

Abstract
Background and Objectives
Microglia, the resident immune cell of the brain and retina, is widespread activated in the white and gray matter (GM) in multiple sclerosis (MS). The objective of this study is to evaluate the presence and number of hyperreflecting foci (HRF), considered clusters of activated and proliferating retinal microglia, and their association with clinical and radiologic disease parameters in relapsing-remitting MS (RRMS).
Methods
At baseline, 80 patients with RRMS underwent optical coherence tomography (OCT) and 3T-MRI (including 3-dimensional T1, fluid-attenuated inversion recovery, and double inversion recovery sequences), closed to their disease onset (6.3 ± 5.1 months). These patients were then clinically and radiologically followed up for a mean of 43 months, evaluating the no evidence of disease activity (NEDA) condition, further divided into clinical (cNEDA) and radiologic (rNEDA). Patients with a clinical history or MRI/OCT findings suggestive of optic neuritis (ON) were excluded from the study.
Results
Compared with healthy controls, the HRF number was significantly higher in the inner nuclear layer (INL) of patients with RRMS (19.55 ± 5.65 vs 13.84 ± 2.57, p < 0.001) and associated with INL volume (β: 1.21, p < 0.001). GM lesion volume significantly correlated with the INL HRF count (p = 0.008). Survival analysis revealed a significant association between INL HRF and both cNEDA (p = 0.017) and rNEDA (p = 0.002).
Discussion
We found a strong association between retinal microglial proliferation and cortical pathology in RRMS, a finding suggesting a possible underlying common immunopathologic mechanism. Furthermore, microglial activation at baseline was observed to predict subsequent inflammatory events, indicating that HRF might be a candidate prognostic biomarker worthy of further investigation.
Classification of Evidence
This study provides Class II evidence that in patients with early RRMS but without ON, the number of HRF on OCT of the retinal inner nuclear layer is associated with cNEDA and rNEDA.
Microglia, the CNS-resident antigen-presenting cells, is increasingly recognized as a key player in the pathogenesis of inflammatory and degenerative diseases.1,2 Evidence suggests that microglia is widespread activated in multiple sclerosis (MS), both in the white matter (WM) and in the gray matter (GM), not only in focal inflammatory lesions but also in the so-called normal-appearing WM and GM.3-6 Indeed, activated microglia is thought to play a pivotal role in CNS inflammation by orchestrating a complex pattern of local modifications, involving all surrounding immune (infiltrating) and glial cells. These include antigen presentation to T cells, release of proinflammatory cytokines in active lesions, increased major histocompatibility complex (MHC)-II and costimulatory molecule expression, T-cell activation, and astrocyte proliferation.7 Moreover, locally released cytokines can induce neuronal changes, including increased MHC-I molecule expression and oxidative stress–induced cell damage. All these modifications reshape brain tissue into an ideal setting for self-reactive B- and T-cell proinflammatory functions.8
Recent in vivo observations have disclosed the presence of high-density activated microglia in the MS GM9 and in the so-called WM slowly enlarging lesions, suggesting a pivotal role of these cells in the so-called silent disability progression.10,11 In addition, postmortem histology provided evidence that microglia can be clustered in 2 different phenotypes. The first, characterized by a high cellular density and elevated expression of both human leukocyte antigen Class II and CD68, is spatially associated with neural cell bodies, whereas the second, having a hyperramified morphology and low P2Y12 expression, is associated with the presence of B cells in the adjacent meninges and significant neural loss.12 These findings suggest that microglial cells play a detrimental role in both acute and chronic immunopathologic processes taking place in the MS brain.
Expanding evidence indicates that the retina can be considered the ideal CNS site for investigation of inflammatory-induced neurodegenerative damage.13 Indeed, the use of optical coherence tomography (OCT) has disclosed single retinal layer degenerative modifications following optic pathway inflammation.14,15 Silent retinal damage driven by activated microglia was hypothesized, and retinal microglia, visualized by OCT scan as hyperreflecting foci (HRF) in the inner retina, was found to be associated with the presence of clinical or radiologic MS activity.16 Moreover, recent in vivo data from myelin oligodendrocyte glycoprotein 35-55 experimental autoimmune encephalomyelitis showed a strong and dynamic microglial activation following optic neuritis (ON).17
The presence of HRF has been reported in a range of retinal and neuro-ophthalmologic diseases; however, the underlying pathology is still a matter of debate because at present no histologic or animal retinal findings are available. Different hypotheses have been proposed, encompassing lipid extravasation acting as a precursor of hard exudates,18 degenerated photoreceptor cells,19 or migrating retinal pigment epithelial cells.20 However, one of the most accredited hypotheses suggests that HRF having specific morphologic features are composed by clusters of activated and proliferating microglia.21 Indeed, in the eyes of patients with diabetes, HRF are initially observed in the inner retina, where microglia is resident.21 With the development of retinal disease and diabetic macular edema, HRF further increase in number and spread toward middle and outer retinal layers, thus mimicking microglial activation and migration from the inner to outer retina described in histopathologic studies.22 Moreover, several observations indicate that microglia, together with Müller cells, drives the inflammatory processes taking place in the retina.23 Finally, a moderate association with HRF and inflammatory markers (i.e., interleukin [IL]-8 and vascular cell adhesion molecule 1) in the aqueous humor has been described.24 Recently, HRF have been reported within the inner retinal layers (IRLs) of patients with Fabry disease.25 In this study, HRF correlated with lyso-Gb3, a serum biomarker of disease severity. These findings suggested that the accumulation of products of degradation of glycosphingolipids and the concomitant capillary dysfunction might appear as pathologically hyperreflective capillary plexus of the inner retina.
To what extend retinal microglia can be considered a biomarker of local pathology or the remote mirror of brain WM or GM inflammation merits investigation. Thus, we designed a longitudinal study on patients with early-onset MS, in which OCT parameters of microstructural peripapillary and macular damage and the number of HRF (proliferating microglia), were correlated with MRI parameters of WM and GM inflammation and deep and cortical GM atrophy. To exclude the possible effect of optic nerve inflammation on retinal microglial activation,17 we excluded subclinical ON, applying both OCT and MRI. Moreover, the putative prognostic role of retinal parameters was explored following clinically and radiologically the patients for a median period of 43 months.
Primary Research Questions Being Addressed in the Study
(1) To evaluate associations between HRF count and both OCT and MRI parameters of inflammatory disease in patients with MS and (2) to evaluate HRF role as a prognostic factor in patients with MS.
