

Abstract

The SARS-CoV-2 main protease is among the most attractive targets for the development of therapeutic interventions for COVID-19. Successful candidate agents will not only possess potent on-target activity versus SARS-CoV-2 Mpro but also minimal polypharmacology versus human cysteine proteases. This Viewpoint explores the activity profile of the first approved SARS-CoV-2 Mpro inhibitor (Nirmatrelvir) versus a panel of cysteine proteases and considers the therapeutic implications of the data.
Keywords: SARS-CoV-2, cysteine proteases, SARS-CoV-2 Mpro, Caspase 1, Nirmatrelvir, polypharmacology
The marshalling of rapid and comprehensive medical advances in response to the COVID-19 pandemic has been astonishing. Among the most promising new tools in the therapeutic armament is Nirmatrelvir (1, PF-07321332) (Figure 1A).1 Nirmatrelvir is a potent inhibitor of the SARS-CoV-2 main protease (Mpro), which is responsible for cleavage of two viral polyproteins (pp1a and pp1ab) into smaller effector protein members of the multiprotein replicase–transcriptase complex (RTC) required for viral RNA replication.2,3 SARS-CoV-2 Mpro is a cysteine protease, a protein class with significant precedent for being druggable as evidenced by multiple published inhibitors including several investigational drugs.
Figure 1.
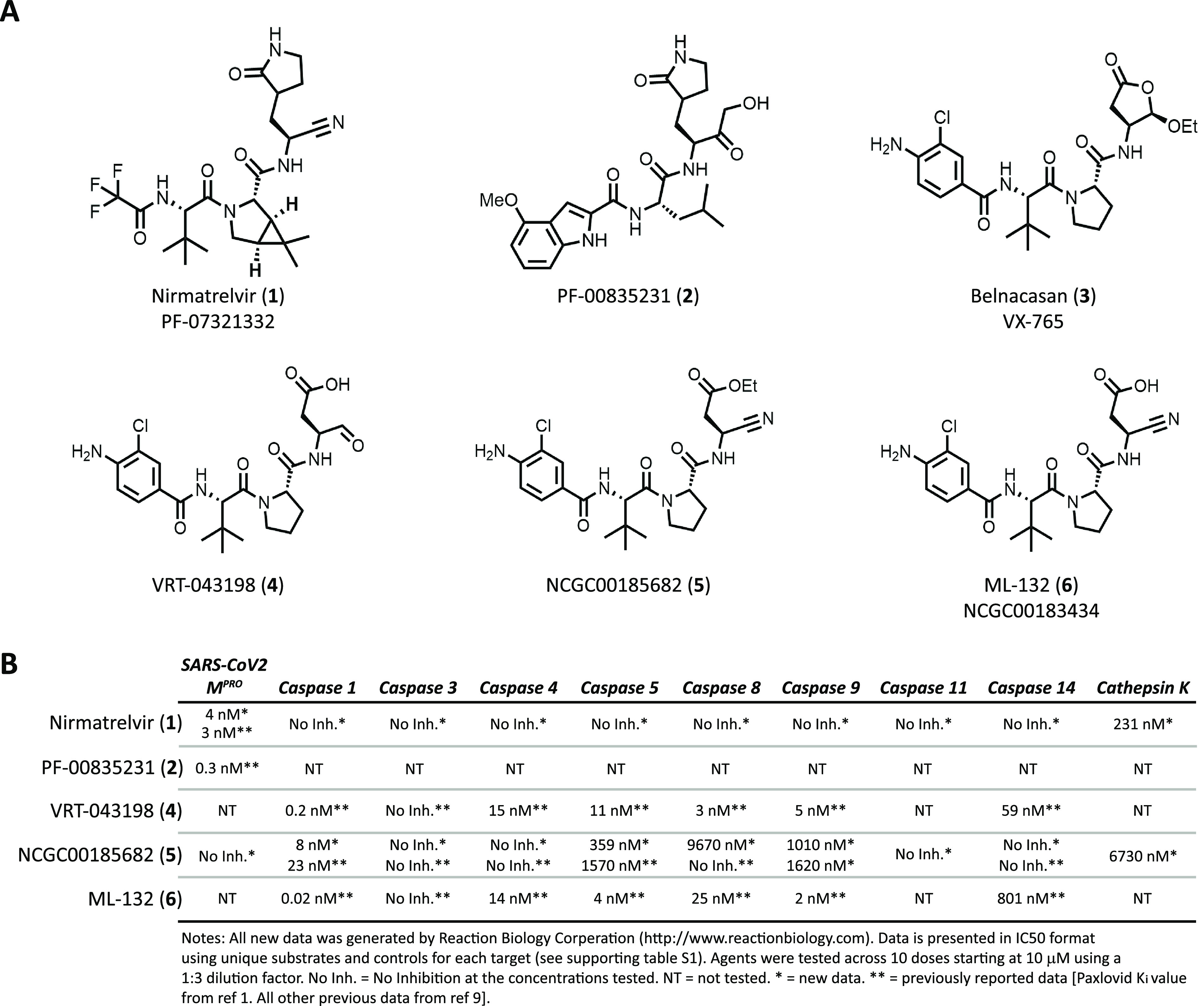
Among the best characterized examples of cysteine protease inhibitors are agents targeting earlier Mpro variants associated with previous SARS outbreaks. Scientists at Pfizer were at the forefront of these efforts with the development of the SARS-CoV-1 Mpro inhibitor PF-00835231 (2) which was later noted to have cross activity versus SARS-CoV-2 Mpro (Ki = 0.271 nM).1,4 Limited oral bioavailability for PF-00835231 sent the Pfizer team back to the drawing board in hopes of identifying an easily administered and efficacious therapy for COVID-19. The extraordinary effort that followed produced Nirmatrelvir which boasted remarkable clinical efficacy when combined with Ritonavir (i.e. Paxlovid) with an 89% reduced risk of hospitalization or death in the EPIC-HR study. Emergency use authorization was granted by the FDA in December of 2021.
Cysteine proteases are a conserved proteolytic enzyme class utilized across the entire spectrum of living organisms.5,6 This includes over 100 characterized cysteine proteases in the human genome. The success and failure of all drugs is manifestly tied to issues of tolerability which, generally speaking, can be improved by eliminating unwanted cross-activity versus related (and unrelated) protein/enzyme classes (i.e., polypharmacology). For Nirmatrelvir this surely involved a nominal level of cross-activity against human proteases including cysteine proteases. Owen et al. provided key selectivity data for Nirmatrelvir by demonstrating a lack of activity in a panel of seven proteases including selected cysteine (3), serine (3), and aspartyl (1) proteases.1
