

Abstract
To explore the use of insertion-duplication mutagenesis (IDM) as a random gene disruption mutagenesis tool for genomic analysis of Streptococcus pneumoniae, a large mutagenic library of chimeric plasmids with 300-bp inserts was constructed. The library was large enough to produce 60,000 independent plasmid clones in Escherichia coli. Sequencing of a random sample of 84 of these clones showed that 85% of the plasmids had inserts which were scattered widely over the genome; 80% of these plasmids had 240- to 360-bp inserts, and 60% of the inserts targeted internal regions of apparent open reading frames. Thus, the library was both complex and highly mutagenic. To evaluate the randomness of mutagenesis during recombination and to test the usefulness of the library for obtaining specific classes of nonessential genes, this library was used to seek competence-related genes by constructing a large pneumococcal transformant library derived from 20,000 mutagenic plasmids. After we screened the mutants exhaustively for transformation defects, 114 competence-related insertion mutations were identified. These competence mutations hit most previously known genes required for transformation as well as a new gene with high similarity to the Bacillus subtilis competence gene comFA. Mapping of the mutation sites at these competence loci showed that the mutagenesis was highly random, with no apparent hot spots. The recovery of a high proportion of competence genes and the absence of hot spots for mutational hits together show that such a transformant library is useful for finding various types of nonessential genes throughout the genome. Since a promoterless lacZ reporter vector was used for the construction of the mutagenic plasmid library, it also serves as a random transcriptional fusion library. Finally, use of a valuable feature of IDM, directed gene targeting, also showed that essential genes, which can be targets for new drug designs, could be identified by simple sequencing and transformation reactions. We estimate that the IDM library used in this study could readily achieve about 90% genome coverage.
Despite many examples of successful identification of specific genes in Streptococcus pneumoniae (pneumococcus), a general genetic strategy for mutagenesis and gene identification in this species has remained elusive. Transposons are valuable mutagens and gene tags but often exhibit some degree of target preference and cannot be used easily to identify essential genes. Since libraries of DNA fragments carrying functional genes from this species in common Escherichia coli plasmid or phage vectors are often incomplete, due at least partly to high expression of pneumococcal genes in E. coli (5, 25, 40), options for genetic complementation approaches are also limited. An alternate method of mutagenesis and gene tagging is insertion-duplication mutagenesis (IDM) (42) with the highly efficient natural genetic transformation system of S. pneumoniae. When chimeric circular donor DNA comprising a portion homologous to the recipient genome and a heterologous region (often a nonreplicative plasmid with a marker) is introduced into competent pneumococcal cells, the heterologous region can be inserted specifically into the homologous region of the chromosome by circular integration, a reciprocal recombination event which creates flanking duplications of the homologous targeting region (17, 30). The resulting recombinant insertion-duplication structure provides both phenotypic and sequence tags at the site of mutation and can cause gene or operon disruption if the targeting fragment incorporated in the chimeric donor is internal. Although similar genetic strategies relying on different DNA delivery systems have been described for species, including Lactobacillus sake (22), Lactobacillus plantarum (21), Lactococcus lactis (2, 6, 20), Streptococcus pyogenes (33), and Bacillus subtilis (10, 16, 27, 31), and although IDM has been exploited for analysis of many individual genes in S. pneumoniae (e.g., references 17, 26, 29, and 37), the potential of IDM for use as a highly random mutagen in S. pneumoniae has not been systematically explored. Recently (19), we showed that targeting by IDM is highly specific, that there is little site bias for recombination when targeting fragments of uniform size are used, and that even with small targeting fragments, enough recombinants can readily be obtained to saturate the genome with mutations. These facts suggested that IDM could be a valuable general tool for genomic analysis, if uniform fragments of appropriate sizes are used for construction of chimeric circular plasmids. Here we describe the construction of a collection of such mutagenic plasmids prepared as clones in an E. coli host by following the quantitative guidelines described previously (19). We evaluated the mutagenic plasmids themselves for size uniformity and randomness of targeted sites by sequencing randomly selected plasmids, and we examined their collective utility as a mutagenic library by characterizing mutants obtained by integration into the pneumococcal chromosome. The results suggest that a high proportion of both dispensable and essential genes can be identified by use of this tool.
MATERIALS AND METHODS
Bacterial strains, media, and plasmid.
Two pneumococcal strains (34) were used: 5MC (CP1500; Mal+ Strr Novr), as source of targeting fragments and the donor for transformation assays, and CP1250 (Mal− Strr Novs Cms Com+), as a recipient strain for mutagenic plasmids. DH10B was used for transformation of mutagenic plasmids into E. coli. An insertion vector, pEVP3 (7), was used for cloning pneumococcal targeting fragments in E. coli. Luria-Bertani (LB) medium (39) was used for culture of E. coli, and CAT complete medium and its modified forms were used for culture of S. pneumoniae (see below for details).
Construction of a random mutagenic library of E. coli plasmid clones.
To prepare purified 250- to 350-bp chromosomal DNA fragments, 1 mg of 5MC DNA in 3.3 ml of sonication buffer (0.3 M sodium acetate, 10 mM Tris-HCl [pH 7.6], 1 mM EDTA, 30% glycerol) was sonicated (Branson model 450 sonifier) at power 2.0 for 210 s at 0°C with a 3-mm-diameter probe (11), with repeated interruptions for cooling on ice. After ethanol precipitation, the DNA was redissolved and resolved by preparative electrophoresis in 2% agarose gels. Gel slices containing unirradiated 300-bp fragments were cut out, and the DNA was purified as recommended for a QIAquick gel extraction kit (Qiagen), to yield 170 μg.
To prepare the termini of the sonicated DNA, 2.3 μg of fragmented DNA was trimmed with T4 polymerase as recommended by the enzyme supplier (Gibco BRL). After the enzyme was heat inactivated, the trimmed DNA was purified by using a QIAquick nucleotide removal kit as recommended by the supplier (Qiagen). To remove terminal phosphate groups, 1 μg of DNA with 30 U of calf intestinal phosphatase (CIP) (New England Biolabs) and in buffer recommended by the supplier was incubated for 1 h at 37.5°C and repurified by using the QIAquick nucleotide removal kit. To verify the effectiveness of T4 DNA polymerase and CIP treatments, a ligation mixture (20 μl) containing a sample (150 ng) of the DNA product, 15% polyethylene glycol (PEG; molecular weight 8,000; Sigma), 150 mM NaCl, and 5 U of T4 ligase, and buffer recommended by the supplier (Gibco BRL) was incubated for 20 h at 16°C.
