

Abstract
Systematic complex genetic interaction studies have provided insight into high-order functional redundancies and genetic network wiring of the cell. Here, we describe a method for screening and quantifying trigenic interactions from ordered arrays of yeast strains grown on agar plates as individual colonies. The protocol instructs users on the trigenic synthetic genetic array analysis technique, τ-SGA, for high-throughput screens. The steps describe construction of the double mutant query strains and the corresponding single mutant control query strains, which are screened in parallel in two replicates. The screening experimental set-up consists of sequential replica pinning steps that enable automated mating, meiotic recombination and successive haploid selection steps for the generation of triple mutants, which are scored for colony size as a proxy for fitness, which enables the calculation of trigenic interactions. The procedure described here was used to conduct 422 trigenic interaction screens, which generated ~460,000 yeast triple mutants for trigenic interaction analysis. Users should be familiar with robotic equipment required for high-throughput genetic interaction screens and be proficient at the command line to execute the scoring pipeline. Large-scale screen computational analysis is achieved using MATLAB pipelines that score raw colony size data to produce τ-SGA interaction scores. Additional recommendations are included for optimizing experimental design and analysis of smaller scale trigenic interaction screens using a web-based analysis system, SGAtools. This protocol provides a resource for those who would like to gain a deeper, more practical understanding of trigenic interaction screening and quantification methodology.
Introduction
Systematic genetic interaction studies in the budding yeast, Saccharomyces cerevisiae (S. cer.), have been instrumental in deciphering the genotype to phenotype relationship and revealing the general principles of genetic networks. A genetic interaction is observed when mutations in different genes combine to generate an unexpected phenotype given the phenotypes caused by the corresponding individual mutations1. There are two broad classes of genetic interactions: negative and positive. The broadest definition of a negative interaction is a case where the fitness of a double mutant is lower than that expected given the fitness of the corresponding single mutants2. Synthetic lethality is an extreme case of a negative genetic interaction and occurs when mutations in two genes combine to give rise to lethality, whereas neither mutant is lethal on its own3,4. Mapping the conservation of synthetic lethal genetic interactions based on a multi-species approach has been harnessed to develop effective therapeutic combinations for such diseases as cancer revealing the strongest interactions between genes with roles in DNA damage checkpoint, cell cycle checkpoint, topoisomerase and chromatin remodeling5. A classic example of a conserved synthetic lethal genetic interaction, which is used therapeutically, involves PARP (poly ADP ribose polymerase) and BRCA1/2. Cells with a defect in BRCA1 or BRCA2, which are involved in the homologous recombination double-stranded DNA break repair are more sensitive to perturbations in the single-strand DNA break repair pathway through base excisions mediated by PARP6. Negative genetic interactions tend to connect functionally related genes, including genes in functionally-related pathways or protein complexes converging on a common essential function7. Genes involved in the same essential pathway or complex may also exhibit a negative genetic interaction, if each mutation partially reduces the activity of the functional module.
A positive genetic interaction is observed when mutations in different genes result in a less severe phenotype than would be predicted based on single mutant phenotypes2. Further, sub-classification into ‘symmetric’ positive genetic interactions is based on observing equivalent single and double mutant loss-of-function phenotypes. This is often observed between genes encoding members of a non-essential protein complex, because the effect of the deletion of one member is not worsened by the deletion of other members. On the other hand, ‘asymmetric’ positive interactions occur when the single mutant phenotype differs from the double mutant. For example, ‘genetic masking’ is observed when the double mutant is not as sick as predicted and equals to the sickest single mutant, whereas ‘genetic suppression’ occurs when the double mutant is more fit than the sickest single mutant. Positive interactions of non-essential genes overlap with protein-protein interactions thus capturing protein complex membership8. However, positive interactions between essential genes do not show significant molecular or functional relationship and reflect a different kind of relationship, which is not captured by the current large-scale data sets or functional standards8.
Previously, we developed the Synthetic Genetic Array (SGA) analysis method to automate yeast genetics for systematic construction of yeast double mutants and subsequent analysis of genetic interactions using colony size as a read-out for cell fitness2,9. Using large-scale SGA analysis we constructed a global digenic interaction network comprised of ~550,000 negative genetic interactions and ~350,000 positive interactions, by testing ~18 million double mutants8. Grouping genes together based on shared patterns or profiles of genetic interactions revealed a global genetic network composed of functionally-enriched gene modules. Network modules were organized in a hierarchical manner and corresponded to specific pathways and protein complexes, biological processes and subcellular compartments, thus providing a global view of the functional organization of a cell. In a systematic analysis of genetic suppression, which used strains harbouring spontaneous suppressor mutations, a spectrum of adaptive mutations, including gain-of-function, a phenotype that can be dominant to that of its wild-type allele, and separation-of-function mutations, which decouple phenotypes of multifunctional proteins, were uncovered10.
While there are 18 million possible gene pairs in yeast, the number of gene triplets is approximately 36 billion. To sample this complex genetic interaction space, we systematically surveyed trigenic interactions by using key features of the global digenic interaction network to select query genes: (1) digenic interaction strength, (2) average number of digenic interactions and (3) digenic interaction profile similarity11. Upon testing ~400,000 double and ~200,000 triple mutants for fitness defects, we identified ~9,500 digenic and ~3,200 trigenic negative interactions. Trigenic interactions were slightly weaker than digenic interactions, but were similarly functionally informative and were statistically overrepresented for genes that were co-expressed, co-annotated to the same Gene Ontology (GO) terms and encoded physically interacting proteins. Despite their functional enrichment, trigenic interactions often bridged more distant biological processes. Based on our statistical extrapolations, we estimate that the global trigenic interaction network is ~100-fold more extensive than the global digenic network, highlighting the potential for complex genetic interactions to impact phenotype and emphasizing the need to gain a deeper understanding of how complex genetic interactions modulate genome-encoded individual variation7,11.
We also used trigenic interaction analysis to explore the evolution of duplicated genes resulting from the whole genome duplication (WGD) event in yeast and understand the factors that lead to duplicate gene retention12. S. cer. arose from WGD approximately 100 million years ago and after massive gene loss it retained 551 pairs of genes. Screening 240 double mutants and their corresponding single mutants, involving pairs of dispensable gene duplicates, generated ~550,000 double and ~260,000 triple mutants and revealed ~4700 negative and ~2500 positive digenic interactions and ~2500 negative and ~2100 positive trigenic interactions. Integrating these interactions into a metric termed ‘trigenic interaction fraction’, which quantified the fraction of negative trigenic relative to the total negative trigenic and digenic interactions, captured the extent of their functional redundancy revealing two paralog classes, a functionally redundant one and another more divergent class. Analysis of position-specific evolutionary rate patterns and in silico computational modeling was consistent with what we termed a ‘functional and structural entanglement’ model of evolution of paralogs in which highly entangled duplicates reverted to a singleton state, those that were minimally entangled and unconstrained diverged, and those with intermediate level of entanglement that were somewhat constrained diversified and evolved paralog specific functions, while retaining functional overlap at steady-state. Since duplicated genes are pervasive throughout evolution comprising ~29-59% of the plant genome13 and up to ~26% of the human genome14, these findings offer important insight into the evolutionary forces that shape genomes.
Development of the protocol
We adapted the SGA methodology15–17 to construct triple mutants and study their complex genetic interactions, which we refer to as trigenic-SGA (τ-SGA)11,12 (Fig. 1). In total we constructed 422 double mutant query strains along with 844 corresponding single mutant query strains. Single and double mutant queries were screened against a diagnostic array of single mutants. The diagnostic array consisted of 1,182 strains comprising of 990 nonessential gene deletion mutants and 192 mutants carrying temperature sensitive alleles of essential genes, covering ~20% of the entire yeast genome. Strains on the array span major biological processes in the cell and are representative of the distribution of single mutant fitness and digenic interaction degree of the entire genome. Using trigenic-SGA, double and single mutant query strains were crossed to the diagnostic array and following a series of replica pinning steps to sequentially select the mutants carrying the desired combination of genetic markers resulted in high-density arrays of triple and double mutants that could be analyzed for genetic interactions (Fig. 1 top). We note that the reduced size of the diagnostic array relative to the genome-wide array enabled all the trigenic interaction screens to be conducted in two replicates to achieve a sufficient level of precision11. We developed a scoring pipeline to quantify trigenic interactions using colony size as a proxy for fitness. Our score compares triple mutant fitness to a model that combines the single and double mutant phenotypes to the expected fitness of the triple mutant, taking any digenic interactions into account (Fig. 1 bottom).
Fig. 1 |. Overview of the trigenic SGA (τ-SGA) screening and quantification methodology.
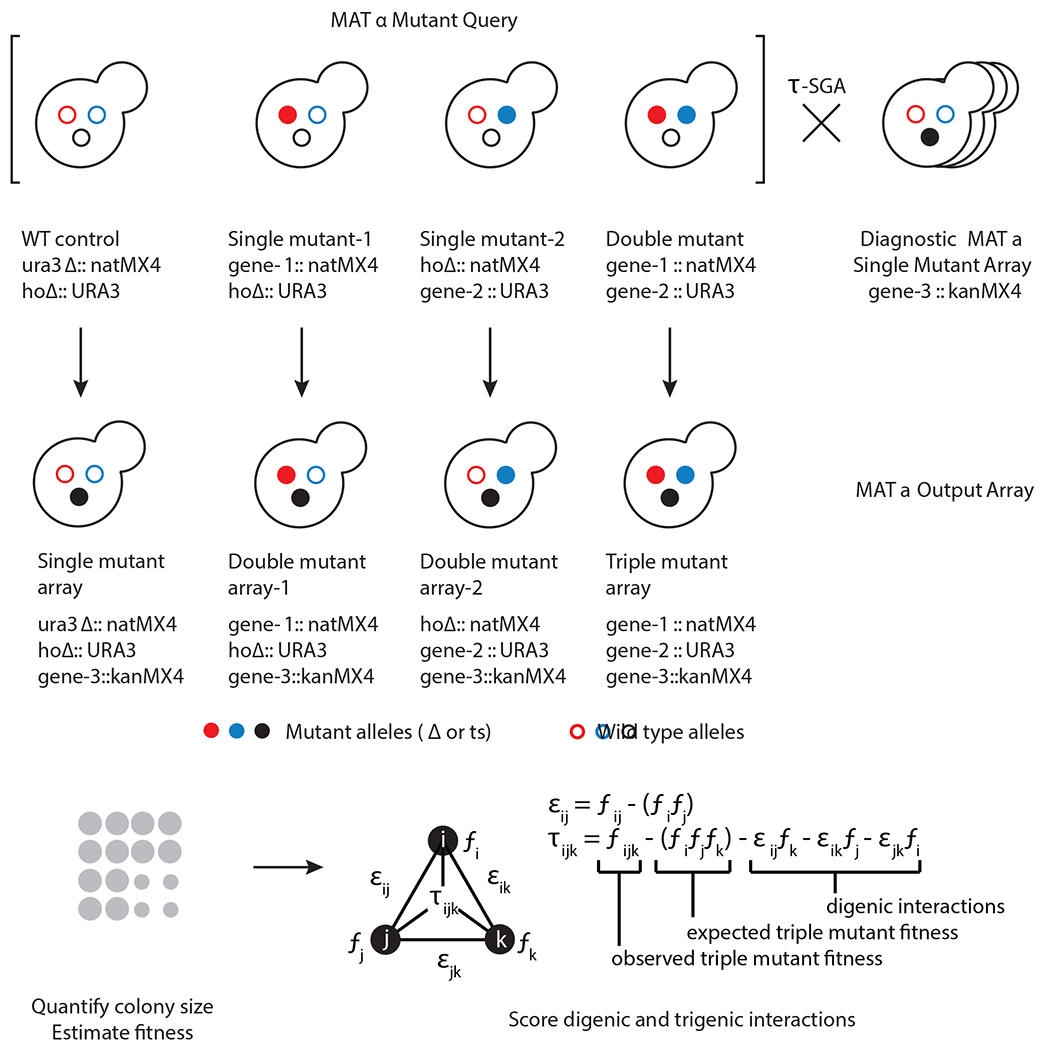
Top, a double mutant query strain with its two corresponding single mutant query strains and a wild-type reference strain are crossed in parallel using τ-SGA automated replica pinning and selection steps to the diagnostic array, which is representative of the genome-wide array collection to generate respective triple, double and single mutants carrying the desired genetic markers. Bottom, τ-SGA scoring pipeline is used to quantify trigenic interactions using colony size as a proxy for fitness accounting for any digenic interactions.
Classes of trigenic interactions
Our quantitative analysis allows us to identify two broad classes of trigenic interactions11,12. ‘Novel’ interactions involve gene triplets that do not show any pairwise digenic interactions between either single mutant query gene and the array gene or exhibit an interaction within the query gene pair itself. In our published survey of trigenic interactions, the ‘novel’ class comprised one-third of all trigenic interactions11. The remaining two-thirds of trigenic interactions defined a ‘modified’ class, in which mutations of a third gene exacerbates or alleviates a pairwise digenic interaction involving at least one of the gene pairs within the triplet11. We evaluate these digenic and trigenic interactions independently based on a previously defined genetic interaction score threshold at an intermediate cut-off: for digenic interactions (|ε| > 0.08, p < 0.05) and trigenic interactions (|τ| > 0.08, p < 0.05). Both ‘modified’ and ‘novel’ trigenic interactions significantly overlapped with known functional standards, such as co-expression, co-annotation to the same biological process and subcellular co-localization. ‘Modified’ rather than ‘novel’ interactions were also enriched for genes encoding proteins that exhibited protein-protein interactions. Thus, while ‘novel’ interactions captured entirely new functional information, ‘modified’ interactions expanded upon the digenic interaction network.
Trigenic interactions uncover novel functional relationships among gene products. These functional relationships can involve a number of different genetic mechanisms, 5 of which we highlighted below.
Functional redundancy in which any of the three genes are sufficient for pathway function (Fig. 2a), such as ODC1, ODC2 and CTP1, which are all members of the multi-gene family of mitochondrial carrier proteins12, or CLN1, CLN2 and CLN3 cyclins involved in G1 to S phase transition, which function by activating CDK CDC2818.
Within functional module redundancy in which the disruption of any combination of two components is tolerated, but a third perturbation results in sickness (Fig. 2b); for instance, OCA4, OCA5 and OCA6, which encode members of the OCA complex, or SET1, SDC1 and SWD1, which encode members of the COMPASS complex, functioning as histone methyltransferase complex controlling the silencing of telomeric regions11.
Distant functional bridging through by-product, when the activity of two genes impinges on a common function and their perturbation activates a compensatory pathway that generates a byproduct that impairs another function, which is supported by a third gene and thus its perturbation results in a loss of viability (Fig. 2c). For instance, perturbations of MDY2 and MTC1 lead to a defect in vesicle trafficking of aromatic amino acid transporters, which appears to lead to a compensatory pathway activation for the de novo production of aromatic amino acids and the downstream kynurenine pathway activation. This de novo pathway activation results in NAD+ accumulation, which may impair telomere capping and sensitize the mdy2Δ mtc1Δ mutants to further defects in DNA damage repair and telomere stability pathways, leading to a negative trigenic interaction for mdy2Δ mtc1Δ mre11Δ 11.
Genetic masking of a multi-member nonessential gene pathway, when the perturbation of each individual gene is detrimental to cell fitness, but a second or a third perturbation does not further reduce cell fitness (Fig. 2d), such as RAD52, RAD51 and RAD54 that function in the same DNA repair pathway and elimination of any one of these gene products is detrimental to the pathway19.
