

Abstract
Cancer cells depend on metabolic reprogramming for survival, undergoing profound shifts in nutrient sensing, nutrient uptake and flux through anabolic pathways, in order to drive nucleotide, lipid, and protein synthesis and provide key intermediates needed for those pathways. Although metabolic enzymes themselves can be mutated, including to generate oncometabolites, this is a relatively rare event in cancer. Usually, gene amplification, overexpression, and/or downstream signal transduction upregulate rate‐limiting metabolic enzymes and limit feedback loops, to drive persistent tumor growth. Recent molecular‐genetic advances have revealed discrete links between oncogenotypes and the resultant metabolic phenotypes. However, more comprehensive approaches are needed to unravel the dynamic spatio‐temporal regulatory map of enzymes and metabolites that enable cancer cells to adapt to their microenvironment to maximize tumor growth. Proteomic and metabolomic analyses are powerful tools for analyzing a repertoire of metabolic enzymes as well as intermediary metabolites, and in conjunction with other omics approaches could provide critical information in this regard. Here, we provide an overview of cancer metabolism, especially from an omics perspective and with a particular focus on the genomically well characterized malignant brain tumor, glioblastoma. We further discuss how metabolomics could be leveraged to improve the management of patients, by linking cancer cell genotype, epigenotype, and phenotype through metabolic reprogramming.
Keywords: epigenetics, glioblastoma, metabolome, mTOR complex, omics
Cancer cells depend on metabolic reprogramming for survival, undergoing profound shifts in nutrient sensing, nutrient uptake, and flux through anabolic pathways to drive nucleotide, lipid, and protein synthesis, and to provide key intermediates needed for those pathways. Here, we provide an overview of cancer metabolism, especially from an omics perspective and with a particular focus on the genomically well characterized malignant brain tumor, glioblastoma. We further discuss how metabolomics could be leveraged to improve the management of patients, by linking cancer cell genotype, epigenotype, and phenotype through metabolic reprogramming.
1. INTRODUCTION: INSIGHTS FROM METABOLOMICS IN BRAIN CANCER
Metabolism is a universal principle of all living organisms, and its reprogramming is a central hallmark of cancer. 1 Rewiring of intracellular metabolism enables cancer cells to adapt “metabolic phenotypes” that maximize tumor growth in rapidly changing conditions, including enabling tumor cells to take up and use glucose, amino acids, lipids, and nucleic acids to drive proliferation and to catalyze the formation of intermediates that buffer reactive oxygen species (ROS) for their survival. 2 Deciphering the molecular underpinnings of metabolic reprogramming in cancer could pave the way for new avenues for diagnostics and therapeutic intervention as well as for the management and clinical follow‐up of cancer patients.
Cancers of the brain have been particularly illuminating in providing insight into altered tumor metabolism. The brain is one of the most metabolically active organs in the body, using glucose, lactate, ketone bodies, fatty acids, and amino acids as fuel sources. 3 The reciprocal interaction among brain constituents including neurons and glial cells (such as astrocytes and oligodendrocyte precursor cells), heavily influences brain metabolic homeostasis. 4 Malignant brain tumors including glioblastoma (GBM) usurp the repertoire of metabolic networks in the brain for supporting their aggressive tumor growth. For example, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors (AMPARs) mediate synaptic contacts on glioma cells and neurons for their proliferation and invasion, 5 and peri‐synaptically located N‐methyl‐d‐aspartate receptors (NMDARs), another type of ionotropic glutamate receptor, facilitate the growth of brain macro‐metastases of breast cancer. 6 Furthermore, a small number of treatment‐resistant glioma stem cells depend on distinct metabolic paths to form a niche within the intricate metabolic network in the brain. 7 Moreover, the unique dependencies in lipid metabolism formed by oncogene amplification in GBM may generate actionable metabolic vulnerabilities. 8 , 9 , 10 , 11
Recent metabolomic approaches involving the systematic measurement of metabolic enzymes and metabolites, have proven to be powerful tools to identify cancer biomarkers as well as drivers of tumorigenesis. 12 Furthermore, advanced technologies for in vivo metabolic analysis have been developed, including isotope‐labeled metabolite tracing and noninvasive metabolic imaging, and these have permitted in vivo measurement of metabolic fluxes and abundances in tumor cells. 13 Interestingly, studies of the metabolomic landscape of cancer have unraveled the reciprocal interaction among each metabolic path, driven by cancer‐specific alterations of genetics and epigenetics.
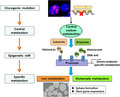
In this review, we primarily consider the metabolic landscape of cancer that has been derived from large‐scale proteomic and metabolomic analyses, to provide a better understanding of the biology of cancer as well as to improve the diagnosis, monitoring, and treatment of cancer. An oncogenic phenotype could be formed by cancer metabolic reprogramming, the importance of which has been clarified by “multi‐omics” research approaches including genetics, epigenetics, transcriptomics, proteomics and metabolomics (Figure 1).
FIGURE 1.
