

Abstract
We recently showed that patients with different chronic pain conditions (such as chronic low back pain, fibromyalgia, migraine and Gulf War illness) demonstrated elevated brain and/or spinal cord levels of the glial marker 18-kDa translocator protein (TSPO), which suggests that neuroinflammation might be a pervasive phenomenon observable across multiple aetiologically heterogeneous pain disorders. Interestingly, the spatial distribution of this neuroinflammatory signal appears to exhibit a degree of disease specificity (e.g. with respect to the involvement of the primary somatosensory cortex), suggesting that different pain conditions may exhibit distinct ‘neuroinflammatory signatures’. To explore this hypothesis further, we tested whether neuroinflammatory signal can characterize putative aetiological subtypes of chronic low back pain patients based on clinical presentation. Specifically, we explored neuroinflammation in patients whose chronic low back pain either did or did not radiate to the leg (i.e. ‘radicular’ versus ‘axial’ back pain).
Fifty-four patients with chronic low back pain, 26 with axial back pain [43.7 ± 16.6 years old (mean ± SD)] and 28 with radicular back pain (48.3 ± 13.2 years old), underwent PET/MRI with 11C-PBR28, a second-generation radioligand for TSPO. 11C-PBR28 signal was quantified using standardized uptake values ratio (validated against volume of distribution ratio; n = 23). Functional MRI data were collected simultaneously to the 11C-PBR28 data (i) to functionally localize the primary somatosensory cortex back and leg subregions; and (ii) to perform functional connectivity analyses (in order to investigate possible neurophysiological correlations of the neuroinflammatory signal). PET and functional MRI measures were compared across groups, cross-correlated with one another and with the severity of ‘fibromyalgianess’ (i.e. the degree of pain centralization, or ‘nociplastic pain’). Furthermore, statistical mediation models were used to explore possible causal relationships between these three variables.
For the primary somatosensory cortex representation of back/leg, 11C-PBR28 PET signal and functional connectivity to the thalamus were: (i) higher in radicular compared to axial back pain patients; (ii) positively correlated with each other; (iii) positively correlated with fibromyalgianess scores, across groups; and finally (iv) fibromyalgianess mediated the association between 11C-PBR28 PET signal and primary somatosensory cortex–thalamus connectivity across groups.
Our findings support the existence of ‘neuroinflammatory signatures’ that are accompanied by neurophysiological changes and correlate with clinical presentation (in particular, with the degree of nociplastic pain) in chronic pain patients. These signatures may contribute to the subtyping of distinct pain syndromes and also provide information about interindividual variability in neuroimmune brain signals, within diagnostic groups, that could eventually serve as targets for mechanism-based precision medicine approaches.
Keywords: glial cells, functional connectivity, chronic pain, neuropathic, inflammation
Alshelh et al. show that patients with chronic low back pain demonstrate different patterns of neuroinflammation depending on their clinical presentation. These neuroinflammatory signatures are accompanied by neurophysiological changes, and correlate with the degree of nociplastic pain.
Introduction
Over the past two decades, preclinical studies have implicated astrocytes and microglia in pain models, suggesting that neuroimmune responses may be key to the development and maintenance of chronic pain.1-7 Both microglia and astrocytes are important in the defence against acute stress by restoring homeostasis,8–10 but their chronic activation poses a threat to the normal functioning of the CNS.11,12 Moreover, in animal models, glial inhibitors prevent, delay, or reverse persistent pain behaviours.13–20 These observations suggest that neuroimmune activation represents a viable target in our search for novel methods of treating chronic pain.
While the role of glia in human pain remains unknown, our group, using integrated PET/MRI, has found elevated levels of 18-kDa translocator protein (TSPO), a marker of glial activation, in the brain and/or spinal cord of patients with chronic low back pain (cLBP),21–23 fibromyalgia,24 migraine25 and Gulf War illness.26 Because TSPO is upregulated in activated astrocytes and microglia,27–29 this body of work suggests that neuroinflammation is likely present in human chronic pain. This thereby adds clinical evidence to the plethora of preclinical studies supporting the exploration of glial cells as possible therapeutic targets for pain.
Interestingly, different patient groups appear to present with seemingly different spatial patterns of TSPO signal elevations, i.e. distinct ‘neuroinflammatory signatures’. For example, we previously reported comparable TSPO signal elevations in the thalamus in two independent cohorts of cLBP patients compared to healthy controls,21,22 whereas in patients with fibromyalgia we observed very little thalamic involvement. Instead, patients with fibromyalgia exhibited cortical TSPO signal elevation that was widespread (possibly reflecting the complex and multi-symptom nature of this disorder) and appeared to be quite similar to that observed in veterans with Gulf War illness (paralleling the similarity in clinical presentation often observed across these two disorders).24,26 Along with the primary somatosensory cortex (S1), we observed elevation in TSPO signal in regions compatible with the lumbar spine cortical representation in cLBP, the face area in migraine and in a large portion of the sensorimotor strip in patients suffering from fibromyalgia, thus paralleling the body distribution of the pain (lumbar, facial and whole-body) reported in these patient groups.21,24,25 Collectively, these studies raise the intriguing possibility that TSPO imaging may be used to objectively characterize subtypes of patient populations based on their clinical presentation, an important step towards the identification of disorder-specific imaging biomarkers, that could eventually serve as targets for mechanism-based precision medicine approaches.
To test the hypothesis that TSPO signal may be used to characterize subtypes of patient populations, we used PET/MRI imaging with 11C-PBR28,30,31 a second-generation TSPO ligand,30,31 to investigate differences in neuroinflammatory signatures within a cohort of patients with cLBP. We explored two subtypes of cLBP: patients with cLBP that radiates to the leg (radicular cLBP, cLBPRAD) and patients with cLBP that does not radiate (axial cLBP, cLBPAX). Typically, cLBPRAD has a neuropathic component explained by damage/presumed damage to the nerve,32 whereas cLBPAX is usually considered non-neuropathic.33 Importantly, pharmacological treatments showing some efficacy in one subtype of cLBP may not work in the other,33 implying different pathomechanisms in patients with different clinical presentation. However, it is currently unknown whether different cLBP subtypes demonstrate distinct neuroimmune patterns. Therefore, in the present study we explored whether cLBPRAD exhibit distinct neuroinflammatory patterns compared to cLBPAX. In particular, based on the above mentioned differential involvement of S1 observed in patient groups with different clinical presentations, we predicted that cLBPRAD would have more pronounced neuroinflammatory signal in the S1 leg area compared to cLBPAX. To relate changes in PET signal to the cortical representations of clinically relevant body regions, we used functional MRI collected simultaneously to the PET to functionally localize the S1 back and leg subregions in these patients. Furthermore, we collected resting-state blood oxygen level-dependent (BOLD) functional MRI data to investigate the possible functional significance of the neuroinflammatory signal. The investigation of functional connectivity in this study was motivated both by preclinical work supporting the occurrence of a bidirectional interplay between glial cells and neurons (as neuroinflammation may affect neuronal communication34 and, contrariwise, neural activity may activate neuroinflammatory cells)35 and by our work linking TSPO signal elevations to alterations in functional connectivity in patients with negative affect comorbid with chronic pain.36
Materials and methods
Patients and study design
Twenty-six patients with cLBPAX [15 females; 43.7 ± 16.6 years old (mean ± standard deviation, SD)] and 28 patients with cLBPRAD (16 females; 48.3 ± 13.2 years old) were identified from two separate protocols. Protocol 1 (10 cLBPAX: 35.1 ± 11.5 years old; 15 cLBPRAD: 47.2 ± 12.2 years old) was a cross-sectional study while Protocol 2 (16 cLBPAX: 49.1 ± 17.3 years old; 13 cLBPRAD: 49.6 ± 14.5 years old) was a randomized, double-blind, placebo-controlled clinical trial testing the effect of a medication (ClinicalTrials.gov identifier: NCT03106740). Only baseline (i.e. pretreatment) data from Protocol 2 were included. Data from Protocol 1 have been included in prior publications.21,22,36,37 However, none of these previous publications investigated differences between cLBPAX and cLBPRAD (i.e. the main question of the present study). Data from Protocol 2 have not previously been published.