Genetic suppression is observed when a double mutant is sick, but a perturbation of a third gene suppresses the negative effect, which could be caused by a third gene whose function becomes toxic in the absence of the other two genes (Fig. 2e). For example, the severe double mutant defect caused by the perturbation of CAC1, a subunit of the CAF-1 (chromatin assembly factor-1) complex and any of the members of the HIRA complex (HIR1, HIR2, HIR3, HPC2, RTT106) involved in nucleosome assembly, as a result of the toxic function acquired by ASF1, another factor involved in chromatin assembly, which is suppressed by ASF1 deletion20. Another example of a mechanism of genetic suppression is the emergence of antibacterial drug resistance in RifR StrR double mutants, which have a fitness defect but can acquire increased fitness through compensatory mutations in genes with roles in transcription and translation21. Altogether, trigenic interactions offer new and functionally important insight into biological mechanisms and processes.
Fig. 2 |. Classes of trigenic interactions.
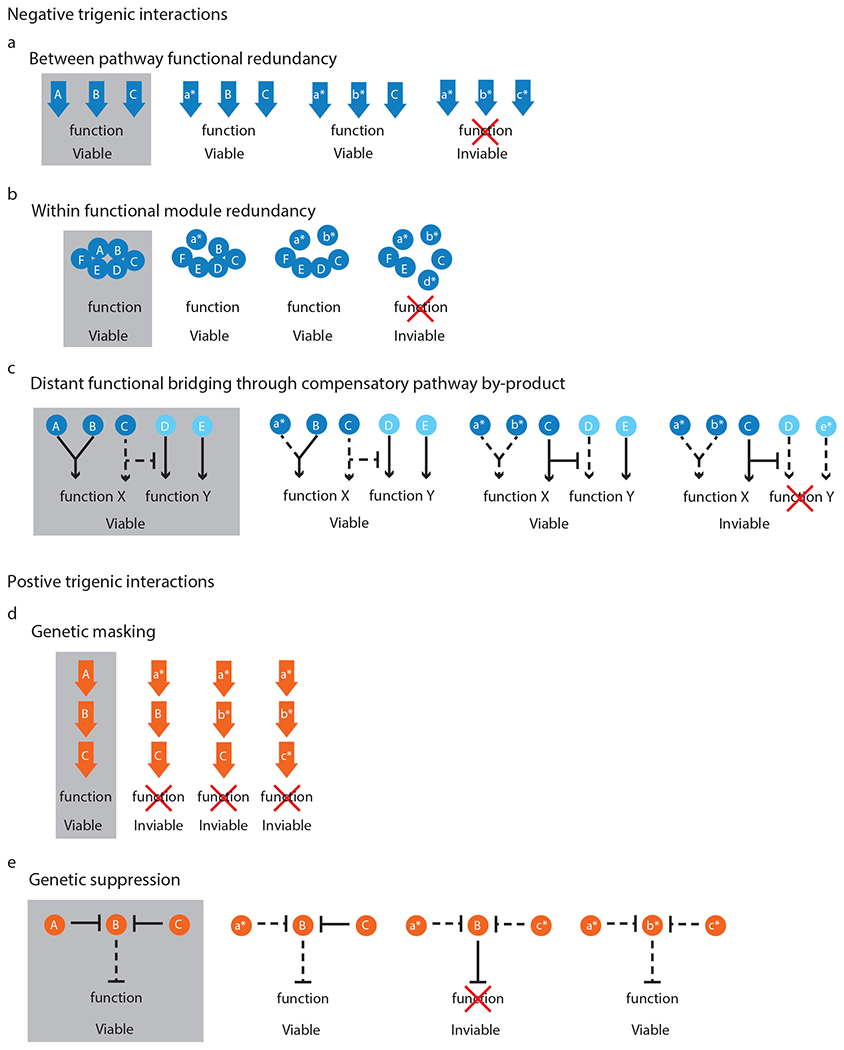
a, Between-pathway functional redundancy, when any of the three genes (A-C) are sufficient for pathway function e.g. ODC1, ODC2 and CTP1 or CLN1, CLN2 and CLN3; b, within functional module redundancy, when a protein complex contains up to three non-essential subunits in which the disruption of any combination of two components is tolerated, but a third perturbation results in sickness (A, B, D) e.g. OCA4, OCA5 and OCA6 or SET1, SDC1 and SWD1; c, distant functional bridging through compensatory pathway by-product, when the activity of two genes (A, B e.g. MDY2, MTC1) impinges on a common function (e.g. trafficking of aromatic amino acid transporters) and their perturbation activates a compensatory pathway (C e.g. de novo production of aromatic amino acids) that generates a by-product (e.g. NAD+) that impairs another function (e.g. telomere capping), which is mediated by another gene (D), which itself is synthetic lethal with another parallel pathway (E e.g. DNA damage repair pathway, MRE11); d, genetic masking of a multi-member pathway, when the perturbation of each individual gene is detrimental to cell fitness, but a second or a third perturbation does not increase the disruptive effect (A-C e.g. RAD52, RAD51 and RAD54); e, genetic suppression is observed when a double mutant is sick (A, C e.g. CAC1 and HIRA complex member), but a perturbation in a third gene (B e.g. ASF1) suppresses the negative effect which could be caused by a third gene (B) whose function becomes toxic in the absence of the other two genes (A, C). An uppercase letter denotes a wild-type allele, a lowercase letter with an asterisk denotes a mutant allele, a solid line denotes an active pathway and dashed line denotes a pathway, which is inactive or reduced in its activity.
Functional information associated with digenic and trigenic interactions offers insight into gene function
Digenic and trigenic interactions provide comparable functional information, when their distributions are compared across different biological processes or common functional standards11. Thus, they can be used to shed light on unknown or poorly characterized genes using ‘guilt by association’ approaches. For example, trigenic interactions for ECM13-YJR115W revealed a role in spindle function and chromosome segregation for this previously poorly characterized pair12. Additionally, trigenic interactions are more functionally diverse than digenic interactions since trigenic interaction profiles are enriched for a broader set of bioprocesses11. For example, MDY2 and MTC1 are involved in the early secretory pathway, which is also consistent with their digenic interaction bioprocess enrichments. Interestingly, their trigenic interactions mediate new connections between the secretory pathway, DNA replication/repair and nutrient sensing bioprocesses. Detailed analysis revealed that the MDY2-MTC1 double mutant is primarily defective in trafficking functions and can affect DNA synthesis and repair indirectly by modulating metabolic intermediates, such as NAD+ levels through the kynurenine pathway. Likewise, MVP1 and MRL1 which are important for sorting proteins to the vacuole, and SEC27 and GET4, which have roles in the endoplasmic reticulum-to-Golgi transport, both show trigenic interaction enrichments for genes involved in DNA replication and repair. Thus, trigenic interactions can uncover new functional connections between components of the cellular machinery.
Application of the method
τ-SGA methodology was developed to study genetic interactions involving perturbations among three different genes. In principle, the method can be further modified to study higher order genetic interactions by expanding the repertoire of genetic selection markers. In the budding yeast, useful markers may include those that confer resistance to additional antibiotics, such as zeocin and hygromycin, or those that complement other auxotrophies (e.g. leucine), potentially enabling analysis of complex genetic interactions up to the 6th order. Other methods have been used to study higher-order genetic interactions, including QTL mapping that identified up to 5 loci that interacted to influence colony morphology using a cross of two genetically diverse yeast22. Another approach involves engineering barcoded pools of cells composed of a defined mutant gene set. This approach was used to explore phenotypes associated with various combinations of up to 5 mutations in 16 ABC transporters23, although theoretically higher-order interactions could also be studied. Furthermore, the interrogation of essential genes using digenic and trigenic-SGA screening is enabled by the construction of temperature sensitive alleles24, DAmP (decreased abundance by mRNA perturbation) alleles25, as well as transcriptionally regulated alleles under control of the tunable estradiol-inducible system26 or CRISPRi (CRISPR interference)27.
The current version of the τ-SGA protocol was optimized for assessing the fitness effect resulting from genetic perturbations in standard conditions. However, the method could be readily adapted to explore condition-specific trigenic interactions, such as those that occur in response to environmental or chemical stressors. Given that the global digenic interaction network analysis showed that ~20% of genes exhibit sparse genetic interaction profiles lacking functional information, conducting screens in genetically and environmentally sensitized backgrounds may uncover higher-order genetic redundancies and shed light on mechanisms of genetic network rewiring7,8. Additionally, this method could be integrated with high-content screening platforms to study the effect of multiple perturbations on protein localization and abundance28–30 or transcriptional regulation31. Finally, a previous cross-study analysis showed that colony size fitness measurements are highly correlated with barcode-based fitness measurements2, suggesting that our trigenic interaction scoring may be applicable to growth measurements other than colony size.
The concepts associated with trigenic SGA analysis can also be extended to other organisms. CRISPR-Cas9 has been an invaluable tool for generating gene knockouts in mammalian cells, and analysis of pools of cells expressing barcoded guide RNA libraries have enabled cataloguing of cell line-specific essential gene sets32. Co-expressing multiple CRISPR-associated nucleases should enable fitness based CRISPR screens to study complex genetic interactions, especially when combined with microfluidic systems33,34. The development of CRISPR-Cas9 strategies coupled with single-cell RNA sequencing, termed Perturb-seq or CROP-seq, should enable complex genetic interaction analysis at the single cell transcriptome resolution in yeast and other organisms, including human cells35–37.
Comparison with other methods
The τ-SGA analysis technique provides a systematic way to construct and quantify trigenic interactions in the budding yeast. Another method, the triple mutant analysis (TMA) approach has also been developed to study complex genetic interactions involving three genes in yeast20,38. As described above, τ-SGA relies on constructing the desired double mutant query strain with the corresponding two single mutant control queries to generate genetically homogeneous arrays of triple and double mutants. The TMA method involves using a single double mutant query to generate mixed arrays of double and triple mutants in addition to homogeneous arrays of triple mutants. In this case, a competition effect may generate false negative digenic interactions and result in trigenic interactions appearing more subtle than they actually are.
A scoring approach called the MinDC (minimum difference comparison) was developed to quantify interactions from TMA screens20,38. MinDC is aligned with the S-score approach39, whereas the τ-SGA score is based on the SGA score2, which includes several additional steps to normalize statistical artifacts associated with plate-based screens. Besides the differences between the baseline approaches detailed previously2, there are also differences in how MinDC and τ-SGA score approach trigenic scoring. To score a triple mutant, the MinDC approach subtracts the S-score of the more severely affected double-mutant (out of a possible three double mutants) from the S-score of the triple mutant. However, the τ-SGA scoring approach subtracts the SGA scores of all three double mutants from the SGA score of the triple mutant. As demonstrated previously11, both MinDC and τ-SGA scores agree on the existence of trigenic interactions in the majority of cases. However, in cases of negative interactions with both double mutants showing a negative score, the MinDC approach incorrectly reports negative interactions that can actually be accounted for by the sum of the double mutant effects11. A similar trend is also seen for positive trigenic interactions. The τ-SGA score, therefore, helps remove a set of false positive trigenic interactions by controlling for the effects of all possible double mutants.
Experimental design
The trigenic-SGA analysis method includes an experimental component for systematic high-throughput triple gene mutant construction and an analysis pipeline for quantifying trigenic interactions. To begin a τ-SGA screen, query mutant strains must be constructed, such that every double mutant query strain, which carries mutations in two genes of interest marked by different selectable markers, has a corresponding pair of single mutant control query strains, carrying a mutation in each gene of interest marked with the relevant selectable markers, along with the second selectable marker inserted into a benign control locus, YDL227C, which encodes the nonfunctional HO-endonuclease. A similarly marked wild-type control query strain must also be constructed. Each query mutant strain of interest (wild-type, single- or double-mutant) is then crossed to an array of single mutants to generate single-, double- and triple mutant arrays (Fig. 3, 4). Robotic replica-pinning of yeast strains onto a series of defined solid media enables the selection of mutants carrying the desired genetic markers.
Fig. 3 |. An overview of the Trigenic Synthetic Genetic Array (τ-SGA) experimental pipeline.
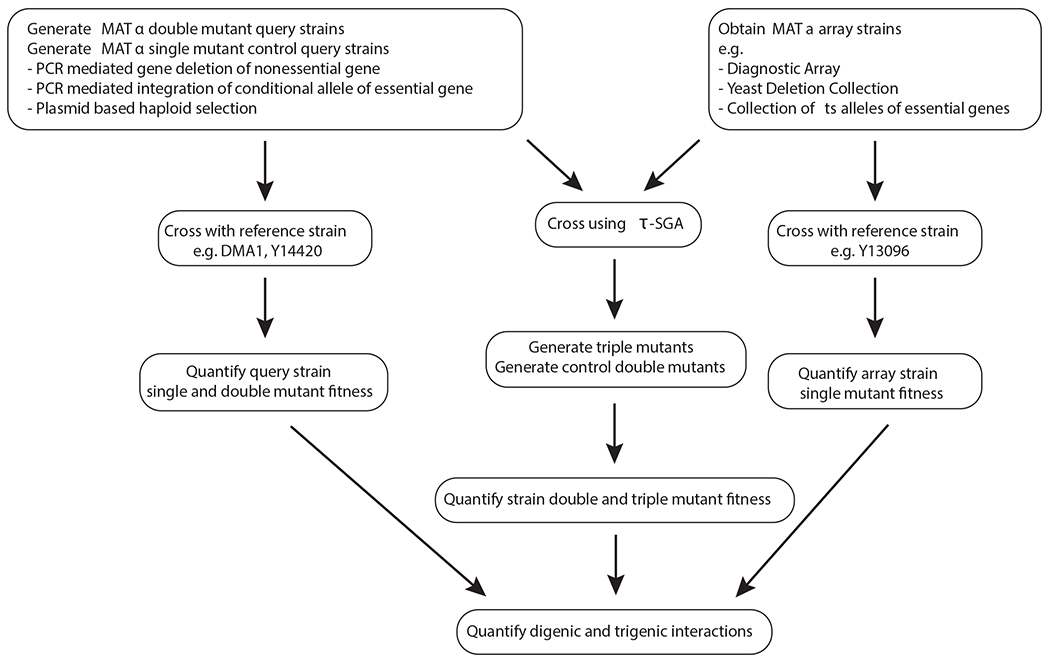
MATα single and double mutant query strains are generated. A diagnostic array of MATa single mutant array strains is constructed. Query strains and array strains are crossed using τ-SGA method to generate double and triple mutants, which are scored for colony size, which is a proxy for fitness. Separately, fitness of the original query strains and array strains is measured. All the resulting fitness values are incorporated into the final quantification of digenic and trigenic interactions.
Fig. 4 |. Trigenic Synthetic Genetic Array (τ-SGA) experimental pipeline.
