

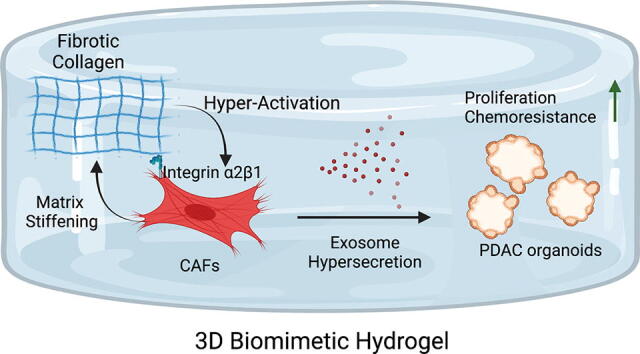
Graphical abstract
Keywords: Pancreatic cancer, Desmoplastic matrix, Cancer-associated fibroblasts, Exosome, Chemoresistance, 3D bio-mimetic model
Highlights
-
•
Pancreatic cancer organoid-stromal fibroblasts co-culture displayed significant chemoresistance in 3D culture system.
-
•
Cancer associated fibroblasts in the physiologically relevant matrix system tended to be more phenotypically activated.
-
•
Increased extracellular matrix stiffness induces hypersecretion of chemoresistance-promoting exosomes in a cancer associated fibroblasts-tumor organoid biomimetic model system.
Abstract
In pancreatic ductal adenocarcinoma (PDAC), the abundant stromal cells which comprise the tumor microenvironment constitute more than 90% of the primary tumor bulk. Moreover, this desmoplastic environment has been found to be three times stiffer than normal pancreas tissue. Despite the importance of studying the desmoplastic environment of PDAC, there is still a lack of models designed to adequately recapitulate this complex stiff microenvironment, a critical hallmark of the disease that has been shown to induce chemoresistance. Here, we present a bio-mimetic, 3-dimensional co-culture system that integrates tumor organoids and host-matching stromal cancer associated-fibroblasts (CAFs) that recapitulates the complex, fibrotic matrix of PDAC using advanced biomaterials. With this model, we show that matrix-activated CAFs are able to “re-engineer” the fibrotic environment into a significantly stiffer environment through lysyl-oxidase dependent crosslinking. Moreover, we show that culture of CAFs in this model leads to an increase of exosomes; extracellular vesicles known to promote chemoresistance. Finally, using previously identified exosome inhibitors, climbazole and imipramine, we demonstrate how abrogation of exosome hypersecretion can reduce matrix stiffness-induced chemoresistance. These data highlight the importance of the development of new models that recapitulate not only the cellular composition found in PDAC tumors, but also the biophysical stresses, like stiffness, that the cells are exposed to in order to identify therapies that can overcome this critical feature which can contribute to the chemoresistance observed in patients. We believe that the 3D bio-mimetic model we have developed will be a valuable tool for the development, testing, and optimization of “mechano-medicines” designed to target the biophysical forces that lead to tumor growth and chemoresistance.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers. Despite recent therapeutic advances in other types of cancers, the five year survival rate for PDAC patients is still only 11% [1], and most patients succumb to their disease within the first year [2]. While late-stage diagnosis, metastasis, and irresectability are contributing factors in PDAC lethality [3], a common thread in all cases is the development of therapeutic resistance, resulting in poor survival outcomes following treatment [4].
For the majority of patients diagnosed with advanced PDAC, chemotherapy such as gemcitabine is considered the standard of care [5]. Though prior research has shown that gemcitabine is more effective against PDAC than alternative anticancer agents, development of gemcitabine resistance was observed in most patients within the first few weeks of treatment [4], [6]. Most research has focused on the roles of aberrant signaling pathways that are found in the epithelial cancer cells in order to elucidate molecular mechanisms which may lead to chemoresistance [6]. However, this research has largely ignored the potential influences of the tumor stromal microenvironment on acquired resistance to treatment.
Unlike other cancer types, PDAC is characterized by a desmoplastic stroma that accounts for up to 90% of the bulk tumor volume [7]. The highly crosslinked extracellular matrix (ECM) contains hyaluronic acid and fibrotic proteins such as collagen-I, laminin and fibronectin [8], which contribute to a significant increase in tumor tissue stiffness compared to a healthy pancreas [9]. Researchers previously hypothesized that the dense and stiff ECM acted as a barrier to block the delivery of therapeutic agents [10], and thus, the predictions were that stromal cells should be ablated in order to improve treatment response. However, when these studies were carried out, it was observed that strategies designed to simply ablate stromal cells actually led to more aggressive tumors [11] and failed to improve survival rate and clinical outcomes in patients [12]. These results suggested that the desmoplastic ECM plays a far more complex role in therapeutic resistance. Some stromal elements can restrain the tumor cells while other elements can promote growth and spread. Thus, models that can recapitulate this complex environment are needed to develop more effective treatment strategies.
Previous studies using traditional 2D models failed to recapitulate a physiologically relevant microenvironment, where the desmoplastic matrix environment acts as “soil” for both the stroma cells and the tumor cells [13]. Our past studies using novel, genetically engineered mouse models (GEMM) of PDAC have shown the ability of the desmoplastic environment to drive molecular mechanisms linked to chemoresistance [14], [15]. However, such in vivo models present an experimental challenge as it is not possible to easily investigate the role that individual cell types, ECM components, or physical properties may play in the system.
In PDAC, cancer associated fibroblasts (CAFs) are known to be important mediators of chemoresistance [16]. They are the cell type most responsible for “engineering” the desmoplastic matrix [16], [17]. While most past studies have identified conventional secreted factors from CAFs which promote tumor growth in PDAC cells [18], our previous data showed how exosomes from CAFs exposed to chemotherapy could transfer chemoresistance to recipient PDAC cells in 2D systems and GEMMs [19]. However, the role of ECM stiffness on the activation of these CAFs, and the roles this plays in exosome hypersecretion, remains unclear.
To address this gap in knowledge, we have developed a Matrigel-based, orthogonally tunable 3-dimensional (3D) culture system to co-culture mouse derived PDAC organoids and host-matching CAFs. This model gives us the ability to grow tumor-derived organoids in an environment that recapitulates the key features of the human disease (e.g. high CAF-to-organoid ratio, high Collagen-I, increased stiffness). Using this system, we found that stiff matrix-activated CAFs readily remodel the desmoplastic matrix through lysyl-oxidase dependent crosslinking. Moreover, our novel system demonstrates how collagen-I and matrix stiffness induce hypersecretion of CAF-derived exosomes and demonstrate how the use of compounds which inhibit exosome secretion can reduce matrix stiffness-induced chemoresistance in this system.
