

Abstract
Here we report a computation-driven chemoenzymatic synthesis and biosynthesis of the natural product deoxyakanthomycin, an atropisomeric pyridone natural product that features a 7-membered carbocycle with five stereocenters, one of which a quaternary center. The one-step synthesis from a biosynthetic precursor is based on computational analysis that predicted a σ-bridged cation mediated cyclization mechanism to form deoxyakanthomycin. The σ-bridged cation rationalizes the observed substrate-controlled selectivity; diastereoselectivity arises from attack of water anti to the σ-bridging, as is generally found for σ-bridged cations. Our studies also reveal a unifying biosynthetic strategy for 2-pyridone natural products that derive from a common o-quinone methide to create diverse structures.
Chemists and nature leverage inherent substrate reactivity to conduct stereoselective catalyst-free reactions – perhaps most notably exemplified by Stork’s synthesis of germine.1,2 More commonly, chemists and nature rely on catalyst development to control stereoselectivity, regioselectivity, and chemoselectivity in synthesis. In synthesis, a plethora of small molecule catalysts that select the reaction outcomes of complex molecules have been designed and implemented.3–5 In comparison, nature evolved a variety of enzymes to impose selectivity via noncovalent interactions. One such family is the recently discovered pericyclases that catalyze pericyclic reactions.6 Such enzymes can selectively generate complex molecular architectures from acyclic intermediates in a single step exemplified by the structural diversity in the 4-hydroxy 2-pyridone family of natural products (Figure 1).7 Previously, we have characterized the enzyme-catalyzed pericyclic reactions that lead to pyridone natural products.8–11 Herein, we describe natural products obtained from the exclusion of enzymes that control these reaction pathways and showcase nature’s use of inherent substrate reactivity to synthesize challenging-to-make molecular architectures. This work establishes a chemoenzymatic synthesis and biosynthesis of the natural product deoxyakanthomycin (1a and 1b).
Keywords: Synthesis, Biosynthesis, Natural Products, Non-Classical Ions, Hyperconjugation, DFT, Computations
Graphical Abstract
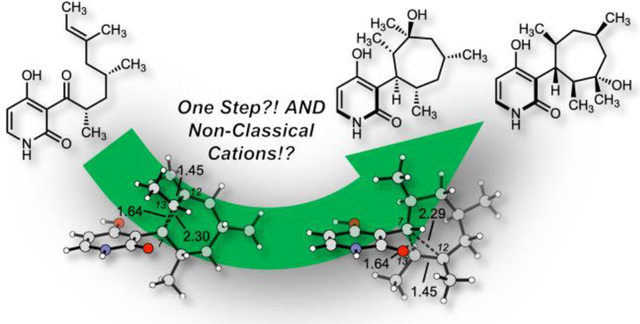
Deoxyakanthomycin (1a, 1b), akanthomycin (2a, 2b), cordypyridone A (3a), B (3b), and C (4) are structurally diverse natural products from the 4-hydroxy-2-pyridone natural product family (Figure 1). These natural products share an identical carbon backbone: the 4-hydroxy 2-pyridone connects at C3 to an 8-unit alkyl chain that is methylated at C8 and C10. Structural diversity arises from the various possible intramolecular cyclizations of the alkyl chain. Deoxyakanthomycin (1a, 1b) and akanthomycin (2a, 2b) were originally isolated in 2002 as mixtures of atropisomers (M and P) and feature a 7-membered carbocycle with 5 stereocenters, one of which at a quaternary carbon C12.12 Despite multiple attempts, there is no total synthesis of deoxyakanthomycin (1a, 1b) or akanthomycin (2a, 2b).7,13 The cordypyridones were originally isolated in 2001 and found to have potent anti-malarial activity.14 Cordypyridone A and B (3a, 3b) are also atropisomeric and feature a vinyl cyclohexyl moiety from a biological Alder-ene reaction. These natural products were synthesized in 2009 in seven steps.15 In 2020, the Baran lab revamped this synthetic route and completed a total synthesis of (–)-maximiscin, a further functionalized derivative of cordypyridone A and B (3a, 3b) that features an shikimic acid derivative linkage at the pyridone nitrogen.16 Lastly, cordypyridone C (4) is a fused tricyclic structure derived from a hetero-Diels Alder cyclization, and features an N-methoxy substituent.14 Structural diversity among these natural products leads to unique bioactivities ranging from potent anti-bacterial, free radical scavengers, or anti-malarial agents.