The first ligation step was designed to saturate pEVP3 ends with pneumococcal DNA fragments in multimeric arrays. The ligation mixture (100 μl) contained 150 ng of dephosphorylated and blunt-ended 250- to 350-bp fragments, 1 μg of linear pEVP3 (gel-fractionated SmaI product), 150 mM NaCl, 20 U of T4 ligase, 15% PEG (molecular weight, 8,000), and buffer recommended by the supplier (Gibco BRL). After 24 h at 16°C, the DNA was purified by precipitation with PEG (36, 39). To release chimeric molecules of the unit size and split any vector-vector joints, the ligated comultimers were cut with SmaI and BssHII sequentially according to the recommendation of the supplier (New England Biolabs). The restriction mixture was loaded on a 0.7% agarose gel. Gel slices containing unirradiated, 6.6-kb fragments were excised after electrophoresis in TAE (39) for 2.5 h at 90 V, and the DNA was purified from the gel. The DNA amount was about 40 ng (4% overall yield). To circularize the chimeric linear molecules, the second ligation step was carried out at 16°C for 24 h in a mixture (100 μl) containing 38 ng of chimeric linear DNA (ratio of j to i, 50 [9]), 5 U of T4 ligase, and the buffer recommended by the supplier (Gibco BRL).
To establish a structured mutagenic library, 100 μl of competent cells (Max Efficiency DH10B; BRL) was transformed with a 1-μl portion of the ligation mixture. To retain maximal library complexity, outgrowth in liquid culture was omitted. Instead, the transformed cells were diluted 10-fold with SOB medium (39) and plated directly on the surfaces of freshly prepared 60-mm-diameter plates containing 3 ml of LB agar (1.5%) with 34 μg of chloramphenicol per ml and a top layer (poured just before plating) of 9 ml of LB agar (1.5%). Multiple rounds of separate transformation reactions were performed. After 40 h at 37°C, the 2.5-mm-diameter colonies were counted and pooled in sets of 100. The yield was 3 × 106 transformants/μg of ligation mixture. Two hundred fourteen such colony pools were collected by adding 1.5 ml of Terrific Broth per pool to the surface of the plate and mixing. A 1/10 portion of each pool was stored at −80°C after glycerol was added to 15% for the library stock, and the remainder of the pool was lysed to obtain a pooled plasmid preparation (90 to 120 different plasmids per pool) by a plasmid mini-prep method with the Promega Wizard column purification system. The yield from a pool was typically 1 to 2 μg of DNA in a 50-μl volume. Eighty-four clones were also individually selected at random for analysis. Finally, the remaining 20 ng of ligation mixture was stored at −20°C.
Transformation of CP1250 for construction of a mutant pneumococcal library.
For transformation, a 2-ml portion of a competent culture of CP1250 (14, 28) was exposed to about 300 ng of DNA from a single plasmid pool. After 45 min at 37°C, the culture was chilled and cells were collected by centrifugation and resuspended in 100 μl of supernatant. Fifty microliters of resuspended cells was spread on a 100-mm-diameter plate filled with CAT complete agar (1.5%) containing 2.5 μg of chloramphenicol per ml. After 20 h at 37°C, transformants were collected by resuspending the cells from about 300 pneumococcal colonies with 1.5 ml of CAT complete medium. Most of the resuspended cells were stored at −80°C, after glycerol was added to 15% and phenol red was added to 2 μg/ml, to form one pool of the mutated pneumococcal library (version 1); a small portion of the remaining cells were replated on chloramphenicol agar to remove residual drug-sensitive cells (nontransformants). After 20 h at 37°C, pools of about 900 colonies from this second plating were stored as described above for the mutated pneumococcal library (version 2).
Screening mutants by competence phenotype.
In the in situ colony competence test (papillation assay), cells (deficient for mal) to be tested for transformability were plated (500 to 5,000 per 100-mm-diameter plate) with 5MC DNA (Mal+) in a competence-supporting medium with a limiting amount of sucrose (to support the growth of small colonies and competence induction) and an excess of maltose (to permit mal+ transformants within those colonies to grow out as large papillations after 2 days of incubation) (18, 28). The plates contained three layers of a medium supporting transformation. The top and bottom (supporting) layers contained (per liter) 15 g of Difco Bacto Agar, CAT base (5 mg of choline, 5 g of tryptone [Difco Laboratories], 10 g of enzymatic casein hydrolysate [ICN Biomedicals], 1 g of yeast extract [Difco], 5 g of NaCl), 16.7 mmol of K2HPO4, 0.75 mmol of CaCl2, 6 mmol of NaOH, 2 g of maltose, and 0.1 g of sucrose. In a middle layer inoculated with cells at 37°C and then solidified at 4°C, 0.45% low-gelling-temperature agarose (FMC Corp.) was substituted for agar and supplemented with 5MC DNA (10 mg/liter) and bovine serum albumin (2 g/liter). After 40 h at 37°C, rare transformant-free (small, nonpapillated) colonies were identified under a dissecting microscope and sampled as possibly transformation defective. As CP1250 itself yielded 98 to 99.5% papillated colonies under these conditions, this initial screen is expected to accomplish a 50-fold enrichment for transformation-defective mutants.
For batch assays of competence in liquid culture, cells from nonpapillated colonies were transferred into 200 μl of CAT base broth (adjusted to 17 mM K2HPO4, 0.2% bovine serum albumin, 0.5 mM CaCl2, 0.02% glucose, and 6 mM NaOH and with 100 ng of 5MC DNA per ml and 10 μg of phenol red [growth indicator] per ml) in a microliter plate well and into a second well supplemented with competence-stimulating peptide (CSP) (200 ng/ml). At 16 h, each active culture was assayed for novobiocin-resistant transformants by spotting 5 μl of culture on the surface of a plate filled with CAT complete agar containing 2.5 μg of novobiocin per ml and, for viable cells, by spotting 5 μl of a 1:1,600 dilution of culture on the surface of a plate filled with CAT complete (CAT base plus 16.7 mmol of K2HPO4 and 0.2% glucose per liter) agar (1.5%) and incubation for 18 h at 37°C. A typical Com+ culture contained 5 × 107 viable cells/ml and 105 Novr transformants/ml. Insertion mutants were scored as transformation defective if they repeatedly yielded ≤5 × 102 Novr cells/ml. A parallel sample of each strain was retained in CAT complete medium at room temperature and plated for single-colony isolation after scoring of the transformation assay. After a second round of competence assays, each confirmed transformation-defective mutant was grown to 108 cells/ml from a single colony and stored with 15% glycerol at −80°C. Mutants were classified as Csp− if competence was restored by supplementation with CSP and as Xfo− if not. For backcrosses, CP1250 was transformed with DNA from each Csp− and Xfo− mutant, with selection for Cmr. The transformation defect was scored as linked to the insertion if none of four transformants tested was transformable.
High-resolution mapping of insertion mutations.
To prepare chromosomal DNA from each pneumococcal mutant, cells from a 10-ml culture (optical density at 550 nm, 0.3) were lysed in a solution containing 1 ml of 50 mM Tris, 10 mM EDTA (pH 8), 1% Triton X-100, and ∼70 μg of RNase A per ml for 10 min at 37°C. After addition of sodium dodecyl sulfate to 1%, phenol-chloroform extraction, and ethanol precipitation (39), the DNA was dissolved in 40 μl of Tris-EDTA and stored at −20°C. The typical yield was 20 μg of DNA.