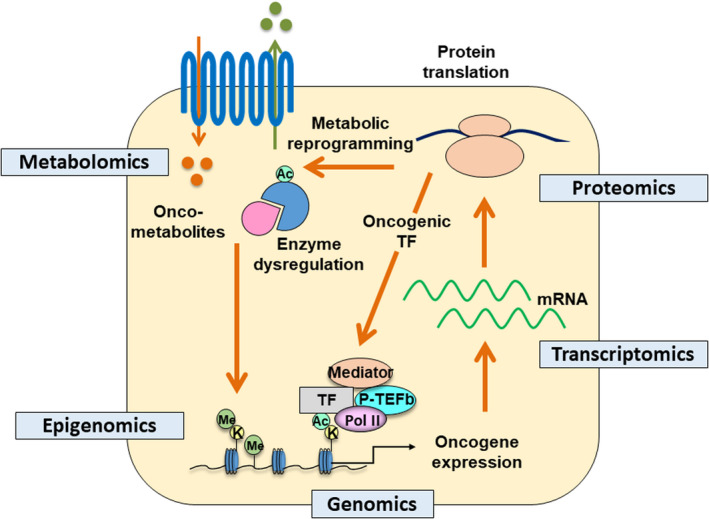
Multi‐omics approaches to study the metabolic landscape of cancer cells. Genotype of the cancer cells is translated into a metabolic phenotype to facilitate cancer cell survival. This circuit can be studied at multiple omics levels including genomics, epigenomics, transcriptomics, proteomics, and metabolomics. Ac, acetyl‐group; K, lysine residues; Me, methyl‐group; Pol II, RNA polymerase II; P‐TEFb, positive transcription elongation factor b; TF, transcription factor
2. DYNAMIC LANDSCAPE OF METABOLIC REPROGRAMMING IN CANCER
2.1. Aberrant oncogenic signaling in cancer metabolic reprogramming
The complexities of neoplastic disease may be understood through the fundamental principles of the hallmarks of cancer. 14 Metabolic reprogramming is one such emerging core hallmark of cancer, 15 , 16 and comprehensive genomic studies are clarifying the regulators of cancer metabolism. 1 Constitutively activating mutations of phosphoinositide 3‐kinase (PI3K)‐Akt‐mechanistic target of rapamycin (mTOR) signaling components are particularly prominent, and occur through several mechanisms including receptor tyrosine kinase (RTK) amplification and mutations, phosphatidylinositol 4,5‐bisphosphate 3‐kinase catalytic subunit alpha isoform (PIK3CA) mutations, and phosphatase and tensin homolog deleted from chromosome 10 (PTEN) loss. 17 , 18 mTOR is a serine/threonine kinase that merges growth factor receptor signaling into cell growth, proliferation and survival through two distinct multiprotein complexes: mTOR complex 1 (mTORC1), a well established protein translation and metabolism regulator, 19 and mTOR complex 2 (mTORC2), which was recently demonstrated to promote tumor growth and chemotherapy resistance in cancer cells independent from canonical Akt signaling. 20
One of the master regulators of cancer metabolism is the oncogenic transcription factor, c‐Myc. 21 c‐Myc expression is regulated at various levels of genes, transcripts, and protein. We showed that aberrant epidermal growth factor receptor (EGFR) signaling in GBM increases the activity of c‐Myc through both the PI3K‐Akt‐mTORC1 pathway, as well as an Akt‐independent activity of mTORC2 (Figure 2). 22 , 23 c‐Myc, in turn, facilitates glucose transport and glycolysis by upregulating such metabolic enzymes as glucose transporter 1 (GLUT1), hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase 1 (PDK1); that is, the mTOR‐Myc axis governs a repertoire of genes to promote the Warburg effect in cancer. c‐Myc may be further involved in supporting cancer cell survival by promoting pentose phosphate pathway (PPP) oxidative and nonoxidative branch enzymes, as well as fatty acid metabolism. 22 , 24 Furthermore, GBM‐related pathways established by The Cancer Genome Atlas (TCGA) such as p53 and retinoblastoma (RB) also couple cancer metabolism to the biological behavior of tumor cells. 25 , 26 Other transcriptional regulators, including peroxisome proliferator‐activated receptor γ coactivator 1‐α (PGC‐1α) and sterol regulatory element‐binding protein (SREBP) converge the neoplastic phenotype of GBM cells into more aggressive behavior via reprogramming of cellular metabolism. 27 , 28
FIGURE 2.
