

SUMMARY
Basal cell carcinomas (BCCs) frequently possess immense mutational burdens; however, the functional significance of most of these mutations remains unclear. Here, we report that loss of Ptch1, the most common mutation that activates upstream Hedgehog (Hh) signaling, initiates the formation of nascent BCC-like tumors that eventually enter into a dormant state. However, rare tumors that overcome dormancy acquire the ability to hyperactivate downstream Hh signaling through a variety of mechanisms, including amplification of Gli1/2 and upregulation of Mycn. Furthermore, we demonstrate that MYCN overexpression promotes the progression of tumors induced by loss of Ptch1. These findings suggest that canonical mutations that activate upstream Hh signaling are necessary, but not sufficient, for BCC to fully progress. Rather, tumors likely acquire secondary mutations that further hyperactivate downstream Hh signaling in order to escape dormancy and enter a trajectory of uncontrolled expansion.
Graphical abstract
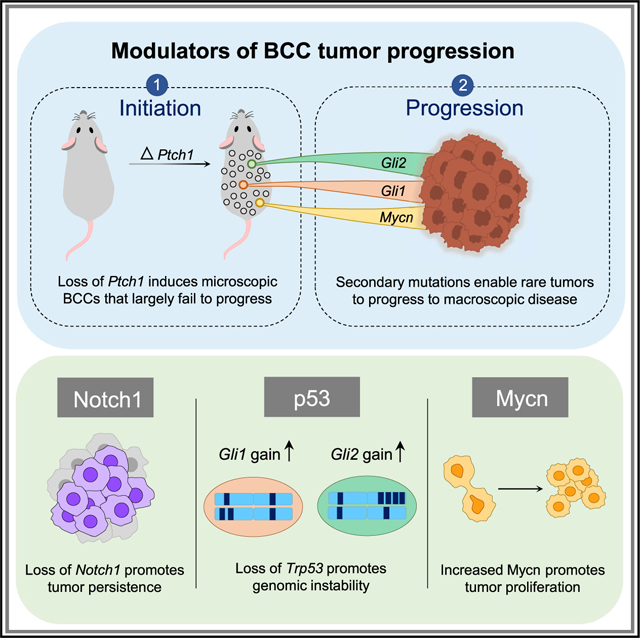
In brief
Trieu et al. generate BCC mouse models in which rare macroscopic tumors form alongside numerous failed microscopic lesions. Successful macroscopic tumors acquire secondary changes that elevate Gli1, Gli2, and/or Mycn levels, causing hyperactivation of downstream Hedgehog (Hh) signaling. Loss of p53 and Notch1 also contributes to tumor progression.
INTRODUCTION
Our skin is exposed to the mutagenic effects of UV radiation on a daily basis. While UV exposure is a major risk factor for skin cancer, recent deep-sequencing studies have revealed that clinically normal, sun-exposed skin can tolerate surprisingly high rates of mutation without forming tumors (Fowler et al., 2021; Lynch et al., 2017; Martincorena et al., 2015). Indeed, previous studies have suggested that keratinocytes that acquire cancer-associated mutations may be actively eliminated from the skin or may persist and carry out normal physiological functions (Brown et al., 2017; Murai et al., 2018; Pineda et al., 2019; Ren et al., 1997). Why certain tumor-initiated cells fail to maintain dysmorphic growth, whereas others display unbridled proliferation, currently remains unclear.
A tumor that arises more frequently than any other is basal cell carcinoma (BCC), the world’s most common cancer (Crowson, 2006; Epstein, 2008). Consistent with the high mutational rates caused by UV exposure, most BCCs arise sporadically in sun-exposed skin; however, Gorlin syndrome patients who inherit one defective copy of PTCH1 are predisposed to forming numerous BCCs (Bonifas et al., 1994; Hahn et al., 1996; Johnson et al., 1996). Although rarely lethal, the ubiquity of BCC, which is diagnosed in over 5 million patients annually, represents a quality-of-life issue for many patients and poses a major burden on our healthcare system (American Cancer Society statistics, 2021).
Dysregulated Hedgehog (Hh) signaling is the key feature that drives all BCCs (Kasper et al., 2012). Normally, Hh signaling is suppressed by PTCH1, whose main function is to inhibit SMO, an upstream activator of the pathway. Upon binding to Hh ligands, PTCH1 is itself inactivated, allowing SMO to signal through GLI transcription factors to induce target gene expression. These targets include those that encode cell cycle regulators, such as MYCN and CCND1, as well as core components of the Hh pathway, such as GLI1, PTCH1, and PTCH2, which provide both positive and negative feedback (Bonifas et al., 2001; Mill et al., 2005; Regl et al., 2002; Tojo et al., 1999).
BCCs are classically driven by mutations that activate upstream Hh signaling, either through loss-of-function mutations in PTCH1 (~70% of tumors) or gain-of-function mutations in SMO (~10%–20% of tumors). However, BCC is also the most highly mutated cancer, with 50–75 mutations/Mb in sporadic tumors and 21–33 mutations/Mb in Gorlin tumors (Atwood et al., 2015; Bonilla et al., 2016; Jayaraman et al., 2014; Sharpe et al., 2015). Not surprisingly, mutations in the Hh pathway (PTCH1, SMO, SUFU, GLI1/GLI2, and MYCN) arise frequently, as do mutations in TP53, Notch signaling (NOTCH1 and NOTCH2), and Hippo signaling (YAP1, PTPN14, and LATS1/LATS2; Bonilla et al., 2016; Kilgour et al., 2021; Di Nardo et al., 2021). These recurrent mutations suggest that tumors initiated by loss of PTCH1 or oncogenic SMO may require additional genetic changes to override tumor-suppressive controls in the skin; however, this remains to be proven.
The immense mutational burden in BCC complicates the ability to distinguish functional mutations from random passenger mutations. To circumvent this problem, we generated simpler BCC mouse models that allow rare macroscopic tumors to form alongside numerous failed microscopic lesions. By comparing tumors that succeed against those that fail, we identify secondary changes that enable nascent tumors to progress to macroscopic disease.
RESULTS
Nascent BCC-like tumors driven by hallmark mutations fail to progress
We and others have previously demonstrated that microscopic BCC-like tumors form efficiently after Ptch1 deletion in hair follicle and surface mechanosensory touch dome (TD) epithelia (Peterson et al., 2015; Sun et al., 2020). To further assess the growth kinetics of these lesions, we analyzed mice expressing tamoxifen (TAM)-inducible Cre recombinase under the control of the Gli1 promoter (Gli1-CreERT2), coupled with homozygous Ptch1 floxed alleles (GP mice) (Ahn and Joyner, 2004; Uhmann et al., 2007). As we previously reported, 5 weeks after TAM administration in GP mice, numerous microscopic lesions arose from Gli1+ stem cells in the hair follicle and TD (Peterson et al., 2015) (Figures 1A–1C). Hair follicle-associated tumors resembled nodular BCC, whereas TD-derived tumors possessed features reminiscent of infundibulocystic BCC and fibroepithelioma of Pinkus.
Figure 1. Microscopic tumors initiated by activation of upstream Hh signaling fail to progress.
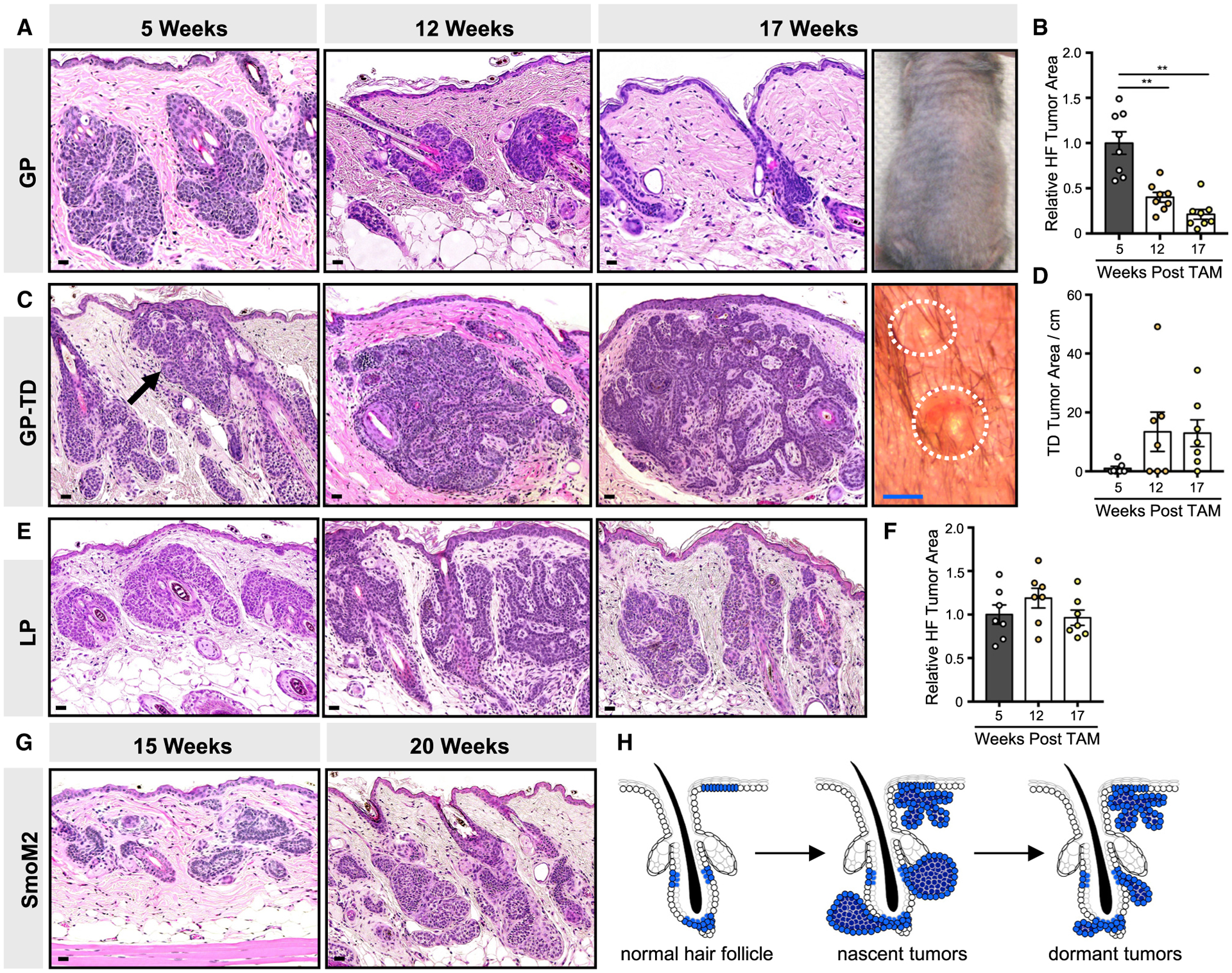
(A) Histology of hair follicle (HF)-associated GP tumors, 5–17 weeks post-TAM. Right photo shows shaved dorsal skin devoid of palpable tumors, 17 weeks post-TAM.
(B) Quantitation of HF-associated tumor area.
(C) Histology of touch dome (TD)-derived GP tumors (arrow), 5–17 weeks post-TAM. Right photo shows view of small TD papules, ~1 mm diameter.
(D) Quantitation of TD-derived tumor area, in arbitrary pixel units per cm.
(E) Histology of HF-derived LP tumors, 5–17 weeks post-TAM.
(F) Quantitation of LP tumors.
(G) Histology of microscopic HF-associated SmoM2 tumors arising from Lrig1+ stem cells (left, 15 weeks post-TAM) or Gli1+ stem cells (right, 20 weeks post-TAM).
(H) Schematic of GP tumor kinetics.