For ligation-mediated PCR (LMPCR), 2 μg of DNA from each mutant was digested with MseI, Csp6I, DpnII, or BfaI (10 U) to produce fragments with sites for linker attachment for 3 to 5 h. To construct linkers with either a 5′-TA-protruding end or a 5′-GATC-protruding end, complementary oligonucleotides (50 pmol/μl) in Tris-EDTA were mixed in a 1.5-ml microcentrifuge tube, incubated for 5 min at 95°C, and annealed. Four hundred to 500 ng of restriction fragments and 25 pmol of an appropriate linker (per 20-μl reaction mixture) were prewarmed to 45°C, mixed with 1 U of T4 DNA ligase (Gibco BRL), and incubated for 2 h at 19°C. For PCR, 1 to 2 μl of an unpurified ligation product (20 to 50 ng of DNA) was added to a 100-μl PCR mixture containing 100 pmol of a pEVP3 primer (2 μl) and 50 pmol of a second primer complementary to the linker with Taq DNA polymerase. PCR was carried out with mineral oil in a model 480 thermal cycler (Perkin-Elmer) with 1 initiation cycle (94°C, 5 min; 58°C, 45 s; 72°C, 1 min, 30 s), 26 expansion cycles (94°C, 1 min; 58°C, 45 s; 72°C, 1 min, 30 s), and 1 final cycle (94°C, 1 min; 58°C, 45 s; 72°C, 12 min). The oligonucleotides used and their positions relative to the SmaI cloning site were DAM138 (5′-CTTCCACAGTAGTTCACCACCTT-3′, nucleotides [nt] 117 to 95), DAM139 (5′-TAGCTCTAGACACGCGTAGCAT-3′), DAM140 (5′-TAATGCTACGCGTGTCTAGAGCTA-3′), DAM141 (5′-TCAATTCAAGCTGGGGATCCATA-3′, nt 69 to 47), DAM142 (5′-CATATGACGTCGACGCGTCTG-3′, nt 50 to 30), and DAM143 (5′-GATCATGCTACGCGTGTCTAGAGCTA-3′).
As an alternative to LMPCR, in some cases the targeting inserts were amplified directly by using flanking vector primers to amplify the inserts from excised plasmids or tandom repeats. DNA (1 μg) from a mutant was transferred to a 100-μl PCR mixture of the same composition as that described above, but with 50 pmol of each primer. The cycling procedure was also the same as that described above, except that at least nine more expansion cycles were used. Primer pairs were either DAM090 (5′-TGTGCTGCAAGGCGATTAAGTT-3′, nt 186 to 165) and DAM091 (5′-TCTATCGATGCATGCCATGGTA-3′, nt 6277 to 6298) or DAM155 (5′-CATCAAGCTTATCGATACCGTCG-3′, nt 6268 to 6290) and DAM156 (5′-TTCAAGCTGGGGATCCATATGA-3′, nt 65 to 44). The nucleotide numbers in parentheses are relative positions when the SmaI site is set as 1 (see Fig. 1).
FIG. 1.

Strategy for construction of a random mutagenic library of insertion plasmids. Elements of pEVP3 indicated are ori, a replication origin active in E. coli but not in S. pneumoniae; cat, the gene for chloramphenicol acetyltransferase, which confers Cmr to both hosts; and ′lacZ, the promoterless gene for β-galactosidase. Small open boxes, 300-bp pneumococcal fragments; horizontal lines, linear pEVP3 molecules; dots, replication origins; vertical line, vector-vector joint within a concatemer of pEVP3 and inserts. conc., concentrations.
PCR products were sequenced by the University of Illinois at Chicago Core DNA Sequencing Facility with the primers shown in Table 2. Most sequences (173 of 193) displayed at least 95% identity to the sequence of a vector or linker region or both. If not, sequencing was repeated until this criterion was satisfied and a junction with pneumococcal DNA could be identified. Sequencing of inserts in 84 randomly chosen mutagenic plasmids was done at The Institute for Genomic Research (TIGR). To map sites targeted for mutation, sequences of the inserts of mutagenic plasmids or sequences adjacent to the insertion in mutant strains were used to search a contig collection for a type 4 strain (1) (obtained from TIGR’s website [40a]) with DNASIS (Hitachi America, Ltd.). For each match (more than 95% identity), the neighboring region was inspected for recognizable features by using BLAST services at the National Center for Biotechnology Information to search GenBank.
TABLE 2.
Determination of junction sequences in insertion mutants
| Method | Restriction site | Linkera | PCR primers | Primer for sequencing | No. of sequences recovered | Avg length of sequence read (bp) |
|---|---|---|---|---|---|---|
| LMPCR | Csp6I | 1 | DAM138, DAM139 | DAM138 or DAM141 | 38 | 330 |
| DpnII | 2 | DAM139, DAM142 | DAM142 | 7 | 320 | |
| MseI | 1 | DAM139, DAM155 | DAM155 | 45 | 190 | |
| BfaI | 1 | DAM139, DAM155 | DAM155 | 3 | 320 | |
| Direct PCRb | DAM090, DAM091 | DAM091 | 20 | 300 | ||
| DAM155, DAM156 | DAM155 | 1 | 300 |
Linker 1, annealed DAM139 and DAM140; linker 2, annealed DAM139 and DAM143.
Direct PCR using total chromosome preparations as templates.
To characterize adjacent deletions, PCR with sequential primer pairs was used to detect the presence or absence of specified portions of donor plasmid and to detect possible “joint fragments” predicted for each recombinant. More precise identification of the deletion junction was accomplished by PCR as described above, after chromosomal DNA primers and vector primers were surveyed for productive pairs. The smallest PCR product from each of four deletion strains was purified with centrifugal filter units and sequenced by the DNA Sequencing Core Facility at Florida State University (Gainesville).
Sampling theory.
We note that the probability (PL) of recovering at least L types of plasmids from a mixture of plasmids of N types when M transformants are examined is the same as a function calculated previously (19) as the probability Pi) to mutate at least i genes out of a total of N genes with a mutagenic library of size n. Thus, by substituting N for N, M for n, L for i, and 1/N for q, the following equation is obtained:
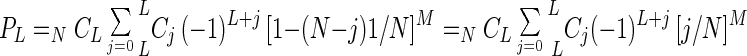 |
where NCL is N!/(N − L)!L!
RESULTS AND DISCUSSION
Design and construction of a random mutagenic plasmid library.
A critical parameter in the design of a plasmid library for insertion mutagenesis via natural transformation is the size of the random targeting fragment placed in the insertion vector: smaller targeting inserts have a higher chance of disrupting targeted genes, but smaller inserts produce sharply lower rates of transformation. Considering this parameter, we showed previously (19) that the smallest practicable insert size for producing the large collection of mutants required for genomic analysis was about 300 bp and estimated that 22,500 plasmids would be required to knock out at least 99.6% of a pneumococcus-sized genome of 1-kb genes. Of course, real genes vary in size. However, a survey of the sizes of all predicted genes in three organisms where complete genome sequences were available (Helicobacter pylori [41]), Methanococcus jannaschii [3], and Haemophilus influenzae [11]) suggested that only about 10% (180 of 1,590, 175 of 1,681, and 187 of 1,740, respectively) of bacterial genes are smaller than 300 bp and thus not directly mutatable by this method and that another 6% below 500 bp in size would be targeted with reduced mutagenic proficiency. An equally critical parameter is uniformity of the targeting fragment sizes because of the steep size dependence of recombination (19).
