

Summary
High-content imaging of tumor organoids (TOs) treated with therapeutic agents provides detailed cell viability readouts at the organoid level. In contrast, most used protocols provide one number per well. While requiring the use of inverted microscopy with an automated stage, this protocol can provide critical information about heterogeneous responses of TOs to various treatments. This protocol describes a technique for culturing and drug testing TOs using fluorescent indicators of cell viability with high reproducibility.
For complete details on the use and execution of this protocol, please refer to Larsen et al. (2021).
Subject areas: Cancer, High Throughput Screening, Microscopy, Organoids
Graphical abstract
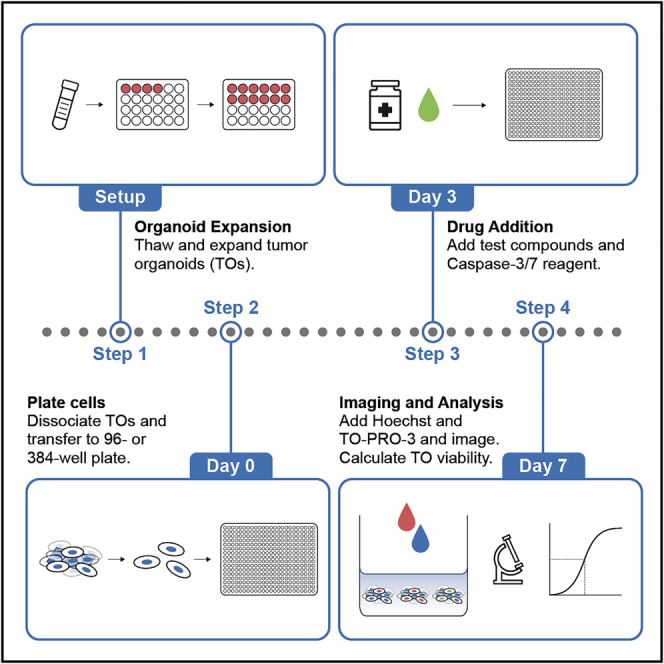
Highlights
-
•
Protocol for screening of compounds in 3D culture using high-content imaging
-
•
This protocol can be used for tumor organoids of any cancer type
-
•
Procedures for routine tumor organoid culture as well as screening
-
•
Optimized for previously cryopreserved and validated tumor organoid cultures
Publisher's note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
High-content imaging of tumor organoids (TOs) treated with therapeutic agents provides detailed cell viability readouts at the organoid level. In contrast, most used protocols provide one number per well. While requiring the use of inverted microscopy with an automated stage, this protocol can provide critical information about heterogeneous responses of TOs to various treatments. This protocol describes a technique for culturing and drug testing TOs using fluorescent indicators of cell viability with high reproducibility.
Before you begin
This protocol describes a method to culture tumor organoids (TOs) and assess response to therapeutic agents using a high-content fluorescent microscopy system. TOs have been utilized commonly for drug development and precision medicine studies. Several groups have shown consistencies between patient response to chemotherapies and patient derived TO cultures (de Witte et al., 2020; Driehuis et al., 2019a, 2019b; Grossman et al., 2021; Narasimhan et al., 2020; Ooft et al., 2019; Wensink et al., 2021).
An important consideration when designing a screen, is that the investigator should choose a panel of compounds appropriate for the TO cancer type and/or genetic background of interest. To ensure best results from drug screen studies, TOs derived from patient specimens should be screened for malignancy through expert pathology review and next-generation sequencing. The protocol described here uses a set timeline of TO growth after seeding and drug incubation (based on previous experience) with the compounds used. TOs at the described seeding density grow in 384-well plates at the specified duration without losing viability. The plate layout described here excludes the edges of the plate to avoid edge effects. It contains seven, 10-fold dilutions in technical triplicates to accommodate 14 compounds per plate.
Institutional permissions
No experiments described here involved the use of live vertebrates or higher invertebrates.
Experimental design
The protocol involves four main steps:
-
1.Thaw and expand TOs of interest.
-
a.Identify TO lines of interest and thaw prior to screening to allow recovery and expansion. TO lines derived from different patients recover and proliferate at varying rates.Note: Previous screening experience with the chosen lines should be used to determine how far in advance a line should be thawed before screening.
-
b.Expand TOs in culture media containing the necessary supplements for optimal growth based on TO cancer type (Larsen et al., 2021). Newly derived TO lines may also be used for screening if properly validated by pathology review and sequencing (to confirm the culture is composed of cancer cells rather than benign).Note: In addition, variables like seeding density, media changes, plate selection, and time of drug treatment may be optimized especially for compounds requiring prolonged exposure. Additionally, setting up positive and negative controls, number of dilutions, replicates, controls, and drugs can be tailored to fit the experimental need.
-
c.Efforts should be made to screen TO lines at the earliest passage number possible.Note: For those interested in minimizing differential clonal expansion, new lines should not be passaged at all.
-
a.
-
2.Preparation of screening libraries.
-
a.Compounds are purchased either individually or as a custom library from a vendor. Compounds may be bought pre-diluted in solvents like DMSO, water, ethanol etc. Alternately, the dry powder can be obtained and dissolved by the user in the recommended solvent and at a desired concentration (e.g., 10 mM).
-
b.These compound solutions should be aliquoted and stored as recommended by the manufacturer (typically at −80°C for long-term storage).
