

Abstract
Cancer progression is driven by both somatic copy number aberrations (CNAs) and chromatin remodeling, yet little is known about the interplay between these two classes of events in shaping the clonal diversity of cancers. We present Alleloscope, a method for allele-specific copy number estimation that can be applied to single-cell DNA and/or ATAC sequencing data, enabling combined analysis of allele-specific copy number and chromatin accessibility. On scDNA-seq data from gastric, colorectal, and breast cancer samples, with validation using matched linked-read sequencing, Alleloscope finds pervasive occurrence of highly complex, multi-allelic copy number aberrations, where cells that carry varying allelic configurations adding to the same total copy number co-evolve within a tumor. On scATAC-seq from two basal cell carcinoma samples and a gastric cancer cell line, Alleloscope detects multi-allelic copy number events and copy neutral loss-of-heterozygosity, enabling dissection of the contributions of chromosomal instability and chromatin remodeling to tumor evolution.
Introduction
Cancer is a disease caused by genetic alterations and epigenetic modifications which, in combination, shape the dysregulated transcriptional programming of tumor cells1, 2. These somatic genomic events lead to a diverse cellular population from which clones with advantageous alterations proliferate and metastasize3. Comprehensive study of cancer requires the integrative profiling of genetic and epigenetic changes at single-cell resolution. We consider here the analysis of two such genomic dimensions – DNA copy number and chromatin accessibility – through massively parallel single-cell sequencing assays.
First, consider copy number aberrations (CNAs), through which we have derived much of our current understanding of the relationship between genome instability and tumor evolution4. Total copy number profiling, which estimates the sum of the copy numbers of the two homologous chromosomes, is inadequate to characterize some types of cancer genomic aberrations. Such events include the pervasive copy-neutral losses of heterozygosity (LOH)5–8 and the intriguing “mirrored events”9, 10, where cells carrying amplification of one haplotype are intermingled with cells carrying amplification of the other haplotype. Although the importance of allele-specific copy number profiling has been emphasized in bulk DNA sequencing analysis5–8, 11, most single-cell studies profile only total copy number due to the difficulty of reliable allele-specific estimation at low per-cell coverage12–19. Recently, Zaccaria and Raphael developed CHISEL10, a method for single-cell allele-specific copy number analysis that relies on externally phased haplotypes. This was the first time that allele-specific of CNAs were reported at single-cell resolution, but the requirements for high sequencing depth and external phasing limit the general applicability of CHISEL. Thus, the prevalence of mirrored subclones and the allelic complexity of CNA regions at single-cell resolution remain largely unexplored.
Now consider the profiling of chromatin accessibility20, 21 in the study of epigenetic plasticity of cancer cells22–26. Because copy number alterations involve large gains and losses of chromatin, we expect a region’s accessibility to be confounded by changes in its underlying copy number. Current single cell transposase-accessible chromatin sequencing (scATAC-seq) analyses estimate the total copy number profile of each cell by smoothing the read coverage across large bins, normalizing to a control cell population23, 25. Yet, the appropriate control is difficult to identify, as chromatin remodeling can lead to broad shifts in coverage that are unrelated to underlying copy number. There is yet no reliable method for total or allele-specific copy number profiling in scATAC-seq data, and thus, CNA and chromatin accessibility are confounded in current analysis pipelines.
Addressing these challenges, we present Alleloscope, a method for allele-specific copy number estimation and multiomic profiling in single cells. Alleloscope can be applied to low coverage scDNA-seq data or to scATAC-seq data with sample-matched bulk DNA sequencing data, and does not require external haplotype phasing. To investigate the allelic complexity of CNAs in cancer at single-cell resolution, we first apply Alleloscope to scDNA-seq data from four gastric cancer samples, four colorectal cancer samples, and two different regions of a breast cancer sample10, 12, 27. For five of the gastrointestinal cancer samples, results are validated by 10x linked-read sequencing which provides accurate phasing information28–30. In these datasets, Alleloscope finds pervasive occurrence of mirrored subclones and other highly complex multi-allelic CNA loci where cells carrying distinct allelic configurations adding to the same total copy number co-evolve within a tumor. The ubiquity of such events in all three cancer types analyzed reveals that they may be a general source of intratumor genetic heterogeneity. We then turn to scATAC-seq data from two basal cell carcinoma samples with paired bulk whole exome sequencing data25 and a complex polyclonal gastric cancer cell line that we also analyzed by scDNA-seq. In these samples, we evaluate the accuracy of Alleloscope in genotyping and subclone assignment for scATAC-seq data and demonstrate its application to the integrative analysis of CNA and chromatin accessibility.
Results
Overview of Alleloscope model and algorithm
We first describe allele-specific copy number estimation (Fig. 1). Integration with scATAC-seq peak signals will be described later. Consider the two parental haplotypes, we call the one with higher count across cells the “major haplotype”, and the other the “minor haplotype”. Note that, within any single cell, it is possible for the major haplotype to have fewer copies than the minor haplotype. For each cell i and genome region r, we define two key parameters: (1) the major haplotype proportion (θir) is the copy number of the major haplotype divided by the total copy number and (2) the total copy fold change (ρir) is the ratio of the total copy number relative to that in normal cells.
Fig. 1: Overview of allele-specific copy number estimation of single-cells with Alleloscope.
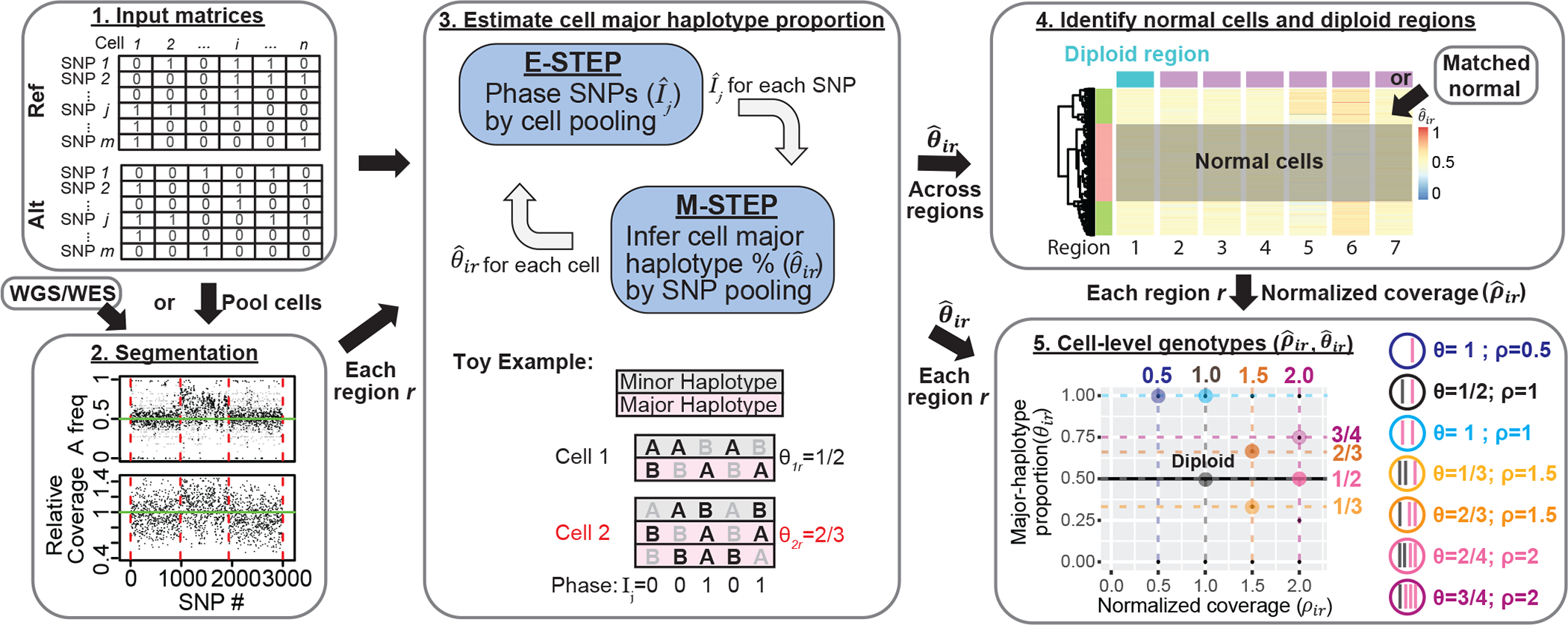
1. The algorithm operates on raw read count matrices for reference allele (Ref) and alternative allele (Alt) computed from single-cell DNA or ATAC sequencing. 2. First, we obtain a segmentation of the genome based on sample-matched whole genome or whole exome sequencing data using FALCON5. If scDNA-seq is available, cells can be pooled to derive a pseudo-bulk. 3. We first define as the major haplotype proportion for cell i for region r derived from the segmentation. Then for each region r, Alleloscope simultaneously phase SNPs () and estimate cell major haplotype proportion () by expectation maximization (EM) algorithm. In the E-step, information is pooled across cells to estimate the phasing of each SNP. In the M-step, information is pooled across all SNPs in the region are pooled to estimate the major haplotype proportion for each cell. The toy example shows a scenario with two cells for a region containing 5 SNPs, with cell 2 carrying an amplification of the major haplotype (in pink). For each cell and each SNP, alleles that are observed in a sequenced read are bolded in black (we assume that only one read is observed, reflecting the sparsity of the data). The true phase (Ij) of the SNPs and the true major haplotype proportion (θir) are shown. 4. After are estimated for each region ’s are pooled across all regions to identify candidate normal cells and candidate normal regions for computing a normalized coverage for region r in cell i. Matched normal can be specified if available. 5. Alleloscope assigns integer allele-specific copy numbers to each cell for each region based on the () pairs.
Alleloscope starts by segmenting the genome into regions of homogeneous population-average allele-specific copy number using the bulk coverage and variant allele frequency (VAF) profiles, derived from pooling reads across cells (Methods). For scATAC-seq data, we recommend that the segmentation be performed on matched bulk or single-cell DNA-seq data, which ensures that the characterized CNA regions are supported by evidence from DNA.