Data are represented as mean ± SEM, with significance calculated by one-way ANOVA. **p < 0.01. Scale bars, 50 μm. Blue scale bar, 1 mm.
To examine the long-term fates of these nascent tumors, we collected serial biopsies up to 17 weeks post-TAM. Unexpectedly, we observed that hair follicle-associated lesions spontaneously regressed over time, leaving behind small residual tumor nests (Figures 1A and 1B). In no instance did we observe macroscopic tumors in any GP mice (Figure 1A). By contrast, TD-derived tumors neither progressed nor regressed between 12 and 17 weeks post-TAM, although we occasionally observed ~1-mm-diameter papules that did not enlarge over time (Figures 1C and 1D). Macroscopic tumors failed to appear, even when we followed GP mice up to 25 weeks post-TAM (Figure S1A).
To determine whether the lack of tumor progression is generalizable to lesions originating from other stem cell populations, we also targeted Lrig1+ hair follicle stem cells for Ptch1 deletion (LP mice) (Powell et al., 2012). Similar to above, we observed nascent microscopic lesions in LP mice but no macroscopic tumors (Figures 1E and 1F). Finally, we assessed tumor formation following overexpression of a constitutively active form of Smo (SmoM2), targeted to either Gli1+ or Lrig1+ stem cells (Mao et al., 2006). Again, abundant microscopic BCC-like tumors emerged but no macroscopic tumors (Figure 1G). Altogether, these findings demonstrate that nascent tumors initiated by either loss of Ptch1 or gain of Smo fail to progress in the most widely studied conditional models of BCC (Figure 1H).
Nascent BCCs become dormant despite constitutively elevated Hh signaling
Sporadic BCCs often arise in aged skin, which undergoes epidermal and dermal changes over time (Rittié and Fisher, 2015). To better characterize our BCC model, we asked whether aging confers a permissive environment for GP tumors to progress. We therefore induced Ptch1 deletion in young or older mice at 4 or 25 weeks of age, respectively, and assessed tumor kinetics relative to animals induced at 8 weeks of age, our standard starting point (Figure 2A). In all cases, we observed abundant microscopic tumors at 5 weeks post-TAM, followed by spontaneous regression of hair follicle-associated tumors at 12–17 weeks post-TAM (Figures 2B–2D). As before, no macroscopic tumors emerged. These findings indicate that the relative age of the tumor (time after initiation), rather than the absolute age of the animal, likely determines regression kinetics in our system.
Figure 2. Microscopic tumors exhibit reduced proliferation over time.
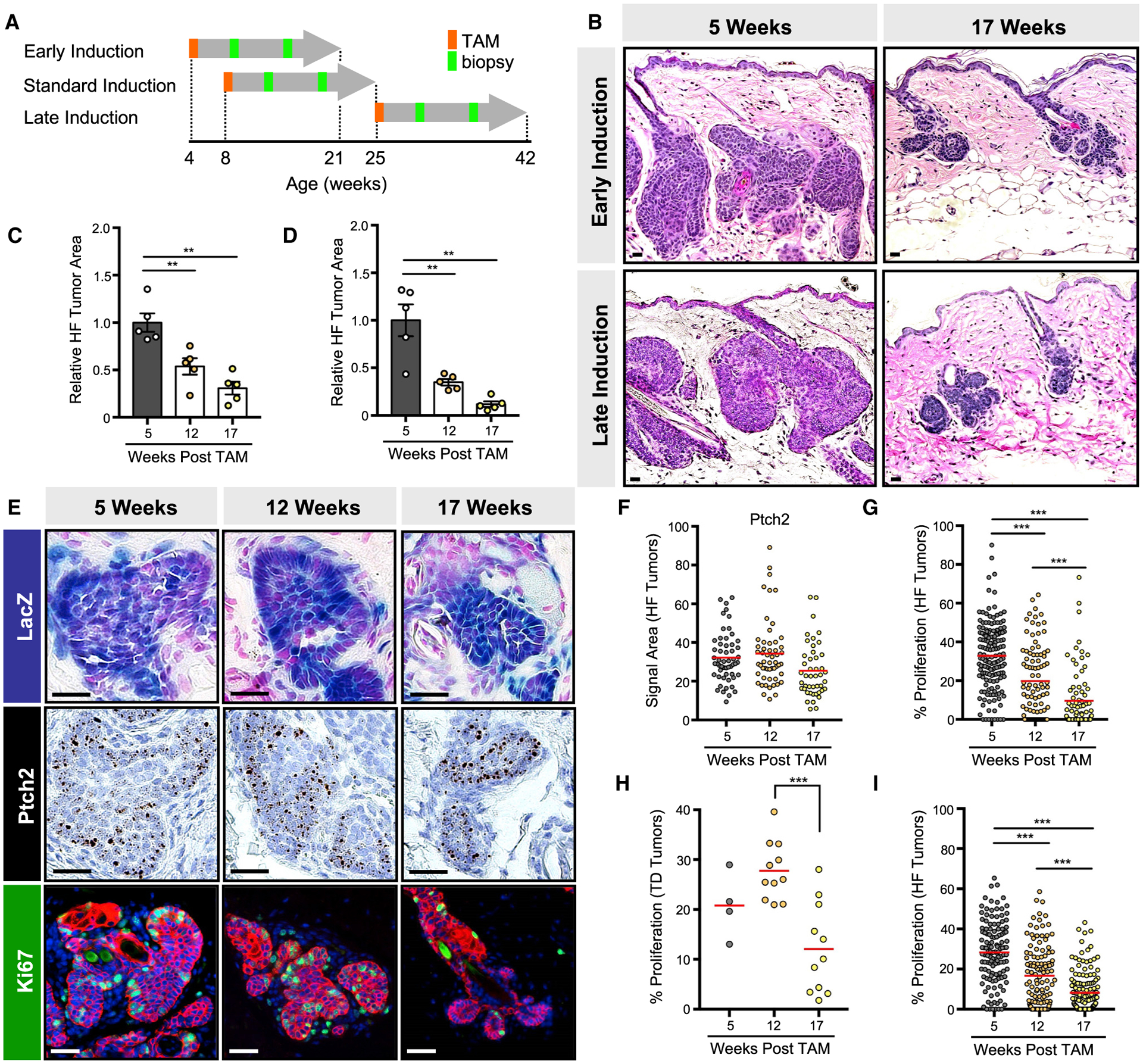
(A) Schematic for TAM induction and skin biopsy in GP mice. Mice were also biopsied at the end of the experiment.
(B) GP tumors regress by 17 weeks post-TAM, regardless of induction scheme.
(C) Quantitation of tumor area in early-induced GP mice.
(D) Same as (C) but for late-induced GP mice.
(E) GP tumors maintain elevated Hh signaling, as assessed by LacZ (top panels) and Ptch2 mRNA (middle panels), but reduce their proliferation over time, as assessed by Ki67 (green, lower panels). Red, keratin 14 (K14).
(F) Quantitation for Ptch2 in HF-associated GP tumors.
(G) Quantitation of proliferation in HF-associated GP tumors.
(H) Same as (G) but for touch-dome-derived GP tumors.
(I) Quantitation of proliferation in LP tumors.
For (E)–(I), analysis was performed on tumors arising from the standard induction scheme. Data are represented as mean ± SEM, with significance calculated by one-way ANOVA. Significance for beeswarm plots was calculated using a linear mixed model. **p < 0.01; ***p < 0.001. Scale bars, 50 μm. See also Figures S1 and S2.
Since BCCs regress in response to pharmacological inhibition of Hh signaling (von Hoff et al., 2009), we next asked whether spontaneous tumor regression occurs due to the inability to maintain high-level Hh signaling. To measure downstream pathway activity, we incorporated a Gli1-responsive β-galactosi-dase (LacZ) allele into GP mice and assessed LacZ activity. Alternatively, we quantitated mRNA in situ for the canonical Hh target gene, Ptch2. In both cases, we found that Hh pathway activity is maintained even in regressed residual tumors (Figures 2E and 2F).
Further characterization of regressing tumors revealed that proliferation is significantly reduced between 12 and 17 weeks post-TAM (Figure 2E), concordant with previous findings in Ptch1-deficient skin lesions (Nitzki et al., 2010). This reduction was detected in both hair follicle- and TD-derived tumors in GP mice (Figures 2G and 2H), as well as in hair follicle-derived tumors in LP and SmoM2 mice (Figures 2I, S1B, and S1C). Notably, regressed GP tumors were not apoptotic and did not express classic markers of senescence, such as p16 and p21 (Figures S2A and S2B). Interventions such as treating the skin with a phorbol ester tumor promoter failed to restore proliferation, while depilation caused hair follicles to enter the anagen growth phase without affecting neighboring regressed lesions (Figures S2C and S2D). These findings indicate that nascent tumors initiated by deletion of Ptch1 eventually become suspended in a dormant state where cells are neither highly proliferative, apoptotic, nor senescent—features that somewhat resemble those of dormant hair follicle stem cells.
Nascent tumors exhibit hair follicle progenitor-like organization and persist upon Notch1 deletion
Given the parallels between spontaneously regressed GP tumors and slow-cycling hair follicle stem cells, we next investigated whether, conversely, nascent proliferating tumors might resemble growing hair follicles. Indeed, we previously reported that BCCs can possess two molecularly distinct cellular sub-compartments: peripheral basal layer cells with high Hh pathway activity and interior suprabasal tumor cells with elevated Notch signaling, as assessed by staining for cleaved Notch1 intracellular domain (NICD) (Eberl et al., 2018) (Figure 3A). Notably, these features are recapitulated in the normal-growing hair follicle bulb, where basal layer matrix progenitor cells exhibit high Hh target gene expression and give rise to NICD+ suprabasal progeny (Figures 3B and 3C).
Figure 3. Notch1-deficient microscopic tumors do not undergo spontaneous regression.
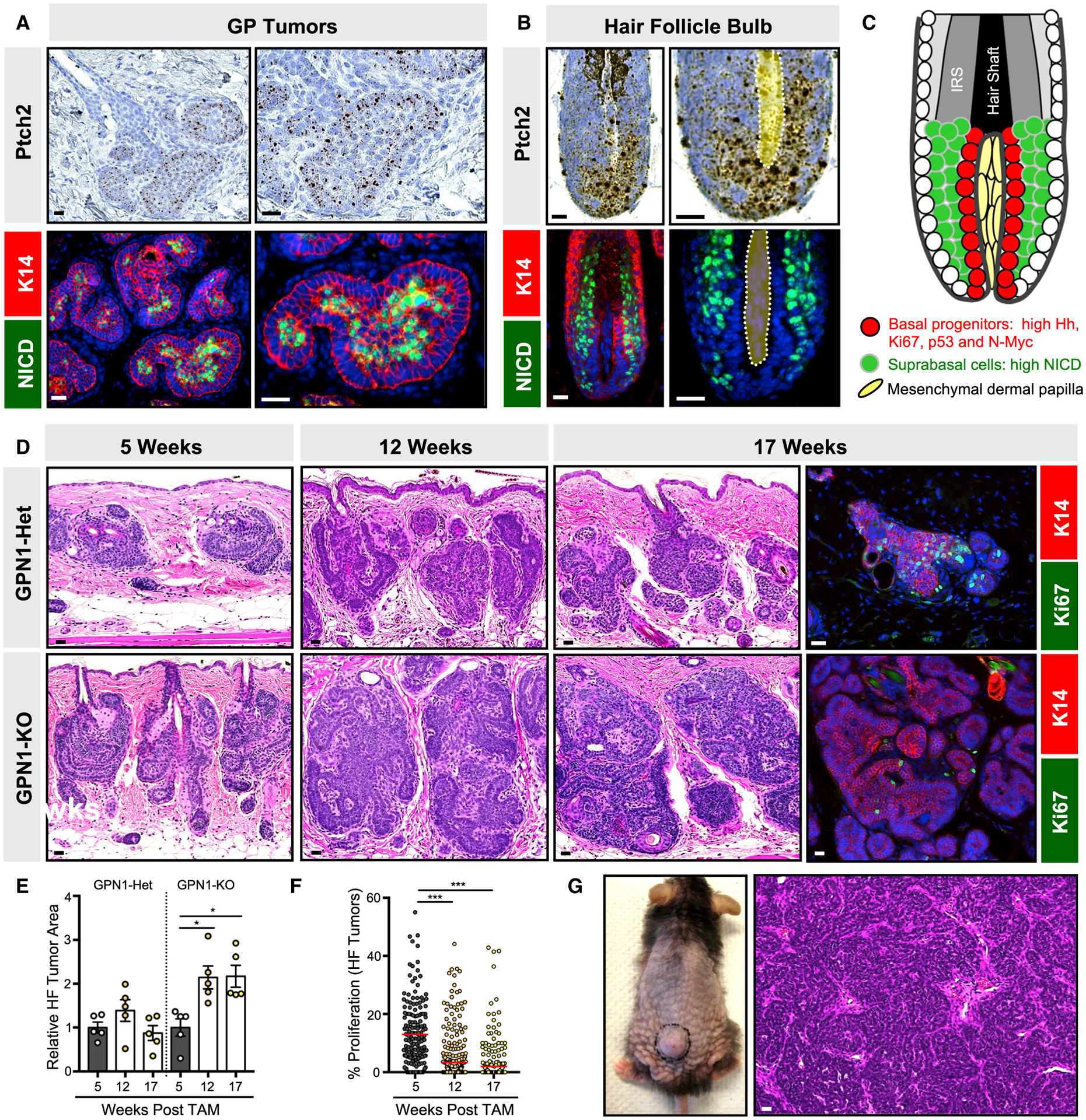
(A) GP tumors have elevated Hh target genes, as assessed by Ptch2 mRNA, in peripheral basal cells (top panels) and high NICD (green) in interior suprabasal cells (lower panels). Red, K14.
