

Abstract
DNA G‐quadruplexes (G4s) are key structures for the development of targeted anticancer therapies. In this context, ligands selectively interacting with G4s can represent valuable anticancer drugs. Aiming at speeding up the identification of G4‐targeting synthetic or natural compounds, we developed an affinity chromatography‐based assay, named G‐quadruplex on Oligo Affinity Support (G4‐OAS), by synthesizing G4‐forming sequences on commercially available polystyrene OAS. Then, due to unspecific binding of several hydrophobic ligands on nude OAS, we moved to Controlled Pore Glass (CPG). We thus conceived an ad hoc functionalized, universal support on which both the on‐support elongation and deprotection of the G4‐forming oligonucleotides can be performed, along with the successive affinity chromatography‐based assay, renamed as G‐quadruplex on Controlled Pore Glass (G4‐CPG) assay. Here we describe these assays and their applications to the screening of several libraries of chemically different putative G4 ligands. Finally, ongoing studies and outlook of our G4‐CPG assay are reported.
Keywords: affinity chromatography, cancer, drug discovery, G-quadruplexes, oligonucleotides
Ligands selectively targeting DNA G‐quadruplexes (G4s) represent valuable targeted anticancer drugs. Aiming at speeding the identification of G4‐targeting ligands, the affinity chromatography‐based assay named G‐quadruplex on Oligo Affinity Support (G4‐OAS) was developed, by synthesizing G4‐forming sequences on commercial polystyrene OAS. Then, due to unspecific binding of hydrophobic ligands on nude OAS, a universal support based on Controlled Pore Glass (CPG) was conceived for both G4‐forming oligonucleotides synthesis and affinity chromatography‐based assay, renamed as G‐quadruplex on Controlled Pore Glass (G4‐CPG) assay.
1. Introduction
G‐quadruplex (G4) structures of DNA represent appealing targets in the context of selective anticancer therapeutic strategies.[ 1 , 2 , 3 ]
G4s are non‐canonical structures of nucleic acids formed by stacking of two or more parallel G‐quartets, that is, cyclic arrays in which four guanines are bound through hydrogen bonds (Figure 1A).[ 2 , 4 , 5 ] In detail, each G‐quartet (also defined G‐tetrad) is stabilized by eight Hoogsteen‐type hydrogen bonds involving, for each guanine, the N1 and the exocyclic NH2 on C2 as H‐bond donors, and the O6 and N7 as H‐bond acceptors. Guanine arrangement in a G‐quartet determines the formation of a cavity in the centre of the planar structure, delimited by guanine carbonyl oxygens, which represents a specific binding site for metal cations (Figure 1A).[ 2 , 4 , 5 ]
Figure 1.