Potent inhibition of SARS-CoV-2 Mpro and lack of activity at other proteases is based on the chemical structure of Nirmatrelvir. Nirmatrelvir’s chemical structure represents a modified peptide sequence designed to enhance binding at the target of interest. The Nirmatrelvir structure incorporates a hybrid proline and Z-L-tert-butyl glycine dimer. A similar structural motif was employed by Vertex Pharmaceuticals in the design of the caspase 1 inhibitor Belnacasan (3, VX-765) (Figure 1A).7,8 Belnacasan is a prodrug that utilizes a cleverly designed 5-ethoxydihydrofuran-2(3H)-one moiety which is susceptible to intracellular esterase cleavage to reveal the active aldehyde drug form VRT-043198 (4). The aldehyde functionality represents a pharmacological warhead that forms a covalent interaction with the active site cysteine residue. Such “warheads” are a common feature shared by most cysteine protease inhibitors. Nirmatrelvir utilizes a nitrile group as a covalent reversible warhead which aided both SARS-CoV-2 Mpro activity as well as improved physicochemical properties that supported oral bioavailability. Previously, Boxer et al. incorporated a similar nitrile functionality onto the Belnacasan peptide scaffold to generate highly active caspase 1 inhibitors NCGC00185682 (5) and ML132 (6, NCGC00183434) (Figure 1A).9
The structural similarity between these agents and Nirmatrelvir certainly allowed for a reasonable anticipation of cross-activity versus caspase 1. Further, previous evaluation of VRT-043198 (4), NCGC00185682 (5), and ML132 (6) across a panel of cysteine proteases highlighted potent activity versus caspases 4, 5, 8, 9, and 14 (Figure 1B). The profile released by Owen et al. included data for only three cysteine proteases (caspase 2, cathepsin B, and cathepsin L). Interestingly, inhibitory activity versus caspase 1 would not necessarily be undesirable. Caspase 1 plays a key role in inflammasome activation and pyroptotic cell death that can be associated with an overactive immune response.10,11 In advanced COVID-19 cases, exacerbated inflammation in the lungs is a significant contributor to respiratory failure and death.12,13 Given Nirmatrelvir’s structural relationship to well-studied caspase 1 inhibitors and its remarkable clinical activity in a setting where dampened inflammation would be therapeutically beneficial, it was of interest to further probe the selectivity of Nirmatrelvir versus a broader panel of human cysteine proteases.