Materials and methods
Mouse colony generation
The methodology utilized to generate a KRASG12D; PTENlox/lox; COX2-overexpression (COE) mouse line was described previously (Hill et. al.) [14]. Briefly, Pdx1-Cre+ mice were crossed with floxed Cox-2 over-expressing (COE) transgenic mice. The Pdx1-Cre+; COE line was further crossed with Pdx1-Cre+; K-RASG12D; PTENlox/lox to produce the target mouse line. Genotyping of the mouse was confirmed through PCR. Animal housing and procedures adhered strictly to Institutional Animal Care and Use Committee (IACUC) standard and were monitored by the USC Department of Animal Resources (USC DAR).
PDAC tumor and host-matching CAF isolation
The cell isolation protocol in the manuscript was adapted from LA Baker et al [20]. In brief, mice were euthanized in a CO2 chamber and subjected to cervical dislocation as secondary confirmation. The mouse was dissected, with the pancreas and associated tumor resected and harvested. The preserved tissue was rinsed with PBS for 3 times and minced. The minced tissue was washed with regular DMEM and digested in 0.125 mg/ml Collagenase XI and Dispase II (MilliporeSigma) at 37 °C for 2 h. The digested tumor was strained through to a 100 µm strainer and further digested by TrypLE for 10 mins after washing with DMEM. The resulting digested sample was cultured either in Matrigel with organoid complete feeding media (CFM) [20] or on regular culture dishes with DMEM + GlutaMax, 5% fetal bovine serum (FBS) plus 100units/mL penicillin, and 100 µg/mL streptomycin (Life Technologies). After 72 h, PDAC tumor organoids formed in the Matrigel cultures, and fibroblasts were able to grow in regular culture dishes. Finally, the 2D culture of fibroblasts was completed using Fluorescence-activated Cell Sorting (FACS) for EpCAM− cells by USC Flow Cytometry Facility.
PDAC organoid culture
When organoids reached confluency, the culture was digested by Gentle Cell Dissociation buffer (STEMCELL Technologies) on ice for 30 mins followed by 5 mins centrifugation. The pellet was resuspended with freshly thawed Matrigel and plated on regular tissue culture plates. After 37 °C incubation for 15 mins, prewarmed CFM media was added to the culture, and the media was replaced every 2–3 days based on growth.
3D gel encapsulation
PDAC organoids were subjected to routine dissociation as described above. The dissociated cell pellet was subjected to 5 mins TrypLE (Thermofisher Scientific LLC) plus 10 µg/mL DNAse treatment. The resulting dissociated pellet was further subjected to 70 µm cell strainer for single cell counting. For co-culture, 2000 organoid cells and 20,000 CAFs were resuspended in 60ul hydrogel mixture. Collagen-I from rat tail (A1048301 from ThermoFisher Scientific, LLC) was obtained in native form. For high collagen-I (1 mg/mL Col-I-MAT) gel, each gel contained 40 µL Matrigel and 20 µL Collagen-I (3 mg/mL stock). For low collagen-I (0.1 mg/mL Col-I-MAT), each gel contained 41.8 µL Matrigel, 2 µL Collagen-I (3 mg/mL stock) and 16.2 µL PBS. Gels were allowed to solidify in non-tissue culture treated cell culture plate in 37 °C incubator for 15mins. After addition of 1 mL co-culture medium (CFM with 5%FBS to ensure survival of fibroblast), a blunt end aspirating pipet tip was used to gently lift the gel from the bottom of the well plate.
Drug treatment for 3D cultures
48 h after the establishment of co-culture, fresh co-culture medium was used to replace the old medium. Another 48 h later, fresh co-culture media containing 500 nM gemcitabine or an equivalent amount of DMSO (vehicle) was added to cell culture. If combination treatment was involved during the experiment, the additional supporting agents (β-Aminopropionitrile (MilliporeSigma) 10 mM, GFOGER (customized peptide from GenScript Inc.) 100 μM, TCI-15 (Tokyo Chemical Industry Co., Ltd) 1 μM, Climbazole (MilliporeSigma) 10 μM, Imipramine (MilliporeSigma) 10 μM) were added at the beginning of the co-culture establishment.
Measurement of organoid size
48 h after vehicle (equivalent amount solvent, no drug), gemcitabine, or any combination therapy treatment, 3D culture samples were visualized under a light microscope (Echo Revolve). A minimum of 3 random bright field images were taken from each gel. Each condition had at least 3 individually cultured gel samples. Diameters of in-focus organoids were measured using ImageJ software. Relative size of organoid was derived by taking the square of the measured diameter from the organoids.
Gel contraction measurement
To measure gel contraction by cultured cells, hydrogel cultures were maintained in corresponding growth medium for 48 or 72 h before the measurement. Concurrently, an empty hydrogel (control, no cells encapsulated) with identical gel components were soaked in the same medium for the same amount of time. At the time of measurement, the culture medium was removed completely, and hydrogels were gently moved to the center of the wells in the culture well plates. Photographs were taken from the top of the well plate. Diameters of hydrogels and corresponding wells were measured by ImageJ software. The size of the hydrogel was derived by taking the ratio of measured gel diameter over the measured well diameter. Relative contraction percentage was calculated based on calculating the area difference with respect to the control gel size. At least 4 individual samples from the same condition were measured.
Cell viability assay
Hydrogel cultures were washed with PBS once. 500 µL of Live/Dead solution containing ethidium homodimer and calcien AM (Life Technologies) in PBS was added to the cell culture and incubated in 37 °C; 5% CO2 for 30 mins. At least 3 independent fluorescence images for each experimental condition were taken using an Echo Revolve microscope through the RFP and GFP channels. Representative overlay images are shown in the figures.
Preservation of hydrogel cultures
The gels were first washed with PBS at the time of preservation. Next, the PBS was replaced with 2% PFA in PBS and incubated in 37 °C incubator for 15 mins. Subsequently, the gel was transferred to a cassette and soaked in 70% ethanol. To complete the preservation process, the samples were submitted to the USC Laboratory Service Core for paraffin embedding and sectioning at 5 µm.