A) Structure and schematic representation of a G‐quartet. B) Schematic representation of unimolecular, bimolecular and tetramolecular G4s formed by stacking of three G‐quartets. C) Examples of three different topologies of unimolecular G4. D) Types of linking loops in a G4. E) The anti/syn conformations of the 2′‐deoxyguanosine. M+ indicates a stabilizing metal cation, for example K+ or Na+. R=1‐β‐D‐2‐deoxyribofuranosyl group. Green circles indicate the 5’‐end.
Several metal cations with different ionic radii can be hosted in this central cavity. Circular dichroism (CD) studies revealed that cations with ionic radii between 1.3 and 1.5 Å, such as K+, Rb+, Sr2+ and Ba2+, stabilize G4 structures better than other ions, i. e. Li+, Na+, Cs+, Mg2+ and Ca2+, due to their ability to perfectly fit between two adjacent G‐quartets, coordinating eight guanine carbonyl oxygens. [6] Smaller cations, such as Li+, or larger cations, for example Cs+, are not well accommodated in the binding site formed by two consecutive G‐quartets. [6] Notably, if G4 and monovalent cation concentrations are low enough, high concentrations of divalent cations such as Ca2+, Co2+, Mn2+, Zn2+, Ni2+ and Mg2+ can induce G4 instability and unfolding. [7] Presumably, the divalent ions interact with guanine‐rich oligonucleotides in two different ways: they can bind to the phosphate groups of the oligonucleotide backbone forming tight ion pairs, thus locally reducing the charge repulsions, and, at higher concentrations, also coordinate in a bidentate manner the 6‐keto and 7‐imino groups of guanines involved in hydrogen bonding, thus disrupting the G4 architecture. [8]
G4 structures are highly polymorphic. The peculiar conformational behaviour of G4s depends on the nature of the associated metal cations, but also on: i) the number of strands involved in the structure, which can be one, two or four, resulting in unimolecular, bimolecular and tetramolecular G4s (Figure 1B); ii) the relative strand orientation, that is, parallel, antiparallel or mixed parallel/antiparallel (Figure 1C), which identifies the different topologies (also referred to as conformations) of G4s (parallel, antiparallel or hybrid, respectively); iii) the type of linking loops, which can be lateral, diagonal or propeller (Figure 1D); and iv) the anti/syn conformation of the guanine residues (Figure 1E).[ 2 , 5 ] Importantly, while nucleobases in B‐DNA are only in the anti‐conformation, in G4 structures guanines can adopt either anti or syn conformation, thus giving rise to a variety of different arrangements within the G‐quartets, which sensibly contribute to increase the topological diversity of G4s.[ 9 , 10 , 11 ] Thus, differently from B‐DNA, in which there are only two different grooves, a major and a minor one, the remarkable variation in the glycosidic torsion angles results in the formation of four grooves of very different size (wide, medium or narrow) in the G4 backbone.[ 5 , 12 , 13 ]
Moreover, a further structural peculiarity of G4s is their ability to stack on top of each other, forming dimeric or higher‐order structures (multimers), featured by peculiar pockets at the interface between two G4 units.[ 14 , 15 , 16 , 17 ]
Their remarkable structural polymorphism is probably the main reason why nature chose G4s – and not the rigid and regular B‐DNA duplex conformation – as key elements for the fine regulation of specific biological mechanisms.[ 2 , 18 ] Notably, the presence of G4 structures has been proved in the genetic material of human cells by using specific antibodies.[ 19 , 20 , 21 ] Indeed, G4s are non‐randomly distributed in the genome, but are mainly located in cancer‐related DNA regions, i. e. at telomeres and oncogene promoters,[ 1 , 22 , 23 , 24 , 25 ] thus validating their biological relevance as well as their role as suitable targets for effective anticancer strategies. Accordingly, in the last two decades novel promising anticancer strategies have emerged involving G4s as privileged targets.[ 25 , 26 ] In detail, a specific drug – or a cocktail of drugs – inducing and stabilizing G4 structures at telomeres and/or at oncogene promoters can negatively affect telomerase activity and/or oncogene transcription, thus specifically blocking uncontrolled cancer cells growth and proliferation.[ 22 , 23 , 24 , 25 , 27 ]
Several studies have fully demonstrated the relationship between the anticancer activity of various ligands and their ability to recognize G4 structures.[ 25 , 26 ] Specific G4 binding modes include stacking on the outer G‐quartets and/or on nucleobases in loops or in the flanking segments, and/or binding at the grooves or loops mediated by hydrogen bonds or electrostatic interactions.[ 28 , 29 , 30 , 31 , 32 ]
In the last two decades intensive research efforts have been devoted to the design and synthesis of potential ligands able to selectively target G4 structures and discriminate them over the other and more abundant form of DNA, that is, the B‐DNA duplex.[ 27 , 29 , 32 , 33 ] Currently, four molecules able to interfere with the in vitro and/or in vivo functions of G4 structures reached advanced clinical trials for anticancer treatments, that is, Quarfloxin (ClinicalTrials.gov Identifier: NCT00780663), CX‐5461 (ClinicalTrials.gov Identifier: NCT02719977), APTO‐253 (ClinicalTrials.gov Identifier: NCT02267863) and pyrvinium pamoate (ClinicalTrials.gov Identifier: NCT05055323), providing proof‐of‐principle for the high anticancer therapeutic potential of targeting G4 structures.[ 26 , 34 , 35 , 36 ] Only for Quarfloxin, Phase II studies have been concluded, while Phase I studies are ongoing for the other three G4 ligands. Particularly, Quarfloxin did not proceed after Phase II trials because of bioavailability issues, but its toxicity profile was very encouraging, suggesting that a future optimization could be the solution to obtain a G4 ligand with more favourable pharmacological properties. [22] In this frame, it is evident that, in order to achieve efficient G4‐targeting ligands approved as innovative anticancer drugs, a massive commitment to produce a larger number of potential G4 ligands, in a combinatorial chemistry perspective, is necessary. However, to be really effective, the effort in preparing huge libraries of new potential G4 ligands has to be coupled with fast and reliable High Throughput Screening (HTS) methods for fishing suitable hit compounds.
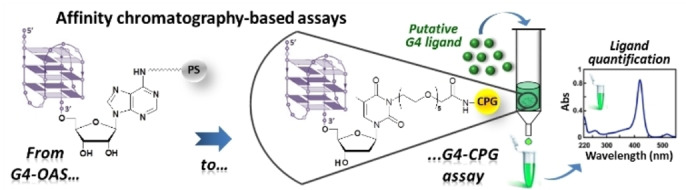
In the search for HTS methods allowing rapid and efficient identification of potential anticancer candidate drugs, our group recently developed an affinity chromatography‐based assay, which we named G‐quadruplex on Oligo Affinity Support (G4‐OAS), for the screening of libraries of putative G4 ligands. [37] In detail, it requires the immobilization of G4‐forming sequences on a polystyrene resin, which is left in contact with solutions at known concentration of the potential ligands, thus allowing the discrimination of high‐affinity ligands, captured by the resin‐bound G4 target, from low‐affinity binders, easily eluted with simple washings. Simple spectrophotometric measurements on the eluted fractions provide the concentration of the unbound ligand and thus the percentage of binding for each sample.
Compared to other methods thus far used to identify G4 ligands – such as Fluorescence Resonance Energy Transfer (FRET)‐based melting, G4‐Fluorescent Intercalator Displacement (FID), Isothermal Titration Calorimetry (ITC), Surface Plasmon Resonance (SPR), NMR, Electrospray Ionisation Mass Spectrometry (ESI‐MS) and small molecule microarray‐based screenings[ 38 , 39 ] – the G4‐OAS assay showed several advantages. First of all, it allows elongating on the support the oligonucleotide sequences without necessarily modifying them to detect ligand‐G4 binding, and to study the ligand‐G4 complexes under pseudo‐physiological conditions, thus better mimicking their possible interactions in cell nuclei. Moreover, all the above methods require expensive equipment along with specific expertise, and/or special ligand/G4 properties (e. g., fluorescence) or a very high amount of ligand/G4, while the G4‐OAS assay proved to be rapid, simple, cheap and reproducible. However, for the identification of G4 ligands, it showed some limitations in the screening of compounds featuring large aromatic cores and low hydrophilicity, which could give unspecific interactions with the polystyrene support.[ 18 , 40 ]
With the aim of addressing this issue, in parallel preserving all the advantages of the method, our studies were then extended to Controlled Pore Glass (CPG) supports. Thus, a suitable variant of the affinity chromatography‐based assay was conceived, named G‐quadruplex on Controlled Pore Glass (G4‐CPG), by ex‐novo designing and synthesizing the proper functionalized solid support on which elongating the G4‐forming oligonucleotides of interest. With this optimization, we developed a universal solid support extremely useful for effective screenings of not only structure‐ but also conformation‐selective G4 ligands, that is, ligands able to discriminate G4s over the duplex DNA but also G4s having different conformations.[ 18 , 41 ]
Herein, we describe the G4‐OAS assay as well as its evolution into the G4‐CPG assay, along with their applications to the screening of several libraries of putative G4 ligands. A summary of all the achieved results concerning different families of potential G4 ligands and their peculiar binding modes is provided, together with ongoing applications and perspectives of our affinity chromatography‐based assays in the context of the identification of G4‐targeting agents.
2. Development of an Affinity Chromatography‐Based Method for the Screening of Potential G4 Ligands: G4‐OAS Assay and its Applications
2.1. Design and set up of the G4‐OAS assay
In a first design of our affinity chromatography‐based method for the identification of G4‐selective ligands, we selected the Oligo Affinity Support (OAS), a commercially available polystyrene/polyethylene glycol copolymer functionalized with a 4,4′‐dimethoxytriphenylmethyl (DMT)‐protected adenosine derivative attached through the adenine exocyclic amino group (Figure 2A, Glen Research Corporation, Sterling, VA, USA). Differently from the supports typically used for oligonucleotide synthesis, this resin allows the solid‐phase assembly of oligonucleotides and also their on‐support deprotection, that is, removal of all the protecting groups from the oligonucleotide backbone, without detaching them from the solid support. Thus, by using standard phosphoramidite chemistry on an automated DNA synthesizer, the selected oligonucleotide sequences can be assembled on the OAS resin and then deprotected by a final standard aq. ammonia treatment. Then, the oligonucleotides, still attached to the resin, are left in contact with suitable pseudo‐physiological aqueous solutions (see below) and subjected to an annealing procedure, comprising a heating and a successive cooling step, thus favouring their correct structuring. After that, the resins carrying the folded oligonucleotides are ready to be used for the affinity chromatography‐based assays.
Figure 2.
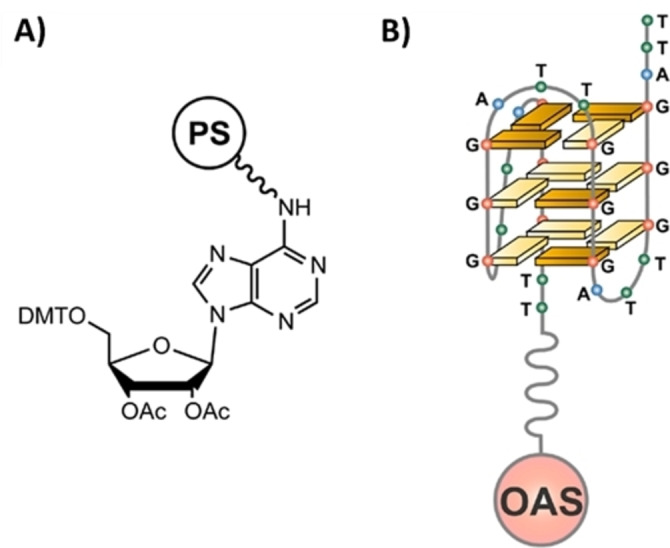
A) Schematic representation of the commercially available Oligo Affinity Support (OAS), a polystyrene/polyethylene glycol (PS)‐based resin functionalized with a 4,4′‐dimethoxytriphenylmethyl (DMT)‐protected adenosine derivative. B) Schematic representation of the OAS resin functionalized with the 26‐mer d[(TTAGGG)4TT] sequence (tel26), a truncation of the human telomeric DNA, folded into a G4 structure in a K+‐containing solution. Ac=acetyl. Reproduced with permission from Ref. [37] Copyright 2014, American Chemical Society.
In our experiments, the OAS resin was functionalized with proper G4‐forming oligonucleotides and then used to set up our assay for the identification of G4‐selective ligands. [37] Particularly, the 26‐mer d[(TTAGGG)4TT], hereafter named tel26, reproducing a sequence from human telomeric DNA, able to fold into a monomeric G4 structure in proper conditions, was chosen as a model oligonucleotide for our binding assay (Figure 2B).
Before performing the binding assay, the tel26‐OAS was suspended in a K+‐containing solution, hereafter named washing solution, and subjected to an annealing procedure, that is, heated at 75 °C for 5 min and then allowed to slowly cool to r. t., in order to promote the G4 folding of the resin‐bound tel26. Higher temperatures typically used for G4 annealing in solution, for example 85–95 °C, were avoided to prevent the detachment of even tiny amounts of the oligonucleotide from the support. [37]
The general protocol adopted for the binding assays consisted in leaving the potential ligand dissolved in the washing solution at a known concentration (generally 60 μm) in contact with a weighed amount (5–20 mg) of the support functionalized with the G4‐forming oligonucleotide of interest (Figure 3). After 4 min incubation on a vibrating shaker, the resin was eluted with defined volumes of the washing solution and all the eluted fractions were then analysed by UV‐Vis measurements, in order to determine the amount of unbound ligand recovered from the solid support. On the other hand, the amount of bound ligand was calculated by subtracting the quantity of ligand eluted upon washings, as derived from direct UV‐Vis measurements, from the ligand initially loaded on the resin. Furthermore, as a control, a direct measurement of the bound ligand was obtained by treating the resin with a releasing solution, which allowed the complete oligonucleotide denaturation and, as a consequence, the quantitative recovery of the bound ligand in solution. In order to induce the correct G4 refolding after this treatment, the support was resuspended in the washing solution and then subjected again to the annealing procedure, as described above (Figure 3). The reversibility of the folding/unfolding processes of unimolecular G4 structures allowed effectively recycling the functionalized resin for several different tests. Therefore, a large number of binding assays can be performed on the same batch of resin without losing in efficiency and reliability of the experiments. Typically, a single batch of resin could be used for the screening of at least 10 ligands – with each ligand analysed in triplicate, so to reach a total number of 30 different experiments – before being discarded.[ 18 , 37 ]
Figure 3.

Schematic representation of the affinity‐chromatography‐based G4‐binding assay. r. t.=room temperature.
This protocol was first tested and optimized on a set of small molecules known for their ability to interact with G4 structures with different affinity and binding modes. In detail, the following molecules were used as models: i) three π–π end‐stackers, that is, the cationic porphyrin 5,10,15,20‐tetrakis(1‐methyl‐4‐pyridinio)porphyrin (TMPyP4), thiazole orange (TO) and the acridine derivative 3,11‐difluoro‐6,8,13‐trimethyl‐8H‐quino[4,3,2‐kl]acridinium methosulfate (RHPS4), ii) two pyrrole‐containing polyamide analogues as representative examples of groove binders, that is, Distamycin and Netropsin, as well as iii) acridine‐9‐carboxylic acid (9‐Acr‐COOH), chosen as negative control, being unable to bind G4 structures (Figure 4). [37]
Figure 4.