An expectation-maximization (EM) algorithm is then applied separately for each region r to phase the SNPs in the region and estimate its major haplotype proportion (θir) for each cell (Fig. 1, Step 3). For each SNP j in region r, let Ij ∈ {0,1} be the indicator of whether the reference allele of j is carried by the major haplotype, see example in Fig. 1. An initial estimate is first derived from the bulk VAF profile. Then, in iteration t of EM, Alleloscope computes by pooling counts across SNPs within the region for cell i, weighted by the current phasing , then updates the estimate of Ij by pooling across cells. The estimates of θir and Ij usually converge within a few iterations. If matched scDNA-seq data are available for a sample sequenced by scATAC-seq, Ij can be estimated from scDNA-seq and then used to compute θir for each cell in the scATAC-seq data, enabling more information sharing between the two data types.
The estimated major haplotype proportions (’s), along with a preliminarily normalized coverage statistic (’s; Methods), are then used to identify a set of normal cells and diploid regions (Fig. 1, Step 4). These control cells and regions are then used to obtain an improved coverage fold-change () for each cell and each region. If cell i’s true allele-specific copy numbers are homogeneous within the given region r, then its true value of (θir, ρir) should belong to the set of canonical points displayed in Step 5 of Fig. 1. Thus, the estimated () are clustered across cells and associated with one of the canonical values to yield the cell-level haplotype profiles for region r. These cell- and region-specific haplotype profiles serve as the basis for clone assignment and subsequent integration with peak signals in scATAC-seq data.
To improve sensitivity for the most difficult scenario of balanced mirrored subclones at small clonal frequencies, Alleloscope allows an additional refinement step to improve the estimation of , see Supplementary Methods.
Validation using matched linked-read sequencing
First, we characterize the allelic heterogeneity of CNA events in 8 gastrointestinal tumors and 2 sections of a breast tumor (Supplementary Table 1) by applying Alleloscope to the scDNA-seq data for these samples. To benchmark accuracy, we performed matched linked-read whole-genome sequencing on five of the gastrointestinal tumor samples that represent different levels of chromosomal instability. In linked-read sequencing, barcoded reads are derived from individual high molecular weight DNA molecules, allowing the reconstruction of haplotypes covering megabases in length28–30. We compared the haplotypes estimated by Alleloscope to the haplotypes obtained from matched linked-read WGS to evaluate phasing accuracy. Then, we used the linked-read haplotypes with Alleloscope to derive gold-standard allele-specific copy number estimates for each cell, which are compared to those derived without the linked-read haplotypes (Fig. 2a). This allows us to compute the sensitivity and specificity of our method (Supplementary Methods).
Fig. 2: Validation of the Alleloscope results on the P5931 gastric cancer patient sample and linked-read sequencing data.
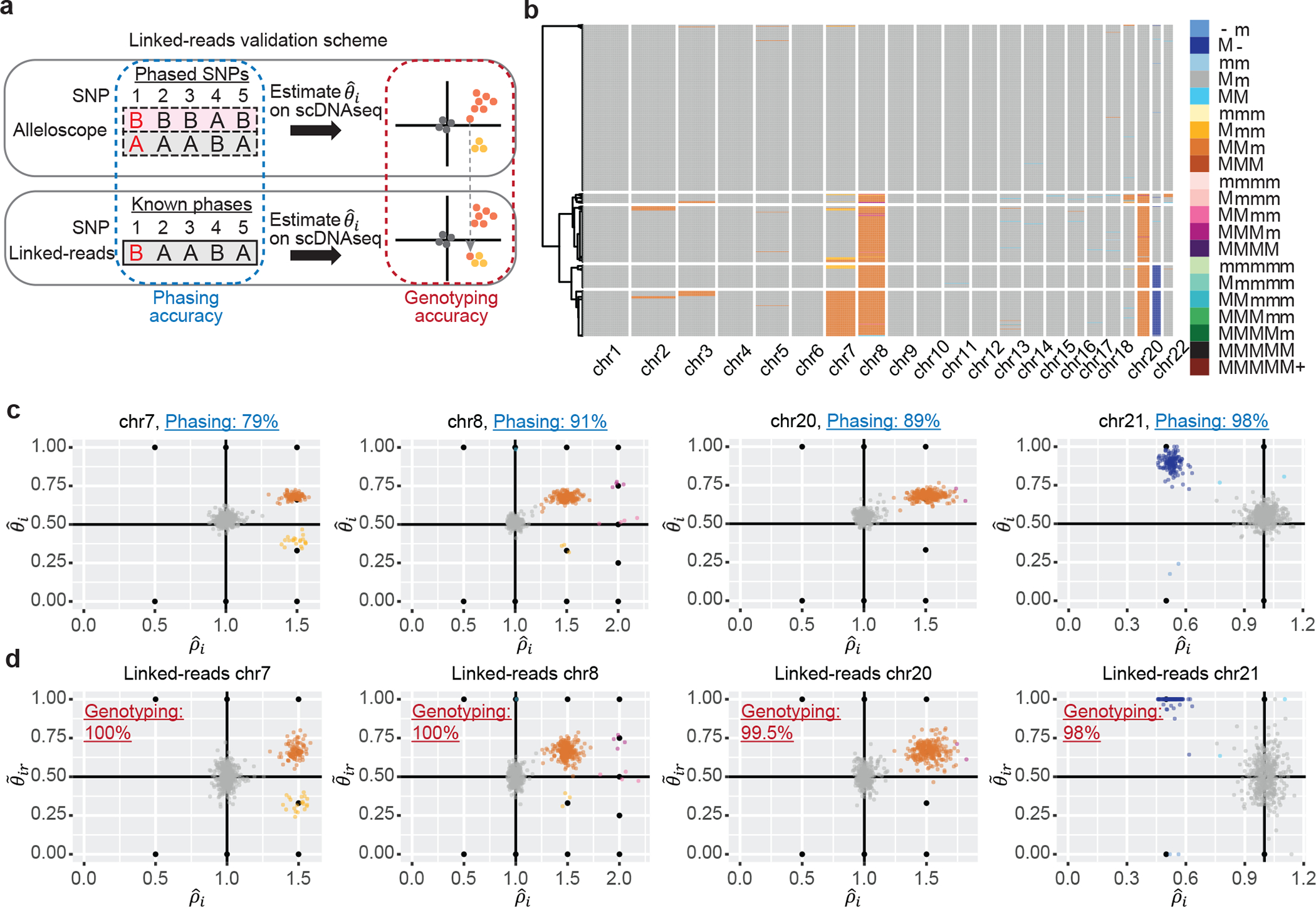
(a) Illustration of the validation scheme using linked-read sequencing data. Phasing accuracy and accuracy in allele-specific copy number state estimation are used to access performance of the method. (b) Hierarchical clustering of cells in the P5931t sample based on allele-specific copy numbers given by Alleloscope, showing normal cells and 4 main clones, as well as a number of small clones marked by highly confident low-frequency mutations. M: Major haplotype, m: minor haplotype. (c) estimated by Alleloscope for four regions, colored by the inferred haplotype profile. Note that clusters fall on canonical points corresponding to discrete allele-specific copy number configurations. Phasing accuracy for each region is shown in the plot title. In the color legend, M and m represent the “Major haplotype” and “minor haplotype” respectively. (d) Similar to (c), with estimated using known SNP phases from matched linked-read sequencing data, colored by the haplotype profiles assigned in (c) using Alleloscope without the given phasing information. Accuracy in allele-specific copy number state estimation (Genotyping accuracy) is labeled in the plots.
As illustration, consider the gastric cancer sample P5931. The heatmap in Fig. 2b gives a bird’s-eye-view of Alleloscope’s estimated profile for this sample, showing it to have a relatively simple genome with few CNAs, which is expected given this tumor’s MSI status. However, detailed inspection of the four chromosomes carrying clear CNA events, chr7, chr8, chr20, and chr21, reveals surprising complexity at the allelic level. For each event, scatter plots of , colored by haplotype-specific CNA state, are shown in Fig. 2c. Reassuringly, clusters align with the canonical values (e.g. (1/2, 1) for diploid, (2/3, 1.5) for 1 copy major haplotype gain). For chromosomes 7, 8, and 21, we find subclonal clusters differentiated by allelic ratios that would be masked if we were to consider only total copy number. Let “M” be major and “m” be minor haplotype. The chromosome 7 amplification exhibits two mirrored subclones with allelic configurations MMm and Mmm. Chromosome 21 exhibits mirrored subclonal deletions (M- and m-). Four subclones are revealed by the chromosome 8 amplification: MMm, Mmm, MMmm, and MMMm.
Comparing against the haplotypes derived from linked-reads, the phasing accuracy is 98% for the chr21 deletion, ~90% for the two clonal amplifications (on chr8 and chr20), and 79% for the subclonal chr7 amplification. Fig. 2d shows scatterplots of against major haplotype proportion computed using linked-read haplotypes (), with cells colored by their state assignment from Fig. 2c (i.e. estimated without the linked-read information). Comparing Fig. 2d to Fig. 2c reveals that Alleloscope’s allele-specific copy number state assignments, derived without external phasing information, are almost exactly concordant (~ 100%) with those derived using linked-read haplotypes. This shows that allele-specific copy number estimation in Alleloscope is, to a degree, robust to errors in phasing.
On the detection of rare mirrored subclones, Alleloscope (with the refinement step) revealed an amplification distinguishing two mirrored subclones each comprising <10 out of 700 cells on chr2 and chr3 of this sample; both events were confirmed by estimation using linked-read haplotypes (Supplementary Fig. 1).
CHISEL is the only other method for allele-specific copy number estimation with scDNA-seq data. Thus, we benchmarked Alleloscope against CHISEL by comparing each method’s estimates under default settings to the same method’s estimates using the true haplotypes provided by linked-read WGS. Thus, each method’s performance is assessed using its own “gold standard” computed with linked-read haplotypes. The sensitivity and specificity of both methods on each of the five samples are given in Table 1. Overall, Alleloscope maintains high sensitivity and specificity across all samples. Varying results obtained using different external phasing datasets and different block sizes for CHISEL suggest that CHISEL’s accuracy depends on choices for these inputs (Supplementary Table 2; Supplementary Fig. 2–3). More discussion on this benchmark study is given in Supplementary Results.
Table 1.