In the present study, we chose to make a library of 20,000 plasmids with 300-bp pneumococcal inserts, in expectation of achieving insertional disruption of nearly all of the genes large enough to target by this method (approximately 90% of total genes). The strategy for constructing the random mutagenic library is outlined in Fig. 1. The insertion vector pEVP3 (7) was chosen because it is a suicide vector in S. pneumoniae but replicates in E. coli (Fig. 1), it carries a robust Cmr marker that is expressed as a single copy independently of target gene transcription, it has essentially a zero background integration rate, and it has a promoterless lacZ gene downstream of cloning sites, allowing transcriptional fusion of a reporter to a targeted gene after integration. To obtain random targeting fragments of uniform size, chromosomal DNA was sonicated and fractionated by gel electrophoresis (data not shown). After treatment with T4 DNA polymerase, quality control showed that more than 70% of the fragments could be incorporated into dimers or multimers by DNA ligase. To block ligation between these fragments during insertion into the vector, the blunt-ended fragments were further treated with alkaline phosphatase. The vector was prepared by digestion with SmaI and gel fractionation to remove residual plasmids and other possible contaminating DNA. Preparative ligation was done in two successive reactions so that intermolecular linkage and circularization could be optimized separately (Fig. 1) to achieve both a high level of insertion and a high yield of circular monomers. Thus, after ligation of linear pEVP3 and the 300-bp fragments at a high concentration of DNA, vector-vector joints were cut with SmaI and “head-insert-tail” monomers were released with BssHII and purified. Self-ligation of the linear monomer at low concentrations of DNA and transformation of E. coli was followed directly by plating on the surfaces of chloramphenicol diffusion plates to prevent overrepresentation of the more rapidly growing clones. With a transformant titer of about 3 × 106/μg of DNA, the mutagenic plasmid library available from a 20-ng ligation reaction mixture was ∼6 × 104 transformants.
Analysis of random samples from the plasmid library.
To evaluate the nature and quality of this insertion plasmid library in detail, 84 randomly selected plasmid clones were subjected to a single round of sequencing reactions, in both directions. Eighty-five percent (72 of 84) of the plasmids carried inserts. The inserts’ median size was 295 bp, with 80% of the inserts being between 240 and 360 bp in length. The smallest and the largest were 220 and 470 bp, respectively. Thus, the DNA manipulation yielded a plasmid library with a low proportion of empty vectors, a very low content of double insertions (none found among 72 sequenced inserts), and a reasonably narrow distribution of insert sizes around the desired size of 300 bp.
Comparison with the working collection of 379 sequence contigs released by TIGR (40a) for a type 4 S. pneumoniae genome allowed further evaluation of the inserts in the sequenced sample of plasmids from the library. All but three could be aligned over the full length with at least one contig. One of the exceptions contained 300 bp of E. coli DNA, while the two others matched capsule synthesis genes of serotype 3, which are expected to be absent from the serotype 4 genome sequenced but present in the library source strain, Rx. The 69 other inserts were scattered widely over 46 contigs. Inspection of the working sequences flanking each insert showed that 42 (of 71 with pneumococcal inserts; 60%) were apparently internal gene fragments and thus likely to be mutagenic for the target gene. Thus, the insertion plasmids constituted a library of high complexity and high mutagenic potential with rather uniformly sized targeting inserts, fulfilling the requirements outlined in the introduction for a library to be a useful random mutagenic tool.
In a study of plasmids targeting various nonessential genes, transformation yields from 100 ng of pEVP3 derivatives carrying 300-bp pneumococcal inserts were reported to be 200 to 400 transformants per ml of competent cell culture (19). For a plasmid with an insert targeting an essential gene, no transformant would be expected at all, as the same study showed little or no background of nonspecific circular integration of this vector. Following the idea that this uniformly efficient and highly specific gene targeting should allow rapid identification of essential genes, transforming activities were determined for small-scale plasmid preparations of the 71 pEVP3 derivatives carrying pneumococcal inserts. Only 12 plasmids had pneumococcal inserts but displayed negligible transforming activity (0 to 1 transformant per ml), while the transforming activities of the other plasmids averaged 120 transformants per ml. Sequence analysis showed that the sites targeted by those 12 inserts were rich in known essential genes (Table 1). Eight of them were classed above as mutagenic. Six were in genes of the translation or transcription apparatus, and two were in unidentified putative genes. Four others overlapped the ends of genes but may have had polar effects on downstream genes in operons required for translation or for membrane synthesis. The high proportion of indispensible functions associated with regions targeted by these null plasmids confirms that this is an effective strategy for identifying essential genes.
TABLE 1.
Lethal insertion targets
| Clonea | Gene(s) targetedb | Function category |
|---|---|---|
| G8 | RNA polymerase, beta | Transcription |
| B11 | Peptide release factor 2 | Translation |
| B4 | tRNA cluster operon | Translation |
| H10 | asp-tRNA synthetase | Translation |
| F7 | leu-tRNA synthetase | Translation |
| B6 | phe-tRNA synthetase | Translation |
| C1 | ser-tRNA synthetase | Translation |
| A2 | Genes encoding ribosome proteins (nusA-IF2 operon) | Translation |
| D6 | Genes of ribosome protein operon (S5, L30, SecY) | Translation |
| A6 | Genes of fatty acid synthesis operon (fabF, fabZ, accC, accD) | Membrane synthesis |
| H9 | Unknown | |
| D2 | Unknown |
Clone containing a lethal mutagenic plasmid. The insert sequences from these plasmids mapped (more than 95% identity) to the following positions on TIGR contigs: G8 (contig 4102, bp 10776 to 11079), B11 (contig 4113, bp 6008 to 6290), B4 (contig 4119, bp 2220 to 2504), H10 (contig 4230, bp 6722 to 6971), F7 (contig 4120, bp 8556 to 8820), B6 (contig 4142, bp 13006 to 13276), C1 (contig 4103, bp 363 to 622), A2 (contig 4151, bp 7602 to 7902), D6 (contig 4127, bp 12011 to 12300), A6 (contig 4200, bp 13076 to 13323), H9 (contig 4105, bp 670 to 932), and D2 (contig 4196, bp 2483 to 2707).
Sequences from G8 to A6 exhibited more than 50% identity to the sequences of the listed proteins or RNA from B. subtilis. Although we did not assign functions to the genes targeted by clones H9 and D2, the H9 sequence was less than 45% identical to the putative aspartate aminotransferase from B. subtilis and the D2 sequence was 35% identical to the putative carbonic anhydrase of Methanobacterium thermoformicicum.
Application of the plasmid library for hunting dispensable genes by construction of a library of pneumococcal mutants.