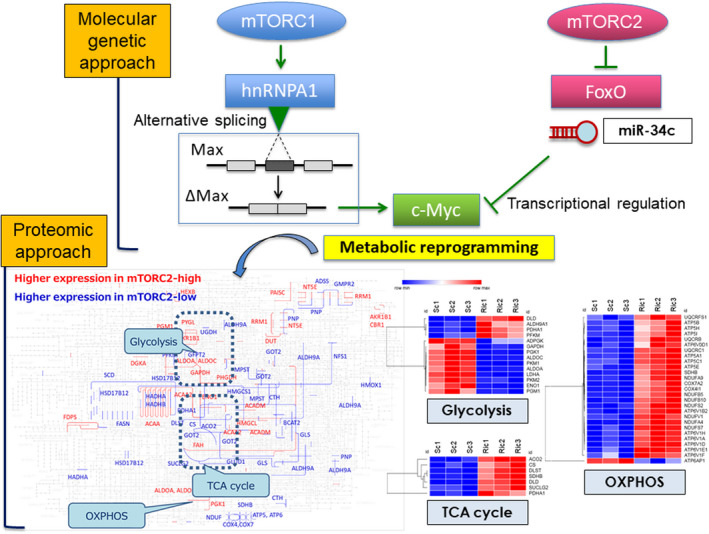
Analysis on mTOR‐dependent cancer metabolism by molecular‐genetic versus proteomics approaches. mTORC1 promotes the glycolytic metabolism by activating hnRNPA1‐dependent alternative splicing of a Myc‐binding partner Delta Max, whereas mTORC2 signaling controls c‐Myc transcription, translation, and protein level through the regulation of FoxO and microRNA. Quantitative proteome (iMPAQT) reveals that mTORC2 governs the Warburg effect in a comprehensive manner, including glycolysis, TCA cycle, and oxidative phosphorylation. FoxO, forkhead box O; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; iMPAQT, in vitro proteome‐assisted MRM for protein absolute quantification; Max, myc‐associated factor X; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid
In addition to reprogramming metabolic circuits, the ability to sense nutrients in the microenvironment is necessary for cancer cells to exploit energy from metabolism. Of note, mTOR complexes play an important role in sensing these nutrients. 29 mTORC1 responds to a range of amino acids and relevant metabolites, including leucine and arginine. 19 Furthermore, we unraveled the novel role of mTORC2 in responding to glucose and acetate in the microenvironment through acetyl‐coenzyme A (acetyl‐CoA)‐mediated acetylation of Rictor, the main component of mTORC2. 30 Using an additional, unbiased proteomics approach, we also showed that mTORC2 could suppress the activity of the cystine‐glutamate antiporter, system Xc transporter‐related protein (xCT), indicating a new role for mTORC2 as a potential regulator of ROS metabolism. 31 This suggests that mTORC2 senses the availability of amino acids including glutamate and cystine, enabling tumor cells to buffer oxidative stress through glutathione, as necessary. These data lead to the proposal that glucose and amino acid metabolism interact in mTOR‐activated cancer cells as dictated by the availability of nutrients. Apart from mTOR‐dependent nutrient sensing, the adenosine monophosphate (AMP)‐activated protein kinase (AMPK) pathway and hexosamine biosynthetic pathway (HBP)‐hypoxia‐inducible factor (HIF) axis are critical sensors of energy and nutrient status in cancer stem cells, 32 and nutrient sensing could therefore be the essential function to maximize the survival of cancer cells in various metabolic niches.
2.2. Comprehensive view of the landscape of metabolic enzymes in cancer
A comprehensive approach for the evaluation of cellular metabolism is now based upon mass spectrometry (MS)‐based proteomics, effectively linking cellular genotype and phenotype. Recent advancement in instrumentation as well as in bioinformatics has made it possible to quantify a repertoire of proteins simultaneously. 33 Following several studies on the large‐scale generation of synthetic peptides, 34 and a more comprehensive project called ProteomeTools, 35 Matsumoto et al. have established an absolute quantitative approach to assess the metabolic landscape of cells. 36 This approach to define human proteomes relies on the generation of more than 18,000 recombinant proteins from human cDNA libraries to obtain proteotypic peptides for most human proteins, and the analytic platform is called iMPAQT (in vitro proteome‐assisted multiple reaction monitoring (MRM) for protein absolute quantification). 36 Despite its mTRAQ approach with limited availability, the platform enables absolute quantification of the human proteome with internal peptide standards at known concentrations. An absolute quantification approach with targeted proteomics including iMPAQT is a powerful tool to reveal the pathogenesis of various human cancer from a metabolic standpoint. In addition to its applicability to unravel a novel metabolic network of glutamine fate in malignant progression of cancer, 37 identification of biomarkers for cancer detection, 38 as well as prediction of the efficacy of anti‐cancer drugs in various types of cancer, 39 , 40 , 41 could be achieved by quantitative targeted proteomics through the detection of key metabolic enzymes. Of interest, a recent study with targeted proteomics revealed the role of ubiquitin‐like protein encoded by “noncoding” RNA with small open reading frames, 42 indicating its potential applicability to broader areas of the physiological and pathological conditions. Considering the multifaceted power of targeted proteomics approaches, we applied the iMPAQT technique to the proteome of GBM cells with genetic manipulation of mTORC2, in order to obtain a comprehensive and dynamic map of the metabolic landscape in cancer cells. The data showed upregulation of essential metabolic enzymes in glycolysis including lactate metabolism, in association with suppression of the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, indicating the intricate involvement of mTORC2 in the Warburg effect (Figure 2). Although the iMPAQT platform is now limited by the inability to generate proteotypic peptides containing predefined post‐translational modifications, the continued development of methods to bypass this and enable the generation of synthetic human proteomes will certainly expand knowledge in protein biology, including the cancer proteome. 43 , 44
2.3. Comprehensive view of the spatio‐temporal regulation of oncometabolites in cancer
Major oncogenic drivers reprogram cancer metabolism by shifting the landscape of metabolic enzymes in cancer (Figure 2), supported by the production of intermediary metabolites, so‐called oncometabolites. 45 Isocitrate dehydrogenase (IDH) mutation in such tumors as glioma and acute myelogenous leukemia (AML) represents the concept of oncometabolites that affect oncogenic signaling by control of global epigenetics, 46 , 47 as well as by mediating the DNA damage response of cancer cells and host immune responses. 48 , 49 Recent reports pinpoint several oncometabolites, including 2‐hydroxyglutarate (2‐HG), glucose, fumarate, succinate, sarcosine, glutamine, asparagine, choline, and lactate, which play a role in the cancers of the brain, prostate, gastrointestinal tract, breast, and endocrine systems. 50 , 51 These metabolites all integrate cell cycle progression and molecular tumorigenesis through metabolic and epigenetic reprogramming.