-
a.
Note: Compounds should be aliquoted also to avoid repeated freeze-thaw cycles. Typically, aliquots of individual compounds will be thawed and pipetted to a 96- or 384-well storage plate in the desired arrangement and stored at −80°C or −20°C for short-term regular use.
-
3.Choice of microscopic equipment and fluorescent compounds.
-
a.High-content, automated fluorescent imaging is necessary for this protocol due to the high number and quality of images required for analysis. It is critical to have clear, in-focus, reliable imaging for use in downstream analysis.
-
b.This protocol describes the use of three fluorescent molecules. Hoechst 33342 - used to identify all cell nuclei, TO-PRO-3 to identify all dead cells, and Caspase-3/7 activated fluorescent reagent to identify apoptotic cells.
-
a.
Note: Alternate fluorescent reagents may be used. It is critical to select fluorescent reagents depending on the microscope filters and the assay performed.
-
4.Choice of image analysis method.
-
a.There are multiple open access options for image analysis; however, this protocol utilizes an ImageXpress Micro Confocal from Molecular Devices with a MetaXpress image analysis module. One such open-source image analysis software alternative is CellProfiler (Carpenter et al., 2006; Stirling et al., 2021).
-
b.Once the choice of image analysis software has been determined, the specific analysis method should be validated. A test set of images including appropriate controls (wells without fluorescent dye) is required to optimize settings for the identification of live and dead cells.
-
a.
CRITICAL: different TO lines with different morphologies may affect image analysis. Results must be reviewed to ensure accuracy.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Y-27632 | Bio-Techne | Cat# 1254 |
| Staurosporine | Selleckchem | Cat# S1421 |
| Bortezomib | Selleckchem | Cat# S1013 |
| DNase | Worthington | Cat# LS006328 |
| Dispase II | STEMCELL Technologies | Cat# 07913 |
| Critical commercial assays | ||
| TO-PRO-3 | Invitrogen | Cat# T3605 |
| Caspase-3/7 | Sartorius | Cat# 4440 |
| Hoechst 33342 | Invitrogen | Cat# H3570 |
| Deposited data | ||
| Drug Screening Source Data | Vivli | Cat# T21.01 |
| Experimental models: Cell lines | ||
| Tumor Organoids | Various | N/A |
| Software and algorithms | ||
| MetaXpress | Molecular Devices | https://moleculardevices.com |
| R | R | https://www.r-project.org/ |
| Other | ||
| GFR Matrigel | Corning | Cat# 356231 |
| TrypLE Express | Gibco | Cat# 12604021 |
| Advanced DMEM/F12 | Life Technologies | Cat# 12634028 |
| DPBS | Thermo Fisher Scientific | Cat# 14190250 |
| AlumaSeal II | Excel Scientific | Cat# AFS25 |
| Bio-Rad hard shell 96-well plate | Bio-Rad | Cat# HSP9601 |
| 384-well Plates | Corning | Cat# 3985 |
| 384-well polystyrene plates | Nunc | Cat# 242757 |
| Breathe-Easy sealing membrane | Sigma-Aldrich | Cat# Z380059 |
| Micro Confocal IXM-C | Molecular Devices | ImageXpressMicro Confocal |
| Countess II | Invitrogen | Cat# AMQAX1000 |
| Integra Viaflo Pipette | INTEGRA | Cat# 4623 |
| Integra Assist Plus Pipetting Robot | INTEGRA | Cat# 4505 |
Materials and equipment
Passaging reagents
| Reagent | Final concentration | Amount |
|---|---|---|
| TrypLE Express solution | 1× | 500 μL/well |
| PBS | 1× | Variable |
| Dispase II (10 mg/mL PBS) | 1× | 250 μL/250 μL growth media |
| DNase I (5 mg/mL PBS) | 10 K.U./mL | 10 μL/10 mL PBS |
| Matrigel | 100% | 50 μL/well |
| TO Culture Media | 1× | 500 μL/well |
| 10 mM Rock Inhibitor Y-27632 | 10 μM | 1 μL/1 mL growth media |
Staining reagents
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F12 | 1× | 7,740 μL |
| TO-PRO™-3 Iodide (642/661) | 900 nM | 7 μL |
| Hoechst 33342 | 7.5 μg/mL | 5.8 μL |
| Total | n/a | 7752.8 μL |
Plating reagents
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | 1× | 80 μL/well |
| Matrigel | 100% | 80 μL/well |
| TO Culture Media | 1× | 500 μL/well |
Integra Assist Plus Pipetting Robot
We use the Integra Assist Plus Pipetting Robot with a Viaflo electronic pipette for most liquid handling steps, including plating TO cells in the drug assay plates, aliquoting media for serial dilutions, generating serial dilutions, and adding staining reagents to the drug assay plates. Use any method of liquid handling with the required level of throughput and precision (Du et al., 2020; Francies et al., 2019).
MetaXpress
Analyze images acquired on the ImageXpress Micro Confocal using the Custom Module Editor tool of MetaXpress (Molecular Devices). This software identifies TOs by clusters of Hoechst staining and identifies live vs dead cells by overlap of either TO-PRO-3 or Caspase-3/7 fluorescent signal with Hoechst. Optimize fluorescence intensity thresholds to identify fluorescent positive cells for each reagent individually.
Alternatives: Image analysis softwares capable of identifying fluorescent positive cells such as ImageJ (Schneider et al., 2012) or CellProfiler may be used to assess TO viability from images generated during the experiment.