(B) In the growing anagen hair follicle, basal matrix progenitor cells directly abutting the dermal papilla (yellow, dotted) express high Ptch2 (top panels), whereas their suprabasal progeny express high NICD (green, bottom panels).
(C) Schematic for basal progenitors (red) and suprabasal progeny (green) in the hair follicle bulb. IRS, inner root sheath.
(D) Histology of HF-derived GP tumors that are either Notch1-heterozygous (GPN1-Het) or deleted (GPN1-knockout [KO]), 5–17 weeks post-TAM. Right panels, tumor proliferation as assessed by Ki67 (green), 17 weeks post-TAM.
(E) Quantitation of GPN1-Het and GPN1-KO tumor area.
(F) Quantitation of GPN1-KO tumor proliferation.
(G) Photo and histology of GPN1-KO macroscopic tumor.
Data are represented as mean ± SEM, with significance calculated by one-way ANOVA. Significance for beeswarm plots was calculated using a linear mixed model. *p < 0.05; ***p < 0.001. Scale bars, 50 μm. See also Figure S3 and Table S1 for animal numbers.
NOTCH1 and NOTCH2 are among the most frequently mutated genes in BCC, and we previously showed that loss of Notch1 enables tumors to persist when Hh signaling is inhibited pharmacologically (Eberl et al., 2018). To test whether Notch also modulates tumor progression, we generated GP mice harboring additional homozygous Notch1 conditional deletion alleles (GPN1 mice) (Yang et al., 2004). In contrast to GP mice, GPN1 animals developed extensive, microscopic hair follicle-associated lesions that did not undergo spontaneous regression (Figures 3D and 3E). In spite of their increased persistence, however, these tumors still reduced their proliferation over time, which was again not reversible by phorbol ester treatment (Figures 3F and S3A). Overall, these findings indicate that losing Notch1 promotes tumor persistence but does not enable these lesions to escape dormancy. Thus, even after developing substantial microscopic tumor burdens that persisted up to 17 weeks post-TAM, GPN1 mice were largely devoid of macroscopic BCC-like tumors, with rare exceptions (Figure 3G; Table S1), which we will discuss in greater detail below.
Loss of p53 is not sufficient to drive BCC tumor progression
TP53 is also commonly mutated in BCC, and loss of Trp53 promotes tumorigenesis in an irradiated model of BCC; however, the mechanism by which p53 modulates BCC progression remains unclear (Ponten et al., 1997; Wang et al., 2011, 2017). Indeed, our previous studies demonstrated that deleting Trp53 affects neither initial tumor formation nor drug-induced regression (Eberl et al., 2018). To examine whether loss of p53 affects later stages of tumor progression, we first confirmed that p53 is highly expressed in basal layer cells in GP tumors, which again mimics the expression pattern seen in basal matrix progenitors in the normal-growing hair follicle (Xue et al., 2019) (Figures 4A–4C).
Figure 4. p53-deficient microscopic tumors largely do not progress.
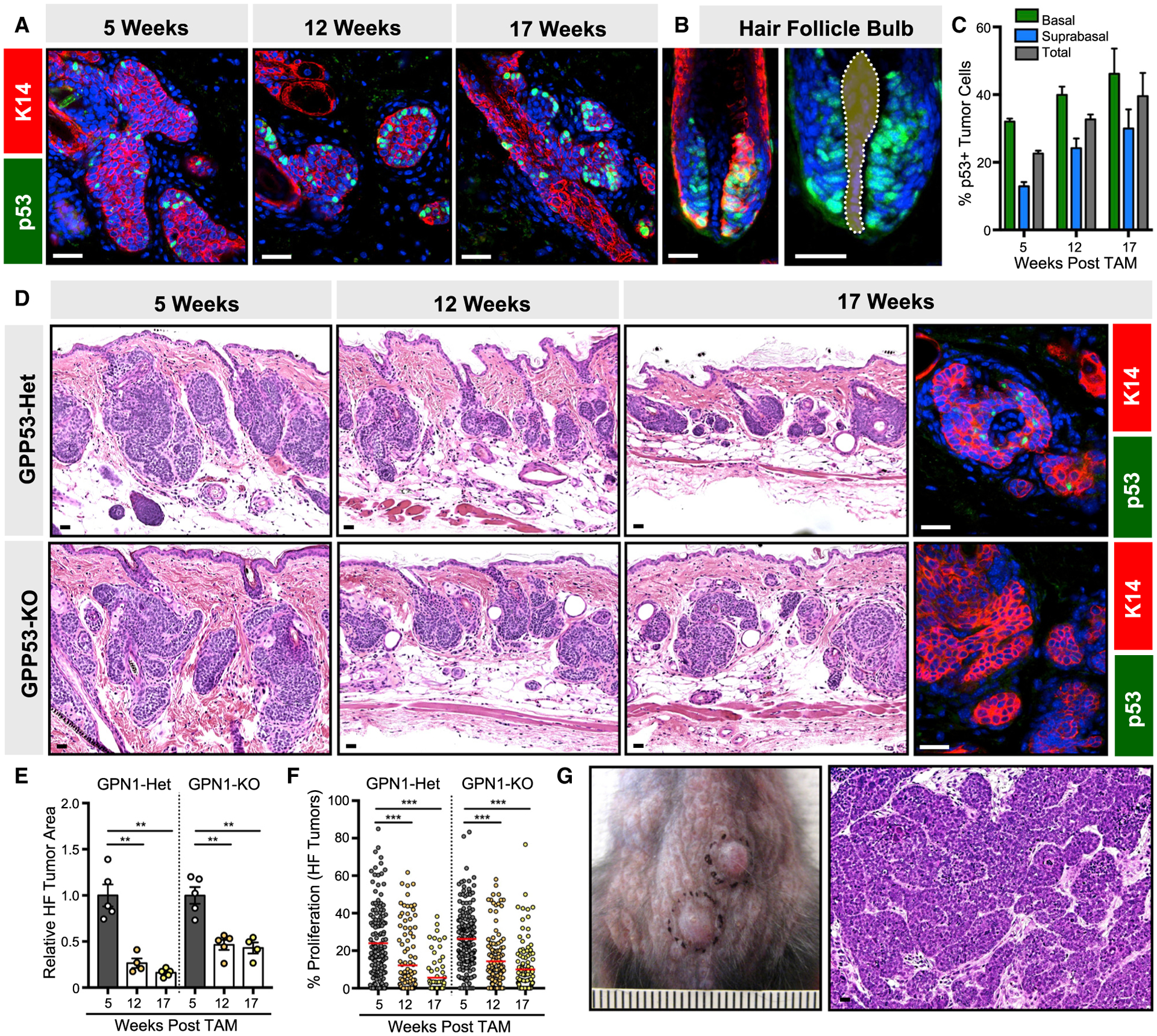
(A) Peripheral basal tumor cells express p53 (green) in GP mice, 5–17 weeks post-TAM.
(B) Basal matrix progenitors directly abutting the dermal papilla (dotted) also express p53 (green) in the growing hair follicle.
(C) Quantitation for p53 in GP basal and suprabasal tumor compartments, 5–17 weeks post-TAM.
(D) Histology of HF-derived GP tumors that are either p53-heterozygous (GPP53-Het) or deleted (GPP53-KO), 5–17 weeks post-TAM. Right panels, validation of p53 loss (green) in GPP53-KO tumors, 17 weeks post-TAM.
(E) Quantitation of GPP53-Het and GPP53-KO tumor area.
(F) Quantitation of GPP53-Het and GPP53-KO tumor proliferation.
(G) Photo and histology of GPP53-KO macroscopic tumors.
Data are represented as mean ± SEM, with significance calculated by one-way ANOVA. Significance for beeswarm plots was calculated using a linear mixed model. **p < 0.01; ***p < 0.001. Scale bars, 50 μm. Ruler marks, 1 mm. See also Table S1 for animal numbers.
We next generated GP mice harboring homozygous Trp53 conditional deletion alleles (GPP53 mice) (Marino et al., 2000) and observed that nearly all microscopic GPP53 lesions still underwent spontaneous regression (Figures 4D and 4E). As seen in GP and GPN1 mice, GPP53 tumors similarly exhibited reduced proliferation over time (Figure 4F), and we confirmed that dormant regressed lesions deleted p53, as expected (Figure 4D). Along with our previously published data (Eberl et al., 2018), these findings suggest that losing p53 does not affect tumor initiation, persistence, dormancy, or drug response. In contrast to GPN1 mice above, GPP53 animals harbored lower microscopic tumor burdens following spontaneous regression. Nonetheless, most GPP53 mice developed at least one macroscopic tumor between 12 and 17 weeks post-TAM (Figure 4G). Below, we explore the pathological and molecular features that distinguish these macroscopic tumors from failed microscopic lesions.
Macroscopic tumors vary histologically
Irradiated Ptch1-heterozygous mice have previously been reported to develop multiple types of skin tumors (Mancuso et al., 2006; Wang et al., 2011). We therefore assessed the histology of macroscopic tumors from GPP53 and GPN1 mice and determined that most tumors can be classified into three categories (Figure 5A): type 1 tumors most resembled human BCC and formed dense basaloid nests with peripheral palisading. Rare type 2 tumors had myoepithelial features, such as expression of α smooth muscle actin (SMA). Finally, type 3 tumors were composed of numerous cell islands with stromal involvement, reminiscent of trichoblastoma. Whereas GPP53 mice mostly developed type 1 tumors, GPN1 mice formed both type 1 and type 3 tumors, with the latter subtype predominating in mice aged beyond 20 weeks post-TAM. In GPP53 mice, type 3 tumors displayed extensive Notch pathway activation (Figure 5A). The incidence and distribution of macroscopic tumor subtypes is summarized in Table S1. For all studies below, we focus exclusively on characterizing type 1 BCC-like tumors.