Noninvasive biomarkers such as oncometabolites could enable oncologists to make more precise predictions of aggressive tumor behavior. 52 In addition to laboratory‐based assays on plasma levels of an mutant IDH‐dependent oncometabolite 2‐HG for assessing minimal residual disease and early recurrence of IDH‐mutated AML, 53 noninvasive and accurate modality of magnetic resonance spectroscopy (MRS) has the potential to detect 2‐HG for specific prediction of IDH‐mutant diffuse gliomas both preoperatively and at the time of suspected tumor recurrence. 54 , 55 In other words, it is essential to examine the landscape of cancer‐related dynamic metabolite shifts for understanding tumor biology, as well as its precise diagnostics. To determine the power of metabolomics approaches for constructing a comprehensive and dynamic map of metabolite production in cancer cells, we analyzed the metabolome of GBM cells before and after genetic manipulation of mTORC2 components. We used the C‐SCOPE package from Human Metabolome Technologies (HMT, Yamagata, Japan), using capillary electrophoresis TOF‐MS (CE‐TOFMS) for cation analysis and CE‐tandem MS (CE‐MS/MS) for anion analysis, as described previously. 56 , 57 Such comprehensive approaches are useful for the identification of specific hubs of metabolites (Figure 3), and we detected the convergence of the path to the production of acetyl‐CoA, which is the substrate closely associated with GBM biology through acetylation of nucleosomal histone tails, as well as nonhistone proteins through a variety of metabolic pathways. 58 Furthermore, combined with iMPAQT proteome data on the expression of metabolic enzymes that showed a significant upregulation of phosphoglycerate dehydrogenase (PHGDH) by mTORC2 (Figure 3), which coordinates serine synthesis and one‐carbon unit fate, 59 our metabolome analysis also identified a shift of metabolites from one‐carbon metabolism, the methyl‐donor S‐adenosylmethionine (SAM), which profoundly affects epigenetic changes including DNA and histone methylation for the survival of cancer cells (Figure 3). 60 , 61 Therefore, by combining proteomic and metabolomic data, we were able to obtain a more accurate spatio‐temporal map of cancer metabolic activity.
FIGURE 3.
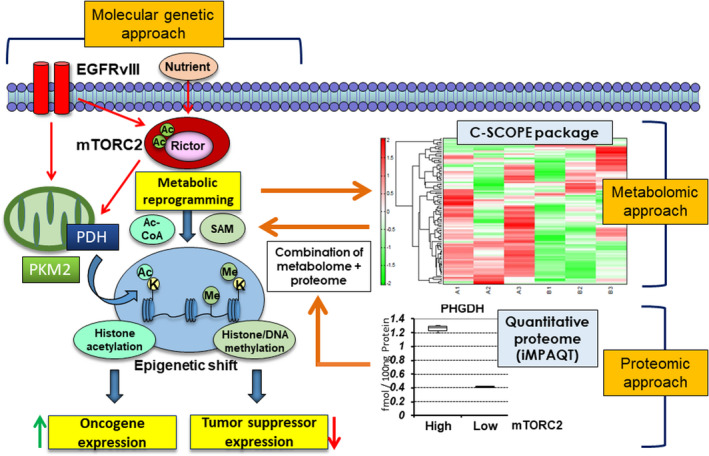
Metabolism‐dependent epigenetic shifts in GBM analyzed by molecular‐genetic versus metabolomic/proteomic approaches. Comprehensive metabolome approaches are useful for the identification of a specific hub of metabolites such as acetyl‐CoA. Furthermore, combined with our iMPAQT proteome data on PHGDH upregulation which coordinates serine synthesis and one‐carbon unit fate, the methyl‐donor SAM was identified as mTORC2 targets, which can profoundly affect DNA and histone methylation status. Ac, acetyl‐group; C‐SCOPE, metabolome analysis by HMT Inc.; EGFRvIII, constitutively active form of EGFR mutant; iMPAQT, in vitro proteome‐assisted MRM for protein absolute quantification; K, lysine residues; PDH, pyruvate dehydrogenase; PHGDH, phosphoglycerate dehydrogenase; PKM2, pyruvate kinase; M2; SAM, S‐adenosylmethionine
3. METABOLISM DRIVES GENOME‐WIDE EPIGENETIC REPROGRAMMING IN CANCER
Cancer metabolism, induced by activated oncogenes (e.g., EGFR, RAS, MYC) and dysregulated tumor suppressor genes (e.g., TP53, RB1), could exert a global shift in the epigenome of cancer cells. 62 , 63 As for malignant brain tumors (diffuse astrocytic and oligodendroglial tumors) and other systemic cancers (AML, cholangiocarcinoma, and chondrosarcoma), IDH mutations connect the genome, metabolome, and epigenome to drive tumor progression. 64 A neomorphic, cancer‐derived mutant IDH enzyme converts α‐ketoglutaric acid (α‐KG) to 2‐HG, which inhibits α‐KG‐dependent dioxygenases including Jumonji (JmjC) domain‐containing histone demethylases and the TET family of 5′‐methylcytosine hydroxylases. This leads to a dynamic and pathognomonic change in the cancer epigenome. 65 IDH mutations therefore integrate metabolism in a distinct subgroup of tumors with CpG island methylator phenotype (CIMP), 66 which inactivates differentiation‐related genes, 67 distorts chromosomal topology with loss of insulator functions, 68 and associates with recurrence and malignant progression of gliomas. 69 Importantly, high prevalence of the IDH hotspot mutations, their occurrence early in tumorigenesis, and the resulting uniform expression of the mutated protein in tumor cells make mutant IDH an appealing therapeutic target. 16 , 70 Equally important is the development of a comprehensive, machine‐learning‐based tumor classifier based on DNA methylome profiling across all entities and age groups, with the potential to fundamentally transform tumor pathology. 71