In both protocols, patients had been diagnosed with cLBP at a minimum of 6 months prior to enrolment, formally confirmed and categorized into cLBPAX or cLBPRAD by a trained nurse practitioner (Protocol 1) or a pain physician (Protocol 2). Patients had an ongoing pain of at least 3 on a 0–10 scale, present for at least 50% of days during a typical week. Patients were excluded for history of major psychiatric illness, neurological illness, cardiovascular disease, peripheral nerve injury, routine use of benzodiazepines to avoid possible binding competition for TSPO (except clonazepam, lorazepam and alprazolam, which have a known low binding affinity for this target38–42) history of substance abuse, current or past history within the last 5 years of major medical illness not affecting the CNS other than chronic pain, change in pain regimen during the enrolment period, epidural steroid injection within 3 (Protocol 1) or 6 weeks (Protocol 2) prior to scanning, inability to communicate in English, and contraindication for PET/MRI scanning (e.g. pacemaker, metallic implants, pregnancy). Protocol 2 had additional inclusion/exclusion criteria, requiring patients to have been on a stable pain treatment for 4 weeks prior to recruitment, and excluding patients receiving new interventions during the enrolment period, routine use of opioids ≥60 mg morphine or contraindication to medication used in the clinical trial.
Both protocols were conducted at the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital. The Institutional Review Board and the Radioactive Drug Research Committee approved these studies. All patients gave written informed consent.
Behavioural visit
All patients participated in a behavioural visit, during which a clinician completed a history and physical examination to assess eligibility and clinically characterize the patients. During this visit, patients completed various questionnaires (see below) and venous blood or saliva was collected for genotyping of the Ala147Thr TSPO polymorphism, which predicts high (Ala/Ala), mixed (Ala/Thr) or low (Thr/Thr) binding affinity to the radioligand.43,44 Patients exhibiting the Thr/Thr genotype, i.e. low-affinity binders, were excluded from any additional study procedures, whereas those with the Ala/Ala or Ala/Thr polymorphisms could proceed to the imaging visit. Additionally, in this visit, patients from Protocol 2 were familiarized with the electrical stimulation (e-stim) protocol to be used during the imaging visit.
Imaging visit
For all eligible patients, brain imaging was performed with Siemens PET/MRI tomographs. Patients from Protocol 1 were imaged using a Siemens 3 T Tim Trio whole-body MRI with a dedicated avalanche photodiode-based brain PET scanner (BrainPET)45 with a spatial resolution of 2–3 mm.46 Patients from Protocol 2 were imaged using a Siemens Biograph mMR scanner, with a spatial resolution of 4–5 mm.47 The dynamic PET data were acquired in list mode and reconstructed with corrections for decay, random coincidences, detector sensitivity and scatter. Up to 15 mCi of 11C-PBR28, produced in-house using a procedure modified from the literature,48 was injected as an intravenous bolus, and dynamic PET were acquired for 90 min as described previously.21,37 Simultaneous with the PET, a 6-min BOLD resting-state functional MRI scan was acquired in each patient (Protocol 1: repetition time/echo time = 2 s/30 ms, flip angle = 90°, voxel size = 3.1 × 3.1 × 3 mm, 37 slices; Protocol 2: repetition time/echo time = 2.3 s/30 ms, flip angle = 90°, voxel size = 3 × 3 × 3 mm, 41 slices), with eyes open. Further, to localize the somatotopic representation in S1 area for the back and leg, BOLD functional MRI scans concurrent with e-stim were performed in a subset of patients (n = 21) from Protocol 2 (repetition time/echo time = 2.3 s/30 ms, flip angle = 90°, voxel size = 3 × 3 × 3 mm, 41 slices). Detailed methods are provided in the Supplementary material.
For anatomical localization, spatial normalization and generation of attenuation correction maps,49 a multi-echo MPRAGE (T1-weighted structural MRI) volume was also acquired (repetition time/echo time 1, 2, 3, 4 = 2530/1.64, 3.5, 5.36, 7.22 ms, flip angle = 7°, voxel size = 1 mm isotropic).
In 23 patients (eight cLBPAX and 15 cLBPRAD), a radial artery catheter was inserted and blood samples were collected at 3–10-s intervals for the first 3 min, followed by samples collected at 5, 10, 20, 30, 50, 70 and 90 min post-11C-PBR28 injection. These data were used to perform full kinetic modelling, in order to validate the semiquantitative ratio metric used in the study (see below). Blood data were excluded from further analyses for one patient due to technical difficulties during sample collection. Detailed methods on blood metabolite analysis are included in the Supplementary material.
Behavioural measures
During either the behavioural visit (Protocol 1) or the imaging visit (Protocol 2), patients completed the PainDETECT50 and Brief Pain Inventory51 to assess components of pain, including intensity, interference and likelihood of a neuropathic component. A subset of patients also completed the American College of Rheumatology Fibromyalgia Survey Criteria52–54 (n = 35), which is traditionally used to differentiate patients with fibromyalgia from those without (survey scores ≥13 and <12, respectively). It can also be used as a continuous measure of symptom severity and to assess the degree of nociplastic pain (i.e. ‘fibromyalgianess’) in individuals who meet criteria for fibromyalgia55 and individuals with other pain disorders who do not.56,57
PET
For all patients from both protocols, PET data were corrected for radioactive decay, dead time, variable detector sensitivity, random coincidences, photon attenuation and scatter using software provided by the manufacturer or developed in house. Attenuation correction was performed using a magnetic resonance-based approach developed in house.49 The PET volumes were reconstructed using a 3D ordinary Poisson ordered subset expectation maximization (OP-OSEM) algorithm provided by the manufacturer and the space–variant point spread function of the BrainPET was modelled as described in Bowen et al.58 To minimize the attenuation–emission mismatch, the MPRAGE volume was co-registered to the reconstructed PET volume corresponding to the 60–90-min frame. Standardized uptake value (SUV) ratio (SUVR) images were generated from data collected over the 60–90-min post-injection 11C-PBR28 PET interval, as previously described.21,37,59 In brief, SUV maps were computed by normalizing radioactivity by injected dose/body weight. The SUV maps were non-linearly transformed to Montreal Neurological Institute (MNI) space (MNI152), applying to these maps the transformation computed from the co-registration of MPRAGE to PET volume and smoothed with an 8-mm full-width at half-maximum Gaussian kernel for consistency with prior studies,24–26,36,37 using tools from FSL (FMRIB Software Library, http://www.fmrib.ox.ac.uk/fsl/), AFNI (Analysis of Functional NeuroImages, http://afni.nimh.nih.gov/afni) and FreeSurfer (http://surfer.nmr.mgh.harvard.edu/). To obtain SUVR maps, SUV maps were intensity-normalized by the mean SUV extracted from the whole-brain (i.e. an average of all brain voxels within the MNI standard template), which showed no significant difference between cLBPAX and cLBPRAD (P = 0.45), indicating that the use of this signal as a normalizing factor did not bias our analyses.
To support the use of SUVR as an outcome metric in the present data, we compared the SUVR against the more quantitative distribution volume (VT) and the ratio of distribution volume (DVR) outcome, determined using kinetic modelling, in a subset of patients (n = 23) from whom arterial plasma data were available (Supplementary material; detailed methods have been described previously37). A radiometabolite-corrected arterial input function was used as the input for traditional two-tissue-compartmental modelling60 and VT was computed via Logan plot analysis, from ‘target regions’ (i.e. regions identified as statistically significant across groups in the voxelwise SUVR analyses in this study; see below) as well as the whole-brain. Then, each target region was divided by whole-brain VT to obtain the DVR. In all evaluated regions, VT was not significantly correlated with SUVR (r ≤ 0.29; P > 0.05); however, SUVR was strongly correlated with DVR in all regions (r ≥ 0.87; P ≤ 0.0001; Supplementary Fig. 1). These results provide further support for the use of SUVR as a viable PET metric in our study.