Immunofluorescence staining and imaging
Slides were deparaffinized in an alcohol gradient and underwent citrate buffer (ABCAM, 20 mM citrate acid in water) mediated antigen retrieval at 97 °C for 15 mins. A blocking buffer containing 5% normal goat serum and 1% BSA was applied to the slides for 1 h before overnight primary antibody incubation at 4 °C. The following day, the slides were washed in tris buffered saline (TBS) and goat anti rabbit secondary antibody (Thermofisher Scientific, Alexa Fluor 647) was applied to the slides for 1hr. At last, the slides were washed, and mounting solution containing DAPI was applied with coverglass (0.1 mm). In the same set of comparisons, all fluorescence images were taken with Echo Revolve using identical exposure times. All images used to prepare figures were processed under same cutoff parameters to ensure fair comparison. For fluorescence signal quantification, ImageJ software was used to derive average intensity using raw images.
Assessment of YAP1 nuclear expression
5 µm-sectioned gel samples were co-stained with Alpha smooth cell actin (aSMA) antibody (Cell Signaling Technology #48938), YAP1 antibody (Novus Biological #NB110-58358), and DAPI. Immunostaining and IF images were taken as described above. Raw fluorescence intensity values of YAP1 at DAPI location of aSMA positive cells were measured by ImageJ (Fig. S1A). Several random images from at least 3 independent gel samples were used in the analysis.
Organoid cyclin D1 quantification
At the end of the hydrogel culture cycle, samples were preserved, 5 µm-sectioned, immunofluorescence labeled, and imaged. Because the cultured organoids processed gland-like morphologies, outer rim cells of each organoid were assessed. Total cyclinD1 (Cell Signaling Technology #55506) positive cell number was divided by total DAPI positive number for each organoid. At least 20 organoids from 3 individually cultured gels for each experimental conditions were analyzed for statistical results.
Lentiviral transduction
CAFs and organoids were dissociated and counted for passaging. For 2D CAF culture, lentivirus encoding an overexpression of mouse shRAB27A was added to the culture medium at multiplicity of infection (MOI) of 20 along with 5 µg/ml polybrene. For organoid cultures, the lentivirus was mixed with the cells and Matrigel during cell passaging at MOI of 20 along with 5 µg/ml polybrene. The next day, fresh medium was added to cell culture. Another 72 h later, puromycin was added to select the successfully transfected cell. ShRNA knockdown experiments were reproduced at least 3 times individually.
Exosome isolation
To isolate exosomes, exosome-free FBS was used to culture the fibroblasts for 48 to 72 h to collect the supernatant. The supernatant was spun for 15 mins at 3000×g to remove the cellular debris. ExoQuick TC (System Bioscience) was added to the supernatant and incubated at 4 °C overnight. Next day, the solution was spun at 1500xg for 30 mins, and the resulting pellet was resuspended in 100 µL PBS and stored at −20 °C for subsequent analysis.
Western blotting
To assess protein expression of RAB27A(Cell Signaling Technologies), equal amount of denatured cell lysate was loaded onto Bolt Bis-Tris plus gel (Thermofisher Scientific) and transferred to 0.2 µm PVDF membrane (BioRad Laboratories). To quantify exosome CD63 (antibody obtained from SystemBiology) in the purified exosomes, loaded volumes of denatured exosomes were adjusted based on counted cell numbers to ensure exosome quantities from equal number of cells were assessed. The blot membrane was blocked with 5% milk in Tris Buffered Saline with 0.1% Tween-20 (TBST) solution and incubated with anti-RAB27A antibody (Cell Signaling Technology #69295) or anti-CD63 antibody (System Biosciences #EXOAB-CD63A-1) overnight at 4 °C. Next day, the membrane was washed and incubated with goat anti rabbit-HRP antibody. To complete the blotting, the membrane was incubated with West Pico Signal (Thermofisher Scientific) and imaged using BioRad Imager system. ImageJ was used to quantify the raw chemiluminescence images.
Rheology
Hydrogel storage moduli (G′) were measured using a discovery hybrid rheometer-2 (DHR-2, TA Instruments) at 37 °C. Rheology frequency sweep experiments were performed under 1% constant strain in the range of 0.1 to 1.0 Hz. Storage modulus of each sample was calculated as an average value of the linear region of the storage curve from the frequency sweep plot. For statistical analysis 3 separate measurements were taken in which 5 samples from each condition were measured. All data were normalized to empty gel condition to ensure consistent comparisons.
Results
Novel 3D co-culture system preserves physiologically relevant state and recapitulates chemoresistance in PDAC
In order to construct a physiologically relevant desmoplastic microenvironment for PDAC organoids, we added a high amount of fibrotic collagen-I into the Matrigel solution, and also utilized a 10-to-1 host-matching CAFs to PDAC organoids ratio to mimic the stromal cell imbalance observed in the human disease [7] (Fig. 1A). After 4 days of co-culture, dissociated PDAC cells readily grew into organoids among the CAFs. More importantly, both cell types expressed corresponding molecular markers (α-Smooth Cell Actin (αSMA) [21] for CAFs and cytokeratin-19 [22] for PDAC cells) specific to their cell types (Fig. 1B). Moreover, we were able to see increased tumor organoid size when comparing organoids co-cultured with CAFs compared to organoid alone cultures in the same gel formulation or with organoid alone cultures in the pure Matrigel (Fig. 1C). As introduced previously, an ideal model to study PDAC should recapitulate resistance to treatment, a hallmark of the disease. 72 h following gemcitabine treatment, PDAC organoids in our co-culture system displayed a significant increase in resistance against gemcitabine as indicated by the resulting organoid sizes (Fig. 1D). A more detailed comparison between vehicle and gemcitabine treatment groups showed that gemcitabine could effectively reduce organoid size and cyclin D1 positivity rate in both culture conditions, but co-cultured organoids were far more resistant (Fig. S1B and C). Because our co-culture system significantly boosted growth and chemoresistance in PDAC organoids in the presence of CAFs, it was speculated that CAFs could be the main reason for the observed phenotype. CAFs’ activation status can be reflected by their ability to contract the collagen-I based hydrogels [23]. Here, we observed that CAFs were able to induce gel contraction in the presence of high amount of Collagen-I (Fig. S1.D), and CAFs had a much stronger ability to modify the surrounding matrix compared to normal fibroblasts (Fig. S1.E). To investigate the role that cell intrinsic factors may play in our model, repeated the aforementioned experiments with normal fibroblasts and normal pancreatic organoids. We found that co-culture with both normal and cancer associated fibroblasts could increase normal pancreas (non-cancer) organoid growth, but we did not observe any chemoresistance in normal organoid co-cultures (Fig. S1.F&G). These data show that the observed chemoresistance in our model was still organoid-intrinsic mutation-dependent instead of being solely caused by the co-culture with CAFs. Using rheology measurements, we confirmed that gel contraction is correlated to stiffness of the resulting hydrogels, and CAFs alone were sufficient to modify the hydrogel stiffness (Fig. S2.A). The above result show that CAFs are highly active in the desmoplastic environment. Therefore, we next investigated whether the fibrotic collagen-I influenced chemoresistance and CAF activation.
