

Abstract
Unhealthy lifestyle factors, such as obesity, disrupt organismal homeostasis to accelerate cancer pathogenesis, in part via metabolic and immune dysregulation. Exercise is a prototypical strategy that maintains and restores homeostasis at organismal, tissue, cellular, and molecular levels with the capacity to prevent or inhibit numerous disease conditions, including cancer. Here we review unhealthy lifestyle factors that contribute to metabolic and immune dysregulation to drive tumourigenesis, focusing on the patient physiology (host) – tissue / tumour microenvironment interaction. We also discuss how exercise may sculpt distant tissue microenvironments to improve tissue function through both metabolic and immune-specific pathways. Finally, we consider future directions that merit consideration in basic and clinical translational exercise studies.
Introduction
Achievement of the World Health Organization’s (WHO) objective of a 25% reduction in cancer mortality rates by 2025 will require, among many elements, effective cancer prevention strategies. The nature of cancer prevention strategies should, in turn, be guided by epidemiological data deciphering the major determinants of cancer pathogenesis. These data provide clear insight: modifiable lifestyle factors are the strongest determinants for the most common cancers, responsible for 35% to 50% of all diagnoses and cancer-related deaths.1,2 Indeed, factors related to host energy balance (e.g., poor diet and inactivity) now directly competes with tobacco smoking as the primary preventable cause of cancer.
From an integrative physiology perspective, phenotypes such as chronic positive energy balance, (i.e., obese phenotype) are associated with aberrant mobilization, recruitment, retention and function of specific cell types and/or molecules collectively dysregulating numerous regulatory networks, including metabolism, hormonal regulation, immunity and oxidative balance at the host, tissue or tumour microenvironment (TME), cellular, and molecular levels.3,4 All regulatory networks, and the interplay between them, play crucial roles in tumourigenesis, however metabolism and immune function are particularly germane.5
Altered cellular metabolism is a hallmark of cancer pathogenesis.5,6 Reprogramming of cancer cell metabolism dramatically alters metabolite influx, pathway regulation, and cell fate decisions that drive tumourigenesis.6 Such reprogramming also alters the extracellular metabolite composition in the surrounding microenvironment, driving phenotypic shifts in stromal cells to further propagate tumour growth.7 Altered or misled immune responses and the inflammatory TME are also established drivers of cancer.5,8 Cancer cell evasion of immunological destruction from T and B lymphocytes, macrophages, and natural killer (NK) cells is a feature of every occult tumour escaping the bonds of immune surveillance.5,9 TME accumulation of innate immune cells such as monocytes, neutrophils and macrophages can further promote tumourigenesis via angiogenesis, hyperproliferation, suppression of NK and T cell cytotoxicity, and metastatic dissemination.5,8,10,11
Regulatory networks, including metabolism and immune function, do not operate in isolation, but instead are highly inter-dependent in order to maintain organismal homeostasis.12 One example of such interplay is the metabolic reprogramming of cancer cells, which induces an acidic, nutrient-depleted TME, driving accumulation of immune suppressive cell (pheno)types, inhibition of tumour antigen presentation and T cell activation which collectively inhibit appropriate antitumour immune responses.6 At the cellular level, the discovery of the intricate link between metabolite and nutrient availability in the TME and alteration of immune-effector function due to metabolic rewiring, further illustrates the interdependence of such regulatory networks in cancer.7,13–16
Effective targeting of metabolic and immune dysregulation in cancer are areas of immense discovery and therapeutic efforts. Work to date has mostly adopted the classic paradigm of targeting single molecules within an individual regulatory network.17–22 Since molecules operate within pathways that interact to form larger regulatory networks and integrative systems, complementary strategies with the capacity to regulate multiple higher order networks may represent an alternative, more effective therapeutic approach. Exercise, defined as structured, repeated and purposeful physical activity with the objective of improving health or cardiorespiratory fitness, is one such strategy. Observational data suggests that exercise may reduce the primary risk of multiple forms of cancer23 as well as the risk of recurrence in certain solid tumours.24 Preclinical studies confirm the biological plausibility of exercise-induced inhibition of tumourigenesis in multiple cancer models.25 The underlying mechanisms of how exercise inhibits or delays tumourigenesis remain elusive, however reprogramming of metabolic and immune dysregulation are likely key facets of its antitumour effects.25,26
In this Review, we provide an overview of how certain unhealthy lifestyle behaviors induce metabolic and immune dysregulation at the level of the host and tissue/TME to drive tumourigenesis, and how exercise may regulate these processes to re-establish homeostasis. Finally, we discuss future directions that merit consideration in basic and clinical translational studies.