The data presented above showed that the present strategy produced a library of insertional plasmids of high complexity with targeting fragments of uniform size. As our previous experiments with small numbers of insertional plasmids had found little site bias in recombination of plasmids bearing uniformly sized targeting fragments (19), this suggested that the mutagenic plasmids made here could produce a highly random collection of mutations. To evaluate the random mutagenic potential of this plasmid library more directly, and also to test its usefulness for discovering specific types of nonessential genes, we decided to use 20,000 insertion plasmids for hunting one class of such genes, those important for competence for genetic transformation. More than a dozen competence genes had previously been identified and shown to be dispensable (4, 34, 35, 43), but it was unknown how many more existed since those genes were found only in piecemeal fashion. If IDM with such a mutagenic plasmid library is a useful random general tool for gene hunting, then a large-scale screen should identify mutations in most of the known competence genes, could identify new competence genes, and would suggest how many pneumococcal competence genes exist in total within the genome of about 2,200 genes (13). Before the plasmid library was used on a large scale for introducing mutations into S. pneumoniae, it was necessary to design a practical strategy, since handling plasmid-carrying E. coli strains separately would require an impractically large effort to complete a genome-saturating screen for competence mutants: 20,000 plasmid preparations, 20,000 pneumococcal transformation reactions, and so on. We adopted the strategy of mixing the E. coli transformant clones in pools before plasmid extraction and then screening the pools of pneumococcal insertion mutants obtained by transformation with the plasmid mixtures. To estimate an appropriate degree of mixing to minimize work while excluding siblings, we worked from the assumption that 1% of pneumococcal genes might be competence related. If 100 clones of the library contained on average 1 plasmid targeting a competence-related gene, most pools of 100 plasmids would yield a mutation-affecting competence, yet accepting only one mutant per pool would eliminate siblings without rejecting many new mutants. Accordingly, we screened 214 pools, each containing about 100 Cmr E. coli transformants from the initial plasmid library selection plates, representing an actual total of 23,400 independent plasmid clones, among which approximately 20,000 plasmids (85%) were expected to have inserts. This number is large enough to achieve at least 98.5% mutagenesis of 1-kb genes (19) but will be less effective among the minority of smaller genes of a real genome. The plasmids from each pool were prepared together (214 plasmid preparations, instead of 23,400 preparations), with a typical yield of 1 μg of plasmid DNA per pool.
Optimal pneumococcal library size.
Construction of a library of pneumococcal mutants required transformation of a recipient strain (CP1250) with the library of insertion plasmids. To determine the number of pneumococcal transformants to collect in order to ensure representation of nearly all the 100 types of plasmids within a single pool, we considered the case of a mixture of N types of plasmids and assumed that the number of plasmids of each type was large (certainly above 107). We then noted that the sampling theory described in the work of Lee et al. (19) can be applied to this case (see Materials and Methods). According to the theory, when N is over 100, sampling of three N plasmids from a mixture of N types of plasmids would ensure representation at least 90% of the plasmid types in the mixture and a sample of four N individuals would represent at least 95% of them (Fig. 2). Given the asymptotic nature of the probability distribution, we chose the sampling value (M) 3 × N so that a minimal amount of work would be invested per new clone recovered. This threefold rule was adopted as a practical guideline for representing at least 90% of previous clones during any mixing step.
FIG. 2.

Dependence of the number of plasmid types (L) represented at least once in a sample on the number of transformants collected (M) after transformation by a pool of 100 plasmids in equimolar concentrations. The probability (PL) of representing at least L plasmid types (Materials and Methods) is shown for several numbers of collected transformants (M).
Construction of a library of pneumococcal transformants.
Each pool of insertion plasmids was used to generate a collection of pneumococcal transformants in strain CP1250 (Mal− Cms Novs) as described in Materials and Methods. For each plasmid pool, about 300 pneumococcal colonies were collected from chloramphenicol plates as a mixed transformant pool. In all, 214 such pneumococcal transformant pools were collected separately. The 66,700 clones within these pools constituted a version 1 library, representing at least 18,400 types of plasmids (for M = 3.3-fold N, the minimum representation fraction [L/N] was 92%; 92% × 20,000 = 18,400). Since chloramphenicol is bacteriostatic, there were some Cms cells within these transformant pools. To remove the residual sensitive cells, each transformant pool was replated at a high dilution, and about 900 of the resulting Cmr colonies were recovered to form a repurified mutant pool. The repurified mutant pools formed a version 2 library, containing cells from a total of 177,000 colonies. As the pooled clone number is 2.7 times the number of mixed clones (66,700), the minimum fraction of the initial mutant types represented is 89%. Thus, the number of plasmid types represented by the version 2 library is at least 16,400 (18,400 × 89%), a number large enough to achieve at least 97.5% mutagenesis of 2,200 1-kb genes (19).
Screening the library for transformation-defective mutants.
The 177,000 Cmr transformants were passed through three successive transformation screens. First, a preliminary screen was applied to about 2,700 clones from each transformant pool by the in situ colony competence test described in Materials and Methods, which monitors Mal+ papillae arising directly within colonies growing in solid transformation medium containing mal+ donor DNA. In all, 544,000 colonies from the mutant library were examined. As the number of colonies is 3.0 times the number of clones in the library (177,000), the minimum representation is 90%. Thus, the number of plasmid types represented in the pneumococcal clones tested was at least 15,000 (16,400 × 0.90), sufficient to achieve at least 97.0% mutagenesis of 2,200 1-kb genes. A total of 6,900 unpapillated colonies (about 32 per pool) were collected for further screening. In the second screen, competence tests of the selected clones during growth in liquid culture (with or without CSP) eliminated many false negatives and classified the transformation defects as CSP complementable (Csp−) or not CSP complementable (Xfo−), leaving 139 Csp− and 202 Xfo− mutants. Third, mutant cultures were retested in triplicate in the same media with or without CSP. Only strains yielding less than 1% of the control transformation levels in all tests were accepted for accession to the mutant collection. Retaining only one of each phenotype (Xfo− or Csp−) per pool, the yield was 90 Csp− and 106 Xfo− mutants. In pools with mutants, the average failure rate for unpapillated colonies was 30% (2,100 of 6,900). Mutants mapped to rarely hit contigs were checked by a transformation backcross with chromosomal DNA and entered in the permanent collection only if the Com− phenotype was linked to the Cmr of the insertion mutation. One-third of the mutants were eliminated by this criterion. Overall, 113 pools yielded no transformation-defective mutants, even after at least 32 unpapillated colonies per pool were screened; a single mutant was recovered from each of 88 pools, and two mutants were recovered from each of 13 pools.
Analysis of the targeted sequences in transformation-defective mutants.
The most direct way to start analysis of the competence mutants is to map them precisely by recovering sequences of the mutated region from each mutant. To do this, we used both LMPCR (32) and direct PCR strategies, as outlined in Fig. 3. As detailed in Table 2, sequences from 93 mutants were recovered by the first approach and sequences from 21 additional mutants were recovered by the second approach.
FIG. 3.

Strategy for recovery of sequences adjacent to an integrated pEVP3 plasmid by LMPCR. In the insertion orientation shown, lacZ is transcribed from the same strand as the target gene. Hatched boxes represent duplications of the target region. In this orientation, the targeted gene is truncated at the right end of the left hatched box. Vertical lines indicate the restriction sites closest to the ends of the plasmid insert for each enzyme. Map positions of primers used for LMPCR are shown by flags. Junction fragments produced by digestion of mutant DNA with one of the enzymes shown were attached to a linker oligonucleotide before PCR amplification with primers complementary to the vector and linker. The combinations of restriction enzymes, linkers, PCR primers, and sequencing primers used are shown in Table 2.