Fig. 1.

Novel 3D co-culture system preserves physiologically relevant state and recapitulates chemoresistance in PDAC. A) Schematic description of the 3D model system. Briefly, both tumor and fibroblast cell were derived from same mouse host. Both cell types were brought to single cell level before being encapsulated into Matrigel and type-I collagen mixture to ensure accurate seeding ratio. B) Co-cultured cells maintain physiologically relevant morphologies and molecular markers. Left panel: representative H&E staining of 4-day co-culture. Right panel: representative epithelial tumor marker cytokeratin 19(green), fibroblast marker alpha smooth cell actin (red) and nuclear (DAPI, blue) staining of a 4-day co-culture sample. C, D) 3D co-culture model boosts tumor organoid growth and chemoresistance. C) Left panel: representative phase contrast images of 4-day organoid cultures in different culture conditions. Right panel: quantitative analysis of relative organoid size in each corresponding culture conditions. D) Left panel: representative phase contrast images of 4-day plus 2-day gemcitabine (500 nM) treated organoid cultures in different culture conditions. Right panel: quantitative analysis of relative organoid size in each corresponding culture conditions. For B&C, One-Way ANOVA with Dunnett’s multiple comparison test was performed. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars represent standard deviation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Desmoplastic collagen-I influences CAF mediated-matrix stiffening and chemo-response in the co-culture system
To investigate how collagen-I affects PDAC cells in co-culture, we meticulously tuned collagen-I concentration from 1 mg/mL to 0.1 mg/mL while maintaining a constant total polymer concentration and initial stiffness. Interestingly, only CAFs in the high collagen-I condition were able to contract and stiffen the hydrogel to achieve a stiff matrix while CAFs in all conditions maintained high viability (Fig. S2.A-C). Although lower collagen-I gel yielded larger organoid size in the co-culture experiment, possibly due to less restriction from gel contraction (Fig. 2A), gemcitabine treatment could yield much smaller and collapsed organoids compared to those in a stiff gel (high collagen-I) condition (Fig. 2B). To further validate our observation in chemoresistance, we analyzed the cyclinD1 ratio in co-cultured organoids to assess relative proliferation rate. As expected, gemcitabine treated organoids cultured in the stiff gels displayed a higher proliferation rate compared to those in soft gels (Fig. 2C). Previous research has shown that stiffened substrates could increase CAF activation through the YAP/TAZ signaling pathway, which can be visualized through nuclear localization of YAP1 [23], [24]. Here, after assessing the nuclear YAP1 intensity, we were able to show that CAFs were more activated in the stiffened high collagen-I matrix compared to those in the soft, low-collagen-I gels (Fig. 2D).
Fig. 2.

Desmoplastic collagen-I influences CAF mediated-matrix stiffening and chemo-response in the co-culture system. A) Left panel, representative microscope image of 4-day organoid-CAF co-culture in high or low collagen-I concentration with constant total polymer content. Right panel, quantitative analysis of relative organoid size in each corresponding culture conditions. B) Left panel: representative microscope images of 4-day plus 2-day gemcitabine (500 nM) treated organoid cultures in different culture conditions. Right panel: quantitative analysis of relative organoid size in each corresponding culture conditions. C) Left panel, representative immuno-fluorescent staining of proliferation marker Cyclin D1 (red) and nuclear staining DAPI (blue) from a total of 6-day co-culture in corresponding conditions. Right panel, quantification of cyclin D1 positive ratio in organoids from corresponding co-culture conditions after gemcitabine treatment. D) Type I collagen directly influences expression of activation marker in CAF. Left panel, representative YAP1 (red) and DAPI (blue) staining images of co-cultured CAFs 48hrs after gemcitabine treatment in corresponding gel conditions. Right panel, quantitative analysis of nuclear YAP1 intensity in corresponding conditions. Two-tail student’s tests were performed. ***p < 0.001, ****p < 0.0001. Error bars represent standard deviation. E) BAPN prevents CAF-mediated gel contraction and stiffening. Left panel, representative photos showing 72 h CAF hydrogel cultures with equal starting size and seeding density. Right panel, quantitative analysis of resulting gel sizes relative to an empty gel soaked in media. F) BAPN and gemcitabine co-treatment reduces resulting organoid in co-culture. Left panel, representative images of 48hr gemcitabine treated co-cultures with or without presence of BAPN. Right panel, quantitative analysis of relative organoid sizes after corresponding treatments. G) Left panel, representative immuno-fluorescent staining of proliferation marker Cyclin D1 (red) and nuclear staining DAPI (blue) from a total of 4-day plus 2-day treated co-culture in corresponding conditions. Right panel, quantification of cyclin D1 positive ratio in organoids from corresponding co-culture conditions after gemcitabine treatment. H) Left panel, representative YAP1 (red) and DAPI (blue) staining images of co-cultured CAFs 48hrs after gemcitabine treatment in corresponding gel conditions. Right panel, quantitative analysis of nuclear YAP1 intensity in corresponding conditions after 48hr gemcitabine treatment. Two-tail student’s tests were performed. **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent standard deviation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Lysyl-oxidase dependent crosslinking in our co-culture system
At this point, we had shown CAFs were more activated in a stiffened high collagen-I matrix, contributing to significantly increased chemoresistance in the co-cultured PDAC organoids. Next, we aimed to further investigate the role of matrix stiffness in the observed phenotypes. Past research has indicated that lysyl-oxidase is mainly responsible for crosslinking the collagen matrix in the desmoplastic environment [25]. To further elucidate the molecular mechanism responsible for the matrix stiffening in our co-culture model, we utilized lysyl oxidase inhibitor β-aminopropionitrile (BAPN) [26]to specifically target the gel stiffening in our co-culture system and assessed how this affected our experimental readouts. Initially, we found that 10 mM BAPN could effectively reduce gel contraction and stiffening by CAFs (Fig. 2E, Fig. S3A), while not affecting their viability (Fig. S3B). As expected, the softened matrix environment restored chemoresistance in co-cultured organoids in terms of organoid size and relative proliferation rate after gemcitabine treatment (Fig. 2 F&G, Fig. S3C). Finally, we were able to show decreased YAP1 nuclear staining intensity after treatment with BAPN (Fig. 2H). The above results established a potentially complicated connection between matrix stiffness, fibroblast activation and chemoresistance. We speculated that the observed chemoresistance was a result of CAF activation from the stiffened matrix. Next, investigated how the activated CAFs influenced chemoresistance in our system.