Chemical structures of some known G4 ligands (TMPyP4, TO, RHPS4, Distamycin and Netropsin) and two molecules unable to bind the G4s (9‐Acr‐COOH and Resveratrol), used as models for the G4‐binding assays. [37]
Particularly, these compounds were used to optimize the G4‐binding assay protocol, in terms of: i) concentration of compound left in contact with the solid support, ii) volumes and composition of both the washing and releasing solutions to be used, iii) reduction of unspecific binding on the nude resin, that is, the OAS resin bearing no oligonucleotides, and iv) reliability of the assay in identifying effective G4 ligands, ranking them based on their relative affinities.
Concerning the concentration of compound to be incubated with the resin, it had to be neither too high, to avoid formation of aggregates and/or precipitation of the tested molecules, which can significantly impair the quantitative UV‐Vis measurements during the assays as well as clog the frit and more generally produce strong unspecific interactions with the assay equipment, nor too low, which could prevent reliable UV‐Vis measurements. A 60 μm compound concentration was found to be a good compromise for both the above known G4 ligands and the novel compounds discussed below.
Different conditions were tested to optimize the washing solution. In all cases a 100 mM KCl solution was adopted, so to mimic the intranuclear environment and guarantee the complete G4 folding, varying the amount of DMSO from 0 to 20 %. Overall, a 15 % DMSO solution proved to be the best compromise for the binding assays in terms of minimization of unspecific interactions with the nude OAS resin and solubility of the ligands, in turn not sensibly affecting the G4 folding and the interaction of the ligands with the G4 structure, as verified by CD measurements in solution. These conditions were then chosen to assess the ability of the model molecules to bind the tel26‐OAS. Notably, in our tests TMPyP4, TO, RHPS4 and Netropsin were efficiently retained by tel26‐OAS (% of bound ligand=93–99 %), Distamycin was only partially retained (% of bound ligand=28 %), whereas 9‐Acr‐COOH was completely released. Remarkably, the retention order perfectly reflected the affinity trend of these ligands for the tel26 G4 structure observed in solution,[ 31 , 42 , 43 , 44 , 45 , 46 ] thus confirming the general reliability of the method. [37]
In parallel, different releasing solutions, containing 1 M LiCl or 2.5 M CaCl2, and/or solutions at different percentages of DMSO (15 or 100 %, v/v), were tested to achieve the fast and complete recovery of the ligands bound to tel26‐OAS, thus allowing to recycle the resin for further binding assays. The best releasing solution was found to be 2.5 M CaCl2/15 % DMSO. For the poor water‐soluble ligands TO and RHPS4 a complete elution was achieved only using 100 % DMSO. [37]
To verify the reliability of the assay in identifying G4 selective ligands, i) a resin carrying a 26‐mer scrambled sequence d[AA(GT)10GAAG], hereafter named scr26 and unable to fold into a G4 structure and ii) a resin carrying a model duplex, indicated as ds‐OAS, obtained by hybridizing the scr26‐OAS with the complementary 24‐mer sequence d[T(CA)10CTT], hereafter named compl24, were prepared and used for the binding assays with RHPS4 and Netropsin, using the same protocol used for tel26‐OAS. No significant binding was found for both ligands to scr26‐OAS. In contrast, both RHPS4 and Netropsin bound ds‐OAS with similar percentages of bound ligand as found for tel26‐OAS, in agreement with their known ability to bind also duplex DNA,[ 47 , 48 , 49 ] thus overall proving the general efficacy of the method for the evaluation of G4‐binding selectivity. [37]
Furthermore, in order to test the possibility of screening a mixture of different small molecules by the G4‐OAS assay, a mixture of TO, Netropsin and 9‐Acr‐COOH was incubated on tel26‐OAS and then a gradient elution was applied to the resin (Figure 5). Indeed, Netropsin and TO were initially fully retained on the resin, while 9‐Acr‐COOH was immediately released by elution with the washing solution. Then, Netropsin was selectively released by elution with the 2.5 M CaCl2/15 % DMSO releasing solution, while TO was finally recovered only upon elution with 100 % DMSO. Notably, these results provided a valuable proof‐of‐principle proving the feasibility and efficacy of the method for a rapid analysis of mixtures of potential ligands endowed with different affinities for the G4 target and non‐overlapped characteristic UV absorption maxima. [37]
Figure 5.

General scheme for the screening of a mixture of multiple small molecules by the G4‐OAS assay.
2.2. Application of the G4‐OAS assay to various libraries of putative G4 ligands
After optimization, the G4‐OAS assay was applied to the screening of two different libraries of novel, putative small molecule G4 ligands.
The first library (Library I) consisted of molecules selected as G4 ligands by virtual screening (VS) using different models of telomeric G4s as targets.[ 50 , 51 , 52 ] Interestingly, 3 out of the 8 VS‐selected molecules, that is, 2, 6 and 7 (Figure 6), proved to be effective (% of bound ligand=83–90 %) and selective ligands of tel26‐OAS, in parallel showing a low affinity for ds‐OAS (% of bound ligand=5–45 %). [37]
Figure 6.

Chemical structures of the best G4 ligands from Library I (2, 6, 7) and II (10B, 4D, 7E, 1F, 2F, 7F, 8F).[ 37 , 40 ]
The second library (Library II) included a focused panel of putative small molecule ligands of G4 grooves. [40] Due to the significant structural differences between G4 and duplex DNA grooves, G4 groove ligands are generally expected to exhibit higher discrimination ability than end‐stacker or intercalating agents in the recognition of G4 versus duplex DNA, thus in principle assuring higher selectivity and consequently lower toxicity. In order to discover selective G4 groove ligands, a high‐speed VS tool, known as Rapid Overlay of Chemical Structures (ROCS), was used to select compounds sharing a three‐dimensional shape similar to Distamycin, which is one of the few well‐characterized G4 groove ligands, in its G4‐bound conformation.[ 31 , 40 ] Thus, based on shape comparison, 60 molecules were selected by VS from a commercially available database consisting of 14,400 compounds.
Then, the VS was coupled to the G4‐OAS assay to experimentally validate the selected 60 molecules as G4 ligands. In particular, careful optimization of the composition of the washing solution (changed to 50 mM KCl/20 % DMSO) allowed enhancing the solubility of the tested molecules and minimizing the unspecific interactions of these compounds with the polystyrene resin. However, even in the presence of 20 % DMSO, about one third of the 60 small molecules showed strong unspecific binding on the nude OAS resin, due to stacking interactions between the polystyrene resin and the aromatic cores of the ligands, making therefore impossible to acquire information on their binding with the G4‐functionalized support. Moreover, in the here used conditions, necessary to ensure the full solubility of the tested compounds, the duplex structure obtained by hybridization of scr26 and compl24 on the support was not very stable, partially releasing the complementary strand in solution. Consequently, we were not able to test the selectivity of the ligands towards tel26‐OAS versus ds‐OAS, but only their preference for tel26‐OAS over scr26‐OAS.
Interestingly, among the ligands soluble in the washing solution, not showing unspecific binding on the nude OAS resin, seven ligands (10B, 4D, 7E, 1F, 2F, 7F, 8F, Figure 6) exhibited good affinity for the tel26‐OAS (% of bound ligand=41–100 %) and also good selectivity over scr26‐OAS (% of bound ligand=27–73 %). [40]
Therefore, these compounds were selected as the best G4 ligands and further studied in solution in their interaction with tel26 G4. Particularly, fluorescence and NMR studies proved that compounds 10B, 7E and 7F bound the telomeric G4 structure in solution with good affinity (Kb∼106 m −1), mainly through interactions at grooves and loops, showing preference for telomeric hybrid G4 over non‐telomeric parallel ones and duplex DNA. Moreover, biological assays demonstrated that these G4 ligands were able to induce a significant DNA damage response at telomeres in the low μm range on human transformed fibroblasts (BJ‐EHLT), cervical epithelial carcinoma (HeLa), osteosarcoma (U2OS) and melanoma (M14) cell lines. [40]
Overall, the G4‐OAS assay applied in tandem with VS tools proved to be a successful strategy to identify novel G4‐targeting chemotypes endowed with a relevant biological activity. [40]
3. Optimization of the G4‐Binding Assays: G4‐CPG Assay and its Applications
3.1. Design and set up of the G4‐CPG assay
Though rapid and simple, a serious limitation emerged when using the G4‐OAS assay, resulting from the intrinsic chemical nature of the OAS, that is, unspecific binding of some lipophilic aromatic ligands on the nude polystyrene resin. [40] Hence, considering that most known G4 ligands are featured by planar aromatic cores, essential for realizing stacking interactions with the outer G‐quartets and/or flanking/loop nucleobases, there was a felt need to provide chemically inert supports, which could be properly functionalized so to assemble the secondary structure‐forming oligonucleotides as well as perform the affinity chromatography‐based binding assays.
The crucial problem of the OAS, which limited the scope of the assay, was addressed moving from polystyrene to CPG supports.[ 18 , 41 ] CPG has two major advantages useful for our purposes: i) its intrinsic chemical inertness, which avoids the undesired stacking interactions with aromatic ligands, and ii) its validated use as the support of choice for solid‐phase oligonucleotide synthesis, allowing to easily customize the synthetic protocols for the G4‐forming oligonucleotides. However, all the commercially available CPG supports are typically functionalized with linkers cleavable under the basic conditions required in the final deprotection step of the oligonucleotide synthesis, and thus are not suitable for our purposes. Indeed, a crucial requirement in our design is the attachment of the first nucleoside through a linker chemically stable to the final deprotection step. In this way, the fully deprotected oligonucleotides remain covalently linked to the support on which the affinity chromatography‐based binding assays can be performed.
To this aim, we conceived a novel functionalization for CPG supports.[ 18 , 41 ] In detail, the commercially available Long Chain AlkylAmine Controlled Pore Glass (Figure 7A, LCAA‐CPG; Link Technologies, Bellshill, UK) was functionalized with an ad hoc synthesized hexaethylene glycol spacer (indicated as DMT‐HEG‐COOH), using N,N′‐dicyclohexylcarbodiimide/1‐hydroxybenzotriazole (DCC/HOBt) as activation method (Figure 7B). [41] After functionalization with the HEG‐based spacer, the DMT group was removed by acidic treatment and the resulting Support 1 reacted with 5′‐O‐DMT, 3′‐O‐acetyl‐thymidine through a Mitsunobu reaction. Standard capping treatments with acetic anhydride in pyridine were subsequently performed to block both the unreacted amino groups on the support after DMT‐HEG‐COOH incorporation and the unreacted hydroxyl groups on Support 1 after the Mitsunobu reaction. Overall, Support 2 was obtained with an average oligonucleotide functionalization of 0.023 meq g−1. [41]
Figure 7.