Methods
Study Design and Participants
Eighty patients with a diagnosis of relapsing-remitting MS (RRMS) achieved at the Multiple Sclerosis Centre, University Hospital of Padua, between January 2014 and June 2020 were recruited in this prospective single-center study. Each patient underwent neurologic examination, lumbar puncture, OCT, and brain and spinal cord MRIs at the time of the diagnosis. Inclusion criteria were the following: (1) diagnosis of RRMS achieved according to the revised 2017 McDonald criteria26; (2) disease duration (defined in this study as the interval between the first clinical symptom attributable to MS and the date of the OCT evaluation) <18 months; and (3) age between 18 and 60 years. Exclusion criteria were the following: (1) systemic and ophthalmologic disorders (i.e., diabetes); (2) diagnosis of progressive MS; (3) steroid therapy in the month before OCT acquisition; (4) previous clinical history of ON; and (5) evidence of subclinical ON (intereye difference in peripapillary retinal nerve fiber layer [RNFL] of >20% and/or optic nerve hyperintensity in ≥2 slices at brain MRI double inversion recovery [DIR] sequence).27
Spectral-domain OCT images of 38 age- and sex-matched healthy volunteers (healthy controls, HCs) without any neurologic or ophthalmologic disease were also examined (HC-OCT). Written informed consent was obtained from each patient and HC.
Spectral-Domain OCT
At baseline, all patients with MS and HCs underwent spectral-domain OCT (Spectralis; Heidelberg Engineering, Carlsbad, CA; Heidelberg Eye Explorer version 1.7.0.0) examination of both eyes in a dark room without the injection of any mydriatic agent. One macular map 6 × 6 mm and 1 peripapillary 3.5-mm ring scan were acquired by 2 operators. The macular map scan, centered on the fovea, consisted of 25 vertical 180° SD-OCT linear scans in the High Resolution Mode (Automatic Real-Time was settled at 49 frames averaged per B-scan). The retinal layering was obtained using the automatic layering of the Spectralis SD-OCT. An early treatment diabetic retinopathy study grid centered onto the fovea subdivided the macular area in 9 parts, according to the incorporated Spectralis software, consisting of a central circular zone with a 1-mm diameter and inner and outer rings of 3- and 6-mm diameter, respectively. The internal and external rings were subdivided into 4 quadrants (superior, inferior, temporal, and nasal). Automatic segmentation software (segmentation technology; Heidelberg Engineering, version 6.3.1.0) was used to identify and measure the volume (for each layer) and thickness (of each quadrant). After the automated segmentation process, all scans and all layers were carefully revised for algorithm failure. The algorithm detects 11 separation markers. The following retinal slabs, automatically obtained by the incorporated algorithm, were measured: full retina (from the internal limiting membrane to Bruch membrane); RNFL; ganglion cell layer (GCL); inner plexiform layer (IPL); inner nuclear layer (INL); outer plexiform layer; outer nuclear layer;; and retinal pigment epithelium. The GCL and IPL were then unified in a unique layer (GCIP). The peripapillary 3.4-mm ring scan was used to measure mean global peripapillary RNFL and mean sectorial peripapillary RNFL thickness (temporal RNFL; supero-temporal RNFL; supero-nasal RNFL; nasal RNFL; infero-nasal RNFL; infero-temporal RNFL). Moreover, mean thickness of the papillomacular bundle (PMB-RNFL) and relationship between the thickness at the nasal level and that of the temporal level were measured. The ring scan was manually superimposed to the optic nerve head. Automatic Real Time was settled at 100 frames. All examinations were checked for sufficient quality using the OSCAR-IB criteria.28 The results are reported in line with the Advised Protocol for OCT Study Terminology and Elements.29
Hyperreflective Intraretinal Foci
In line with recent publications,16,30 the analysis of the central linear scan of the macular map, crossing the fovea, was considered for HRF counting. HRF were counted in the area included between 2 perpendicular lines to Bruch membrane traced at 1,500 μm both temporally and nasally from the center of the fovea. HRF were defined as isolated, small-size (<30 μm), punctiform elements with moderate reflectivity (similar to that of the nerve fiber layer) but without any back shadowing. The count was performed in the GCIP and INL separately; results were reported also as inner retinal HRF count (i.e., GCIP HRF count + INL HRF count). The presence of HRF was rated by 2 independent blind observers (M.P. and T.T.). Figure 1 summarizes the HRF count protocol. The intergrader agreement was at least substantial for all measurements (intraclass correlation coefficient, 0.84). In case of a mismatch between the 2 observers, a consensus was always obtained.
Figure 1. HRF Protocol.

Macular scans and HRF visualization in RRMS (upper image) and HC (lower image); INL foci are indicated by yellow arrows and ganglion cell and inner plexiform layer (GCIP) HRF by blue arrows. HRF were counted in the area included between 2 perpendicular lines to Bruch membrane traced at 1,500 μm both temporally and nasally from the center of the fovea. HRF were defined as isolated, small size (<30 μm), punctiform elements with moderate reflectivity but without any back shadowing. HC = healthy control; HRF = hyperreflecting foci; INL = inner nuclear layer; RRMS = relapsing-remitting multiple sclerosis.
MRI Data Acquisition
MRI was achieved on a 3.0 T scanner (Ingenia, Philips Medical Systems, Best, the Netherlands) with 33-mT/m power gradient and a 32-channel head coil. No major hardware upgrades occurred during the study and bimonthly quality assurance sessions for measurement stability. The MRI protocol included the following sequences: (1) 3-dimensional (3D) T1 MPRAGE: repetition time (RT) = 7.8 milliseconds, echo time (ET) = 3.6 milliseconds; 180 contiguous axial slices with the off-center positioned on zero with thickness of 1.0 mm; flip angle = 8°; matrix size = 220 × 220; field of view (FOV) 220 × 220 × 180 mm3; (2) 3D-fluid-attenuated inversion recovery (FLAIR): RT = 4,800 milliseconds, ET = 310 milliseconds, inversion time (IT = 1,650 milliseconds; 365 contiguous axial slices with thickness of 1.0 mm; matrix size 256 × 256; and FOV = 256 × 256 × 182 mm3; (3) 3D-DIR: RT = 55,000 milliseconds, ET = 284 milliseconds, IT = 2,550 milliseconds; contiguous axial slices with thickness of 1.0 mm; matrix size 212 × 212; and FOV = 256 × 256 × 182 mm3.
