glutaminyl cyclase-mediated toxicity of pyroglutamate-beta amyloid induces striatal neurodegeneration. BMC Neurosci. 2013;14:108. doi: 10.1186/1471-2202-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hampel H, Vassar R, De Strooper B, Hardy J, Willem M, Singh N, et al. The β-secretase BACE1 in Alzheimer’s disease. Biol Psych. 2021;89:745–56. doi: 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 20.Becker-Pauly C, Pietrzik CU. The metalloprotease meprin beta is an alternative beta-secretase of APP. Front Mol Neurosci. 2016;9:159. doi: 10.3389/fnmol.2016.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sevalle J, Amoyel A, Robert P, Fournie-Zaluski MC, Roques B, Checler F. Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem. 2009;109:248–56. doi: 10.1111/j.1471-4159.2009.05950.x. [DOI] [PubMed] [Google Scholar]
- 22.Valverde A, Dunys J, Lorivel T, Debayle D, Gay AS, Lacas-Gervais S, et al. Aminopeptidase A contributes to biochemical, anatomical and cognitive defects in Alzheimer’s disease (AD) mouse model and is increased at early stage in sporadic AD brain. Acta Neuropathol. 2021;141:823–39. doi: 10.1007/s00401-021-02308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonyan A, Schlenzig D, Schilling S, Naumann M, Sharoyan S, Mardanyan S, et al. Concerted action of dipeptidyl peptidase IV and glutaminyl cyclase results in formation of pyroglutamate-modified amyloid peptides in vitro. Neurochem Int. 2018;113:112–9. doi: 10.1016/j.neuint.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Bien J, Jefferson T, Causevic M, Jumpertz T, Munter L, Multhaup G, et al. The metalloprotease meprin beta generates amino terminal-truncated amyloid beta peptide species. J Biol Chem. 2012;287:33304–13. doi: 10.1074/jbc.M112.395608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frerker N, Wagner L, Wolf R, Heiser U, Hoffmann T, Rahfeld JU, et al. Neuropeptide Y (NPY) cleaving enzymes: structural and functional homologues of dipeptidyl peptidase 4. Peptides. 2007;28:257–68. doi: 10.1016/j.peptides.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 26.Valverde A, Dunys J, Lorivel T, Debayle D, Gay AS, Caillava C, et al. Dipeptidyl peptidase 4 contributes to Alzheimer’s disease-like defects in a mouse model and is increased in sporadic Alzheimer’s disease brains. J Biol Chem. 2021;297:100963. doi: 10.1016/j.jbc.2021.100963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angelopoulou E, Piperi C. DPP-4 inhibitors: a promising therapeutic approach against Alzheimer’s disease. Ann Transl Med. 2018;6:255. doi: 10.21037/atm.2018.04.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Q, Cheng J, Cordato D, Gao J. Can dipeptidyl peptidase-4 inhibitors treat cognitive disorders? Pharm Ther. 2020;212:107559. doi: 10.1016/j.pharmthera.2020.107559. [DOI] [PubMed] [Google Scholar]
- 29.Yaribeygi H, Sathyapalan T, Sahebkar A. Molecular mechanisms by which GLP-1 RA and DPP-4i induce insulin sensitivity. Life Sci. 2019;234:116776. doi: 10.1016/j.lfs.2019.116776. [DOI] [PubMed] [Google Scholar]
- 30.Cynis H, Schilling S, Bodnar M, Hoffmann T, Heiser U, Saido TC, et al. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim Biophys Acta. 2006;1764:1618–25. doi: 10.1016/j.bbapap.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Schilling S, Appl T, Hoffmann T, Cynis H, Schulz K, Jagla W, et al. Inhibition of glutaminyl cyclase prevents pGlu-Abeta formation after intracortical/hippocampal microinjection in vivo/in situ. J Neurochem. 2008;106:1225–36. doi: 10.1111/j.1471-4159.2008.05471.x. [DOI] [PubMed] [Google Scholar]