About 45% of the mutants (50 of 114) carried partial deletions of plasmid and target DNAs as indicated by the failure of LMPCR to amplify one end of the expected product of circular integration. To analyze the structure of these deletion integration events further, we amplified sequential portions of vector DNA in 33 randomly chosen mutated strains. We found multiple vector endpoints, representing losses of vector DNA ranging from 1 to 5 kb. Four of the 33 strains which mapped to a 4-kb region within the comAB locus were further analyzed to define the chromosomal deletion precisely by sequencing across the junctions; chromosomal deletions extended 1.2 to 2.4 kb from the point of insertion, and sequences interacting for the illegitimate recombination events generating these deletions contained small (3 to 6-bp) regions of sequence identity at the point of crossover (data not shown).
The recovered sequences, averaging 270 bp per mutation, established a tag for mapping mutations to known sequences. Although the sequence of a full pneumococcal genome is not yet available, the 379 working TIGR contigs covering about 90% of the genome of a type 4 strain allowed mapping of all mutations (see Materials and Methods). The recovered sequences matched regions of most of the nonessential competence operons previously identified, the comAB (43), cilB (dal, homologue to dprA [15]), cilC (ccl, homologue to comC in B. subtilis), cilD (cgl, homologue to comG in B. subtilis and comY in S. gordonii [4, 24, 35]), and cilE (cel, homologue to comE in B. subtilis [35]) loci. In addition, several insertion tags matched a gene previously unknown in S. pneumoniae that has high similarity to the B. subtilis competence gene comFA (23); we designate the locus cfl (ComF like) here. As can be seen in Fig. 4, all insertion-duplications mapped within previously known competence operons or the newly identified operon, cfl. While most insertion-deletions were also within these operons, 10 insertion-deletions outside (but less than 3 kb from) the known operons were oriented such that the deletion extended toward the competence genes. So, linkage of these flanking insertion-deletions to competence defects seems likely to reflect deletion of part of the nearby competence genes, which was confirmed in a few specific cases by mapping the deletion junctions (data not shown). Thus, as expected, the distribution of insertion-duplications on the map defines the extent of competence genes or operon units while insertion-deletions map to a wider neighborhood around such units. Virtually all insertional hits recovered were different, and they were widely scattered (Fig. 4), demonstrating a high complexity in the mutant library. As the six loci contain 13 genes in 12 kb, the 104 mutations mapped within these loci represent an average of approximately 8 hits per gene and 8.5 hits per kb of target DNA. These values are not far from the degree of saturation predicted above for the number of different mutagenic plasmids that were screened (15,000 plasmids), namely, approximately 7.5 hits per gene. Indeed, most genes that were hit once were hit several times, in good accord with the estimated degree of saturation for this library. The random distribution of insertion sites within these loci implies that the targeting inserts of the mutagenic plasmids were essentially random, as expected for sheared DNA, and that they directed integration with approximately equal efficiencies during recombination, as predicted from previous work (19).
FIG. 4.

Distribution of insertions in competence mutants at six loci. Locations, orientations, and structures of plasmid insertions in 114 transformation-defective mutants. Numbers of mutants were 42 at comAB, 38 at cgl, 20 at cel, 4 at cfl, 9 at ccl, and 1 at dal. Mutations are represented above the maps by a pentagon showing the position of the targeting fragment deduced from a sequenced junction. Pentagons pointing to the right indicate vector insertion in the orientation shown in Fig. 3. Open pentagons, insertion-deletions; filled pentagons, insertion-duplications. Putative genes with high levels of similarity to known genes in other organisms are given the prefix “h” (homologue of); those with no significant homology are given “orf” designations. Index genes for these loci are located in TIGR’s data release of 1997 as follows: cclA (contig 4291, bp 1992 to 1333), celA (contig 4139, bp 5654 to 5004), cflA (contig 4155, bp 6454 to 5156), cglA (contig 4194, bp 17495 to 16554), comA (contig 4105, bp 4693 to 6852), and dalA (contig 4179, bp 2168 to 1308). (celB1 and celB2 are fused in strain Rx [35] to form a full-length homologue of B. subtilis comEC.)
Certain competence genes were not expected to appear in the present screen. recA, for example, is strictly required for transformation, but recA mutants are somewhat impaired for growth in some media and nonviable in others. recP (38) and cilA are important for transformation, but strains with deletions of these genes exhibit residual levels of transformation up to 10% of normal, a phenotype that would have been rejected by our screening criteria. Also, any gene that is duplicated would be missed. In contrast, the comCDE locus, required for the production and sensing of competence pheromone, could be expected to be included in the competence mutant collection since in transformation experiments with one of these competence genes with a 390-bp (comD) internal fragment, the transforming activity (∼200/ml) was quite similar to those (∼400/ml) of other targeting fragments of similar sizes (300 to 400 bp) (19), and comD mutants are absolutely defective in competence induction (34). Yet, the comCDE operon was not represented in the mutant collection at all. For this case, we think that the cause is probably an unknown screening bias. Nonetheless, the overall results suggest strongly that the pneumococcus map is nearly saturated for the class of transformation genes that are dispensable for growth but required for transformation.
IDM as a gene knockout tool for genomic analysis.
We estimate that about 90% of individual genes in a typical bacterial genome can be mutated with a large library of the type analyzed here. This knockout power would not be improved much by reducing the targeting fragment length, because the yield of the pneumococcal transformation step would decrease prohibitively for significantly smaller inserts (19). Within this 90% maximum mutation limit, we estimate that we knocked out about 88% (97.5% × 90%) of the genes of the pneumococcal genome since the pneumococcal library (version 2) has at least 97.5% mutagenic power for 1-kb genes and the proportion of apparent mutatable genes is about 90% of total genes. We also estimate in the same way that we collected 87% (97% × 90%) of potential competence-related mutants with sequence tags by following several simple quantitative guidelines to avoid overrepresentation of any single clone in the mutant collection. (The estimated percentage yield of competence mutants should be reduced if screening bias is considered, although the degree of bias is difficult to estimate.)
The mutations produced by this method may not be gene specific. It is well known that chimeric donor DNA gives rise both to insertion-duplication products and to insertion-deletions (8, 18). As either type of mutation may affect the activities of multiple linked genes simultaneously, by polar effect or by deletion, both will consequently require that interesting mutants be exploited by further analysis to determine the specific genes responsible for a scored phenotype. On the other hand, these effects also mean that genes small enough to be immune to direct disruption by insertion-duplication events may be revealed by the effects of deletion or polar mutations.
Possible uses of the mutagenic plasmid library and the pneumococcal transformant library.