Figure 5. Characterization of GPP53 and GPN1 macroscopic tumors.
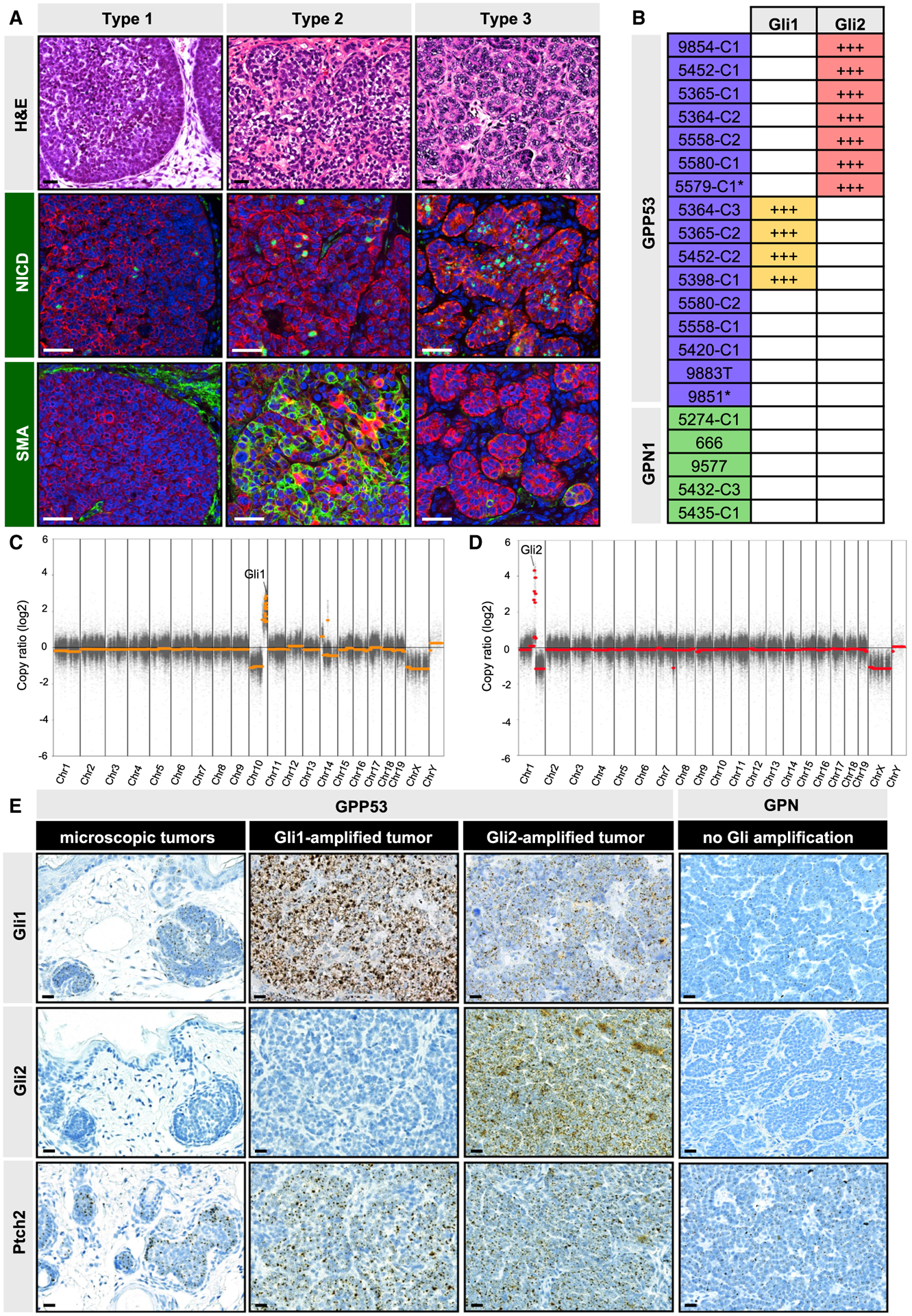
(A) Histologic classification of macroscopic tumors (top panel) and immunohistochemistry for NICD (green, middle panels) and α smooth muscle actin (SMA) (green, lower panels). Red, K14 staining. All examples shown are from GPP53 mice.
(B) Table showing tumors with somatic Gli1 or Gli2 copy number gains (+++, amplified). Asterisk, GPP53-Het tumor.
(C) Representative DNA copy number plot of a GPP53 tumor that amplified a region of chromosome 10 containing Gli1.
(D) Same as (C) but of a different GPP53 tumor that amplified a region of chromosome 1 containing Gli2.
(E) mRNA in situ staining for Gli1 (top panels), Gli2 (middle panels), and Ptch2 (lower panels) in GPP53 tumors (microscopic or Gli1- or Gli2-amplified) and in macroscopic GPN1 tumors.
Scale bars, 50 μm. See also Figures S4 and S5.
A subset of macroscopic tumors acquire downstream Hh pathway hyperactivation
What enables rare macroscopic tumors to “break through,” when millions of other Ptch1-deleted cells in the skin fail to progress? Since the inability to maintain high-level proliferation appears to be a common roadblock for nascent tumors arising in GP, LP, GPN1, GPP53, and SmoM2 mice, we reasoned that macroscopic tumors likely acquire mutations that confer sustained replicative ability. To identify these somatic changes, we performed whole-exome sequencing (WES) on 16 macroscopic GPP53 tumors and 5 GPN1 tumors, along with matched normal control tissue.
Although overall mutational burdens varied widely among tumors, the dominant genomic alterations were somatic DNA copy number changes, with far fewer single-nucleotide variations and insertion/deletions (Figures S4A and S4B; Data S1). In particular, we detected two recurrent amplifications: 7/16 GPP53 tumors acquired copy number gains in regions of chromosome 1 encompassing Gli2, while 4/16 GPP53 tumors acquired gains in regions of chromosome 10 encompassing Gli1 (Figures 5B–5D, S5A, and S5B). These amplifications were often accompanied by smaller copy number changes on the same chromosome, and notably, no tumor exhibited amplification of both Gli1 and Gli2. Since these genes encode the key transcriptional mediators of Hh signaling, we next validated that GPP53 tumors with amplified chromosome 1 possessed increased Gli2 mRNA, whereas tumors with amplified chromosome 10 had increased Gli1 mRNA (Figure 5E). As expected, regressed microscopic lesions presumably lacking these mutations had lower levels of both transcripts and displayed less Hh pathway activation (Figure 5E). Overall, our findings are consistent with previous studies showing that forced overexpression of either transcription factor induces BCC formation (Grachtchouk et al., 2000; Nilsson et al., 2000). Unlike these overexpression systems, however, a key distinction here is that GPP53 tumors spontaneously acquired Gli amplifications, which are similarly detected in 8% of human BCCs (Bonilla et al., 2016). These findings suggest that a subset of tumors initiated by loss of Ptch1 acquire secondary mutations to further hyperactivate downstream Hh signaling in order to drive progression.
Macroscopic tumors converge upon Mycn upregulation
Although we observed recurrent Gli amplifications, 5/16 macroscopic GPP53 tumors and 5/5 macroscopic GPN1 tumors did not possess either mutation (Figure S5A). Working from a list of 70 commonly mutated genes in BCC compiled by Villani et al. (2021), from previous studies (Bonilla et al., 2016; Jayaraman et al., 2014), and from the Catalog of Somatic Mutations in Cancer (COSMIC) database, we further noted that, among the five GPP53 tumors without Gli amplification, three tumors had copy number gains in either Yap1 or Kif7 or copy number loss of Ptch2 (Figures S5A and S5B). These changes may potentially explain how 3/5 GPP53 tumors progressed to macroscopic disease without amplifying Gli (Adolphe et al., 2014; Debaugnies et al., 2018; Li et al., 2012; Maglic et al., 2018).
For the remaining tumors lacking these mutations, we decided to take a different tack to understand how they overcame dormancy. We reasoned that all type 1 macroscopic tumors, regardless of mutational status, must share certain downstream outputs that set them apart from failed microscopic lesions. For instance, we confirmed that all GPP53 and GPN1 macroscopic tumors are highly proliferative (Figure 6A). We also determined that all type 1 tumors have reduced Notch signaling (Figure 6A). Finally, we noted that all macroscopic tumors, regardless of genotype, Gli mutation status, or Hh pathway activity, possess high levels of Mycn (Figure 6A).
Figure 6. Macroscopic GPP53 and GPN1 tumors share common features with human BCCs.
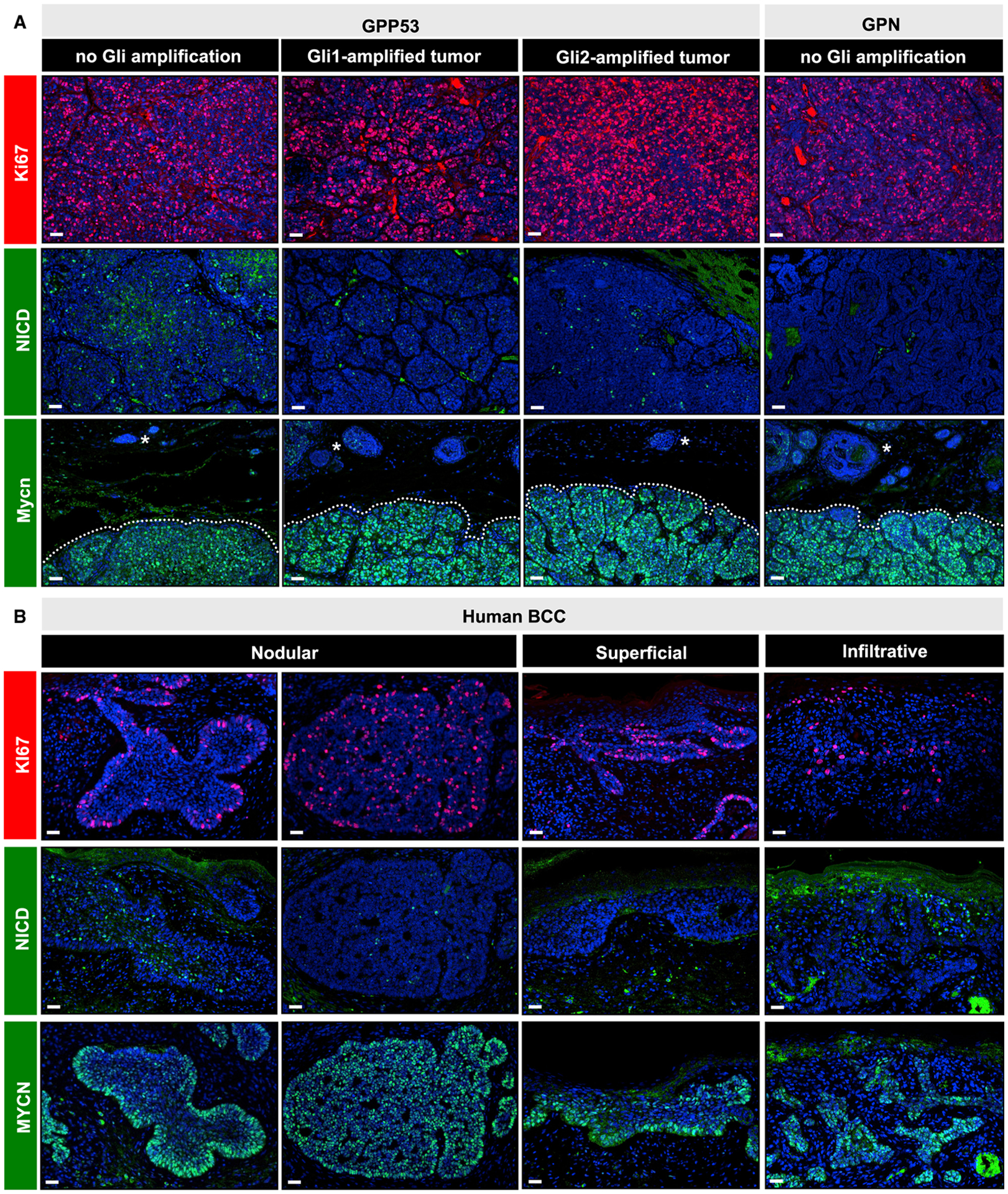
(A) Immunohistochemistry for Ki67 (red, top panels), NICD (green, middle panels), and Mycn (green, lower panels) in GPP53 tumors, sub-divided by Gli amplification status (non-amplified, Gli1-amplified, or Gli2-amplified), and in macroscopic GPN1 tumors lacking Gli amplification. Regardless of genotype, all macroscopic tumors (dotted) express higher levels of Mycn relative to adjacent microscopic lesions (asterisks).