Tissue culture assay plates
There are many options for tissue culture plates. When selecting a plate for high-content imaging it is important to choose a plate with a clear, flat bottom manufactured for imaging. It is also important to consider the type of plate (96- vs 384-well plate) best suited for the experiment.
Alternatives: Corning brand plates were used for this study. Suitable alternative assay plates are the CellStar μClear in 384-well plate (Greiner Bio-One, Cat #781090) and 96-well plate formats (Greiner Bio-One, Cat #655090).
Step-by-step method details
Thawing of TOs
Timing: 1 h
TOs of interest were thawed as follows:
-
1.
Thaw Matrigel on ice at 4°C. Matrigel must be always maintained at 4°C.
Note: Proper thawing of Matrigel can take several hours depending on volume. Be sure to plan and thaw an appropriate volume before beginning.
-
2.
Prepare 9 mL of Advanced DMEM/F12 in one low-binding 15-mL tube and keep it on ice.
-
3.
Pre-warm one 24-well plate for each line by placing it inside the 37°C incubator for at least 15 min.
-
4.
Prepare a single use aliquot of TO growth media supplemented with 10 μM ROCK inhibitor. Unused TO growth media without ROCK inhibitor can be stored for 1–2 weeks at 4°C. You will need 500 μL of media per well. ROCK inhibitor can be used to prevent apoptosis in TOs when passaging (Driehuis et al., 2019b). In this protocol, ROCK inhibitor is only required when cells are newly plated. It is not required in subsequent media changes.
-
5.
Identify the sample to be thawed and locate the cryovial from the LN2 tank.
-
6.
Remove the cryovial(s) from the LN2 tank and place on dry ice.
-
7.
Thaw the cryovial content by incubating it in the water bath at 37°C. Keep the cryovial in the water bath until a little piece of ice is left (approximately 1–3 min).
-
8.
Take the cryovial from the water bath, spray it with 70% ethanol and transfer it to the biosafety cabinet.
-
9.
Add the content of the cryovial (1 mL) to the prepared 9 mL of Advanced DMEM/F12 on ice.
-
10.
Centrifuge the TO-containing, 15-mL tube at 200 g for 5 min at 4°C.
-
11.
Check for the TO pellet and remove the supernatant. TO pellets and tubes should be kept on ice at this point to prevent premature crosslinking of Matrigel when resuspending the pellet.
-
12.
Resuspend thawed TOs in the requisite volume (50 μL per well) of Matrigel. It is critical to avoid adding bubbles to the Matrigel. Mix the TO pellet with Matrigel slowly and without fully compressing the plunger of the pipette to prevent bubbles. To avoid premature crosslinking of Matrigel, ensure the 15-mL tube is cooled to 4°C.
Note: The number of cells per cryovial should be known at the time of cryopreservation and upon thawing the cell pellet should be resuspended in a volume of Matrigel to plate the cells at a density of 100,000 cells/well. If the number of cells per cryovial is unknown, count the cells to determine the volume of Matrigel needed.
-
13.
Pipette 50 μL of the TO-Matrigel mix to the center of each well of the pre-warmed 24-well plate (Figure 1A). Avoid touching the bottom of the well with the pipette tip and do not dispense to the second stop of the pipette to help prevent bubbles in the dome.
-
14.
Carefully place the 24-well plate with the TOs in the incubator for 15 min to let the Matrigel polymerize.
-
15.
Once the Matrigel domes have polymerized, add 500 μL of TO growth media supplemented with 10 μM ROCK inhibitor to each well.
-
16.
If few wells are plated, add 500 μL of PBS to the wells surrounding those containing TOs to help reduce media evaporation.
Figure 1.

Evaluating organoid proliferation by light microscopy
(A) Matrigel domes should be positioned in the center of the well. This image shows a 50 μL dome of Matrigel in a 24-well plate.
(B) Time-lapse brightfield imaging of TO growth in a Matrigel dome. Scale bar: 500 μm.
Culture and expansion of TOs
Recently thawed TO lines may need to be expanded prior to drug screening. TO lines derived from different patients will recover and proliferate at varying rates. TO lines are expanded in culture media containing the necessary supplements for optimal growth based on TO cancer type. If TOs are healthy, abundant, and no cancer-associated fibroblasts (CAFs) are present visually by brightfield microscopy, propagate the TOs by obtaining a single-cell suspension. If CAFs are present, refer to troubleshooting problem 6 for a strategy to reduce the number of CAFs in the culture. Sterilize the hood and equipment before beginning the following procedure. TOs should be seeded uniformly by making sure the cell/Matrigel mixture is mixed homogeneously before pipetting the Matrigel dome. Once TOs reach an appropriate density they should be passaged (Figure 1B). TOs may need to be passaged before the dome is densely populated if the TOs grow too large and cells begin to die in the middle of the TO (evidenced by very dark centers), if cells adhere to the bottom of the well and the Matrigel dome begins to loosen from the well bottom, or if the TOs begin to grow out of the Matrigel and risk floating in the media.
-
17.
Thaw Matrigel on ice at 4°C. Matrigel must be always maintained at 4°C.
Note: Properly thawing Matrigel can take several hours depending on the volume. Be sure to plan and thaw an appropriate volume prior to beginning.
-
18.