To satisfy this curiosity, we submitted Nirmatrelvir to a commercially available panel14 of 20 human cysteine proteases as well as SARS-CoV-2 Mpro and SARS-CoV-2 PLpro. We included NCGC00185682 (5) in this submission as an active control. The results confirmed Nirmatrelvir’s strong activity versus SARS-CoV-2 Mpro (IC50 = 4 nM) while demonstrating an extraordinary level of selectivity versus the remainder of the panel with submicromolar activity being noted for only cathepsin K (IC50 = 231 nM) (Figure 1B) (table S1). Importantly, these data also showed good alignment with our previously reported profile of NCGC00185682 (5) lending confidence in the overall results. The clinical implications of the cross-activity of Nirmatrelvir versus cathepsin K is not clear. Cathepsin K is primarily expressed in osteoclasts and inhibitors of this enzyme have been explored for the treatment of osteoporosis. There is no evidence that cathepsin K participates in the human viral or inflammation response and the potency differential may be wide enough to nullify any phenotypic contribution to the therapeutic index of Nirmatrelvir. Therefore, based on these data, its reasonable to conclude that Nirmatrelvir’s clinical benefit is associated solely with inhibition of SARS-CoV-2 Mpro and the accompanying antiviral activity.
In time, optimization details will reveal how Nirmatrelvir achieves its remarkable selectivity. Until then we are left to speculate on the role of the lactam, the 6,6-dimethyl-3-azabicyclo[3.1.0]hexane, or the trifluoroacetamide as structural features that hone the drug’s binding to only SARS-CoV-2 Mpro. What is not left to conjecture is the remarkable skill, effort, and professionalism demonstrated by the Nirmatrelvir team to whom we all owe a debt of gratitude.
Acknowledgments
This work was supported by the Division of Preclinical Innovation, National Center for Advancing Translational Research and the Center for Cancer Research, National Cancer Institute.
Glossary
Abbreviations
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- COVID-19
coronavirus disease 2019
- SARS-CoV-2 Mpro
severe acute respiratory syndrome coronavirus 2 main protease
- EPIC-HR
evaluation of protease inhibition for COVID-19 in high-risk patients
- FDA
U.S. Food and Drug Administration
- RTC
replicase-transcriptase complex
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00065.
Profile of Nirmatrelvir (PF-07321332) and NCGC00185682 versus a panel of 23 cysteine proteases (PDF)
This work was supported by the intramural program of NCATS Project 1ZIATR000043-07.
The authors declare no competing financial interest.
Supplementary Material
References
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J. Jr.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J. 3rd; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S.; Dixit R.; Pandey K. C. Cysteine proteases: modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107. 10.3389/fphar.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente X. S.; Sánchez L. M.; Overall C. M.; López-Otín C. Human and mouse proteases: a comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- Wannamaker W.; Davies R.; Namchuk M.; Pollard J.; Ford P.; Ku G.; Decker C.; Charifson P.; Weber P.; Germann U. A.; Kuida K.; Randle J. C. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J. Pharmacol. Exp. Ther. 2007, 321, 509–516. 10.1124/jpet.106.111344. [DOI] [PubMed] [Google Scholar]
- Yang X. M.; Downey J. M.; Cohen M. V.; Housley N. A.; Alvarez D. F.; Audia J. P. The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J. Cardiovasc. Pharmacol. 2017, 22, 574–578. 10.1177/1074248417702890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer M. B.; Quinn A. M.; Shen M.; Jadhav A.; Leister W.; Simeonov A.; Auld D. A.; Thomas C. J. A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety. ChemMedChem. 2010, 5, 730–738. 10.1002/cmdc.200900531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L.; Eigenbrod T.; Muñoz-Planillo R.; Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune response and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao E. A.; Rajan J. V.; Aderem A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A. C.; Soares V. C.; de Azevedo-Quintaniha I. G.; Dias S. dS. G.; Fintelman-Rodrigues N.; Sacramento C. Q.; Mattos M.; de Freitas C. S.; Temerozo J. R.; Teixeira L.; Hottz E. D.; Barreto E. A.; Pão C. R. R.; Palhinha L.; Miranda M.; Bou-Habib D. C.; Bozza F. A.; Bozza P. T.; Souza T. M. L. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discovery 2021, 7, 43. 10.1038/s41420-021-00477-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan P.; Shen M.; Yu Z.; Ge W.; Chen K.; Tian M.; Xiao F.; Wang Z.; Wang J.; Jia Y.; Wang W.; Wan P.; Zhang J.; Chen W.; Lei Z.; Chen X.; Luo Z.; Zhang Q.; Xu M.; Li G.; Li Y.; Wu J. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. 10.1038/s41467-021-25629-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaction Biology.https://www.reactionbiology.com/ (accessed 2022).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.