(B) Same markers as in (A), probing human BCCs sub-divided by histological subtype. Two representative examples of nodular BCCs with variable marker expression are shown.
Scale bars, 50 μm.
We next extended these analyses to human BCCs of different subtypes. Indeed, we observed high proliferation and reduced Notch in most tumors, although a subset possessed NICD+ cells in the suprabasal compartment, as we have previously noted (Eberl et al., 2018) (Figure 6B). In addition, all tumors had increased MYCN, which was either uniformly high throughout the tumor or enriched at the basal periphery, as has also previously been reported (Figure 6B) (Brandl et al., 2019; Freier et al., 2006). Altogether, we conclude that three characteristics—high proliferation, reduced Notch, and high MYCN—are often seen in BCC. These features are also shared by all type 1 macroscopic tumors in our system, irrespective of mutational status.
MYCN overexpression promotes key features of tumor progression
MYCN amplification occurs in 12% of human BCCs, while focal mutations that lead to protein stabilization have been detected in 30% of these tumors (Bonilla et al., 2016; Freier et al., 2006). As noted above, nascent GP lesions resemble growing hair follicles, and consistent with this theme, we observed enriched Mycn protein and RNA in the basal layer of both early tumors and hair follicle-matrix progenitors (Figures 7A and S6A–S6C). In contrast, expression of other Myc family members (Myc and Mycl) was not as highly enriched in these compartments (Figures S6A–S6C). In GP microscopic tumors, cells with high Mycn are more likely to be proliferative (Figures 7B and 7C), suggesting a role in cell-cycle regulation (Ellis et al., 2019).
Figure 7. MYCN overexpression promotes tumor progression.
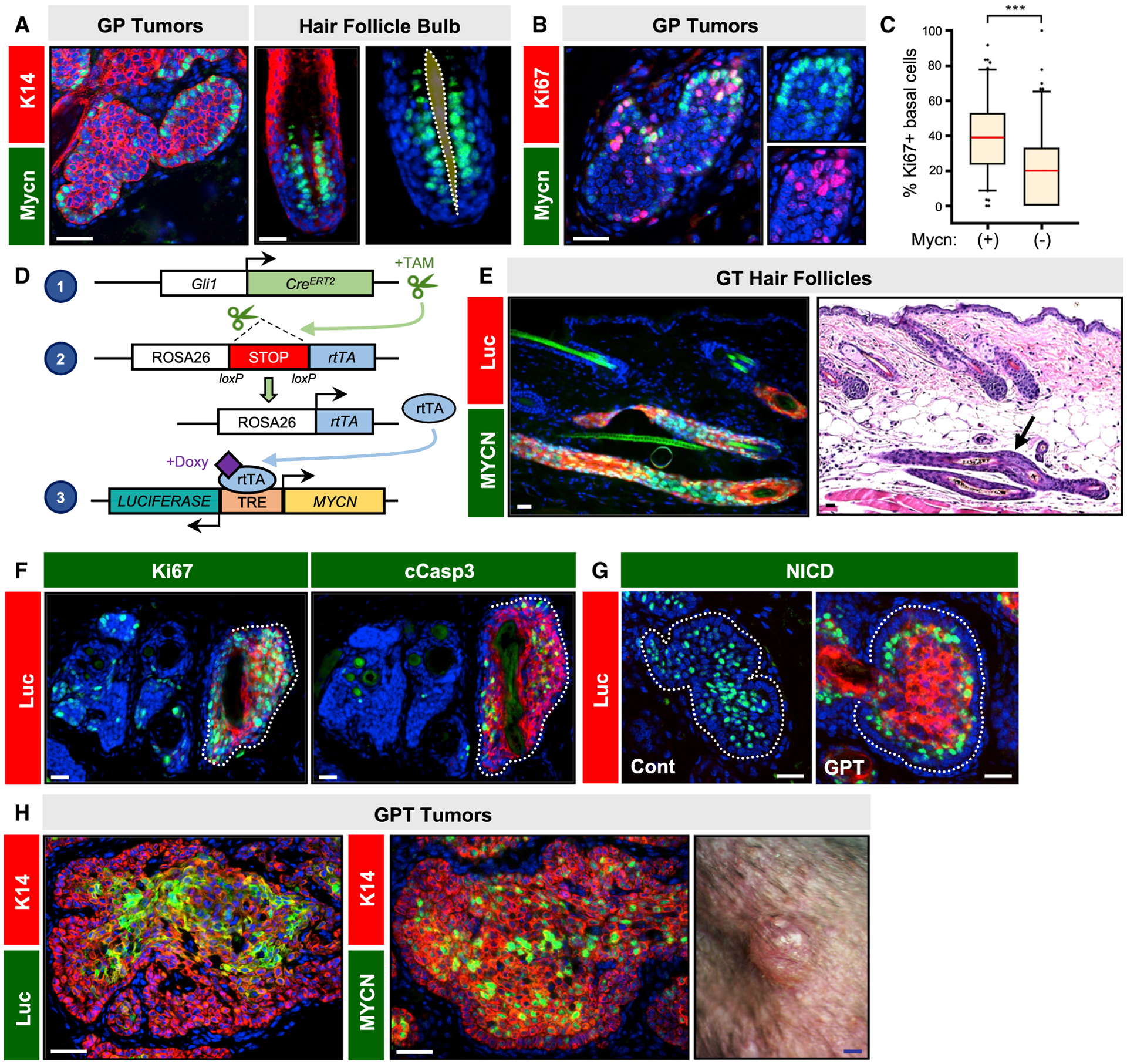
(A) Peripheral basal tumor cells and basal matrix progenitors in anagen hair follicles express Mycn (green). Dotted region, mesenchymal dermal papilla.
(B) Co-localization of Mycn (green) and Ki67 (red) in microscopic GP tumor, 5 weeks post-TAM.
(C) Quantitation showing that Mycn+ basal tumor cells are more likely to be proliferative.
(D) Schematic for MYCN/Luciferase overexpression system.
(E) Histology of dysmorphic anagen hair follicles (arrow) expressing MYCN (green) and Luciferase (red) in GT mice.
(F) Microscopic GPT tumors expressing MYCN/Luciferase (red, dotted) display increased Ki67 (green, left panel) and cleaved caspase 3 (green, right panel).
(G) Microscopic GP tumors possess extensive suprabasal NICD expression (left panel, green), whereas GPT tumors expressing MYCN/Luciferase (right panel, red) re-localize NICD to suprabasal tumor cells just inside of the basal layer.
(H) Immunohistochemistry showing expression of Luciferase (green, left photo) and exogenous MYCN/endogenous Mycn (green, middle photo) in macroscopic GPT tumor (right photo).
p values were calculated by a Wilcoxon rank-sum test with continuity correction. ***p < 0.001. Scale bars, 50 μm. Blue scale bar, 1 mm. See also Figure S6 and Tables S1 and S2 for animal numbers.
To directly test the role of Mycn in our system, we generated mice expressing Gli1-CreERT2, coupled with Cre-inducible reverse Tet transactivator (rtTA), and a bidirectional tetracycline-responsive element (TRE)-driven MYCN/luciferase (GT mice) (Swartling et al., 2010) (Figure 7D). In this system, GT mice are first injected with TAM to activate rtTA expression, which subsequently drives MYCN/luciferase overexpression in the presence of doxycycline (DOXY). Following 12–20 weeks of continuous DOXY treatment, GT mice formed dysmorphic anagen hair follicles but no tumors (Figure 7E).
Having validated this system, we next incorporated these genetic elements into our Ptch1-deficient model (GPT mice). Tumors were initiated by TAM and allowed to grow for 5 weeks before mice were shifted onto DOXY-chow to activate MYCN expression for an additional 12 weeks. We noted that transgene expression was localized primarily to the tumor suprabasal compartment (Figures 7F and 7G), which may either reflect biased TRE promoter activity or inward movement of transgene-expressing basal layer tumor cells. Regardless, MYCN overexpression induced massive proliferation in nascent tumors (Figure 7F). Strikingly, we also observed that Notch activation was re-localized away from the tumor suprabasal compartment to cells residing just inside of the basal layer, possibly reflecting early suprabasal cells that express lower levels of the transgene (Figure 7G). These results indicate that MYCN overexpression is sufficient to promote key features of tumor progression—increased proliferation and reduced Notch—that we observed in human BCC and mouse type 1 BCC-like tumors.
Despite these findings, most GPT mice did not form macroscopic tumors, likely because MYCN overexpression caused increased apoptosis (Figure 7F). In 4/16 GPT mice, however, we observed small palpable tumors, which contained transgene-expressing basaloid cells (Figure 7H). We did not allow these lesions to continue growing due to frequent gastrointestinal-related morbidity in GPT mice. Similar to GPN1 and GPP53 macroscopic tumors, these rare GPT tumors likely also acquired additional somatic mutations that enabled them to progress. Collectively, our findings argue that loss of Ptch1 by itself is not sufficient for full BCC progression and that secondary mutations—resulting in loss of Notch1 or p53, increased Gli, and/or gain of Mycn—contribute functionally to the critical transition from microscopic to macroscopic disease.
DISCUSSION
Our skin is the most highly mutated organ, and mutations in cancer-associated genes, such as NOTCH1/2, TP53, and RAS, are frequently detected in photoaged epithelia (Fowler et al., 2021; Lynch et al., 2017; Martincorena et al., 2015; Ren et al., 1997). Previously, we and others have observed that mutant cells with aberrant Notch1 or p53 can persist long term in the epidermis without forming tumors (Figures S6D and S6E; Table S2) (Murai et al., 2018; Veniaminova et al., 2019). Targeted expression of oncogenic Kras in hair follicle stem cells also causes only temporary tissue disruption that normalizes over time (Brown et al., 2017; Pineda et al., 2019). Along a similar vein and consistent with previous findings (Nitzki et al., 2010), we report here that loss of Ptch1 or gain of Smo, both hallmark mutations in BCC, induces nascent tumors that do not progress.
Given the high failure rate for tumor-initiated cells in the skin, it is likely that both cell-intrinsic and cell-extrinsic factors suppress tumor progression. Indeed, an idiosyncratic feature of the skin is the periodic phases of hair growth and regression, with the anagen growth phase favoring tumor formation and the telogen resting phase associated with tumor regression (Grachtchouk et al., 2011; Mancuso et al., 2006; Oro and Higgins, 2003; Pineda et al., 2019; White et al., 2014). In our GP model, nascent BCC-like tumors share some resemblance to growing hair follicles, including basal layer enrichment for multiple factors—high Hh pathway activation, proliferation, p53, and Mycn—as well as suprabasal activation of Notch signaling (Figure 3C). Thus, it is conceivable that early tumor growth and spontaneous regression are linked to the hair cycle. However, since regressed tumors in our model remain dormant even during subsequent anagen, other factors likely cause tumor exhaustion over time.