Comparison between Alleloscope and CHISEL using two benchmark strategies.
| Dataset | Method | Sensitivity | Specificity |
|---|---|---|---|
|
| |||
| Benchmark with matched linked-read sequencing data | |||
|
| |||
| P5915 | Alleloscope | 0.9373 | 0.9915 |
| CHISEL (before correction) | 0.7284 | 0.9757 | |
| CHISEL (after correction) | 0.7786 | 0.9722 | |
| P5931 | Alleloscope | 0.9402 | 0.9986 |
| CHISEL (before correction) | 0.7520 | 0.9434 | |
| CHISEL (after correction) | 0.0112 | 0.9508 | |
| P6198 | Alleloscope | 0.9433 | 0.9666 |
| CHISEL (before correction) | 0.9397 | 0.9311 | |
| CHISEL (after correction) | 0.9700 | 0.9359 | |
| P6335 | Alleloscope | 0.9671 | 0.9906 |
| CHISEL (before correction) | 0.7858 | 0.9873 | |
| CHISEL (after correction) | 0.8404 | 0.9943 | |
| P6461 | Alleloscope | 0.8638 | 0.9904 |
| CHISEL (before correction) | 0.7932 | 0.9624 | |
| CHISEL (after correction) | 0.8143 | 0.9411 | |
|
| |||
| Benchmark with subsampling | |||
|
| |||
| 50% subsample | Alleloscope | 0.9261 | 0.9905 |
| CHISEL (before correction) | 0.9267 | 0.9383 | |
| CHISEL (after correction) | 0.9496 | 0.9446 | |
| 25% subsample | Alleloscope | 0.8249 | 0.9525 |
| CHISEL (before correction) | 0.6515 | 0.6412 | |
| CHISEL (after correction) | 0.6533 | 0.6475 | |
Performance assessment via data downsampling and simulations
We also compared Alleloscope and CHISEL on the breast cancer sample that was analyzed by Zaccaria and Raphael10. This sample does not have true phasing information, but was sequenced at much higher coverage, and thus, we downsampled the reads to 50% and 25% of the original depth and compared the estimates obtained from the downsampled datasets to those obtained from the original dataset (Supplementary Results and Methods). At 25% of the original depth, Alleloscope has specificity of 82% as compared to CHISEL’s 65%, and sensitivity of 95% as compared to CHISEL’s 64% (Table 1) with the detailed heatmaps of the genome-wide profiles shown in Supplementary Fig. 4.
Finally, we used simulations to investigate the performance of Alleloscope over a grid of experimental parameters (number of cells, total per cell coverage, and total coverage at heterozygous SNP sites, Supplementary Fig. 5). Across settings, Alleloscope attains higher accuracy for deletions than for amplifications, which is expected given that the change in both coverage ratio and major haplotype proportion () is bigger for deletions. SNP phasing within deletions (and other LOH events) is accurate across the board, but for amplifications it becomes inaccurate at 10% and 5% clonal frequencies. CNA state assignment accuracy is, to some extent, robust to phasing error, and increases steadily with coverage, number of heterozygous SNPs, and number of cells.
Pervasive occurrence of subclonal allele-specific CNAs
Alleloscope allows a comprehensive survey of the landscape of single-cell allele-specific copy number profiles for the samples analyzed (Supplementary Table 1, Supplementary Figs. 6–12). First consider copy-neutral LOH events, which are well-known drivers of cancer evolution that can only be identified through allele-specific copy number analysis. Figure 3a shows an example of a sample (P6198) which, according to 10x CellRanger, has whole genome duplication, but several copy-neutral regions as marked. Consider chromosome 5, which seems to be copy neutral for its entire length, yet bulk VAF clearly reveals a LOH in the q arm (Fig. 3b). Concordantly, Alleloscope reveals a cluster centered at (ρ, θ) = (1,1), the canonical point for copy-neutral LOH, for this region (Fig. 3b). All copy neutral LOH events identified by Alleloscope in this sample (chr5, chr11, chr16) are validated by matched linked-read WGS (Supplementary Fig. 13).
Fig. 3: Across multiple cancer types, Alleloscope detects loss-of-heterozygosity events and multi-allelic copy number aberrations, delineating complex subclonal structure which are invisible to total copy number analysis.
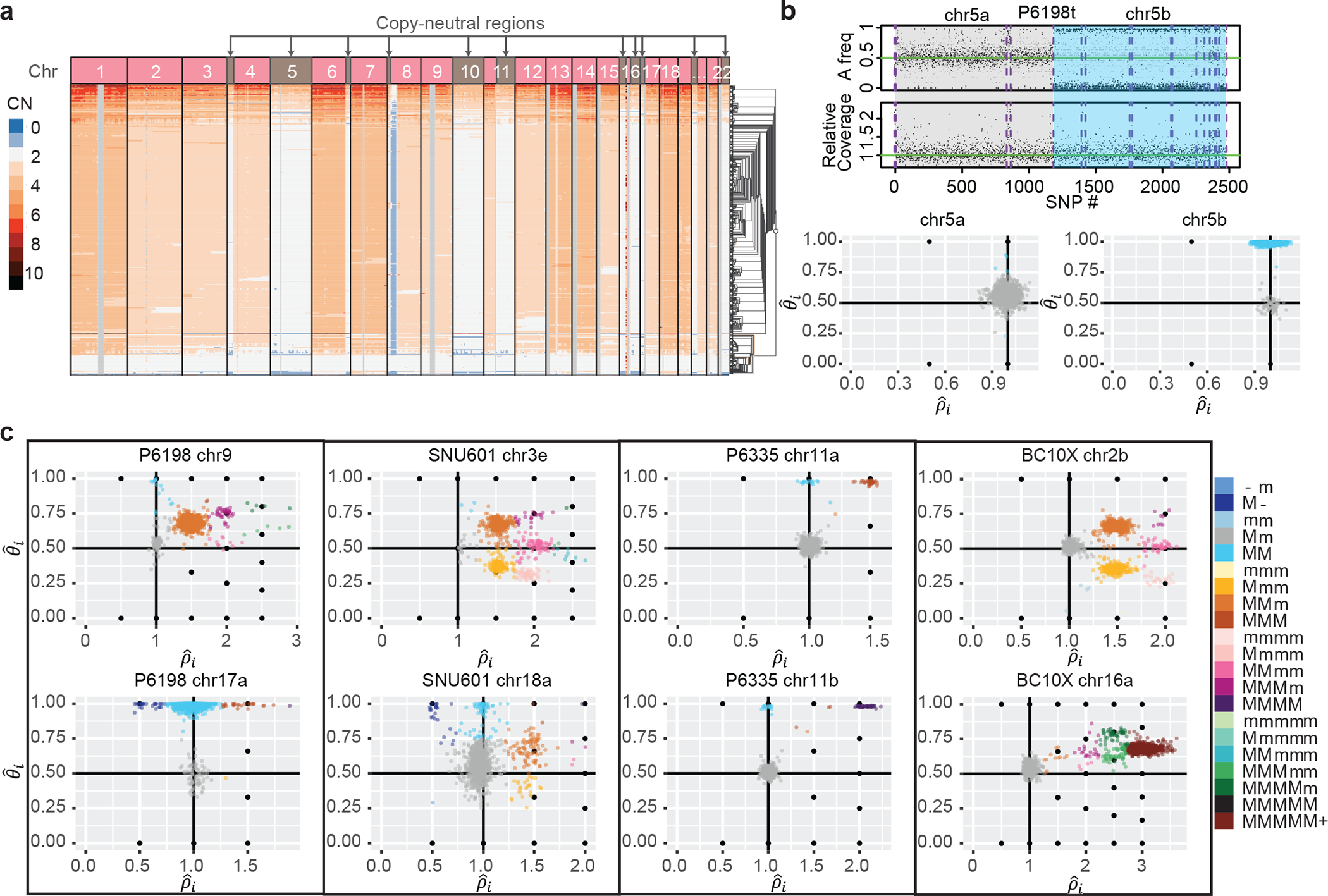
(a) The Cell Ranger hierarchical clustering result for P6198t with copy-neutral regions labeled (total 512 cells). (b) Top: FALCON segmentation of P6198 chr5 into two regions with different allele-specific copy number profiles. Bottom: Detailed haplotype profiles of the two regions from Alleloscope, showing that the first region is diploid across cells and the second region has a loss-of-heterozygosity for a subpopulation of cells. The a and b following the chromosome number denote two ordered segments. (c) Single-cell allele-specific estimates , colored by assigned haplotype profiles, for select regions in the samples P6198t (metastasized colorectal cancer sample), SNU601 (gastric cancer cell line), P6335 (colorectal cancer sample), and BC10X (breast cancer cell line). In the color legend, M and m represent the “Major haplotype” and “minor haplotype” respectively. The lower-case letters following the chromosome number in the titles denote the ordered genomic segments.
Beyond copy-neutral LOH, we observe across samples a high prevalence of complex subclonal CNAs. Prototypical examples from P6198, SNU601, P6335 and BC10X are shown in Fig. 3c. In some regions, such as P6198 chromosome 9, SNU601 3q, BC10× 2q and 16p, allelic ratios differentiate as many as seven subclonal clusters. Many such clusters vary only in allelic dosage, not in total copy number. We find instances of minor subclones carrying deletion of one haplotype coevolving with larger subclones carrying amplification of the other haplotype; such cases would be missed by bulk-tissue analyses or by single-cell analyses using total coverage alone.
Recurrent chromosomal alterations affecting both haplotypes and producing gradients in haplotype dosage are common across the samples. Consider, for example, the region on chromosome 9 of P6198 (Fig. 3c), which reveals 7 subclones: besides the normal cell cluster and the dominant tumor cell cluster with profile MMm, there is a small cluster with copy neutral LOH (MM), two small clusters with 4 total copies (MMmm, MMMm) and two more with 5 total copies (MMMmm, MMMMm). This produces clones carrying major haplotype ratios that vary across the gradient . Other examples of such complexity include chromosome 3q of SNU 601 and chromosome 2q of BC10x, also shown in Figure 3c. Interrogating the evolutionary route by which such diversity was achieved, Alleloscope reveals that a whole genome doubling was likely in the histories of BC10x and P6198, but not in the history of SNU601 (Supplementary Fig. 9&12). Thus, the subclones at 2q in BC10x and 3q in SNU601 were likely achieved through different evolutionary routes: In BC10x, whole-genome doubling produced the MMmm subclone, from which the other subclones likely followed through successive loss and gene conversion events. On the contrary, the clusters on 3q of SNU601 were most likely produced by successive amplifications starting from the diploid genome. Larger studies involving more samples would be needed to fully elucidate the evolutionary dynamics of these complex regions.