Figure 1.

4-hydroxy 2-pyridone natural products (1-6) with identical carbon backbones and divergent molecular architectures all derive from a common biosynthetic intermediate, QM 7.
We set out to uncover how the unique structures of 1a, 1b, 2a, and 2b form. Based on our previous work on pyridoxatin (5a and 5b), asperpyridone A (6), and related pyridones that are structurally identical except for lacking the methyl group at C12, we predict that the biosynthesis of the cordypyridones derive from a common o-quinone methide intermediate (7, Figure 1).8,9,11 This intermediate undergoes either an enzymatic Alder-ene reaction to form 3a and 3b or hetero-Diels–Alder reaction to form 4 and 6 (Figure 1).8,9,11 Therefore, we propose that 1a, 1b, 2a, and 2b derive from a common quinone methide intermediate that instead of undergoing a pericyclic reaction, undergoes a cationic cyclization followed by hydration to form the 7-membered carbocycle. Our proposal is different from Clardy’s original biosynthetic proposal12 in which akanthomycin (2) is formed by an oxidative ring expansion reaction from cordypyridone A and B (3a and 3b). Of note, the C12 desmethyl derivative of akanthomycin has never been isolated. This indicates that this is likely cation driven as the presence of a C12 methyl group is necessary to form a tertiary carbocation.
We calculated the proposed cationic cyclization pathway to form 1a, 1b, 2a, and 2b (Figure 2A and S1) from 7 at the ωB97X-D-CPCM(H2O)/def2-QZVPP//ωB97X-DCPCM(H2O)/6–311+G(d,p) level of theory. These calculations also give evidence to whether the stereochemistries at positions C7 and C12, are substrate-controlled (nonenzymatic) or enzyme-controlled. We employed a protonated model system to best aid in locating transient cationic intermediates. These simplistic model calculations do not account for the barrier to form this protonated intermediate, but such a species should be energetically accessible in a neutral aqueous solution or the enzyme environment. Calculations indicate the protonated (Z) o-QM 7a or (E) o-QM 7b will readily undergo a cationic cyclization via TS-1 or TS-2, respectively, with a Gibbs free energy of activation (ΔG‡) of 5.3 or 5.1 kcal·mol–1 to form the cyclization adducts 8a and 8b (Figure 2A). These energetic barriers indicate that if both the protonated (Z) and (E) o-QM 7 are in solution, a mixture of atropisomers of 8 will rapidly form.
Figure 2.

A. Calculated reaction scheme to form atropisomeric deoxyakanthomycin hydrates (1a’ and 1b’). B. sdx gene cluster from Sarocladium oryzae. C. Biosynthetic pathway to form deoxyakanthomycin 1. D. Standards of 12 and 2a/2b (i, ii) and enzymatic reaction of 12 with a SDR (iii) and chemical reaction of 12 with NaBH4 (iv) to form deoxyakanthomycin.