An important question is how metabolism and epigenetics are reprogrammed in IDH‐wildtype tumors, especially the most malignant GBM. Genetic mutations of the histone H3 gene itself (e.g., H3K27M, H3G34R/V/D) has been reported to globally shift the epigenetic status of histone protein (e.g., H3K27me3, H3K36me3) in certain types of malignant brain tumors. 72 H3K27me3 is particularly important in the biology and diagnostics of brain neoplasms as its methylation status shifts in various types of tumors. 73 Our studies demonstrated that metabolic reprogramming, potentially thorough the aforementioned EGFR‐mTOR axis, could significantly affect the metabolism‐dependent epigenome in cancer cell by regulating key metabolic enzymes as well as multiple intermediary metabolites. 62 Essential histone modifications are represented by acetylation on the N‐terminal lysine tail of histones that facilitates an open chromatin configuration to promote gene expression and histone methylation induced by methyltransferases with SAM for gene silencing. In EGFR‐mutant GBMs, we and others found that aberrant EGFR signaling and downstream PI3K‐Akt‐mTOR activation could modulate the enhancer landscape of GBM (represented by H3K4me1 and H3K27ac), facilitating tumorigenesis through a SOX9‐ and FOXG1‐dependent transcriptional network. 74 Additionally, our comprehensive metabolomic analyses demonstrated that the two mTOR complexes (mTORC1 and mTORC2) synergistically drive global histone methylation and tumor cell survival. 60 Of note, other metabolic regulators, including HIF‐1α, could regulate the epigenome‐modifying proteins such as histone methyltransferase mixed‐lineage leukemia 1 (MLL1) in the maintenance of GBM stem cells. 75 Alteration in the expression of epigenetic‐modifying genes themselves including lysine/arginine methyltransferases, as well as acetyltransferases/deacetylases, contributes to GBM pathogenesis. 76 Future studies are needed to unravel the mechanisms by which cancer cells survive in various niches through EGFR/mTOR‐ and other metabolic regulator‐dependent dynamic shifts in their epigenetic landscapes.
4. ALL ROADS LEAD TO METABOLISM: METABOLISM AS AN ONCOGENIC PHENOTYPE
Cancer mutations reprogram intracellular metabolism through facilitated expression of metabolic enzymes as well as intermediary metabolites, which subsequently shifts the epigenome of cancer cells. Intriguingly, the epigenetic reprogramming via metabolic change exerts its oncogenic effect by modulating cancer‐specific metabolism, meaning that cancer metabolism could be reprogrammed, both genetically and epigenetically. We recently unraveled cancer‐specific metabolism that is epigenetically driven by global shifts in the histone landscape through metabolic reprogramming. 77 , 78 One of the major marks in actively transcribed promoters is acetylation at the ninth lysine residue of histone H3 (H3K9ac). We recently demonstrated that GBM cells with activated EGFR‐mTORC2 signaling increased H3K9ac through metabolic reprogramming in cooperation with histone‐modifying enzymes including pyruvate dehydrogenase (PDH) and class IIa histone deacetylases (HDACs) (Figure 4). 77 Comprehensive studies with RNA‐seq and ChIP‐seq analyses revealed that the mTORC2‐dependent increase in H3K9ac was uniquely induced at the promoter of iron metabolism genes including ferritin, transferrin receptor, divalent metal transporter 1, and hepcidin (Figure 4). 77 The mechanisms by which intracellular iron accumulation leads to cell survival await investigation, 79 but our data and others suggest that epigenetic regulation of iron metabolism is essential for the induction of stemness in cancer cells (Figure 4). 80 Of note, comprehensive metabolomic analyses revealed that, in addition to histone acetylation, DNA methylation and histone methylation (H3K27me3) could contribute to tumor aggressiveness by rewiring intracellular metabolic pathways such as those for glutamine, methionine, and ROS metabolism, leading to the maintenance of cancer stem cells. 81 Furthermore, other epigenetic modulators of metabolism include a member of the sirtuin families, which was found mutated in different human cancer, suggesting its tumor suppressive function. 82 , 83 Therefore, epigenetic regulation of metabolism could be a prevalent phenomenon in cancer, supported by EGFR/mTOR‐signaling as well as other epigenetic regulators. These findings lead to the fascinating proposal that oncogene signaling first reprograms far reaching phenomena such as central carbon metabolism, which eventually governs each specific, effector metabolism to adapt to a variety of environments through a genome‐wide epigenetic shift (Figure 4).
FIGURE 4.