Analysis of scATAC-seq with matched bulk WGS
To demonstrate the analysis of scATAC-seq data with Alleloscope, we first consider two basal cell carcinoma samples with matched whole-exome sequencing (WES) data31. Using the matched WES data, the genome of each sample was first segmented into regions of homogeneous bulk copy number (Fig. 4a, middle panel shows the segmentation for SU008). For each cell in the scATAC-seq data, Alleloscope then estimates the allele-specific copy number in each region. Scatterplots of for five example CNA regions and one control region (chr12) from SU008 are shown in Fig. 4a. For this sample, a standard peaks-based Uniform Manifold Approximation and Projection (UMAP) embedding separates the cells confidently into three clusters: 308 tumor cells, 259 fibroblasts and 218 endothelial cells. Density contours for each cell type are shown in the - scatterplots (Fig. 4a). For each CNA region, Alleloscope’s -plot clearly separates the tumor cells from the normal fibroblast and endothelial cell populations, with the tumor cells clustered around canonical points. This indicates that Alleloscope can accurately distinguish amplifications and loss-of-heterozygosity events in low-coverage scATAC-seq data. In particular, Alleloscope differentiated the cells that carry the putative copy-neutral LOH events in regions 4a, 6b, and 15b through shifts in major haplotype proportion. Note that normal cells, which are not expected to carry chromosome-scale CNAs, exhibit chromosome-scale shifts in total coverage due to broad chromatin remodeling but no significant difference in , as exemplified by the chr6b region. Even when comparing one normal cell type (fibroblasts) against another (endothelial cells), total coverage can exhibit chromosome-scale shifts (Supplementary Fig. 14; Supplementary Methods). This underscores the fact that relying solely on shifts in coverage, without complementary changes in major haplotype proportion, is unreliable for copy number estimation in scATAC-seq data.
Fig. 4: Alleloscope multiomic analysis of scATAC-seq data of a basal cell carcinoma sample (SU00825).
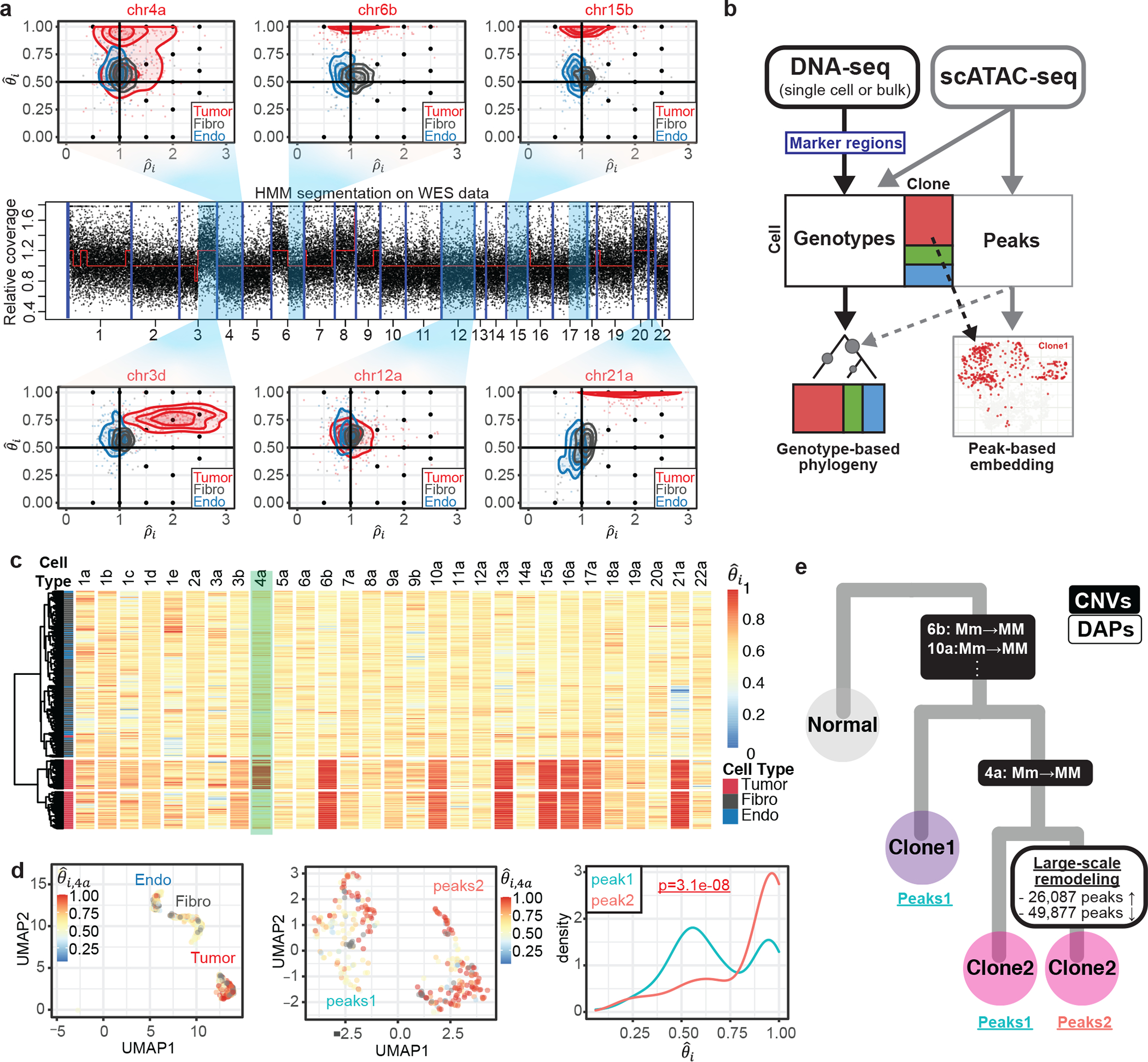
(a) Genotype profiles for six example regions for cells in scATAC-seq data. The regions are taken from segmentation of matched whole exome sequencing (WES) data. Each dot represents a cell-specific () pair. Cells are colored by annotation derived from peak signals25, Tumor: tumor cells, Fibro: fibroblasts, Endo: Endothelial cells]. Density contours are computed for each cell type (tumor, fibroblasts, endothelial) separately and shown by color on the plot. The lower-case letters following the chromosome number in the titles denote the ordered genomic segments. (b) Pipeline for multi-omics analysis integrating allele-specific copy number estimates and chromatin accessibility peak signals on ATAC-seq data. (c) Hierarchical clustering of cells by major haplotype proportion () allows the separation of tumor cells from normal cells, as well as the differentiation of a subclone within the tumor cells. The marker region on chr4a separating the two tumor subclones is highlighted. (d) Integrated visualization of chr4a major haplotype proportion () and genome-wide peak profile. Left: UMAP projection of the 788 cells in the dataset by their genome-wide peak profile, colored by . The cell type annotation (endothelial, fibroblasts, and tumor cells) is labeled in the plot. Middle: UMAP projection of only the 308 tumor cells by their genome-wide peak profile shows two well-separated clusters: peaks1 and peaks2. Right: Density of values for the peaks1 and peaks2 subpopulations. (e) Intratumor heterogeneity of SU008 is shaped by a subclonal LOH of chr4a followed by subsequent genome-wide chromatin remodeling leading to three subpopulations: Clone 1 which does not carry the chr4a LOH (peaks cluster 1), Clone 2 carrying the chr4a LOH (peaks cluster 1), and remodeled clone 2 (peaks cluster 2).
By assigning allele-specific CNA profiles to single cells in scATAC-seq data, Alleloscope allows the integrative analysis of copy number and chromatin accessibility as follows (Fig. 4b): The scATAC-seq data, paired with bulk or single-cell DNA sequencing data, allows us to detect subclones. In parallel, a peak-by-cell matrix can be generated following standard pipelines. Then, subclone memberships or CNA profiles can be visualized on the low-dimensional embedding of the peak matrix, and, conjointly, peak and transcription factor motif enrichment can be quantified at splits along the clonal tree. Alleloscope also allows identification of significantly enriched/depleted peaks after accounting for copy number differences, thus delineating events that are uniquely attributable to chromatin remodeling.
Hierarchical clustering using major haplotype proportion identifies the tumor cells from the normal cells for both SU006 (Supplementary Fig. 15) and SU008, and clearly delineates a subclone in SU008 marked by a copy-neutral LOH event on chr4a (Fig. 4c). Focusing on SU008, we call the cell lineage that carries the chr4a LOH event clone-2, and the other lineage clone-1. In parallel, clustering by peaks cleanly separates the tumor cells from the epithelial cells and fibroblasts (Fig. 4d: left), and further, demarcates two distinct clusters in the tumor cells (peaks-1 and peaks-2) (Fig. 4d: middle). Coloring by chr4a major haplotype proportion () on the peaks-derived UMAP shows that the LOH in this region is carried by almost all of the cells in peaks-2 but only a subset of the cells in peaks-1 (Fig. 4d: middle). This can also be clearly seen in the density of (Fig. 4d: right): While is heavily concentrated near 1 for peaks-2, it is bimodal for peaks-1. Because clone-1 and clone-2 are differentiated by a copy-neutral event, this separation by peaks into two clusters is not driven by broad differences in total copy number. Since clone-2 is split into two groups of distinct peak signals, we infer that the chromatin remodeling underlying the divergence of the peaks-2 cells is likely to have occurred in the clone-2 lineage, after the chr4a LOH event (Fig. 4e). In this way, Alleloscope analysis of this scATAC-seq dataset allowed us to overlay two subpopulations defined by peak signals with two subpopulations defined by a subclonal copy-neutral LOH.
Integrative analysis of a complex polyclonal population
The gastric cancer cell line SNU601 exhibits complex subclonal structure, as evidenced by multiple multiallelic CNA regions (chr3e and chr18a shown in Fig. 3c). In addition to scDNA-seq, we also performed scATAC-seq on this sample to profile the chromatin accessibility of 3,515 cells at mean coverage of 73,845 fragments per cell. This allows us to compare the allele-specific copy number profiles obtained by scATAC-seq with those given by scDNA-seq and integrate the two data types in a multi-omic characterization of this complex tumor cell population.