Exosome secretion mediates chemoresistance in the biomimetic co-culture system
Our previous study showed CAFs under stressful conditions hyper-secrete chemoresistance-promoting exosomes [19]. Surprisingly, we also found our CAFs increased CD63-positive exosome secretion when cultured in 3D hydrogels compared to CAFs cultured on traditional 2D petri dish (Fig. S4A). Therefore, we hypothesized that exosomes played a critical role in the chemoresistance phenotype we observed in our co-culture system. To test our hypothesis, we treated our co-cultured organoids with previously identified exosome inhibitors, climbazole (Fig. S4B) and imipramine [27], [28]. As speculated, treatment with climbazole or imipramine alone could slow the growth of co-cultured organoids (Fig. 3A). Moreover, co-treatment of climbazole or imipramine with gemcitabine effectively eliminated more organoids than gemcitabine alone (Fig. 3A). To further elucidate the role of exosomes from each cell type in our system, we aimed to intrinsically knockdown the exosome biosynthesis in our cultured cells. Previous research indicated that RAB27A serves as an essential factor for exosome biogenesis [29]. With this information, we used lentivirus expressing shRAB27A RNA to knockdown RAB27A in each or both types of co-cultured cells to observe the resulting effect on chemoresistance. After validating the knockdown efficiency of the shRNA expression system (Fig. 3B), we found that despite the previously identified increase in CAF-derived exosomes, knockdown of RAB27A in both organoids and CAFs was required for gemcitabine sensitization (Fig. 3C, Fig. S4C)), and transfer of conditioned media from co-culture of intact cells could mitigate the effect of RAB27A knockdown (Fig. 3D, Fig. S4D). At last, we validated the effect of RAB27A knockdown and conditioned media rescue through cyclinD1 analysis (Fig. 3E).
Fig. 3.

Exosome secretion mediates chemoresistance in the biomimetic co-culture system. A) Exosome inhibitor drugs slowed organoid growth and abolished chemoresistance in the co-culture model. Upper panel, representative microscope image of 4-day organoid-CAF co-culture treated with vehicle (DMSO), climbazole (20 μM) or Imipramine (10 μM) and corresponding quantitative analysis of resulting organoid sizes. Lower panel, representative microscope image of organoid-CAF 48hr-gemcitabine treated co-culture along with or without climbazole (20 μM) or Imipramine (10 μM) and corresponding quantitative analysis of resulting organoid sizes. B) Representative Western blot image showing RAB27A knockdown in CAFs 1 week after lentiviral infection. C) RAB27A knockdown in both CAF and tumor organoids are required to decrease chemoresistance in the co-culture system. Quantitative analysis of organoid size in co-cultures in corresponding knockdown conditions. D) Conditioned media from non-transfected co-culture cells rescued chemosensitivity by RAB27A knockdown. Quantitative analysis of organoid sizes in co-cultures after gemcitabine treatment with or without an addition of conditioned media from a regular 4-day co-culture. E) Left panel, representative immuno-fluorescent staining of proliferation marker Cyclin D1 (red) and nuclear staining DAPI (blue) from a total of 4-day plus 2-day gemcitabine or gemcitabine plus conditioned media treated co-culture in corresponding conditions. Right panel, quantification of cyclin D1 positive ratio in organoids from corresponding co-culture conditions after gemcitabine treatment. For A, C and E, One-Way ANOVA with Dunnett’s multiple comparison test was performed. For D, two-tailed student’s test was performed. ***p < 0.001, ****p < 0.0001.Error bars represent standard deviation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Discussion
Lack of an appropriate, tunable in vitro model has been a major obstacle which has impeded the development of therapies that can target matrix-mediated chemoresistance in PDAC. This model provides an opportunity to elucidate how key components of desmoplastic tumor microenvironment (TME) form a complex interactive network with cancer cells to promote growth and chemoresistance in PDAC through exosome release.
Throughout this brief report, we showed how our 3D culture system recapitulated multiple physiologically relevant factors of PDAC. While it is clear all cell components in the TME play an important role in chemoresistance [30], our model’s utility comes from its ability to further elucidate the role of CAF activation in matrix-mediated chemoresistance.
CAF-mediated matrix remodeling is an intricate process that is still poorly understood in regards to its exact mechanism of action and impacts on tumor progression [31]. In breast cancer, researchers found that fibroblasts near tumor cells could realign fibers surrounding the tumor, increasing fibroblast activation [32]. It has been hypothesized that covalent crosslinking of collagen fibers participates in CAF-mediated matrix remodeling in PDAC [33]. However, due to the complicated stromal environment, the mechanism by which matrix remodeling influences chemoresistance had yet to be fully investigated in the past studies.
In this brief report, we showed that desmoplastic collagen-I provided adhesion sites for the cultured fibroblasts, allowing a transduction of the elastic force from the matrix through integrins. We believe the sensing of the stiffness through the matrix was the critical step to further activate the CAFs. Moreover, the amount of existing collagen-I seemed to be critical for CAF sensing and activation. Interestingly, the CAFs did not show a significant difference in cell spreading morphology when cultured with matrices of varying collagen concentration, over a five-fold range (between a 0.1 to 0.5 mg/mL collagen-I level (Fig. S2B)). Precise and uniform sensing of stiffness was achieved by another tweak of the system: allowing the gels to float in the media (see Materials and Methods). As a result, it appears that the encapsulated cells sense relatively uniform stiffness throughout the gels. We believe that allowing the gels to stick to the bottom would generate an unrealistic stiffness gradient in which the gel part closer to the bottom would be much stiffer, and the material stiffness of the hydrogels would become partially irrelevant to cultured cells [34].