MRI Data Processing
T1-weighted images were processed following these procedures: correction for magnetic field inhomogeneity, performed with advanced normalization tools (ANTs) N4 Bias Field Correction tool,31 brain extraction, performed with ANTs Brain Extraction tool,32 brain segmentation, performed with FSL FAST33 tool, and lesion filling, performed with FSL lesion-filling tool.34 The FLAIR images were processed following these steps: correction for magnetic field inhomogeneity, performed with ANTs N4BiasFieldCorrection31 tool, and brain extraction, performed with FSL BET tool.35 The processed FLAIR image was then registered to the processed T1-weighted image using ANTs Registration SyN Quick32 tool, and the estimated transformation was applied to the WM lesion mask. DIR images were corrected for magnetic field inhomogeneity with ANTs N4BiasFieldCorrection31 tool and was then registered to the processed T1-weighted image using FSL flirt tool.36 Thus, the GM lesion mask, designed in the DIR space, was registered to the same space applying the transformation estimated in the previous step. To evaluate the role of the WM lesions on the optic radiations (ORs), we used a previously described atlas,37 which includes the tracts of the OR, defined in the MNI152 space. To conduct a subject-level analysis, the MNI152 brain image was registered to the processed T1-weighted image as described elsewhere.38 After having applied the estimated transformations to the OR tracts, an expert neuroradiologist fixed the lower thresholds for the left OR tract to 0.7 and for the right OR to 0.55. Then, we quantified global and normalized WM and GM lesion volume (WMLV and GMLV) as well as WMLV and their normalized values in both ORs.
Clinical and Radiologic Follow-up
All patients were followed up clinically (neurologic examination and Expanded Disability Status Scale [EDSS], done every 6 months) and radiologically (brain MRI). Brain MRI was performed after 6 months (including postcontrast T1 sequence) and then annually or in the case of clinical relapse (also in this last event with postcontrast T1 sequence). The following clinical and radiologic variables were taken into account during the follow-up: (1) disease-modifying therapies (DMTs, divided into first- and second-line DMTs), (2) time to switching from first- to second-line treatment because of evidence of disease activity, (3) clinical relapses (namely, time to first clinical relapses and annual relapse rate), (4) new T2/FLAIR lesions, and (5) gadolinium-enhancing lesions (namely, number and time to). In the event of a relapse, defined as the occurrence of new symptoms or exacerbation of existing symptoms that lasted for 24 hours or longer, in the absence of concurrent illness or fever, and occurring 30 days or more after a previous relapse, a further clinical evaluation was performed. Clinical no evidence of disease activity (cNEDA) was defined as the absence of relapses and sustained progression of disability (1-step EDSS progression for EDSS ≤5.5 or 0.5-step EDSS progression for EDSS >5.5 confirmed at 2 consecutive examinations done every 6 months over a period of 12 months). Radiologic NEDA (rNEDA) was defined as the absence of new gadolinium-enhancing lesion and new-enlarging WM lesion. Finally, the NEDA condition was defined as the concomitance of both cNEDA and rNEDA.
Standard Protocol Approvals, Registrations, and Patient Consents
The study was conducted in agreement with the Declaration of Helsinki and approved by the local Ethic Committee (Comitato Etico per la Sperimentazione Clinica dell’Azienda Ospedaliera di Padova, prot.n. 17760). Written informed consent was obtained from each patient and HC.
Statistical Analysis
Data were reported as mean (±SD) or median (range) for continuous variables and number (percentage) for categorical variables. Group differences between patients with RRMS and OCT HCs were tested using the χ2 test for sex and the t test for age. Generalized estimating equation (GEE) models were performed to take into account the correlation between observations from the same patient (right and left eyes). In the GEE models, the dependent variables were assumed to be Gaussian or Poisson according to the nature of the variables, and an unstructured correlation matrix was used as correlation structure. As the primary outcome, we investigated the association of the layers with the HRF count, and the association of each specific quadrant with the HRF count was analyzed as the secondary outcome.
The Kaplan-Meier method was used to estimate the survival curve for time to first relapse or MRI modification; Cox-regression analyses were used to assess the influence of HRF on the outcome's measures during follow-up. We found cutoff values of HRF using time-dependent receiver operating characteristic (ROC) curves with the Liu approach, and stability was guaranteed by a 100-times bootstrap resampling with replacement; we compared survival curves using the log-rank test. A p value of less than 0.05 was considered statistically significant. Stata 15.1 (StataCorp., College Station, TX, 2017) was used for all the analysis.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Baseline Characteristics of the Study Population
At baseline, the 80 patients with RRMS enrolled in the study had a median EDSS score of 2.0 (range 1.0–4.0) and a mean disease duration of 6.3 ± 5.1 months (range 0–18). Notably, 51 patients (63.8%) had a disease duration <6 months and 69 (86.3%) <12 months. OCT data were obtained from all patients, of whom 86.3% (69) were treatment naive, and 11 (14.7%) had been treated for a very short time with DMT (mean of 4.5 months; 7 with first-line and 4 with second-line DMTs). The signal strength in macular scan of 3 eyes from 2 patients with MS was not optimal (<15), and thus, scans were not further considered in the analysis, whereas signal strength in peripapillary scans of 15 eyes from 15 patients with MS and 3 eyes from 3 HCs was discarded for the same reason. MRI data were collected from 72 (90%) patients. Clinical and demographic characteristics of patients and HCs are summarized in Table 1.
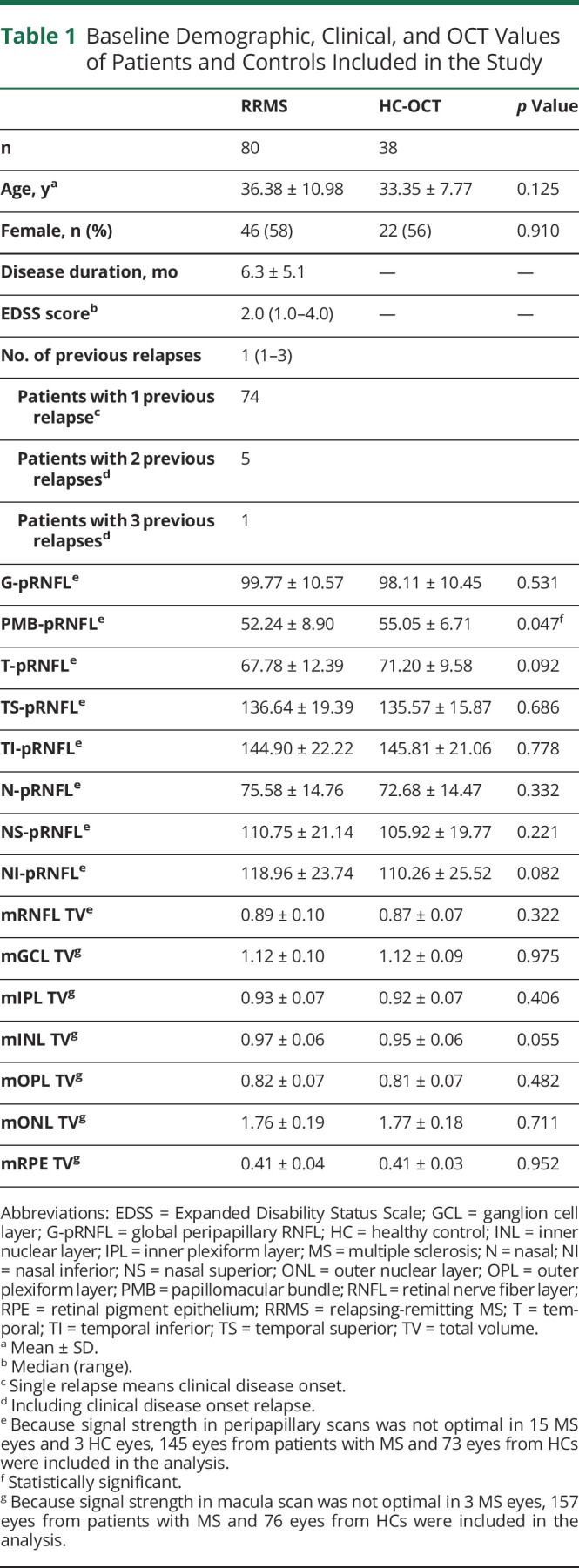
Table 1.