Pre-warm a 24-well plate in the incubator. If passaging multiple TO lines, warm one 24-well plate per line.
-
19.
Gently remove the media from the TOs by pipetting it out or aspirating.
-
20.
Add 500 μL of TrypLE to each well.
-
21.
Disrupt the TO-containing Matrigel domes by pipetting the TrypLE up and down.
Note: Be careful to only use the force of pipetting to disrupt the Matrigel dome rather than scraping the plate with the pipette tip.
-
22.
Pipette the TOs into a 15-mL, low-binding centrifuge tube.
-
23.
Rinse the wells of the 24-well plate with TrypLE to collect any remaining TOs and collect into the 15-mL tube.
-
24.
Place the tube in the 37°C water bath for 10 min.
-
25.
Pipette the supernatant up and down to further break up the TOs.
-
26.
Spin the cells at 200 g for 5 min in a 4°C centrifuge.
Note: The TOs should form a compact pellet if all Matrigel has been dissolved and there is not excessive DNA causing cell clumps (Figure 2). If a loose pellet is observed, refer to the troubleshooting tips at the end of this article.
-
27.
Carefully pipette out and discard the supernatant.
-
28.
Re-suspend the cell pellet in 1 mL PBS.
-
29.Mix well and count cells using Countess II or an alternate instrument such as a hemocytometer.
-
a.Mix equal volumes of cell suspension and trypan blue to assess viability.
-
b.Use viable cell numbers to determine plating density.Note: Each well should contain 20–30,000 fully dissociated cells.
-
i.If there are clumps of cells, DNase may be needed. Refer to troubleshooting problems 3 and 4.
-
i.
-
a.
-
30.
Centrifuge the cell-containing tube at 200 g for 5 min at 4°C.
-
31.
Remove the supernatant.
-
32.
Add the requisite volume (50 μL per well) of Matrigel to the single cell/TO pellet to be plated.
-
33.
Gently pipette the Matrigel up-and-down to incorporate the pelleted cells/TOs until the mixture appears homogeneous. Take great care to prevent bubbles from forming in the mixture. Incorporation of air bubbles in the domes may lead to future detachment or rupturing of the Matrigel.
-
34.
Remove the pre-warmed plate from the incubator and use a 200 μL pipet, set to a 50 μL volume, to gently pipette the Matrigel/cell mixture up and down to re-homogenize.
-
35.
Carefully dispense 50 μL of Matrigel/cell mixture in the center of a desired well in the warmed 24-well plate, taking care not to touch the bottom of the plate with the tip-and more importantly-not to incorporate bubbles into the domes.
Note: Utilize the most central wells in the plate to prevent media evaporation.
-
36.
Repeat until all desired wells are filled.
-
37.
Cover the plate with the lid and return the plate to the 37°C incubator for at least 15 min to allow the domes to polymerize prior to adding media to the plate.
-
38.
After the incubation period, visually inspect the plate to ensure that the domes have polymerized.
Note: Domes should not move or vibrate when the plate is moved.
-
39.
While the Matrigel domes are polymerizing, prepare TO growth media containing ROCK inhibitor at a final concentration of 10 μM. Once Matrigel domes have polymerized, gently pipette 500 μL of pre-warmed (37°C) TO growth media into each well.
Note: To prevent detachment, take care not to dispense the media directly onto the domes. This can be achieved by directing the flow of media onto the wall of each well.
-
40.
Return the plate to a 37°C incubator.
Figure 2.
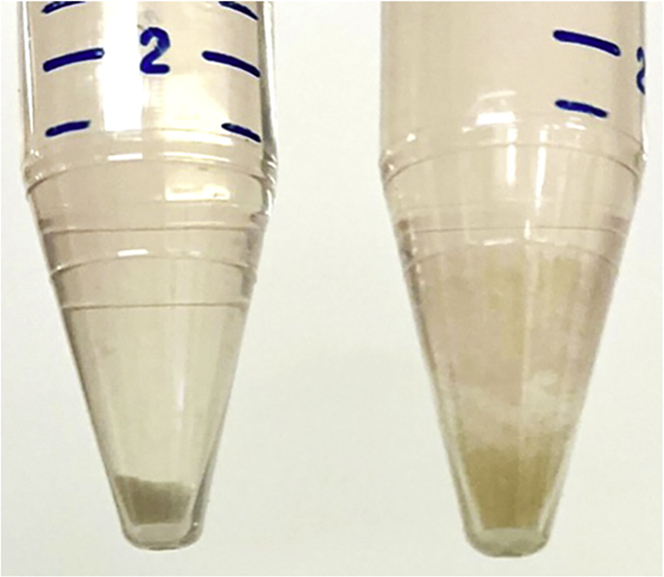
Evaluating dissociation of organoid cultures and cell pellets
Cell pellets after centrifugation of TrypLE dissociated TO cultures. Upon complete digestion of Matrigel (left) cell pellets are compact in nature with clear demarcation between pellet and supernatant as compared to incomplete digestion (right).
Plating of the TOs in 384-well plates for drug screening
This describes the process for plating a single cell suspension from TOs grown in 24-well plates to 384-well plates for drug screening.
-
41.
Prepare a single-cell suspension of cultured TOs as described in the previous section following steps 17–27.
-
42.
Re-suspend the cell pellet in 1 mL cold TO growth media.
-
43.Mix well and count cells using Countess II or an alternate instrument such as a hemocytometer.