First, we segmented the genome and estimated the allele-specific copy number profiles of single cells at each segment for both the scATAC-seq and scDNA-seq data, following the procedure in Fig. 1 with some modifications due to the lack of normal cells to use as control for this sample (Methods). Fig. 5a shows the normalized total coverage, pooled across cells from scDNA-seq. Fig. 5b shows -scatterplots for five example CNA regions in scDNA-seq and scATAC-seq. Compared to the scATAC-seq data, the scDNA-seq data has about 8-fold higher total read coverage and 7-fold higher heterozygous site coverage per cell. Thus, whereas subclones corresponding to distinct haplotype profiles are cleanly separated in the scDNA-seq data, they are much more diffuse in the scATAC-seq data. Yet, cluster positions in scATAC-seq roughly match those in scDNA-seq.
Fig. 5: Alleloscope analysis of scDNA-seq and scATAC-seq data reveals complex subclonal heterogeneity in the SNU601 gastric cancer cell line.
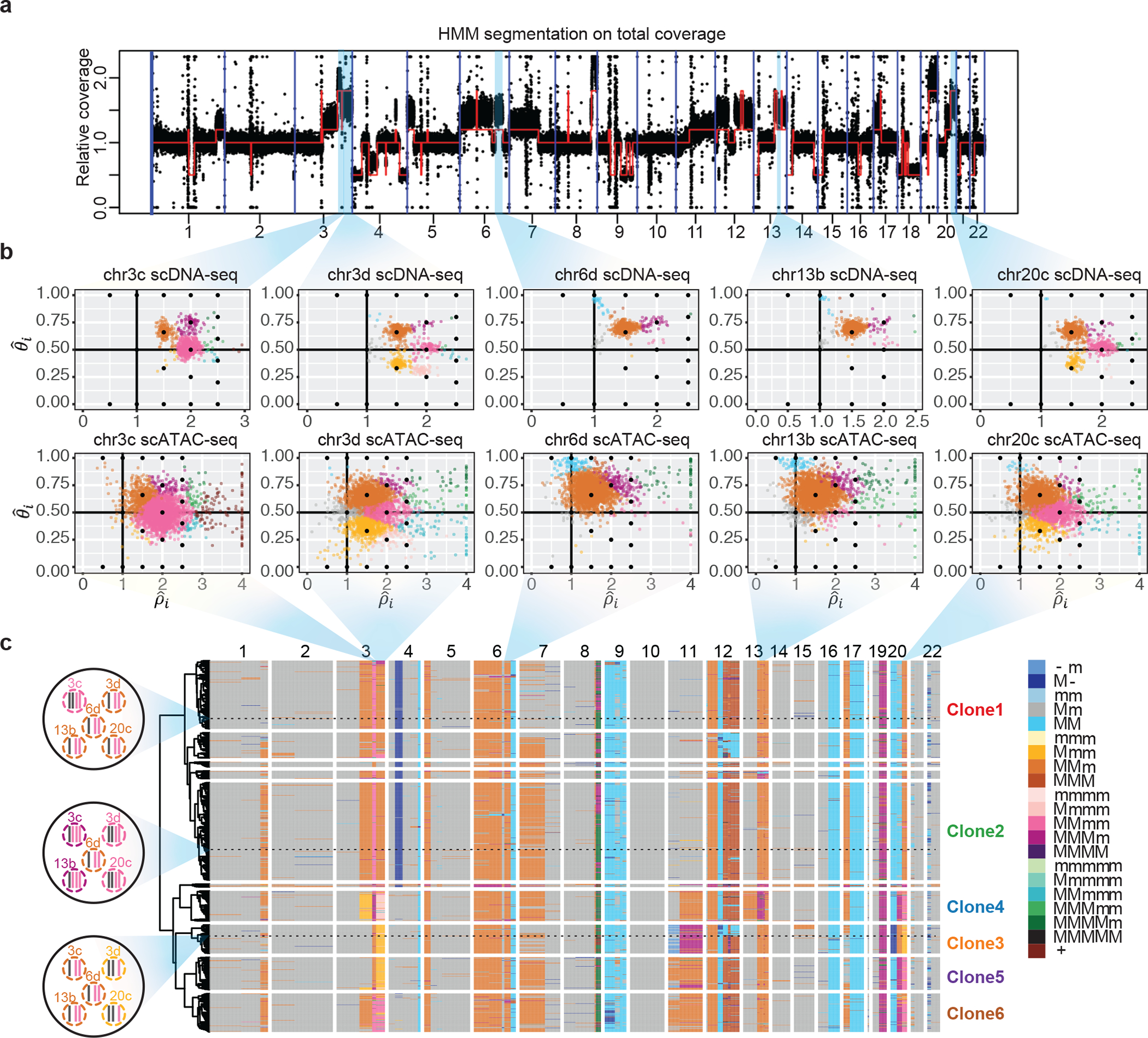
(a) Genome segmentation using HMM on the pooled total coverage profile computed from scDNA-seq data. (b) Single-cell allele-specific copy number profiles () for five regions in scDNA-seq and scATAC-seq data. Cells are colored by haplotype profiles according to legend in Fig. 5c. (c) Tumor subclones revealed by hierarchical clustering of allele-specific copy number profiles from the scDNA-seq data. Genotypes of the five regions shown in Fig. 5b, for three example cells, are shown in the left. The haplotype structures for the 5 regions in Fig. 5b of three cells randomly chosen from Clone 1, 2, and 3, are shown to the left of the heatmap. In the color legend, M and m represent the “Major haplotype” and “minor haplotype” respectively. The six clones selected for downstream analysis in scATAC-seq data are labeled in the plot.
Fig. 5c shows the hierarchical clustering of cells from scDNA-seq based on their allele-specific copy number profiles, revealing the subclonal structure and the co-segregating CNA events that mark each subclone. For each cell in each region, Alleloscope also produces a confidence score for its copy number state assignment (Supplementary Fig. 16). Based on visual examination of the confidence scores at the marker regions in both the ATAC and DNA sequencing data sets, we chose 6 subclones for further investigation (Clones 1–6 labeled at the right of the heatmap). We manually reconstructed the probable evolutionary tree relating these 6 clones to abide by the following three rules:
Parsimony: The tree with the least number of copy number events is preferred.
Monotonicity: For a multi-allelic region with escalating amplifications (e.g. Mm, MMm, MMMm), the states were reached in monotonic order (e.g. Mm→ MMm→ MMMm) unless a genome doubling event occurred (in which case a sequence such as Mm→ MMmm→MMm would be allowed).
Irreversibility of LOH: Once a clone completely loses an allele (i.e. copy number of that allele becomes 0), it can no longer gain it back.
The evolutionary tree, thus derived, is shown in Fig. 6b. The mirrored-subclonal amplifications on chr3q, the deletion on chr4p, and the multiallelic amplification on chr20q allowed us to infer the early separation of clones 3–6 from clones 1–2. Subclones 3–6 are confidently delineated by subsequent amplifications on chr3q, chr20q, chr11, chr13, and chr17. Note that high chromosomal instability led to concurrent gains of 1q and 7p in both the Clone 1–2 and Clone 3–6 lineages. We also observed a large number of low-frequency but high-confidence CNA events indicating that ongoing chromosomal instability in this population is spawning new sporadic subclones that have not had the chance to expand.
Fig. 6: Integrative analysis of allele-specific copy number and chromatin accessibility for SNU601 ATAC sequencing data.
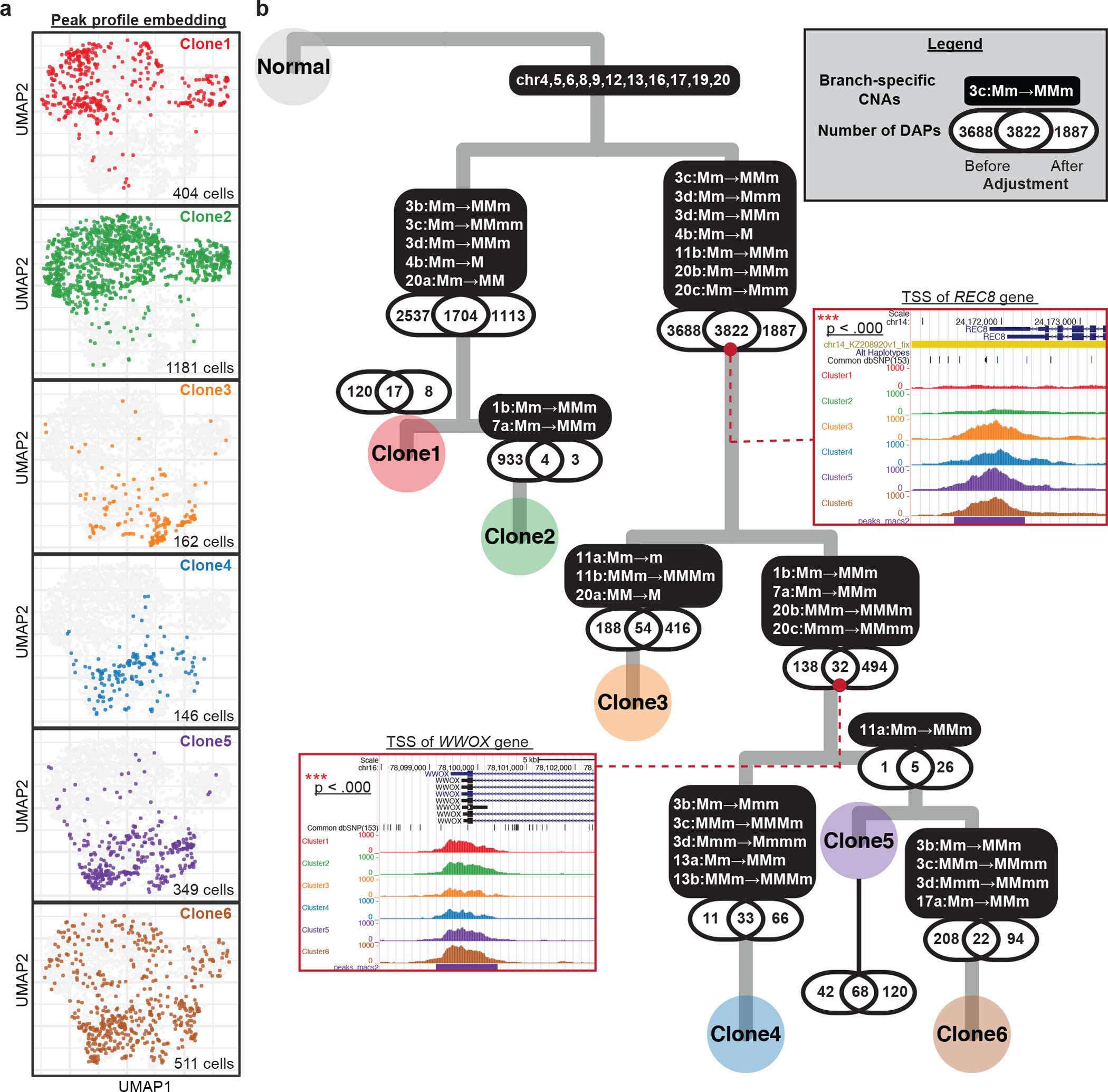
(a) UMAP projection of genome-wide scATAC-seq peak profile on 2,753 cells. The same group of cells were clustered into one of the six subclones based on their allele-specific copy number profiles across the 10 selected regions. Cells in different subclones are labeled with different colors, using the same color scheme as that for the subclone labels in Fig. 4c. The number of cells colored in each UMAP is shown at the bottom-right corners. (b) A highly probable lineage history of SNU601, with copy number alterations (CNAs) and differentially accessibility peaks (DAPs) marked along each branch. P-values of the tests for association between DAPs and CNAs are shown along each branch. For two example DAP genes, pooled peak signals for each subclone are shown as inset plots.