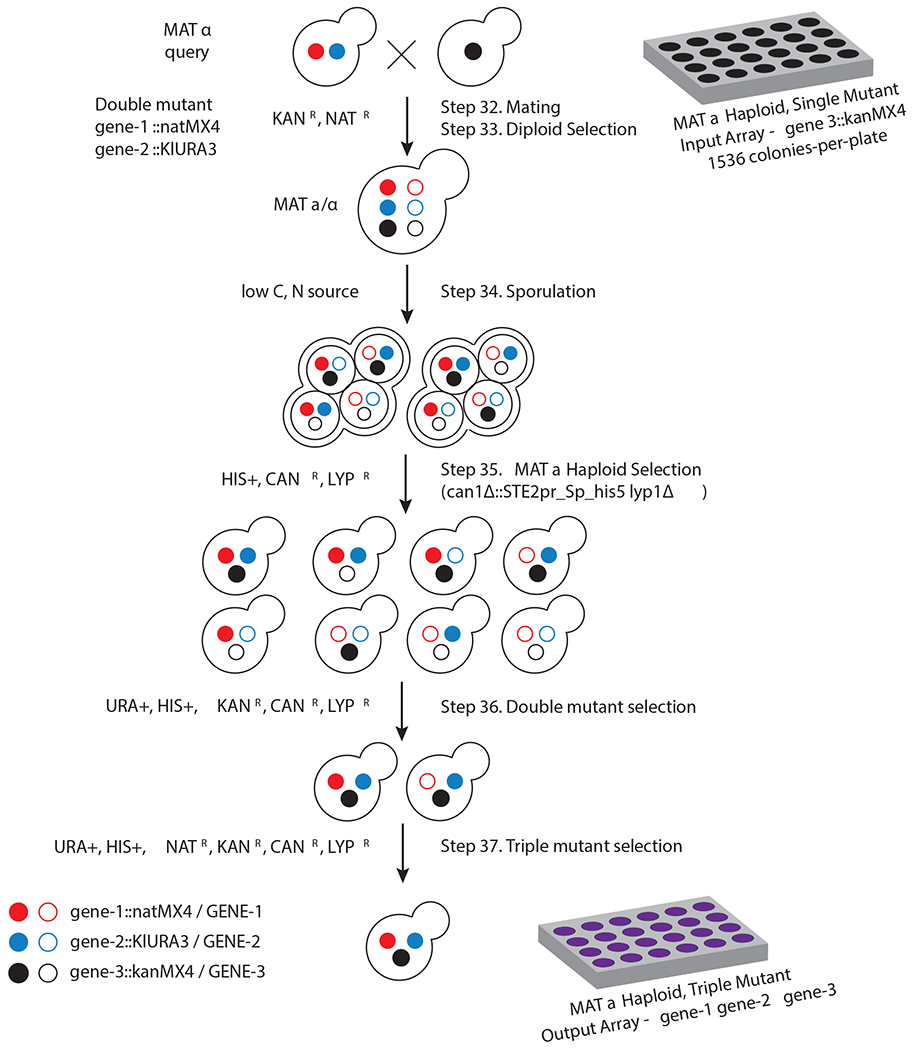
A MATα query double mutant strain harboring loss of function mutations in two different genes of interest linked to dominant selectable markers (natMX4 and KIURA3), which confer resistance to the antibiotic nourseothricin and the ability to grow in media lacking uracil (filled red and blue circles, respectively). The arginine and lysine permeases, CAN1 and LYP1, which confer sensitivity to canavanine and thialysine, are also deleted in the query strain and are used to select against diploids following the sporulation step. Typically, in a τ-SGA screen the query strain is crossed to an ordered diagnostic array of MATa non-essential gene deletion strains and ts alleles of essential genes (‘array’ mutants) linked to a dominant selectable marker, kanMX4, which confers geneticin resistance (filled black circle). The resulting heterozygous diploids are replica-pinned to a medium low in carbon and nitrogen sources to induce meiosis. The resulting sporulated mix is then replica-pinned to a synthetic medium depleted for histidine, but containing canavanine and thialysine to allow for the selective germination of MATa haploid meiotic progeny. This selective germination is possible because the SGA reporter, STE2pr_Sp_his5, in which the STE2 MATa-specific promoter (STE2pr) controls the expression of the Schizosaccharomyces pombe his5 gene, replaces the CAN1 gene. The MATa haploid progeny is then transferred to the selective medium lacking uracil and containing geneticin to select for mutants harboring one of the query strain mutations and an array mutation. In the final step, the selected haploids are transferred to the medium lacking uracil and containing geneticin and nourseothricin to select for all three markers.
Query strains for trigenic-SGA are constructed using PCR-mediated deletion of a nonessential gene(s) or integration of a conditional allele(s) of interest and subsequent tetrad analysis or plasmid-based haploid selection. Every double mutant query strain, harbouring mutations in two different genes of interest is marked with NATR and URA+ (geneA::natMX4 geneB::KlURA3). Two single mutant control query strains are also constructed, carrying one of the genes of interest along with the marker of the second gene in a benign HO locus (geneA::natMX4 hoΔ::KlURA3 and hoΔ::natMX4 geneB::KlURA3), as well as a wild-type control with both markers inserted in benign loci (ura3Δ::natMX4 hoΔ::KlURA3, available from our group, Table 1). Constructing single mutant query strains in the same genetic background harboring the same genetic markers allows for the single, double and triple mutants to be assessed under the same conditions eliminating any conditional effects that may otherwise confound the analysis.
Table 1.
Strains and plasmids. Unless otherwise stated, all the strains and plasmids are available from the Boone lab.
| Strain | Genotype (S288c) | Comments |
|---|---|---|
| Y7092 | MATα can1Δ::STE2pr-Sp_his5 lyp1Δ ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Haploid background strain for SGA query strain construction through PCR-mediated gene deletion. |
| Y7091 | MATa can1Δ::STE2pr-Sp_his5 lyp1Δ ura3Δ0 leu2Δ 0 his3Δ1 met15Δ0 | Haploid background strain for SGA query strain construction by crossing strains. |
| Y14476 | MATa/α can1Δ::STE2pr-Sp_his5/can1Δ::STE2pr-Sp_his5 lyp1Δ/lyp1Δ his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 met15Δ0/met15Δ0 LYS2+/LYS2+ | Diploid background strain for SGA query strain construction through PCR-mediated gene deletion. |
| Y9687 | MATa/α can1Δ::STE2pr-Sp_his5/+; lyp1Δ::STE3pr-LEU2/+; his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0met15Δ0/met15Δ0 LYS2+/+ | The background strain for SGA query strain construction through PCR-mediated gene integration of a conditional allele. |
| Y14391 | MATα hoΔ::natMX4 can1Δ::STE2pr-Sp his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | SGA query strain carrying hoΔ::natMX4 for constructing single mutant control strain by PCR-mediated gene deletion or crossing with other strains to screen for digenic interactions. |
| Y14120 | MATa hoΔ::KlURA3 can1Δ::STE2pr-Sp his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | SGA query strain carrying hoΔ::KlURA3 for constructing single mutant control strain for crossing with other strains. |
| Y13096 | MATa ura3Δ::natMX4 hoΔ::KlURA3 can1Δ::STE2pr-Sp_his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 LYS2+) | Reference query strain for obtaining single mutant array fitness for τ-SGA procedure. |
| DMA1 | MATa his3Δ1::kanMX4 leu2Δ0 ura3Δ0 met15Δ0 | Reference strain for crossing with a collection of query strains to obtain query strain fitness for τ-SGA procedure. |
| Y14420 | MATa his3Δ1 leu2Δ0 ura3Δ0::kanMX4 met15Δ0 | Reference strain for crossing with a collection of query strains to obtain query strain fitness for τ-SGA procedure. |
| Yeast Deletion Collection | MATa his3Δ 1 leu2Δ0 met15Δ0 ura3Δ0 | The collection of MATa deletion strains is available for purchase as 96-well agar plates from Invitrogen, American Type Culture Collection, EUROSCARF; or 96-well agar plates and 96-well plate of frozen stock from Open Biosystems. |
| Collection of ts alleles of essential genes | MATα his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | The collection of MATa strains of temperature sensitive alleles. |
| p4339 | pCRII-TOPO, natMX4 | Plasmid to amplify natMX49. |
| p5749 | pFA6a-GST-KlURA3 | Plasmid to amplify K. lactis URA344. |
| p6981 | pBlueScript II (+), CEN6/ARSH4, STE3pr-hphR, GAL1pr-KAR1, LEU2 | Plasmid for haploid selection. |
A subset of query strains and the array of interest (we recommend using the diagnostic array on 4 plates) are thawed from glycerol stocks and spotted onto solid medium. The array strain arrangement for the mating step should be in a 1536 colony-per-plate format. Each query strain is then grown in liquid medium overnight to saturation. A query lawn is generated by spreading the liquid saturated culture onto solid rich media plates. The query lawn is then replica-pinned onto the next set of plates with rich media, on top of which the array strains are replica-pinned, thereby mating the two strain types. Diploids are selected by replica-pinning the mated mix onto medium that selects for the query and array mutations. One of the genetic markers in the query strain is sufficient for diploid selection, and the protocol involves selection of the NATR and KANR diploids. Transferring the diploids onto low carbon and nitrogen medium induces meiosis. Haploids are germinated by replica-pinning onto medium which selects for haploidMATa specific progeny (HIS+) and counterselects for diploids using SGA markers based on resistance to canavanine (CANR) and thialysine (LYPR). The next step selects for haploids carrying one of the query strain markers and the array strain marker, URA+ and KANR followed by the final pinning step that selects for haploids with all three markers: NATR, URA+ and KANR. Thus, all three screens (single, double and triple mutant) are conducted in media with exactly the same composition. Each screen against the diagnostic array requires 4 plates. The BioMatrix carousel robot can accommodate 112 source and 112 destination plates, which is sufficient to screen one wild-type control query strain along with 9 sets of double mutants with two corresponding single mutant control query strains. Thus, each replica-pinning step requires 112 destination plates. This type of a robot can operate without supervision and thus has the capacity of 112 daytime and 112 nighttime sets of plates for a total of 18 sets of trigenic SGA screens per batch.
Upon completion of the screen, the resulting final plates are imaged using a plate imaging system and processed for colony size measurements, which serve as a proxy for fitness (Fig. 5). Colony segmentation is integrated into the MATLAB routines for high-throughput screen scoring (Fig. 6) and SGAtools for small-scale screen (Fig. 7) quantification. Digenic interactions (SGA score) and trigenic interactions (τ-SGA score) are computed using the aforementioned algorithms to identify double and triple mutant effects on fitness that deviate from the expectation. In particular, the τ-SGA score identifies all instances in which the triple mutant fitness cannot be explained by the product of single mutant fitness estimates accounting for all pairwise digenic interactions between the query and the array genes, and within the query gene pair itself. The digenic and trigenic interaction data is clustered to reveal biological insight using Cluster 3.0 and visualized using JavaTreeView or the recently updated TreeView 3.040, which integrates both clustering and heatmap generation into one user-friendly application (Fig. 8a). Further downstream analysis can include overlap of genetic interactions with functional standards, such as a protein-protein interaction standard (Fig. 8b). Another useful analysis involves assessing trigenic interaction profiles using Spatial Analysis of Functional Enrichment (SAFE)41, which is based on the global digenic interaction network topology and enables visualization of the subcellular compartments, bioprocesses, and protein complexes that underlie the genetic interactions (Fig. 8c).
Fig. 5 |. An example of a trigenic interaction.
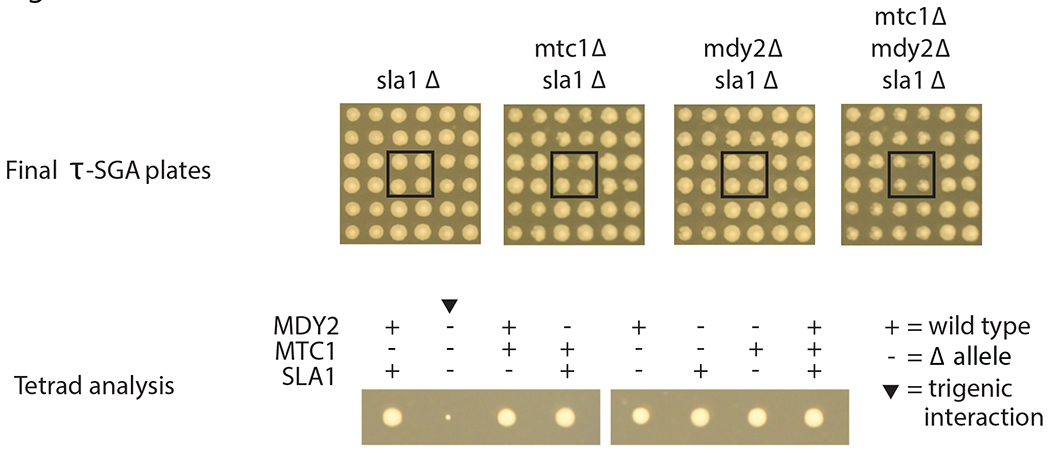
Top, sample cropped images of final τ-SGA plates. The deletion of the array gene (sla1Δ) alone or in combination with either one of the query genes (mtc1Δ or mdy2Δ) does not result in any observable fitness defects. The triple mutant exhibits a severe growth defect, and as such a strong negative trigenic interaction. Each mutant is represented in quadruplicate on the array and is highlighted with a black box. Bottom, tetrad analysis confirmation for the negative trigenic interaction of mtc1Δ mdy2Δ sla1Δ.
Fig. 6 |. Schematic of the steps involved in processing and scoring high-throughput trigenic interaction screens using τ-SGA scoring pipeline.
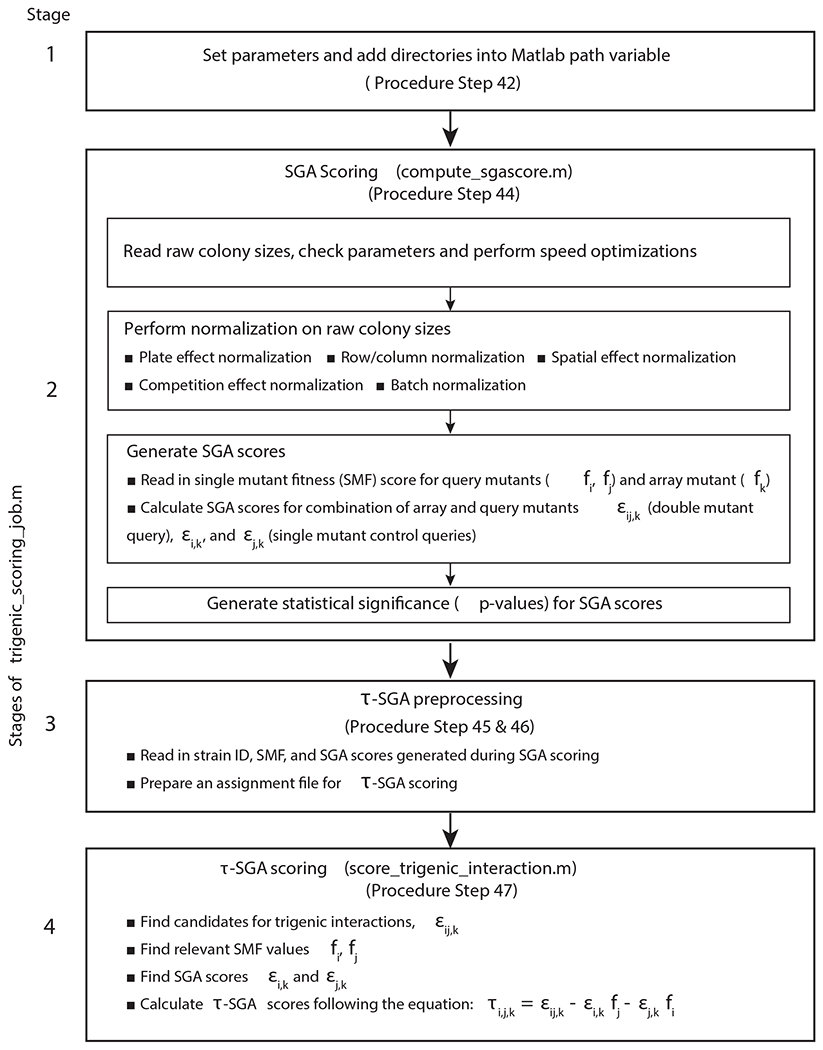
Sequential stages of the main script trigenic_scoring_job.m, which defines parameters and then invokes compute_sgascore.m to generate and output SGA scores. Using these SGA scores and an assignment file designed for the specific query screens in the dataset, the script then computes the τ-SGA trigenic interaction scores. It should be noted that the trigenic scoring equation used in Stage 4 is slightly different from the one in Fig. 1, and the former can be directly derived from the latter as shown previously9.