Functional MRI
Data from both resting-state and S1 ‘functional localizer’ scans were preprocessed using a combination of tools from FSL, AFNI and FreeSurfer software packages. Data were corrected for slice-timing, head motion and B0 field inhomogeneities and, for the e-stim scans, frame displacement-based motion outlier detection was applied. Data from both scans underwent brain extraction, co-registration to the MPRAGE, spatial smoothing with a 6-mm Gaussian kernel and high-pass temporal filtering (cut-off frequency = 0.008 Hz). Non-linear transformation to MNI space was used to spatially normalize the contrast of parameter estimates and associate variance images for both resting-state and e-stim scans (see below). To reduce physiological noise in the resting-state BOLD functional MRI data, MPRAGE images were segmented in probabilistic maps of grey matter, white matter, and CSF using SPM 12 (http://www.fil.ion.ucl.ac.uk/spm/). To minimize potential partial volume effects, white mattter and CSF masks were thresholded at 90% and eroded by one voxel. BOLD data were masked with white mattter- and CSF-inclusive masks and denoised with principal component analysis.61
Statistical analysis
Group differences were assessed with Student’s t-tests for continuous variables (age, clinical variables) and chi-square (χ2) tests for categorical variables (sex, genotype) using Statistica (TIBCO Software Inc., v.13). Main group analyses compared all cLBPAX with all cLBPRAD, statistically correcting for the factor ‘Protocol’, thus taking advantage of the larger sample size achieved when combining data. PET analyses also corrected for genotype to account for differences in binding affinity.43,44
Group PET analyses were performed using two strategies. Given our specific focus on S1 (the region where we predicted group differences in 11C-PBR28 signal, as mentioned above) and thalamus (the region consistently associated with 11C-PBR28 signal elevation in cLBP in our previous analyses), we first performed region of interest analyses using the S1 and thalamus clusters from our previous analysis21 as our two a priori regions of interest. Mean 11C-PBR28 signal extracted from each of these two regions was compared between cLBPAX and cLBPRAD, using two separate general linear models and an alpha level corrected for two comparisons (0.05 / 2 = 0.025). Next, a whole-brain voxelwise analysis was performed to evaluate the presence of group differences in the 11C-PBR28 signal beyond the boundaries of the a priori regions of interest, as well as to localize any effects observed in the region of interest analyses with higher spatial accuracy. Voxelwise ordinary least squares analysis was performed with FSL’s FEAT GLM tool (www.fmrib.ox.ac.uk/fsl, version 5.0.10), a cluster-forming threshold of Z = 3.1, and a cluster size significance threshold of P = 0.05 to correct for multiple comparisons. To understand the potential clinical significance of S1 neuroinflammation, the S1 11C-PBR28 signal was correlated with PainDetect and Fibromyalgia Survey Scores, as these were the two behavioural measures that were significantly different across groups. For these analyses, we used Pearson’s correlation coefficient, with an alpha level corrected for two comparisons (0.05 / 2 = 0.025).
To estimate brain responses to e-stim for S1 localization, general linear modelling was performed on the preprocessed functional MRI data. The stimulation period, as well as anticipatory cues (Supplementary material), were modelled for each of the three body parts as explanatory variables in first-level analyses, including six motion parameters (three rotations and three translations) and frames flagged as motion outliers as covariates. Resultant outputs such as parameter estimates and their variances, spatially normalized to MNI152, were then passed up to a one-sample mixed effects analysis (FLAME1), to identify mean S1 back and leg activations, across the entire group of participants. To be maximally sensitive to S1 back and leg regions, which are known to be localized in the most dorsal portions of the postcentral gyrus and in the paracentral lobule,62 these analyses were performed restricting the search area to a mask covering only the portions of the ‘postcentral gyrus’ label from the Harvard–Oxford Cortical Atlas superior to Z = 54 mm. In addition, using the same approach, we compared differences in S1 activations between cLBPAX and cLBPRAD, in an exploratory analysis. These analyses were also performed with FSL’s FEAT GLM tool, a cluster-forming threshold of Z = 2.3, and a cluster size significance threshold of P = 0.05 to correct for multiple comparisons. This cluster-forming threshold was used in this analysis to measure S1 activations from the leg that were not evident at a higher threshold (Z = 3.1). Indeed, the use of a cluster-forming threshold of P = 0.01 (which corresponds to Z = 2.3) with FSL FLAME1 provides an acceptable false error rate of around 5% (particularly in event-related designs).63
Because S1 demonstrated statistically significant differences in both region of interest and whole-brain voxelwise analyses, and largely overlapped the somatotopic representation of S1 localized with back/leg stimuli (see the ‘Results’ section), we performed seed-based functional connectivity analyses using the S1 cluster from the results of the voxelwise PET group analysis (see the ‘Results’ section). Further, because our previous studies demonstrated consistent elevations in thalamic 11C-PBR28 signal in cLBP patients compared to controls,21,22 our functional connectivity analysis was restricted to a search space comprising the thalamic labels from the Harvard–Oxford Subcortical Atlas (Center for Morphometric Analyses, http://www.cma.mgh.harvard.edu/fsl_atlas.html), to determine whether any thalamic regions showed stronger functional connectivity with S1 in cLBPRAD compared to cLBPAX. This analysis was also performed with FSL’s FEAT GLM tool and FLAME1 (www.fmrib.ox.ac.uk/fsl, version 5.0.10), a cluster-forming threshold of Z = 3.1 and a cluster size significance threshold of P = 0.05 to correct for multiple comparisons. To explore its clinical significance and relationship with neuroinflammation, S1-thalamus functional MRI connectivity strength was correlated with the S1 11C-PBR28 PET signal and the Fibromyalgia Survey Scores (the only behavioural measure significantly correlated with S1 11C-PBR28 PET signal; see the ‘Results' section). Again, for this analysis, we used a Pearson’s correlation and an alpha level corrected for two comparisons (0.05 / 2 = 0.025).
For visualization purposes, as well as for correlation analyses (see below), mean PET signal (SUVR) and mean functional MRI values (contrast of parameter estimates) were extracted from the significant clusters identified in the voxelwise PET and functional MRI analyses, and split in anatomically separate subregions using labels from the Harvard–Oxford Cortical Structural Atlas (Center for Morphometric Analyses, http://www.cma.mgh.harvard.edu/fsl_atlas.html), whenever applicable.
As both S1 11C-PBR28 signal and S1-thalamus connectivity correlated with each other and with Fibromyalgia Survey Scores (see ‘Results' section), we performed mediation analyses in a subset of patients with available survey scores (n = 35) to explore possible causal relationships between variables. We designed six mediation models using the Preacher and Hayes Indirect Mediation Analysis tool for SPSS,64 version 24 (IBM Corp., Armonk, NY), with the following independent (IV), mediator (M) and dependent variables (DV): Model 1, IV = S1 11C-PBR28 signal, M = Fibromyalgia Survey Scores, DV = S1-thalamus connectivity; Model 2, IV = Fibromyalgia Survey Scores, M = S1 11C-PBR28 signal, DV = S1-thalamus connectivity; Model 3, IV = S1-thalamus connectivity, M = Fibromyalgia Survey Scores, DV = S1 11C-PBR28 signal; Model 4, IV = Fibromyalgia Survey Scores, M = S1-thalamus connectivity, DV = S1 11C-PBR28 signal; Model 5, IV = S1-thalamus connectivity, M = S1 11C-PBR28 signal, DV = Fibromyalgia Survey Scores; and Model 6, IV = S1 11C-PBR28 signal, M = S1-thalamus connectivity, DV = Fibromyalgia Survey Scores. Unstandardized regression coefficients in this mediation model and bootstrapped 95% confidence intervals (CIs) for total and indirect effects of the independent variable on the dependent variable through mediator (5000 bootstrap samples) were estimated. The indirect (i.e. mediation) effect was considered statistically significant if the bias-corrected 95% CI did not include zero.
Data availability
Data will be made available upon reasonable request.
Results
Patient sample characteristics
Demographic and other key characteristics for all patients are displayed in Table 1. There was no significant difference in sex, age, TSPO polymorphism, injected dose, specific activity or injected mass between the cLBPAX and cLBPRAD groups (P > 0.05).
Table 1.