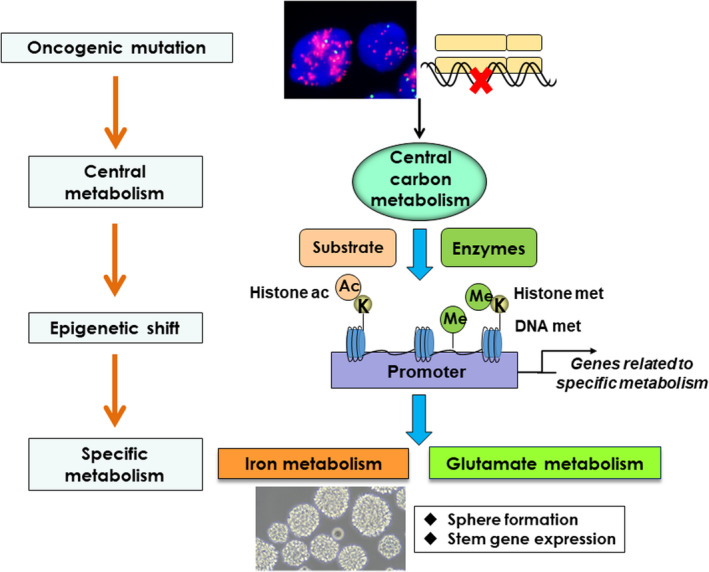
Epigenetic regulations of cancer‐prone metabolism as a central oncogenic phenotype. Oncogene signaling reprograms central carbon metabolism at the outset. The effect of carbon metabolism reprogramming is far reaching, and globally shifts the epigenome of cancer cells including histone modifications and DNA methylation. Genome‐wide epigenetic changes eventually govern each specific, effector metabolism for tumor cell survival through the induction of cancer cell stemness. Ac, acetyl‐group; ac, acetylation; K, lysine residues; Me, methyl‐group; met, methylation
5. CONCLUSION AND FUTURE PERSPECTIVE: INSIGHTS FROM MULTI‐OMICS ANALYSES IN CANCER
Cancer development, progression, and therapy response are profoundly influenced by intracellular metabolism and the exogenous microenvironment. This variably shifts the epigenetic landscape, including DNA methylation and histone modifications. Interestingly, multi‐omics analyses of cancer cells revealed that all pathways including genomics, epigenomics, transcriptomics, and proteomics converge on cancer metabolism, which is potentially the most important executioner of the oncogenic phenotype. Importantly, cancer research is moving from a genotype‐based static picture to a dynamic view in which genotype and tissue context interact to define the metabolic repertoire of tumor cells. 84 A complete “metabolic catalog” of human cancer should also be established through the development of the Cancer Cell Line Encyclopedia (CCLE), integrating quantitative analyses of 225 metabolites in 928 cell lines from more than 20 cancer types by liquid chromatography–mass spectrometry (LC‐MS). 85 This effort, associated with unbiased and quantitative approaches of proteome and metabolome, enables association analyses linking the cancer metabolome to genetic alterations, epigenetic features, and gene dependencies. Cooperative, multidisciplinary, and translational approaches will be needed to translate metabolic insights into better treatments for cancer patients.
CONFLICTS OF INTEREST
PSM is a co‐founder of Boundless Bio, Inc. He has equity in the company and chairs the scientific advisory board, for which he is compensated. PSM is also a consultant for Sage Therapeutics, Asteroid Therapeutics, and Autobahn Therapeutics, and scientific co‐founder and consultant for Pretzel Therapeutics, Inc. WKC is co‐founder of Interleukin Combinatorial Therapeutic, Inc., InVaMet, Inc., and io0, LLC. KM and NS do not have any conflict of interest.
ACKNOWLEDGMENTS
We thank the Department of Neurosurgery, Tokyo Women’s Medical University for biospecimen and biorepository support. This work is supported by Japan Society for the Promotion of Science KAKENHI Grant JP19K07649 (KM). PSM is supported in part by grants U24CA264379 and RO1 CA238249 from the National Institutes of Health (NIH) and a grant from the National Brain Tumor Society.
Masui K, Cavenee WK, Mischel PS, Shibata N. The metabolomic landscape plays a critical role in glioma oncogenesis. Cancer Sci. 2022;113:1555–1563. doi: 10.1111/cas.15325
Funding information
National Institutes of Health, (grant/award number: “RO1 CA238249,” “U24 CA264379”) Japan Society for the Promotion of Science, (Grant / Award Number: ‘JP19K07649’)
REFERENCES
- 1. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 2013;104:275‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bi J, Chowdhry S, Wu S, et al. Altered cellular metabolism in gliomas — an emerging landscape of actionable co‐dependency targets. Nat Rev Cancer. 2020;20:57‐70. [DOI] [PubMed] [Google Scholar]
- 4. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte‐neuron metabolic cooperation. Cell Metab. 2011;14:724‐738. [DOI] [PubMed] [Google Scholar]
- 5. Venkataramani V, Tanev DI, Strahle C, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573:532‐538. [DOI] [PubMed] [Google Scholar]
- 6. Zeng Q, Michael IP, Zhang P, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573:526‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA. 2011;108:16062‐16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo D, Reinitz F, Youssef M, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/ SREBP‐1/LDLR‐dependent pathway. Cancer Discov. 2011;1:442‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Villa GR, Hulce JJ, Zanca C, et al. An LXR‐cholesterol axis creates a metabolic co‐dependency for brain cancers. Cancer Cell. 2016;30:683‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bi J, Ichu TA, Zanca C, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab. 2019;30:525‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bi J, Khan A, Tang J, et al. Targeting glioblastoma signaling and metabolism with a re‐purposed brain‐penetrant drug. Cell Rep. 2021;37:109957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaushik AK, DeBerardinis RJ. Applications of metabolomics to study cancer metabolism. Biochim Biophys Acta Rev Cancer. 2018;1870:2‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim MM, Parolia A, Dunphy MP, et al. Non‐invasive metabolic imaging of brain tumours in the era of precision medicine. Nat Rev Clin Oncol. 2016;13:725‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 15. Masui K, Cavenee WK, Mischel PS. Cancer metabolism as a central driving force of glioma pathogenesis. Brain Tumor Pathol. 2016;33:161‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masui K, Onizuka H, Cavenee WK, et al. Metabolic reprogramming in the pathogenesis of glioma: update. Neuropathology. 2019;39:3‐13. [DOI] [PubMed] [Google Scholar]
- 17. Brennan CW, Verhaak RGW, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valvezan AJ, Manning BD. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab. 2019;1:321‐333. [DOI] [PubMed] [Google Scholar]