Unhealthy lifestyle factors, metabolic and immune dysregulation, and cancer
As reviewed previously,27–31 obesity – the phenotypic manifestation of chronic excess nutritional intake in the context of insufficient physical activity – provides a prototypical example of how environmental and lifestyle factors drive dysregulation of the immune-metabolism axis at the organismal, TME, and cellular levels to facilitate tumourigenesis.28
Briefly, aberrant availability of key metabolic growth factors such as glucose, insulin, insulin-like growth factor (IGF-1), and leptin stimulate chronic activation of numerous growth factor signaling pathways that promote cell growth, survival and proliferation, which coupled with increased concentrations of mutagenic substances (e.g. increased reactive oxygen species) and epigenetic shifts in gene regulation, collectively lower the barrier for cells to undergo oncogenic transformation, as well as drive progression following transformation.2,29 Obesity also drives inflammation and immune dysregulation.27,28,30,31 The outgrowth and hypertrophy of adipocytes in various tissue and fat depots lead to hypoxia, adipocyte stress and death.32 This stimulates the production of pro-inflammatory mediators and tissue-specific recruitment and accumulation of innate immune cells (e.g. neutrophils, monocytes, and macrophages), promoting chronic activation of cellular proliferation and survival pathways, providing an alternative driver of tumourigenesis.27,28
The TME in obese states is also immunosuppressed.15 For example, in obese mouse models of breast cancer, myeloid-derived suppressor cells (MDSCs) in the TME upregulate the immune checkpoint molecule programmed death-ligand 1 (PD-L1), induced by intratumoural (IFN)-γ, resulting in impaired CD8+ T cell function.33 Furthermore, CD4+ and CD8+ T cells across a variety of animal models display upregulation of programmed cell death receptor-1 (PD-1) expression and impaired proliferative responses in the obese state, as well as CD8+ T cell exhaustion (higher frequency of PD-1, Tim3 and Lag3 and decreased Ki67+ cells) in the TME.34 Recent discoveries also reveal metabolic and immune crosstalk in obesity. Obesity-induced increased circulating 27-hydroxycholesterol (27HC) induces accumulation of polymorphonuclear-neutrophils and gammadelta (γδ)-T cells in the lung, facilitating metastatic seeding in mouse models of breast cancer.35
In related work, obesity-driven breast cancer growth occurred in conjunction with increased adipocyte release of leptin, a potent regulator of energy balance, which activated signal transducer and activator of transcription 3 (STAT3) in tumour infiltrating CD8+ T cells, increasing intracellular fatty acid oxidation and inhibiting glycolysis, resulting in dampened effector functions.36 Finally, in a murine melanoma model, Michelet et al. reported obesity enhanced natural killer (NK) cell lipid accumulation, resulting in intracellular metabolic paralysis through interference of the mammalian target of rapamycin (mTOR) - peroxisome proliferator-activated receptor (PPAR) pathways, and loss of cytotoxic function which accelerated melanoma growth.37
Of interest, several additional host-related acute perturbations such as surgery,38 acute myocardial infarction,39 and heart failure40–42 accelerate tumourigenesis through immune and/or metabolic dysregulation. Although the extent and temporal effects of these events on reprogramming of immuno-metabolic regulation and tumourigenesis is obviously distinct from the chronic nature of obesity and other lifestyle factors, these emerging data further highlight the importance of host response in cancer pathogenesis.