In mice, the absence of macroscopic BCCs has been a major shortcoming of both the conditional Ptch1-deletion and SmoM2-overexpression models, confirming that tumors do not progress, regardless of how upstream Hh signaling becomes activated or which Cre driver is utilized (Brandes et al., 2020; Grachtchouk et al., 2003; Kasper et al., 2011; Mao et al., 2006; Peterson et al., 2015; Wang et al., 2017; Youssef et al., 2010). Indeed, previous studies have shown that nascent BCC-like tumors induced by non-targeted Ptch1 deletion similarly regress due to macrophage-induced tumor differentiation (Nitzki et al., 2010). During postnatal brain development, Ptch1-heterozygous mice also exhibit transient hyperplasia in the external granular layer, a site of medulloblastoma formation, that subsequently regresses (Thomas et al., 2009). These data suggest that lesions initiated by loss of Ptch1 or gain of Smo likely encounter steep obstacles across multiple organ systems. Since BCC mouse models likely do not recapitulate the full set of mutations seen in human tumors, this may explain why animal models often fail to yield macroscopic tumors.
While our findings in mice indicate that Ptch1 loss alone is insufficient for inducing macroscopic tumors, Gorlin patients can develop numerous BCCs, seemingly arguing that loss of heterozygosity of a single tumor suppressor, in most cases PTCH1, can lead to palpable tumors. However, although the average mutational burden in Gorlin BCCs is lower than in sporadic BCCs (Chiang et al., 2018; Sharpe et al., 2015), the mutational load in Gorlin tumors is still higher than that of many internal tumors. In addition, TP53 mutations are present in ~40% of Gorlin BCCs, similar to sporadic tumors (Chiang et al., 2018). Finally, Gorlin BCCs can manifest a variety of histologic subtypes (Rehefeldt-Erne et al., 2016), and Gorlin patients can also develop basaloid follicular hamartoma (BFH), a benign hair follicle tumor (Besagni et al., 2021; Chikeka et al., 2021; Ponti et al., 2018; Requena and Sangueza, 2017). While the connection between BFH and BCC remains unclear, some have speculated that these neoplasms are variations of the same disease (Ponti et al., 2018). Indeed, previous studies have shown that downstream Hh signaling strength can modulate BFH versus BCC tumor phenotype in mice (Grachtchouk et al., 2003, 2011). Altogether, these data suggest that secondary somatic mutations may play functional roles in modulating tumor phenotype in Gorlin patients, possibly during the transition from BFH-like lesions to BCC.
In contrast to Ptch1-conditional mice, Ptch1-heterozygous animals form macroscopic BCCs following irradiation, but how these tumors progress remains unknown (Aszterbaum et al., 1999; Chaudhary et al., 2015; Wang et al., 2017). Transgenic overexpression of either GLI1 or Gli2 from an exogenous promoter also induces macroscopic BCC-like tumors (Grachtchouk et al., 2000; Nilsson et al., 2000). While these studies demonstrate that high-level downstream Hh pathway activity can induce tumorigenesis, GLI amplification is not regarded as a primary driver of human BCC. Indeed, nearly all human BCCs possess canonical mutations in upstream Hh pathway components, including tumors that possess GLI amplifications (Bonilla et al., 2016). This suggests that constitutive activation of upstream Hh signaling, mediated by primary cilia, may be critical for unlocking the full potential of secondary mutations that activate downstream components of the pathway. In line with this, GPP53 tumors in our study acquired Gli amplifications spontaneously, without additional experimental manipulation, again arguing that Ptch1-deficient lesions must hyperactivate both upstream and downstream Hh signaling in order to progress.
It is important to note that the prevalence of Gli amplification in our GPP53 model is higher than in human BCCs, where analogous amplifications are seen in 8% of tumors and may drive resistance to SMO antagonists (Atwood et al., 2015; Bonilla et al., 2016; Buonamici et al., 2010; Sharpe et al., 2015). Since our mice are not exposed to UV radiation, this likely affects the spectrum of mutations that arises in our models. Apart from direct Gli amplification, there are likely many ways by which BCCs can hyperactivate downstream Hh signaling. For instance, Gli activity can be enhanced by aPKC, transforming growth factor β (TGFβ), and AP-1 (Atwood et al., 2013; Yao et al., 2020), while the phosphatidylinositol 3-kinase (PI3K)/Akt and epidermal growth factor (EGF) pathways can cooperate with canonical Hh signaling to promote downstream activity (Chow et al., 2021; Eberl et al., 2012; Kim et al., 2016).
Regardless of Gli amplification status, all type 1 macroscopic tumors upregulated Mycn, which plays critical roles in many tumors, including nervous system cancers, prostate cancer, and retinoblastoma (Rickman et al., 2018; Wu et al., 2017). We further demonstrated that MYCN overexpression induces certain features of tumor progression, including increased proliferation and reduced differentiation. Notably, Mycn is a downstream target of Hh signaling (Hatton et al., 2006; Mill et al., 2005) (Figure S6F), yet this gene is still frequently mutated in BCC (Bonilla et al., 2016; Freier et al., 2006). This further supports the view that neither loss of Ptch1 nor gain of Smo is sufficient for tumor progression. Rather, mutations that augment MYCN levels may provide a secondary boost for tumors to sustain high-level proliferation while limiting differentiation. Collectively, these data indicate that MYCN is likely situated at a critical nexus for driving BCC progression and possibly drug resistance.
In summary, by utilizing BCC models that largely fail to progress, we identify genetic factors that enable rare tumors to succeed. Our findings suggest that BCC formation is guided by the acquisition of somatic secondary mutations. Activation of upstream Hh signaling, via loss of Ptch1 or gain of Smo, initiates nascent tumors that eventually regress and become dormant unless additional mutations are acquired. Concomitant loss of Notch1 prevents tumor regression, and loss of p53 likely increases genomic instability; however, neither is sufficient for macroscopic tumors to form. Ultimately, tumor-initiated cells must acquire additional changes that either directly or indirectly cause exuberant downstream Hh pathway activation in order to progress. These findings may explain why some BCCs persist after pharmacological inhibition of upstream Hh signaling, while highlighting the possibility that essential downstream factors, such as MYCN, may represent critical targets for therapy.
Limitations of the study
While our study suggests a role for secondary mutations in driving BCC progression, mouse BCC-like tumors likely do not recapitulate the full panoply of mutations seen in human BCC, since mice are not typically exposed to UV radiation. Thus, other genetic and epigenetic changes may contribute to the progression of human tumors. Mutations within intergenic regions of the chromosome are not detected by WES and may also affect tumor progression in our system. While all BCC-like tumors in our study exhibited increased Mycn, it remains unclear how this occurs in tumors lacking Gli amplification. Aside from Hh signaling, other pathways, such as PI3K/Akt, have been reported to modulate Mycn expression or stability (Liu et al., 2021). Loss of Trp53 may also be necessary for overriding cellular controls that guard against excessive Mycn and induce cell death. Finally, the mechanisms underlying tumor dormancy and spontaneous regression remain unclear. Given the parallels between nascent BCC formation and hair follicle regeneration, a deeper understanding into how hair follicles normally develop, cycle, and regress may inspire additional novel insights into the biology of these tumors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sunny Wong (sunnyw@umich.edu).
Materials availability
All reagents generated in this study are available from the lead contact.
Data and code availability
WES data generated for this study are available through the NCBI SRA Run Selector (BioProject: PRJNA782990).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the Lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Unless otherwise indicated, 8 week-old mice were induced with a single intraperitoneal injection of tamoxifen (TAM) at 5 mg/40 grams body weight. GPT mice were induced with TAM and later transferred to 200 mg/kg doxycycline-containing chow for 12 weeks. For animal strain information, please see the Key resources table. All studies were performed on mice of both genders in a mixed genetic background, using littermate animals for comparisons whenever possible. All mice were maintained in specific pathogen free housing and were used in accordance with regulations established by the University of Michigan Unit for Laboratory Animal Medicine.
KEY RESOURCES TABLE
| REAGENTS or RESCOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-cleaved caspase-3 | Cell Signaling | Cat # 9661 |
| Chicken anti-K14 | Biolegend | Cat # 906004 |
| Mouse anti-Ki67 | BD Biosciences | Cat # 550609 |
| Rabbit anti-Ki67 | Cell Signaling | Cat # 12202S |
| Goat anti-luciferase | Novus | Cat # NB100-1677SS |
| Rabbit anti-NICD | Cell Signaling | Cat # 4147P |
| Rabbit anti-N-Myc | Cell Signaling | Cat # D4B2Y |
| Mouse anti-p16 | Invitrogen | Cat # 1E12E10 |
| Rabbit anti-p21 | Cell Signaling | Cat # 2947T |
| Rabbit anti-p53 | Novocastra | Cat # NCL-p53-CM5p |
| Rabbit anti-smooth muscle actin | Cell Signaling | Cat # 19245T |
| Biological samples | ||
| Mouse tissue samples, obtained in accordance with guidelines established by the University of Michigan Unit for Laboratory Animal Medicine | This manuscript | Study protocol # PRO00010041 |
| Human BCC samples, obtained with informed consent and approved by the Institutional Review Board at the University of Michigan Medical School | This manuscript, University of Michigan Departments of Dermatology and Pathology | Study protocol # HUM00042233, HUM00075822, HUM00051875 |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline chow (200 mg/kg) | BioServ Inc | Cat # S3888 |
| Nair hair removal lotion | Nair | Cat # B001E6OAM8 |
| Nuclear fast red | Sigma | Cat # N3020-100ML |
| Tamoxifen | Sigma | Cat # T5648-1G |
| X-Gal | Roche | Cat # 10651745001 |
| Critical commercial assays | ||
| DNeasy Blood & Tissue Kit | Qiagen | Cat # 69504 |
| RNAscope 2.5 HD Reagent Kit-BROWN | ACD (RNAscope) | Cat #322310 |
| RNAscope 2.5 Pretreat Reagents-H202 and Protease Plus | ACD (RNAscope) | Cat # 322330 |
| RNAscope Target Retrieval | ACD (RNAscope) | Cat # 322000 |
| RNAscope Wash Buffer | ACD (RNAscope) | Cat #310091 |
| TSA Fluorescein Plus Kit | Akoya Biosciences | Cat # NEL741E001KT |
| Deposited data | ||
| WES data | This manuscript | BioProject: PRJNA782990 |
| Experimental models: Organisms/strains | ||
| Mouse: Gli1tm3(cre/ERT2)Alj (Gli1-CreERT2) | The Jackson Laboratory (Ahn and Joyner, 2004) | Cat # 007913 |
| Mouse: Lrig1tm1.1(cre/ERT2)Rjc (Lrig1-CreERT2) | The Jackson Laboratory (Powell et al., 2012) | Cat # 018418 |
| Mouse: Ptch1tm1Hahn (Ptch1flox) | The Jackson Laboratory (Uhmann et al., 2007) | Cat # 012457 |
| Mouse: Gt(ROSA)26Sortm1(Smo/EYFP)Amc/J (SmoM2) | The Jackson Laboratory (Mao et al., 2006) | Cat # 005130 |
| Mouse: Notch1tm2Rko/GridJ (Notch1flox) | The Jackson Laboratory (Yang et al., 2004) | Cat # 007181 |
| Mouse: Trp53tm1Brn (p53flox) | The Jackson Laboratory (Marino et al., 2000) | Cat # 008462 |
| Mouse: Gt(ROSA)26Sortm1(rtTA,EGFP)Nagy | The Jackson Laboratory (Belteki et al., 2005); Laboratory of Dr. Anj Dlugosz | Cat # 005572 |
| Mouse: TRE-MYCN/Luciferase | Laboratory of Dr. William Weiss (Swartling et al., 2010) | N/A |
| Oligonucleotides | ||
| In situ probe: mGli1 | ACD (RNAscope) | Cat #311001 |
| In situ probe: mGli2 | ACD (RNAscope) | Cat # 405771 |
| In situ probe: mPtch2 | ACD (RNAscope) | Cat #435131 |
| In situ probe: mMycn | ACD (RNAscope) | Cat #477151 |
| In situ probe: mMycl | ACD (RNAscope) | Cat # 552711 |
| In situ probe: mMyc | ACD (RNAscope) | Cat # 413451 |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| AxioVision, Version 4 | Carl Zeiss | https://www.micro-shop.zeiss.com/en/us/ |
| GraphPad Prism 7 | GraphPad Prism Software | https://www.graphpad.com/scientific-software/prism/ |
| Burrows-Wheeler Aligner | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| GATK | DePristo et al., 2011 | https://gatk.broadinstitute.org/hc/en-us |
| MuTect | Cibulskis et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutect |
| Strelka | Saunders et al., 2012 | https://github.com/Illumina/strelka/blob/v2.9.x/docs/userGuide/README.md |
| Control-FREEC | Boeva et al., 2012 | http://boevalab.inf.ethz.ch/FREEC/ |
| CNVKit | Talevich et al., 2016 | https://cnvkit.readthedocs.io/en/stable/ |
| ANNOVAR | Wang et al., 2010; Yang and Wang, 2015 | https://annovar.openbioinformatics.org/en/latest/ |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
Human samples
De-identified human BCC samples embedded in paraffin were obtained through study protocol HUM00042233, HUM00075822 and HUM00051875, in accordance with procedures approved by the Institutional Review Board at the UM Medical School.