Although we used the screen for competence-defective mutants in the library of pneumococcal transformants as a model, the same library can be used for tagging genes specifically induced during competence by using a β-galactosidase activity assay, as demonstrated previously with a much less comprehensive pEVP3 fusion library (35). In that case, noninduced genes important for genetic transformation would be missed but the requirement for a disruptive insertion would be relieved entirely. The saturation statistics for this case would be the same as those for a transposon. Therefore, this pneumococcal library could in principle be used for hunting many kinds of nonessential genes, by either testing mutant phenotypes or observing the transcriptional reporter, as long as convenient assays or screening conditions for a relevant phenotype are available.
Integration of mutagenic plasmids delivered by the natural transformation pathway has one especially strong advantage in contrast to transposon mutagenesis: directed gene targeting. Using this feature, we showed in a pilot experiment that essential genes can be identified efficiently. As automated sequencing becomes cheaper and more accessible, it seems likely that a simple expansion of this approach—to include one-pass sequencing and simple transformation assays of several thousand plasmids from a library similar to the one described here—could readily be used to identify most essential genes of this naturally transformable, pathogenic organism. Since a significant proportion of bacterial genes are still of totally unknown function, this could provide a direct guide to vital new aspects of the biology of S. pneumoniae and thus to new potential targets for therapeutic interventions. The scale and yield of such a search can be estimated roughly from the present experience as follows. Among 1,000 plasmids, we expect to find approximately 850 inserts in total, 500 mutagenic inserts, and targets in 140 essential genes. Taking redundancy into account, 15,000 insertion plasmids should cover about 97.5% of the essential genes in the accessible size range. Since these inserts together represent twofold coverage of the genome size, such an effort would probably require about 1/5 of the amount of brute-force sequencing needed for a complete genome sequence and thus perhaps 1/10 of the total cost of such an effort (since there would be no labor-intensive closure phase) (12).
In summary, we report construction of a large mutagenic library of chimeric plasmids which is both highly complex and highly mutagenic. A pilot experiment demonstrated one application of this mutagenic library, i.e., for finding essential genes in S. pneumoniae. We also derived comprehensive transformant libraries by circular integration in a pneumococcal strain and showed that insertion mutagenesis is a good tool for genomic analysis, since it hit most known competence genes repeatedly and mutated an additional competence gene not previously identified by random or specific mutation methods. These results show that such libraries can be used for hunting other classes of nonessential genes and for defining essential genes, both at high levels of redundancy.
ACKNOWLEDGMENTS
This work was supported in part by the U.S. National Science Foundation (award MCB-9722821 to D.A.M.). Some of the sequencing of S. pneumoniae was accomplished with support from TIGR/NIAID/MGRI.
Assistance with mutant screening by B. Goldfarb and C. Varga is gratefully acknowledged. Assistance with transformation assays by W. Raegert and C. Yuan and preparation and sequencing of plasmids by Matt Cotton and Terry Utterbach (TIGR) are also gratefully acknowledged. Finally, the generosity of TIGR in making available genome sequences prior to publication (40a) is also gratefully acknowledged.
REFERENCES
- 1.Aaberge I S, Eng J, Lermark G, Løvik M. Virulence of Streptococcus pneumoniae in mice: a standardized method for preparation and frozen storage of the experimental bacterial inoculum. Microb Pathog. 1995;18:141–152. doi: 10.1016/s0882-4010(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 2.Biswas I, Gruss A, Ehrlich S D, Maguin E. High-efficiency gene inactivation and replacement system for gram-positive bacteria. J Bacteriol. 1993;175:3628–3635. doi: 10.1128/jb.175.11.3628-3635.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bult C J, et al. Complete genome sequence of the methanogenic archaeon Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 4.Campbell E A, Choi S Y, Masure H R. A competence regulon in Streptococcus pneumoniae revealed by genomic analysis. Mol Microbiol. 1998;27:929–939. doi: 10.1046/j.1365-2958.1998.00737.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen J D, Morrison D A. Cloning of Streptococcus pneumoniae DNA fragments in E. coli requires vectors protected by strong transcriptional terminators. Gene. 1987;55:179–187. doi: 10.1016/0378-1119(87)90278-2. [DOI] [PubMed] [Google Scholar]
- 6.Chopin M C, Chopin A, Rouault A, Galleron N. Insertion and amplification of foreign genes in the Lactococcus lactis subsp. lactis chromosome. Appl Environ Microbiol. 1989;55:1769–1774. doi: 10.1128/aem.55.7.1769-1774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claverys J P, Dintilhac A, Pestova E V, Martin B, Morrison D A. Construction and evaluation of new drug-resistance cassettes for gene disruption mutagenesis in Streptococcus pneumoniae, using an ami test platform. Gene. 1995;164:123–128. doi: 10.1016/0378-1119(95)00485-o. [DOI] [PubMed] [Google Scholar]
- 8.Claverys J P, Lefèvre J C, Sicard A M. Transformation of Streptococcus pneumoniae with Streptococcus pneumoniae-lambda phage hybrid DNA: induction of deletions. Proc Natl Acad Sci USA. 1980;77:3534–3538. doi: 10.1073/pnas.77.6.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dugaiczyk A, Boyer H W, Goodman H M. Ligation of EcoRI endonuclease-generated DNA fragments into linear and circular structures. J Mol Biol. 1975;96:171–184. doi: 10.1016/0022-2836(75)90189-8. [DOI] [PubMed] [Google Scholar]
- 10.Duncan C H, Wilson G A, Young F E. Mechanism of integrating foreign DNA during transformation of Bacillus subtilis. Proc Natl Acad Sci USA. 1978;75:3664–3668. doi: 10.1073/pnas.75.8.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleischmann R D, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 12.Fraser C M, Fleischmann R D. Strategies for whole microbial genome sequencing and analysis. Electrophoresis. 1997;18:1207–1216. doi: 10.1002/elps.1150180803. [DOI] [PubMed] [Google Scholar]
- 13.Gasc A M, Kauc L, Barraillé P, Sicard M, Goodgal S. Gene localization, size, and physical map of the chromosome of Streptococcus pneumoniae. J Bacteriol. 1991;173:7361–7367. doi: 10.1128/jb.173.22.7361-7367.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Håvarstein L S, Coomaraswamy G, Morrison D A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc Natl Acad Sci USA. 1995;92:11140–11144. doi: 10.1073/pnas.92.24.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karudapuram S, Zhao X, Barcak G J. DNA sequence and characterization of Haemophilus influenzae dprA+, a gene required for chromosomal but not plasmid DNA transformation. J Bacteriol. 1995;177:3235–3240. doi: 10.1128/jb.177.11.3235-3240.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khasanov F K, Zvingila D J, Zainullin A A, Prozorov A A, Bashkirov V I. Homologous recombination between plasmid and chromosomal DNA in Bacillus subtilis requires approximately 70 bp of homology. Mol Gen Genet. 1992;234:494–497. doi: 10.1007/BF00538711. [DOI] [PubMed] [Google Scholar]