- 20. Masui K, Harachi M, Cavenee WK, et al. mTOR complex 2 is an integrator of cancer metabolism and epigenetics. Cancer Lett. 2020;478:1‐7. [DOI] [PubMed] [Google Scholar]
- 21. Stine ZE, Walton ZE, Altman BJ, et al. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Masui K, Tanaka K, Akhavan D, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c‐Myc. Cell Metab. 2013;18:726‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Babic I, Anderson ES, Tanaka K, et al. EGFR mutation‐induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013;17:1000‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Huang Z, Wu Q, et al. MYC‐regulated mevalonate metabolism maintains brain tumor‐initiating cells. Cancer Res. 2017;77:4947‐4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mai WX, Gosa L, Daniels VW, et al. Cytoplasmic p53 couples oncogene‐driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat Med. 2017;23:1342‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mammucari C, Porcelli AM, Stoll EA, et al. Metabolic reprogramming in glioma. Front Cell Dev Biol. 2017;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bruns I, Sauer B, Burger MC, et al. Disruption of peroxisome proliferator‐activated receptor γ coactivator (PGC)‐1α reverts key features of the neoplastic phenotype of glioma cells. J Biol Chem. 2019;294:3037‐3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis CA, Brault C, Peck B, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid‐ and oxygen‐deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34:5128‐5140. [DOI] [PubMed] [Google Scholar]
- 29. Harachi M, Masui K, Okamura Y, et al. mTOR complexes as a nutrient sensor for driving cancer progression. Int J Mol Sci. 2018;19:3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Masui K, Tanaka K, Ikegami S, et al. Glucose‐dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc Natl Acad Sci USA. 2015;112:9406‐9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gu Y, Albuquerque CP, Braas D, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine‐glutamate antiporter xCT. Mol Cell. 2017;67:128‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robles‐Flores M, Moreno‐Londoño AP, Castañeda‐Patlán MC. Signaling pathways involved in nutrient sensing control in cancer stem cells: an overview. Front Endocrinol. 2021;12:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanash S, Taguchi A. The grand challenge to decipher the cancer proteome. Nat Rev Cancer. 2010;10:652‐660. [DOI] [PubMed] [Google Scholar]
- 34. Kusebauch U, Campbell DS, Deutsch EW, et al. Human SRMAtlas: a resource of targeted assays to quantify the complete human proteome. Cell. 2016;166:766‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zolg DP, Wilhelm M, Schnatbaum K, et al. Building ProteomeTools based on a complete synthetic human proteome. Nat Methods. 2017;14:259‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsumoto M, Matsuzaki F, Oshikawa K, et al. A large‐scale targeted proteomics assay resource based on an in vitro human proteome. Nat Methods. 2017;14:251‐258. [DOI] [PubMed] [Google Scholar]
- 37. Kodama M, Oshikawa K, Shimizu H, et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat Commun. 2020;11:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chu HW, Chang KP, Hsu CW, et al. Identification of salivary biomarkers for oral cancer detection with untargeted and targeted quantitative proteomics approaches. Mol Cell Proteomics. 2019;18:1796‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakano D, Kawaguchi T, Iwamoto H, et al. Effects of canagliflozin on growth and metabolic reprograming in hepatocellular carcinoma cells: Multi‐omics analysis of metabolomics and absolute quantification proteomics (iMPAQT). PLoS One. 2020;15:e0232283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshino H, Yamada Y, Enokida H, et al. Targeting NPL4 via drug repositioning using disulfiram for the treatment of clear cell renal cell carcinoma. PLoS One. 2020;15:e0236119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitazawa M, Hatta T, Sasaki Y, et al. Promotion of the Warburg effect is associated with poor benefit from adjuvant chemotherapy in colorectal cancer. Cancer Sci. 2020;111:658‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nita A, Matsumoto A, Tang R, et al. A ubiquitin‐like protein encoded by the “noncoding” RNA TINCR promotes keratinocyte proliferation and wound healing. PLoS Genet. 2021;17:e1009686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Perez‐Riverol Y, Vizcaíno JA. Synthetic human proteomes for accelerating protein research. Nat Methods. 2017;14:240‐242. [DOI] [PubMed] [Google Scholar]
- 44. Barber KW, Muir P, Szeligowski RV, et al. Encoding human serine phosphopeptides in bacteria for proteome‐wide identification of phosphorylation‐dependent interactions. Nat Biotechnol. 2018;36:638‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652‐3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han S, Liu Y, Cai SJ, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020;122:1580‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Montalban‐Bravo G, DiNardo CD. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018;14:979‐993. [DOI] [PubMed] [Google Scholar]
- 48. Sulkowski PL, Oeck S, Dow J, et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature. 2020;582:586‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Friedrich M, Sankowski R, Bunse L, et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH‐mutant gliomas. Nat Cancer. 2021;2:723‐740. [DOI] [PubMed] [Google Scholar]