- 32.Jawhar S, Wirths O, Schilling S, Graubner S, Demuth HU, Bayer TA. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J Biol Chem. 2011;286:4454–60. doi: 10.1074/jbc.M110.185819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat Med. 2008;14:1106–11. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 34.Hartlage-Rubsamen M, Morawski M, Waniek A, Jager C, Zeitschel U, Koch B, et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Abeta deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011;121:705–19. doi: 10.1007/s00401-011-0806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cynis H, Scheel E, Saido TC, Schilling S, Demuth HU. Amyloidogenic processing of amyloid precursor protein: evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-beta. Biochemistry. 2008;47:7405–13. doi: 10.1021/bi800250p. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Wang YN, Wu H. Glutaminyl cyclase, diseases, and development of glutaminyl cyclase inhibitors. J Med Chem. 2021;64:6549–65. doi: 10.1021/acs.jmedchem.1c00325. [DOI] [PubMed] [Google Scholar]
- 37.Cynis H, Hoffmann T, Friedrich D, Kehlen A, Gans K, Kleinschmidt M, et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol Med. 2011;3:545–58. doi: 10.1002/emmm.201100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–15. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 39.Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett. 1996;215:173–6. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- 40.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature. 2012;485:651–5. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Z-W, Au DF, Cruceta L, Vugmeyster L, Qiang W. N-terminal modified Aβ variants enable modulations to the structures and cytotoxicity levels of wild-type Aβ fibrils through cross-seeding. ACS Chem Neurosci. 2020;11:2058–65. doi: 10.1021/acschemneuro.0c00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu ZW, Cruceta L, Zhang S, Sun Y, Qiang W. Cross-seeded fibrillation induced by pyroglutamate-3 and truncated Abeta40 variants leads to Abeta40 structural polymorphism modulation and elevated toxicity. ACS Chem Neurosci. 2021;12:3625–37. doi: 10.1021/acschemneuro.1c00341. [DOI] [PubMed] [Google Scholar]
- 43.Lopez-Noguerola JS, Giessen NME, Ueberuck M, Meissner JN, Pelgrim CE, Adams J, et al. Synergistic effect on neurodegeneration by N-truncated Abeta4-42 and pyroglutamate Abeta3-42 in a mouse model of Alzheimer’s disease. Front Aging Neurosci. 2018;10:64. doi: 10.3389/fnagi.2018.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wittnam JL, Portelius E, Zetterberg H, Gustavsson MK, Schilling S, Koch B, et al. Pyroglutamate amyloid β (Aβ) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J Biol Chem. 2012;287:8154–62. doi: 10.1074/jbc.M111.308601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camargo LC, Schoneck M, Sangarapillai N, Honold D, Shah NJ, Langen KJ, et al. PEAbeta triggers cognitive decline and amyloid burden in a novel mouse model of Alzheimer’s disease. Int J Mol Sci. 2021;22:7062. doi: 10.3390/ijms2213706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouter Y, Dietrich K, Wittnam JL, Rezaei-Ghaleh N, Pillot T, Papot-Couturier S, et al. N-truncated amyloid beta (Abeta) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013;126:189–205. doi: 10.1007/s00401-013-1129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youssef I, Florent-Béchard S, Malaplate-Armand C, Koziel V, Bihain B, Olivier J-L, et al. N-truncated amyloid-β oligomers induce learning impairment and neuronal apoptosis. Neurobiol Aging. 2008;29:1319–33. doi: 10.1016/j.neurobiolaging.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 48.He W, Barrow CJ. The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry. 1999;38:10871–7. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- 49.Jawhar S, Wirths O, Bayer TA. Pyroglutamate Abeta—a hatchet man in Alzheimer disease. J Biol Chem. 2011;286:38825–32. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bayer TA, Wirths O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014;127:787–801. doi: 10.1007/s00401-014-1287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayer TA. N-truncated Aβ starting at position four—biochemical features, preclinical models, and potential as drug target in Alzheimer’s disease. Front Aging Neurosci. 2021;13:710579. doi: 10.3389/fnagi.2021.710579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borzì AM, Condorelli G, Biondi A, Basile F, Vicari ESD, Buscemi C, et al. Effects of vildagliptin, a DPP-4 inhibitor, in elderly diabetic patients with mild cognitive impairment. Arch Gerontol Geriatr. 