

Abstract
Introduction
Neuroimaging heterogeneity in dementia has been examined using single modalities. We evaluated the associations of magnetic resonance imaging (MRI) atrophy and flortaucipir positron emission tomography (PET) clusters across the Alzheimer's disease (AD) spectrum.
Methods
We included 496 Alzheimer's Disease Neuroimaging Initiative participants with brain MRI, flortaucipir PET scan, and amyloid beta biomarker measures obtained. We applied a novel robust collaborative clustering (RCC) approach on the MRI and flortaucipir PET scans. We derived indices for AD‐like (SPARE‐AD index) and brain age (SPARE‐BA) atrophy.
Results
We identified four tau (I–IV) and three atrophy clusters. Tau clusters were associated with the apolipoprotein E genotype. Atrophy clusters were associated with white matter hyperintensity volumes. Only the hippocampal sparing atrophy cluster showed a specific association with brain aging imaging index. Tau clusters presented stronger clinical associations than atrophy clusters. Tau and atrophy clusters were partially associated.
Conclusions
Each neuroimaging modality captured different aspects of brain aging, genetics, vascular changes, and neurodegeneration leading to individual multimodal phenotyping.
Keywords: Alzheimer's disease, amyloid beta, heterogeneity, mangetic resonance imaging, positron emission tomography, prognosis, tau
1. INTRODUCTION
Alzheimer's disease (AD) is the most common cause of dementia. However, most subjects with dementia have multiple contributing pathologies. 1 , 2 In addition, multiple deposition patterns have been described for neurofibrillary tangles, such as hippocampal sparing and limbic predominant patterns. 3 , 4 , 5 , 6 This heterogeneity is in line with the different clinical presentations related to AD neuropathological changes. 5 , 7 , 8 This heterogeneity is likely the result of individual genetic susceptibility (with apolipoprotein E [APOE] explaining the largest heritability), 9 diverse environmental risk factors encountered during the lifetime, comorbidities, and the protective effect of the cognitive reserve. 10 , 11 , 12 , 13
Structural magnetic resonance imaging (MRI) quantifies gray matter thickness or volume independently of the etiology. Gray matter analyses have identified various brain atrophy patterns in AD patients, which are associated with different rates of clinical progression. These different patterns are likely the results of differences in regional pathology distribution and variable presence of co‐pathologies. 6 , 14 , 15 , 16 MRI scans also quantify white matter hyperintensities (WMH), an imaging marker of small vessel disease that increases with aging and in vascular cognitive impairment. WMHs present a higher burden in AD dementia subjects. 17 , 18 , 19 , 20 One advantage and limitation of structural MRIs is that they quantify brain changes resulting from the multiple pathologies present in the brain, which are therefore not specific. Recently, in vivo molecular biomarkers that bind specific protein deposits have been developed. 21 These positron emission tomography (PET) ligands can specifically quantify different proteins deposited in the brain. Amyloid beta (Aβ) PET has been available for almost two decades, whereas tau tracers have been introduced more recently and are being characterized. Tau neurofibrillary tangles, measured by flortaucipir PET scans, are more closely correlated with cognitive changes in neuropathologic studies, 22 and recent imaging studies have confirmed this association. 23 These imaging modalities capture disease processes at different stages with tau accumulation preceding brain atrophy. 24
Advanced analytical techniques can be applied to these images to discover heterogeneous patterns across the AD continuum using data‐driven clustering techniques. 6 These approaches have identified MRI atrophy patterns 6 , 10 and, more recently, tau‐derived imaging clusters. 25 However, to our knowledge, the relationship between data‐driven clusters derived independently for structural MRI and flortaucipir PET scans have not been studied in the same subjects. To evaluate the heterogeneity across the complete AD spectrum, we studied Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with pathological Aβ biomarker values independently of their cognitive status. We hypothesize that there will be (1) an association between flortaucipir PET and MRI‐derived clusters, albeit with an incomplete overlap in pattern captured with both imaging modalities, due to their different specificities, (2) genetic factors and WMH will modify the associations between the clusters differently because they might exert a stronger effect on specific brain changes, and (3) tau‐derived clusters will have a stronger association with clinical progression based on the previous region of interest–derived findings. To evaluate these hypotheses, we analyzed flortaucipir PET binding and MRI‐measured gray matter atrophy in a large clinical cohort using an advanced unsupervised clustering machine learning method. 26
2. METHODS
2.1. Participants and clinical testing
See ADNI cohort goals in the supporting information. A total of 496 ADNI participants with flortaucipir PET scans, MRI scans, and cerebrospinal fluid (CSF) Aβ1‐42 or florbetapir PET Aβ testing during the same study visit were included (Table 1). Participants had repeated neuropsychological and clinical assessments yearly. Median follow‐up was 105.7 weeks (interquartile range [IQR]: 0 to 361.3 weeks). Further details on the clinical core, recruitment, and diagnostic methods have been previously published, 27 , 28 and details can be found at http://adni.loni.usc.edu/. We included in this study cognitively unremarkable (CU) participants with negative Aβ biomarker values (Aβ–) as a reference. CU, mild cognitive impairment (MCI), and dementia participants with positive Aβ biomarker values (Aβ+) were included in the clustering analyses. All the data is available at http://adni.loni.usc.edu/. All participants provided informed consent.
TABLE 1.