Baseline Demographic, Clinical, and OCT Values of Patients and Controls Included in the Study
HRF Are Increased in the INL of Patients With MS and Associate With INL Volume
The exclusion of subclinical and clinical ON was further confirmed by the absence of a relevant difference in all morphologic OCT parameters between RRMS and HC. Indeed, in the peripapillary scan, only PMB was slightly reduced in patients with RRMS compared with HCs (52.24 ± 8.9 μm vs 55.05 ± 6.71 μm, p = 0.047) (Table 1), whereas among macular volume parameters, only INL total volume was slightly increased in RRMS compared with HC (0.97 ± 0.06 mm3 vs 0.95 ± 0.06 mm3, p = 0.055) (Figure 1B). Peripapillary RNFL thicknesses and macular volumes are shown in Table 1 and further detailed in eTable 1 (links.lww.com/NXI/A721).
Even in the absence of both layer volume and thickness modifications, the HRF count was significantly higher in the INL of patients with RRMS (19.55 ± 5.65) than in HCs (13.84 ± 2.57, p < 0.001) (Figure 2A). Moreover, INL volume was associated with the HRF count in the INL (β: 1.21, p < 0.001) (Figure 2B, Table 2). On the other hand, the GCIP HRF count did not differ between HC and RRMS (Figure 1C), and no association between HRF GCIP count and GCIP volume was observed in RRMS (Figure 1D). In addition, the IRL HRF count was increased in RRMS compared with HC (29.55 ± 7.50 vs 23.07 ± 3.66, p < 0.001) and was associated with IRL volume (β: 1.04, p < 0.001) (Table 2). The association between HRF INL count and INL volume was confirmed in all sectors (both inner and outer ring sectors), but temporal outer ring (eTable 2, links.lww.com/NXI/A721).
Figure 2. HRF Are Increased in the INL of Patients With RRMS and Associate With Macular INL Volume.

(A) HRF were significantly increased in the INL of RRMS. (B) INL HRF count associated with INL volume (volumes were clustered in quartiles, from lowest values, Q1, to highest, Q4). (C) The HRF count in the GCIP did not differ between RRMS and HC and (D) was not associated with GCIP volume. Each dot represents 1 eye; however, the shown p values refer to the analysis with generalized estimating equation models (see Statistical Analysis in Methods). HC = healthy control; HRF = hyperreflecting foci; INL = inner nuclear layer; OCT = optical coherence tomography; RRMS = relapsing-remitting multiple sclerosis.
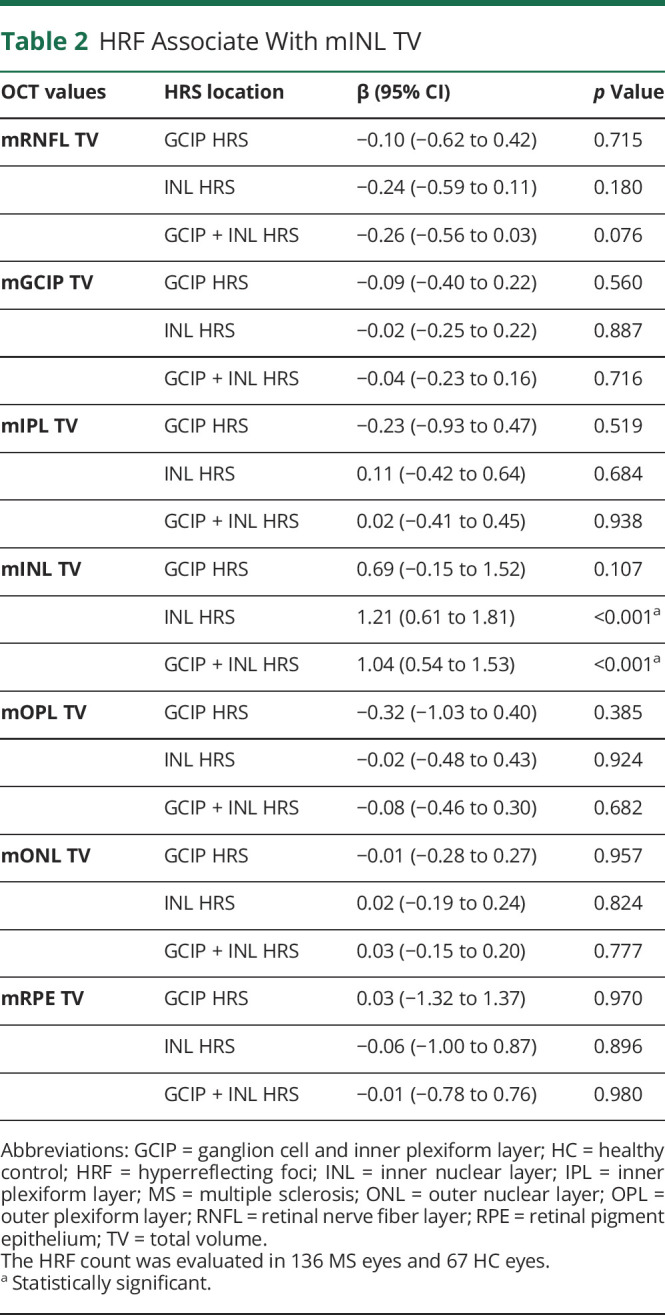
Table 2.