Participant characteristics
| Protocol 1 | Protocol 2 | Combined | ||||
|---|---|---|---|---|---|---|
| Radicular (n = 15) | Axial (n = 10) | Radicular (n = 13) | Axial (n = 16) | Radicular (n = 28) | Axial (n = 26) | |
| Age, years, | 49.6 ± 14.5 | 49.1 ± 17.3 | 49.6 ± 14.5 | 49.1 ± 17.3 | 48.3 ± 13.2 | 43.7 ± 16.6 |
| Sex, female:male, n | 8:5 | 10:6 | 8:7 | 5:5 | 16:12 | 15:11 |
| TSPO polymorphism | 10H; 3M | 12H; 4M | 13H; 2M | 8H; 2M | 23H; 5M | 20H; 6M |
| Body mass index, kg/m2 | 27.4 ± 5.6 | 23.6 ± 3.9 | 28.7 ± 4.3 | 24.0 ± 4.0 | 28.1 ± 4.9** | 23.9 ± 3.9 |
| Injected dose, MBq | 403.3 ± 45.4 | 432.1 ± 45.5 | 515.6 ± 41.1 | 531.0 ± 32.9 | 455.4 ± 71.2 | 492.9 ± 61.7 |
| Specific activity, GBq/μmol | 91.7 ± 29.4 | 67.4 ± 23.8 | 43.3 ± 15.0 | 41.3 ± 14.9 | 69.2 ± 34.0 | 51.4 ± 22.5 |
| Injected mass, μg | 2.4 ± 1.0 | 3.0 ± 2.3 | 5.5 ± 2.6 | 6.0 ± 2.7 | 3.8 ± 2.5 | 4.8 ± 2.9 |
| PainDetect (0–38) | 10.6 ± 4.5 (n = 5) | 9.6 ± 4.7 (n = 10) | 13.4 ± 4.7** | 7.6 ± 4.2 | 12.6 ± 4.7** (n = 18) | 8.4 ± 4.4 (n = 26) |
| Brief Pain Inventory (0–10) | 1.1 ± 0.6 (n = 12) | 1.0 ± 0.3 (n = 4) | 3.9 ± 1.8 | 3.2 ± 1.8 | 2.6 ± 1.2 (n = 25) | 2.8 ± 1.9 (n = 20) |
| Fibromyalgia Survey Scores | 8.6 ± 5.5 (n = 3) | 5.9 ± 2.0 (n = 3) | 8.7 ± 3.9 | 6.1 ± 2.9 | 8.6 ± 4.0* (n = 16) | 6.1 ± 2.7 (n = 19) |
| Back e-stim—Pain intensity | – | – | 25.6 ± 33.9 (n = 9) | 29.5 ± 36.5 (n = 12) | – | – |
| Right leg e-stim—Pain intensity | – | – | 28.1 ± 31.3 (n = 9) | 33.5 ± 29.6 (n = 12) | – | – |
| Left leg e-stim—Pain intensity | – | – | 39.4 ± 27.8 (n = 9) | 26.0 ± 36.5 (n = 12) | – | – |
Values in bold represents axial significantly different from radicular. TSPO polymorphism: H = high; M = mixed. *P < 0.05. **P < 0.01.
There was, however, a significant difference in body mass index across groups (cLBPAX: 23.9 ± 3.88; cLBPRAD: 28.1 ± 4.94; P = 0.001). Both cLBPRAD and cLBPAX demonstrated similar clinical pain intensity, as measured using the Brief Pain Inventory (P = 0.26). As expected by clinical subtyping, cLBPRAD reported significantly higher PainDetect scores, indicative of a more likely neuropathic component, than cLBPAX (P = 0.004) and higher Fibromyalgia Survey Scores (P = 0.02). All patients reported having perceived the electrical stimuli. Back, right leg and left leg stimuli were rated at 27.6 ± 34.4 (mean ± SD), 30.9 ± 29.7 and 32.4 ± 32.5, respectively, on a 0–100 pain intensity numerical rating scale. There was no significant difference in pain ratings between cLBPRAD and cLBPAX in any body region (P > 0.05; Table 1).
PET imaging results
When evaluating regions of interest from our previous 11C-PBR28 PET study in patients with cLBP,21 cLBPRAD demonstrated significantly elevated 11C-PBR28 PET signal compared to cLBPAX in S1 [F(1,50) = 5.7, P = 0.04, corrected], but no significant difference was observed in the thalamus [F(1,50) = 1.2, P = 0.57, corrected; Fig. 1]. In addition, the whole-brain voxelwise group comparison revealed 11C-PBR28 PET signal elevations in cLBPRAD compared to cLBPAX in S1 (in a cluster localized largely overlapping the one identified in our prior study, used in this study as our a priori S1 region of interest), as well as in the intraparietal sulcus, left and right white mattter and the posterior cingulate cortex (Fig. 2A and B). To explore the clinical significance of S1 neuroinflammation, the 11C-PBR28 PET signal in S1 was assessed for correlation with neuropathic and fibromyalgia symptom measures as both showed a significant difference across groups. S1 11C-PBR28 PET signal displayed a significant positive correlation with Fibromyalgia Survey Scores (r = 0.43, P = 0.026, corrected) but no significant correlation with PainDetect scores (r = 0.25, P = 0.11, corrected).
Figure 1.

Region of interest analyses. Group differences in 11C-PBR28 signal in a priori regions of interest. A priori regions were selected as they demonstrated 11C-PBR28 PET SUVR elevations in chronic low back pain patients compared to healthy controls.21 Average ± SD SUVR extracted showing differences between cLBPRAD and cLBPAX (adjusted for scanner and genotype). *Significant difference between groups (P < 0.05). Triangle denotes data from Protocol 1, circle denotes data from Protocol 2. The range of the y-axis is set depending on the distribution of individual data-points.
Figure 2.

Voxel-wise group differences in 11C-PBR28 signal. (A) Maps displaying areas with significantly elevated 11C-PBR28 SUVR in cLBPRAD compared to cLBPAX in a voxelwise analysis, adjusted for protocol and genotype. (B) Average ± SD SUVR extracted from several clusters identified as statistically significant in the voxelwise SUVR analysis from A (adjusted for scanner and genotype). The range of the y-axis is set depending on the distribution of individual data-points. (C) BOLD functional MRI localizing the somatotopic representation of S1 area for the Back+Leg and the overlap between Back+Leg e-stim and 11C-PBR28 SUVR signal in cLBPRAD > cLBPAX. IPS = intraparietal sulcus; PCC = posterior cingulate cortex; WM = white matter. Triangle denotes data from Protocol 1, circle denotes data from Protocol 2.
Functional MRI results
As shown in Fig. 2C, e-stim of back and legs revealed the expected dorsal S1 functional activations. Notably, the portion of the postcentral gyrus commonly activated by both back and right leg demonstrated a distinct overlap with the S1 area as identified in the PET analyses, indicating that the S1 neuroinflammatory signal in cLBPRAD was indeed localized to the representation of back and leg. There was no significant difference in voxelwise functional activation in cLBPAX compared to cLBPRAD. Because S1 demonstrated a significantly elevated 11C-PBR28 signal in both region of interest and whole-brain voxelwise analyses and largely overlapped the somatotopic representation of S1 localized with back/leg e-stim, we focused on this region for further analyses. The S1 cluster identified in the voxelwise group differences was used as a seed to compare connectivity to the thalamus between cLBPAX and cLBPRAD. cLBPRAD had stronger S1 connectivity to the right thalamus (in regions compatible with the ventral lateral posterior nucleus (VLp) and ventral posterior lateral nucleus compared to cLBPAX (Fig. 3A). The mean S1 connectivity values (Z-score) from this region is displayed in Fig. 3B. No thalamic nuclei were identified with stronger S1 connectivity in cLBPAX than cLBPRAD.
Figure 3.

Thalamic voxelwise group difference in connectivity with S1. (A) Volumetric maps displaying areas within the thalamus with significantly elevated connectivity with S1 (seed region of interest displayed on top left in green) in cLBPRAD compared to cLBPAX in a thalamic specific voxelwise analysis. (B) Average ± SD connectivity scores extracted from statistically significant cluster in the voxelwise connectivity analysis from A. Triangle denotes data from Protocol 1, circle denotes data from Protocol 2. Data adjusted for protocol. The range of the y-axis is set depending on the distribution of individual data-points. VLp = ventral lateral posterior nucleus; VPL = ventral posterior lateral nucleus.
To test the hypothesis that higher S1 connectivity to the ‘neuroinflammation-prone’ thalamus is accompanied by higher S1 neuroinflammatory signal, S1-thalamus connectivity was regressed against S1 11C-PBR28 PET signal. S1-thalamus connectivity was also regressed against the fibromyalgianess scores, as this was the only behavioural measure significantly correlated with S1 11C-PBR28 PET signal. S1-thalamus connectivity displayed significant positive correlation with S1 11C-PBR28 PET signal (r = 0.43, P = 0.004, corrected; Fig. 4) and Fibromyalgia Survey Scores (r = 0.57, P = 0.002, corrected; Fig. 5).
Figure 4.