Fig. 7 |. Schematic of the steps for processing and scoring small-scale trigenic interaction screens using SGAtools platform.
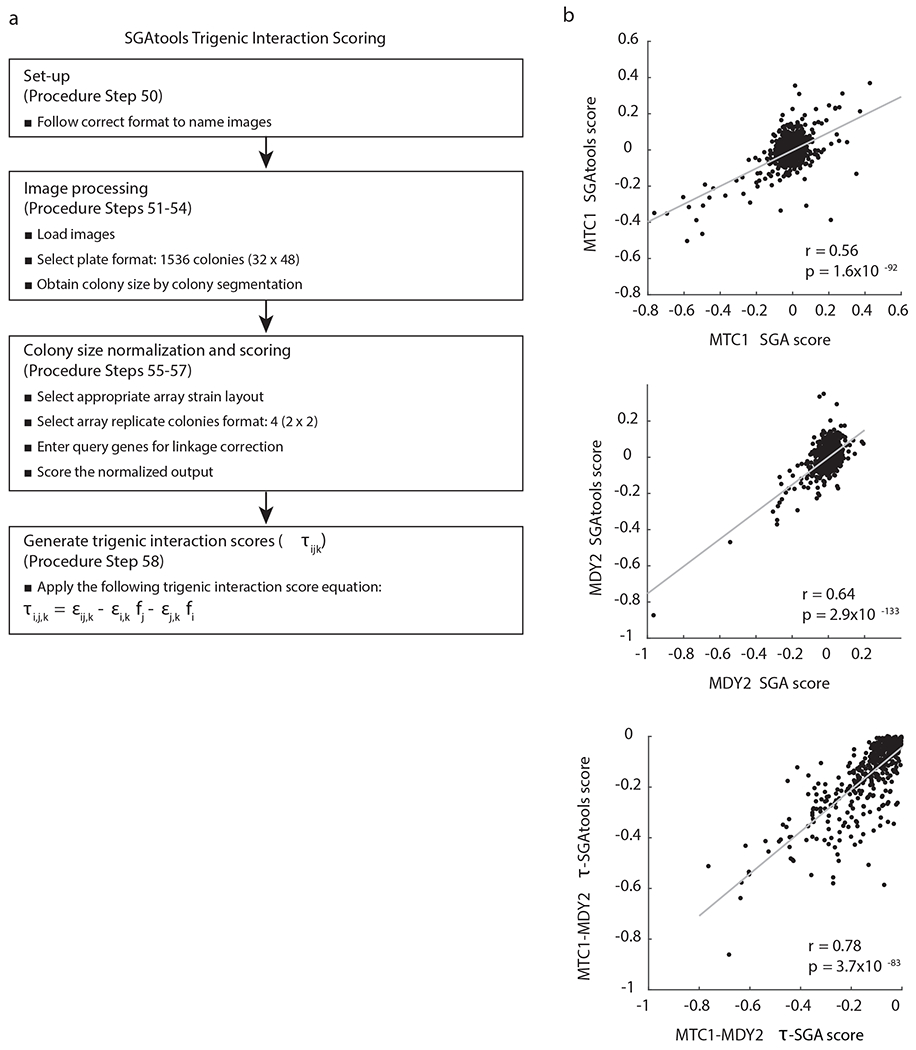
a, Files are named using the appropriate format, followed by image processing to segment colonies and obtain normalized colony sizes, which are then scored for trigenic interactions by applying the trigenic interaction score equation. It should be noted that the equation for scoring trigenic interactions slightly differs from Fig. 1, and the former can be directly derived from the latter as shown previously9. b, Scatter plot comparing digenic and trigenic interactions scores derived from SGAtools and a previous study11 for the following query strains: mtc1Δ, mdy2Δ and mtc1Δ mdy2Δ. r denotes the Pearson correlation coefficient.
Fig. 8 |. Representative examples of clustering and visualization of τ-SGA scores.
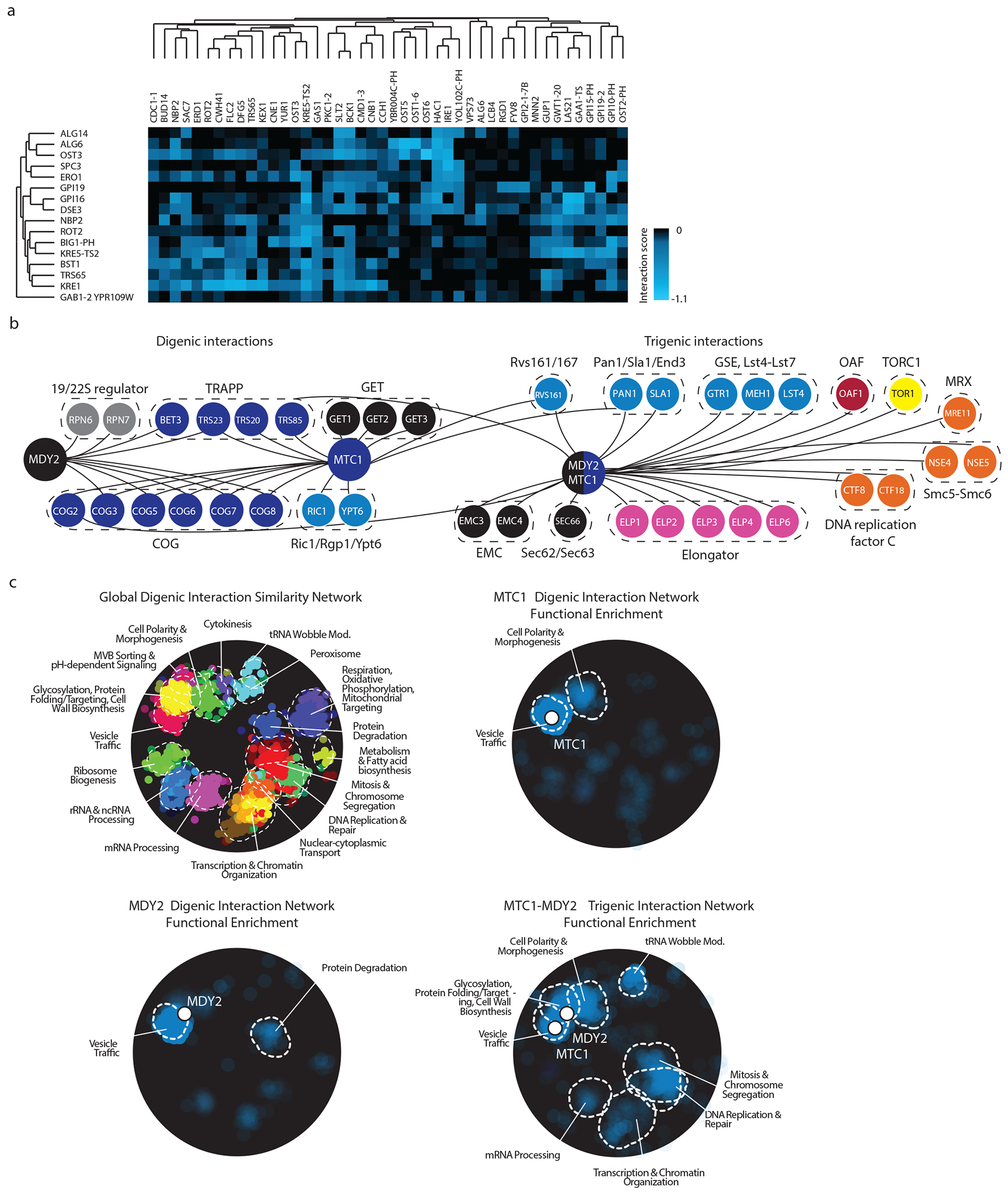
a, A sample visualization of the .cdt file generated from the output of sample data as described in steps 44 - 50. Genes associated with query strains are on the y-axis and array strains are on the x-axis. b, Representative digenic and trigenic interactions for the single mutant query strains MDY2, MTC1 and their corresponding double mutant query strain MDY2-MTC1 as visualized by Cytoscape and overlapped with representative protein complexes. c, Enrichment of digenic and trigenic interactions within the global digenic interaction profile similarity network annotated by SAFE bioprocess terms. Panels b and c have been adapted from a previous study11.
Limitations
Trigenic-SGA is useful for understanding genetic redundancies of a pair of singleton genes or duplicated genes. Cases of even higher-order genetic interactions would not be uncovered using trigenic-SGA. Some query strains, including those with mating, sporulation, or cell wall defects may not survive all the selection steps of τ-SGA. Query strains with a modest to moderate growth defect are expected to reveal richer digenic and trigenic interaction profiles relative to query strains that have a wild-type growth rate.
It is advisable to confirm trigenic and digenic interactions using random spore or tetrad analyses, especially if detailed follow-up experiments are planned. The diagnostic array comprises ~20% of the genome and thus would not capture, for example, all members of a protein complex. If interactions are detected with genes encoding protein complex members, it is often useful to perform directed tests with mutant strains encoding other complex members, not present on the diagnostic array.
Required Expertise
To conduct trigenic SGA screens, the user should be familiar with handling the BioMatrix robot and standard yeast genetics techniques. To run the τ-SGA scoring pipeline, the user should be familiar with the MATLAB (The Mathworks Inc. Natick, MA, USA) coding environment. Some familiarity with the web interface of SGAtools and thecellmap.org as well as knowledge of Cluster 3.0, JavaTreeView, TreeView 3.0, Cytoscape and SAFE for data visualization and downstream analyses are also expected.
Materials
Biological Materials
Yeast strains which are used throughout this protocol are listed in Table 1.
Reagents
Media reagents
Bacto-Yeast Extract (Fisher, cat. no. DF0127-08-0)
Bacto-Peptone (Fisher, cat. no. DF0118081)
Bacto-Agar (Fisher, cat. no. DF0140083)
Dextrose ((D-Glucose) Anhydrous (Granular Powder/Certified ACS); Fisher, cat. no. D16-10)
Yeast nitrogen base without amino acids (Fisher, BD Difco, cat. no. DF0919-07-3)
Yeast nitrogen base without amino acids and ammonium sulfate (Fisher, BD Difco, cat. no. DF0335-15-9)
Monosodium glutamic acid (L-Glutamic acid monosodium salt hydrate; Sigma, G5889-1KG)
Potassium acetate (BioShop, cat. no. POA301)
Adenine (Adenine hemisulfate salt; Sigma, cat. no. A9126-100G)
Alanine (L-Alanine; Fisher BioReagents, cat. no. BP369-100)
Arginine (L-Arginine; Fisher, cat. no. ICN10073680)
Asparagine (L(+)-Asparagine monohydrate; Fisher, cat. no. AC175271000)
Aspartic acid (L-Aspartic acid; Fisher BioReagents, cat. no. BP374-100)
Cysteine (L-Cysteine hydrochloride monohydrate; Fisher BioReagents, cat. no. BP376-100)
Glutamic acid (L-Glutamic acid monosodium salt hydrate; Sigma, cat. no. G1626)
Glutamine (L-Glutamine; Fisher BioReagents, cat. no. BP379-100)
Glycine (BioShop, cat. no. GLN001.1)
Histidine (L-Histidine; Sigma, cat. no. H8000-500G)
Inositol (Fisher, cat. no. DF0164-15-5)
Isoleucine (L-Isoleucine; Fisher BioReagents, cat. no. BP384-100)
Leucine (L-Leucine; Sigma, cat. no. L8000-250G)
Lysine (L-Lysine monohydrochloride; Sigma, cat. no. L5626-500G)
Methionine (L-Methionine; Fisher BioReagents, cat. no. BP388-100)
P-aminobenzoic acid (4-aminobenzoic acid sodium salt; Acros, cat. no. AC226641000)
Phelylalanine (L-Phelylalanine; Sigma, cat. no. P2126-100G)
Proline (L-Proline; Sigma, cat. no. P5607-100G)
Serine (L-Serine; Fisher, cat. no. AC132661000)
Threonine (L-Threonine; Fisher BioReagents, cat. no. BP394-100)
Tryptophan (L-Tryptophan; Sigma, cat. no. T0254-100G)
Tyrosine (L-Tyrosine; Sigma, cat. no. T8566-100G)
Uracil (Fisher, cat. no. AC157301000)
Valine (L-Valine; Fisher BioReagents, cat. no. BP397-100)
Canavanine (L-canavanine sulfate salt; Sigma, cat. no. C9758-5G)
Thialysine (S-[2-aminoethyl]-L-cysteine hydrochloride; Sigma, cat. no. A2636-5G)
clonNAT (Nourseothricin, Werner BioAgents, cat. no. CAS 96736-11-7)
G418 (Geneticin; ThermoFisher, cat. no. 11811098)
Query strain construction reagents
Expand High Fidelity PCR System (Roche, cat. no. 11732641001)
DMSO (Sigma, cat. no. D5879-1L)
Lithium acetate (Sigma, cat. no. L4158-1kg)
PEG3350 (BioShop, cat. no. PEG335.1) CRITICAL It is important to use PEG3350 since transformations using PEG4000 result in cell fusion, which is not evident with PEG335042.
ssDNA (Deoxyribonucleic acid, single stranded from salmon testes; Sigma, cat. no. D9156)
Galactose (D-(+)-Galactose >98%; Sigma, cat. no. G0625)
Hygromycin (Wisent, cat. No. 400-141-UG)
Glycerol (BioShop, cat. no. GLY002)
Equipment
Robotic pinning – high throughput
BM6-SC1 Robot with Carousel (S&P Robotics, www.sprobotics.com)
Nunc OmniTray Single-Well Plates (Thermo Scientific, N242811) should be used to prepare medium for all replica pinning steps on a BM6-SC1 Robot.
Robotic pinning – medium throughput
BM6-BC Benchtop robot (S&P Robotics, www.sprobotics.com)
Robotic pinning – low throughput
ROTOR benchtop robot (Singer Instruments, UK, www.singerinst.co.uk)
RePads (Singer Instruments, UK, www.singerinst.co.uk)
PlusPlates (Singer Instruments, UK, www.singerinst.co.uk)
Incubators
Incubators at 22°C, 26°C and 30°C (as needed)
Shaking incubators at 22°C and 30°C (as needed)
Plate imaging system
High-resolution digital imaging system, e.g., SPImager (S&P Robotics, www.sprobotics.com)
Generating query strains
Thermocycler (e.g. BioRad, DNA Engine Tetrad2)
Tetrad dissector (e.g. Singer Instruments, UK, www.singerinst.co.uk, SporePlay)
96-Well Assay Plates (Axygen, P-96-450R-C-S)
AlumaSeal II aluminum sealing tape (Excel Scientific, AF100)
Data analysis
Hardware: 64-bit computer running Linux with at least 4 GB of RAM. The trigenic scoring pipeline will also run on Mac OS, but will not run on a Windows OS without minor updates to the source code.