A) Commercially available Long Chain AlkylAmine Controlled Pore Glass (LCAA‐CPG) solid support. B) Functionalization of LCAA‐CPG with 5′‐O‐DMT, 3′‐O‐acetyl‐thymidine through the selected hexaethylene glycol spacer. Ac=acetyl; DCC=N,N′‐dicyclohexylcarbodiimide; DEAD=diethyl azodicarboxylate; DMF=N,N‐dimethylformamide; DMT=4,4′‐dimethoxytriphenylmethyl; HOBt=1‐hydroxybenzotriazole; Ph=phenyl; TCA=trichloroacetic acid; THF=tetrahydrofuran. Adapted with permission from Ref. [41] Copyright 2018, Elsevier.
More specifically, hexaethylene glycol was chosen as the spacer for: i) its good solubility in both apolar and polar solvents, making it suitable for both oligonucleotide synthesis, carried out in organic solvents, and affinity chromatography‐based assays, performed in aqueous solutions, and ii) its length and flexibility, which ensure minimization of steric effects of the CPG support during both the synthesis and binding assays and guarantee proper conformational freedom to the covalently attached oligonucleotide, which can adopt its preferred secondary structure as if in solution.
On the other hand, a pyrimidine (and not a purine, as in the commercially available OAS) nucleoside was chosen as the first monomer on the support, considering that pyrimidines are less prone than purines to produce additional stacking interactions with both the tested ligands and the oligonucleotides, which could affect the ligand binding as well as the oligonucleotide conformation, and hence the overall results of the binding assays.
Thus, the synthesis of several secondary structure‐forming oligonucleotides was performed on Support 2 by using standard phosphoramidite chemistry on an automated DNA synthesizer along with on‐support deprotection carried out after each synthesis. Particularly, for the deprotection step, two different protocols were tested: i) treatment with 30 % ammonium hydroxide at r. t. for 24 h or ii) 30 % ammonium hydroxide/40 % methylamine (AMA) 1 : 1, v/v at r. t. for 2 h. The best results in terms of final functionalization yield were obtained with the AMA deprotection. [41]
Overall, our optimized protocol for the on‐support oligonucleotide synthesis and deprotection allowed obtaining supports with 0.011–0.013 meq g−1 final oligonucleotide functionalization. This proved to be a suitable functionalization for the successive binding assays, that is, neither too low, thus allowing the use of small amounts of support for the binding tests, nor too high, thus avoiding steric hindrance between contiguous oligonucleotide molecules which can impair the binding assay, thus altering the results.[ 18 , 41 ]
In detail, in addition to the hybrid tel26 G4‐forming oligonucleotide, we extended our assay to the 22‐mer d(AGGGAGGGCGCTGGGAGGAGGG) named c‐kit1, originating from the c‐kit oncogene promoter and folding into a parallel G4. [53]
Moreover, considering that in our first studies on OAS we proved that bimolecular duplexes obtained by hybridization on the support were not sufficiently stable for the binding assays, a unimolecular hairpin duplex, named ds27, was conceived and synthesized on Support 2, to be used as a control in the evaluation of G4 versus duplex selectivity. [41] Particularly, the model duplex consisted of the self‐complementary oligonucleotide d(CGCGAATTCGCG), known as the Dickerson sequence, chosen for being the best well‐known model of B‐DNA duplex, [54] in which the two 12‐mer complementary tracts were connected by a TTT loop. As a control, a CD analysis was performed to compare the stability and conformation of ds27 hairpin duplex in comparison with the Dickerson bimolecular duplex, which validated ds27 as a good and stable model of B‐DNA duplex, preserving its overall conformation but showing higher thermal stability.[ 41 , 55 ]
After oligonucleotide synthesis and deprotection, adopting the optimized protocol described above, the same molecules used to validate the G4‐OAS assay, that is, TMPyP4, TO, RHPS4, Distamycin, Netropsin and 9‐Acr‐COOH (Figure 4), were investigated, applying the general procedure described in Figure 3, to evaluate the efficiency and reliability of the G4‐CPG assay in comparison with the G4‐OAS assay. To this set of ligands, Resveratrol (Figure 4) was added as additional low affinity G4‐binder,[ 56 , 57 ] and, more importantly, a focused set of compounds chosen from the 60 VS‐selected molecules – previously analysed on the G4‐OAS and excluded since strongly and unspecifically retained by the nude OAS – was also tested.[ 41 , 55 ]
Additionally, compared to the G4‐OAS assay, some minor modifications to the washing solution were introduced to further optimize it. Indeed, instead of the previously used K+‐containing aq. buffer/DMSO mixtures, a 50 mM KCl/10 % DMSO/10 % CH3CH2OH solution was here used. A small percentage of ethanol was introduced in the washing solution so to fully avoid undesired absorption of the tested ligands on the assay equipment, that is, the polypropylene column and the frit bearing the functionalized supports. However, the total percentage of organic solvent was unaltered (20 %) to guarantee the solubility of the tested ligands without perturbing the correct folding of the support‐bound oligonucleotide, as verified by CD analysis of our model oligonucleotides in solution.[ 18 , 41 , 55 ]
Using these conditions, the binding assays were repeated on the nude and functionalized OAS resins for all the tested compounds and performed in parallel on the nude and the new functionalized CPG supports. No change was observed, within the experimental error, in the amounts of released/bound ligand on the nude and functionalized OAS resins compared to our previous data obtained with 100 mM KCl/15 % DMSO or 50 mM KCl/20 % DMSO.[ 41 , 55 ]
Notably, our results proved that the nude CPG had low‐to‐null unspecific interactions with all the tested model ligands, in contrast with the previously used OAS. Indeed, all the investigated ligands were generally recovered quantitatively from nude CPG, requiring smaller volumes of the washing solution compared to nude OAS. In addition, highly specific binding was maintained for those ligands which are well validated G4 and/or duplex binders, displaying in our tests binding data with a trend respecting the order of binding affinities determined in solution, thus confirming the general reliability of our method.[ 18 , 41 , 55 ]
Moreover, UV controls showed that only 0.1–0.5 % of the nucleotide material was released from the oligonucleotide‐bound CPG support upon oligonucleotide annealing at 75 °C, in contrast to the 0.5–1.0 % oligonucleotide loss observed for the same thermal treatment on OAS resins.
Overall, for its lower unspecific binding and higher thermal stability, the same batch of functionalized CPG could be used for typically more than 50 experiments, that is, about twice the number of binding assays generally carried out on the OAS resin, moreover offering much cleaner and more reliable results.[ 18 , 41 , 55 ]
Additionally, a fundamental requirement for an effective affinity chromatography‐based assay is that the oligonucleotide linked to the support maintains its native conformation, as if in solution. This is especially true when the immobilized targets are G4 structures featured by a notable structural polymorphism. Thus, in order to unravel the real conformations adopted by the oligonucleotides immobilized on the CPG supports, we used a fluorescent core‐extended naphthalene diimide (cex‐NDI, Figure 8A), recently designed to provide different fluorescence responses upon interaction with different secondary structure‐forming oligonucleotides. [58] In detail, upon interaction with DNA in solution, cex‐NDI produced a dramatic fluorescence enhancement when bound to hybrid G4s, a weaker emission was in turn observed when bound to parallel G4s and an even weaker fluorescence intensity was found in the binding to duplex DNA structures. This peculiar behaviour was explained considering the different binding modes of cex‐NDI to the different DNA structures tested: this ligand interacts with the grooves of hybrid G4s, is an end‐stacker of parallel G4s and typically binds duplex DNA via intercalation (Figure 8B).[ 58 , 59 ] Incubating the tel26‐, c‐kit1‐ and ds27‐CPG supports – as examples of hybrid G4, parallel G4 and duplex DNA, respectively – with cex‐NDI and analysing the resulting samples by confocal microscopy, we found the highest values of fluorescence intensity for tel26‐CPG, an intermediate value for c‐kit1‐CPG and the lowest values for the ds27‐CPG (Figure 8C and D), fully confirming that, under the G4‐CPG assay conditions, tel26, c‐kit1 and ds27 sequences, linked to the CPG supports, were folded into a hybrid G4, a parallel G4 and a hairpin duplex, respectively, as in solution. This can be mainly attributed to i) the long and flexible hexaethylene glycol spacer attached to the CPG support, able to guarantee proper flexibility and distance of the oligonucleotides from the solid support, so that the oligonucleotides can be fully embedded in the washing solution as if in solution, and ii) the optimal CPG pore size selection (pore diameter=1000 Å), which is a good compromise between support mechanical stability (large‐pore supports are more fragile) and minimization of the local crowding around the oligonucleotides attached to the solid support.
Figure 8.