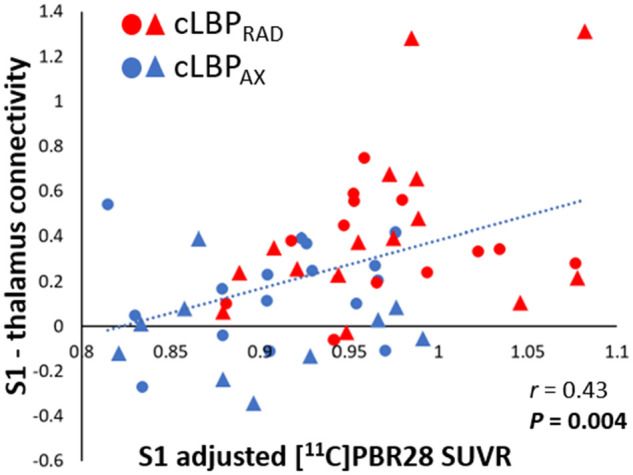
S1 11C-PBR28 signal correlates with S1-thalamus connectivity. The 11C-PBR28 SUVR signal was extracted from the S1 cluster that was significant in the PET group comparisons (Fig. 2). Connectivity scores were extracted from the thalamic cluster that was significant in the S1 connectivity analyses (Fig. 3). All data have been adjusted for protocol and genotype. Triangle denotes data from Protocol 1, circle denotes data from Protocol 2. The range of the y-axis is set depending on the distribution of individual data-points.
Figure 5.
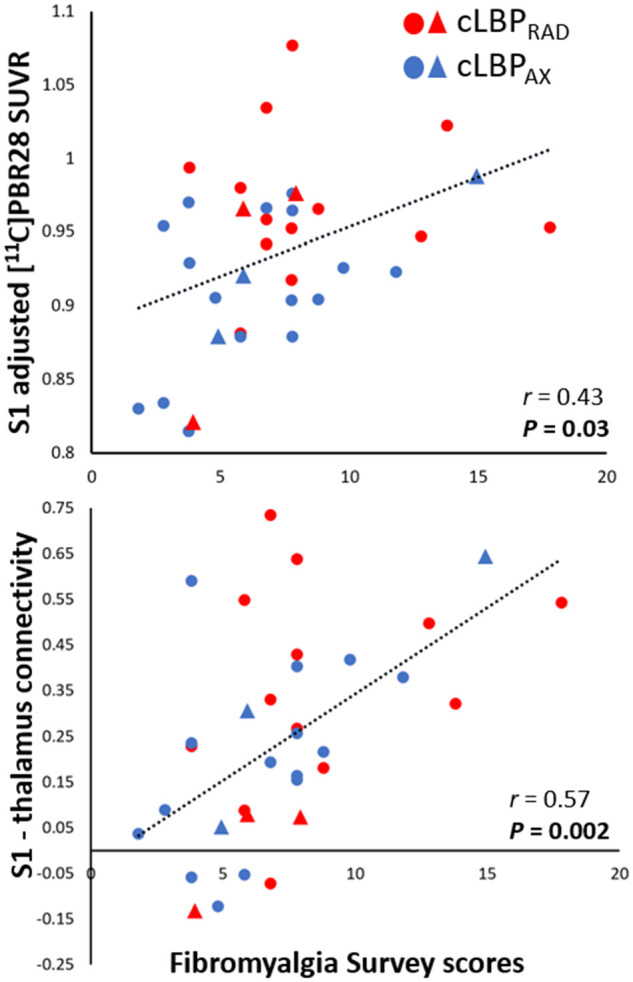
11C-PBR28 signal in the S1 and S1-thalamus connectivity correlations with Fibromyalgia Survey Scores. Top: Average SUVR was extracted from S1 (see Fig. 4 caption) and plotted against Fibromyalgia Survey Scores (data have been adjusted for scanner and genotype). Bottom: S1-thalamus connectivity values were extracted (see Fig. 4 caption) and plotted against Fibromyalgia Survey Scores (data have been adjusted for scanner). Triangle denotes data from Protocol 1, circle denotes data from Protocol 2. The range of the y-axis is set depending on the distribution of individual data-points.
Mediation between Fibromyalgia Survey Scores, PET signal and functional connectivity
As the Fibromyalgia Survey Scores, S1 11C-PBR28 PET signal, and S1-thalamus connectivity were cross-correlated (Figs 4 and 5), we ran six bootstrapped mediation models to investigate whether one variable mediated the relationship between the other two. Of these six models, Model 1 (IV = S1 11C-PBR28 PET signal; M = Fibromyalgia Survey Scores; DV = S1-thalamus connectivity) reached statistical significance. This model revealed that the strength of the association between the S1 11C-PBR28 PET signal and S1-thalamus connectivity (path c; β ± standard error: Model 1: 1.54 ± 0.74) was significantly reduced after accounting for the effects of the mediator, Fibromyalgia Survey Scores (path c′; Model 1: 0.59 ± 0.07). The bias-corrected 95% CIs for the indirect effect of S1 11C-PBR28 PET signal on S1-thalamus connectivity through Fibromyalgia Survey Scores (Model 1: path a × b; β = 0.95 ± 0.37) yielded a lower limit of 0.35 and an upper limit of 1.82. Thus, as the 95% CI range contains zero, Fibromyalgia Survey Scores significantly mediate the association between S1 11C-PBR28 PET signal and S1-thalamus connectivity (Fig. 6).
Figure 6.

Fibromyalgia Survey Scores mediate the relationship between S1-thalamus connectivity and S1 11C-PBR28 signal. A bootstrapped mediation analysis revealed that Fibromyalgia Survey Scores significantly mediated the relationship between S1-thalamus connectivity and S1 11C-PBR28 signal. Values within parentheses represent bootstrap standard errors for each path. *P < 0.05; **P < 0.01.
Discussion
Our investigations provide compelling evidence of neuroinflammatory and functional connectivity differences in subtypes of cLBP. Compared to cLBPAX, cLBPRAD patients showed elevated levels of TSPO, a neuroinflammatory marker, as measured with 11C-PBR28 PET. TSPO signal elevations were observed in several brain structures including S1, a statistically significant region in both region of interest and voxelwise analyses and overlapping functionally localized S1 representations of the back/leg. Compared to cLBPAX, cLBPRAD also demonstrated increased S1 functional connectivity to the thalamus, as measured with resting-state BOLD functional MRI. Indeed, S1 TSPO signal and S1-thalamus functional connectivity were significantly correlated, an association that was statistically mediated by the levels of ‘fibromyalgianess’, a measure of nociplastic pain.
While this study is the first to report neuroinflammatory differences between subtypes of cLBP, our results conform to a growing body of evidence suggesting that neuroinflammation might present at least partially distinct spatial patterns of signal distribution in different pain conditions.21,23–26 For instance, in patients with widespread pain (fibromyalgia) we observed TSPO signal elevations in large portions of S1,24 whereas only ventral or dorsal portions of this region were involved in migraineurs25 and cLBP,21 compatible with head/face and back/leg representations, respectively.62 These observations led us to hypothesize that, at least within this brain area, neuroinflammatory responses might present a somatotopic organization, paralleling the body distribution of the pain reported in each condition. In support of such hypothesis, in the present study we were able to directly show TSPO signal elevations in a portion of the postcentral gyrus overlapping with a functionally localized representation of back and leg in cLBPRAD patients (who report pain in back and leg), compared to cLBPAX patients (who present pain only in the back), while thalamic signal was comparable across groups. It should be noted that when we22 recently investigated a mixed group of cLBP patients that included patients with/without leg symptoms, our results replicated the thalamic, but not cortical, TSPO signal elevations observed in our initial study (which included only patients with both back and leg symptoms).21 One possible reason for this discrepancy is that inflammation in regions processing only back (or, perhaps, only leg) information might be too weak to be reliably detected, whereas inflammation in regions linked to processing of both leg and back pain may yield a stronger signal (hence the higher signal in cLBPRAD compared to cLBPAX in our study).