Demographics and clinical characteristics of clustered participants (Aβ positive participants) and cognitively unremarkable participants
| Flortaucipir tau Aβ+ clusters | Structural MRI atrophy Aβ+ clusters | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | CN Aβ–reference (n = 214) | I (n = 181) | II (n = 75) | III (n = 16) | IV (n = 10) | P‐value | Limbic predominant (n = 93) | Hippocampal sparing (n = 91) | Diffuse (n = 98) | P‐value | Missing (%) |
| Age (years) | 71.2 [(67.2)–(76.7)] | 75.58 [(70.5)–(80.2)] | 76.98 [(71.7)–(82.6)] | 74.59 [(70.9)–(78.8)] | 62.48 [(60.6)–(65.1)] | <.0001 | 71.4 [(67.1)–(75.7)] | 76.4 [(70.8)–(81.4)] | 79.8 [(74.22)–(83.5)] | <.0001 | 0.2% |
| Race (% White) | 89.3% | 88.4% | 94.7% | 100% | 100% | .83 | 87.1% | 92.3% | 93.9% | .84 | 1.4% |
| Education (years) | 18 [(16)–(18)] | 16 [(14)–(18)] | 16 [(15)–(18)] | 16 [(14.8)–(18)] | 15.5 [(14.3)–(16.8)] | .024 | 16 [(14)–(18)] | 16 [(15)–(18)] | 16 [(14)–(18)] | .013 | 0% |
| ADAS‐Cog13 | 7.3 [(5.0)–(10.7)] | 9.0 [(5.7)–(13.3)] | 20 [(11.8)–(25.7)] | 31 [(23.2)–(39.8)] | 31.8 [(24.9)–(40.8)] | <.0001 | 9 .0 [(5.3)–(14.8)] | 9.0 [(5.8)–(14.3)] | 20.7 [(12.8)–(29.1)] | <.0001 | 1.4% |
| Florbetapir composite SUVR | 0.73 [(0.71)–(0.75)] | 0.87 [(0.81)–(0.98)] | 1 [(0.94)–(1.06)] | 1.02 [(0.9)–(1.13)] | 1.16 [(1.11)–(1.17)] | <.0001 | 0.9 [(0.83)–(1)] | 0.86 [(0.8)–(0.97)] | 1.01 [(0.93)–(1.11)] | <.0001 | 26.4% |
| CSF Aβ1‐42 (pg/ml) | 1519.8 [(1226.4)–(1899.0)] | 767.1 [(623.4)–(906.9)] | 643.3 [(549.6)–(797.9)] | 525.9 [(451.4)–(698.3)] | 445.9 [(425.0)–(518.5)] | <.0001 | 759.5 [(611.3)–(886.5)] | 720.8 [(584.4)–(858.8)] | 632.6 [(512.7)–(844.2)] | <.0001 | 36.1% |
| WMH volume (cc) | 1.6 [(0.6)–(3.7)] | 2.4 [(1.0)–(7.4)] | 3.8 [(1.6)–(8.0)] | 3.1 [(2.2)–(7.6)] | 2.7 [(1.6)–(3.6)] | <.0001 | 1.7 [(0.7)–(3.6)] | 2.9 [(1.4)–(8.1)] | 4.8 [(2.0)–(11.7)] | <.0001 | 3.4% |
| SPARE‐AD | –1.8 [(–2.5)–(–1.1)] | ‐1.5 [(–2.1)–(–0.63)] | 0.34 [(–0.7)–(1.5)] | 2.1 [(0.35)–(2.7)] | 0.98 [(–0.58)–(3.6)] | <.0001 | –1.5 [(–2.3)–(–0.73)] | –1.4 [(–2.0)–(–0.55)] | 0.9 [(–0.59)–(2.2)] | <.0001 | 6.9% |
| SPARE‐BA (years) | 71.2 [(65.4)–(76.4)] | 74.3 [(69.4)–(81.2)] | 79.0 [(72.9)–(84.2)] | 76.1 [(72.3)–(81.9)] | 73.8 [(66.6)–(74.6)] | <.0001 | 70.3 [(65.2)–(73.0)] | 76.9 [(71.9)–(82.0)] | 81.8 [(75.8)–(84.9)] | <.0001 | 7.1% |
| Flortaucipir Braak I (SUVRs) | 1.6 [(1.5)–(1.8)] | 1.8 [(1.5)–(2.0)] | 2.6 [(2.4)–(2.9)] | 3.1 [(2.5)–(3.9)] | 2.6 [(2.5)–(3.3)] | <.0001 | 1.8 [(1.6)–(2.3)] | 1.8 [(1.6)–(2.1)] | 2.5 [(2.0)–(3.0)] | <.0001 | 0.4% |
| Flortaucipir Braak III‐IV (SUVRs) | 1.4 [(1.3)–(1.5)] | 1.4 [(1.4)–(1.6)] | 1.7 [(1.6)–(2.0)] | 2.8 [(2.5)–(3.3)] | 2.9 [(2.6)–(3.4)] | <.0001 | 1.5 [(1.4)–(1.6)] | 1.5 [(1.4)–(1.6)] | 1.7 [(1.51)–(2.24)] | <.0001 | 0.4% |
| Flortaucipir Braak V–VI (SUVRs) | 1.5 [(1.4)–(1.5)] | 1.5 [(1.4)–(1.7)] | 1.6 [(1.5)–(1.8)] | 2.5 [(2.0)–(2.7)] | 3.3 [(2.7)–(4.0)] | <.0001 | 1.5 [(1.4)–(1.7)] | 1.6 [(1.5)–(1.7)] | 1.68 [(1.51)–(1.95)] | <.0001 | 0.4% |
| APOE ε4 (% present) | 23.7% | 43.8% | 72.6% | 57.1% | 62.5% | <.0001 | 61.5% | 41.2% | 55.4% | <.0001 | |
| Sex (% male) | 39.3% | 48.1% | 49.3% | 37.5% | 60% | .25 | 25.8% | 63.7% | 55.1% | <.0001 | 0% |
| Clinical diagnosis | |||||||||||
| CU | 100% | 66.9% | 26.7% | 18.8% | 0% | 63.4% | 63.4% | 59.3% | 31.6% | <.0001 | 0% |
| MCI | 0% | 26% | 46.7% | 25% | 30% | 32.3% | 32.3% | 30.8% | 31.6% | ||
| Dementia | 7.2% | 26.7% | 56.2% | 70% | 4.3% | 4.3% | 9.9% | 36.7% | |||
Abbreviations: Aβ, amyloid beta; ADAS‐Cog13, Alzheimer's Disease Assessment Scale–13‐item cognitive subscale; APOE, apolipoprotein E; CSF, cerebrospinal fluid; CU, cognitively unremarkable; MCI, mild cognitive impairment; SPARE‐AD, Spatial Pattern of Abnormality for Recognition of Early Alzheimer's Disease; SPARE‐BA, Spatial Pattern of Atrophy for Recognition of Brain Aging (SPARE‐BA); SUVR, standardized uptake value ratio; WMH, white matter hyperintensities.
2.2. MRI and PET acquisition and processing
3T sagittal magnetization‐prepared rapid gradient echo scans for each subject were selected at the same clinical visit as the flortaucipir scan and were segmented and parcellated with FreeSurfer (v 5.3). 29 WMH volume was estimated as previously described. 30 Additional details for the imaging processing can be found on the ADNI website (http://www.adni‐info.org/).
The Spatial Pattern of Abnormality for Recognition of Early Alzheimer's Disease 31 (SPARE‐AD) index is an imaging signature used to estimate AD‐like atrophy patterns in the brain and has been previously validated. 32 , 33 The Spatial Pattern of Atrophy for Recognition of Brain Aging (SPARE‐BA) model 34 is a multivariate pattern regression model based on support vector regression to predict individualized brain, capturing typical versus atypical or advanced brain aging, for each participant. 34 The brain age gap was calculated as the difference between SPARE‐BA and age. See supporting information for additional details.
For the florbetapir composite score, means from the gray matter in subregions were extracted within four large regions (frontal, anterior/posterior cingulate, lateral parietal, lateral temporal), 35 , 36 and weighted means for each of the four main regions were created. A composite was used as a reference region, based on the whole cerebellum, brain stem/pons, and eroded subcortical white matter.
See supporting information and the ADNI website (http://adni.loni.usc.edu/) for information on flortaucipir and florbetapir PET scan processing.