- 17.Lacks S A. Cloning and expression of pneumococcal genes in Streptococcus pneumoniae. Microb Drug Res. 1997;3:327–337. doi: 10.1089/mdr.1997.3.327. [DOI] [PubMed] [Google Scholar]
- 18.Lacks S A, Greenberg B. Competence for deoxyribonucleic acid uptake and deoxyribonuclease action external to cells in the genetic transformation of Diplococcus pneumoniae. J Bacteriol. 1973;114:152–163. doi: 10.1128/jb.114.1.152-163.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee M S, Seok C, Morrison D A. Insertion-duplication mutagenesis in Streptococcus pneumoniae: targeting fragment length is a critical parameter in use as a random insertion tool. Appl Environ Microbiol. 1998;64:4796–4802. doi: 10.1128/aem.64.12.4796-4802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leenhouts K J, Kok J, Venema G. Campbell-like integration of heterologous plasmid DNA into the chromosome of Lactococcus lactis subsp. lactis. Appl Environ Microbiol. 1989;55:394–400. doi: 10.1128/aem.55.2.394-400.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leer R J, Christiaens H, Verstraete W, Peters L, Posno M, Pouwels P. Gene disruption in Lactobacillus plantarum strain 80 by site-specific recombination: isolation of a mutant strain deficient in conjugated bile salt hydrolase activity. Mol Gen Genet. 1993;239:269–272. doi: 10.1007/BF00281627. [DOI] [PubMed] [Google Scholar]
- 22.Leloup L, Ehrlich S D, Zagorec M, Moreldeville F. Single-crossover integration in the Lactobacillus sake chromosome and insertional inactivation of the ptsI and lacL genes. Appl Environ Microbiol. 1997;63:2117–2123. doi: 10.1128/aem.63.6.2117-2123.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Londono-Vallejo J A, Dubnau D. comF, a Bacillus subtilis late competence locus, encodes a protein similar to ATP-dependent RNA/DNA helicases. Mol Microbiol. 1993;9:119–131. doi: 10.1111/j.1365-2958.1993.tb01674.x. [DOI] [PubMed] [Google Scholar]
- 24.Lunsford R D, Roble A G. comYA, a gene similar to comGA of Bacillus subtilis, is essential for competence-factor-dependent DNA transformation in Streptococcus gordonii. J Bacteriol. 1997;179:3122–3126. doi: 10.1128/jb.179.10.3122-3126.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin B, Alloing G, Boucraut C, Claverys J-P. The difficulty of cloning Streptococcus pneumoniae mal and ami loci in Escherichia coli: toxicity of malX and amiA gene products. Gene. 1989;80:227–238. doi: 10.1016/0378-1119(89)90287-4. [DOI] [PubMed] [Google Scholar]
- 26.Mejean V, Claverys J-P, Vasseghi H, Sicard A M. Rapid cloning of specific DNA fragments of Streptococcus pneumoniae by vector integration into the chromosome followed by endonucleolytic excision. Gene. 1981;15:289–293. doi: 10.1016/0378-1119(81)90139-6. [DOI] [PubMed] [Google Scholar]
- 27.Michel B, Niaudet B, Ehrlich S D. Intermolecular recombination during transformation of Bacillus subtilis competent cells by monomeric and dimeric plasmids. Plasmid. 1983;10:1–10. doi: 10.1016/0147-619x(83)90052-5. [DOI] [PubMed] [Google Scholar]
- 28.Morrison D A, Lacks S A, Guild W R, Hageman J M. Isolation and characterization of three new classes of transformation-deficient mutants of Streptococcus pneumoniae that are defective in DNA transport and genetic recombination. J Bacteriol. 1983;156:281–290. doi: 10.1128/jb.156.1.281-290.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison D A, Trombe M-C, Hayden M K, Waszak G, Chen J-D. Isolation of transformation-deficient Streptococcus pneumoniae mutants defective in control of competence using insertion-duplication mutagenesis with the erythromycin resistance determinant of pAMβ1. J Bacteriol. 1984;159:870–876. doi: 10.1128/jb.159.3.870-876.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortier-Barriere I, Humbert O, Martin B, Prudhomme M, Claverys J-P. Control of recombination rate during transformation of Streptococcus pneumoniae: an overview. Microb Drug Res. 1997;3:233–242. doi: 10.1089/mdr.1997.3.233. [DOI] [PubMed] [Google Scholar]
- 31.Niaudet B, Goze A, Ehrlich S D. Insertional mutagenesis in Bacillus subtilis: mechanism and use in gene cloning. Gene. 1982;19:277–284. doi: 10.1016/0378-1119(82)90017-8. [DOI] [PubMed] [Google Scholar]
- 32.Palittapongarnpim P, Chomyc S, Fanning A, Kunimoto D. DNA fingerprinting of Mycobacterium tuberculosis isolates by ligation-mediated polymerase chain reaction. Nucleic Acids Res. 1993;21:761–762. doi: 10.1093/nar/21.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Casal J, Price J A, Maguin E, Scott J R. An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol Microbiol. 1993;8:809–819. doi: 10.1111/j.1365-2958.1993.tb01628.x. [DOI] [PubMed] [Google Scholar]
- 34.Pestova E V, Håvarstein L S, Morrison D A. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol Microbiol. 1996;21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- 35.Pestova E V, Morrison D A. Isolation and characterization of three Streptococcus pneumoniae transformation-specific loci by use of a lacZ reporter insertion vector. J Bacteriol. 1998;180:2701–2710. doi: 10.1128/jb.180.10.2701-2710.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pheiffer B H, Zimmerman S B. Polymer-stimulated ligation: enhanced blunt- or cohesive-end ligation of DNA or deoxyribooligonucleotides by T4 DNA ligase in polymer solutions. Nucleic Acids Res. 1983;11:7853–7871. doi: 10.1093/nar/11.22.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puyet A, Greenberg B, Lacks S A. Genetic and structural characterization of EndA, a membrane-bound nuclease required for transformation of Streptococcus pneumoniae. J Mol Biol. 1990;213:727–738. doi: 10.1016/S0022-2836(05)80259-1. [DOI] [PubMed] [Google Scholar]
- 38.Rhee D K, Morrison D A. Genetic transformation in Streptococcus pneumoniae: molecular cloning and characterization of recP, a gene required for genetic recombination. J Bacteriol. 1988;170:630–637. doi: 10.1128/jb.170.2.630-637.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 40.Stassi D L, Lacks S A. Effect of strong promoters on the cloning in Escherichia coli of DNA fragments from Streptococcus pneumoniae. Gene. 1982;18:319–328. doi: 10.1016/0378-1119(82)90170-6. [DOI] [PubMed] [Google Scholar]
- 40a.The Institute for Genomic Research Website. 21 November 1997, sequence release date. [Online.] http://www.tigr.org. [19 March 1999, last date accessed.]
- 41.Tomb J F, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 42.Vasseghi H, Claverys J P, Sicard A M. Mechanism of integrating foreign DNA during transformation of Streptococcus pneumoniae. In: Polsinelli M, Mazza G, editors. Transformation—1980. Oxford, United Kingdom: Cotswold Press, Ltd.; 1981. pp. 137–153. [Google Scholar]
- 43.Zhou L, Hui F M, Morrison D A. Competence for genetic transformation in Streptococcus pneumoniae: organization of a regulatory locus with homology to two lactococcin A secretion genes. Gene. 1995;153:25–31. doi: 10.1016/0378-1119(94)00841-f. [DOI] [PubMed] [Google Scholar]