Matlab R2019b (version 9.7) with the following toolboxes: Image processing and Statistics toolbox; any Matlab release starting with R2011a (version 7.12) will be sufficient.
SGA scoring pipeline (available on GitHub: https://github.com/csbio/SGA_Public). The release for trigenic-SGA interaction scoring is available at https://github.com/csbio/SGA_Public/releases/tag/tau_score_v1.1.0.
Input raw colony size data (should conform to the 9-column input format detailed in SGA scoring pipeline file formats: https://github.com/csbio/SGA_Public/blob/master/Column_Key.md).
SGAtools for small-scale screen analysis (http://sgatools.ccbr.utoronto.ca/).
Cluster 3.0 to cluster (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm), and Treeview to visualize the clustering results (http://jtreeview.sourceforge.net/). A recently updated version of Java TreeView, called Treeview 3.0, has also been developed that integrates clustering and heatmap visualization into a single user-friendly application (http://doi.org/10.5281/zenodo.1303402).
Cytoscape to visualize the digenic and trigenic interaction networks (https://cytoscape.org/).
Global digenic interaction network (http://thecellmap.org).
-
Trigenic interaction datasets can be browsed interactively here: http://boonelab.ccbr.utoronto.ca/supplement/kuzmin2018/supplement.html http://boonelab.ccbr.utoronto.ca/paralogs/
They were also deposited in the DRYAD Digital Repository (doi:10.5061/dryad.tt367) and (doi:10.5061/dryad.g79cnp5m9), respectively.
Spatial Analysis of Functional Enrichment (SAFE).
Reagent Setup
CRITICAL
Unless otherwise stated, solutions can be stored at room temperature (RT, 20-25°C) for 1-2 months and agar plates with various media can be stored for 1-2 months at 4°C.
Canavanine
Prepare a stock solution by dissolving canavanine in water to a concentration of 100 mg/ml, filter-sterilize and store aliquots at 4°C for 1-2 weeks. For frequent use, 50 ml aliquots are recommended.
Thialysine
Prepare a stock solution by dissolving thialysine in water to a concentration of 100 mg/ml, filter-sterilize and store aliquots at 4°C for 1-2 weeks. For frequent use, 50 ml aliquots are recommended.
clonNAT
Prepare the stock solution by dissolving clonNAT in water to a concentration of 100 mg/ml, filter-sterilize and store aliquots at 4°C for at least 1 year. For frequent use, 50 ml aliquots are recommended.
G418
Prepare the stock solution by dissolving G418 in water to a concentration of 200 mg/ml, filter-sterilize and store aliquots at 4°C. For frequent use, 50 ml aliquots are recommended. Geneticin is very stable and does not display any reduction of activity after it has been added to agar plates and stored for at least 2 months at 4 °C.
Hygromycin
Prepare the stock solution by dissolving hygromycin in water to a concentration of 50 mg/ml, filter-sterilize and store aliquots at 4°C for 1-2 weeks. For frequent use, 10 ml aliquots are recommended.
Glucose
Prepare a stock solution 40% w/v in water. Aliquots of 500 ml are recommended.
Galactose
Prepare a stock solution 40% w/v in water. Aliquots of 500 ml are recommended.
Glycerol
Prepare a stock solution 40% v/v in water. To generate glycerol frozen stock of strains, mix equal parts (1:1) 40% glycerol and yeast culture. Aliquots of 200 ml are recommended.
Complete synthetic medium amino acid supplement powder
Mix 3 g adenine, 2 g alanine, 2 g arginine, 2 g asparagine, 2 g aspartic acid, 2 g cysteine, 2 g glutamic acid, 2 g glutamine, 2g glycine, 2 g histidine, 2 g inositol, 2 g isoleucine, 10 g leucine, 2 g lysine, 2 g methionine, 0.2 g p-aminobenzoic acid, 2 g phelylalanine, 2 g proline, 2 g serine, 2 g threonine, 2 g tryptophan, 2 g tyrosine, 2 g uracil, 2 g valine. Invert end to end for 15 min. Store the contents in a dark bottle at RT. Use 2 g of mixture to make 1 L of synthetic complete medium. Agar plates containing synthetic medium should be stored in the dark, because they are sensitive to light. Plates stored at 4°C are stable for at least 1 month.
Drop-out (DO) amino acid supplement powder
Prepare a drop-out (DO) mixture by excluding the desired amino acid from the complete synthetic medium amino acid supplement powder. Use 2 g of DO mixture to make 1 L of synthetic medium.
Sporulation amino acid supplement powder
Mix 2 g histidine, 10 g leucine and 2 g uracil. Invert end to end for 15 min. Store the contents in a dark bottle at room temperature.
Mating medium (Yeast Extract Peptone Dextrose, YEPD)
Prepare 1% Bacto-yeast extract (w/v), 2% Bacto-peptone (w/v), 2% glucose (w/v), 2% Bacto-agar (w/v) by mixing 10 g Bacto yeast extract, 20 g Bacto peptone, 20 g Bacto agar, 120 mg Adenine with 950 ml water. Autoclave and cool to ~ 65°C. Supplement with 2% glucose by adding 50 ml of 40% glucose.
Array propagation medium (YEPD+G418)
Prepare 1% Bacto-yeast extract (w/v), 2% Bacto-peptone (w/v), 2% glucose (w/v), 2% Bacto-agar (w/v) by mixing 10 g Bacto yeast extract, 20 g Bacto peptone, 20 g Bacto agar, 120 mg Adenine with 950 ml water. Autoclave and cool to ~ 65°C. Supplement with 2% glucose by adding 50 ml of 40% glucose and 200 μg/ml G418.
Plasmid-based haploid selection medium (YEPD+Hygromycin)
Prepare 1% Bacto-yeast extract (w/v), 2% Bacto-peptone (w/v), 2% glucose (w/v), 2% Bacto-agar (w/v) by mixing 10 g Bacto yeast extract, 20 g Bacto peptone, 20 g Bacto agar, 120 mg Adenine with 950 ml water. Autoclave and cool to ~ 65°C. Supplement with 2% glucose by adding 50 ml of 40% glucose and 220 μg/ml hygromycin.
Plasmid-based haploid selection plasmid counterselection medium (SGMSG – (Arg,Lys,Ura) + (canavanine,thialysine,clonNAT))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% galactose, 50 μg/ml of canavanine, 50 μg/ml thialysine and and 100 μg/ml clonNAT. Stir solution for 15 min and pour into plates. Since ammonium sulfate interferes with the activity of the antibiotics, such as clonNAT, it cannot be used as the nitrogen source in this medium, and instead MSG is used as an alternative nitrogen source.
Diploid selection medium (YEPD+(G418, clonNAT))
Prepare 1% Bacto-yeast extract (w/v), 2% Bacto-peptone (w/v), 2% glucose (w/v), 2% Bacto-agar (w/v) by mixing 10 g Bacto yeast extract, 20 g Bacto peptone, 20 g Bacto agar, 120 mg Adenine with 950 ml water. Autoclave and cool to ~ 65°C. Supplement with 2% glucose by adding 50 ml of 40% glucose, 200 μg/ml G418 and 100 μg/ml clonNAT.
Enriched sporulation medium
Mix 0.1 g of sporulation amino acid supplement powder, 10 g potassium acetate, 1 g yeast extract, 0.5 g glucose, 20 g agar in 1 L of water. Autoclave and cool to ~ 65°C. Supplement with G418 at a final concentration of 50 μg/ml to prevent contamination. Enriched sporulation medium induces sporulation and ensures that cells grow slowly generating enough cells to be transferred in the next replica pinning step onto the haploid medium. A low dose of G418 (50 μg/mL) lowers the risk of contamination and is not strong enough to result in any G418 resistance selection.
Sporulation medium
Mix 0.1 g of sporulation amino acid supplement powder, 10 g potassium acetate, 1 g yeast extract, 20 g agar in 1 L of water. Autoclave and cool to ~ 65°C. This sporulation medium is optimal for query strain construction.
MATa haploid selection medium (SD – (His, Arg, Lys) + (canavanine, thialysine))
Mix 6.7 g yeast nitrogen base without amino acids, 2 g DO amino acid supplement powder (drop out His, Arg, Lys) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine and 50 μg/ml thialysine. Stir solution for 15 min and pour into plates. Since this medium does not contain antibiotics, G418 and clonNAT, yeast nitrogen base (which contains ammonium sulfate) can be used as a nitrogen source.
MATa double and triple mutant haploid selection medium (SDMSG – (His, Arg, Lys, Ura) + (canavanine, thialysine, G418))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out His, Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine and 200 μg/ml G418. Stir solution for 15 min and pour into plates. Ammonium sulfate interferes with the activity of the antibiotics, such as G418; thus, MSG is used as an alternative nitrogen source.
MATa triple mutant haploid selection medium (SDMSG – (His, Arg, Lys, Ura) + (canavanine, thialysine, G418, clonNAT))
Mix 6.7 g yeast nitrogen base without amino acids, 2 g DO amino acid supplement powder (drop out His, Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine, 200 μg/ml G418 and 100 μg/ml clonNAT. Stir solution for 15 min and pour into plates. MSG instead of ammonium sulfate is used as a nitrogen source, because the latter interferes with the activity of the antibiotic.
Additional Random Spore Media
MATa single mutant haploid selection medium (SD – (His, Arg, Lys, Ura) + (canavanine, thialysine))
Mix 6.7 g yeast nitrogen base without amino acids, 2 g DO amino acid supplement powder (drop out His, Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine and 50 μg/ml thialysine. Stir solution for 15 min and pour into plates.
MATa single mutant haploid selection medium (SDmsg – (His, Arg, Lys) + (canavanine, thialysine, clonNAT))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out His, Arg, Lys) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine and 100 μg/ml clonNAT. Stir solution for 15 min and pour into plates.
MATa single mutant haploid selection medium (SDmsg – (His, Arg, Lys) + (canavanine, thialysine, G418))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out His, Arg, Lys) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine and 200 μg/ml G418. Stir solution for 15 min and pour into plates.
MATa double mutant haploid selection medium (SDmsg – (His, Arg, Lys, Ura) + (canavanine, thialysine, clonNAT))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out His, Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine and 100 μg/ml clonNAT. Stir solution for 15 min and pour into plates.
MATa double mutant haploid selection medium (SDmsg – (His, Arg, Lys) + (canavanine, thialysine, clonNAT, G418))
Mix 1.7 g yeast nitrogen base without amino acids and ammonium sulfate, 1 g monosodium glutamic acid (MSG), 2 g DO amino acid supplement powder (drop out His, Arg, Lys, Ura) and 200 ml water in a 250-mL glass bottle. In a separate 2-L Erlenmeyer flask combine 20 g Bacto-Agar and 750 mL water. Cool both to ~ 65°C. Supplement with 2% glucose (w/v), 50 μg/ml of canavanine, 50 μg/ml thialysine, 200 μg/ml G418 and 100 μg/ml clonNAT. Stir solution for 15 min and pour into plates.
Construction of array strain library
Select the subset of strains from the collection of deletion mutants of non-essential genes or temperature sensitive alleles of essential genes to construct the diagnostic array or use the genome wide array of deletion mutants of non-essential genes or temperature sensitive alleles of essential genes. Prepare 1536-colony format array for the screens from 384-colony format array, which itself is constructed from 96-colony format yeast deletion collection or a collection of temperature-sensitive mutants. Maintain the array on YEPD+G418 and store in 4°C for weeks at a time. The diagnostic and genome-wide arrays have a border consisting of a control strain (his3Δ1::kanMX4), as well as known auxotrophs and sterile mutants for quality control. The genome-wide deletion mutant array lacks 432 slow growing strains, because of their slower growth rate compared to other strains. It is advisable to assess these slow growing strains separately in an unbiased manner, providing sufficient amount of time for them to grow without prolonging unnecessarily the growth of the fast-growing strains. In total the 384-format diagnostic array contains 1,182 unique strains on 4 plates11, the genome-wide deletion mutant43 array contains 4,294 unique strains on 14 plates, and the genome-wide collection of temperature sensitive alleles24 of essential genes contains 1,016 unique strains carrying temperature sensitive alleles and 201 unique nonessential gene deletion mutant strains, which are distributed on 4 plates.
Equipment Set-up
Preparing yeast agar plates
CAUTION Differences in dimensions necessitate that Nunc OmniTrays are used for preparing medium for all replica pinning steps on a BioMatrix Colony Arrayer robot and PlusPlates for the ROTOR benchtop robot. CAUTION For every pinning step OmniTrays are filled with 40 mL and PlusPlates with 50 mL of medium per plate. To make a query lawn, use a pipette to spread the yeast culture on agar plates that have been dried on the bench for 3 days after pouring. Using dry agar plates will ensure that your lawns set evenly. For all other pinning steps, dry the agar plates for 2 days after they were poured. Take care not to overdry plates and leave them sufficiently moist to ensure optimal colony transfer and prevent colonies from mixing with one another and create smears.
Thawing out yeast mutant arrayed collections
Glycerol frozen stock in 96-well plate format is thawed at room temperature two plates at a time. ROTOR benchtop robot and 96-format long RePads are used to transfer thawed culture onto agar containing PlusPlates. Aluminum sealing tape is used to seal the 96-well plates, which are immediately returned to dry ice and stored in −80°C. CRITICAL Defrosting a few plates at a time ensures that the yeast culture does not have time to settle and is frozen within the shortest period of time.
Robotics
For high-throughput pinning, we recommend several machines that were designed specifically for replica pinning yeast arrays. The S&P Robotics BM6-SC1 Robot with a Carousel enables the replica pinning of 112 plates per run for fully automated screening. It also has a colony picking function and thus a re-arraying capability to construct custom arrays, such as the diagnostic array. All the screens described in this protocol have been conducted with this type of a robot. A slightly less expensive version from S&P Robotics is BM6-BC. This benchtop robot lacks a carousel and thus has a lower capacity of 48 plates for replica pinning of 24 plates per run, however it still has a colony picking function. It is a suitable option for labs conducting screens at a lower throughput while still requiring an automated platform. The Singer RoToR benchtop robot provides another option, it can be loaded with a maximum of 4 plates, for the replica pinning of 2 plates with plastic pins, which enables rapid replica pinning and re-loading of plates.
Pinning
When using the ROTOR benchtop robot, RePads can be reused, if washed in bleach and sterilized by UV exposure or autoclaved.
Preparing for analysis
For large-scale screen analysis, MATLAB routines that score raw colony size data to produce SGA and τ-SGA scores are available at https://github.com/csbio/SGA_Public. For smaller scale screen analysis, a web-based analysis system, SGAtools (http://sgatools.ccbr.utoronto.ca/)45, is available for scoring genetic interactions, including quantifying colonies on agar plates, normalizing systematic effects, and calculating fitness scores relative to a control experiment. Visualization tools for genetic interaction data are accessible from a web database (http://thecellmap.org)46.
Procedure
Query strain construction
CRITICAL
Construct the following query strains for the τ-SGA procedure using one of the methods described below.
Double mutant query strain: gene1::natMX4 gene2::KlURA3
Single mutant control query strain 1: gene1::natMX4 hoΔ:KlURA3
Single mutant control query strain 2: hoΔ::natMX4 gene2::KlURA3
Double mutant query strains will be screened against the diagnostic array to generate triple mutants and single mutant control query strains will be screened to produce double mutants (Fig. 1, 3). To obtain the single and double mutant fitness of query strains, additional wild-type strains, such as DMA1 and Y14420 (Table 1), should be screened against the query strain set (Fig. 3). Screening a wild-type strain, such as Y13096 (Table 1), will enable the scoring of single mutant fitness of array strains (Fig. 3).