HRF Associate With mINL TV
HRF Associate With Radiologically Active Disease Activity at Baseline
To further investigate HRF relevance, we explored their associations with brain MRI parameters of inflammation or degeneration (eFigure 1, links.lww.com/NXI/A721). Inasmuch as most of our patients had only a single clinical episode, no association was found between number relapse and HRF count (HRF count in the INL: p = 0.9; HRF count in the GCIP: p = 0.2). However, gadolinium-enhancing lesions in the brain associated with a higher INL HRF count (18.7 ± 5.4 vs 21.8 ± 5.8, p = 0.026), but not with the GCIP HRF count (9.7 ± 2.9 vs 10.8 ± 3.2, p = 0.103) (Figure 3A), whereas the presence of gadolinium-enhancing lesions in the spinal cord did not (INL, GCIP, and IRL HRF: p = 0.812, p = 0.627, and p = 0.948, respectively).
Figure 3. HRF Associated With Brain Inflammatory Parameters.

(A) Both IRL and INL HRF count are increased in the presence of brain gadolinium-enhancing lesion. (B) Both INL and IRL HRF count associated with GMLV. Each dot represents 1 eye; however, the shown p values refer to the analysis with generalized estimating equation models (see Statistical Analysis in Methods). GMLV = gray matter lesion volume; HRF = hyperreflecting foci; INL = inner nuclear layer; IRL = inner retinal layer; MS = multiple sclerosis.
WMLV and global cortical thickness were not associated with the HRF count in the GCIP (p values 0.431 and 0.359, respectively), INL (p values 0.148 and 0.362, respectively), and IRL (p values 0.107 and 0.226, respectively). Cortical lesions were found to be associated with the IRL HRF count (27.5 ± 5.7 vs 30.9 ± 9.1, p = 0.021) and mildly with the GCIP and INL HRF count (p value: 0.096 and 0.084, respectively). In line with these findings, GMLV correlated significantly with the INL HRF count (p = 0.008) and IRL HRF count (0.001), but weakly with the GCIP (p = 0.053) (Figure 3B).
HRF count in the GCIP correlated inversely with the volume of lateral geniculate nucleus (LGN) (β: −0.002, p = 0.003). The associations between HRF count and thalamic volume, LGN volume, and cortical precentral and pericalcarine thickness, as well as with OR WMLV, are detailed in eTable 2 (links.lww.com/NXI/A721).
HRF Associated With the Radiologic Activity During the Follow-up
Longitudinal data were available from 79 (98.8%) patients that were followed for 43 ± 25 months. Treatments are reported in eTable 3 (links.lww.com/NXI/A721). Survival analysis revealed that cNEDA was not associated with the HRF count (Figure 4A), whereas rNEDA associated with both INL and IRL HRF count (p values: 0.002 and 0.024, respectively) (Figure 4B and eFigure 2). Finally, only INL HRF count associated with NEDA (p = 0.017) (Figure 4C). The predictive value of HRF was explored using time-dependent ROC curves with the Liu approach, identifying a cutoff value of 32 for IRL HRF. Both radiologic NEDA (p = 0.0072) and NEDA (p = 0.0155) significantly differ in line with this cutoff (eFigure 2).
Figure 4. INL HRF Count Associated With NEDA Condition.
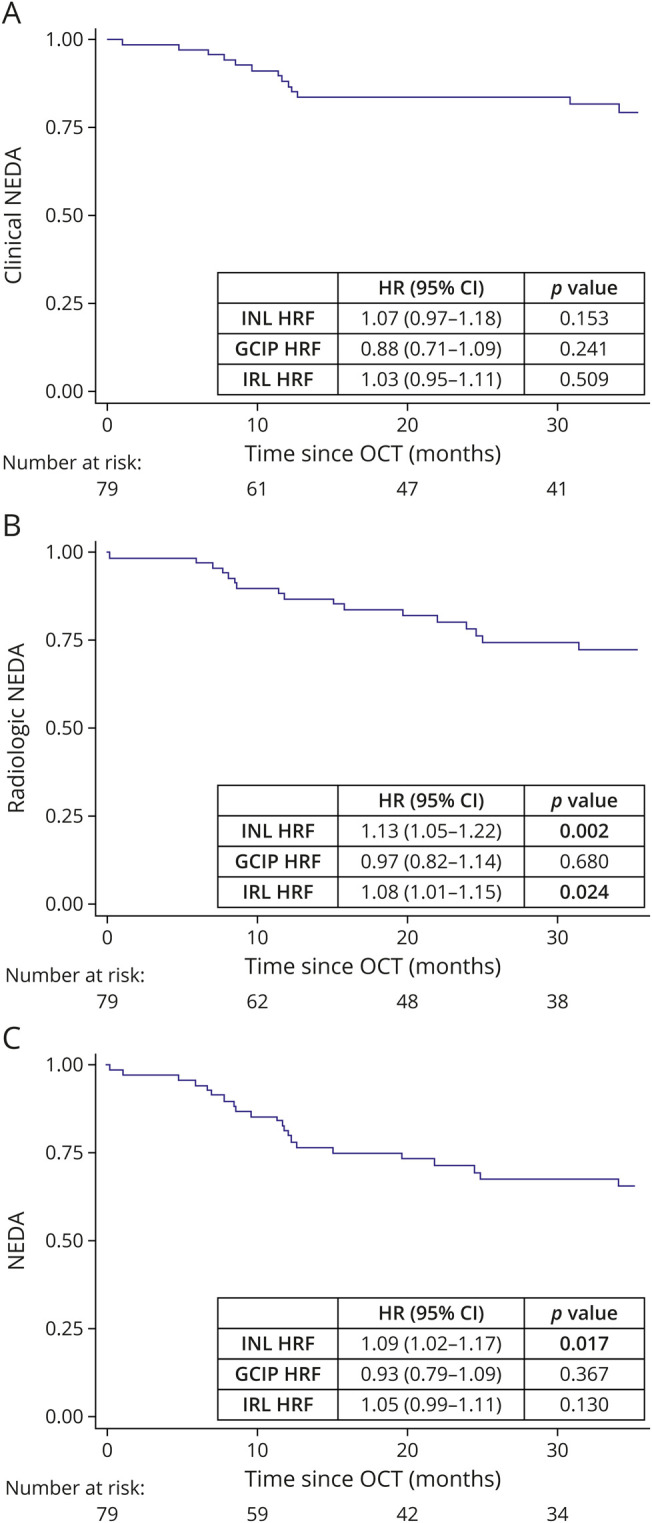
(A) Clinical NEDA was not associated with the HRF count. (B) Radiologic NEDA associated with both INL and IRL HRF count. (C) NEDA condition associated with the INL HRF count. HRF = hyperreflecting foci; INL = inner nuclear layer; IRL = inner retinal layer; NEDA = no evidence of disease activity; OCT = optical coherence tomography.