While most studies suggest that CAF activation relies mostly on paracrine secretion from tumor cells [35], we believe there are other important mechanisms underlying this transformation. Here we identified a key role of the desmoplastic matrix in CAF hyperactivation. We postulate that this process uniquely relies on both biochemical and biomechanical interactions in a sequential manner. First, in a relatively soft matrix, the presence of a high amount of fibrotic collagen-I was required to ‘awaken’ the CAFs. After sensing the presence of the proteins, CAFs were able to quickly modify the matrix environment (Fig. 2) by covalently crosslinking the collagen-I fibers (Fig. S2A & Fig. 3). The crosslinking caused stiffening of the matrix, which consequently further activated CAFs through mechano-sensing, resulting in a ‘hyper-activated’ state indicated by both matrix contraction (Fig. S2) and elevated presence of nuclear YAP1 (Fig. 2D).
These findings support studies that found that type I collagen plays a critical role in the stiffening and mechanosensing of pancreatic tumors [36]. Likewise, in other cancer types, researchers have found that high collagen-I deposition increased cancer risk [37]. It’s important to note that simply eradicating these CAFs loses some of the growth restraining aspects of CAFs [11], [36]. Thus, using models to elucidate ways to block the mechanosensing, could yield better effects compared to attempting to ablate these cells.
After confirming that CAF-mediated matrix stiffening influenced chemoresistance in our co-cultures, we successfully identified that the process was LOX dependent (Fig. 3). Past studies have largely focused on the effect of LOX inhibition on FAK/Src signaling within tumor cells [38]. However, data also show that increased LOX family activity correlated with increased collagen deposition and increased chemoresistance in pancreatic mouse models [39]. Furthermore, targeting LOX in Pdx1-Cre KrasG12D/+ Trp53R172H/+ (KPC) mice suppressed tumor progression, and interestingly, this was associated with decreased collagen deposition [40]. While these studies did not specifically focus on CAFs as ours did, they lend support to the notion that the mechanism through which LOX inhibition reduces chemoresistance is fundamentally CAF-mediated. When strategies that target the LOX axis have failed to show improvement in PDAC mouse models [40] or patients [41], it has been suspected to be because these compounds were not capable of reversing type I collagen crosslinking [40]. Our model provides data to support this hypothesis. Furthermore, it highlights the potential of our model to identify potential therapeutic compounds that target matrix re-arrangement to overcome microenvironment-mediated chemoresistance since we can utilize CAF activation and collagen crosslinking among our many experimental readouts.
Tumor and CAF-derived exosomes are critical mediators of factors that can drive all stages of tumorigenesis [42], [43], [44]. In our previous work, we discovered that CAFs exposed to chemotherapy hypersecrete exosomes which promote tumor growth and chemoresistance in recipient cancer cells [19]. Here, we advance this research and show that culturing CAFs in a desmoplastic environment that recapitulates the human disease also causes them to hypersecrete exosomes compared to 2D culture (Fig. S4A). Consistent with our previous findings, systematic inhibition of exosome release achieved chemo-sensitization [19]. Imipramine and climbazole inhibited matrix-mediated organoid growth and chemoresistance by reducing exosome secretion (Fig. S4C-E). Our data support the investigation of novel compounds that inhibit exosome release to potentially overcome matrix-mediated chemoresistance.
The results generated by our novel 3D model point to several future directions for elucidating the molecular mechanisms underlying pancreatic tumor aggressiveness in stiff environments. First, it would be important to investigate how matrix stiffness affects mechano-sensing in CAFs through integrins, well characterized cell-ECM mechanoreceptors known to play a role in CAF activation [45]. Our biomimetic model gives us an opportunity to investigate how various integrins may be differentially regulated on organoids compared to CAFs in response to external forces. This could lead to targeted therapies designed to target a specific set of integrins. Furthermore, the inhibition of certain integrins may have an effect on YAP activation and subsequent CAF-activation based exosome secretion. Although this CAF-specific mechanism has not been fully elucidated, ECM stiffness was found to induce exosome secretion in breast cancer cells through a YAP pathway dependent mechanism in breast cancer [46]. Evidence to confirm that a similar mechanism may be at play in the pancreas has not been uncovered, but recent work showing how extracellular vesicles from pancreatic cancer stem cells modulate YAP activity in PDAC cells hints that exosomes may modulate cells in the TME through the same mechanism [47].
Finally, we believe our model provides the opportunity to develop potential prognostic biomarkers identified from CAF-derived exosomes. We are still in the early stages of elucidating the role that CAF-derived exosomes play in PDAC progression. Moreover, the role that the composition of the ECM itself has on the contents of exosomes remains to be fully investigated. A recent study shows that stroma density constitutes an independent prognostic marker in PDAC patients treated with adjuvant chemotherapy [48]. It is possible that the rate of CAF-exosome secretion may also change based on ECM density/stiffness and could serve as an independent prognostic factor. The literature showing the growing number of exosomal molecules which could serve as potential biomarkers supports this hypothesis [49].
Conclusion
Our novel biomimetic model illustrates how a stiffened desmoplastic matrix hyperactivates CAFs leading to the hypersecretion of chemoresistance-promoting exosomes in PDAC. This tunable model will allow us to study several potential mechanisms to overcome matrix-mediated chemoresistance. Future studies will focus on validation of these mechanisms in vivo and a deeper deciphering of the role of exosome crosstalk in the tumor stroma.
Funding
This research was funded by The Ellison Institute of Transformative Medicine of USC and the Stephenson Family and the Stephenson Family Personalized Medicine Center.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of The University of Southern California (20909-CR004).