Exercise-induced regulation of immuno-metabolism in normal tissues
Exercise is a potent challenge to homeostasis that engages numerous regulatory systems at the organismal, tissue, and cellular level but in contrast to unhealthy lifestyle factors stimulates physiological (e.g. favorable) adaptation to promote enhanced performance and function.43,44 The complex and highly coordinated response underlying exercise-induced physiological adaptation is extensively reviewed in prior work.44,45 In brief, exercise stimulates inter-organ communication characterized by complex interplay between organs such as skeletal muscle, heart, bone, liver and adipose tissue. Inter-organ communication is regulated through paracrine and endocrine signaling facilitating subsequent reprogramming of multiple regulatory systems, including metabolism and immunity.26,46 For instance, in their accompanying review, Murphy et al. overview how exercise is a major regulator of host metabolism in health and disease states. Over time, the cumulation of these organismal-level adaptations, which are the product of integrated cell and tissue-specific adjustments across a range of tissues, establish a higher homeostatic ‘set point’, fostering enhanced performance, and tolerance to system stress.47
A fundamental question in exercise biology is whether the beneficial adaptations observed in organs directly engaged in, and responsible for the cardiovascular / respiratory response to exercise (e.g. heart, lung, vascular, skeletal muscle), organs that enable the convective delivery of oxygen from the environment to the skeletal muscle mitochondria, termed the oxygen cascade.48 To support the exercise response, other non-cardiovascular tissues organs play a central role including the brain (e.g. central nervous system control), liver (e.g. release of glucose), and adipose tissue (e.g. release of free fatty acids), adrenal glands (e.g. release of adrenaline and cortisol) and pancreas (e.g. release of glucagon).26,44,45 While the contribution of multiple organs to the exercise response is well documented, understanding of how chronic exercise alters the biology of these and other distal tissues at the cellular and molecular level is almost exclusively confined to skeletal muscle.44,49 However, several emergent findings provide initial exciting insights that exercise not only significantly regulates biological processes in distal tissues/organs but modulation of metabolic or immune regulatory pathways play a central role (Fig. 1). We selected three tissue-specific examples to illustrate these effects. These tissues were selected as they are highly susceptible to malignancy (e.g. liver, colon) with observational data suggesting that exercise may reduce the primary risk of cancer in these organs23, or are known mediators of the systemic milieu and TME (e.g. bone marrow)11. We also recognize that skeletal muscle is a mediator of the systemic milieu and has been linked to cancer pathogenesis, which has been reviewed elsewhere.50,51
Figure 1:
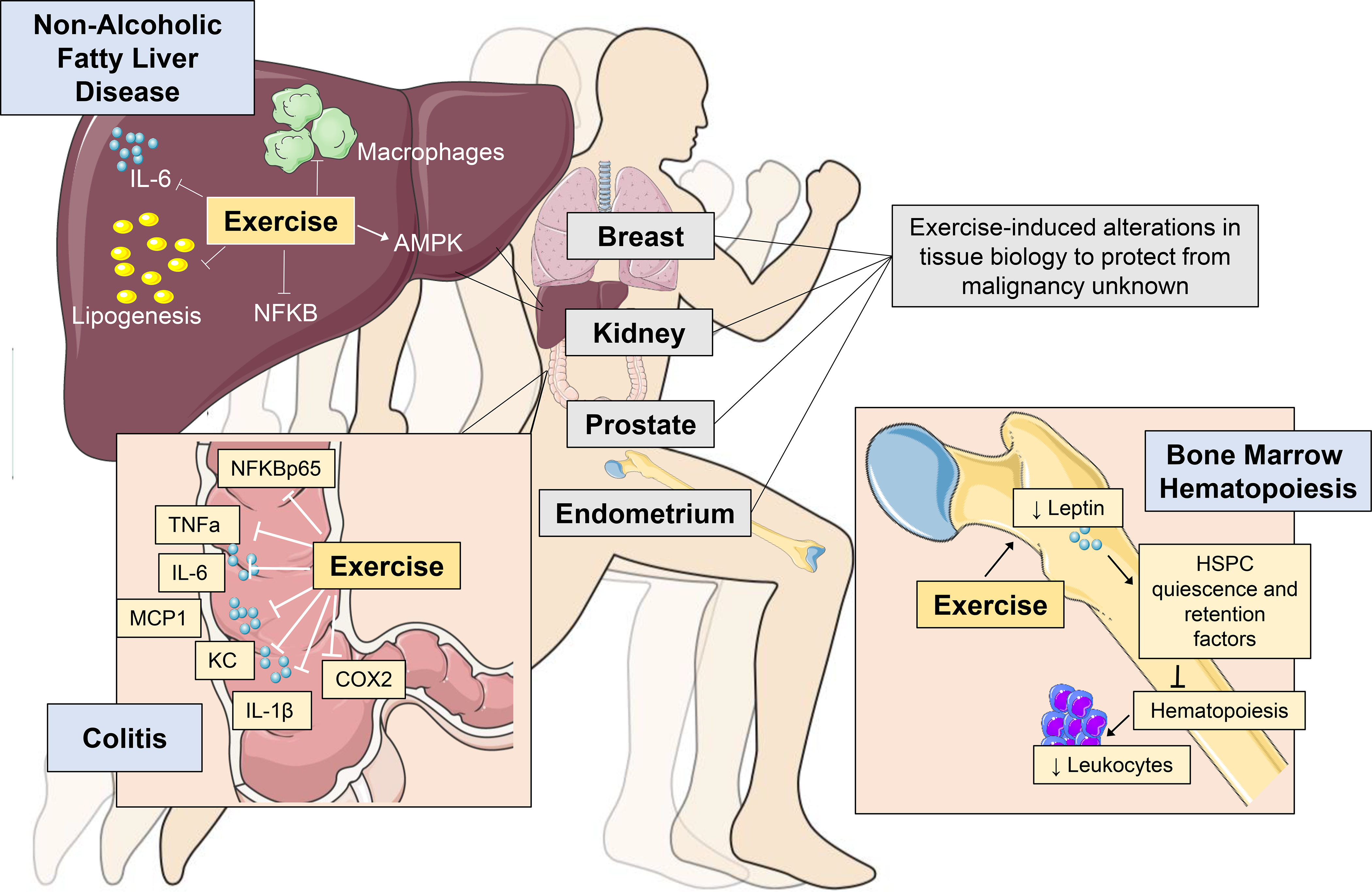
Exercise-induced protection from tissue-specific perturbations in organs involved in cancer regulation or prone to malignancy. Blue boxes contain illustrative examples in the liver, colon and bone marrow. Grey boxes indicate examples of tissues where data to support exercise-induced regulation of tissue biology in the absence of frank malignancy is currently lacking. NAFLD: non-alcoholic fatty liver disease; AMPK: adenosine monophosphate kinase; NFκB: nuclear factor kappa-light-chain-enhancer of activated B cells; TNFα: tumour necrosis factor alpha; IL: interleukin: IL; KC: keratinocyte chemoattractant; MCP-1: monocyte chemoattract protein; COX2: cyclooxygenase-2; HSPC: hematopoietic stem and progenitor cell; Cxcl12: C-X-C motif chemokine 12.
Liver.