Discussion
Microglia probably plays complex and multifaceted roles in MS-related pathology, as indicated by their presence throughout all stages of lesion formation and evolution.9 Indeed, it is found at the border of slowly expanding lesions and are diffusely proliferating throughout the cortex, where associate with synaptic loss.39 Many recently discovered MS risk genes are highly expressed in microglia.40 In the context of WM lesions, microglia shows specific transcriptome profiles, which partially overlap with those observed in other neurodegenerative disorders. Moreover, on the base of gene expression in inflammatory lesions, microglia can be clustered in 2 different subsets, reflecting probably different effector functions.11 Thus, the elucidation of microglial proliferation and activation in vivo may help to clarify its role as driver of CNS inflammation.
The retina shares many structural and functional features with the CNS. Indeed, the retina has a blood-retina barrier (BRB), which acts similarly to the blood-brain barrier and makes retinal microenvironment a specific interstitial space in terms of soluble molecules and ions. Moreover, the immunologic trafficking into and out from the retina parallels CNS dynamics, suggesting the presence of a retinal glymphatic system, prone to react to systemic or local pathologic processes.41 This reactivity might explain the modification of retinal layer thickness during CNS inflammation or therapies acting on the BRB24,42 and the effect of disease-modifying therapies in reducing INL thickness.38 The recognition of the INL as an inflammatory site supports the view of its thickening as inflammatory lesion.43,44
We have previously described an association between increased HRF count in the inner retina and evidence of disease activity.16 This observation indicated the need to investigate the relationship between HRF and structural changes of retinal layers in early phases of RRMS with no evidence of clinical or subclinical ON, in agreement with a recently proposed protocol.27 Indeed, acute ON is followed by retrograde degeneration that induces significant retinal inner layer modification, including INL thinning. Because no data exist on HRF during or following ON, we excluded the possibility of an increased HRF count in the context of the so-called retrograde maculopathy.45 In MS, the identification of asymptomatic ON is highly debated. A cutoff of 5 μm for pRNFL was established in a large cohort of RRMS.46 However, it has to be pointed out that retinal atrophy might not be determined by ON exclusively. Indeed, transsynaptic degeneration can induce pRNFL thinning14,15; in addition, primary retinal pathology might be an independent factor of retinal atrophy. In line with this consideration, the above-mentioned cutoff had a moderate specificity (65%–75%)46,47 and positive predictive value (53%–72%).46,47 Also in our cohort, this cutoff would have excluded 23% of HC and 19% of RRMS (specificity 77%, positive predictive value 62%); therefore, we choose a more conservative cutoff.47 Moreover, no relevant difference between morphologic OCT parameters was observed between RRMS and HC, and no pathologic optic nerve signal in DIR sequences was observed.
We found that, even in the presence of normal macular INL volume, HRF accumulate in the INL of RRMS and their count strongly and directly associates with INL volume, thus further confirming HRF inflammatory pathogenesis (as foci of activated microglia). Because the HRF count in the GCIP did not associate with GCIP volume, microglial activation in the INL of patients with MS should be considered a specific and early immunopathologic phenomenon. Our findings are in line and expand previous observations on the association of HRF with INL microcystic macular edema,48 a condition in which INL volume increases because of both an impairment of Müller cells to maintain retinal fluid homeostasis and an increased BRB permeability induced by microglial production of proinflammatory cytokines (IL-1 and IL-6) and inducible nitric oxide synthase.49 All together, these findings suggest that retinal activated microglia and Müller cells constitute a glial-pathologic network that determines BRB dysfunction and INL modifications. Because we excluded patients with MS with clinical or subclinical ON, retinal microglial activation was not driven by optic nerve inflammation, but probably by local immunopathologic mechanism, thus suggesting that the retina is also a primary pathologic site in MS. In line with this hypothesis, microvascular damage driven by endothelial glycosphingolipids deposition in Fabry might activate microglia, which, in turn, determines a disruption of the BRB, determining the capillary dysfunction observed in Fabry.25 In this view, HRF might thus represent a link between microglial activation, BRB dysfunction, and retinal layer osmotic swelling. Future studies involving OCT-angiography and histologic findings might shed more light on HRF pathogenesis.
An interesting finding of our work consists in the association of INL HRF with cortica (but not WM) inflammation. This finding suggests that the retina and GM might share common immunopathogenic mechanisms. Indeed, histologic studies support a marginal role for T cells in retinal inflammatory pathology, as indicated by the absence of intense CD4 and CD8 T-cell infiltration, a characteristic feature of GM inflammation. On the other hand, the association between GCIP HRF and lateral geniculate body volume might suggest an association between microglial activation in the GCIP due to retrograde transsynaptic degeneration. However, the lack of association between GCIP volume and HRF count in early disease phases may suggest that neuronal loss occurs later, after microglial activation, as observed in the animal models and in the cortex of patients with progressive MS.12
Finally, we investigated whether inner retinal HRF were associated with a peculiar disease course. Survival analysis revealed that the INL HRF count was significantly associated with both rNEDA and NEDA conditions, but not with cNEDA, a finding that can be explained with the characteristics of our RRMS cohort, which included very early treated patients. As indicated in eFigure 2 (links.lww.com/NXI/A721), a cutoff for global HRF count could be obtained for the identification of patients at risk of losing NEDA condition in the following years. Although our results further confirm the strong association between HRF count and disease severity, we are aware that these findings need to be confirmed in longitudinal multicenter studies and larger cohorts of patients.
In conclusion, we have demonstrated that HRF associates with INL volume, further characterizing the local inflammatory process in RRMS. The strong association between retinal and cortical pathology might suggest common and possibly shared microglia-related mechanisms of damage. Finally, the HRF count at baseline predicts the additional inflammatory events observed during the follow-up, indicating that they should be further explored as candidate prognostic biomarkers in MS.
Glossary
- 3D
3 dimensional
- ANT
advanced normalization tool
- BRB
blood-retina barrier
- cNEDA
clinical NEDA
- DMT
disease-modifying therapy
- EDSS
Expanded Disability Status Scale
- ET
echo time
- FLAIR
fluid-attenuated inversion recovery
- FOV
field of view
- GEE
generalized estimating equation
- GCL
ganglion cell layer
- GM
gray matter
- GMLV
GM lesion volume
- HC
healthy control
- HRF
hyperreflecting foci
- IL
interleukin
- INL
inner nuclear layer
- IPL
inner plexiform layer
- IRL
inner retinal layer
- IT
inversion time
- LGN
lateral geniculate nucleus
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- NEDA
no evidence of disease activity
- OCT
optical coherence tomography
- ON
optic neuritis
- OR
optic radiation
- PMB
papillomacular bundle
- rNEDA
radiologic NEDA
- RNFL
retinal nerve fiber layer
- ROC
receiver operating characteristic
- RRMS
relapsing-remitting MS
- RT
repetition time
- WM
white matter
- WMLV
WM lesion volume
Appendix. Authors
Contributor Information
Marta Pengo, Email: marta.pengo4@gmail.com.