Non-Essential Gene Deletion Mutant SGA Query Strain Construction by PCR-Mediated Gene Deletion Timing variable; 10-18 d
Use p5749 to PCR amplify K. lactis URA3 or p4339 to PCR amplify natMX4 (Table 1), to mark gene deletions, with K. lactis URA3 or natMX4, respectively. Design a primer with 55 bp of homology specific to the 5’ region of your favourite gene (YFG), including the start codon followed by a marker specific forward primer (MX4-F or KlURA3-F see Table 2) and a primer specific to the 3’ region of YFG, including the stop codon, followed by marker specific reverse primer (MX4-R or KlURA3-R, Table 2).
-
Set up a 100 μL PCR reaction with the following components:
Reagent Amount Final concentration H2O 74.2 μL - 10x buffer 10 μL 1x 10 mM dNTPs 2 μL 200 μM 50 μM forward primer, 4 μL 2 μM 50 μM reverse primer 4 μL 2 μM p4339 DNA template 0.1 μg in 0.5 μL H2O 1 ng/μL DMSO 5 μL 5% vol/vol 5U/μL Taq DNA Polymerase 0.3 μL 0.015 U/ μL Total 100 μL - Initiate the amplification of the cassette with a 5 min denaturation at 95°C, followed by 30 cycles of: 95°C for 30 s, 55°C for 30 s, 68°C for 2 min; terminate the reaction with a 10 min extension at 68°C and hold at 4°C, if necessary. PAUSE POINT Store PCR products at −20°C.
Transform 20-40 ul PCR product by standard LiAc transformation42 into Y7092, the MATα standard SGA starting strain. Use Y7091, the MATa version of Y7092, for crosses with other strains of interest and Y6964, MATa/α heterozygous for SGA markers for query construction in a diploid. Select for transformants on appropriate selection medium following 3-4 day incubation at 30°C.
Confirm correct integration of the marker by PCR using primers that anneal within the selectable marker and either 200 bp upstream or 200 bp downstream of the deleted gene of interest. Confirm MATα mating type and marker segregation.
Upon successful construction of single mutants, cross them to select for diploids on SDMSG – Ura + clonNAT for 2-3 days at 30°C, sporulate on sporulation medium for 7-14 days at 22°C and conduct tetrad analysis to isolate the desired double mutants.
Prepare glycerol stocks of the query strains in 20% (vol/vol) glycerol and freeze at −80°C.
Table 2.
Primer sequences for natMX4 and KlURA3 amplification
| Primer | Sequence (5’-3’) | Comments |
|---|---|---|
| MX4-F | ACATGGAGGCCCAGAATACCCT | Forward amplification primer of MX4 series cassettes |
| MX4-R | CAGTATAGCGACCAGCATTCAC | Reverse amplification primer of MX4 series cassettes |
| HO MX4-F | CATATCCTCATAAGCAGCAATCAATTCTATCTATACTTTAAAATGacatggaggcccagaatacc | Forward primer for deleting HO (uppercase) and marking it with MX4 series cassettes (lowercase) |
| HO MX4-R | TTACTTTTATTACATACAACTTTTTAAACTAATATACACATTTTAcagtatagcgaccagcattc | Reverse primer for deleting HO (uppercase) and marking it with MX4 series cassettes (lowercase) |
| K1URA3-F | cggagacaatcatatgggag | Forward amplification primer of K. lactis URA3 |
| KlURA3-R | tctggaggaagtttgagagg | Reverse amplification primer of K. lactis URA3 |
| HO KlURA3-F | CATATCCTCATAAGCAGCAATCAATTCTATCTATACTTTAAAATGcggagacaatcatatgggag | Forward primer for deleting HO (uppercase) and marking it with KlURA3 (lowercase) |
| HO KlURA3-R | TTACTTTTATTACATACAACTTTTTAAACTAATATACACATTTTAtctggaggaagtttgagagg | Reverse primer for deleting HO (uppercase) and marking it with KlURA3 (lowercase) |
| MX4-KlURA3-F | ACATGGAGGCCCAGAATACCCTCCTTGACAGTCTTGACGTGCGCAGCTCAGGGGCcggagacaatcatatgggag | Forward primer for marker-switching kanMX4 (uppercase) to KlURA3 (lowercase) |
| MX4-KlURA3-R | CAGTATAGCGACCAGCATTCACATACGATTGACGCATGATATTACTTTCTGCGCAtctggaggaagtttgagagg | Reverse primer for marker-switching kanMX4 (uppercase) to KlURA3 (lowercase) |
Conditional Allele of Essential Gene SGA Query Strain Construction by Two-Step PCR-Mediated Integration Timing variable; 11–21 d
7. PCR amplify a conditional temperature-sensitive (ts) allele of interest using two pairs of oligonucleotide primers. The first pair of primers is designed to amplify the conditional allele, including 200 bp downstream of its stop codon and an additional 25 bp of sequence complementary to the 5’ end of the preferred marker: natMX4 or K. lactis URA3. A second pair of primers is used to amplify the preferred marker: natMX4 or K. lactis URA3 such that the reverse 3’ primer contains 45 bp of complementary sequence immediately downstream of the gene of interest.
8. Combine PCR products and co-transform into the SGA background strain, Y7092 (Table 1).
9. Select transformants on YEPD + clonNAT and incubate at the permissive temperature ~22 °C for 3-5 days.
10. Verify the integration of the conditional allele by replica plating on YEPD and YEPD + clonNAT and incubating at the restrictive temperature for 1-2 days. Test on rich medium lacking clonNAT to ensure that the ts phenotype is linked to the gene of interest rather than the natMX4 selectable marker.
11. Following the successful construction of single mutants, cross them to select for diploids on SDMSG – Ura + clonNAT for 2-3 days at 22°C, sporulate on sporulation medium for 7-14 days at 22°C and conduct tetrad analysis to isolate the desired double mutants.
12. Prepare glycerol stocks of the SGA query strains in 20% glycerol (vol/vol) and freeze at - 80°C.
Double mutant query strain construction by using plasmid-based haploid selection Timing variable; 12-20 d
13. Construct the gene-1 single mutant strains according to steps 1-6 or 7-12, such that they are of the following genotype: MATα gene-1::natMX4 can1Δ::STE2pr-Sp_his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0.
14. Transform gene-1 single mutant strains with plasmid p6981 (see Table 1 for plasmid details), which will be used for the downstream MATα haploid selection, select transformants on SD-Leu solid medium.
15. Construct gene-2 single mutant strains according to steps 1-6 or 7-12 such that they are of the following genotype: MATα gene-2Δ::KlURA3 his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 by marker-switching the kanMX4 cassette to K. lactis URA3 in the yeast deletion collection strains (see Table 2 for primer details). The primer sequences that are used to amplify the KlURA3 cassette contain 55 bp homology to the 5’ region of the kanMX4 cassette and 55 bp homology to 3’ of the kanMX4 cassette.
16. Array the resulting gene-1 and gene-2 strains in 96-colony format on YEPD + clonNAT and SD - Ura OmniTrays, respectively.
17. Mate strains by pinning gene-1 and gene-2 strains on YEPD for 1 day at RT.
18. Select diploids on SDMSG – (Leu, Ura) + clonNAT for 2-3 days at RT.
19. Then transfer diploids by patching onto sporulation medium containing plates and incubate for 7-14 days at 22°C.
20. Selectively germinate MATα meiotic progeny using plasmid-based STE3pr-hphR marker contained in p6981 by patching the sporulated mix onto YEPD + hygromycin and incubating for 2 days at RT.
21. Transfer the haploid mix by patching onto YEPD to facilitate plasmid loss. Incubate 1 day at RT.
22. To ensure plasmid loss, induce GAL1pr-KAR1, which is on the same plasmid to counter select for query strains lacking the plasmid, which is accomplished by streaking the haploid mix onto SGMSG - (Arg, Lys, Ura) + (canavanine, thialysine, clonNAT), 3 days, RT. Final double mutant strains are of the following genotype: MATα gene-1::natMX4 gene-2::KlURA3 can1Δ::STE2pr-Sp_his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0.
23. Prepare glycerol stocks of the query strains in 20% (vol/vol) glycerol and freeze at −80°C.
Array strain construction Timing 4 d
24. Use the Colony Arrayer to select the subset of array strains from the collection of deletion mutants of non-essential genes or temperature sensitive alleles of essential genes to construct the diagnostic array on 4-plates in 384-format. Alternatively, use the genome wide non-essential collection of deletion mutants or temperature sensitive alleles of essential genes. Incubate the plates with mutant strains for 2 days at room temperature. CRITICAL STEP Colonies exhibit a larger growth along the edges than in the middle of a plate, due to a greater access to nutrients. This increase in colony size does not reflect greater fitness or a positive genetic interaction. Thus, to eliminate false positives, a control strain is grown along the border of the array (two top and bottom rows and two leftmost and rightmost columns in the 1536-format). The border colonies are excluded from the analysis. The control border strain, his3Δ1::kanMX4, can be obtained from the yeast deletion collection. It complements the histidine auxotrophy in the SGA query strain, which harbors the STE2pr_ Sp his5 reporter enabling it to survive all the steps of the τ-SGA procedure.
25. Replicate each 384-format array in quadruplicate onto a single plate to generate a 1536-density array. Incubate the cells for 1 day at room temperature.
26. Replicate the 1536-format array into two copies on separate plates to generate both a working and a master copy. Incubate the cells for 1 day at room temperature.
27. Use the working copy of the 1536-format array to replicate as many copies as necessary. Each copy of the array can be used up to 4 times for replicating or mating.
Query strain lawn preparation Timing 6 d
28. Streak out query strains from glycerol stock (from step 1-23) on agar plates containing YEPD + clonNAT. Incubate for 2–3 days at 30°C. CRITICAL STEP Incubation temperature varies depending on the array that you are using. Since the diagnostic array and some of the query strains are composed of strains that harbor temperature sensitive alleles, you should use 22°C for all steps of the τ-SGA procedure, except for the final step when the plates should be incubated at a semi-permissive temperature, such as 26°C. For an array and query strains composed exclusively of non-essential genes you can use 30°C incubation temperatures throughout.
29. Inoculate 5 mL of YEPD liquid medium with a single colony. Incubate for 2 days at 30°C in a shaking incubator, 250 rpm.
30. Spread 800 μL of the saturated liquid culture on an OmniTray containing YEPD solid medium. Repeat to prepare a total of one query strain lawn for one miniaturized screen with 4 diagnostic array plates. Allow the lawns to dry, and then incubate for 2 days at 30°C. CRITICAL STEP For spreading a query lawn, use agar plates that have been drying for 3 days on the bench after pouring. Use a 1 mL pipette to spread the lawn on dry agar plates to ensure that your lawns set evenly, which is important for the mating step query strain transfer. ?Troubleshooting
Conducting the τ-SGA procedure Timing 17 d
31. Mate the query strain with the mutant array by pinning the query strain from the lawn to fresh YEPD plates and then the mutant array (from step 24-27) on top of the query. Incubate at room temperature, 1 day. One query lawn provides a sufficient source of query strains for mating with 4 array plates. CRITICAL STEP Dry the agar plates for 2 days on the bench after pouring for all the pinning steps to prevent colonies from mixing with one another.
32. Select for diploids by pinning the resulting MATa/α diploid zygotes onto YEPD+(G418, clonNAT). Incubate at 30°C, 2 days. ?Troubleshooting
33. Sporulate the diploids by pinning the resulting MATa/α KANR NATR diploids onto the enriched sporulation agar plates. Incubate at 22°C, 7 days. PAUSE POINT Following the incubation time of any of the steps 31 – 35 the user can store plates for several days at 4°C and continue the procedure at a later date.
34. Select for MATa meiotic haploid progeny by pinning the spores onto SD – (His, Arg, Lys) + (canavanine, thialysine). Incubate at 30°C, 2 days.
35. Select for MATa URA+ KANR meiotic haploid progeny by pinning the spores onto SDMSG – (His,Arg,Lys,Ura) + (canavanine,thialysine,G418). Incubate at 30°C, 2 days.
36. Select for MATa NATR URA+ KANR meiotic haploid progeny by pinning the mix of double and triple mutant haploids onto SDMSG – (His, Arg, Lys,Ura) + (canavanine, thialysine, G418, clonNAT). Incubate at 30°C, 2 days. The resulting output array consists of a collection of yeast triple mutant strains in which each KANMX4-marked array mutant is also deleted for the query genes of interest, which are marked with KlURA3, NATMX4. CRITICAL STEP The sequence of steps for the selection of triple mutant haploids is the following: (URA+ and KANR) followed by (NATR, URA+ and KANR) and is our preferred time- and cost-effective approach that produces evenly round colonies useful for quantification. It involves pinning the haploid mix initially onto medium that selects for two and then all three selectable markers and reduces the progeny first by 4-fold and then by 2-fold. Alternatively, you can sequentially pin haploids onto the medium that first selects for one, then two and then three selectable markers: (KANR), (URA+ and KANR) followed by (URA+, NATR and KANR), which reduces the number of haploid progeny by 2-fold each time generating evenly round colonies. The section option contains an additional step and thus it is not preferred due to requiring extra 2 days of growth and additional medium. Selecting initially single, and then triple mutants: (KANR), (URA+, NATR and KANR), reduces the haploid progeny first by 2-fold and then by 4-fold and produces uneven colonies that are difficult to score, and thus we do not recommend this approach.
37. Image the double mutant array using a high-resolution digital imaging system, such as the one developed by S&P Robotics, Inc. (Toronto, ON). For Canon EOS Rebel T3i EOS 600D, the following settings are recommended: inactivated flash; P = Programmed Automatic; AWB = Auto White Balance, ◢L = Quality setting: Large (smooth); ISO 400 = Light sensitivity is set to 400.
38. Repeat each double-mutant and triple-mutant screen along with the single-mutant screen twice to obtain a sufficient level of precision for trigenic interaction scoring11. Conducting these three types of screens in the same batch minimizes noise by allowing for the effective normalization of potential sources of systematic bias.
39. Quantify genetic interactions by processing the images to distinguish colonies from the background and measuring their area in pixels. Correct the raw colony sizes for systematic effects and quantify genetic interactions using the single and double mutant control screens as references. Large-scale high-throughput screens using the τ-SGA analysis pipeline (Steps 42-48) and smaller scale screens can be analyzed using SGAtools (Steps 49-58). CRITICAL STEP Visually inspect all plate images and ensure that gene linkage, as evident by reduced colony sizes, is observed for both query genes and τ-SGA marker genes. ?Troubleshooting
Confirming genetic interactions Timing variable: 4-7 d
-
40. Confirm the resulting genetic interactions using random spore analysis. Mate strains of interest, select for diploids and sporulate, after which resuspend a small amount of spores (~ on the tip of a 200 ul pipette tip) in 1 mL of sterile water and mix well. Then, plate the following volumes of suspension on the specified media:
Volume of suspension (μL) Media composition: 20 SD – (His, Arg, Lys) + (canavanine, thialysine) 40 SD – (His, Arg, Lys, Ura) + (canavanine, thialysine) 40 SDMSG – (His, Arg, Lys) + (canavanine, thialysine, G418) 40 SDMSG – (His, Arg, Lys) + (canavanine, thialysine, clonNAT) 80 SDMSG – (His, Arg, Lys, Ura) + (canavanine, thialysine, clonNAT) 80 SDMSG – (His, Arg, Lys, Ura) + (canavanine, thialysine, G418) 80 SDMSG – (His, Arg, Lys) + (canavanine, thialysine, G418, clonNAT) 160 SDMSG – (His, Arg, Lys, Ura) + (canavanine, thialysine, G418, clonNAT) Incubate at 30°C for 2-3 days and score the meiotic progeny by comparing the number of colonies on single selection media to the number of colonies on the double and triple selection media plates. The expected number of meiotic progeny on each medium should be equal. Petri dishes, which are 6 cm wide can be filled with ~ 8 mL of solid media and are suitable for conducting this assay. PAUSE POINT The sporulation mix can be stored for several days/weeks at 4°C, if necessary.