-
a.Mix equal volumes of cell suspension and trypan blue to assess viability.
-
b.Use viable cell numbers to determine plating density.
-
a.
Note: The following calculations are for plating 2,000 cells per well in all wells of columns 2–23 of a 384-well plate. Adjust cell numbers and media/Matrigel volumes as needed for alternate plate layouts. Final volume per well is 20 μL of a 70% media / 30% Matrigel mix with a concentration of 100 cells/μL.
-
44.
Determine the volume needed for 790,000 cells and transfer cells to a new 15-mL, low-binding centrifuge tube.
-
45.
Aliquot as many batches of cells as there are drug assays (plates) to set up.
-
46.
Bring the volume of media to 5,530 μL using ice cold TO growth media. The growth media should be the same type as the TOs are cultured in normally.
-
47.
Keep the 15-mL tubes of cells on ice. This is important so that the Matrigel does not polymerize when added to the cell suspension.
-
48.
Add 2,370 μL of Matrigel to each cell suspension and mix thoroughly by pipetting.
Note: Matrigel is highly viscous. Pipette slowly and ensure the appropriate volume is transferred.
-
49.
Plate cells using a liquid handling machine, in this case we used the Integra Assist Plus Pipetting Robot with Integra Voyager electronic pipette. Diluting the Matrigel to 30% in cold media allows for repeat dispensing of the TOs to the plate.
Note: If Matrigel is solidifying in the pipette tips, change tips as needed.
-
50.
After the Integra has finished loading the plate, check the plate to ensure that all wells have received cells. If any do not, manually add 20 μL of cell suspension to the well.
Note: This should only be an issue if the target height of the pipette is incorrect. The dispensing pipette tip should be close enough to the well bottom that the cell suspension touches but the tip itself should not touch the well bottom. The target height for the Integra Assist Plus is 0.6 mm above the well bottom in this system. Settings may vary with different plate brands and pipette types. Adjust the target height as needed to ensure consistent and accurate pipetting.
-
51.
Pipette 80 μL of PBS to each well of columns 1 and 24 so that there are no empty wells. If a different plate layout is used, add PBS to all wells immediately surrounding those with cells.
-
52.
Cover the plate with a Breathe-Easy sealing membrane, replace the plastic plate cover, and move the plate to the incubator. Breathe-Easy sealing membranes help to prevent evaporation and may mitigate some edge effects.
Preparation of the drug library
-
53.
Locate the appropriate drug aliquots needed for the drug library. All drug aliquots should be stored as recommended by the manufacturer. Most will be stored at −80°C. Thaw the drug aliquots at room temperature.
Note: Before thawing, make sure each drug is at the correct concentration needed.
-
54.
While the drugs are thawing, put a Bio-Rad Hard shell 96-well plate in the biosafety cabinet and label with the project name, date, expiration date (1 month after preparation date) and drug volume.
Note: For a typical drug assay, 20 μL of drug in each well is sufficient.
-
55.
Once the drugs are thawed, pipet the appropriate volume of drug in the correct well. Be sure to carefully match each drug location with the appropriate drug library plate map.
Note: If the drug library is to be used more than once, mark the side of the plate with a tally to keep track of the freeze thaw count and expiration date.
-
56.
When all drugs are aliquoted in the correct wells, tightly seal an AlumaSeal II on the top of the plate.
-
57.
When done, store the drug library at −20°C or −80°C depending on vendor recommendation.
Preparation of serial dilutions
-
58.Determine how much of each reagent is needed based on the number of 384-well assay plates that require compound addition.
-
a.Reagents needed:
-
i.Growth media.
-
ii.Caspase-3/7.
-
iii.DMSO.
-
iv.Appropriate drug aliquots at the correct concentration.
-
v.Staurosporine.
-
i.
-
a.
Note: 13.5 mL of growth media per 384-well assay plate will be needed.
One 384-well setup plate is enough for every two 384-well assay plates 50-mL conical tubes may be used to mix growth media with IncuCyte® Caspase-3/7 Green Apoptosis Assay Reagent (Essen BioScience #4440). Media of the same type can be added to the same 50-mL conical tube.
-
59.
Pipette the appropriate amount of growth media into 50-mL conical tubes for all 384-well assay plates. Growth media should be the same media type in which the TO line is normally grown and should be consistent with the media used to plate the TO lines in the drug assay plate.
-
60.
Once the appropriate amounts of media aliquots are made, add Caspase-3/7 reagent to media at a 2× dilution of 2.5 μM (e.g., 6.75 μL / 13.5 mL growth media).
-
61.
After diluting Caspase-3/7 reagent in media, aliquot enough of the mixture to use for controls. For each assay plate, take 500 μL of Caspase-3/7 and media mixture for 0.1% DMSO vehicle control wells and 500 μL of the mixture for 10 μM staurosporine control wells. Controls will be diluted 2× in the final assay plate. Generate enough 2× concentration controls for all assay plates as described below.
Note: 0.2% DMSO vehicle control: Add 1.0 μL of DMSO to 500 μL of Caspase-3/7 and media mix.
20 μM staurosporine: Add 1.0 μL of 10 mM staurosporine to 500 μL of Caspase-3/7 and media mix.
Note: High-quality controls are necessary to assess the reliability of the assay. Quality control metrics such as Z′-factor and coefficient of variance are important to determining the quality of each assay (Figure 3).
-
62.