RESEARCH CONTEXT
Systematic Review: We searched PubMed using the terms “Alzheimer's disease,” “PET,” “MRI,” and since January 1, 1990. Unsupervised clustering of positron emission tomography and magnetic resonance imaging in the same participants and differential association with genetics, brain aging, and vascular pathology has not been explored.
Interpretation: We identified a differential contribution of genetics and white matter hyperintensities to tau and atrophy clusters, respectively. We also included our machine learning‐based indices to evaluate Alzheimer's disease (AD) and brain aging patterns. We showed how one of the atrophy clusters (hippocampal sparing) had less AD‐like and more brain aging atrophy patterns.
Future Directions: Our data‐driven approach demonstrated the importance of multimodal imaging to capture pathological aspects of neurodegeneration. Future extensions include additional biomarker modalities to provide sophisticated personalized predictions of individuals’ brain injury. This approach will inform and enrich future clinical trials.
2.3. Cerebrospinal fluid collection and Aβ1‐42 measurements
CSF samples were obtained in the morning after an overnight fast and processed as previously described 37 , 38 (http://adni.loni.usc.edu/). Roche Elecsys Aβ1‐42 CSF immunoassay measurements were performed at the University of Pennsylvania/ADNI biomarker laboratory following the Roche Study protocol. 22 The cutoff for abnormal Aβ1‐42 was 977 pg/ml. 39 Measurements performed during the same ADNI yearly visit were selected (12 days median time interval between CSF draw and PET scans).
2.4. Robust collaborative clustering approach
The robust collaborative clustering (RCC) approach exploits correlations between the subjects and the features while reducing the influence of noise and outliers, thus achieving robustness against noise in the data. 26 , 40 RCC uses the matrix tri‐factorization technique to simultaneously cluster subjects and features into heterogeneous groups. The grouping information of features facilitates the clustering of subjects and vice versa. RCC views the matrix tri‐factorization as a dictionary learning procedure and further integrates sparsity regularization into the system for data denoising to solve the optimization problem. 26 RCC improves both the clustering of subjects and the features (here, regional MRI volumes and flortaucipir standardized uptake value ratios [SUVRs], respectively) by incorporating the interaction of subject clustering and feature clustering. The RCC algorithm was implemented in MATLAB, in which the matrix tri‐factorization module used the toolbox. 41 See supporting information for additional details. RCC can be downloaded from https://github.com/UTHSCSA‐NAL/RCC‐Code.git. Clusters are presented in Figures 1 and 2.
FIGURE 1.
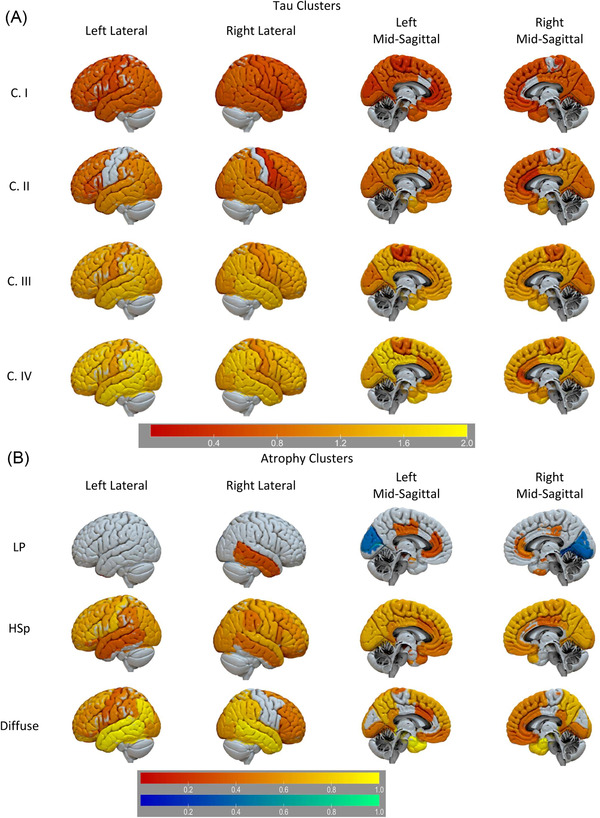
Flortaucipir tau PET (A) and MRI atrophy (B) clusters. A, Regions with greater (red‐yellow) flortaucipir PET SUVRs compared to the Aβ– cognitively unremarkable (CU) participants (reference). B, Regions with greater (red‐yellow) and lower (blue‐green) MRI‐defined atrophy compared to the Aβ– CU participants (reference). Color‐scale reflects log‐scales t‐score. Aβ, amyloid beta; HSp, hippocampal sparing; LP, limbic predominant; MRI, magnetic resonance imaging; PET, positron emission tomography; SUVR, standardized uptake value ratio
FIGURE 2.
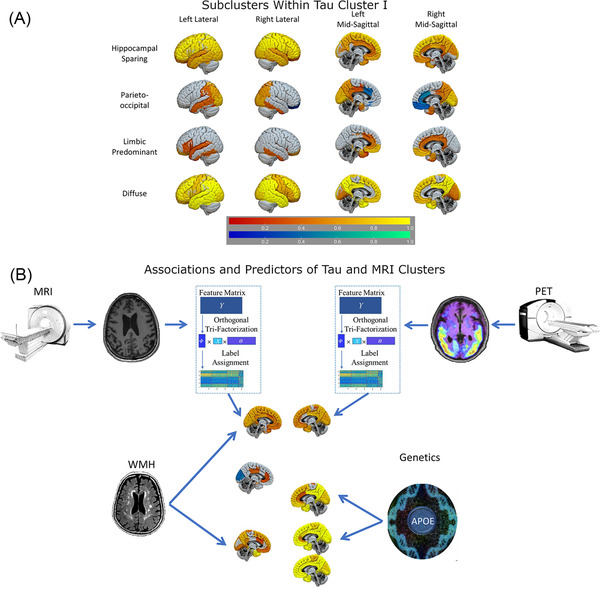
Flortaucipir tau PET subclusters and cluster modeling. A, Subclusters originated from further clustering of tau cluster I. Regions with greater (yellow‐red) and lower (blue‐green) AV‐1451 PET binding compared to the Aβ‐posterior cluster (reference). Color‐scale reflects log‐scales t‐score. B, Multimodal evaluation of biomarker heterogeneity in subjects across the Alzheimer's disease spectrum. Neuroimaging modalities present different associations with vascular and genetic risk factors Aβ, amyloid beta; MRI, magnetic resonance imaging; PET, positron emission tomography
2.5. Statistical analysis
The demographic table includes median and IQR values. Group comparisons were performed applying Kruskal‐Wallis analyses for quantitative variables and Chi‐square tests for categorical variables in Table 1. Power transformations were used in further analyses to normalize the distribution of quantitative variables as needed. Post hoc comparisons between groups were computed using Tukey's honest significance differences. Clinical progression from CU to MCI/dementia and a second model for MCI to dementia was evaluated using Cox proportional hazards models that include sex and age as predictors, together with the imaging clusters, and we present hazard ratio (HR) values. Longitudinal changes in Alzheimer's Disease Assessment Scale–13‐item cognitive subscale (ADAS‐Cog13) were evaluated using linear mixed‐effects models with imaging clusters, clinical diagnosis, sex, and age as fixed effects. Subjects and time were included as random effects (nlme R package). Associations between atrophy and tau clusters were evaluated with multinomial logistic regression models; we evaluated if each of the clusters within a modality predicted the clusters in the other imaging modality (e.g., if the diffuse atrophy cluster was associated with a greater probability of belonging to tau cluster II versus cluster I, compared to the other two atrophy clusters). We included APOE ε4 presence and WMH volume as predictors. We used a backward selection model to include all predictors to select the best model. Participants with a missing value in a variable included in a specific model were excluded from that particular analysis. Imaging t‐test analyses were implemented using the MATLAB library package. P‐values <.05 (two‐sided) were considered statistically significant. Bonferroni‐Holm multiple comparison correction was applied. Analyses were performed using R version 4.1.0.