- 50. Yang M, Soga T, Pollard PJ, et al. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012;2:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khatami F, Aghamir SMK, Tavangar SM. Oncometabolites: a new insight for oncology. Mol Genet Genomic Med. 2019;7:e873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dando I, Pozza ED, Ambrosini G, et al. Oncometabolites in cancer aggressiveness and tumour repopulation. Biol Rev Camb Philos Soc. 2019;94:1530‐1546. [DOI] [PubMed] [Google Scholar]
- 53. Mcgehee E, Rakheja D, Oliver D, et al. The importance of plasma D‐2HG measurement in screening for IDH mutations in acute myeloid leukaemia. Br J Haematol. 2016;173:323‐326. [DOI] [PubMed] [Google Scholar]
- 54. Suh CH, Kim HS, Jung SC, et al. 2‐Hydroxyglutarate MR spectroscopy for prediction of isocitrate dehydrogenase mutant glioma: a systemic review and meta‐analysis using individual patient data. Neuro Oncol. 2018;20:1573‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou M, Zhou Y, Liao H, et al. Diagnostic accuracy of 2‐hydroxyglutarate magnetic resonance spectroscopy in newly diagnosed brain mass and suspected recurrent gliomas. Neuro Oncol. 2018;20:1262‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ooga T, Sato H, Nagashima A, et al. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol Biosyst. 2011;7:1217‐1223. [DOI] [PubMed] [Google Scholar]
- 57. Ohashi Y, Hirayama A, Ishikawa T, et al. Depiction of metabolome changes in histidine‐starved Escherichia coli by CE‐TOFMS. Mol Biosyst. 2008;4:135‐147. [DOI] [PubMed] [Google Scholar]
- 58. Harachi M, Masui K, Cavenee WK, et al. Protein acetylation at the interface of genetics, epigenetics and environment in cancer. Metabolites. 2021;11:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pacold ME, Brimacombe KR, Chan SH, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one‐carbon unit fate. Nat Chem Biol. 2016;12:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harachi M, Masui K, Honda H, et al. Dual regulation of histone methylation by mTOR complexes controls glioblastoma tumor cell growth via EZH2 and SAM. Mol Cancer Res. 2020;18:1142‐1152. [DOI] [PubMed] [Google Scholar]
- 61. Serefidou M, Venkatasubramani AV, Imhof A. The impact of one carbon metabolism on histone methylation. Front Genet. 2019;10:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masui K, Harachi M, Cavenee WK, et al. Codependency of metabolism and epigenetics drives cancer progression: a review. Acta Histochem Cytochem. 2020;53:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dai Z, Ramesh V, Locasale JW. The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet. 2020;21:737‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu Y, Yang C. Oncometabolites in cancer: current understanding and challenges. Cancer Res. 2021;81:2820‐2823. [DOI] [PubMed] [Google Scholar]
- 65. Xu W, Yang H, Liu Y, et al. Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of α‐ketoglutarate‐dependent dioxygenases. Cancer Cell. 2011;19:17‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Klughammer J, Kiesel B, Roetzer T, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med. 2018;24:1611‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. 2021;18:645‐661. [DOI] [PubMed] [Google Scholar]
- 71. Capper D, Jones DTW, Sill M, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555:469‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kitahama K, Iijima S, Sumiishi A, et al. Reduced H3K27me3 levels in diffuse gliomas: association with 1p/19q codeletion and difference from H3K27me3 loss in malignant peripheral nerve sheath tumors. Brain Tumor Pathol. 2021;38:23‐29. [DOI] [PubMed] [Google Scholar]
- 74. Liu F, Hon GC, Villa GR, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell. 2015;60:307‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Heddleston JM, Wu Q, Rivera M, et al. Hypoxia‐induced mixed‐lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012;19:428‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kunadis E, Lakiotaki E, Korkolopoulou P, et al. Targeting post‐translational histone modifying enzymes in glioblastoma. Pharmacol Ther. 2021;220:107721. [DOI] [PubMed] [Google Scholar]
- 77. Masui K, Harachi M, Ikegami S, et al. mTORC2 links growth factor signaling with epigenetic regulation of iron metabolism in glioblastoma. J Biol Chem. 2019;294:19740‐19751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Onizuka H, Masui K, Amano K, et al. Metabolic reprogramming drives pituitary tumor growth through epigenetic regulation of TERT. Acta Histochem Cytochem. 2021;54:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hirayama T. Development of chemical tools for imaging of Fe(II) ions in living cells: a review. Acta Histochem Cytochem. 2018;51:137‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schonberg DL, Miller TE, Wu Q, et al. Preferential iron trafficking characterizes glioblastoma stem‐like cells. Cancer Cell. 2015;28:441‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Z, Yip LY, Lee JHJ, et al. Methionine is a metabolic dependency of tumor‐initiating cells. Nat Med. 2019;25:825‐837. [DOI] [PubMed] [Google Scholar]
- 82. Kugel S, Feldman JL, Klein MA, et al. Identification of and molecular basis for SIRT6 loss‐of‐function point mutations in cancer. Cell Rep. 2015;13:479‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Etchegaray JP, Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol Cell. 2016;62:695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chowdhry S, Zanca C, Rajkumar U, et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature. 2019;569:570‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li H, Ning S, Ghandi M, et al. The landscape of cancer cell line metabolism. Nat Med. 2019;25:850‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]