A) Chemical structure of cex‐NDI. B) Schematic picture of the fluorescent behaviour of cex‐NDI when bound to hybrid G4s, producing a dramatic fluorescence enhancement, and parallel G4s or duplex DNA with a weaker fluorescence emission. C) Representative confocal images of tel26‐, c‐kit1‐ and ds27‐functionalized CPG supports after incubation with the cex‐NDI. Merged images of cex‐NDI fluorescence and transmitted light. Scale bars correspond to 100 μm. D) Mean fluorescence intensity values (±S.D.) taken from different edge glass beads‐containing regions of each image acquired for tel26‐, c‐kit1‐ and ds27‐functionalized CPG supports. p‐values have been calculated using the Student's t‐test (****p<0.0001). Adapted with permission from Ref. [41] Copyright 2018, Elsevier.
Overall, all the above results proved that our novel G4‐functionalized CPG‐based assay is a useful tool to quickly and efficiently identify not only structure‐selective but even conformation‐selective ligands, which may represent effective candidate drugs in anticancer targeted therapies.[ 18 , 41 ]
3.2. Application of the G4‐CPG assay to various libraries of putative G4 ligands
After full optimization and validation of the method, six different libraries of synthetic or natural compounds have been investigated by the G4‐CPG assay thus far.
First, a focused library of analogues, sharing a furobenzoxazine naphthoquinone core and differing for the pendant groups on the N‐atom of the oxazine ring, was evaluated (Library III). [60] In detail, these compounds were selected as analogues of the anticancer compound 4 (Figure 9), previously selected by VS of a 6,000 compounds library included in a commercially available database using as the target the tetramolecular, parallel G4 of d(TGGGGT) sequence.[ 50 , 61 ] Considering the promising DNA damage ability of compound 4 on BJ‐EHLT cell line, and aiming at developing more potent and selective ligands efficiently targeting multiple G4 structures, we hence focused on a small library of its structural analogues selected by a similarity approach from the ZINC database collection of commercially available compounds (see: https://zinc15.docking.org) setting the similarity threshold to 70 %. [60]
Figure 9.
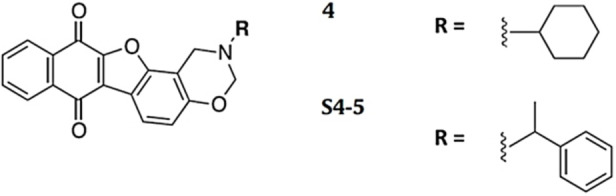
Chemical structures of the best G4 ligand (S4‐5) from Library III and its parent compound (4). [60]
By this approach, ten compounds – named S4‐1–S4‐10 – were selected and then evaluated for their binding to multiple G4s by the G4‐CPG assay. Indeed, aiming at expanding the use of the G4‐CPG assay to a wide variety of topologically different G4 structures, we extended the panel of cancer‐related CPG support‐bound oligonucleotides, already including tel26, c‐kit1 and the control ds27, to the following sequences, all forming mainly parallel G4s: i) the 33‐mer d(TGGGGAGGGTGGGGAGGGTGGGGAAGGTGGGGA), originating from the c‐myc promoter and hereafter named c‐myc,[ 62 , 63 ] ii) the 21‐mer d(CGGGCGGGCGCGAGGGAGGGG) from the c‐kit promoter, hereafter named c‐kit2, [53] iii) the 24‐mer d(AAGGGGAGGGGCTGGGAGGGCCCGGA) from the regulatory region of the hTERT gene, hereafter named hTERT1.[ 64 , 65 ] In detail, for the latter oligonucleotide sequences elongated on the CPG support, as well as for all the sequences added in successive analyses, the correct functioning of the G4‐functionalized supports was evaluated by using TO and Resveratrol as model ligands, obtaining in all cases affinity data in full agreement with what previously found through solution studies. After this validation, the binding assays on the novel G4‐functionalized supports were performed for the new libraries of putative G4 ligands.
Notably, for S4‐1–S4‐10, as well as for all the libraries investigated below, no unspecific binding on the nude CPG was observed, thus fully confirming the higher reliability of the results obtained by the G4‐CPG than G4‐OAS assay.
S4‐1–S4‐10 were all able to bind both the telomeric and extra‐telomeric G4s (% of bound ligand=67–94 %). In this series, S4‐5 (Figure 9), having null affinity for ds27‐CPG, emerged as the most promising analogue, also compared to its parent compound 4. [60] Indeed, the higher G4 versus duplex DNA selectivity of S4‐5 compared to 4 was also confirmed by biophysical analyses in solution. Additionally, molecular docking and NMR studies proved that S4‐5 interacted with the grooves of the target G4s, providing a possible explanation of its high G4 versus duplex selectivity. Finally, biological assays demonstrated that S4‐5 produced effective DNA damage in BJ‐EHLT cells, showing marked antiproliferative efficacy in the low μm range, also being less cytotoxic than the parent compound 4 on the normal counterpart of BJ‐EHLT cells, that is, BJ‐hTERT. [60]
Successively, a focused library of novel differently functionalized monomeric and dimeric naphthalene diimides (NDIs) – a well‐known class of G4‐binding ligands[ 30 , 59 , 66 , 67 , 68 ] – was designed, synthesized and evaluated by the G4‐CPG assay (Library IV). [66] Monomers included tri‐ and tetrasubstituted NDIs, decorated with N,N‐dimethylpropylamine groups and characterized by alkyl chains carrying a terminal amino group or diethylene glycol chains on the naphthalene aromatic core. Dimers differed for the nature of the linker, that is, an oligoethylene glycol chain or a seven‐carbon atoms chain. These compounds were able to interact with all the G4 targets bound to CPG supports, thus showing multi‐targeting ability, and particularly, monomeric NDI‐5 (Figure 10) was found to be the most promising G4 ligand (% of bound ligand=99 %), also due to its very good ability to discriminate duplex DNA (% of bound ligand=30 %) (Figure 11A).
Figure 10.
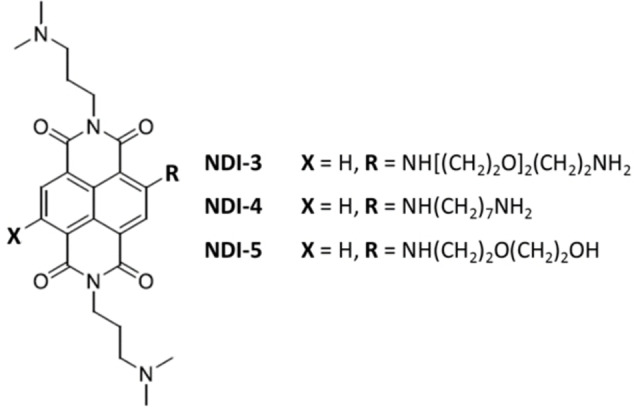
Chemical structures of the best G4 ligands from Library IV–V.[ 66 , 67 ]
Figure 11.

A) Summary of the binding assay data for NDI‐5 on nude and functionalized CPG supports. Bound ligand calculated as a difference from the unbound ligand and expressed as % of the amount initially loaded on the support. The errors associated with the % are within ±2 %. B) Representative merged images of immunofluorescence of BJ‐EHLT cancer cell line treated with NDI‐5; H2AX (DNA damage marker) spots are green, TRF1 (telomeric marker) spots are red and nuclei are blue. Enlarged views of Telomere Induced Foci (TIFs) are reported on the right smaller panels. C) Job plot analysis for the binding of NDI‐5 to tel26 G4. D) CD spectra of tel46 G4 in the absence and presence of increasing amounts of NDI‐5. Black arrows indicate intensity changes of the specific bands upon increasing NDI‐5 concentration. E) ITC binding isotherm for titration of tel46 G4 with NDI‐5. F) Binding mode of NDI‐5 to tel46 G4 obtained by molecular docking. A), B) and C) are adapted with permission from Ref. [66] under CC‐BY 4.0, Copyright 2020 by the authors; D), E) and F) are adapted with permission from Ref. [67] Copyright 2020 Elsevier.
In parallel, the biological assays proved that NDI‐3, NDI‐4 and NDI‐5 (Figure 10) were the compounds with the highest anticancer activity, showing: i) IC50 value of 79 nm on HeLa cancer cells, ii) ability to selectively target cancer (BJ‐EHLT) vs. normal (BJ‐hTERT) cells, and iii) ability to induce DNA and telomeric damage in BJ‐EHLT (Figure 11B). Thus, taking into account the results of both the G4‐CPG assay and the biological data, NDI‐5 emerged as the best G4 selective ligand and was further investigated by CD and fluorescence spectroscopy. CD titrations and CD‐melting experiments further corroborated the NDI‐5 ability to preferentially affect and stabilize G4 structures (ΔTm=+15 °C) compared to duplex DNA. Interestingly, NDI‐5 was also able to induce G4 formation in the absence of metal cations. Furthermore, fluorescence experiments proved that NDI‐5 formed peculiar 1 : 3 complexes with both hybrid and parallel G4s (Figure 11C). [66] Finally, NMR studies provided insights into the interaction of NDI‐5 with G4s of different topologies, proving that NDI‐5 mainly targets the 5′‐end quartet of hybrid G4s, while it binds both the 3′‐ and 5′‐end quartets of parallel G4s. [30]
In parallel, the same compounds of the previous NDI library, as well as additional newly functionalized NDIs, were evaluated as putative ligands of multimeric G4 structures (Library V). [67] The G4‐CPG assay was thus extended to the screening of longer oligonucleotide sequences able to fold into more than one G4 unit, which can particularly better mimic the effective folding of telomeric DNA. In detail, the 46‐mer sequence d[AGGG(TTAGGG)7], hereafter named tel46, was chosen as the model of multimeric G4 structures, endowed with the ability to fold into two consecutive G4s.[ 14 , 69 , 70 , 71 , 72 ]
Also for the compounds included in Library V, the G4‐CPG assay proved NDI‐5 as the best ligand in terms of affinity for the target of choice (% of bound ligand=92 %) and dimeric G4 versus duplex DNA selectivity (Figure 11A). [67] Thus, it was further investigated in its binding to tel46 G4 in solution studies by CD, ITC, fluorescence spectroscopy, gel electrophoresis, as well as by molecular docking. NDI‐5 proved to strongly interact (Kb∼107 m −1) and stabilize (ΔTm=+12 °C) the hybrid folding of tel46 G4 by CD and CD‐melting experiments (Figure 11D). ITC and fluorescence‐based Job plot analyses proved that its interaction was associated with two binding events: the first one involving three molecules of NDI‐5 and the second one additional six molecules (Figure 11E). Moreover, NDI‐5 was also able to induce tel46 G4 antiparallel folding in the absence of metal cations, as proved by CD and gel electrophoresis analyses. Finally, docking studies proved that NDI‐5 mainly binds the pocket at the interface between two G4 units, thus validating it as an appealing ligand for multimeric G4s (Figure 11F). [67]
In addition to the three libraries of synthetic compounds above described, we evaluated thus far also three libraries of natural compounds. Indeed, in the search for selective G4 ligands, natural compounds have been overlooked, though representing very appealing candidates due to the high structural diversity of their scaffolds. [73] First, we investigated a unique high diversity in‐house library composed of ca. 1,000 individual natural products, isolated mainly from indigenous plants collected in biodiversity‐rich countries, especially in tropical and subtropical areas, and enlarged with their semi‐synthetic and synthetic derivatives. [32]
This library was firstly screened by a docking‐based VS using both tel26 and c‐myc G4 models as targets, aiming at identifying natural compounds targeting the G4 grooves and endowed with the highest structural diversity. 28 putatively selective G4 ligands (Library VI) were identified by VS and then experimentally screened by exploiting the G4‐CPG assay, which proved Bulbocapnine, Chelidonine, Ibogaine, Rotenone and Vomicine (Figure 12) as promising G4 ligands in terms of affinity (% of bound ligand=14–44 %) and selectivity versus the duplex DNA (% of bound ligand=6–16 %). These five compounds were further analysed in solution by CD and fluorescence spectroscopy, which confirmed the selective interaction of these compounds with G4s (ΔTm=+1–13 °C, Kb∼106 m −1) over the duplex DNA.
Figure 12.