3. RESULTS
3.1. Tau cluster patterns
We identified four tau clusters (Table 1). Figure 1 compares the flortaucipir SUVRs from the four Aβ+ clusters versus the Aβ– CU reference participants. The tau clusters (I–IV) presented a gradient of increasing binding. Florbetapir composite score values and flortaucipir SUVRs were lower in cluster I than in clusters II–IV (Figure S1 and Table S5 in supporting information). There were no differences in florbetapir SUVRs among clusters II–IV. Conversely, all tau clusters showed differences in the averaged Braak V–VI area SUVRs and only clusters III and IV showed similar values in averaged Braak III–IV area SUVRs. Clusters II–IV showed no differences in flortaucipir Braak I area SUVRs (Figure S1 and Table S5). Cluster I, the largest cluster, was subclustered in a sensitivity analysis, identifying limbic predominant, parieto‐occipital, hippocampal sparing, and diffuse subclusters (Figure 2).
3.2. Cross‐sectional characteristics of the tau clusters
APOE ε4 prevalence was higher in the tau clusters I–III than the Aβ– CU group (Table S4 in supporting information). Within the tau clusters, there was a progressive increase of cognitively impaired participants and ADAS‐Cog13 scores (Figure 3, Tables S4 and Table S5) in supporting information. Tau clusters I and II participants were older than the Aβ– CU group, whereas the tau cluster IV participants were younger. SPARE‐AD was higher in clusters II–IV compared to the reference group (P‐value < .0001) and cluster I (P‐value < .0001). The brain age gap was higher in clusters II–IV compared to the reference group (P‐value < .025) and in cluster IV compared to the other tau clusters (P‐value < .006; Figure 4A).
FIGURE 3.
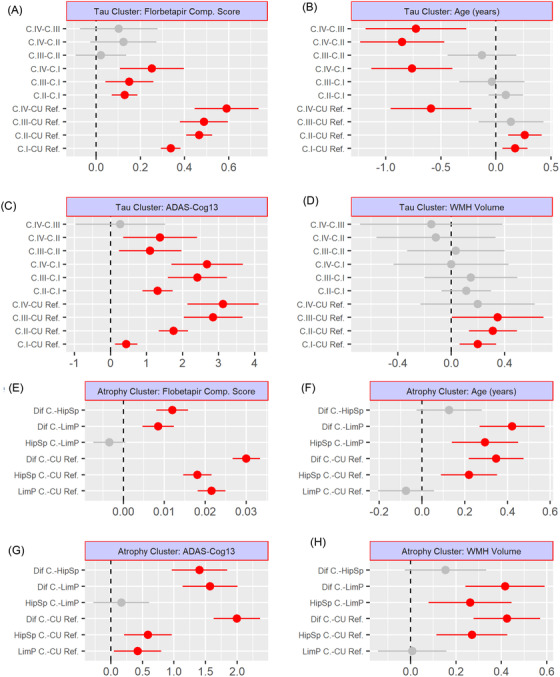
Post hoc comparison among tau (A–D) and atrophy (E–H) clusters. Florbetapir composite score (A, E), age (B, F), ADAS‐Cog13 (C, G), and WMH volume (D, H). ADAS‐Cog13, Alzheimer's Disease Assessment Scale, 13‐item Cognitive subscale; C., cluster; CU, cognitively unremarkable; Dif., diffuse; HSp, hippocampal sparing; LP, limbic predominant; Ref., reference (CU amyloid beta–negative participants); WMH, white matter hyperintensity
FIGURE 4.
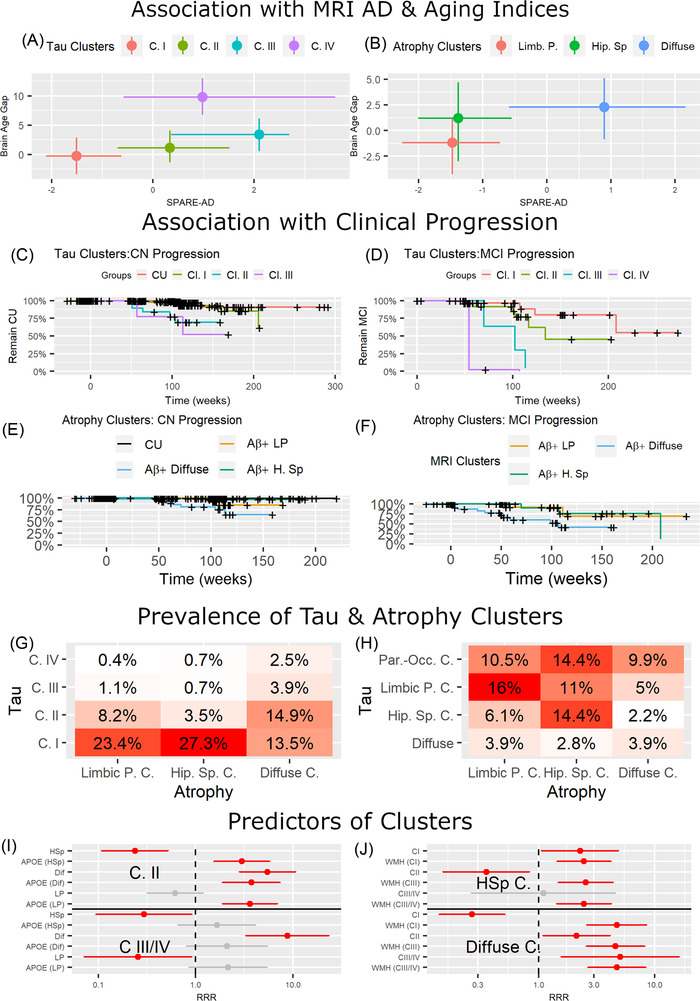
Biomarker and clinical association of imaging clusters. A, Plot showing the median and interquartile range of Spatial Pattern of Abnormality for Recognition of Early Alzheimer's disease 31 (SPARE‐AD) (x‐axis) and brain age gap (y‐axis) for the four flortaucipir clusters. B, Clinical progression from cognitively unremarkable (CU) to mild cognitive impairment (MCI)/dementia for tau clusters. C, Clinical progression from MCI to dementia for tau clusters. D, Plot showing the median and interquartile range of SPARE‐AD (x‐axis) and brain age gap (y‐axis) for the three magnetic resonance imaging (MRI) clusters. E, Clinical progression from CU to MCI/dementia for atrophy clusters. F, Clinical progression from MCI to dementia for atrophy clusters. G, Prevalence for flortaucipir and MRI cluster combinations. H, I, Relative risk ratio (RRR) of multinomial logistic regression models evaluating the association between atrophy and tau clusters. In (H), multinomial logistic regression models with limbic predominant cluster as reference and white matter hyperintensities (WMH) volume and tau clusters as predictors (on the y‐axis). In (I), multinomial logistic regression models with tau cluster I as reference and apolipoprotein E ε4 and MRI clusters as predictors (on the y‐axis). The red color indicates significant associations. Gray color indicates non‐significant associations. X‐scale is log‐scaled. J, Prevalence for flortaucipir subcluster and MRI cluster combinations. HSp, hippocampal sparing; LP, limbic predominant
3.3. Longitudinal characteristics of tau clusters
Tau clusters II (HR = 15.9, P‐value = .0001) and III (HR = 17.0, P‐value = .0009) were associated with faster progression from CU to MCI or dementia compared to the CU Aβ– reference group (cluster IV included no CU participants; Figure 4C,D). Tau clusters II (HR = 6.2, P‐value = .005) and III (HR = 8.0, P‐value = .013) remained significantly associated with progression when we evaluated progression from CU to MCI using the tau cluster I as reference. Tau clusters III (HR = 24.8, P‐value = .002) and IV (HR = 294.4, P‐value = .002) were associated with faster progression from MCI to dementia than cluster I. Clusters II–IV showed a more rapid cognitive decline than the CU Aβ– group and tau cluster I. Conversely, cluster I presented no longitudinal ADAS‐Cog 13 score differences (Table 2).