After generating controls, use the remaining media / Caspase-3/7 mix for the generation of drug compound dilutions. Integra Assist Plus system distributes media to 384-well setup plates.
-
63.
Add 25–30 mL of media/Caspase-3/7 mix to the reservoir (or whatever remains if only one assay plate is used/remains).
-
64.
Choose and run the Integra program depending on how many assay plates will have compounds added. Media for one assay plate will be distributed to columns 2–9 of the setup plate and media for the second assay plate will be distributed to columns 17–24 of the setup plate.
Note: 90 μL of media/Caspase-3/7 mix will be pipetted into columns 2–8 and 17–23, and 98 μL of media/Caspase-3/7 mix will be pipetted into columns 9 and 24.
-
65.
Pipette 2 μL of 10 mM drug stock to columns 9 and/or 24 to generate a 200 μM dilution of each compound.
Note: These volumes assume that a high concentration of 10 μM is desired as the highest dose. Adjust as needed.
-
66.Once all drugs have been added, generate a serial dilution.
-
a.Using an 8-channel, 10-μL pipet, set pipet to 10 μL and pipet the media/drug mix, in column 9, up-and-down 10 times to mix the drug.
-
b.Take 10 μL and transfer to column 8 to generate a 10-fold dilution.
-
c.Mix and again transfer 10 μL to column 7.
-
d.Repeat for the next column until the drugs have been diluted throughout all columns.
-
e.Repeat this process for columns 17–24 if needed.
-
f.The serial dilution can also be done using the Integra Assist Plus system.
-
a.
-
67.Distribute 50 μL of control mixes to columns 1 and 16. Be sure to use controls diluted in the correct media type if multiple media types are needed for multiple TO lines.
-
a.Alternate 0.2% DMSO and 20 μL staurosporine controls every other row in rows A-H.
-
a.
-
68.
Transfer 20 μl of controls and each drug dilution to the appropriate well in the 384-well cell plate using an Integra Assist Plus program or by hand.
Figure 3.

High-content imaging controls and quality metrics
(A) Brightfield and fluorescent images of experiment positive and negative controls. Hoechst is used to identify the nuclei of all cells, Caspase-3/7 activated reagent is used to identify apoptotic cells, and TO-PRO-3 is used to identify all dead cells. Scale bar: 200 μm.
(B) Viability values from positive and negative control well replicates are used to assess assay quality by Z′-factor (left) and coefficient of variance (right). Each point represents the Z′-factor or coefficient of variance for each experiment plate from the 351-compound screen described in the companion publication (Larsen et al., 2021).
Fluorescent dye addition to TOs
Fluorescent dyes are added 2 h prior to imaging. In this study we imaged the drug assay plates 72 h post-drug addition.
-
69.
Pre-warm an aliquot of Advanced DMEM/F-12 to room temperature. Determine how much media is needed based on how many wells of the 384-well drug assay plate are to be treated.
Note: If following the standard 384-well layout of all wells in columns 2–23, use 7,740 μL. For custom layouts use 20 μL per well + 700 μL dead volume.
-
70.
Add Hoechst 33342 and TO-PRO-3 to each aliquot of media. A 3× concentration of fluorescent dye solution is made to compensate for the 40 μL volume from cell plating and drug addition currently in each well of the drug assay plate. The concentration of TO-PRO-3 is 900 nM for a final concentration of 300 nM. The concentration of Hoechst is 7.5 μg/mL for a final concentration of 2.5 μg/mL when added to the TO plate.
Note: If following the standard 384-well layout of all wells in columns 2–23 with 7,740 μL media, use 7 μL of 1 mM stock of TO-PRO-3 and 5.8 μL of 10 mg/mL stock of Hoechst 33342.
-
71.
Dispense the total volume of fluorescent dye-containing media to a reservoir and distribute 20 μL to each well using The Integra Assist Plus Pipetting Robot or by hand using a multi-channel pipette.
-
72.
Return plate to incubator for 2 h before imaging.
Imaging the drug assay
-
73.
An IXM Confocal microscope was used to image the 384-well plates. Alternative high-content automated imaging instruments may be used.
Note: The default wavelengths to be acquired in these protocols are brightfield, DAPI, Cy5, and FITC.
-
74.Design an acquisition protocol suitable for imaging TOs in 3-dimensional space with the following considerations:
-
a.Z-series that will encompass most TOs without excessively increasing imaging time by including too many steps.
-
b.Exposure times for each individual fluorophore that maximize signal to noise ratio without saturating the signal.
-
c.Include multiple sites per well to ensure enough TOs are imaged for analysis. A greater number of TOs can improve Z′-factor; however, experiments with as few as 20 TOs per well can consistently produce Z′-factor scores greater than 0.5.
-
d.Magnification for individual cell clarity; a 10× objective is recommended for resolution and throughput.
-
a.
Note: Microscope environmental controls should be used if available for long imaging times to maintain 5% CO2 and 37°C.
-
75.
Test the acquisition settings in multiple wells. Once satisfied, save the acquisition settings to be used for all plates.
-
76.
Run the acquisition protocol on all plates, ensuring that the timing between dye addition and imaging is 2 h.
-
77.
Discard completed plates properly by sealing and disposing in a biohazard waste appropriate for chemotherapy or other compounds used.