Chemical structures of the best G4 ligands from Library VI. [32]
Then, molecular dynamics simulations proved that all these compounds specifically interacted with G4 structures mainly as groove binders, while they were unable to firmly bind the DNA duplex. Finally, biological assays indicated Chelidonine and Rotenone as the most active anticancer compounds, with IC50 values of 0.64 and 0.15 μm, respectively, on BJ‐EHLT cancer cells, and also showing a good selectivity over normal BJ‐hTERT cells. [32]
Encouraged by these results and aiming at identifying even more effective and selective G4 ligands, ongoing studies, [74] including the G4‐CPG assay, additional biophysical techniques, molecular dynamics and biological tests, are focused on a set of structural analogues of the above five hit natural compounds, particularly including the natural alkaloids Canadine, D‐Glaucine and Dicentrine and the flavonoids Deguelin and Millettone (Library VII, Figure 13).[ 75 , 76 , 77 , 78 ]
Figure 13.

Chemical structures of Canadine, D‐Glaucine, Dicentrine, Deguelin and Millettone from Library VII, and of J10, the best G4 ligand from Library VIII.[ 33 , 74 ]
Finally, a set of in‐house natural compounds isolated from Juncaceae (Library VIII) was evaluated by the G4‐CPG assay, consisting of dihydrophenanthrene, benzocoumarin and dihydrodibenzoxepin derivatives named J1–J10. [33] Interestingly, we found that while natural dihydrophenanthrenes had low affinity for G4s (% of bound ligand=0–16 %), molecules based on benzocoumarin and dihydrodibenzoxepin scaffolds were relatively good binders (% of bound ligand=7–53 %), with one dihydrodibenzoxepin derivative, that is, J10 (Figure 13), emerging as the best G4 ligand in the series also showing no affinity for the duplex model. Its ability to bind G4 structures discriminating duplex DNA models was corroborated by CD and fluorescence data, which also confirmed the slight preference of J10 for parallel G4s (ΔTm=+8 °C, Kb∼106 m −1). A clue for the found G4 versus duplex selectivity was provided by molecular docking studies, which emphasized the ability of J10 to bind to the grooves of the target G4s. Finally, biological assays showed that J10 had a significant antiproliferative activity by inducing apoptosis on cancer cells, particularly showing IC50 value of ∼60 μm on human leukemia cell line (Jurkat) and no relevant effects on normal cells (healthy human fibroblasts, HDF). [33]
4. Overview of the Features of the Identified G4 Ligands
By analysis of both molecular modelling and experimental data acquired for all the above libraries the following outcomes can be inferred, which are helpful for the future design of novel synthetic or semi‐synthetic derivatives of organic/natural compounds as selective G4‐targeting ligands. In detail, it appeared that ligands featured by non‐extended aromatic moieties and overall bent conformations, such as those belonging to Library I–II–III–VI–VII–VIII, prefer to interact with the grooves and/or loops of the G4 targets by hydrophobic interactions and/or stacking with the loop nucleobases, and additionally by forming hydrogen bonds and/or electrostatic interactions with loop/groove residues. On the other hand, ligands endowed with a completely planar, extended aromatic core, such as NDIs of Library IV–V, showed stacking binding mode on the outer G‐quartets of monomeric G4s or at the interface between two consecutive G4 units of dimeric G4 models, with their pendant groups pointing either into the grooves or in the opposite direction, thus interacting with the target by hydrogen bonds and/or electrostatic interactions either with the grooves/loops or the flanking residues, respectively.
Generally, synthetic ligands exhibited higher affinity for the G4 targets than the natural compounds, whereas the latter ones showed slightly higher G4 versus duplex DNA selectivity. Among all the investigated series, NDIs emerged as the strongest G4 ligands, while the alkaloid, dihydrodibenzoxepin and furobenzoxazine naphthoquinone derivatives proved to be the most selective G4 ligands. Accordingly, NDIs were the most active G4 ligands among the investigated ones on cancer cells, even if also the other classes of studied compounds were found to be promising anticancer agents with mechanism of action well correlated to G4 targeting in cells.
5. Summary and Outlook
In the context of discovering new candidate drugs for anticancer therapies based on small molecules selectively targeting in vivo cancer‐related G‐quadruplex structures, we developed an affinity chromatography‐based assay (G4‐CPG assay) using a universal Controlled Pore Glass (CPG) support ad hoc prepared by us, on which both the solid‐phase synthesis of the G4‐forming oligonucleotides of interest and the screening of putative G4 ligands can be performed. The protocol of this assay was optimized in terms of: i) amount and functionalization of the solid support as well as concentration of the putative ligands to be incubated with, ii) volumes and composition of the used washing/releasing solutions, and iii) reduction of unspecific binding on the nude support, guaranteeing clean and reliable results as well as a high number of re‐using cycles of the G4‐functionalized supports.
Thus, the G4‐CPG assay was applied to the effective screening of different libraries of putative G4 ligands, including both synthetic, rationally designed molecules, and natural compounds and/or their semi‐synthetic analogues. Notably, the G4‐CPG assay associated with preliminary VS proved to be a successful HTS strategy to identify, even starting from very large libraries, new bioactive chemotypes able to target several G4 structures with high affinity and discriminate them over the duplex DNA. Then, the best G4 ligands, as determined by the G4‐CPG assay, can be evaluated for their anticancer activity by in vitro bioscreenings on different cancer cell lines and healthy control cells. Finally, or in parallel with the biological experiments, an in‐depth biophysical characterization in solution of the interaction of the most active, less toxic compounds with G4 models can be performed. This workflow proved to be successful in identifying several promising high‐affinity G4‐binders, starting from very different classes of synthetic or natural compounds, quickly and reliably selected as hit compounds selectively targeting cancer‐related G4 structures and representing starting points for the development of new anticancer lead compounds in a drug optimization process.
By using the G4‐CPG assay, not only small molecule‐based ligands can be studied in their interaction with G4‐forming sequences, but also additional potential interacting agents, such as other biological macromolecules. Indeed, in addition to the evaluation of novel classes of small molecules and analogues of the most promising ones selected thus far, ongoing studies in our laboratories are focused on the use of the G4‐CPG assay to identify G4‐binding proteins from nuclear extracts of different cancer and healthy cells. We indeed previously proved the feasibility of using the G4‐CPG assay also with mixtures of small molecules. However, using UV‐Vis analysis to monitor the binding event, the molecules in the mixture need to have different affinities for the G4 targets and UV‐Vis spectra with well‐separated absorption maxima, and thus the G4‐CPG assay could be applied only to specific mixtures of compounds. To overcome this drawback, we are now extending our binding detection system also to advanced mass spectrometry techniques, which can be profitably exploited to identify the isolated proteins by standard proteomic protocols.
More generally, the G4‐CPG assay allows the identification of all the different types of G4‐interacting agents currently searched in the G4 research field: i) ligands able to interact with all monomeric G4s, in a multi‐targeting approach, and discriminate them over the duplex DNA, ii) ligands interacting with a specific monomeric G4 topology, discriminating other G4 conformations as well as duplex DNA, and iii) ligands specifically interacting with the interface region of dimeric G4s, discriminating both monomeric G4s and duplex DNA.
Noteworthy, in addition to the above described G4‐forming sequences, that is, tel26, tel46, c‐myc, c‐kit1, c‐kit2 and hTERT, ongoing studies are directed to extend the assay to a large variety of biologically relevant DNA and/or RNA sequences, of human or viral origin. The goal is to obtain a large panel of support‐bound oligonucleotides – elongated on our CPG support simply using standard phosphoramidite chemistry on an automated DNA/RNA synthesizer – forming different biologically representative secondary structures. Accordingly, our G4‐CPG assay can be extended to any DNA/RNA sequence involved in the regulation of mechanisms related to a specific disease, aiming at discovering targeted drugs for the most various diseases. Finally, the method could be in principle easily automated to make the identification of putative candidate drugs even more rapid.
Conflict of interest
The authors declare no conflict of interest.
6.
Biographical Information
Chiara Platella is currently recipient of a postdoctoral fellowship granted by the Italian Association for Cancer Research (AIRC), under the supervision of Prof. Daniela Montesarchio. She graduated with honours in Chemical Sciences in 2015 and received her Ph.D. in Chemical Sciences in 2019 at Federico II University of Naples. During Ph.D. and post‐doc activities, she carried out part of her research at the Slovenian NMR Centre of Ljubljana, under the supervision of Prof. Janez Plavec. Her main interests are focused on the design, synthesis and evaluation of ligands of G‐quadruplex structures as potential drugs in targeted anticancer therapies.