41. Alternatively, confirm the resulting genetic interactions using tetrad analysis (Fig. 5)47. CRITICAL STEP Using SDMSG complete medium for tetrad dissections should most closely resemble the final selection medium of trigenic-SGA screens. It is also more sensitive than rich YEPD medium for observing subtle growth defects.
High-throughput analysis of trigenic interactions
CRITICAL
For large datasets with hundreds of screens, proceed to high-throughput analysis of trigenic interactions, which is described in steps 42 – 48 (Fig 6). This type of analysis includes batch effect correction since a colony size signature is present in screens resulting from non-biological sources of variation due to them being conducted by the same person, using the same equipment, on the same day. Thus, when a dataset is composed of multiple batches, it is imperative to normalize for batch effect to minimize the number of false positives. In addition, it is not feasible to analyze screens one by one when working with a large dataset and an automated computational pipeline is necessary.
Data Analysis: Setup Timing < 1 min
- 42. Open Matlab, create a script that defines relevant parameters and calls the required functions to generate τ-SGA scores. The provided trigenic_scoring_job.m script (with very few modifications) can be used as a basis for this script (this will be referred to as the “main script” hereafter). The necessary parameters can be divided into three categories:
-
Mandatory parameters: these provide the paths (locations) of required files.<inputfile>: raw colony size data to score (9 column format as described in Data Analysis).<outputfile>: path of the output file (file name does not need an extension).<smfitnessfile>: path to query/array mutant fitness file.<linkagefile>: path to the linkage definition file.<coord_file>: path to the coordinate map file.<removearraylist>: path to the file with a list of known bad array strains (to be removed from the output scores).SGA scoring package has the necessary data included for the last four parameters and therefore those can be borrowed from there for the scoring too.
-
Overridable parameters: have default values in the SGA scoring package, but you will need to specify explicitly how they differ from their default values in your setting.<border_strain_orf>: the ID of the strain found on the border; the default value is ‘YOR202W_dma1’.<wiid_type>: the ID of the wild-type strain. The default value is ‘URA3control_sn4757’, which is for double mutant scoring, and therefore must be changed for triple mutant scoring from the main script.<skip_perl_step>: skips a preprocessing step. The default value is false, however, if a file has been scored before, setting this to true will save time during preprocessing as it avoids unnecessary repetition of preprocessing.
-
Optional parameters:<skip_linkage_detection>: set to true to skip linkage detection altogether (default is false).<skip_linkage_mask>: set to true to replace linkage colonies after corrections (default is false).<skip_wt_remove>: set this to true to include WT query strain/s in the output (default is false).<random_seed>: set to an integer number for reproducibility purposes (using the same number next time will reproduce the previous result).If the user modifies trigenic_scoring_test.m to use as the main script, the parameters <inputfile> and <outputfile> must be modified. <wild_type> and <border_strain_orf> need modification if and only if they are different for your array design. All other parameters and the source code can be used as is.
-
43. Download the SGA scoring pipeline from https://github.com/csbio/SGA_Public and extract it into a local directory. The extracted directory is named SGA_Public-master and all the scripts necessary to generate the τ-SGA scores reside inside this directory. Add the location of SGA_Public-master inside the main script using addpath( (<location>) and then add the necessary subdirectories by calling the function add_SGAPATH().
Data Analysis: compute SGA scores Timing variable, ~1.5h for the example dataset
-
44. Once step 42 and 43 are done, add the line compute_sgascore to your main script. This will invoke the SGA scoring script compute_sgascore.m which will generate SGA scores for all query-array combinations. compute_sgascore.m reads in input data, performs preprocessing, normalizations and eventually calculates SGA scores (also assigns significance to the scores). For a detailed understanding of normalization and SGA score, please consult the SGA scoring manuscript2.
For advanced users, we summarize in Box 1 the steps of compute_sgascore.m to provide a walkthrough of the script; others can skip and proceed with step 45.
CRITICAL STEP compute_sgascore.m generates a log file with the same name of the output file plus ‘.log’ appended to it. This file outputs a summary of different steps of SGA scoring to help ensure quality control and facilitate troubleshooting.
Box 1. The steps of the compute_sgascore.m scoring script.
Stage 1: Preprocessing (start - line 331).
This section accomplishes the following steps:
Create the mentioned log file.
Define the values of the parameters undefined in the main script.
Read the raw data from <inputfile> and load the data into a Matlab struct named <sgadata>; this is done using load_raw_sga_data_withbatch.m.
Perform speed optimizations and output some quality control metrics.
Filter colonies (a combination of colonies from query strain gene linkage groups) to exclude from the normalization and filtering steps.
CRITICAL STEP Investigate the log file to see whether the parameters are set as expected. Also, confirm that the border array strain matches the expected percentage of strains (if it is very different from the expected percentage, you should check to be sure the data has been interpreted correctly).
CRITICAL STEP By navigating to the directory, where the log file is located, you will also see two files ending in ‘numeric’ and ‘orfidmap’. If you want to run the scoring script again, you can now set <skip_perl_step> to true to save some time.
Stage 2: Normalization and Filtering (line 333 - line 457).
Several normalization and filtering steps are applied to the raw data prior to the calculation of interaction scores. The scoring script (compute_sgascore.m) makes use of several other supporting scripts in this step.
Apply plate effect normalizations (apply_plate_normalization.m) to account for plate-to-plate variance.
Filter abnormally large colonies (more than 1.5 times the median colony size).
Calculate colony residuals (calculate_colony_residuals.m). Downstream normalization/filtering is performed on the residuals.
Normalize out the spatial gradient pattern in colony size using a 2D smoothing filter (apply_spatial_normalization.m).
Estimate and correct row-column effects on a plate-by-plate basis using a linear LOWESS smoothing (apply_rowcol_normalization.m).
Perform competition correction (due to local competition for nutrients between neighboring colonies) to normalize the larger colonies associated with small neighbors (apply_competition_correction.m).
Carry out Jackknife variance filtering (apply_jackknife_correction.m) to filter out an outlier colony in each group of replicate double mutants in cases where one is an extreme outlier.
Remove some triple-specific false positive interactions (mostly known slow growers) from the analysis.
Perform batch normalization (batch_correction_wrapper.m) to remove batch effects (screens completed together share a common non-biological signature).
Stage 3: Fitness and interaction scoring (line 459 - end).
This section accomplishes all of the following steps:
Read fitness data from <smfitnessfile> and for strains with unavailable fitness data and estimate single mutant fitness from the normalized data.
Fit a model between the single mutant fitness and normalized colony size and use the model to transform the normalized colony data to SGA scores (interactions). Also, derive an accurate estimate of the variance of each SGA score.
Calculate statistical significance (p-value) associated with each SGA score.
Write the SGA scores into a 12 column format (as described in the SGA scoring pipeline output file formats) to <outputfile> (‘.txt’) and the list of strains to a file with the same name but an extension of ‘.orf’.
CRITICAL STEP Check the log file for the ‘Fitness file report’ to examine if you have many missing values (NaN) in the file. This could indicate that the script is not getting matches for many of the strains in the fitness input <smfitnessfile>. In this case, you may have to generate or provide a new fitness file corresponding to the array and queries in your raw data.
Data analysis: compute τ-SGA scores Timing variable, < 10 mins for the example dataset
45. Use the files generated in step 44 (‘.txt’ and ‘.orf’) to read the SGA scores and fitness into a Matlab struct named <sga> (load_sga_epsilon_from_scorefile.m.)
-
46. Specify the path of an assignment file, assignments = ‘assignment_file_170328.csv’ (located inside the SGA_Public-master/refdata directory) to score trigenic interactions. The provided assignment file has 8 different columns:
StrainID1: strain ID of the first single mutant.
ORF1: ORF of the first single mutant.
StrainID2: strain ID of the second single mutant.
ORF2: ORF of the second single mutant.
DMstrainID: strain ID for the double mutant query (ORFs are ORF1 and ORF2).
SM1strainID: strain ID for the single mutant (one of the ORFs will be YDL227C/HO).
SM2strainID: strain ID for the other single mutant.
Annotation: to which class this interaction should be assigned.
CRITICAL STEP The assignment file heavily depends on the queries that are constructed for your setting and therefore is subject to change. If a different set of queries is used, a new accompanying assignment file should be created.
47. Call the script score_trigenic_interaction.m using the struct <sga> from step 45 and <assignments> from step 46 as input. The script finds the trigenic candidates and uses the corresponding SGA scores to compute τ-SGA scores; it then stores the output in another Matlab struct named <sga_tripie>. ?Troubleshooting
-
48. Print the SGA scores (epsilon for double mutants) and τ-SGA scores (tau for triple mutants) into an output file using print_trigenic.m. The format of the output file is as shown below:
Query Strain ID: the name of the double mutant query.
Array Strain ID: the name of the single mutant array.
Adjusted genetic interaction score (epsilon or tau): updated interaction scores (from step 47).
P-value: the statistical significance of the interaction score (from step 44).
Double/triple mutant fitness: observed fitness of double/triple mutants (from step 44).
Double/triple mutant fitness standard deviation: standard deviation of observed fitness (from step 45).
Small-scale analysis of trigenic interactions using SGAtools
For an analysis of individual custom screens, proceed to small-scale analysis of trigenic interactions, which is described in steps 49 – 58 (Fig. 7). The batch effect is more difficult to detect across a small number of screens. Therefore, for small scale screens, it is highly recommended that a ‘matched’ wild-type control screen is performed in parallel with each set of double and single mutant query screens. In this manner, potential batch effects should be shared across screens and thus normalized out in smaller-scale scoring process.
Data Analysis: Setup Timing < 5 min
49. Open SGAtools: http://sgatools.ccbr.utoronto.ca/ in Google Chrome or Mozilla Firefox browsers.
- 50. Name your screen images using the appropriate format.
- General naming: YourName_ScreenType_queryORF_ArrayPlate#_comments.jpg
- Single mutant control screen: YourName_ctrl_YEL021W_1_trigenicsga.jpg
- Double mutant screen: YourName_dm_queryORF_1_trigenicsga.jpg
- Triple mutant screen: YourName_tm_queryORF1_1_trigenicsga_queryORF2.jpg Name the triple mutant screen in the same manner as the double mutant screen, but note the second query ORF in the comments. CRITICAL STEP Since the file name contains only the first query ORF, you will remove the linkage group for the second query ORF manually at a later stage.
Data Analysis: Process images and obtain colony sizes in pixels Timing 10-15 sec per image
51. Select ‘Image Analysis’ at the top of the page. CRITICAL STEP Analyze one double or triple mutant array screen at a time paired with a single mutant control screen that was screened in the same batch to reduce noise. Thus, for a trigenic interaction screen, run SGAtools for a total of 6 times: two replicates of each double mutant screen and the triple mutant screen.
52. Input screen images. Under ‘Data Input’, ‘Plate image(s)’ click on ‘Select image(s)’ and navigate to the desired directory to select both the control single mutant array and either the double or triple mutant array screen images, click ‘Open’. Trigenic SGA screens are conducted in 1536—colonies-per-plate format. Thus, ‘Plate format’ should say ‘1536 colonies (32 x 48)’. Leave ‘Options’ at default.
53. Ensure the loading of correct files. Under ‘Loaded image file(s)’ ensure that ‘Screen type,’ ‘Query name,’ and ‘Array plate id’ were identified correctly. Click on ‘Process images’.
54. Hover over and visually inspect the black and white images of the arrays to verify the proper segmentation of the colonies. The ‘Status’ of both images should say ‘Passed’. At the top of the page click ‘Normalize and score’.
Data Analysis: Normalize colony sizes and score digenic and trigenic interactions Timing 2-5 sec
55. Select the format of the screened array. Under ‘Data input’, ‘Plate layout’, check ‘Define the map of row/column coordinates to gene names,’ in the ‘-Predefined arrays-’ drop down menu choose the name of your array and ‘All plates’ to score the entire screen or individual plates to score one of the plates. We recommend using the diagnostic array for trigenic interaction screens as previously described11, which is named ‘Triple-Array-Elena-mini1200-v1-1536’. If you are using the genome wide non-essential gene deletion array, then select ‘sga-array-ver2-1536’ and for the array of essential temperature sensitive mutants, select ‘ts-array-v7-1536’. Other array formats are also available. CRITICAL STEP Ensure you are using the correct array version.
56. Select format of replicate colonies and filter out genes linked to the query gene to remove false positives. Under ‘Options’, ‘Replicates’ select ‘4 (2x2)’ because each mutant is represented in quadruplicate in a 1536-array format. Under ‘Linkage correction’ check ‘Filter out genes linked to the query’. Leave ‘Linkage cutoff at default. Manually enter ‘Linkage genes’ associated with query genes and SGA marker genes of the screens you are analyzing: CAN1, LYP1, YEL021W, YDL227C, query ORF-1, query ORF-2. CAN1, LYP1 are genetic markers in all the SGA query strains (canlΔ::STE2pr-Sp_his5 lyp1Δ); YEL021W, YDL227C are query genes in the Y13096, which is used for all single mutant control screens (ura3Δ::natMX4 hoΔ::KlURA3); query gene-1, query gene-2 are genes in the query strain and represent a gene of interest and YDL227C in the double mutant control screen (e.g. YJL123C, YDL227C for mtc1Δ::natMX4 hoΔ::KlURA3) or two genes of interest the triple mutant screen (e.g. YJL123C, YOL111C for mtc1Δ::natMX4 mdy2Δ::KlURA3). Under ‘Score results’ check ‘Score the normalized output’. Click ‘Normalize and score’.
57. Download the resulting data by clicking ‘Download’ and select ‘Excel format’.
-
58. Score trigenic interactions as described previously11 using the following approach for each set of replicate screens:
Where τi,j,k is the trigenic interaction score after accounting for single and double mutant effects, εij,k is the raw trigenic interaction score measured for triple mutants between the double mutant query and an array k, εi,k and εj,k are digenic interaction scores measured for double mutants between the single mutant control query and an array, and fi and fj are single mutant fitness estimates. It should be noted that the trigenic scoring equation depicted here is slightly different from the one in Fig. 1, and the former can be directly derived from the latter as shown previously9. We recommend to experimentally estimate the fitness of query strains and substitute them into this equation by crossing them to DMA1 and Y14420 strains (Table 1). If the genetic markers and strain backgrounds are equivalent to the previous studies then one can rely on the published values8,11. Average the adjusted trigenic interaction scores τi,j,k between replicates to obtain the final adjusted trigenic interaction scores τi,j,k. Generally, SGAtools scores below −0.3 can be clearly observed on the plate, representing cases of synthetic lethality or strong synthetic sick fitness defects and scores below −0.18 are similar to the cut-off used in a large scale SGA analysis 45. The resulting trigenic interactions that are scored by SGAtools are well correlated to those scored by the high-throughput scoring approach described above (Fig. 7b). A case study for quantifying trigenic interactions for MTC1-MDY2 query strain screens with its corresponding MTC1 and MDY2 digenic interactions, which includes plate images and the resulting scores are available at https://github.com/ElenaK35/TrigenicSGA_CaseStudy
Data Analysis: Visualization and downstream analysis tools for trigenic interactions data Timing variable, ~1 hr
CRITICAL Upon the completion of the high-throughput analysis of trigenic interactions, which is described in steps 42 – 48 or the smaller scale analysis of trigenic interactions, which is described in steps 49 – 58, proceed to data visualization and downstream analysis tools.