CRediT authorship contribution statement
Weikun Xiao: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. Mahsa Pahlavanneshan: Formal analysis, Methodology, Writing – review & editing. Chae-Young Eun: Formal analysis, Methodology. Xinyu Zhang: Formal analysis, Methodology. Charlene DeKalb: Methodology. Bayan Mahgoub: Writing – review & editing. Hanaa Knaneh-Monem: Writing – review & editing. Sana Shah: Writing – original draft, Writing – review & editing. Alireza Sohrabi: Formal analysis, Methodology, Writing – review & editing. Stephanie K. Seidlits: Writing – review & editing. Reginald Hill: Conceptualization, Formal analysis, Supervision, Writing – review & editing
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
USC Pancreas Research Team: Bo Han, Heinz-Josef Lenz, Department of Medicine, Keck School of Medicine of USC; Steven Grossman, Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA, USA, Keck School of Medicine of USC; Veronica Wendy Setiawan, Department of Population and Public Health Sciences, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA, Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA, USA, Keck School of Medicine of USC.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mbplus.2022.100111.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Society A.C. American Cancer Society; Atlanta: 2022. Cancer Facts and Figures. [Google Scholar]
- 2.Vincent A., Herman J., Schulick R., Hruban R.H., Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/s0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Labori K.J., Katz M.H., Tzeng C.W., Bjørnbeth B.A., Cvancarova M., Edwin B., Kure E.H., Eide T.J., Dueland S., Buanes T., Gladhaug I.P. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma – A population-based cohort study. Acta Oncol. 2016;55(3):265–277. doi: 10.3109/0284186X.2015.1068445. [DOI] [PubMed] [Google Scholar]
- 4.Amrutkar M., Gladhaug I. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers. 2017;9(12):157. doi: 10.3390/cancers9110157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.-L., Choné L., Francois E., Artru P., Biagi J.J., Lecomte T., Assenat E., Faroux R., Ychou M., Volet J., Sauvanet A., Breysacher G., Di Fiore F., Cripps C., Kavan P., Texereau P., Bouhier-Leporrier K., Khemissa-Akouz F., Legoux J.-L., Juzyna B., Gourgou S., O’Callaghan C.J., Jouffroy-Zeller C., Rat P., Malka D., Castan F., Bachet J.-B. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018;379(25):2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z., Li Y., Ahmad A., Banerjee S., Azmi A.S., Kong D., Sarkar F.H. Pancreatic cancer: understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011;8(1):27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 7.Kadaba R., Birke H., Wang J., Hooper S., Andl C.D., Di Maggio F., Soylu E., Ghallab M., Bor D., Froeling F.EM., Bhattacharya S., Rustgi A.K., Sahai E., Chelala C., Sasieni P., Kocher H.M. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013;230(1):107–117. doi: 10.1002/path.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merika E.E., Syrigos K.N., Saif M.W. Desmoplasia in Pancreatic Cancer. Can We Fight It? Gastroenterol. Res. Practice. 2012;2012:1–10. doi: 10.1155/2012/781765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nabavizadeh A., Payen T., Iuga A.C., Sagalovskiy I.R., Desrouilleres D., Saharkhiz N., Palermo C.F., Sastra S.A., Oberstein P.E., Rosario V., Kluger M.D., Schrope B.A., Chabot J.A., Olive K.P., Konofagou E.E. Noninvasive Young's modulus visualization of fibrosis progression and delineation of pancreatic ductal adenocarcinoma (PDAC) tumors using Harmonic Motion Elastography (HME) in vivo. Theranostics. 2020;10(10):4614–4626. doi: 10.7150/thno.37965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimou A., Syrigos K.N., Saif M.W. Overcoming the stromal barrier: technologies to optimize drug delivery in pancreatic cancer. Ther. Adv. Med. Oncol. 2012;4(5):271–279. doi: 10.1177/1758834012446008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhim A., Oberstein P., Thomas D., Mirek E., Palermo C., Sastra S., Dekleva E., Saunders T., Becerra C., Tattersall I., Westphalen C.B., Kitajewski J., Fernandez-Barrena M., Fernandez-Zapico M., Iacobuzio-Donahue C., Olive K., Stanger B. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakim N., Patel R., Devoe C., Saif M.W. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas (Fairfax) 2019;3(1):e1–e4. doi: 10.17140/POJ-3-e010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schober M., Jesenofsky R., Faissner R., Weidenauer C., Hagmann W., Michl P., Heuchel R., Haas S., Löhr J.-M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers (Basel) 2014;6(4):2137–2154. doi: 10.3390/cancers6042137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill R., Li Y., Tran L.M., Dry S., Calvopina J.H., Garcia A., Kim C., Wang Y., Donahue T.R., Herschman H.R., Wu H. Cell intrinsic role of COX-2 in pancreatic cancer development. Mol. Cancer Ther. 2012;11(10):2127–2137. doi: 10.1158/1535-7163.MCT-12-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gifford J.B., Huang W., Zeleniak A.E., Hindoyan A., Wu H., Donahue T.R., Hill R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016;15(5):1043–1052. doi: 10.1158/1535-7163.MCT-15-0774. [DOI] [PubMed] [Google Scholar]
- 16.Von Ahrens D., Bhagat T.D., Nagrath D., Maitra A., Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017;10 doi: 10.1186/s13045-017-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen M.F.B., Mortensen M.B., Detlefsen S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016;22:2678. doi: 10.3748/wjg.v22.i9.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meads M.B., Gatenby R.A., Dalton W.S. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat. Rev. Cancer. 2009;9:665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 19.Richards K.E., Zeleniak A.E., Fishel M.L., Wu J., Littlepage L.E., Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36(13):1770–1778. doi: 10.1038/onc.2016.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker L.A., Tuveson D.A. Springer; New York: 2019. Methods in Molecular Biology 117–133. [DOI] [PubMed] [Google Scholar]
- 21.Shinde A.V., Humeres C., Frangogiannis N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. BBA. 2017;1863(1):298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain R., Fischer S., Serra S., Chetty R. The Use of Cytokeratin 19 (CK19) Immunohistochemistry in Lesions of the Pancreas, Gastrointestinal Tract, and Liver. Appl. Immunohistochem. Mol. Morphol. 2010;18(1):9–15. doi: 10.1097/PAI.0b013e3181ad36ea. [DOI] [PubMed] [Google Scholar]