Biographical Information
Ettore Napolitano obtained his degree with honors in Chemistry and Pharmaceutical Technologies at Federico II University of Naples in 2019 as student of the year. During his master thesis at the Institute of Protein Biochemistry (IBP) of the National Research Council (CNR), he learned the main methodologies employed in helicase biological studies, focusing on G‐quadruplex‐resolving helicases. He started his Ph.D. in Chemical Sciences in 2019 under the supervision of Prof. Daniela Montesarchio, focusing on the development of aptamer‐based biosensors for the early detection of tumours and inflammations. He is currently working on oligonucleotide aptamers labelled with fluorescent probes for theranostics.

Biographical Information
Claudia Riccardi is recipient of a postdoctoral fellowship granted by AIRC (Italian Association for Cancer Research) under the supervision of Prof. Daniela Montesarchio. She obtained her degree with honours in Chemistry and Pharmaceutical Technologies at Federico II University of Naples in 2013, then her Ph.D. in Chemical Sciences in 2017, and continued with fellowships granted by AIRC and Fondazione Umberto Veronesi. Her activities involved the design, synthesis and characterization of Ru(III)‐ and Pt(II)‐complexes and benzodifuran derivatives as anticancer compounds, as well as G‐quadruplex‐forming aptamers with anticancer or anticoagulant properties. She also studied nanosystems incorporating metal‐based drugs and/or aptamers for theranostics.

Biographical Information
Domenica Musumeci received her Ph.D. in Chemical Sciences in 2001 with a thesis on polyoxygenated steroids. From 2002 to 2009 she worked on peptide nucleic acids, nucleopeptides and oligonucleotide aptamers for the Italian National Research Council at IBB‐CNR. In 2012 she became Assistant Professor at the Department of Chemical Sciences (Federico II University, Naples), where from January 2021 she's Associate Professor of Organic Chemistry. Her main scientific interests deal with oligonucleotide aptamers, especially those adopting G‐quadruplex structures, for biomedical applications; nucleopeptides for nucleic acids recognition or self‐assembly (drug/gene delivery tools); potential anticancer compounds based on G‐quadruplex binders or Ru(III)/Pt(II) complexes.

Biographical Information
Daniela Montesarchio is Professor of Organic Chemistry at Federico II University of Naples, where she obtained her Ph.D. in Chemical Sciences in 1993. After a postdoctoral experience in McGill University, Montreal, she came back to Federico II University, where in 1994 she became Assistant Professor, in 2005 Associate Professor and in 2021 Full Professor. Her research interests are devoted to the design and synthesis of hybrid systems at the interface between chemistry and biology, including oligonucleotide, nucleoside analogues, aptamers, peptido‐ and glycomimetics, as well as metal‐based drugs [Ru(III) and Pt(II) complexes] for innovative therapeutic and/or diagnostic applications.