We now turn to scATAC-seq data, focusing on the 10 marker regions which, together, distinguish Clones 1–6: chr1b, 3b-d, 4b, 7a, 11b, 13b, and 20b-c. The values allow direct assignment of allele-specific copy number states to each cell for each region, with posterior confidence score. We next performed subclone assignment to each cell via a Bayesian mixture model that pools information across the 10 regions (Methods). Despite the low accuracy in per-region genotyping, when information is pooled across the 10 marker regions, 81.6% of the 2,753 filtered cells can be assigned to a subclone with >95% posterior confidence (Supplementary Fig. 17). These subclone assignments for each cell, and cell-level haplotype profiles for each region, can now be integrated with peak-level signals. Following the scheme in Fig. 4b, we computed the UMAP coordinates for the scATAC-seq cells based on their peak profiles (Fig. 6a). UMAP scatterplots colored by clone assignment show that the 6 clones exhibit marked differences in their chromatin accessibility profiles (Fig. 6a). We expect some of these peak-level differences to be driven by CNAs.
To delineate the peaks that differ between clones, and to identify peak differences that are not accountable by CNAs, we developed a statistical test based on a generalized log-likelihood ratio with adjustment parameters computed from the copy number profile of each of the two subclones (Fig. 6b, Methods). The same test, but neglecting coverage adjustment, was also performed for comparison. Along each branch, three numbers are shown —1. DAPs identified only before adjustment; 2. DAPs identified both before and after adjustment; 3. DAPs identified only after adjustment (Fig. 6b). For the smaller subclones (Clone 3,4,5), low coverage limits the detection power and thus limits the DAP counts in both categories. Yet, juxtaposing DAP and CNA events along the tumor phylogeny yields insights: Along most lineages, adjustment for CNA-induced shifts in coverage removes a large number of peaks whose differential-accessibility signal is explainable by the underlying change in copy numbers. This argues for the importance of CNAs as a mechanism underlying subclonal differences in chromatin accessibility in this tumor.
Nevertheless, along some branches we find a large number of DAPs remaining after adjustment, and thus must be due to other mechanisms. Notably, CNA adjustment also identifies new DAPs whose signals were obscured by a change in copy number in the opposite direction. Two example DAPs that remain significant after adjustment are shown as insets in Fig. 6b, with full list given in Supplementary Table 3. The first example is a peak at the transcription start site (TSS) of the REC8 gene, which is located on chr14 where no apparent CNAs were observed across the six major subclones. The TSS of REC8 is open in clones 3–6 but closed in clones 1–2 (p-value<0.0001). REC8 is a gene encoding a meiosis-specific cohesion component that is normally suppressed in mitotic proliferation, whose role in cancer has recently been subject to controversy: While Yu et al.32 found the expression of this gene to suppress tumorigenicity in a gastric cancer cell line, McFarlane et al.33 postulated that it may be broadly activated in some cancers where it generates LOH by reductional segregation. The opening of the TSS of REC8, stably maintained in Clones 3–6, suggests that meiotic processes may underlie the increased chromosomal instability of this multiclonal lineage. The second example is a peak at the TSS of the WWOX gene, located on chr16, which is significantly depleted in Clone 3 (p-value<0.0001). Although chr16 has LOH across all tumor cells, there are no detectable subclonal differences, and thus the decrease in accessibility at WWOX for subclone 3 is not attributable to a copy number event. Because WWOX is a well-known tumor suppressor whose down-regulation is associated with more advanced tumors34, 35, its decrease in accessibility suggests a more aggressive phenotype for Clone 3. These two examples demonstrate how Alleloscope can be used to dissect the roles of CNA and chromatin-level changes in the identification of gene targets for follow-up study.
Discussion
Despite recent advances in the application of single-cell sequencing to cancer, we are still far from understanding the diversity of genomes that are undergoing selection at the single-cell level. Notably, little is yet known about the intratumor diversity of allelic configurations within CNA regions, and to what extent cell-to-cell heterogeneity in chromatin accessibility can be attributed to copy number events. Here, we have developed Alleloscope, a method for allele-specific copy number estimation that can be applied to single-cell DNA and ATAC sequencing data. Through a combination of matched linked-reads whole genome sequencing, downsampling-based benchmark experiments, and simulations, we assessed the accuracy of Alleloscope and benchmarked against CHISEL, which is currently the only other method for scDNA-seq allele-specific copy number profiling. Detailed discussion about the two methods can be found in Supplementary Results.
We applied Alleloscope to a panel of breast, colorectal, and gastric tumors and cancer cell lines, where it revealed a yet under-explored level of allelic heterogeneity within hypermutable CNA regions. We observed multiple instances of convergent evolution involving recurrent CNA events affecting the same region, some verified by linked read sequencing. In accordance with the findings in Watkins et al.36, we found that chromosomal instability drives the formation of subclones not only in primary tumors but also after metastasis. Alleloscope was also applied to a scDNA-seq dataset15 generated from a different sequencing protocol (Supplementary Fig. 18; Supplementary Methods), further demonstrating the general applicability of the approach.
Having established the allelic complexity of CNAs at single-cell resolution, we next applied Alleloscope to scATAC-seq data to perform integrative study of copy number change and chromatin remodeling. First, we considered the analysis of a public dataset consisting of two basal cell carcinoma samples25. Here we showed that Alleloscope can detect amplifications, deletions, and copy-neutral LOH events accurately in scATAC-seq data, and was able to find a subclone delineated by a copy-neutral LOH event. Juxtaposing this subclone assignment with peak signals allowed us to detect a wave of genome-wide chromatin remodeling in the lineage carrying the LOH. Next, we applied Alleloscope to a complex polyclonal gastric cancer cell line with matched scDNA-seq data. By overlaying peak signals with subclones delineated by varying allele-specific copy number states, we were able to identify differentially accessible peaks that are not accountable by copy number differences. Focusing on subclone-enriched peaks that are significant after copy number adjustment allowed us to prioritize genes for follow-up.
Alleloscope can potentially be extended for single-cell data of other modalities, for example snmC-seq37 and scRNA-seq data, to investigate the relationships between clonal evolution, chromatin remodeling, methylation, and transcriptome. To guide experimental design for single-cell omics sequencing protocols, we investigated the performance of Alleloscope under different experimental parameters. Among the parameters, coverage at heterozygous SNP sites is an especially important consideration, especially for scATAC-seq and scRNA-seq where shifts in total coverage can be an unreliable proxy for changes in DNA copy number. Most of the current scRNA-seq technologies only sequence either the 3’ or 5’ end of the mRNA transcripts, which limits the number of heterozygous SNP sites covered by reads. The latest developments in single-cell long read sequencing38–40 and single-cell multimodal sequencing41 bring forth new analysis opportunities with this method.
Methods
ScDNA-seq datasets and pre-processing
The ten 10x scDNA-seq datasets analyzed in this study are summarized in Supplementary Table 1. P5931, P5847 and P6461 scDNA-seq data were generated using the method described in the previous study27. We applied the Cell Ranger DNA pipeline (https://support.10xgenomics.com/single-cell-dna/software/overview/welcome; beta version: 6002.16.0) for sample demultiplexing, read alignment, CNA calling and visualization. Most data were aligned to the GRCh38 reference genome except for the two processed BC10x datasets (GRCh37). For the tumor samples with a matched normal sample, the GATK HaplotypeCaller42 was used to call heterozygous SNPs on the matched normal samples. Otherwise, SNPs were retrieved on the tumor sample themselves. Next, we applied VarTrix (https://github.com/10XGenomics/vartrix) to efficiently generate SNP-by-cell matrices for both reference and alternative alleles of the identified SNPs.
To select high-quality SNPs, we filtered out the SNPs with <5 reads for P5846 and P5847, <10 reads for P5915 and P5931, <15 reads for P6335 and P6461, <20 reads for P6198, SNU601, and the BC10X samples based on the number of SNP detected for each sample. Additionally, SNPs located in the centromeres and telomeres were excluded. To exclude cells that are undergoing apoptosis or mitosis, noisy cells labeled by the Cell Ranger tool were removed.
Single-cell ATAC datasets, sequencing and preprocessing
The scATAC-seq datasets analyzed in this study are summarized in Supplementary Table 4. We used the following procedure to generate the SNU601 scATAC-seq dataset. About 400,000 cells were washed with RPMI media and centrifuged (400g for 5 min at 4°C) twice. The supernatant was removed and resuspended with chilled PBS + 0.04% BSA solution. The resuspended pellet was centrifuged (400g for 5min at 4°C), and the supernatant was removed again. Then 100 μL of chilled Lysis Buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Nonidet P40 Substitute, 0.1% Tween-20 and 0.01% digitonin) was added and carefully mixed 10 times. The tube was incubated on ice for 7 min. After incubation, 1 mL of chilled Wash Buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA and 0.1% Tween-20) was added and mixed 5 times followed by centrifugation of nuclei (500g for 5 min at 4°C). After removing the supernatant, nuclei were resuspended in chilled Nuclei Buffer (10X Genomics), filtered by Flowmi Cell Strainer (40uM) and counted using a Countess II FL Automated Cell Counter. Then the nuclei were immediately used to generate scATAC-seq library.