- 59. Install Cluster 3.0 on your system. Then use <sga_triple> from step 47 to generate the cluster using generate_fg_clustergram.m. This creates two different types of clusters:
- A clustergram with data ordered by chromosome position (.pcl)
- A clustergram in which the data has been clustered on both the query and array dimensions (.cdt).
- 60. Download and unzip the Treeview software on your machine, use Linux terminal to get inside the unzipped directory, and run the software. Once Treeview is running, use ‘File -> Open’ and visualize the output from either or both files generated in step 59. To run the software from Linux terminal, use:
$cd <Tree-view directory>
$java –jar TreeView.jar &
61. SGAtools scored screens can also be clustered using Cluster 3.0 and visualized with Java TreeView or the recently developed version of Java TreeView, called Treeview 3.040, which integrates clustering and heatmap visualization into a single user-friendly application (Fig. 8a).
62. SGAtools includes its own additional data visualization tools. Click ‘Data analysis’ to view the scored colonies spatially on a given plate, to visualize the shape of genetic interaction score distribution and use g:Profiler for Gene Ontology enrichment analysis: https://biit.cs.ut.ee/gprofiler/gost.
63. Plot the resulting genetic network using Cytoscape48,49. Overlap the digenic and trigenic interactions with functional standards to gain further insight into gene function. For example, to identify protein complex membership among genetic interactors, overlap the resulting digenic or trigenic interactions with an available protein complex standard2 (Fig. 8b).
64. The similarity of genetic interaction profiles (the set of all the interactions of a particular gene) is highly predictive of gene function by “guilt-by-association”. The reference digenic interaction profile similarity network8 was constructed by computing pair-wise Pearson correlation coefficients between all query and array digenic interaction profiles and visualized by Cytoscape force-directed network layout, which pulls highly correlated genetic interaction profiles towards each other and less correlated profiles further away from each other. Annotate your resulting digenic and trigenic interactions using an open source tool for biological network annotation, such as SAFE (Functional annotations based on the Spatial Analysis of Functional Enrichment)41, to find statistically significant functional enrichments for a given gene list, such as trigenic interactions, using the global genetic interaction network topology based on the overlap with a direct neighborhood (Fig. 8c) using our online tool http://thecellmap.org/46. A Cytoscape plug-in for SAFE is also available for custom network annotation50. This can also be integrated with the high-throughput trigenic interaction scoring procedure to analyze trigenic and digenic interaction screens as described in the previous section.
Anticipated Results
Trigenic-SGA analysis technique generates high-density arrays of yeast single-, double- and triple-mutant colonies on solid growth media using automated replica pinning selection steps (Fig. 1, 3–5). Screening the complete sets of such screens in the same batch improves signal-to-noise ratio by normalizing systematic biases, which affect colony growth on a plate. Screening against the diagnostic array, which covers 1/5 of the yeast genome consisting of nonessential gene deletion mutants and essential gene mutants carrying temperature sensitive alleles and captures major biological processes in the cell, reveals functionally informative digenic and trigenic interaction profiles.
The high-throughput τ-SGA score generates trigenic interaction scores by integrating a control single mutant screen, two double mutant screens, each carrying individual query mutations, and a single triple mutant screen against the respective double mutant query strain (Fig. 6). The SGA pipeline generates scores for the double and triple mutant screens and includes additional linkage filtering that reflects query strain design and quality control of the screen which removes additional auxotrophs. This procedure is then followed by a series of postprocessing steps to translate the resulting raw scores into final adjusted trigenic interactions (i.e. τ-SGA score) by subtracting the digenic interaction effects scaled by the third mutation from the raw triple mutant scores to account for all cases in which two of the genes are not independent. Importantly, computing the scores from two replicate screens with four colonies per screen results in sufficient precision to confidently call trigenic interactions. Smaller-scale trigenic interaction screens are scored with the use of SGAtools similarly to the high-throughput procedure (Fig. 7). Batch matched single mutant control screens are compared individually to both double mutant screens and the triple mutant screen.
The resulting digenic and trigenic interaction data are analyzed using clustering approaches (Cluster 3.0) and visualized using Java TreeView or the updated TreeView 3.0 (Fig. 8a). GO enrichment analysis is conducted using tools, such as gProfiler or SAFE, the latter of which is specifically designed for spatial enrichment analysis of biological networks. By making use of the global genetic interaction reference network topology, SAFE is useful for identifying clusters of genes reflecting protein complexes and pathways, biological processes and subcellular compartments that are statistically overrepresented within the digenic and trigenic interaction profiles (Fig. 8b&c).
Timing
Steps 1-23, query strain construction: 10-20 d
Steps 24-27, array strain construction: 4 d
Steps 26-30, query strain lawn preparation: 6 d
Step 31, mating query strain with the array strains, 1 d
Step 32, diploid selection, 2 d
Step 33, sporulation, 7 d
Step 34, MATa haploid selection, 2 d
Step 35, MATa, URA+, KANR haploid selection, 2 d
Steps 36, MATa, NATR, URA+, KANR haploid selection, 2 d
Steps 37-39, final trigenic SGA plate imaging
Steps 40-41, confirmations of trigenic interactions by random spore or tetrad analyses
Steps 42-43, setting up MATLAB parameters and working directory, < 1min
Step 44, computing SGA scores, ~ 1.5 hr
Steps 45-48, computing τ-SGA scores, variable < 10 min (for the example data set)
Steps 49-50, setting up SGAtools and file naming, < 1 min Steps 51-54, image processing, 10-15 sec per image
Steps 55-58, normalization and scoring of digenic and trigenic interactions, 2-5 sec
Steps 59-64, visualization and analysis for trigenic interaction data, ~ 1 hr
Troubleshooting
Troubleshooting advice for individual steps can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 30 | Poor query strain lawn growth | Very slow growing query strain that would provide insufficient transfer for mating | Grow the query strain lawn for an additional day |
| 32 | Poor diploid selection | Transfer of the query lawn onto mating plates is done for too many plates at a time | Mate 4 plates at a time rather than pinning the query lawn onto the mating plates for the entire carousel |
| 39 | Absence of query gene linkage group at the final step | Query gene is positioned in a recombination hot spot locus | Confirm the query gene by PCR to ensure that it is of the correct genotype and proceed as usual |
| 47 | Showing message ‘This structure has been scored for trigenics. Skipping…’ | There has been a previous attempt to generate τ-SGA scores. | Repeat step 4 and then try again to regenerate τ-SGA scores. |
| 47 | For the triple mutants, SGA scores and τ-SGA scores are the same. | The assignment file is not in the right format or does not match the queries used in input data. | Follow the instructions to create an assignment file that is relevant for the used input data (examine the sample data and assignment file, if necessary). |
Acknowledgements
This work was primarily supported by the National Institutes of Health (R01HG005853) (C.B., B.J.A., and C.L.M.), Canadian Institutes of Health Research (FDN-143264 and FDN-143265) (C.B. and B.J.A.), National Institutes of Health (R01HG005084 and R01GM104975) (C.L.M.) and the National Science Foundation (DBI\0953881) (C.L.M.). Computing resources and data storage services were partially provided by the Minnesota Supercomputing Institute and the UMN Office of Information Technology, respectively. Additional support was provided by Natural Science and Engineering Research Council of Canada Postgraduate Scholarship-Doctoral PGS D2 (E.K.), University of Toronto Open Fellowship (E.K.), U of Minnesota Doctoral Dissertation Fellowship (B.V). C.B is a fellow of the Canadian Institute for Advanced Research (CIFAR).
Footnotes
Competing interests
Authors declare no competing interests.
Code availability
Scripts for trigenic-SGA scoring pipeline are available on GitHub at https://github.com/csbio/SGA_Public. The release for trigenic-SGA interaction scoring is available at https://github.com/csbio/SGA_Public/releases/tag/tau_score_v1.1.0.
Data availability
A sample dataset for quantifying MTC1-MDY2 trigenic interactions and the corresponding MTC1 and MDY2 digenic interactions is available at https://github.com/ElenaK35/TrigenicSGA_CaseStudy. τ-SGA scores, which are included for comparison, are from a previous study11. Trigenic interaction datasets can be browsed interactively here: http://boonelab.ccbr.utoronto.ca/supplement/kuzmin2018/supplement.html http://boonelab.ccbr.utoronto.ca/paralogs/
They were also deposited in the DRYAD Digital Repository (doi:10.5061/dryad.tt367) and (doi:10.5061/dryad.g79cnp5m9), respectively.
References
- 1.Bateson WRSE, Punnett RC & Hurst CC Reports to the Evolution Committee of the Royal Society, Report II, (Harrison and Sons, London, 1905). [Google Scholar]
- 2.Baryshnikova A, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods 7, 1017–1024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novick P & Botstein D Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell 40, 405–416 (1985). [DOI] [PubMed] [Google Scholar]
- 4.Bender A & Pringle JR Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol Cell Biol 11, 1295–1305 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srivas R, et al. A Network of Conserved Synthetic Lethal Interactions for Exploration of Precision Cancer Therapy. Mol Cell 63, 514–525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361, 123–134 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Costanzo M, et al. Global Genetic Networks and the Genotype-to-Phenotype Relationship. Cell 177, 85–100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costanzo M, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (2001). [DOI] [PubMed] [Google Scholar]
- 10.van Leeuwen J, et al. Exploring genetic suppression interactions on a global scale. Science 354(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuzmin E, et al. Systematic analysis of complex genetic interactions. Science 360(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzmin E, et al. Exploring whole-genome duplicate gene retention with complex genetic interaction analysis. Science 368, 1446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowers JE, Chapman BA, Rong J & Paterson AH Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Dehal P & Boore JL Two Rounds of Whole Genome Duplication in the Ancestral Vertebrate. PLoS Biol 3, e314 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzmin E, Costanzo M, Andrews B & Boone C Synthetic Genetic Arrays: Automation of Yeast Genetics. Cold Spring Harb Protoc 2016, pdb top086652 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Kuzmin E, Costanzo M, Andrews B & Boone C Synthetic Genetic Array Analysis. Cold Spring Harb Protoc 2016, pdb prot088807 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Kuzmin E, et al. Synthetic genetic array analysis for global mapping of genetic networks in yeast. Methods Mol Biol 1205, 143–168 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Richardson HE, Wittenberg C, Cross F & Reed SI An essential G1 function for cyclin-like proteins in yeast. Cell 59, 1127–1133 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Sugawara N, Wang X & Haber JE In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell 12, 209–219 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Haber JE, et al. Systematic triple-mutant analysis uncovers functional connectivity between pathways involved in chromosome regulation. Cell Rep 3, 2168–2178 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moura de Sousa J, Balbontin R, Durao P & Gordo I Multidrug-resistant bacteria compensate for the epistasis between resistances. PLoS Biol 15, e2001741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor MB & Ehrenreich IM Genetic interactions involving five or more genes contribute to a complex trait in yeast. PLoS Genet 10, e1004324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Celaj A, et al. Highly Combinatorial Genetic Interaction Analysis Reveals a Multi-Drug Transporter Influence Network. Cell Syst 10, 25–38 e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, et al. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol 29, 361–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan Z, et al. Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat Methods 5, 719–725 (2008). [DOI] [PubMed] [Google Scholar]
- 26.McIsaac RS, et al. Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic Acids Res 41, e57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith JD, et al. Quantitative CRISPR interference screens in yeast identify chemicalgenetic interactions and new rules for guide RNA design. Genome Biol 17, 45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chong YT, et al. Yeast Proteome Dynamics from Single Cell Imaging and Automated Analysis. Cell 161, 1413–1424 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Mattiazzi Usaj M, et al. Systematic genetics and single-cell imaging reveal widespread morphological pleiotropy and cell-to-cell variability. Mol Syst Biol 16, e9243 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Styles EB, et al. Exploring Quantitative Yeast Phenomics with Single-Cell Analysis of DNA Damage Foci. Cell Syst 3, 264–277 e210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottert H, Mattiazzi Usaj M, Rosebrock AP & Andrews BJ Reporter-Based Synthetic Genetic Array Analysis: A Functional Genomics Approach for Investigating Transcript or Protein Abundance Using Fluorescent Proteins in Saccharomyces cerevisiae. Methods Mol Biol 1672, 613–629 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Tsherniak A, et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonatopoulos-Pournatzis T, et al. Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nat Biotech (2020). [DOI] [PubMed] [Google Scholar]
- 34.Mair B, et al. High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat Biomed Eng 3, 796–805 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Datlinger P, et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McFaline-Figueroa JL, et al. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat Genet 51, 1389–1398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Replogle JM, et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat Biotech (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braberg H, et al. Quantitative analysis of triple-mutant genetic interactions. Nature protocols 9, 1867–1881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins SR, Schuldiner M, Krogan NJ & Weissman JS A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol 7, R63 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keil C, Leach RW, Faizaan SM, Bezawada S, Parsons L, Baryshnikova A . Treeview 3.0 (beta 1) - Visualization and analysis of large data matrices. [Google Scholar]
- 41.Baryshnikova A Systematic Functional Annotation and Visualization of Biological Networks. Cell Syst 2, 412–421 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Gietz RD, Schiestl RH, Willems AR & Woods RA Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Sung MK, Ha CW & Huh WK A vector system for efficient and economical switching of C-terminal epitope tags in Saccharomyces cerevisiae. Yeast 25, 301–311 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Wagih O, et al. SGAtools: One-stop analysis and visualization of array-based genetic interaction screens. Nucleic Acids Research 41, W591–596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Usaj M, et al. TheCellMap.org: A Web-Accessible Database for Visualizing and Mining the Global Yeast Genetic Interaction Network. G3 (Bethesda) 7, 1539–1549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amberg DC, Burke DJ & Strathern JN Tetrad dissection. CSH Protoc 2006(2006). [DOI] [PubMed] [Google Scholar]
- 48.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baryshnikova A Exploratory Analysis of Biological Networks through Visualization, Clustering, and Functional Annotation in Cytoscape. Cold Spring Harb Protoc 2016(2016). [DOI] [PubMed] [Google Scholar]
- 50.Baryshnikova A Spatial Analysis of Functional Enrichment (SAFE) in Large Biological Networks. Methods Mol Biol 1819, 249–268 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A sample dataset for quantifying MTC1-MDY2 trigenic interactions and the corresponding MTC1 and MDY2 digenic interactions is available at https://github.com/ElenaK35/TrigenicSGA_CaseStudy. τ-SGA scores, which are included for comparison, are from a previous study11. Trigenic interaction datasets can be browsed interactively here: http://boonelab.ccbr.utoronto.ca/supplement/kuzmin2018/supplement.html http://boonelab.ccbr.utoronto.ca/paralogs/
They were also deposited in the DRYAD Digital Repository (doi:10.5061/dryad.tt367) and (doi:10.5061/dryad.g79cnp5m9), respectively.