2019;84:103896. doi: 10.1016/j.archger.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Lues I, Weber F, Meyer A, Buhring U, Hoffmann T, Kuhn-Wache K, et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimers Dement. 2015;1:182–95. doi: 10.1016/j.trci.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheltens P, Hallikainen M, Grimmer T, Duning T, Gouw AA, Teunissen CE, et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alz Res Ther. 2018;10:107. doi: 10.1186/s13195-018-0431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vijverberg EGB, Axelsen TM, Bihlet AR, Henriksen K, Weber F, Fuchs K, et al. Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD—VIVIAD. Alz Res Ther. 2021;13:142. doi: 10.1186/s13195-021-00882-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirschner DA, Abraham C, Selkoe DJ. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc Natl Acad Sci USA. 1986;83:503–7. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, et al. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olofsson A, Sauer-Eriksson AE, Ohman A. The solvent protection of alzheimer amyloid-beta-(1-42) fibrils as determined by solution NMR spectroscopy. J Biol Chem. 2006;281:477–83. doi: 10.1074/jbc.M508962200. [DOI] [PubMed] [Google Scholar]
- 59.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, et al. Structural conversion of neurotoxic amyloid-[beta]1-42 oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–7. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bakrania P, Hall G, Bouter Y, Bouter C, Beindorff N, Cowan R, et al. Discovery of a novel pseudo β-hairpin structure of N-truncated amyloid-β for use as a vaccine against Alzheimer’s disease. Mol Psychiatry. 2021. 10.1038/s41380-021-01385-7. [DOI] [PubMed]
- 61.Antonios G, Borgers H, Richard BC, Brauss A, Meissner J, Weggen S, et al. Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci Rep. 2015;5:17338. doi: 10.1038/srep17338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antonios G, Saiepour N, Bouter Y, Richard BC, Paetau A, Verkkoniemi-Ahola A, et al. N-truncated Abeta starting with position four: early intraneuronal accumulation and rescue of toxicity using NT4X-167, a novel monoclonal antibody. Acta Neuropathol Commun. 2013;1:56. doi: 10.1186/2051-5960-1-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bouter Y, Lopez Noguerola JS, Tucholla P, Crespi GA, Parker MW, Wiltfang J, et al. Abeta targets of the biosimilar antibodies of bapineuzumab, crenezumab, solanezumab in comparison to an antibody against N-truncated Abeta in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol. 2015;130:713–29. doi: 10.1007/s00401-015-1489-x. [DOI] [PubMed] [Google Scholar]
- 64.Wirths O, Erck C, Martens H, Harmeier A, Geumann C, Jawhar S, et al. Identification of low molecular weight pyroglutamate A{beta} oligomers in Alzheimer disease: a novel tool for therapy and diagnosis. J Biol Chem. 2010;285:41517–24. doi: 10.1074/jbc.M110.178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acero G, Garay C, Venegas D, Ortega E, Gevorkian G. Novel monoclonal antibody 3B8 specifically recognizes pyroglutamate-modified amyloid beta 3-42 peptide in brain of AD patients and 3xTg-AD transgenic mice. Neurosci Lett. 2020;724:134876. doi: 10.1016/j.neulet.2020.134876. [DOI] [PubMed] [Google Scholar]
- 66.Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, et al. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol. 2013;183:369–81. doi: 10.1016/j.ajpath.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Janssens J, Hermans B, Vandermeeren M, Barale-Thomas E, Borgers M, Willems R, et al. Passive immunotherapy with a novel antibody against 3pE-modified Abeta demonstrates potential for enhanced efficacy and favorable safety in combination with BACE inhibitor treatment in plaque-depositing mice. Neurobiol Dis. 2021;154:105365. doi: 10.1016/j.nbd.2021.105365. [DOI] [PubMed] [Google Scholar]