Silvia Miante, Email: silvia.miante@gmail.com.
Silvia Franciotta, Email: silvia.franciotta@gmail.com.
Marta Ponzano, Email: ponzano.marta@gmail.com.
Tommaso Torresin, Email: tommaso.torresin@unipd.it.
Francesca Bovis, Email: francesca.bovis@gmail.com.
Francesca Rinaldi, Email: dr.francesca.rinaldi@gmail.com.
Paola Perini, Email: dr.paola.perini@gmail.com.
Martina Saiani, Email: martina.saiani@tiscali.it.
Monica Margoni, Email: monica.margoni@phd.unipd.it.
Alessandra Bertoldo, Email: alessandra.bertoldo@unipd.it.
Maria Pia Sormani, Email: mariapia.sormani@unige.it.
Elisabetta Pilotto, Email: elisabetta.pilotto@unipd.it.
Edoardo Midena, Email: edoardo.midena@unipd.it.
Paolo Gallo, Email: paolo.gallo@unipd.it.
Study Funding
This project was supported by 2 grants (“Roche per la Ricerca 2016” and “Progetto di Eccellenza 2020, Department of Neuroscience, University of Padua”).
Disclosure
M. Pengo, S. Miante, and S. Franciotta received travel grant form Biogen Idec, Novartis, Sanofi Aventis, and Teva. M. Ponzano, F. Bovis, E. Pilotto, and E. Midena have nothing to disclose. F. Rinaldi serves as an advisory board member of Biogen Idec and has received funding for travel and speaker honoraria from Merck Serono, Biogen Idec, Sanofi Aventis, Teva, and Bayer Schering Pharma. P. Perini has received funding for travel and speaker honoraria from Merck Serono, Biogen Idec, Sanofi Aventis, and Bayer Schering Pharma and has been consultant for Merck Serono, Biogen Idec, and Teva. M. Margoni and M.P. Sormani received personal fees from Merck Serono, Biogen, Novartis, Genzyme, Teva, Synthon, and Roche. M. Margoni reports grants and personal fees from Sanofi Genzyme, Merck Serono, Novartis, and Almirall. She was awarded a MAGNIMS-ECTRIMS fellowship in 2020. P. Gallo has been a consultant for Bayer Schering, Biogen Idec, Genzyme, Merck Serono, and Novartis; has received funding for travel and speaker honoraria from Merck Serono, Biogen Idec, Sanofi Aventis, Novartis Pharma, Bayer Schering Pharma, and Teva; has received research support from Bayer, Biogen Idec/Elan, Merck Serono, Genzyme, and Teva; and has received research grant from the University of Padova, Veneto Region of Italy, the Italian Association for Multiple Sclerosis, the Italian Ministry of Public Health. These competing of interests do not alter our adherence to PLoS One policies on sharing data and materials. M. Puthenparampil received travel grant and speaker honoraria from Novartis, Genzyme, Biogen Idec, Teva, and Sanofi Aventis; he has been consultant for Genzyme, Novartis, and Biogen. Go to Neurology.org/NN for full disclosures.
References
- 1.Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441-468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spangenberg EE, Green KN. Inflammation in Alzheimer's disease: lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61:1-11. doi: 10.1016/j.bbi.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucchinetti CF, Popescu BFG, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365(23):2188-2197. doi: 10.1056/nejmoa1100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bø L, Vedeler CA, Nyland H, Trapp BD, Mørk SJ. Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler. 2003;9(4):323-331. doi: 10.1191/1352458503ms917oa. [DOI] [PubMed] [Google Scholar]
- 5.van Horssen J, Singh S, van der Pol S, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation. 2012;9:156. doi: 10.1186/1742-2094-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain. 2017;140(7):1900-1913. doi: 10.1093/brain/awx113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong Y, Yong VW. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat Rev Neurol. 2019;15(12):704-717. doi: 10.1038/s41582-019-0253-6. [DOI] [PubMed] [Google Scholar]
- 8.Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. 2018;8:1905. doi: 10.3389/fimmu.2017.01905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Poel M, Ulas T, Mizee MR, et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun. 2019;10(1):1139. doi: 10.1038/s41467-019-08976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653-666. doi: 10.1002/ana.25463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Absinta M, Maric D, Gharagozloo M, et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature. 2021;597(7878):709-714. doi: 10.1038/s41586-021-03892-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Olst L, Rodriguez-Mogeda C, Picon C, et al. Meningeal inflammation in multiple sclerosis induces phenotypic changes in cortical microglia that differentially associate with neurodegeneration. Acta Neuropathol. 2021;141(6):881-899. doi: 10.1007/S00401-021-02293-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.London A, Benhar I, Schwartz M. The retina as a window to the brain - from eye research to CNS disorders. Nat Rev Neurol. 2013;9(1):44-53. doi: 10.1038/nrneurol.2012.227. [DOI] [PubMed] [Google Scholar]
- 14.Gabilondo I, Martínez-Lapiscina EH, Martínez-Heras E, et al. Trans-synaptic axonal degeneration in the visual pathway in multiple sclerosis. Ann Neurol. 2014;75(1):98-107. doi: 10.1002/ana.24030. [DOI] [PubMed] [Google Scholar]
- 15.Puthenparampil M, Federle L, Poggiali D, et al. Trans-synaptic degeneration in the optic pathway. A study in clinically isolated syndrome and early relapsing-remitting multiple sclerosis with or without optic neuritis. PLoS One. 2017;12(8):e0183957. doi: 10.1371/journal.pone.0183957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilotto E, Miante S, Torresin T, et al. Hyperreflective foci in the retina of active relapse-onset multiple sclerosis. Ophthalmology. 2020;127(12):1774-1776. doi: 10.1016/J.OPHTHA.2020.03.024. [DOI] [PubMed] [Google Scholar]
- 17.Cruz-Herranz A, Oertel FC, Kim K, et al. Distinctive waves of innate immune response in the retina in experimental autoimmune encephalomyelitis. JCI Insight. 2021;6(11):e149228. doi: 10.1172/JCI.INSIGHT.149228. [DOI] [Google Scholar]
- 18.Bolz M, Schmidt-Erfurth U, Deak G, Mylonas G, Kriechbaum K, Scholda C. Optical coherence tomographic hyperreflective foci: a morphologic sign of lipid extravasation in diabetic macular edema. Ophthalmology. 2009;116(5):914-920. doi: 10.1016/J.OPHTHA.2008.12.039. [DOI] [PubMed] [Google Scholar]
- 19.Framme C, Wolf S, Wolf-Schnurrbusch U. Small dense particles in the retina observable by spectral-domain optical coherence tomography in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51(11):5965-5969. doi: 10.1167/IOVS.10-5779. [DOI] [PubMed] [Google Scholar]