Acknowledgements
C. P. was supported by an AIRC fellowship for Italy. C. R. was supported by a fellowship from Fondazione Umberto Veronesi. D. M. thanks AIRC for financial support (IG2020 n. 25046).
C. Platella, E. Napolitano, C. Riccardi, D. Musumeci, D. Montesarchio, ChemistryOpen 2022, 11, e202200090.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Varshney D., Spiegel J., Zyner K., Tannahill D., Balasubramanian S., Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spiegel J., Adhikari S., Balasubramanian S., Trends Chem. 2020, 2, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hänsel-Hertsch R., Simeone A., Shea A., Hui W. W. I., Zyner K. G., Marsico G., Rueda O. M., Bruna A., Martin A., Zhang X., Adhikari S., Tannahill D., Caldas C., Balasubramanian S., Nat. Genet. 2020, 52, 878–883. [DOI] [PubMed] [Google Scholar]
- 4. Platella C., Riccardi C., Montesarchio D., Roviello G. N., Musumeci D., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1429–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collie G. W., Parkinson G. N., Chem. Soc. Rev. 2011, 40, 5867–5892. [DOI] [PubMed] [Google Scholar]
- 6. Włodarczyk A., Grzybowski P., Patkowski A., Dobek A., J. Phys. Chem. B 2005, 109, 3594–3605. [DOI] [PubMed] [Google Scholar]
- 7. Hardin C. C., Perry A. G., White K., Biopolymers 2000, 56, 147–194. [DOI] [PubMed] [Google Scholar]
- 8. Lee J. S., Nucleic Acids Res. 1990, 20, 6057–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cang X., Šponer J., Cheatham T. E., Nucleic Acids Res. 2011, 39, 4499–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. del Villar-Guerra R., Trent J. O., Chaires J. B., Angew. Chem. Int. Ed. 2018, 57, 7171–7175; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7289–7293. [Google Scholar]
- 11. Webba da Silva M., Trajkovski M., Sannohe Y., Hessari N. M., Sugiyama H., Plavec J., Angew. Chem. Int. Ed. 2009, 48, 9167–9170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9331–9334. [Google Scholar]
- 12. Wang Y., Patel D. J., J. Mol. Biol. 1995, 251, 76–94. [DOI] [PubMed] [Google Scholar]
- 13. Kang C., Zhang X., Ratliff R., Moyzis R., Rich A., Nature 1992, 356, 126–131. [DOI] [PubMed] [Google Scholar]
- 14. Petraccone L., Top. Curr. Chem. 2013, 330, 23–46. [DOI] [PubMed] [Google Scholar]
- 15. Petraccone L., Spink C., Trent J. O., Garbett N. C., Mekmaysy C. S., Giancola C., Chaires J. B., J. Am. Chem. Soc. 2011, 133, 20951–20961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moccia F., Riccardi C., Musumeci D., Leone S., Oliva R., Petraccone L., Montesarchio D., Nucleic Acids Res. 2019, 47, 8318–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riccardi C., Napolitano E., Musumeci D., Montesarchio D., Molecules 2020, 25, 5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.C. Platella, Towards DNA-targeting magic bullets: searching for potential conformation-selective G-quadruplex ligands, PhD Thesis 2018. University of Naples Federico II, Department of Chemical Sciences.
- 19. Biffi G., Di Antonio M., Tannahill D., Balasubramanian S., Nat. Chem. 2014, 6, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biffi G., Tannahill D., McCafferty J., Balasubramanian S., Nat. Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henderson A., Wu Y., Huang Y. C., Chavez E. A., Platt J., Johnson F. B., Brosh R. M., Sen D., Lansdorp P. M., Nucleic Acids Res. 2014, 42, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balasubramanian S., Hurley L. H., Neidle S., Nat. Rev. Drug Discovery 2011, 10, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han H., Hurley L. H., Trends Pharmacol. Sci. 2000, 21, 136–142. [DOI] [PubMed] [Google Scholar]
- 24. Rezler E. M., Bearss D. J., Hurley L. H., Curr. Opin. Pharmacol. 2002, 2, 415–423. [DOI] [PubMed] [Google Scholar]
- 25. Marchetti C., Zyner K. G., Ohnmacht S. A., Robson M., Haider S. M., Morton J. P., Marsico G., Vo T., Laughlin-Toth S., Ahmed A. A., Di Vita G., Pazitna I., Gunaratnam M., Besser R. J., Andrade A. C. G., Diocou S., Pike J. A., Tannahill D., Pedley R. B., Evans T. R. J., Wilson W. D., Balasubramanian S., Neidle S., J. Med. Chem. 2018, 61, 2500–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu H., Di Antonio M., McKinney S., Mathew V., Ho B., O'Neil N. J., Dos Santos N., Silvester J., Wei V., Garcia J., Kabee F., La D., Soriano P., Banáth J., Chiu D. S., Yap D., Le D. D., Ye F. B., Zhang A., Thu K., Soong J., Lin S.-C., Tsai A. H. C., Osako T., Algara T., Saunders D. N., Wong J., Xian J., Bally M. B., Brenton J. D., Brown G. W., Shah S. P., Cescon D., Mak T. W., Caldas C., Stirling P. C., Hieter P., Balasubramanian S., Aparicio S., Nat. Commun. 2017, 8, 14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendes E., Aljnadi I. M., Bahls B., Victor B. L., Paulo A., Pharmaceuticals 2022, 15, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trajkovski M., Morel E., Hamon F., Bombard S., Teulade-Fichou M. P., Plavec J., Chem. Eur. J. 2015, 21, 7798–7807. [DOI] [PubMed] [Google Scholar]
- 29. Kotar A., Wang B., Shivalingam A., Gonzalez-Garcia J., Vilar R., Plavec J., Angew. Chem. Int. Ed. 2016, 55, 12508–12511; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12696–12699. [Google Scholar]
- 30. Platella C., Trajkovski M., Doria F., Freccero M., Montesarchio D., Plavec J., Nucleic Acids Res. 2020, 48, 12380–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pagano B., Fotticchia I., De Tito S., Mattia C. A., Mayol L., Novellino E., Randazzo A., Giancola C., J. Nucleic Acids 2010, 2010, 247137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Platella C., Ghirga F., Zizza P., Pompili L., Marzano S., Pagano B., Quaglio D., Vergine V., Cammarone S., Botta B., Biroccio A., Mori M., Montesarchio D., Pharmaceutics 2021, 13, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Platella C., Capasso D., Riccardi C., Musumeci D., DellaGreca M., Montesarchio D., Org. Biomol. Chem. 2021, 19, 9953–9965. [DOI] [PubMed] [Google Scholar]
- 34. Carvalho J., Mergny J. L., Salgado G. F., Queiroz J. A., Cruz C., Trends Mol. Med. 2020, 26, 848–861. [DOI] [PubMed] [Google Scholar]
- 35. Local A., Zhang H., Benbatoul K. D., Folger P., Sheng X., Tsai C. Y., Howell S. B., Rice W. G., Mol. Cancer Ther. 2018, 17, 1177–1186. [DOI] [PubMed] [Google Scholar]
- 36. Schultz C. W., McCarthy G. A., Nerwal T., Nevler A., DuHadaway J. B., McCoy M. D., Jiang W., Brown S. Z., Goetz A., Jain A., Calvert V. S., Vishwakarma V., Wang D., Preet R., Cassel J., Summer R., Shaghaghi H., Pommier Y., Baechler S. A., Pishvaian M. J., Golan T., Yeo C. J., Petricoin E. F., Prendergast G. C., Salvino J., Singh P. K., Dixon D. A., Brody J. R., Mol. Cancer Ther. 2021, 20, 2166–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Musumeci D., Amato J., Randazzo A., Novellino E., Giancola C., Montesarchio D., Pagano B., Anal. Chem. 2014, 86, 4126–4130. [DOI] [PubMed] [Google Scholar]
- 38. Felsenstein K. M., Saunders L. B., Simmons J. K., Leon E., Calabrese D. R., Zhang S., Michalowski A., Gareiss P., Mock B. A., Schneekloth J. S., ACS Chem. Biol. 2016, 11, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaumot J., Gargallo R., Curr. Pharm. Des. 2012, 18, 1900–1916. [DOI] [PubMed] [Google Scholar]
- 40. Musumeci D., Amato J., Zizza P., Platella C., Cosconati S., Cingolani C., Biroccio A., Novellino E., Randazzo A., Giancola C., Pagano B., Montesarchio D., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1341–1352. [DOI] [PubMed] [Google Scholar]
- 41. Platella C., Musumeci D., Arciello A., Doria F., Freccero M., Randazzo A., Amato J., Pagano B., Montesarchio D., Anal. Chim. Acta 2018, 1030, 133–141. [DOI] [PubMed] [Google Scholar]
- 42. Martino L., Pagano B., Fotticchia I., Neidle S., Giancola C., J. Phys. Chem. B 2009, 113, 14779–14786. [DOI] [PubMed] [Google Scholar]
- 43. Monchaud D., Allain C., Bertrand H., Smargiasso N., Rosu F., Gabelica V., De Cian A., Mergny J. L., Teulade-Fichou M. P., Biochimie 2008, 90, 1207–1223. [DOI] [PubMed] [Google Scholar]
- 44. Salvati E., Leonetti C., Rizzo A., Scarsella M., Mottolese M., Galati R., Sperduti I., Stevens M. F. G., D'Incalci M., Blasco M., J. Clin. Invest. 2007, 117, 3236–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ward B., Rehfuss R., Goodisman J., Dabrowiak J. C., Biochemistry 1988, 27, 1198–1205. [DOI] [PubMed] [Google Scholar]
- 46. Ferreira R., Aviñó A., Mazzini S., Eritja R., Molecules 2012, 17, 7067–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng M. K., Modi C., Cookson J. C., Hutchinson I., Heald R. A., McCarroll A. J., Missailidis S., Tanious F., Wilson W. D., Mergny J. L., J. Med. Chem. 2008, 51, 963–975. [DOI] [PubMed] [Google Scholar]
- 48. Ren J., Chaires J. B., Biochemistry 1999, 38, 16067–75. [DOI] [PubMed] [Google Scholar]
- 49. Pagano B., Virno A., Mattia C. A., Mayol L., Randazzo A., Giancola C., Biochimie 2008, 90, 1224–1232. [DOI] [PubMed] [Google Scholar]
- 50. Cosconati S., Marinelli L., Trotta R., Virno A., Mayol L., Novellino E., Olson A. J., Randazzo A., J. Am. Chem. Soc. 2009, 131, 16336–16337. [DOI] [PubMed] [Google Scholar]
- 51. Saverio F., Leva D., Zizza P., Cingolani C., Angelo C. D., Pagano B., Amato J., Salvati E., Sissi C., Pinato O., J. Med. Chem. 2013, 56, 9646–9654. [DOI] [PubMed] [Google Scholar]
- 52. Cosconati S., Rizzo A., Trotta R., Pagano B., Iachettini S., De Tito S., Lauri I., Fotticchia I., Giustiniano M., Marinelli L., J. Med. Chem. 2012, 55, 9785–9792. [DOI] [PubMed] [Google Scholar]
- 53. Kuryavyi V., Phan A. T., Patel D. J., Nucleic Acids Res. 2010, 38, 6757–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Drew H. R., Wing R. M., Takano T., Broka C., Tanaka S., Itakura K., Dickerson R. E., Proc. Nat. Acad. Sci. 1981, 78, 2179–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Platella C., Musumeci D., Amato J., Randazzo A., Pagano B., Montesarchio D., in Method for the Preparation of a Low Unspecific Binding-Support for Affinity Chromatography and/or on-Line Synthesis of Oligonucleotides, 2018, EP 3 378 556 A1. [Google Scholar]
- 56. Platella C., Guida S., Bonmassar L., Aquino A., Bonmassar E., Ravagnan G., Montesarchio D., Roviello G. N., Musumeci D., Fuggetta M. P., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2843–2851. [DOI] [PubMed] [Google Scholar]
- 57. Platella C., Raucci U., Rega N., D'Atri S., Levati L., Roviello G. N., Fuggetta M. P., Musumeci D., Montesarchio D., Int. J. Biol. Macromol. 2020, 151, 1163–1172. [DOI] [PubMed] [Google Scholar]
- 58. Zuffo M., Doria F., Botti S., Bergamaschi G., Freccero M., Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1303–1311. [DOI] [PubMed] [Google Scholar]
- 59. Platella C., Gaglione R., Napolitano E., Arciello A., Pirota V., Doria F., Musumeci D., Montesarchio D., Int. J. Mol. Sci. 2021, 22, 10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Amato J., Platella C., Iachettini S., Zizza P., Musumeci D., Cosconati S., Pagano A., Novellino E., Biroccio A., Randazzo A., Pagano B., Montesarchio D., Eur. J. Med. Chem. 2019, 163, 295–306. [DOI] [PubMed] [Google Scholar]
- 61. Pagano B., Amato J., Iaccarino N., Cingolani C., Zizza P., Biroccio A., Novellino E., Randazzo A., ChemMedChem 2015, 10, 640–649. [DOI] [PubMed] [Google Scholar]
- 62. Simonsson T., Pribylova M., Vorlickova M., Biochem. Biophys. Res. Commun. 2000, 278, 158–166. [DOI] [PubMed] [Google Scholar]
- 63. Sun D., Hurley L. H., J. Med. Chem. 2009, 52, 2863–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Palumbo S. M. L., Ebbinghaus S. W., Hurley L. H., J. Am. Chem. Soc. 2009, 131, 10878–10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chaires J. B., Trent J. O., Gray R. D., Dean W. L., Buscaglia R., Thomas S. D., Miller D. M., PLoS One 2014, 9, e115580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Platella C., Pirota V., Musumeci D., Rizzi F., Iachettini S., Zizza P., Biroccio A., Freccero M., Montesarchio D., Doria F., Int. J. Mol. Sci. 2020, 21, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pirota V., Platella C., Musumeci D., Benassi A., Amato J., Pagano B., Colombo G., Freccero M., Doria F., Montesarchio D., Int. J. Biol. Macromol. 2020, 166, 1320–1334. [DOI] [PubMed] [Google Scholar]
- 68. Platella C., Napolitano E., Riccardi C., Musumeci D., Montesarchio D., J. Med. Chem. 2021, 64, 3578–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou C. Q., Liao T. C., Li Z. Q., Gonzalez-Garcia J., Reynolds M., Zou M., Vilar R., Chem. Eur. J. 2017, 23, 4713–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Petraccone L., Trent J. O., Chaires J. B., J. Am. Chem. Soc. 2008, 130, 16530–16532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Petraccone L., Garbett N., Chaires J. B., Trent J. O., Biopolymers 2010, 93, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cummaro A., Fotticchia I., Franceschin M., Giancola C., Petraccone L., Biochimie 2011, 93, 1392–1400. [DOI] [PubMed] [Google Scholar]
- 73. Platella C., Mazzini S., Napolitano E., Mattio L. M., Beretta G. L., Zaffaroni N., Pinto A., Montesarchio D., Dallavalle S., Chem. Eur. J. 2021, 27, 8832–8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.C. Platella et al. Unpublished results.
- 75. Correché E. R., Andujar S. A., Kurdelas R. R., Lechón M. J. G., Freile M. L., Enriz R. D., Bioorg. Med. Chem. 2008, 16, 3641–3651. [DOI] [PubMed] [Google Scholar]
- 76. Kang H., Jang S. W., Pak J. H., Shim S., Mol. Cell. Biochem. 2015, 403, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu Y., Zhang H., Jin X., Xiang J. F., Tang Y. L., Chinese J. Pharmacol. Toxicol. 2011, 25, 543–546. [Google Scholar]
- 78. Ito C., Itoigawa M., Kojima N., Tan H. T. W., Takayasu J., Tokuda H., Nishino H., Furukawa H., Planta Med. 2004, 70, 585–588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.