TABLE 2.
Association of cluster and clinical diagnosis with longitudinal ADAS‐Cog13 scores
| Cognitively unremarkable Aβ negative reference | |||||
|---|---|---|---|---|---|
| Flortaucipir clusters | MRI clusters | ||||
| Variables | Coefficient (S.E.) | P‐value | Variables | Coefficient (S.E.) | P‐value |
| C. I | 0.1 (0.12) | .37 | C. LP | 0.2 (0.19) | .35 |
| C. II | 1.0 (0.17) | <.0001 | C. HSp | 0.1 (0.16) | .52 |
| C. III | 3.1 (0.32) | <.0001 | C. Diffuse | 0.9 (0.19) | <.0001 |
| C. IV | 4.5 (0.51) | <.0001 | |||
| MCI | 0.2 (0.14) | .13 | MCI | 0.4 (0.17) | .009 |
| Dementia | 1.0 (0.19) | <.0001 | Dementia | 1.8 (0.23) | <.0001 |
| Aβ positive reference | |||||
|---|---|---|---|---|---|
| C. II | 0.9 (0.18) | <.0001 | C. HSp | ‐0.1 (0.24) | .81 |
| C. III | 3.1 (0.36) | <.0001 | C. Diffuse | 0.8 (0.26) | .0021 |
| C. IV | 4.8 (0.59) | <.0001 | |||
| MCI | 0.2 (0.17) | .16 | MCI | 0.5 (0.22) | .022 |
| Dementia | 1.1 (0.24) | <.0001 | Dementia | 2 (0.3) | <.0001 |
Abbreviations: Aβ, amyloid beta; ADAS‐Cog13, Alzheimer's Disease Assessment Scale–13‐item cognitive subscale; HSp, hippocampal sparing; LP, limbic predominant; MCI, mild cognitive impairment; MRI, magnetic resonance imaging; S.E., standard error.
Notes: Reference group for the analyses was the cognitively unremarkable Aβ negative participants on the top. Bottom half of the table had tau cluster I and limbic predominant clusters as references for the tau and MRI analyses, respectively.
3.4. Atrophy clustering patterns
We identified three atrophy clusters among the Aβ+ participants (Table 1). A limbic predominant atrophy cluster showed greater cingulate and right hippocampus, inferior and middle temporal lobe atrophy than the CU Aβ– group (Figure 1). The diffuse atrophy cluster showed greater parieto–occipital–temporal atrophy. The “hippocampus sparing” atrophy cluster showed a similar atrophy pattern except for greater atrophy in the temporal lobe.
3.5. Cross‐sectional characteristics of atrophy clusters
The limbic predominant cluster participants had a higher prevalence of APOE ε4 than the hippocampal sparing cluster participants (Table 1 and Table S6 in supporting information). The three clusters had higher ADAS‐Cog13 and flortaucipir SUVR binding in all the Braak staging‐defined areas than the CU Aβ– reference group (Figure 3, Figure S1, and Table S7 in supporting information). The diffuse atrophy cluster had higher ADAS‐Cog13 scores, florbetapir composite scores, and flortaucipir SUVRs in all the Braak staging‐defined areas than the two other atrophy clusters. The diffuse atrophy cluster also included the largest number of cognitively impaired participants. The hippocampal sparing atrophy cluster participants were older and had higher WMH volumes than the limbic predominant atrophy cluster and the CU Aβ– reference group participants.
When we evaluated the SPARE‐AD and brain age gap, we observed that the diffuse atrophy cluster showed higher SPARE‐AD values than the hippocampal sparing (P < .0001) and limbic predominant atrophy clusters (P < .0001; Figure 4B). When we evaluated the SPARE‐BA mismatch, both the diffuse (P < .0001) and the hippocampal sparing atrophy cluster (P = .0004) showed larger values than the limbic predominant atrophy cluster.
3.6. Longitudinal characteristics of atrophy clusters
Only the diffuse atrophy cluster was associated with faster progression from CU to MCI/dementia (HR = 16.6, P‐value < .0001) compared to the CU Aβ– group and from MCI to dementia compared to the limbic predominant atrophy cluster (HR = 4.4, P‐value = .042; Figure 4E,F). The diffuse atrophy cluster remained significantly associated with clinical progression (HR = 6.1, P‐value = .022) when we evaluated progression from CU to MCI including using the limbic predominant cluster as reference. Similarly, only the diffuse atrophy cluster presented a faster ADAS‐cog13 decline (Table 2).
3.7. Comparison of atrophy and tau cluster membership
The overlap between the different atrophy and tau clusters is summarized in Tables S8‐S11 in supporting information and Figure 4G‐I. APOE ε4 presence was a predictor of tau cluster II. Conversely, WMH volume was a significant predictor of atrophy clusters. We observed no interactions between the variables (APOE and WMH) and the imaging clusters (data not shown).