- 23.Calvo F., Ege N., Grande-Garcia A., Hooper S., Jenkins R.P., Chaudhry S.I., Harrington K., Williamson P., Moeendarbary E., Charras G., Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013;15(6):637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elosegui-Artola A., Andreu I., Beedle A.E.M., Lezamiz A., Uroz M., Kosmalska A.J., Oria R., Kechagia J.Z., Rico-Lastres P., Le Roux A.-L., Shanahan C.M., Trepat X., Navajas D., Garcia-Manyes S., Roca-Cusachs P. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell. 2017;171(6):1397–1410.e14. doi: 10.1016/j.cell.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Chen W., Yang A., Jia J., Popov Y.V., Schuppan D., You H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology. 2020;72(2):729–741. doi: 10.1002/hep.31236. [DOI] [PubMed] [Google Scholar]
- 26.A. Bondareva, et al., The Lysyl Oxidase Inhibitor, β-Aminopropionitrile, Diminishes the Metastatic Colonization Potential of Circulating Breast Cancer Cells, PLoS One 4 (2009) e5620. doi:10.1371/journal.pone.0005620. [DOI] [PMC free article] [PubMed]
- 27.Datta A., Kim H., McGee L., Johnson A.E., Talwar S., Marugan J., Southall N., Hu X., Lal M., Mondal D., Ferrer M., Abdel-Mageed A.B. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018;8(1) doi: 10.1038/s41598-018-26411-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catalano M., O'Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J. Extracell. Vesicles. 2020;9:1703244. doi: 10.1080/20013078.2019.1703244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostrowski M., Carmo N.B., Krumeich S., Fanget I., Raposo G., Savina A., Moita C.F., Schauer K., Hume A.N., Freitas R.P., Goud B., Benaroch P., Hacohen N., Fukuda M., Desnos C., Seabra M.C., Darchen F., Amigorena S., Moita L.F., Thery C. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010;12(1):19–30. doi: 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 30.Lunardi S., Muschel R.J., Brunner T.B. The stromal compartments in pancreatic cancer: are there any therapeutic targets? Cancer Lett. 2014;343:147–155. doi: 10.1016/j.canlet.2013.09.039. [DOI] [PubMed] [Google Scholar]
- 31.DeLeon-Pennell K.Y., Barker T.H., Lindsey M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020;91–92:1–7. doi: 10.1016/j.matbio.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.W.-H. Jung, et al., Force-dependent extracellular matrix remodeling by early-stage cancer cells alters diffusion and induces carcinoma-associated fibroblasts, Biomaterials 234 (2020) 119756. doi:10.1016/j.biomaterials.2020.119756. [DOI] [PubMed]
- 33.Xiao Q., Ge G. Lysyl Oxidase, Extracellular Matrix Remodeling and Cancer Metastasis. Cancer Microenviron. 2012;5:261–273. doi: 10.1007/s12307-012-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tusan C.G., Man Y.-H., Zarkoob H., Johnston D.A., Andriotis O.G., Thurner P.J., Yang S., Sander E.A., Gentleman E., Sengers B.G., Evans N.D. Collective Cell Behavior in Mechanosensing of Substrate Thickness. Biophys. J. 2018;114(11):2743–2755. doi: 10.1016/j.bpj.2018.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biffi G., et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discovery. 2019;9:282–301. doi: 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.S. Bhattacharjee, et al., Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts, J. Clin. Invest. 131. doi:10.1172/JCI146987 (2021). [DOI] [PMC free article] [PubMed]
- 37.Martin L.J., Boyd N.F. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008;10(S1) doi: 10.1186/bcr1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Setargew Y.F.I., Wyllie K., Grant R.D., Chitty J.L., Cox T.R. Targeting Lysyl Oxidase Family Meditated Matrix Cross-Linking as an Anti-Stromal Therapy in Solid Tumours. Cancers. 2021;13:491. doi: 10.3390/cancers13030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Calvé B., Griveau A., Vindrieux D., Maréchal R., Wiel C., Svrcek M., Gout J., Azzi L., Payen L., Cros J., de la Fouchardière C., Dubus P., Guitton J., Bartholin L., Bachet J.-B., Bernard D. Lysyl oxidase family activity promotes resistance of pancreatic ductal adenocarcinoma to chemotherapy by limiting the intratumoral anticancer drug distribution. Oncotarget. 2016;7(22):32100–32112. doi: 10.18632/oncotarget.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller B.W., et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.A.B. Benson, 3rd et al., A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma, Oncologist 22 (2017) 241-e215. doi:10.1634/theoncologist.2017-0024. [DOI] [PMC free article] [PubMed]
- 42.Costa-Silva B., Aiello N.M., Ocean A.J., Singh S., Zhang H., Thakur B., Becker A., Hoshino A., Mark M.T., Molina H., Xiang J., Zhang T., Theilen T.-M., García-Santos G., Williams C., Ararso Y., Huang Y., Rodrigues G., Shen T.-L., Labori K.J., Lothe I.M.B., Kure E.H., Hernandez J., Doussot A., Ebbesen S.H., Grandgenett P., Hollingsworth M., Jain M., Mallya K., Batra S.K., Jarnagin W., Schwartz R., Matei I., Peinado H., Stanger B.Z., Bromberg J., Lyden D. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015;17(6):816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melo S.A., Luecke L.B., Kahlert C., Fernandez A.F., Gammon S.T., Kaye J., LeBleu V.S., Mittendorf E.A., Weitz J., Rahbari N., Reissfelder C., Pilarsky C., Fraga M.F., Piwnica-Worms D., Kalluri R. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.K. Ray, Pancreatic cancer: Pancreatic cancer exosomes prime the liver for metastasis, Nat. Rev. Gastroenterol. Hepatol. 12 (2015) 371. doi:10.1038/nrgastro.2015.93. [DOI] [PubMed]
- 45.Jang I., Beningo K. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers (Basel) 2019;11(5):721. doi: 10.3390/cancers11050721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patwardhan S., Mahadik P., Shetty O., Sen S. ECM stiffness-tuned exosomes drive breast cancer motility through thrombospondin-1. Biomaterials. 2021;279 doi: 10.1016/j.biomaterials.2021.121185. [DOI] [PubMed] [Google Scholar]
- 47.C.F. Ruivo, et al., Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network (EVNet) to fuel tumour progression, Gut (2022). doi:10.1136/gutjnl-2021-324994. [DOI] [PMC free article] [PubMed]
- 48.Wang L.M., Silva M.A., D’Costa Z., Bockelmann R., Soonawalla Z., Liu S., O’Neill E., Mukherjee S., McKenna W.G., Muschel R., Fokas E. The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget. 2016;7(4):4183–4194. doi: 10.18632/oncotarget.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li C., Teixeira A.F., Zhu H.J., Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol. Cancer. 2021;20:154. doi: 10.1186/s12943-021-01463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.