Strikingly, the cyclization adducts 8a and 8b feature a non-classical σ-bridged cation in which two electrons are delocalized over three carbon atoms (C7, C12, and C13). Such a bonding motif was first proposed by Winstein in his studies of the norbornyl cation.17 In such structures, σ-bridging leads to cation stabilization through hyperconjugation. This is the first instance of such a structure in natural product biosynthesis outside of terpene biosynthesis.2,18–20 Structurally, we see elongation of the C7–C13 σ-bond from a typical C–C bond length of 1.54 Å to 1.64 Å and σ-bridging at the adjacent carbon with a C7–C12 distance of ~ 2.30 Å. The C7–C12 distance is well within the cutoff for van der Waals interactions (~ 2.30 Å versus 3.40 Å). The C7–C12 σ-bridging interaction is elongated when compared to the C7–C13 σ-bond due to the asymmetrical substitution pattern. Formally, C12 has an extra electron-donating alkyl group compared to C13 with a hydrogen substituent. This indicates that the C7–C13 interaction is to be stronger than the C7–C12 interaction to best stabilize the bridged cation. The best evidence to validate the computational result proposing a σ-bridged cation is the stereochemistry of 1 and 2. These natural products 1 and 2 have only been isolated with C12 in the (S)-configuration. If this reaction were to proceed via this proposed σ-bridged cation, then one would expect diastereoselectivity in the hydration of the cation. This diastereoselectivity arises from attack of water anti to the σ-bridging, as is generally found for σ-bridged cations. Based on the original isolation paper, akanthomycin was isolated as a single diastereomer which validates the proposal of σ-bridged cations.
To validate that this is substrate controlled stereoselectivity, we calculated the anti- and syn- hydration reaction transition states (TS-3 and TS-4 versus TS-5 and TS-6, Figure 2A and S1). The anti-hydration transition states are low in energy and preferred over the syn-hydration transition states by >1 kcal·mol–1 (Figure S1). TS-3 and TS-4 lead to atropisomeric deoxyakanthomycin hydrates (1a’ and 1b’), which upon deprotonation form 1a and 1b. The atropisomeric deoxyakanthomycin hydrates are unable to interconvert between each species due to the hindered rotation C3–C7 bond; the calculated barrier of rotation is greater than 35 kcal·mol–1 (Figure S2). The computational studies predict that akanthomycin and deoxyakanthomycin can form non-enzymatically from the common biosynthetic quinone methide intermediate. This proposed reaction cascade forms three stereocenters, one of which is a quaternary carbon, all in one-pot without the need of a catalyst.
To identify the biosynthetic gene cluster of 1 and verify the proposed reaction scheme, we searched and screened the homologous biosynthetic gene clusters of pyridoxatin in the National Center for Biotechnology Information (NCBI) fungal genome database as reported in our previous study11 as the original producing strain was unavailable. This led us to identify the putative biosynthetic gene cluster (Sdx) of 1 in the genome of Sarocladium oryzae, which encodes a polyketide synthase-nonribosomal peptide synthetase (PKS–NRPS) (SdxC), a partnering enoylreductase (ER) (SdxD), a ring-expansion P450 (SdxA), a putative N-hydroxylation P450 (SdxB), a short-chain dehydrogenase/reductase (SDR) (SdxG) and a putative O-methyltransferase (OMT) (SdxI) (Figure 2B). We then heterologously expressed the PKS–NRPS SdxC, the ER SdxD, and the ring expansion P450 SdxA in Aspergillus nidulans 1145 ΔEMΔST,21 which led to the production of ketone 12 (2 mg·L−1). Compound 12 is structurally confirmed to be the dephenylated 2-pyridone ketone (Table S3 and Figures S3-7). To probe the proposed mechanism of the formation of 1 from the quinone methide 7, we purified soluble SDR homologs to SdxG, including PdxG, AdxG and EpiG and performed the in vitro reaction using 12 as the substrate with NADPH as the reducing cofactor. In good agreement with the calculation data, deoxyakanthomycin 1a and 1b predominantly formed after overnight incubation and proceeded to completion within 72 h (Figure 2C, iii, and Figure S8, 9). To exclude the participation of the SDR in the carbocycle formation, we performed reduction of 12 with excess NaBH4 and quenched the reaction mixture with excess water. This nonenzymatic reaction similarly formed deoxyakanthomycin 1a and 1b (Figure 2C, iv). In trace iv, a peak with MS corresponding to the alcohol species 13 is present, which was also present in the enzymatic in vitro reactions with shorter reaction times (Figure S9). In summary, deoxyakanthomycin 1a and 1b form upon incubation of ketone 12 with a ketone-reducing enzyme or via chemical reaction with NaBH4.