ScATAC-seq library was generated using the Chromium Single Cell ATAC Library & Gel Bead Kit (10X Genomics) following the manufacturer’s protocol. We targeted 3000 nuclei with 12 PCR cycles for sample index PCR. Library was checked by 2% E-gel (Thermofisher Scientific) and quantified using Qubit (Thermofisher Scientific). Sequencing was performed on Illumina NextSeq500 using NextSeq 500/550 High Output Kit v2.5 (Illumina).
Raw sequencing reads of the SNU601 scATAC-seq sample was de-multiplexed with the 10x Genomics Cell Ranger ATAC Software (v.1.2.0; https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/algorithms/overview) and aligned to the GRCh38 reference genome. The aligned scATAC-seq data of the two pre-treatment basal cell carcinoma samples (BCC; SU006 and SU008) were retrieved from GSE12978525. To obtain all potential SNPs for the SU006 and SU008 samples, GATK Mutect243 was used to call single-nucleotide variants (SNVs) on the bam files deduplicated by the Picard toolkits of both the t-cell dataset and the tumor dataset from the same tumor. All SNVs from the paired tumor-normal datasets were combined and the read counts of these SNPs were quantified for each cell in the tumor scATAC-seq dataset. For the SNU601 scATAC-seq data, we instead quantified the read counts of the two alleles of the SNPs more reliably called from the paired normal scDNA-seq data. Then the SNP-by-cell matrices for both reference and alternative alleles were generated using Vartrix for all the scATAC-seq datasets. To select potential germline SNPs, we further filtered out the SNVs <5 reads for the SU008 sample and <10 reads for the SU006 sample. SNPs with extreme VAF values <0.1 or >0.9 were also excluded for both samples. Because we used the phasing information from the paired scDNA-seq data to assist the estimation of the haplotype structures for the SNU601 scATAC-seq data, we did not filter out SNPs with extreme VAF values for the scATAC-seq data. To improve the quality of the downstream analysis for SNU601 scATAC-seq data, we filtered out cells <5 reads and the SNPs <5 reads.
Linked-read sequencing and data processing
The five samples with the linked-read sequencing data were acquired as surgical resections following informed consent under an approved institutional review board protocol from Stanford University. Samples were subjected to mechanical and enzymatic dissociation as previously described, followed by cryopreservation of dissociated cells33.
Cryofrozen cells were rapidly thawed in a bead bath at 37 °C. Cell counts were obtained on a BioRad TC20 cell counter (Biorad, Hercules, CA) using 1:1 trypan blue dilution. Between 1.5–2.5 million total cells were washed twice in PBS. Centrifugation was carried out at 400g for 5 minutes. PBS was removed and cell pellets were frozen at −80°C. DNA extraction was carried out on cell pellets following thawing using either MagAttract HMW DNA Kit (P5931) or AllPrep DNA/RNA Mini Kit (Qiagen Inc., Germantown, MD, USA) as per manufacturer’s protocol. Quantification was carried out using Qubit (Thermofisher Scientific).
Sequencing libraries were prepared from DNA using Chromium Genome Reagent Kit (v2 Chemistry) (10X Genomics, Pleasanton, CA, USA) following manufacturer’s instructions. Sequencing was performed using Illumina HiSeq or NovaSeq sequencers using 150×150 bp paired end sequencing and i7 index read of 8 bp. Long Ranger (10X Genomics; v2.2.0) was used to align reads, call and phase SNPs, indels and structural variants.
Segmentation
The first step of Alleloscope is to segment the genome into regions with different CNA profiles. The appropriate segmentation algorithm depends on what samples are available. First, matched bulk DNA sequencing data (WGS/WES) or pseudo-bulk data from scDNA-seq data can be segmented using FACLON5, a segmentation method that jointly models the bulk coverage and bulk VAF profiles, if a matched normal sample is available. To accommodate segments from rare subclones, methods that integrate shared cellular breakpoints in CNA detection for scDNA-seq44 can improve sensitivity. If a normal sample is unavailable, Alleloscope instead uses an HMM-based segmentation method. The HMM method, which operates on the binned counts of pooled cells, assumes a Markov transition matrix on four hidden states representing deletion, copy-neutral state, single-copy amplification and double-copy amplification: , where t = 1 × 10−6 by default. Emission probabilities follow a normal distribution with means {1.8, 1.2, 1, 0.5} and standard deviation 0.2. The segmentation plots in the study were generated by the HMM algorithm. With the paired sample, the P6198 tumor sample was segmented using FALCON on SNPs >30 reads with default parameters. Besides FALCON and HMM, users can choose the methods that work best for specific datasets. For example, ASCAT11 on the P6198 sample yields similar results (Supplementary Fig. 9).
Whole-exome sequencing (WES) data processing
The WES data of the two paired tumor-normal samples (SU006 and SU008) were obtained from PRJNA53334131. Raw fastq files were aligned to the GRCh37 reference genome using bwa-mem45 with duplicate reads removed using the Picard toolkits46. The copy number calls of paired normal-tumor samples were obtained using Varscan247. Then the HMM algorithm was applied for segmentation.
SNP phasing and single-cell allele profile estimation per region
For each region r after segmentation, an expectation-maximization (EM)-based method is used to iteratively phase each SNP and estimate cell-specific allele-specific copy number states. Recall that “Major haplotype” is defined as the haplotype with higher aggregate copy number in the sample. Let Ij indicate whether the reference allele of SNP j is located on the major haplotype and θir denote major haplotype proportion of cell i for region r. The complete log likelihood of the model is
where and are the observed read counts for the reference and alternative alleles of cell i on SNP j. In the E-step, we calculate the conditional expectation of the hidden variable Ij.
where is the parameter from the tth iteration. In the M-step, is updated by solving
An initial estimate is derived from the bulk VAF profile, and the two steps are iterated until convergence. For the SNU601 scATAC-seq dataset, the phases estimated in the paired scDNA-seq dataset (’s) were directly applied to estimate the ’s of the cells in the scATAC-seq data. To reduce noise from low-coverage SNPs, we only used cells with ≥ 20 reads covering the identified SNPs for each region.
Selecting normal cells and normal regions for single-cell coverage normalization
To compute the relative coverage change for each cell in region ), normal cells and diploid regions are required for normalization. The ’s estimated above are used to identify normal cells and diploid regions. To identify normal cells, all cells were first clustered by hierarchical clustering. For each cell cluster, we compute a score dc that is the sum, across regions, of a per-region metric quantifying the mean deviation of from 0.5 (the normal value):
where Sc is the set of cells in cluster c, and nc = |Sc|. We then identify the cluster c* = argmincΔc. All cells in c* are considered normal.
Putative diploid regions are identified using both and coverage. First, Alleloscope computes
where high values of is evidence for allelic imbalance of cluster c in region r. Because regions with both haplotypes equally amplified can also have small dcr, we also compute an adjusted coverage ratio (; defined as below),
where Nir is the total read count for cell i and region r, and Ni is the total read count for cell i across regions. Lr is length of the region r and L is total length of the genome. For region r, cells with values larger than the 99th percentile are assigned to the 99th percentile value. We then compute the cluster mean for each region r,
Alleloscope identifies potential diploid regions for each cluster by first ranking by dcr to identify the clusters with smallest dcr, and among these, identify clusters with low mcr. These regions are proposed as candidate diploid regions for visual inspection before it is used to compute normalized coverage ratios for each cell in the cluster.
Because coverage on scATAC-seq data is confounded by epigenetic signals, chromosome 22 for SU008 and chromosome 18 for SU006 were selected manually as normal regions based on their VAF and relative coverage values in the matched WES data. Cells were considered as normal or tumor cells based on peak-based clustering. For the SNU601 scATAC-seq dataset, chromosome 10 was selected as the normal region based on the above procedure applied to the matched scDNA-seq data.
Cell-level CNA state estimation
The cell-level allele-specific copy number profiles are defined by both relative coverage change ( and major haplotype proportion () for region r. After the normal cells and control region are identified, cell-specific relative coverage change in region r is calculated as
where Nir is total read counts in region r and Ni0 is total read counts in a reference region for cell i. N0r is a vector denoting total read counts in region r of all identified normal cells and N00 is a vector denoting total read counts in the same reference region of all identified normal cells. For P6461 with no diploid region, was multiplied by 2 for the abnormal cells. Because SNU601 is a tumor cell line with no normal cells in the dataset, for its scDNA-seq data, N0r and N00 were calculated from the cells in the P6198 sample instead. For SNU601 scATAC-seq data, we aligned the distribution of the values in the paired scDNA-seq data to the distribution of the values for each region to get the normalized in the scATAC-seq data. The normalized values for the scATAC-seq data were computed by
Cells with extreme values (above 99th percentile and below 1 percentile) were excluded for each region. With the () pairs, cells in the scDNA-seq data can be classified into haplotype profile g with the expected (ρg, θg) values based on minimum distance. With noisier signals in the scATAC-seq data, the haplotype structures identified in the paired scDNA-seq data are used to guide the genotyping for each region. For region r, the posterior probability that cell i carries haplotype profile g is
where gr denotes the haplotype profile observed in region r in the paired scDNA-seq data and denotes the prior probability that a randomly sampled cell carrying the gr haplotype profile. A uniform prior was used for . In the formula,
where nir is total read count in region r for cell i. For scATAC-seq data, the haplotype profile of cell i in region r was estimated by maximizing the above posterior probability.