METHOD DETAILS
Immunofluorescence
Skin biopsies were fixed in 3.7% formalin overnight for paraffin embedding. For frozen sections, samples were fixed in 3.7% paraformaldehyde at 4°C for 1 h, rinsed in PBS, sunk in 30% sucrose overnight and embedded into OCT. Frozen sections were probed with antibodies against the following antigens: p21 (1:100, Cell Signaling) and p16 (1:100, Invitrogen). Paraffin sections were antigen-retrieved by boiling slides in 1 mM EDTA, pH 8.0, for 10 min, and probed with antibodies against the following antigens: K14 (1:1,000, Biolegend), Ki67 (1:100, Cell Signaling and BD Biosciences), α smooth muscle actin (1:500, Cell Signaling), cleaved caspase-3 (1:100, Cell Signaling) and luciferase (1:1000, Novus). Staining was amplified using the TSA Fluorescein Plus kit for antibodies against the following targets: p53 (1:5000, Novocastra), NICD (1:500, Cell Signaling) and Mycn (1:500, Cell Signaling). Amplification was performed for 2 min, 4 min and 10 min, respectively, following manufacturer’s instructions. Image processing was performed using Adobe Photoshop with the Auto-Blend feature applied to maximize image sharpness across focal planes.
RNA in situ hybridization
RNA in situ staining was performed using the RNAscope 2.5 brown kit (ACD). Paraffin slides were antigen-retrieved by boiling in RNAscope retrieval buffer for 15 min, treated with protease for 30 min and incubated with target probes at 40°C for 2 h. Probe detection was performed according to manufacturer’s instructions. Paraffin slides were counterstained with hematoxylin. Please see the Key resources table for information on RNAscope probes.
LacZ visualization
Frozen sections were incubated at 37°C in 1 mg/mL X-gal dissolved in 5 mM potassium ferrocyanide and 5 mM potassium ferricy-anide for 30 min, and counterstained with nuclear fast red.
Phorbol ester treatment
100 μL of 12-O-Tetradecanoylphorbol-13-acetate (TPA, Sigma) dissolved at a concentration of 0.25 mg/mL in ethanol was topically applied onto shaved GP and GPN1 mice for 2 consecutive days. Two days after treatment, dorsal skin was collected.
DNA extraction
DNA was harvested using the DNeasy Blood & Tissue kit (Qiagen). Tumor and paired liver samples (15–25 mg) were minced in ATL buffer containing proteinase K and incubated overnight at 56°C. The next day, digested tumor and liver samples were processed according to the manufacturer’s instructions.
Whole exome sequencing (WES)
WES was performed on macroscopic GPP53 and GPN1 tumors, and on matched liver samples. All sequencing and analyses were performed by Novogene. Briefly, 1 μg of DNA was used for library preparation using the Agilent SureSelectXT Mouse All Exon kit. Fragmentation was performed to generate 180–280 bp fragments and assessed on the Agilent Bioanalyzer 2100 system for quality control. Captured libraries were sequenced using Illumina NovaSeq 6000. Reads were aligned with Burrows-Wheeler Aligner (BWA; v0.7.17) using the mm10 reference genome. Conversion to BAM files was performed using Picard (v2.18.9). Single-nucleotide variants (SNVs) and InDels were identified by GATK (v4.0), followed by ANNOVAR to annotate variants. Somatic SNVs and InDels were identified by MuTect and Strelka, respectively. Somatic copy number variants (CNVs) were called by Control-FREEC (v11.4), using the setting minCNAlength parameter = 2. Low confidence CNV changes annotated as “genomic superduplications” with CNV = 1 or 3 were omitted from analyses. CNV plots were generated using CNVkit with default settings.
QUANTIFICATION AND STATISTICAL ANALYSIS
Tumor measurements
Tumor area was quantitated from 3 representative fields per sample, and an overall average was calculated for each animal and time point. Final values were normalized relative to that of tumors, 5 weeks post-TAM. TD tumors were quantitated by inspecting ~1 cm of skin H&E histology, and normalizing to the length of the section. Cell proliferation was quantitated from 3 representative fields per sample and expressed as the percentage of Ki67 + tumor cells/K14 + total tumor cells. p53 + tumor cells were counted at the tumor periphery (basal) or interior (suprabasal) compartments. Three random fields were assessed for each sample, and a single average was calculated for each animal and compartment. Similar methods were used to quantitate Mycn and Ki67 overlap.
Quantitating in situ Intensity
The Color Threshold function in ImageJ was used to highlight areas of staining and to filter out background. The total signal area was then measured using the Analyze Particles function. For Ptch2, the total signal area was divided by the total number of tumor cells to obtain the average signal per cell. For Mycn, Mycl and Myc, the signal area was quantitated separately for the basal and suprabasal compartments.
Statistics
For comparisons between two groups, an unpaired t test was performed to calculate statistical significance. A Wilcoxon rank sum test with continuity correction was used to calculate p values for non-parametric data depicted by box and whisker plot. For comparisons with greater than two groups, one-way ANOVA with posthoc test (Tukey’s method) was performed. For beeswarm plots, statistical significance was calculated using a linear mixed model using the Ime4, ImerTest and emmeans packages on R.
Supplementary Material
Highlights.
Upstream Hh pathway activation is necessary, but not sufficient, for BCCs to progress
Failed nascent tumors become dormant and may spontaneously regress over time
Secondary mutations enable rare tumors to overcome barriers to progression
Increased Mycn promotes BCC progression
ACKNOWLEDGMENTS
We are grateful to Dr. S.X. Atwood (UC Irvine) and Dr. J.T. Seykora (University of Pennsylvania) for helpful discussions, Dr. W.A. Weiss (UCSF) for sharing mice, and Linguo Hao (Novogene) for project management. S.Y.W. acknowledges the support of the LEO Foundation (LF18017), the American Cancer Society (RSG-18-065-01-TBG and TLC-21-161-01-TLC), the Donald & Patricia Roof Fund for Skin Cancer Research, and the NIH (R01AR065409 and R56AR075638). K.G.T. acknowledges the support of the NIH (F31CA 254080). A.A.D. acknowledges the support of the NIH (R01CA087837). The authors also acknowledge support from the UM Skin Biology and Disease Resource-based Center (P30AR075043) and NCI Cancer Center support grant (P30CA046592).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110779.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adolphe C, Nieuwenhuis E, Villani R, Li ZJ, Kaur P, Hui CC, and Wainwright B (2014). Patched1 and Patched2 redundancy plays a key role in regulating epidermal differentiation. J. Invest Dermatol 134, 1981–1990. [DOI] [PubMed] [Google Scholar]
- Ahn S, and Joyner AL (2004). Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118, 505–516. [DOI] [PubMed] [Google Scholar]
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, and Epstein EH Jr. (1999). Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat. Med 5, 1285–1291. [DOI] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, and Oro AE (2013). GLI activation by atypical protein kinase Cl/gamma regulates the growth of basal cell carcinomas. Nature 494, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang AL, et al. (2015). Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, and Nagy A (2005). Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 33, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besagni F, Dika E, Ricci C, Misciali C, Veronesi G, Corti B, Gurioli C, and Neri I (2021). Basaloid follicular hamartomas in pediatric Basal Cell Nevus Syndrome: a diagnostic challenge. J. Dermatol 48, 1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, and Barillot E (2012). Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifas JM, Bare JW, Kerschmann RL, Master SP, and Epstein EH Jr. (1994). Parental origin of chromosome 9q22.3-q31 lost in basal cell carcinomas from basal cell nevus syndrome patients. Hum. Mol. Genet 3, 447–448. [DOI] [PubMed] [Google Scholar]
- Bonifas JM, Pennypacker S, Chuang PT, McMahon AP, Williams M, Rosenthal A, de Sauvage FJ, and Epstein EH Jr. (2001). Activation of expression of hedgehog target genes in basal cell carcinomas. J. Invest. Dermatol 116, 739–742. [DOI] [PubMed] [Google Scholar]
- Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, et al. (2016). Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet 48, 398–406. [DOI] [PubMed] [Google Scholar]
- Brandes N, Mitkovska SH, Botermann DS, Maurer W, Müllen A, Scheile H, Zabel S, Frommhold A, Heß I, Hahn H, et al. (2020). Spreading of isolated Ptch mutant basal cell carcinoma precursors is physiologically suppressed and counteracts tumor formation in mice. Int. J. Mol. Sci 21, 9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl L, Hartmann D, Kirchner T, and Menssen A (2019). Expression of n-MYC, NAMPT and SIRT1 in basal cell carcinomas and their cells of origin. Acta Derm Venereol. 99, 63–71. [DOI] [PubMed] [Google Scholar]
- Brown S, Pineda CM, Xin T, Boucher J, Suozzi KC, Park S, Matte-Martone C, Gonzalez DG, Rytlewski J, Beronja S, et al. (2017). Correction of aberrant growth preserves tissue homeostasis. Nature 548, 334–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, et al. (2010). Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl Med 2, 51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary SC, Tang X, Arumugam A, Li C, Srivastava RK, Weng Z, Xu J, Zhang X, Kim AL, McKay K, et al. (2015). Shh and p50/Bcl3 signaling crosstalk drives pathogenesis of BCCs in Gorlin syndrome. Oncotarget 6, 36789–36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang A, Jaju PD, Batra P, Rezaee M, Epstein EH Jr., Tang JY, and Sarin KY (2018). Genomic stability in syndromic basal cell carcinoma. J. Invest. Dermatol 138, 1044–1051. [DOI] [PubMed] [Google Scholar]
- Chikeka I, Chang LW, Collins MK, Pugliano M, Ho J, House N, and Kazlouskaya V (2021). Basaloid follicular hamartoma: an additional criterion of nevoid basal cell carcinoma syndrome. Am. J. Dermatopathol 44, 66–69. [DOI] [PubMed] [Google Scholar]
- Chow RY, Levee TM, Kaur G, Cedeno DP, Doan LT, and Atwood SX (2021). MTOR promotes basal cell carcinoma growth through atypical PKC. Exp. Dermatol 30, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowson AN (2006). Basal cell carcinoma: biology, morphology and clinical implications. Mod. Pathol 19, S127–S147. [DOI] [PubMed] [Google Scholar]
- Debaugnies M, Sánchez-Danés A, Rorive S, Raphaël M, Liagre M, Parent MA, Brisebarre A, Salmon I, and Blanpain C (2018). YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 19, e45809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo L, Pellegrini C, Stefani AD, Ricci F, Fossati B, Regno LD, Carbone C, Piro G, Corbo V, Delfino P, et al. (2021). Molecular alterations in basal cell carcinoma subtypes. Sci. Rep 11, 13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer HC, Solca F, et al. (2012). Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumor-initiating pancreatic cancer cells. EMBO Mol. Med 4, 218–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl M, Mangelberger D, Swanson JB, Verhaegen ME, Harms PW, Frohm ML, Dlugosz AA, and Wong SY (2018). Tumor architecture and Notch signaling modulate drug response in basal cell carcinoma. Cancer Cell 33, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SJ, Gomez NC, Levorse J, Mertz AF, Ge Y, and Fuchs E (2019). Distinct modes of cell competition shape mammalian tissue morphogenesis. Nature 569, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein EH (2008). Basal cell carcinomas: attack of the hedgehog. Nat. Rev. Cancer 8, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JC, King C, Bryant C, Hall MWJ, Sood R, Ong SH, Earp E, Fernandez-Antoran D, Koeppel J, Dentro SC, et al. (2021). Selection of oncogenic mutant clones in normal human skin varies with body site. Cancer Discov. 11, 340–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier K, Flechtenmacher C, Devens F, Hartschuh W, Hofele C, Lichter P, and Joos S (2006). Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol. Rep 15, 1141–1145. [PubMed] [Google Scholar]
- Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, Hui CC, and Dlugosz AA (2000). Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet 24, 216–217. [DOI] [PubMed] [Google Scholar]
- Grachtchouk M, Pero J, Yang SH, Ermilov AN, Michael LE, Wang A, Wilbert D, Patel RM, Ferris J, Diener J, et al. (2011). Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J. Clin. Invest 121, 1768–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grachtchouk V, Grachtchouk M, Lowe L, Johnson T, Wei L, Wang A, de Sauvage F, and Dlugosz AA (2003). The magnitude of hedgehog signaling activity defines skin tumor phenotype. EMBO J. 22, 2741–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. (1996). Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851. [DOI] [PubMed] [Google Scholar]
- Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alboran IM, Olson JM, and Eisenman RN (2006). N-myc is an essential downstream effector of shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 66, 8655–8661. [DOI] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, and Kolodney MS (2014). Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Invest Dermatol 134, 213–220. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr., et al. (1996). Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671. [DOI] [PubMed] [Google Scholar]
- Kasper M, Jaks V, Are A, Bergström Å, Schwäger A, Barker N, and Toftgård R (2011). Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl. Acad. Sci. USA 108, 4099–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper M, Jaks V, Hohl D, and Toftgård R (2012). Basal cell carcinoma - molecular biology and potential new therapies. J. Clin. Invest 122, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgour JM, Jia JL, and Sarin KY (2021). Review of the molecular genetics of basal cell carcinoma; inherited susceptibility, somatic mutations, and targeted therapeutics. Cancers 13, 3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AL, Back JH, Zhu Y, Tang X, Yardley NP, Kim KJ, Athar M, and Bickers DR (2016). AKT1 activation is obligatory for spontaneous BCC tumor growth in a murine model that mimics some features of basal cell nevus syndrome. Cancer Prev. Res. (Phila) 9, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZJ, Nieuwenhuis E, Nien W, Zhang X, Zhang J, Puviindran V, Wainwright BJ, Kim PC, and Hui CC (2012). Kif7 regulates Gli2 through Sufu-dependent and -independent functions during skin development and tumorigenesis. Development 139, 4152–4161. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chen SS, Clarke S, Veschi V, and Thiele CJ (2021). Targeting MYCN in pediatric and adult cancers. Front Oncol. 10, 623679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Lynch CNS, Craythorne E, Liakath-Ali K, Mallipeddi R, Barker JN, and Watt FM (2017). Spatial constraints govern competition of mutant clones in human epidermis. Nat. Commun 8, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglic D, Schlegelmilch K, Dost AF, Panero R, Dill MT, Calogero RA, and Camargo FD (2018). YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 37, e98642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Leonardi S, Tanori M, Pasquali E, Pierdomenico M, Rebessi S, Majo VD, Covelli V, Pazzaglia S, and Saran A (2006). Hair cycle-dependent basal cell carcinoma tumorigenesis in Ptc1-neo67/+ mice exposed to radiation. Cancer Res. 66, 6606–6614. [DOI] [PubMed] [Google Scholar]
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, and McMahon AP (2006). A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 66, 10171–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Loo PV, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill P, Mo R, Hu MC, Dagnino L, Rosenblum ND, and Hui CC (2005). Shh controls epithelial proliferation via independent pathways that converge on N-myc. Dev. Cell 9, 293–303. [DOI] [PubMed] [Google Scholar]
- Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, Nagy T, Cagan A, Goulding D, Klein AM, et al. (2018). Epidermal tissue adapts to restrain progenitors carrying clonal p53 mutations. Cell Stem Cell 23, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Unden AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, and Toftgard R (2000). Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing Gli-1. Proc. Natl. Acad. Sci. U S A 97, 3438–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzki F, Zibat A, König S, Wijgerde M, Rosenberger A, Brembeck FH, Carstens PO, Frommhold A, Uhmann A, Klingler S, et al. (2010). Tumor stroma-derived Wnt5a induces differentiation of basal cell carcinoma of ptch-mutant mice via CaMKII. Cancer Res. 70, 2739–2748. [DOI] [PubMed] [Google Scholar]
- Oro AE, and Higgins K (2003). Hair cycle regulation of Hedgehog signal reception. Dev. Biol 255, 238–248. [DOI] [PubMed] [Google Scholar]
- Peterson SC, Eberl M, Vagnozzi AN, Belkadi A, Veniaminova NA, Verhaegen ME, Bichakjian CK, Ward NL, Dlugosz AA, and Wong SY (2015). Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 16, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CM, Gonzalez DG, Matte-Martone C, Boucher J, Lathrop E, Gallini S, Fons NR, Xin T, Tai K, Marsh E, et al. (2019). Hair follicle regeneration suppresses Ras-driven oncogenic growth. J. Cell Biol 218, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten F, Berg C, Ahmadian A, Ren ZP, Nister M, Lundeberg J, Uhlen M, and Ponten J (1997). Molecular pathology in basal cell cancer with p53 as a genetic marker. Oncogene 15, 1059–1067. [DOI] [PubMed] [Google Scholar]
- Ponti G, Manfredini M, Pastorino L, Maccaferri M, Tomasi A, and Pellacani G (2018). PTCH1 germline mutations and the basaloid follicular hamartoma values in the tumor spectrum of basal cell carcinoma syndrome (NBCCS). Anticancer Res. 38, 471–476. [DOI] [PubMed] [Google Scholar]
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, Hintner H, Quinn AG, Frischauf AM, and Aberger F (2002). Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene 21, 5529–5539. [DOI] [PubMed] [Google Scholar]
- Rehefeldt-Erne S, Nägeli MC, Winterton N, Felderer L, Weibel L, Hafner J, and Dummer R (2016). Nevoid basal cell carcinoma syndrome: report from the Zurich nevoid basal cell carcinoma syndrome cohort. Dermatology 232, 285–292. [DOI] [PubMed] [Google Scholar]
- Ren ZP, Ahmadian A, Ponten F, Nister M, Berg C, Lundeberg J, Uhlen M, and Ponten J (1997). Benign clonal keratinocyte patches with p53 mutations show no genetic link to synchronous squamous cell precancer or cancer in human skin. Am. J. Pathol 150, 1791–1803. [PMC free article] [PubMed] [Google Scholar]
- Requena L, and Sangueza O (2017). Basaloid Follicular Hamartoma. In Cutaneous Adnexal Neoplasms, Requena L and Sangueza O, eds. (Springer; ), pp. 457–468. [Google Scholar]
- Rickman DS, Schulte JH, and Eilers M (2018). The expanding world of N-MYC-driven tumors. Cancer Discov. 8, 150–163. [DOI] [PubMed] [Google Scholar]
- Rittié L, and Fisher GJ (2015). Natural and sun-induced aging of human skin. Cold Spring Harb Perspect. Med 5, a015370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Sequin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, et al. (2015). Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 27, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Are A, Annusver K, Sivan U, Jacob T, Dalessandri T, Joost S, Fullgrabe A, Gerling M, and Kasper M (2020). Coordinated hedgehog signaling induces new hair follicles in adult skin. eLife 9, e46756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S, et al. (2010). Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 24, 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. Plos Comput. Biol 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E, Norris MD, Haber M, Ellis T, Wainwright B, et al. (2009). Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene 28, 1605–1615. [DOI] [PubMed] [Google Scholar]
- Tojo M, Mori T, Kiyosawa H, Honma Y, Tanno Y, Kanazawa KY, Yokoya S, Kaneko F, and Wanaka A (1999). Expression of sonic hedgehog signal transducers, patched and smoothened, in human basal cell carcinoma. Pathol. Int 49, 687–694. [DOI] [PubMed] [Google Scholar]
- Uhmann A, Dittmann K, Nitzki F, Dressel R, Koleva M, Frommhold A, Zibat A, Binder C, Adham I, Nitsche M, et al. (2007). The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood 110, 1814–1823. [DOI] [PubMed] [Google Scholar]
- Veniaminova NA, Grachtchouk M, Doane OJ, Peterson JK, Quigley DA, Lull MV, Pyrozhenko DV, Nair RR, Patrick MT, Balmain A, et al. (2019). Niche-specific factors dynamically regulate sebaceous gland stem cells in the skin. Dev. Cell 51, 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani R, Murigneux V, Alexis J, Sim SL, Wagels M, Saunders N, Soyer HP, Parmentier L, Nikolaev S, Fink JL, et al. (2021). Subtype-specific analyses reveal infiltrative basal cell carcinomas are highly interactive with their environment. J. Invest Dermatol 141, 2380–2390. [DOI] [PubMed] [Google Scholar]
- von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. (2009). Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med 361, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Wang GY, Wang J, Mancianti ML, and Epstein EH Jr. (2011). Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GY, Wood CN, Dolorito JA, Libove E, and Epstein EH Jr. (2017). Differing tumor-suppressor functions of Arf and p53 in murine basal cell carcinoma initiation and progression. Oncogene 36, 3772–3780. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Khuu JK, Dang CY, Hu J, Tran KV, Liu A, Gomez S, Zhang Z, Yi R, Scumpia P, et al. (2014). Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell Biol 16, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Jia D, Bates B, Basom R, Eberhart CG, and MacPherson D (2017). A mouse model of MYCN-driven retinoblastoma reveals MYCN-independent tumor reemergence. J. Clin. Invest 127, 888–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Luis BS, and Lane DP (2019). Intratumour heterogeneity of p53 expression; causes and consequences. J. Pathol 249, 274–285. [DOI] [PubMed] [Google Scholar]
- Yang H, and Wang K (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Klein R, Tian X, Cheng HT, Kopan R, and Shen J (2004). Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev. Biol 269, 81–94. [DOI] [PubMed] [Google Scholar]
- Yao CD, Haensel D, Gaddam S, Patel T, Atwood SX, Sarin KY, Whitson RJ, McKellar S, Shankar G, Aasi S, et al. (2020). AP-1 and TGFß co-operativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat. Commun 11, 5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Keymeulen AV, Lapouge G, Beck B, Michaux C, Achouri Y, Sotiropoulou PA, and Blanpain C (2010). Identification of the cell lineage at the origin of basal cell carcinoma. Nat. Cell Biol 12, 299–305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WES data generated for this study are available through the NCBI SRA Run Selector (BioProject: PRJNA782990).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the Lead contact upon request.