From these computational and experimental results, we propose that the biosynthesis of akanthomycin (2a/2b) occurs by an enzymatic reduction of the ketone 12 to the corresponding alcohol 13 (Figure 2D). This alcohol 13 can slowly dehydrate in solution to form a mixture of the (E) and (Z) o-quinone methide species 7. Once the quinone methide 7 forms, it can rehydrate to reform the alcohol 13 or cyclize to form the σ-bridged cation 8a and 8b which is selectively hydrated to exclusively form atropisomeric deoxyakanthomycin 1a and 1b. Lastly, we predict that deoxyakanthomycin 1a and 1b can be N-hydroxylated by the putative enzyme SdxB to form akanthomycin 2a and 2b. These results showcase how nature can construct a variety of natural product scaffolds from a common intermediate. When nature needs to construct a pyran or cyclohexane, a pericyclase is used. Otherwise, substrate-controlled reactivity is utilized to create difficult-to-synthesize 7-membered rings. This discovery refines our understanding of 2-pyridone natural product biosynthesis and unifies all known biosynthetic routes to go through a reactive o-quinone methide intermediate.
Supplementary Material
ACKNOWLEDGMENT
CSJ is grateful for generous funding from The Saul Winstein Fellowship, UCLA Chemistry & Biochemistry Excellence in Research Fellowship, The University of California Los Angeles Graduate Education Dissertation Year Fellowship, and The Senior Foote Fellowship.
Funding Sources
This work was supported by the NIH grant 1R01AI141481 to Y.T. and K.N.H.
ABBREVIATIONS
- QM
quinone methide
- TS
transition state
- PKS–NRPS
polyketide synthase-nonribosomal peptide synthetase
- ER
enoylreductase
- SDR
short-chain dehydrogenase/reductase
- OMT
O-methyltransferase
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Computational and experimental methods, characterization of key chemical structures, molecular coordinates, and calculated energies of all chemical structures (PDF)
REFERENCES
- (1).Stork G; Yamashita A; Hanson RM; Phan L; Phillips E; Dubé D; Bos PH; Clark AJ; Gough M; Greenlee ML; et al. Synthetic Study toward Total Synthesis of (±)-Germine: Synthesis of (±)-4-Methylenegermine. Org. Lett. 2017, 19 (19), 5150–5153. 10.1021/acs.orglett.7b02434. [DOI] [PubMed] [Google Scholar]
- (2).Tantillo DJ Importance of Inherent Substrate Reactivity in Enzyme-Promoted Carbocation Cyclization/Rearrangements. Angew. Chemie Int. Ed 2017, 56 (34), 10040–10045. 10.1002/anie.201702363. [DOI] [PubMed] [Google Scholar]
- (3).Mohr JT; Krout MR; Stoltz BM Natural Products as Inspiration for the Development of Asymmetric Catalysis. Nature 2008, 455 (7211), 323–332. 10.1038/nature07370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Houk KN; Cheong PH-Y Computational Prediction of Small-Molecule Catalysts. Nature 2008, 455 (7211), 309–313. 10.1038/nature07368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).MacMillan DWC The Advent and Development of Organocatalysis. Nature 2008, 455 (7211), 304–308. 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- (6).Jamieson CS; Ohashi M; Liu F; Tang Y; Houk KN The Expanding World of Biosynthetic Pericyclases: Cooperation of Experiment and Theory for Discovery. Nat. Prod. Rep. 2019, 36 (5), 698–713. 10.1039/c8np00075a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jessen HJ; Gademann K 4-Hydroxy-2-Pyridone Alkaloids: Structures and Synthetic Approaches. Nat. Prod. Rep. 2010, 27 (8), 1168–1185. 10.1039/B911516C. [DOI] [PubMed] [Google Scholar]