- 68.Frost JL, Liu B, Kleinschmidt M, Schilling S, Demuth HU, Lemere CA. Passive immunization against pyroglutamate-3 amyloid-beta reduces plaque burden in Alzheimer-like transgenic mice: a pilot study. Neurodegen Dis. 2012;10:265–70. doi: 10.1159/000335913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frost JL, Liu B, Rahfeld JU, Kleinschmidt M, O’Nuallain B, Le KX, et al. An anti-pyroglutamate-3 Abeta vaccine reduces plaques and improves cognition in APPswe/PS1DeltaE9 mice. Neurobiol Aging. 2015;36:3187–99. doi: 10.1016/j.neurobiolaging.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piechotta A, Parthier C, Kleinschmidt M, Gnoth K, Pillot T, Lues I, et al. Structural and functional analyses of pyroglutamate-amyloid-beta-specific antibodies as a basis for Alzheimer immunotherapy. J Biol Chem. 2017;292:12713–24. doi: 10.1074/jbc.M117.777839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crehan H, Liu B, Kleinschmidt M, Rahfeld JU, Le KX, Caldarone BJ, et al. Effector function of anti-pyroglutamate-3 Abeta antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alz Res Ther. 2020;12:12. doi: 10.1186/s13195-019-0579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hettmann T, Gillies SD, Kleinschmidt M, Piechotta A, Makioka K, Lemere CA, et al. Development of the clinical candidate PBD-C06, a humanized pGlu3-Abeta-specific antibody against Alzheimer’s disease with reduced complement activation. Sci Rep. 2020;10:3294. doi: 10.1038/s41598-020-60319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384:1691–704. doi: 10.1056/NEJMoa2100708. [DOI] [PubMed] [Google Scholar]
- 74.Lowe SL, Willis BA, Hawdon A, Natanegara F, Chua L, Foster J, et al. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimers Dement. 2021;7:e12112. doi: 10.1002/trc2.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 76.Duyckaerts C, Braak H, Brion JP, Buée L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015;129:749–56. doi: 10.1007/s00401-015-1390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 2012;11:669–78. doi: 10.1016/S1474-4422(12)70142-4. [DOI] [PubMed] [Google Scholar]
- 78.Fleisher AS, Pontecorvo MJ, Devous MD, Sr, Lu M, Arora AK, Truocchio SP, et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77:829–39. doi: 10.1001/jamaneurol.2020.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, et al. Multisite study of the relationships between antemortem [(11)C]PIB-PET centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement. 2019;15:205–16. doi: 10.1016/j.jalz.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pontecorvo MJ, Keene CD, Beach TG, Montine TJ, Arora AK, Devous MD, Sr, et al. Comparison of regional flortaucipir PET with quantitative tau immunohistochemistry in three subjects with Alzheimer’s disease pathology: a clinicopathological study. EJNMMI Res. 2020;10:65. doi: 10.1186/s13550-020-00653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sabri O, Sabbagh MN, Seibyl J, Barthel H, Akatsu H, Ouchi Y, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11:964–74. doi: 10.1016/j.jalz.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 82.Thal DR, Beach TG, Zanette M, Lilja J, Heurling K, Chakrabarty A, et al. Estimation of amyloid distribution by [(18)F]flutemetamol PET predicts the neuropathological phase of amyloid β-protein deposition. Acta Neuropathol. 2018;136:557–67. doi: 10.1007/s00401-018-1897-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ackley SF, Zimmerman SC, Brenowitz WD, Tchetgen Tchetgen EJ, Gold AL, Manly JJ, et al. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021;372:n156. doi: 10.1136/bmj.n156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feng J, Zhang Z, Wallace MB, Stafford JA, Kaldor SW, Kassel DB, et al. Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem. 2007;50:2297–2300. doi: 10.1021/jm070104l. [DOI] [PubMed] [Google Scholar]
- 85.Carrillo DR, Parthier C, Janckel N, Grandke J, Stelter M, Schilling S, et al. Kinetic and structural characterization of bacterial glutaminyl cyclases from Zymomonas mobilis and Myxococcus xanthus. Biol Chem. 2010;391:1419–28. doi: 10.1515/BC.2010.130. [DOI] [PubMed] [Google Scholar]