Validations using matched linked-read sequencing data for each region
We performed validations on five samples using matched linked-read sequencing data. Two strategies were used for the validations. First, to assess the phasing accuracy of region r, the largest haplotype block within region r in the matched linked-read sequencing data was selected for comparison, ensuring that the included SNPs are phased with respect to one another. Then the estimates for SNPs within region r in the scDNA-seq data were converted to binary indicator
Using the major/minor haplotype setting, all SNPs in the largest haplotype block in the matched linked-read sequencing data were placed accordingly to retried Ij as the gold standard. Comparing the binary estimates and Ij, the phasing accuracy for region r was computed as follows:
Secondly, we evaluated the accuracy for cell-level CNA state estimation for region r. To do this, for each cell in region r we compared the estimated haplotype profiles () to the haplotype profiles (θir) obtained by plugging in known phases provided by the matched linked-read sequencing data as the gold standard. Similarly, the haplotype block with the largest number of SNPs was used here. The θir’s, used as the gold standard, were retrieved by directly plugging in known phases Ij, provided by the matched linked-read sequencing data (explained in the first strategy). Then the accuracy for cell-level CNA state estimation of region r is computed as
Cell Lineage Reconstruction
For scDNA-seq data, cell-specific haplotype profiles from Alleloscope across the genome are used to reconstruct cell lineage trees. The “Gower’s distance” is calculated using “cluster” R package on the nominal haplotype profiles between cells. The ‘pheatmap’ R package was used to perform hierarchical clustering on the distance using the “ward.D2” method. Because fewer SNPs lead to higher variance in ’s, we by default included segments with more than 2,000 SNPs identified. For visualization, each segment was plotted with its length proportional to 5,000,000 bins, and the heights of the clustering tree were log-transformed.
To explore tumor subclonal structure in the two BCC scATAC-seq datasets, we instead clustered the cells using values, which are orthogonal to the CNA signals based on total coverage, across the segments with more than 500 SNPs. Then, hierarchical clustering is performed on the Euclidean distance using the “ward.D2” method.
Because the subclones for the SNU601 sample were identified first from the scDNA-seq data, we adopted a supervised strategy to assign each cell in the SNU601 scATAC-seq dataset into different subclones. First, we identified 10 marker regions-- chr1b, 3b-d, 4b, 7a, 11b, 13b, and 20b-c that differentiate the cells into the six major subclones based on the subclone specific copy number profiles from the scDNA-seq data. Combining the haplotype profiles across the ten regions for each cell enables assignment of the cells into one of the six subclones with high confidence. The posterior probability of cell i coming from clone k was
where k ∈ {1 ~ 6} for the six clones and πk is the prior probability that a randomly sampled cell coming from the kth clone, which was set to uniform in the analysis. In the formula,
where x indexes the ten marker regions; and are the estimated major haplotype proportion and relative coverage for cell i in the scATAC-seq data; θkx and ρkx are the “known values” for specific haplotype profiles for clone k derived from the paired scDNA-seq data; and nix is the number of total read counts in the xth marker region for cell i. Each cell was assigned into one of the six subclones by maximizing the posterior probability with the confidence score being the posterior probability of the assigned clone.
ScATAC-seq data analysis
To investigate the relationships between allele-specific CNAs and chromatin accessibility, we processed the peak signals in addition to the allele-specific CNAs for each cell in the scATAC-seq data. For the two public BCC samples, the peak-by-cell matrices were obtained from GSE129785. We log-transformed the count matrices, selected the peaks in >10% cells, regressed out cell total coverage for each peak by linear regression, and projected the cells onto the UMAP plot using genome-wide peak signals48. The cell type identify for each cluster was retrieved from the previous study25. To further explore intratumor heterogeneity, we selected the cells labeled as tumor cells, repeated the pre-processing steps described above, and projected the tumor cells onto the UMAP plot.
For the SNU601 scATAC-seq dataset, scATAC-pro49 was used to call peaks and generate the peak-by-cell matrix. We first filtered out the cells that have proportions of fragments on the detected peaks <0.4 and total peaks outside of the range 15,000~100,000, and filtered out the peaks observed in less than 10% of cells. After the matrix was log-transformed, we regressed out cell total coverage for each peak by linear regression. Using Louvain clustering, two clusters with extreme total fragment counts were removed. After cell filtering, we repeated the process and projected the cells onto the UMAP plot. The cells are then colored by the clonal assignment based on the DNA information.
Differentially accessible peaks (DAPs) identification with copy number adjustment for scATAC-seq data
To identify differentially accessible peaks (DAPs) after accounting for copy number differences between two clones, we developed a statistical test based on a generalized log-likelihood ratio (GLLR) statistic. We first define some necessary terms: Given a segmentation of the genome, for a given clone c, let θc = {(LR, ZR)} be the copy number profile, which can be expressed as a vector of region lengths (LR) and average copy numbers (ZR) across all regions R. For peak k, we define function fk (θc to be the proportion of genomic DNA in the peak region based on the copy number profile of clone c, computed as follows:
where r (k) is the region r to which peak k belongs and lr is the length of peak k.
For peak k and clone c, let Yck be the total read count across the cells in the clone, and let Nc be the total read count across all peaks summed across the cells in the clone. Ycg can be modeled by Binomial sampling,
where, pck is the relative probability of detecting reads in the peak k region after adjusting for the copy numbers. Between clones with different copy number profiles, differences in pck is evidence for chromatin remodeling. Thus, pairwise comparisons were performed for each branch (clone versus clone ) and for each peak, using the generalized likelihood ratio test with the hypothesis and .
We compute the GLLR test statistic as below,
Which follows the Chi-squared distribution (see Supplementary Methods for derivation of MLE). Using the GLRT, a peak is considered as a DAP if its FDR adjusted p-value<0.01 for the pairwise clonal comparison.
Simultaneously, the GLRT under the binomial model without copy number adjustment was also performed for comparison:
The same criterion was used to identify DAPs for each pairwise clonal comparison (FDR adjusted p-values<0.01). To identify genes potentially regulated by DAPs, we searched the genes with TSS within ± 2,000 bp distance of each DAP.
Supplementary Material
Acknowledgements
The work is supported by the National Institutes of Health [P01HG00205ESH to B.T.L., S.M.G. and H.P.J., 5R01-HG006137–07 and 1U2CCA233285–01 to C-Y.W. and to N.R.Z., 1R35HG011292–01 to B.T.L.]. Additional support to HPJ came from the Research Scholar Grant, RSG-13–297-01-TBG from the American Cancer Society, Clayville Foundation and the Gastric Cancer Foundation.
Footnotes
Competing interests
The authors declare no competing interests.
Code availability
Alleloscope is available on GitHub at https://github.com/seasoncloud/Alleloscope. And as a compute capsule on Code Ocean (https://doi.org/10.24433/CO.2295856.v1).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The patient scDNA-seq and linked-read sequencing data generated for this study are available under dbGAP identifier phs001711. The scATAC-seq dataset is available in the National Institute of Health’s SRA repository under accession PRJNA674903. There are no restrictions on data availability or use. The other patient scDNA-seq data were obtained from dbGAP under accession phs001818.v3.p127 and phs00171112. The cell line scDNA-seq dataset was from the Sequence Read Archive (SRA) under accession PRJNA498809. The public scATAC-seq data and whole exome sequencing data were obtained from the SRA under accession PRJNA53277425 and PRJNA53334131.
Reference
- 1.Baylin SB & Jones PA A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandoval J & Esteller M Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 22, 50–55 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrell RA, McGranahan N, Bartek J & Swanton C The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Bell JM, Zavala NA, Ji HP & Zhang NR Allele-specific copy number profiling by next-generation DNA sequencing. Nucleic Acids Res 43, e23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favero F et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 26, 64–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ha G et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res 24, 1881–1893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen R & Seshan VE FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44, e131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamal-Hanjani M et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zaccaria S & Raphael BJ Characterizing allele- and haplotype-specific copy numbers in single cells with CHISEL. Nat Biotechnol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Loo P et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A 107, 16910–16915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andor N et al. Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genom Bioinform 2, lqaa016 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakker B et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 17, 115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garvin T et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods 12, 1058–1060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim C et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893 e813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laks E et al. Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing. Cell 179, 1207–1221 e1222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navin N et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Velazquez-Villarreal EI et al. Single-cell sequencing of genomic DNA resolves sub-clonal heterogeneity in a melanoma cell line. Commun Biol 3, 318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corces MR et al. The chromatin accessibility landscape of primary human cancers. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Granja JM et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litzenburger UM et al. Single-cell epigenomic variability reveals functional cancer heterogeneity. Genome Biol 18, 15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sathe A et al. The cellular genomic diversity, regulatory states and networking of the metastatic colorectal cancer microenvironment. bioRxiv (2020). [Google Scholar]
- 28.Bell JM et al. Chromosome-scale mega-haplotypes enable digital karyotyping of cancer aneuploidy. Nucleic Acids Res 45, e162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greer SU et al. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Med 9, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng GX et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34, 303–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu J et al. REC8 functions as a tumor suppressor and is epigenetically downregulated in gastric cancer, especially in EBV-positive subtype. Oncogene 36, 182–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McFarlane RJ & Wakeman JA Meiosis-like Functions in Oncogenesis: A New View of Cancer. Cancer Res 77, 5712–5716 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Aqeilan RI et al. Loss of WWOX expression in gastric carcinoma. Clin Cancer Res 10, 3053–3058 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Baryla I, Styczen-Binkowska E & Bednarek AK Alteration of WWOX in human cancer: a clinical view. Exp Biol Med (Maywood) 240, 305–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watkins TBK et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo C et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta I et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lebrigand K, Magnone V, Barbry P & Waldmann R High throughput error corrected Nanopore single cell transcriptome sequencing. Nat Commun 11, 4025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh M et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun 10, 3120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu C, Preissl S & Ren B Single-cell multimodal omics: the power of many. Nat Methods 17, 11–14 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Poplin R et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv, 201178 (2018). [Google Scholar]
- 43.Benjamin D et al. Calling somatic snvs and indels with mutect2. BioRxiv, 861054 (2019). [Google Scholar]
- 44.Wang R, Lin DY & Jiang Y SCOPE: A Normalization and Copy-Number Estimation Method for Single-Cell DNA Sequencing. Cell Syst 10, 445–452 e446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McInnes L, Healy J & Melville J Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426 (2018). [Google Scholar]
- 49.Yu W, Uzun Y, Zhu Q, Chen C & Tan K scATAC-pro: a comprehensive workbench for single-cell chromatin accessibility sequencing data. Genome Biol 21, 94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The patient scDNA-seq and linked-read sequencing data generated for this study are available under dbGAP identifier phs001711. The scATAC-seq dataset is available in the National Institute of Health’s SRA repository under accession PRJNA674903. There are no restrictions on data availability or use. The other patient scDNA-seq data were obtained from dbGAP under accession phs001818.v3.p127 and phs00171112. The cell line scDNA-seq dataset was from the Sequence Read Archive (SRA) under accession PRJNA498809. The public scATAC-seq data and whole exome sequencing data were obtained from the SRA under accession PRJNA53277425 and PRJNA53334131.