- (8).Ohashi M; Liu F; Hai Y; Chen M; Tang M; Yang Z; Sato M; Watanabe K; Houk KN; Tang Y SAM-Dependent Enzyme-Catalysed Pericyclic Reactions in Natural Product Biosynthesis. Nature 2017, 549, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cai Y; Hai Y; Ohashi M; Jamieson CS; Garcia-Borras M; Houk KN; Zhou J; Tang Y Structural Basis for Stereoselective Dehydration and Hydrogen-Bonding Catalysis by the SAM-Dependent Pericyclase LepI. Nat. Chem. 2019, 11 (9), 812–820. 10.1038/s41557-019-0294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang Z; Jamieson CS; Zhao Y-L; Li D; Ohashi M; Houk KN; Tang Y Enzyme-Catalyzed Inverse-Electron Demand Diels–Alder Reaction in the Biosynthesis of Antifungal Ilicicolin H. J. Am. Chem. Soc. 2019, 141 (14), 5659–5663. 10.1021/jacs.9b02204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ohashi M; Jamieson CS; Cai Y; Tan D; Kanayama D; Tang M-C; Anthony SM; Chari JV; Barber JS; Picazo E; et al. An Enzymatic Alder-Ene Reaction. Nature 2020, 586 (7827), 64–69. 10.1038/s41586-020-2743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wagenaar MM; Gibson DM; Clardy J Akanthomycin, a New Antibiotic Pyridone from the Entomopathogenic Fungus Akanthomyces Gracilis. Org. Lett. 2002, 4 (5), 671–673. 10.1021/ol016737q. [DOI] [PubMed] [Google Scholar]
- (13).Fotiadou AD; Zografos AL Accessing the Structural Diversity of Pyridone Alkaloids: Concise Total Synthesis of Rac-Citridone A. Org. Lett. 2011, 13 (17), 4592–4595. 10.1021/ol2017802. [DOI] [PubMed] [Google Scholar]
- (14).Isaka M; Tanticharoen M; Kongsaeree P; Thebtaranonth Y Structures of Cordypyridones A−D, Antimalarial N-Hydroxy- and N-Methoxy-2-Pyridones from the Insect Pathogenic Fungus Cordyceps Nipponica. J. Org. Chem. 2001, 66 (14), 4803–4808. 10.1021/jo0100906. [DOI] [PubMed] [Google Scholar]
- (15).Jones IL; Moore FK; Chai CLL Total Synthesis of (±)-Cordypyridones A and B and Related Epimers. Org. Lett. 2009, 11 (23), 5526–5529. 10.1021/ol9023437. [DOI] [PubMed] [Google Scholar]
- (16).McClymont KS; Wang F-Y; Minakar A; Baran PS Total Synthesis of (−)-Maximiscin. J. Am. Chem. Soc. 2020, 142 (19), 8608–8613. 10.1021/jacs.0c03202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Winstein S; Trifan DS The Structure of the Bicyclo[2,2,1]2-Heptyl (Norbornyl) Carbonium Ion. J. Am. Chem. Soc. 1949, 71 (8), 2953. 10.1021/ja01176a536. [DOI] [Google Scholar]
- (18).Tantillo DJ The Carbocation Continuum in Terpene Biosynthesis—Where Are the Secondary Cations? Chem. Soc. Rev. 2010, 39 (8), 2847–2854. 10.1039/B917107J. [DOI] [PubMed] [Google Scholar]
- (19).Hong YJ; Tantillo DJ Quantum Chemical Dissection of the Classic Terpinyl/Pinyl/Bornyl/Camphyl Cation Conundrum - The Role of Pyrophosphate in Manipulating Pathways to Monoterpenes. Org. Biomol. Chem. 2010, 8 (20), 4589–4600. 10.1039/c0ob00167h. [DOI] [PubMed] [Google Scholar]
- (20).Hong YJ; Tantillo DJ Biosynthetic Consequences of Multiple Sequential Post-Transition-State Bifurcations. Nat. Chem. 2014, 6 (2), 104–111. 10.1038/nchem.1843. [DOI] [PubMed] [Google Scholar]
- (21).Liu N; Hung Y-S; Gao S-S; Hang L; Zou Y; Chooi Y-H; Tang Y Identification and Heterologous Production of a Benzoyl-Primed Tricarboxylic Acid Polyketide Intermediate from the Zaragozic Acid A Biosynthetic Pathway. Org. Lett. 2017, 19 (13), 3560–3563. 10.1021/acs.orglett.7b01534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.