The hippocampal sparing atrophy cluster was associated with a lower relative risk ratio (RRR) of belonging to tau clusters II‐IV and a higher RRR for tau PET cluster I. In contrast, the diffuse atrophy cluster was associated with a greater RRR of belonging to tau clusters II–IV and lower RRR with tau cluster I.
When we evaluated the subclusters within tau cluster I (Figure 4J), the tau limbic predominant subcluster was mainly composed of limbic predominant atrophy cluster participants (50%), and the hippocampal sparing tau subcluster mainly consisted of hippocampal sparing atrophy cluster participants (63.4%). The hippocampal sparing atrophy cluster was evenly distributed between the tau hippocampal sparing and parieto‐occipital subclusters (33.8% each).
4. DISCUSSION
To our knowledge, this is the first article that presents structural MRI and flortaucipir PET scan clustering across the biomarker‐defined AD continuum. Our data‐driven clustering approach identified partially overlapping tau and atrophy clusters. MRI atrophy‐defined clusters represented three different spatial atrophy patterns, whereas flortaucipir binding‐defined clusters identified progressive tau accumulation. Spatial distribution‐based flortaucipir subclusters were identified within the least severe tau cluster (cluster I). Tau clusters showed a stronger association with cross‐sectional and longitudinal clinical measures than MRI atrophy‐defined clusters. Finally, associations between atrophy‐defined and flortaucipir binding‐defined clusters were differentially modulated by APOE genotype and WMH volume (Figure 2). Our results imply that stratification approaches across different imaging modalities capture various aspects of genetic, neurodegenerative, and vascular processes and how genetic and vascular lesions modulate the association between neuroimaging modalities. These findings have implications for designing personalized multimodal imaging‐based approaches for individualized prognosis and stratification in the era of precision medicine.
4.1. Tau clusters
Tau clusters showed progressive accumulating tau deposition. Clusters I and II showed higher tracer binding in the medial and lateral temporal areas. Clusters III and IV had the highest flortaucipir binding and only showed statistical differences in flortaucipir SUVRs in Braak areas V and VI. Cluster IV included younger participants than all the other clusters. A higher tau deposition in younger cognitively impaired subjects has previously been reported in autopsy studies and a previous flortaucipir clustering article. 25 , 42 This higher flortaucipir binding likely represents a combination of faster deposition in young‐onset subjects, greater cognitive reserve, and a lower degree of co‐pathologies. This is supported by a similar performance on the ADAS‐Cog13 in tau clusters III and IV. Tau cluster IV showed similar SPARE‐AD scores as clusters II and III, indicating a similar degree of AD‐like brain atrophy but a higher brain age gap than the other tau clusters indicating accelerated brain aging.
Braak area I showed an early celling effect and no differences among the four clusters (Figure S1). Conversely, Braak areas V–VI differed among all the clusters, whereas Braak areas III–IV were similar in the most severe clusters (III and IV). These findings have implications when evaluating summary measures for tau imaging biomarkers.
WMH load was higher in tau clusters I–III compared to the Aβ– CU group. This is consistent with previous findings of larger WMH volumes in AD subjects. 20 , 34 , 43 Tau cluster IV showed no statistical differences in WMH volume compared to the Aβ– CU reference group. This could be a combination of lower statistical power due to sample size. The florbetapir composite score showed no differences in tau clusters II–IV, indicating an earlier ceiling effect for Aβ compared to tau. 24
Tau cluster I showed similar clinical progression rates during follow‐up in terms of progression to MCI/dementia or ADAS‐Cog 13 changes as the Aβ– CU reference group. Therefore, this cluster likely represents an early stage in the AD continuum in which biomarker changes are present but not leading to a significantly faster decline. On the other hand, tau clusters II–IV showed a more rapid clinical progression and greater ADAS‐Cog13 increase.
Our sensitivity analysis of the largest tau cluster, cluster I, identified subclusters that had different topographical distributions; a limbic predominant, a hippocampal sparing, a parieto‐occipital, and a diffuse tau PET clusters with similar distributions have been recently described, but their relation to MRI‐based atrophy clusters remained unknown in that study. 25 Our analysis shows that differences among these clusters are present in the earliest disease stages. The results by Vogel et al. 25 also showed that in the highest/most severe SusTain stages there was a convergence of the tau deposition patterns into a single severe cluster. This was an exploratory analysis. Future studies with longer follow‐up and larger sample will be needed to evaluate their clinical implications.
4.2. Association of the atrophy clusters
The limbic predominant, hippocampal sparing, and diffuse atrophy patterns have been described in autopsy and imaging studies. 3 , 16 Our study showed that the hippocampal sparing atrophy cluster had lower APOE ε4 prevalence and greater WMH volume than the limbic predominant atrophy cluster. However, the hippocampal sparing and the limbic predominant atrophy clusters had similar Braak area‐defined flortaucipir SUVRs, florbetapir composite, and ADAS‐Cog13 scores. Hippocampal sparing participants were older, and their atrophy also matched the one associated with pathological brain aging, 17 rather than the one observed in AD, 31 , 34 as demonstrated by the SPARE‐AD and brain age gap analyses. Brain aging patterns have been associated with a predominant frontoparietal pattern and less of a medial temporal pattern, more characteristic of AD dementia. 31 , 33 These would indicate that age‐related mechanisms other than AD pathology might cause atrophy and associated cognitive impairment. 1 , 44 , 45 Previous reports showed that age‐related pathologies could impact the regional neurodegeneration patterns and coexist with AD, 17 , 19 with increased WMH in diffuse AD atrophy patterns. 14 Our analyses indicate greater WMH importance in participants with a neocortical atrophy pattern, who were also older. Previous studies have described the largest burden of WMH volumes in limbic predominant cases with intermediate burden in hippocampal sparing cases. 46 One difference is that the study by Ferreira et al. 46 included participants with dementia, whereas participants in our limbic predominant and hippocampal sparing groups mainly included CU and MCI participants. WMHs explained a greater variance of the MRI than the tau cluster grouping. They were associated with the hippocampal sparing and diffuse atrophy clusters when evaluating the association with the tau clusters. This indicates that WMHs are related to the imaging clusters (and patterns of atrophy), but not with the tau clusters (burden of tau deposition). WMHs represent a combination of axonal loss resulting from demyelination, Wallerian degeneration, and small vessel disease related to brain atrophy, but not conditioned by the tau burden based on our results. 19 , 43 , 47
4.3. Comparison of structural MRI and flortaucipir clusters
The hippocampal sparing atrophy cluster was associated with tau cluster I, which had the lowest flortaucipir binding. The lower Aβ biomarker values indicate that this atrophy cluster is most likely the result of non‐AD pathology. Therefore, this atrophy pattern is probably unrelated to the hippocampal sparing tau distribution subtype described by neuropathological studies. 3
Conversely, tau clusters II–IV were associated with an increased presence of a diffuse atrophy cluster. This indicated that increasing tau pathology is associated with a classic diffuse AD pattern. This atrophy cluster showed the largest atrophy in the temporal lobe and an overall distribution matching Braak's neurofibrillary tangle staging. 48 This is consistent with neuropathological studies showing that AD pathology (mainly tau) explains the largest proportion of cognitive impairment in subjects with dementia.