Radicular back pain is typically caused by damage to the dorsal root ganglion/roots causing inflammation and/or irritation, most commonly between L4 and S1, inducing pain that follows a dermatomal pattern to the lower extremity (i.e. thigh, calf and/or foot).32 Conversely, axial pain can be caused by damage such as muscle strain, facet joints and/or disc degeneration, and the pain is mostly localized within the lower back region.33 As such, cLBPRAD is typically considered a chronic pain condition with a neuropathic component (a result of damage or presumed damage to the nerve), while cLBPAX is more likely to be non-neuropathic in nature. Treatment for cLBP varies depending on the clinical presentation, as some pharmacological treatments may not work in all subtypes of cLBP, reflecting the mix of aetiologies and symptoms that a cLBP diagnosis subsumes.33 That different subtypes of cLBP have neuroinflammatory and neural signatures, as evidenced in this study, further supports that different clinical presentations may be accompanied by distinct neuroimmune mechanisms.
Our observation that S1-thalamus connectivity was linked to higher S1 TSPO signal is notable. The thalamus is a critical structure that transmits ascending nociceptive information to various parts of the cortex, including S1, through direct connections65,66 and has been found in multiple studies by our group to show consistent TSPO signal elevations in cLBP patients compared to healthy controls.21,22 While the mechanisms mediating the relationship between functional connectivity and inflammation remains unknown, one possibility is that elevated S1-thalamus connectivity in some patients (cLBPRAD) may serve as a ‘vehicle’ for neuroinflammation to spread ‘trans-synaptically’67 from the thalamic ‘neuroinflammatory hub’ to the cortex. Indeed, microglial activation can be observed remotely from the location of the original pathological event, spreading along the affected neural pathways.67 Notably, this trans-synaptic neuroinflammatory spread can be driven by alterations in neuronal input. For instance, in a rat model of Huntington’s disease, neuronal hyperexcitation in the striatum of the basal ganglia (through the removal of inhibitory GABAergic input) was shown to trigger trans-synaptic microglial activation in the thalamus.68 Furthermore, in rats, c-fibre stimulation in the sciatic nerve causes a connexin dephosphorylation in the spinal cord and an increase in the number of astrocyte gap junctions, a rise in astrocytic intracellular calcium concentrations within seconds and microglial activation within minutes.35,69 These activated glial cells may then release excessive amounts of glutamate, causing excitotoxity and, more pertinently, sensitizing the neural pathways.70 For example, capsaicin-induced sensitization of the primate spinothalamic tract was exacerbated by infusion of glutamate receptor agonists.71 As such, continuous or aberrant excitatory input from the thalamus to S1 in cLBPRAD may lead to neurogenic neuroinflammation (neuroinflammation due to aberrant neuronal activation) in S1. Interestingly, the thalamic nuclei in which we found an increased connectivity with S1 in cLBPRAD compared to cLBPAX largely overlaps the ventral posterior lateral nucleus, which transmits sensory information from the body to S1. Hence, it is possible that in some patients continuous excitatory input may be transmitted from the periphery, thereby causing neurogenic neuroinflammation.
Another means by which changes in functional connectivity may influence glial activity is through promoting stripping of dysfunctional synapses.72,73 Microglial cells express a variety of receptors for neurotransmitters, neuropeptides and neuromodulators that allow these cells to respond to neuronal activity.73 Cell culture studies have shown that stimulation of these receptors activates microglia,74 which can then remove dysfunctional synapses in the brain by engulfing presynaptic inputs.73 For example, the complement component 1q protein, the protease enzyme and the inflammatory cytokine tumour necrosis factor-α (TNF-α) all mediate synaptic stripping and, remarkably, are all upregulated by microglia in neuropathic pain models.75
Conversely, the association between S1 neuroinflammation and S1-thalamus connectivity may reflect the effects of glial cells on neuronal communication. Preclinical models have shown that glial cells can modulate neuronal activity by expressing receptors that alter synaptic function, such as fraktaline receptors (a transmembrane chemokine), which increase pro-inflammatory cytokines when activated and inhibit pro-inflammatory cytokines when attenuated.34,76,77 These changes in cytokine concentration modulate presynaptic neurotransmitter release, and may contribute to changes in functional connectivity. Furthermore, in mice models of optic nerve crush, resident microglia, mediated by complement proteins (not neuronal activity), engulf synaptic material at distal targets,78 which may modulate neuronal communication. Further, in our current mediation analysis, we found that our chosen measure of nociplastic pain, Fibromyalgia Survey Scores, mediated the relationship between functional connectivity and 11C-PBR28 PET, when functional connectivity was a dependent variable (Model 1). Therefore, our data also suggest that neuroinflammation might precede and perhaps modulate functional connectivity in this cohort, potentially as a function of the degree of nociplastic pain. Nonetheless, a broader interpretation of the mediational role of nociplastic pain warrants further investigation and validation, particularly because the fibromyalgianess data were available only in a subset of the participants evaluated in this study.
While it is possible that neurogenic inflammation is driving the difference between cLBP subtypes (mediated by the degree of nociplastic pain), other mechanisms of action must not be ignored. For example, peripheral activation of the immune response can transport cytokines such as TNF-α into the spinal cord to activate glial cells.35 In the chronic constriction injury model of sciatic neuropathy, TNF-α was transported in sensory fibres from the dorsal root ganglion to the spinal cord.79 Furthermore, lumbar spine compression in mice increased the blood–brain barrier permeability in the spinal cord and in the brain, allowing the increased entry of TNF-α and other immune cells into the brain.80 In this study, patients with cLBPRAD had a significantly higher neuropathic component than cLBPAX as measured by PainDetect. This suggests peripheral nerve involvement, which may drive recruitment of immune cells into the brain, thus activating the neuroinflammatory response.
Several caveats should be considered when interpreting the results of our study. For instance, the cross-sectional nature of our study makes it impossible to resolve the causality between neuroinflammation, alterations of functional connectivity and nociplastic pain. Preclinical studies and longitudinal analyses may further enhance our understanding of the relationship between the three parameters. Moreover, our data were collected using two distinct protocols and scanners. Nonetheless, the differences between cLBPRAD and cLBPAX in the imaging and clinical variables observed in this analysis were still evident when protocols were split (Table 1 and Supplementary Table 1). The consistency across protocols increased our confidence that the results obtained in the full datasets are not reflective of artefacts, but rather are indicative of genuine neuroimmune differences across cLBP subtypes.
It should also be noted that our results used SUVR images that only enable semiquantitative analyses as opposed to other commonly adopted alternatives (such as volume of distribution, VT) due to the limited number of patients with arterial blood sampling. Nonetheless, we have previously utilized SUVR for quantification of 11C-PBR28 PET data (using either whole-brain or localized regions) in patients with cLBP,21,22 fibromyalgia,36 Gulf War illness26 and other conditions. The validity of SUVR as an outcome measure for 11C-PBR28 PET is supported by a growing number of studies. For instance, studies of neurodegenerative disorders have demonstrated statistically significant, reproducible and regionally specific SUVR elevations in structures where neurodegeneration is known to occur, such as primary motor cortex (M1) and corticospinal tracts in amyotrophic lateral sclerosis37,59,81,82 and primary lateral sclerosis,83 or the basal ganglia in Huntington’s disease,84 or again in temporoparietal regions in Alzheimer’s disease.85 Not only are SUVR elevations co-localized with the areas known to be pathological, they can be proportional to disease severity. In amyotrophic lateral sclerosis, for instance, SUVR in M1 was found to be (i) positively correlated to clinical severity (upper motor neuron burden); (ii) positively correlated with the levels of myo-inositol (another putative marker of neuroinflammation), measured using MRI; and (iii) negatively correlated with measures of structural integrity (cortical thickness, measured using morphometric analyses from structural MRI and fractional anisotropy, measured using diffusion tensor imaging).81,82,86 Collectively these data support the validity of SUVR as a measure for TSPO binding in certain populations.
In conclusion, our data support the existence of different ‘neuroinflammatory signatures’ in patients with different clinical presentation, and that S1 neuroinflammatory signal is more pronounced in patients with higher ‘nociplastic’ pain. Further, because S1 TSPO signal was correlated with S1-thalamus connectivity, our data support an association between changes in neuroinflammation and neuronal communication, possibly indicating that the observed alterations reflect neurogenic neuroinflammation. Future preclinical studies will be necessary to determine the underlying mechanisms of these relationships and to determine whether neuroinflammation and related connectivity changes may contribute to the subtyping of distinct pain syndromes and also provide information about interindividual variability in neuroimmune brain signals, within diagnostic groups, that could eventually serve as targets for mechanism-based precision medicine approaches.