Expected outcomes
As described in Larsen et al., vehicle and staurosporine controls should exhibit large dynamic ranges across proliferation, apoptosis, and viability. Technical replicates should have strong pearson correlations and low standard errors. Please refer to Larsen et al., supplementary information for representative data sets and summary statistics for large-scale screens performed in colon and gastric TOs.
Quantification and statistical analysis
-
1.
Confocal images can be analyzed using the MetaXpress software (Molecular Devices) custom module editor feature to design an analysis module that identifies TOs by clusters of Hoechst 33342 staining, individual cells by Hoechst 33342 staining, and dead/dying cells by either TO-PRO-3 or Caspase-3/7 staining overlapping with Hoechst 33342.
-
2.
The parameters for cell segmentation should be set to distinguish individual nuclei based on Hoechst, TO-PRO-3, and Caspase-3/7 staining. Each of these dyes localizes to the nucleus and so the size parameters should match the size of a nucleus in the range of ∼5–20 μm. Intensity thresholds should be optimized based on vehicle and positive controls (e.g., 10 μM staurosporine treatment) and can vary by exposure time. Be sure to test the settings on multiple TO lines and cancer types as these can vary.
-
3.
The percentage of viable cells per TO is calculated based on the image analysis described above.
-
4.
TOs with fewer than three cells by Hoechst 33342 staining, TOs in the top one percent by area, and wells with fewer than 20 TOs detected can be excluded from analysis to ensure only TOs are used for viability calculations and those wells with too few TOs may have an air bubble or similar artifact disrupting calculations.
-
5.
The mean viability for all TOs at a given drug concentration can be used to generate dose-response curves (Figure 4) and to calculate AUC. AUC can be calculated using the computeAUC function using settings for ‘‘actual’’ AUC of the R Package PharmacoGx (v1.17.1).
Alternatives: An alternative software to MetaXpress is CellProfiler. A CellProfiler pipeline can be designed to count the number of cells stained with Hoescht per image. The IdentifyPrimaryObjects module can be used to segment individual cells using global minimum cross-entropy thresholding. Cells can be assigned to TOs by merging and expanding individual cell segmentations within 15 pixels (∼20 μm). All “TOs” with fewer than 3 cells can be excluded from downstream analysis, mirroring the MetaXpress pipeline, in order to remove cells that were dead when plated and never formed a TO.
Alternatives: Train a neural network to assess viability from brightfield (Larsen et al., 2021).
Figure 4.

Quantifying drug effect via high-content imaging dose-response studies
(A) Representative brightfield and fluorescent images of TOs after 72 h of treatment with bortezomib. Hoechst is used to identify the nuclei of all cells, Caspase-3/7 activated reagent is used to identify apoptotic cells, and TO-PRO-3 is used to identify all dead cells. Scale bar: 200 μm.
(B) Dose-response curves of mean ± SEM of technical triplicate wells of the percent viability for all individual TOs after 72 h of treatment with bortezomib. Curves are shown for TO viability indicated by Caspase-3/7 activated reagent (left) and TO-PRO-3 (right).
Limitations
This method can be limited by poorly growing TO lines. Some primary TO lines can be difficult to expand and this method cannot be performed with too few cells. Since the analysis method calculates viability at the TO level, TOs with very few cells can result in decreased dynamic range and inflated viability values.
Another limitation is the time it takes to image a plate. A full 384-well plate can take up to an hour to image at a time. This limits the number of plates that can be assayed at any given time depending on the number of wells per experiment, how many microscopes are available for use, the number of fluorescent channels, Z-steps, etc.
Lastly, TOs at times can exhibit baseline autofluorescence in channels measuring caspase or live and dead cells. In these instances, it is necessary to exclude measurements in these channels or to use alternative vital dye fluorophores.
Troubleshooting
Problem 1
TOs grow slowly. This can result in very small TOs of only a few cells at the time of imaging.
Potential solution
If TOs are slow growing, proceed to propagation using Dispase II or a shorter (5 min) incubation time with TrypLE. This will digest the Matrigel matrix but leave the TOs intact.
Problem 2
TOs are too big and will not dissociate with TrypLE incubation. This results in inaccurate counts and the potential for large TOs to die during the assay unrelated to drug effects.
Potential solution
It is recommended to leave the TOs for approximately 10 min but if they are still not dissociating and have not decreased in size, leaving them in the water bath for an additional 2–5 min can help. After a sufficient waiting time, be sure to physically break TOs using a pipette.
Problem 3
TOs have large clumps after the TrypLE centrifugation step and do not form a compact cell pellet (Figure 2).
Potential solution
It is recommended to use DNase if there are large clumps. Use 10 μL of DNase and fill the 15-mL tube with the pellet with 10 mL of PBS. Spin the cells at 200 g for 5 min in a 4°C centrifuge. Wash one more time with PBS.
Problem 4
Tumor organoids have large clusters even after DNase treatment.
Potential solution
Place a 70-μm cell strainer on a new 15-mL, low-binding centrifuge tube and allow the cell suspension to pass through the strainer to eliminate large cell clusters. Collect any cells remaining in the previous 15-mL, low-binding tube by washing with 1 mL Advanced DMEM/F-12 and transferring through the 70-μm strainer. This will also rinse the strainer of cells stuck to the filter. Centrifuge the cell-containing tube at 200 g for 5 min at 4°C. Pipet out the supernatant and add any media/reagent required for your following procedure.
Problem 5
Some TO lines can exhibit autofluorescence.