The limbic predominant atrophy cluster was associated with an increased risk of being in tau cluster II and a lower risk of being a member of tau clusters III/IV, indicating that it corresponds to an intermediate stage along the AD pathology continuum. Brain bank studies and initial ADNI neuropathology studies have shown multiple pathologies in the brain of aging individuals with cognitive impairment. 1 , 44 , 49 Therefore, it is not unexpected that there is incomplete overlap between a modality that captures deposition of a single misfolded protein (flortaucipir) versus a downstream imaging modality that captures various brain insults (brain MRI).
When the fit of the different longitudinal models was compared, tau clusters showed a lower Akaike information criterion (AIC; better fit) for the ADAS‐Cog13 and the MCI to dementia progression models (Table S12 in supporting informaiton). In the model evaluating progression of CU participants, the model that evaluated the MRI clusters had a lower AIC (although value was close to PET clusters).
4.4. Strengths and limitations
Previous studies evaluated neuroimaging heterogeneity in AD patterns using a hypothesis‐driven study design that classified individual MRI scans into predefined subtypes based on pathological or cognitive profiles demonstrating a rough typical versus atypical AD presentation. 50 , 51 , 52 , 53 These studies suffer from various limitations, including using a priori groups and thresholds, which could not discover patterns beyond the prior definitions of the groups using arbitrary thresholds to define these groups. Therefore, increasing attention has been given to unsupervised clustering approaches on regional neuroimaging features, mainly MRI, to uncover the heterogeneity in AD 6 by finding groups with similar neuroimaging patterns. From a methodological perspective, approaches mainly differ in (1) clustering techniques, (2) validation methods, and (3) features used. Our method defines the groups at an individual level while at the same time fully exploiting correlations between the features, grouping them in covariance patterns, increasing clustering robustness. Compared to classic clustering methods (k‐means and k‐medoids), RCC exploits differences between features and uses the differences between heterogeneous groups of samples to learn a discriminative low dimensional representation. Compared to previous matrix tri‐factorization techniques, 26 , 54 RCC uses adaptive sparsity regularization to make the matrix tri‐factorization performance robust against noise. After denoising via sparsity regularization, the collaborative clustering procedure is more robust, leading to more accurate grouping of both samples and features for clinical analysis. We also included machine learning‐derived brain indices to assess the degree of AD‐like and brain aging atrophy. 31 , 32 , 34
Our paper is among the first to define AD spectrum subtypes using an advanced imaging machine learning method. 6 , 25 , 55 , 56 Most data‐driven neuroimaging studies on AD subtyping and related methodological development have focused on MRI data. 6 The number of studies performing primary clustering on flortaucipir scans is more limited. 25 A difference in our study is that we performed simultaneous clustering of structural MRI data and flortaucipir PET scans.
Limitations of the study include the relatively short clinical follow‐up, the large proportion of CU participants, the small number of participants in tau clusters III and IV, and the limited representation of minority and underserved populations. In addition, unsupervised learning techniques like clustering aim to discover and measure the differences between the data samples, based on which the underlying data groups are identified. However, in the neuroimaging applications, the clustering method does not differentiate whether such differences come from different stages or from different subtypes. As a result, the clustering results can reflect both stages and subtypes. Adjusting for averaged Braak area flortaucipir scores led to a three cluster solution which also was based on severity (data not shown).
5. CONCLUSION
The unsupervised clustering approach identified an incomplete overlap between the neuroimaging patterns. These results indicate that each neuroimaging modality captures different aspects of heterogeneity across the Aβ cognitive impairment spectrum. Moreover, APOE genotype and WMH differentially modified the association of the clusters and brain aging patterns were only associated with the brain atrophy patterns, not tau. Finally, tau clusters showed a stronger association with clinical progression. Atrophy clusters identified different atrophy patterns, likely reflecting different patterns of co‐pathology, which is consistent with MRI capturing downstream changes of multiple brain pathologies. Our analyses point to the complexity of numerous pathologies that can present different distributions with additive deleterious effects on the brain and are captured in various degrees by different neuroimaging modalities. Future studies should assess TDP‐43 and α‐synuclein pathology, which cannot be evaluated with current neuroimaging tools, and additional dementia risk factors. Precise determination of injury patterns across complementary biomarker data offers the opportunity to phenotype these subjects and identify how different contributing factors affect their brains. This phenotypic characterization has implications for clinical trial recruitment and stratification and individual subjects’ prognosis.
CONFLICTS OF INTEREST
Dr. Shaw provides quality control oversight for the Roche Electrosys immunoassay platform as part of the ADNI‐3 study. Dr. Weiner has served on the Scientific Advisory Boards for Alzheon, Inc., Accera, Merck, Nestle (Nolan), PCORI (PPRN), Eli Lilly, Delfino Logic Ltd. (for Merck), Dolby Ventures, Brain Health Registry, and ADNI. He served on the editorial boards for Alzheimer's & Dementia and MRI. He has provided consulting and/or acted as a speaker/lecturer to Synarc, Pfizer, Accera, Inc., Alzheimer's Drug Discovery Foundation (ADDF), Merck, BioClinica, Eli Lilly, Howard University, Guidepoint, Denali Therapeutics, Nestle/Nestec, GLG Research, Atheneum Partners, BIONEST Partners, American Academy of Neurology (AAN), and Society for Nuclear Medicine and Molecular Imaging (SNMMI). Other authors report no competing interests.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
Jon B. Toledo is supported by the Edmond J. Safra Fellowship in Movement Disorders. Michel J. Grothe is supported by the “Miguel Servet” program (CP19/00031) and a research grant (PI20/00613) of the Instituto de Salud Carlos III‐Fondo Europeo de Desarrollo Regional (ISCIII‐FEDER). This study was supported in part by the National Institute of Health (NIH) grant P30AG066546 (South Texas Alzheimer's Disease Research Center) and grant numbers 5R01HL127659, 1U24AG074855 and the San Antonio Medical Foundation grant SAMF–1000003860. Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Toledo JB, Liu H, Grothe MJ, et al., & for the Alzheimer's Disease Neuroimaging Initiative* . Disentangling tau and brain atrophy cluster heterogeneity across the Alzheimer's disease continuum. Alzheimer's Dement. 2022;8:e12305. 10.1002/trc2.12305
Contributor Information
Jon B. Toledo, Email: jtoledoatucha@ufl.edu.
Mohamad Habes, Email: habes@uthscsa.edu.