Supplementary Material
Acknowledgements
We would like to thank Mackenzie Hymen for assistance with electrical stimulation equipment; Courtney Bergan for assistance with patient recruitment, data acquisition and logistical support; Grae Arabasz, Regan Butterfield and Shirley Hsu and the A.A. Martinos Radiochemistry team for producing and administering the radioligand.
Funding
This work was supported by National Institutes of Health (NIH) 1R21NS087472-01A1 (M.L.L.), 1R01NS095937-01A1 (M.L.L.), 1R01NS094306-01A1 (M.L.L.) and Department of Defense (DoD) W81XWH-14–1-0543 (M.L.L.).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- BOLD
blood oxygen level-dependent
- cLBP
chronic low back pain
- cLBPAX/RAD
axial/radicular chronic low back pain
- e-stim
electrical stimulation
- S1
primary somatosensory cortex
- SUV
standardized uptake value
References
- 1.Taylor AM, Mehrabani S, Liu S, Taylor AJ, Cahill CM. Topography of microglial activation in sensory‐ and affect‐related brain regions in chronic pain. J Neurosci Res. 2017;95(6):1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mannelli LDC, Pacini A, Bonaccini L, Zanardelli M, Mello T, Ghelardini C. Morphologic features and glial activation in rat oxaliplatin-dependent neuropathic pain. J Pain. 2013;14(12):1585–1600. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto K, Kume K, Ohsawa M. Role of microglia in mechanical allodynia in the anterior cingulate cortex. J Pharmacol Sci. 2017;134(3):158–165. [DOI] [PubMed] [Google Scholar]
- 4.Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. . P2X 4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. [DOI] [PubMed] [Google Scholar]
- 5.Watkins LR, Hutchinson MR, Johnson KW. Commentary on Landry et al.: “Propentofylline, a CNS glial modulator, does not decrease pain in post-herpetic neuralgia patients: In vitro evidence for differential responses in human and rodent microglia and macrophages”. Exp Neurol. 2012;234(2):351–353. [DOI] [PubMed] [Google Scholar]
- 6.Calvo M, Bennett DL. The mechanisms of microgliosis and pain following peripheral nerve injury. Exp Neurol. 2012;234(2):271–282. [DOI] [PubMed] [Google Scholar]
- 7.Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012;11(7):629–642. [DOI] [PubMed] [Google Scholar]
- 8.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20(3):269–287. [DOI] [PubMed] [Google Scholar]
- 9.Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. [DOI] [PubMed] [Google Scholar]
- 10.Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol Rev. 2014;94(4):1077–1098. [DOI] [PubMed] [Google Scholar]
- 11.Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26(16):4308–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji R-R, Donnelly CR, Nedergaard M. Astrocytes in chronic pain and itch. Nat Rev Neurosci. 2019;20(11):667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledeboer A, Sloane EM, Milligan ED, et al. . Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115(1–2):71–83. [DOI] [PubMed] [Google Scholar]
- 14.Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306(2):624–630. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Chen X, Zhang C, Zhang Y, Yao W. An update on reactive astrocytes in chronic pain. J Neuroinflamm. 2019;16(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain (London, England). 2007;11(2):223–230. [DOI] [PubMed] [Google Scholar]
- 17.Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7(11):816–822. [DOI] [PubMed] [Google Scholar]
- 18.LeBlanc BW, Zerah ML, Kadasi LM, Chai N, Saab CY. Minocycline injection in the ventral posterolateral thalamus reverses microglial reactivity and thermal hyperalgesia secondary to sciatic neuropathy. Neurosci Lett. 2011;498(2):138–142. [DOI] [PubMed] [Google Scholar]
- 19.Guasti L, Richardson D, Jhaveri M, et al. . Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol Pain. 2009;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padi SS, Kulkarni SK. Minocycline prevents the development of neuropathic pain, but not acute pain: Possible anti-inflammatory and antioxidant mechanisms. Eur J Pharmacol. 2008;601(1–3):79–87. [DOI] [PubMed] [Google Scholar]
- 21.Loggia ML, Chonde DB, Akeju O, et al. . Evidence for brain glial activation in chronic pain patients. Brain. 2015;138(Pt 3):604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torrado-Carvajal A, Toschi N, Albrecht DS, et al. . Thalamic neuroinflammation as a reproducible and discriminating signature for chronic low back pain. Pain. 2021;162(4):1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albrecht DS, Ahmed SU, Kettner NW, et al. . Neuroinflammation of the spinal cord and nerve roots in chronic radicular pain patients. Pain. 2018;159(5):968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albrecht DS, Forsberg A, Sandström A, et al. . Brain glial activation in fibromyalgia–A multi-site positron emission tomography investigation. Brain Behav Immun. 2019;75:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albrecht DS, Mainero C, Ichijo E, et al. . Imaging of neuroinflammation in migraine with aura: A [11C] PBR28 PET/MRI study. Neurology. 2019;92(17):e2038–e2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alshelh Z, Albrecht DS, Bergan C, et al. . In-vivo imaging of neuroinflammation in veterans with Gulf War illness. Brain Behav Immun. 2020;87:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): Molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernstadt H, Wang S, Lim G, Mao J. Spinal translocator protein (TSPO) modulates pain behavior in rats with CFA-induced monoarthritis. Brain Res. 2009;1286:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei X-H, Wei X, Chen F-Y, et al. . The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci. 2013;33(4):1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown AK, Fujita M, Fujimura Y, et al. . Radiation dosimetry and biodistribution in monkey and man of 11C-PBR28: A PET radioligand to image inflammation. J Nucl Med. 2007;48(12):2072–2079. [DOI] [PubMed] [Google Scholar]
- 31.Briard E, Zoghbi SS, Imaizumi M, et al. . Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J Med Chem. 2008;51(1):17–30. [DOI] [PubMed] [Google Scholar]
- 32.Patel EA, Perloff MD. Radicular pain syndromes: Cervical, lumbar, and spinal stenosis. Semin Neurol. 2018;38(6):634–639. [DOI] [PubMed] [Google Scholar]
- 33.Urits I, Burshtein A, Sharma M, et al. . Low back pain, a comprehensive review: Pathophysiology, diagnosis, and treatment. Curr Pain Head Rep. 2019;23(3):23. [DOI] [PubMed] [Google Scholar]
- 34.Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkühler J. Selective activation of microglia facilitates synaptic strength. J Neurosci. 2015;35(11):4552–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: Inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15(1):43–53. [DOI] [PubMed] [Google Scholar]
- 36.Albrecht D, Kim M, Akeju O, et al. . The neuroinflammatory component of negative affect in patients with chronic pain. Mol Psychiatry. 2021;26(3):864–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albrecht DS, Normandin MD, Shcherbinin S, et al. . Pseudoreference regions for glial imaging with (11)C-PBR28: Investigation in 2 clinical cohorts. J Nucl Med. 2018;59(1):107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clow A, Glover V, Sandler M. Triazolam, an anomalous benzodiazepine receptor ligand: In vitro characterization of alprazolam and triazolam binding. J Neurochem. 1985;45(2):621–625. [DOI] [PubMed] [Google Scholar]
- 39.Gehlert DR, Yamamura HI, Wamsley JK. Autoradiographic localization of ‘peripheral-type’ benzodiazepine binding sites in the rat brain, heart and kidney. Naunyn-Schmiedeberg's Arch Pharmacol. 1985;328(4):454–460. [DOI] [PubMed] [Google Scholar]
- 40.Canat X, Carayon P, Bouaboula M, et al. . Distribution profile and properties of peripheral-type benzodiazepine receptors on human hemopoietic cells. Life Sci. 1993;52(1):107–118. [DOI] [PubMed] [Google Scholar]
- 41.Wamsley JK, Longlet LL, Hunt ME, Mahan DR, Alburges ME. Characterization of the binding and comparison of the distribution of benzodiazepine receptors labeled with [3H]Diazepam and [3H]Alprazolam. Neuropsychopharmacology. 1993;8(4):305–314. [DOI] [PubMed] [Google Scholar]
- 42.Kalk NJ, Owen DR, Tyacke RJ, et al. . Are prescribed benzodiazepines likely to affect the availability of the 18 kDa translocator protein (TSPO) in PET studies? Synapse (New York, NY). 2013;67(12):909–912. [DOI] [PubMed] [Google Scholar]