Potential solution
In the case of autofluorescence it is critical to have multiple readouts. Testing and having available alternate reagents with different fluorescent wavelengths is useful. Another alternative is using a biochemical reagent such as Promega CellTiter Aqueous or CellTiter-Glo and assessing viability with a microplate reader such as Molecular Devices’ SpectraMax iD3.
Problem 6
Newly derived TO cultures are contaminated with CAFs.
Potential solution
CAFs have the potential to overgrow the TO culture and can be reduced by gravity separation. Allow the TOs to grow to >100 μm in diameter before passaging. This will allow the TOs to separate from the CAFs by gravity. TOs can be released from the Matrigel by incubating the TO culture in Dispase II to a final concentration of 1.5 U/mL at 37°C for 1–2 h. Once TOs have released from the dissociated Matrigel, collect all cells/wells of the culture to a 15-mL tube and allow the TOs to settle to the bottom of the tube before removing the media containing CAFs with a serological pipette. Wash with PBS 2–3 times before replating in Matrigel as any residual Dispase II can degrade the new Matrigel domes.
Resource availability
Lead contact
Any further communication, including those related to reagents or resource sharing, may be directed to and fulfilled by the lead contact Ameen A. Salahudeen (ameen@tempus.com).
Materials availability
There are restrictions to the availability of materials, including models, due to their proprietary nature. Requests are subject to review by Tempus.
Acknowledgments
We wish to thank Amrita Iyer, Matthew Kase, and Alexandria Bobe for critical review of this manuscript. We also wish to thank Evan Gendell and Anna Murnighan for the design and generation of the graphical abstract.
Author contributions
Conceptualization, A.A.S. and B.M.L.; methodology, A.A.S., A.C., B.M.L., and J.M.S.; writing - original draft, A.A.S., A.C., B.M.L., and J.M.S.
Declaration of interests
A.A.S. and B.M.L. are employees and shareholders of Tempus Labs. A.C. and J.M.S. are employees of Tempus Labs. A.A.S. and B.M.L. are inventors on patents related to organoid technology. A.A.S. holds a faculty position at the University of Illinois at Chicago.
Data and code availability
Drug screening source data are available under data use terms through Vivli accession number T21.01.
References
- Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte C.J., Espejo Valle-Inclan J., Hami N., Lohmussaar K., Kopper O., Vreuls C.P.H., Jonges G.N., van Diest P., Nguyen L., Clevers H., et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 2020;31:107762. doi: 10.1016/j.celrep.2020.107762. [DOI] [PubMed] [Google Scholar]
- Driehuis E., Spelier S., Beltran Hernandez I., de Bree R., M Willems S., Clevers H., Oliveira S. Patient-derived head and neck cancer organoids recapitulate EGFR expression levels of respective tissues and are responsive to EGFR-targeted photodynamic therapy. J. Clin. Med. 2019;8:1880. doi: 10.3390/jcm8111880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E., van Hoeck A., Moore K., Kolders S., Francies H.E., Gulersonmez M.C., Stigter E.C.A., Burgering B., Geurts V., Gracanin A., et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. U S A. 2019;116:26580–26590. doi: 10.1073/pnas.1911273116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Li X., Niu Q., Mo X., Qui M., Ma T., Kuo C.J., Fu H. Development of a miniaturized 3D organoid culture platform for ultra-high-throughput screening. J. Mol. Cell Biol. 2020;12:630–643. doi: 10.1093/jmcb/mjaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francies H.E., Barthorpe A., McLaren-Douglas A., Barendt W.J., Garnett M.J. Drug sensitivity assays of human cancer organoid cultures. Methods Mol. Biol. 2019;1576:339–351. doi: 10.1007/7651_2016_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman J.E., Muthuswamy L., Huang L., Akshinthala D., Perea S., Gonzalez R.S., Tsai L.L., Cohen J., Bockorny B., Bullock A.J., et al. Organoid sensitivity correlates with therapeutic response in patients with pancreatic cancer. Clin. Cancer Res. 2021;28:708–718. doi: 10.1158/1078-0432.ccr-20-4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen B.M., Kannan M., Langer L.F., Leibowitz B.D., Bentaieb A., Cancino A., Dolgalev I., Drummond B.E., Dry J.R., Ho C.S., et al. A pan-cancer organoid platform for precision medicine. Cell Rep. 2021;36:109429. doi: 10.1016/j.celrep.2021.109429. [DOI] [PubMed] [Google Scholar]
- Narasimhan V., Wright J.A., Churchill M., Wang T., Rosati R., Lannagan T.R.M., Vrbanac L., Richardson A.B., Kobayashi H., Price T., et al. Medium-throughput drug screening of patient-derived organoids from colorectal peritoneal metastases to direct personalized therapy. Clin. Cancer Res. 2020;26:3662–3670. doi: 10.1158/1078-0432.ccr-20-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooft S.N., Weeber F., Dijkstra K.K., McLean C.M., Kaing S., van Werkhoven E., Schipper L., Hoes L., Vis D.J., van de Haar J., et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019;11:eaay2574. doi: 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics. 2021;22:433. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensink G.E., Elias S.G., Mullenders J., Koopman M., Boj S.F., Kranenburg O.W., Roodhart J.M.L. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis Oncol. 2021;5:30. doi: 10.1038/s41698-021-00168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Drug screening source data are available under data use terms through Vivli accession number T21.01.