REFERENCES
- 1. Toledo JB, Cairns NJ, Da X, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun. 2013;1:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murray ME, Graff‐Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer's disease: clinical, neuroimaging and pathological features. Brain. 2015;138:2732‐2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Habes M, Grothe MJ, Tunc B, McMillan C, Wolk DA, Davatzikos C. Disentangling heterogeneity in Alzheimer's disease and related dementias using data‐driven methods. Biol Psychiatry. 2020;88:70‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimer's Dement. 2017;13:870‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168‐174. [DOI] [PubMed] [Google Scholar]
- 10. Ferreira D, Nordberg A, Westman E. Biological subtypes of Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2020;94:436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397:1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer's dementia attributed to age‐related neuropathologies. Ann Neurol. 2019;85:114‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stern Y, Arenaza‐Urquijo EM, Bartres‐Faz D, et al. Whitepaper: defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer's Dement. 2020;16:1305‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong A, Toledo JB, Honnorat N, et al. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer's disease: links to cognition, progression and biomarkers. Brain. 2017;140:735‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferreira D, Nedelska Z, Graff‐Radford J, et al. Cerebrovascular disease, neurodegeneration, and clinical phenotype in dementia with Lewy bodies. Neurobiol Aging. 2021;105:252‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whitwell JL, Dickson DW, Murray ME, et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer's disease: a case‐control study. Lancet Neurol. 2012;11:868‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Habes M, Erus G, Toledo JB, et al. White matter hyperintensities and imaging patterns of brain ageing in the general population. Brain. 2016;139:1164‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Habes M, Erus G, Toledo JB, et al. Regional tract‐specific white matter hyperintensities are associated with patterns to aging‐related brain atrophy via vascular risk factors, but also independently. Alzheimers Dement (Amst). 2018;10:278‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Habes M, Sotiras A, Erus G, et al. White matter lesions: spatial heterogeneity, links to risk factors, cognition, genetics, and atrophy. Neurology. 2018;91:e964‐e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kandel BM, Avants BB, Gee JC, et al. White matter hyperintensities are more highly associated with preclinical Alzheimer's disease than imaging and cognitive markers of neurodegeneration. Alzheimers Dement (Amst). 2016;4:18‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saint‐Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez‐Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener. 2017;12:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jack CR, Jr. , Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27:871‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu H, Li H, Habes M, et al. Robust collaborative clustering of subjects and radiomic features for cancer prognosis. IEEE Trans Biomed Eng. 2020;67:2735‐2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aisen PS, Petersen RC, Donohue MC, et al. Clinical core of the Alzheimer's disease neuroimaging initiative: progress and plans. Alzheimer's Dement. 2010;6:239‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Veitch DP, Weiner MW, Aisen PS, et al. Understanding disease progression and improving Alzheimer's disease clinical trials: recent highlights from the Alzheimer's Disease Neuroimaging Initiative. Alzheimer's Dement. 2019;15:106‐1052. [DOI] [PubMed] [Google Scholar]
- 29. Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72:578‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carmichael O, Mungas D, Beckett L, et al. MRI predictors of cognitive change in a diverse and carefully characterized elderly population. Neurobiol Aging. 2012;33:83‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davatzikos C, Xu F, An Y, Fan Y, Resnick SM. Longitudinal progression of Alzheimer's‐like patterns of atrophy in normal older adults: the SPARE‐AD index. Brain. 2009;132:2026‐2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Da X, Toledo JB, Zee J, et al. Integration and relative value of biomarkers for prediction of MCI to AD progression: spatial patterns of brain atrophy, cognitive scores, APOE genotype and CSF biomarkers. NeuroImage Clin. 2014;4:164‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Habes M, Janowitz D, Erus G, et al. Advanced brain aging: relationship with epidemiologic and genetic risk factors, and overlap with Alzheimer disease atrophy patterns. Transl psychiatry. 2016;6:e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Habes M, Pomponio R, Shou H, et al. The Brain Chart of Aging: machine‐learning analytics reveals links between brain aging, white matter disease, amyloid burden, and cognition in the iSTAGING consortium of 10,216 harmonized MR scans. Alzheimer's Dement. 2021;17:89‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal‐mediated beta‐amyloid deposition in elderly subjects. Brain. 2009;132:1310‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 2011;121:597‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer's disease or dementia using novel Elecsys Abeta(1‐42), pTau and tTau CSF immunoassays. Sci Rep. 2019;9:19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu H, Grothe MJ, Rashid T, et al. ADCoC: Adaptive distribution modeling based collaborative clustering for disentangling disease heterogeneity from neuroimaging data. IEEE Transactions on Emerging Topics in Computational Intelligence. 2022:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y, Ngom A. The non‐negative matrix factorization toolbox for biological data mining. Source Code Biol Med. 2013;8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aoyagi A, Condello C, Stohr J, et al. Abeta and tau prion‐like activities decline with longevity in the Alzheimer's disease human brain. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gouw AA, Seewann A, Vrenken H, et al. Heterogeneity of white matter hyperintensities in Alzheimer's disease: post‐mortem quantitative MRI and neuropathology. Brain. 2008;131:3286‐3298. [DOI] [PubMed] [Google Scholar]
- 44. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain. 2013;136:2697‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Josephs KA, Whitwell JL, Weigand SD, et al. TDP‐43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127:811‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferreira D, Shams S, Cavallin L, et al. The contribution of small vessel disease to subtypes of Alzheimer's disease: a study on cerebrospinal fluid and imaging biomarkers. Neurobiol Aging. 2018;70:18‐29. [DOI] [PubMed] [Google Scholar]
- 47. Leys D, Pruvo JP, Parent M, et al. Could Wallerian degeneration contribute to “leuko‐araiosis” in subjects free of any vascular disorder? J Neurol Neurosurg Psychiatry. 1991;54:46‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain. 2018;141:2181‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferreira D, Verhagen C, Hernandez‐Cabrera JA, et al. Distinct subtypes of Alzheimer's disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci Rep. 2017;7:46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Risacher SL, Anderson WH, Charil A, et al. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology. 2017;89:2176‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Byun MS, Kim SE, Park J, et al. Heterogeneity of regional brain atrophy patterns associated with distinct progression rates in Alzheimer's disease. PLoS One. 2015;10:e0142756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Young AL, Marinescu RV, Oxtoby NP, et al. Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat Commun. 2018;9:4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu X, Shen F, Yang Y, Zhang D, Tao Shen H, Song J. Matrix tri‐factorization with manifold regularizations for zero‐shot learning. IEEE Conference on Computer Vision and Pattern Recognition; 2017:3798‐807.
- 55. Sun N, Mormino EC, Chen J, Sabuncu MR, Yeo BTT. Alzheimer's Disease Neuroimaging I . Multi‐modal latent factor exploration of atrophy, cognitive and tau heterogeneity in Alzheimer's disease. Neuroimage. 2019;201:116043. [DOI] [PubMed] [Google Scholar]
- 56. Ossenkoppele R, Lyoo CH, Sudre CH, et al. Distinct tau PET patterns in atrophy‐defined subtypes of Alzheimer's disease. Alzheimer's Dement. 2020;16:335‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information