- 43.Owen DR, Howell OW, Tang SP, et al. . Two binding sites for [3H]PBR28 in human brain: Implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metabol. 2010;30(9):1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owen DR, Yeo AJ, Gunn RN, et al. . An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metabol. 2012;32(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolb A, Wehrl HF, Hofmann M, et al. . Technical performance evaluation of a human brain PET/MRI system. Eur Radiol. 2012;22(8):1776–1788. [DOI] [PubMed] [Google Scholar]
- 46.Catana C, Benner T, van der Kouwe A, et al. . MRI-assisted PET motion correction for neurologic studies in an integrated MR-PET scanner. J Nucl Med. 2011;52(1):154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delso G, Fürst S, Jakoby B, et al. . Performance measurements of the Siemens mMR integrated whole-body PET/MR scanner. J Nucl Med. 2011;52(12):1914–1922. [DOI] [PubMed] [Google Scholar]
- 48.Imaizumi M, Kim HJ, Zoghbi SS, et al. . PET imaging with [11C]PBR28 can localize and quantify upregulated peripheral benzodiazepine receptors associated with cerebral ischemia in rat. Neurosci Lett. 2007;411(3):200–205. [DOI] [PubMed] [Google Scholar]
- 49.Izquierdo-Garcia D, Hansen AE, Forster S, et al. . An SPM8-based approach for attenuation correction combining segmentation and nonrigid template formation: Application to simultaneous PET/MR brain imaging. Journal of nuclear medicine: Official publication. Soc Nucl Med. 2014;55(11):1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freynhagen R, Baron R, Gockel U, Tölle TR. Pain DETECT: A new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opinion. 2006;22(10):1911–1920. [DOI] [PubMed] [Google Scholar]
- 51.Cleeland C. Measurement of pain by subjective report. Adv Pain Res Ther. 1989;12:391–403. [Google Scholar]
- 52.Wolfe F, Clauw DJ, Fitzcharles MA, et al. . Fibromyalgia criteria and severity scales for clinical and epidemiological studies: A modification of the ACR Preliminary Diagnostic Criteria for Fibromyalgia. J Rheumatol. 2011;38(6):1113–1122. [DOI] [PubMed] [Google Scholar]
- 53.Wolfe F, Clauw DJ, Fitzcharles MA, et al. . The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010;62(5):600–610. [DOI] [PubMed] [Google Scholar]
- 54.Wolfe F. Fibromyalgianess. Arthritis Rheum. 2009;61(6):715–716. [DOI] [PubMed] [Google Scholar]
- 55.Ellingsen DM, Beissner F, Moher Alsady T, et al. . A picture is worth a 1000 words: Linking fibromyalgia pain widespreadness from digital pain drawings with pain catastrophizing and brain cross-network connectivity. Pain. 2021;162(5):1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brummett CM, Janda AM, Schueller CM, et al. . Survey criteria for fibromyalgia independently predict increased postoperative opioid consumption after lower-extremity joint arthroplasty: A prospective, observational cohort study. Anesthesiology. 2013;119(6):1434–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harper DE, Sayre K, Schrepf A, Clauw DJ, Aronovich S. Impact of fibromyalgia phenotype in temporomandibular disorders. Pain Med. 2021;22(9):2050–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bowen SL, Byars LG, Michel CJ, Chonde DB, Catana C. Influence of the partial volume correction method on (18)F-fluorodeoxyglucose brain kinetic modelling from dynamic PET images reconstructed with resolution model based OSEM. Phys Med Biol. 2013;58(20):7081–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zürcher NR, Loggia ML, Lawson R, et al. . Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: Assessed with [(11)C]-PBR28. NeuroImage Clin. 2015;7:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujita M, Imaizumi M, Zoghbi SS, et al. . Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage. 2008;40(1):43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Behzadi Y, Restom K, Liau J, Liu TT. A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage. 2007;37(1):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penfield W, Boldrey E. Somatic motor and sensory representation in the cerebral cortex of man as studied by electrical stimulation. Brain. 1937;60(Pt 4):389–443. [Google Scholar]
- 63.Eklund A, Nichols TE, Knutsson H. Cluster failure: Why fMRI inferences for spatial extent have inflated false-positive rates. Proc Natl Acad Sci U S A. 2016;113(28):7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Preacher KJ, Hayes AF. Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods. 2008;40(3):879–891. [DOI] [PubMed] [Google Scholar]
- 65.Ab Aziz CB, Ahmad AH. The role of the thalamus in modulating pain. Malays J Med Sci. 2006;13(2):11–18. [PMC free article] [PubMed] [Google Scholar]
- 66.Yen C-T, Lu P-L. Thalamus and pain. Acta Anaesthesiol Taiwanica. 2013;51(2):73–80. [DOI] [PubMed] [Google Scholar]
- 67.Banati RB, Cagnin A, Brooks DJ, et al. . Long-term trans-synaptic glial responses in the human thalamus after peripheral nerve injury. Neuroreport. 2001;12(16):3439–3442. [DOI] [PubMed] [Google Scholar]
- 68.Töpper R, Gehrmann J, Schwarz M, Block F, Noth J, Kreutzberg GW. Remote microglial activation in the quinolinic acid model of Huntington's disease. Exp Neurol. 1993;123(2):271–283. [DOI] [PubMed] [Google Scholar]
- 69.Li WE, Nagy JI. Activation of fibres in rat sciatic nerve alters phosphorylation state of connexin-43 at astrocytic gap junctions in spinal cord: Evidence for junction regulation by neuronal–glial interactions. Neuroscience. 2000;97(1):113–123. [DOI] [PubMed] [Google Scholar]
- 70.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neugebauer V, Chen P-S, Willis WD. Role of metabotropic glutamate receptor subtype mGluR1 in brief nociception and central sensitization of primate STT cells. J Neurophysiol. 1999;82(1):272–282. [DOI] [PubMed] [Google Scholar]
- 72.Paolicelli RC, Bolasco G, Pagani F, et al. . Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. [DOI] [PubMed] [Google Scholar]
- 73.Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: New roles for the synaptic stripper. Neuron. 2013;77(1):10–18. [DOI] [PubMed] [Google Scholar]
- 74.Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. [DOI] [PubMed] [Google Scholar]
- 75.Ward H, West SJ. Microglia: Sculptors of neuropathic pain? R Soc Open Sci. 2020;7(6):200260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ren K, Dubner R. Activity-triggered tetrapartite neuron–glial interactions following peripheral injury. Curr Opin Pharmacol. 2016;26:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: Dynamic mediators of synapse development and plasticity. Trends Immunol. 2015;36(10):605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Norris GT, Smirnov I, Filiano AJ, et al. . Neuronal integrity and complement control synaptic material clearance by microglia after CNS injury. J Exp Med. 2018;215(7):1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shubayev VI, Myers RR. Axonal transport of TNF-α in painful neuropathy: Distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001;114(1–2):48–56. [DOI] [PubMed] [Google Scholar]
- 80.Pan W, Kastin AJ, Bell RL, Olson RD. Upregulation of tumor necrosis factor α transport across the blood–brain barrier after acute compressive spinal cord injury. J Neurosci. 1999;19(9):3649–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alshikho MJ, Zürcher NR, Loggia ML, et al. . Glial activation colocalizes with structural abnormalities in amyotrophic lateral sclerosis. Neurology. 2016;87(24):2554–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alshikho MJ, Zürcher NR, Loggia ML, et al. . Integrated magnetic resonance imaging and [11C]‐PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol. 2018;83(6):1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paganoni S, Alshikho MJ, Zürcher NR, et al. . Imaging of glia activation in people with primary lateral sclerosis. NeuroImage Clin. 2018;17:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lois C, González I, Izquierdo-García D, et al. . Neuroinflammation in Huntington’s disease: New insights with 11C-PBR28 PET/MRI. ACS Chem Neurosci. 2018;9(11):2563–2571. [DOI] [PubMed] [Google Scholar]
- 85.Lyoo CH, Ikawa M, Liow J-S, et al. . Cerebellum can serve as a pseudo-reference region in Alzheimer disease to detect neuroinflammation measured with PET radioligand binding to translocator protein. J Nucl Med. 2015;56(5):701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ratai E-M, Alshikho MJ, Zürcher NR, et al. . Integrated imaging of [(11)C]-PBR28 PET, MR diffusion and magnetic resonance spectroscopy (1)H-MRS in amyotrophic lateral sclerosis. NeuroImage Clin. 2018;20:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available upon reasonable request.