- 20.Uji A, Murakami T, Nishijima K, et al. Association between hyperreflective foci in the outer retina, status of photoreceptor layer, and visual acuity in diabetic macular edema. Am J Ophthalmol. 2012;153(4):710-717, 717.e1. doi: 10.1016/J.AJO.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Vujosevic S, Bini S, Torresin T, et al. Hyperreflective retinal spots in normal and diabetic eyes: B-scan and en face spectral domain optical coherence tomography evaluation. Retina. 2017;37(6):1092-1103. doi: 10.1097/IAE.0000000000001304. [DOI] [PubMed] [Google Scholar]
- 22.Zeng HY, Green WR, Tso MOM. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126(2):227-232. doi: 10.1001/ARCHOPHTHALMOL.2007.65. [DOI] [PubMed] [Google Scholar]
- 23.Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. 2011;8(1):1-19. doi: 10.1186/1742-2094-8-173/FIGURES/9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Li J, Chen K, Wang J, Sheng M, Li B. Association between inflammatory factors in the aqueous humor and hyperreflective foci in patients with intractable macular edema treated with antivascular endothelial growth factor. Dis Markers. 2021;2021:5552824. doi: 10.1155/2021/5552824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atiskova Y, Rassuli R, Koehn AF, et al. Retinal hyperreflective foci in Fabry disease. Orphanet J Rare Dis. 2019;14(1):296. doi: 10.1186/S13023-019-1267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162-173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 27.Petzold A, Wattjes MP, Costello F, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol. 2014;10(8):447-458. doi: 10.1038/nrneurol.2014.108. [DOI] [PubMed] [Google Scholar]
- 28.Tewarie P, Balk L, Costello F, et al. The OSCAR-IB consensus criteria for retinal OCT quality assessment. PLoS One. 2012;7(4):e34823. doi: 10.1371/journal.pone.0034823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aytulun A, Cruz-Herranz A, Aktas O, et al. APOSTEL 2.0 recommendations for reporting quantitative optical coherence tomography studies. Neurology. 2021;97(2):68-79. doi: 10.1212/WNL.0000000000012125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frizziero L, Parrozzani R, Midena G, et al. Hyperreflective intraretinal spots in radiation macular edema on spectral domain optical coherence tomography. Retina. 2016;36(9):1664-1669. doi: 10.1097/IAE.0000000000000986. [DOI] [PubMed] [Google Scholar]
- 31.Tustison NJ, Avants BB, Cook PA, et al. N4ITK: improved N3 bias correction. IEEE Trans Med Imaging. 2010;29(6):1310-1320. doi: 10.1109/TMI.2010.2046908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage. 2011;54(3):2033-2044. doi: 10.1016/j.neuroimage.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Brady M, Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging. 2001;20(1):45-57. doi: 10.1109/42.906424. [DOI] [PubMed] [Google Scholar]
- 34.Battaglini M, Jenkinson M, De Stefano N. Evaluating and reducing the impact of white matter lesions on brain volume measurements. Hum Brain Mapp. 2012;33(9):2062-2071. doi: 10.1002/hbm.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith SM. Fast robust automated brain extraction. Hum Brain Mapp. 2002;17(3):143-155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17(2):825-841. doi: 10.1016/S1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 37.Rojkova K, Volle E, Urbanski M, Humbert F, Dell'Acqua F, Thiebaut de Schotten M. Atlasing the frontal lobe connections and their variability due to age and education: a spherical deconvolution tractography study. Brain Struct Funct. 2016;221(3):1751-1766. doi: 10.1007/s00429-015-1001-3. [DOI] [PubMed] [Google Scholar]
- 38.Knier B, Schmidt P, Aly L, et al. Retinal inner nuclear layer volume reflects response to immunotherapy in multiple sclerosis. Brain. 2016;139(11):2855-2863. doi: 10.1093/BRAIN/AWW219. [DOI] [PubMed] [Google Scholar]
- 39.Geloso MC, D'Ambrosi N. Microglial pruning: relevance for synaptic dysfunction in multiple sclerosis and related experimental models. Cells. 2021;10(3):686. doi: 10.3390/cells10030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen K, Reichelt M, Kyauk RV, et al. Multiple sclerosis risk gene Mertk is required for microglial activation and subsequent remyelination. Cell Rep. 2021;34(10):108835. doi: 10.1016/j.celrep.2021.108835. [DOI] [PubMed] [Google Scholar]
- 41.Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016-1024. doi: 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreur V, Altay L, van Asten F, et al. Hyperreflective foci on optical coherence tomography associate with treatment outcome for anti-VEGF in patients with diabetic macular edema. PLoS One. 2018;13(10):e0206482. doi: 10.1371/JOURNAL.PONE.0206482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saidha S, Sotirchos ES, Ibrahim MA, et al. Microcystic macular oedema, Thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol. 2012;11(11):963-972. doi: 10.1016/S1474-4422(12)70213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knier B, Schmidt P, Aly L, et al. Retinal inner nuclear layer volume reflects response to immunotherapy in multiple sclerosis. Brain. 2016;139(11):2855-2863. doi: 10.1093/brain/aww219. [DOI] [PubMed] [Google Scholar]
- 45.Abegg M, Dysli M, Wolf S, Kowal J, Dufour P, Zinkernagel M. Microcystic macular edema: retrograde maculopathy caused by optic neuropathy. Ophthalmology. 2014;121(1):142-149. doi: 10.1016/j.ophtha.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 46.Nolan-Kenney RC, Liu M, Akhand O, et al. Optimal intereye difference thresholds by optical coherence tomography in multiple sclerosis: an international study. Ann Neurol. 2019;85(5):618-629. doi: 10.1002/ANA.25462. [DOI] [PubMed] [Google Scholar]
- 47.Nolan RC, Galetta SL, Frohman TC, et al. Optimal inter-eye difference thresholds in retinal nerve fiber layer thickness for predicting a unilateral optic nerve lesion in multiple sclerosis. J Neuroophthalmol. 2018;38(4):451-458. doi: 10.1097/WNO.0000000000000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burggraaff MC, Trieu J, de Vries-Knoppert WAEJ, Balk L, Petzold A. The clinical spectrum of microcystic macular edema. Invest Ophthalmol Vis Sci. 2014;55(2):952-961. doi: 10.1167/iovs.13-12912. [DOI] [PubMed] [Google Scholar]
- 49.Petzold A. Retinal glymphatic system: an explanation for transient retinal layer volume changes? Brain. 2016;139(11):2816-2819. doi: 10.1093/brain/aww239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.