Non-alcoholic fatty liver disease (NAFLD), a product of chronic metabolic dysregulation, is characterized by accumulation of triglycerides within hepatocytes (i.e. hepatic steatosis) with accompanying hepatic inflammation, and increases the risk for hepatocellular carcinoma.52 In a diet-induced mouse model of NAFLD, four weeks of exercise (voluntary wheel running) inhibited hepatic steatosis development compared to control.53 Correlative studies revealed that in conjunction with inhibition of lipogenesis, exercise increased liver-specific adenosine monophosphate kinase (AMPK) activation (p-AMPK-α/AMPK-α), which is a key metabolic sensor and regulator, decreasing anabolic (e.g. lipid synthesis) and increasing catabolic (i.e. lipid oxidation) processes.54 These metabolic alterations occurred alongside reductions in hepatic inflammation, as assessed by reduced gene expression of the pro-inflammatory cytokine Il6 and the Adgre1 (the gene encoding the macrophage marker F4/80), and reduced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation (pNF-kB p65/NF-kB p65).
Colon.
The second example links exercise with attenuation of a tissue-specific inflammatory condition - ulcerative colitis - a major risk factor for colorectal cancer.55 Using the dextran sulfate sodium (DSS)-induced rat model of chronic colitis, Qin et al. found exercise treatment (i.e., swimming 1 or 1.5 h.d, 5 d.wk for 7 wks) inhibited colon shortening, colon barrier disruption and splenomegaly compared to control in a dose dependent manner.56 Exercise also attenuated DSS-induced decreases in colon crypt depth, secretion of pro-inflammatory cell mediators (neutrophils, tumour necrosis factor (TNF)α+ and IFNγ+ T cells), and reduced colon-specific ex vivo production of proinflammatory cytokines such as TNFα, interleukin (IL)-1β, IL-6, monocyte chemoattract protein (MCP)-1, and keratinocyte chemoattractant (KC), as well as reduced expression of colon-specific nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) p65 and cyclooxygenase-2 (COX2).
Bone.
Frodermann and colleagues57 conducted one of the most in-depth investigations of exercise regulation of distant organ function. The bone marrow is the primary site for hematopoiesis. In cancer, hematopoiesis is elevated, with a shift toward production of myelopoietic cells;58 elevated circulating myeloid cells also correlate with poor outcomes in a variety of cancers.11,59 In multiple mouse models, Frodermann et al. found that exercise (6 weeks of voluntary wheel running) promoted hematopoietic stem and progenitor cell (HSPC) quiescence compared to control, reducing bone marrow myelopoiesis and systemic monocytosis.
The underlying mechanism was identified as exercise-induced reductions in leptin levels in adipose tissue, blood, and bone marrow, decreasing leptin-receptor signaling in bone marrow stromal cells, increasing expression of genes involved in myeloid cell retention, such as Cxcl12, and lowering HSPC proliferation. Within the HSPC compartment, exercise induced HSPC quiescence, which was associated with epigenetic changes in bone marrow progenitors, reducing chromatin accessibility in lineage marker-negative, Sca1+/c-Kit+ (LSK) progenitor cells, which persisted up to three weeks following exercise cessation. These findings reveal exercise imprints sustained, learned responses in bone marrow progenitors and induces long-term hematopoietic reprogramming. In final experiments, the authors demonstrated that despite a reduction in chronic myelopoiesis, exercise improved emergency hematopoiesis in models of sepsis, demonstrating enhanced host response to immune challenge.
Collectively, these exemplars provide direct evidence that exercise reprogramming of tissue-specific immunometabolic regulation facilitates enhanced resistance to tissue-specific events linked to tumourigenesis. Whether such effects extend to regulation of the TME is discussed in the next section.
Exercise-induced regulation of immuno-metabolism in the TME
Exercise alters numerous specific cell types and/or molecules that regulate systemic metabolic and immune function. For example, exercise increases glucose uptake and decreases circulating insulin, IGF1 and glucose,60 as well as decreases circulating myeloid cells and increases circulating NK cell number and cytotoxic function.61,62 Similar changes also occur in patients with cancer.63–66 Although overly simplistic, such metabolic and immune reprogramming of the systemic milieu might consequently alter the nature and strength of signaling at the tissue and cellular levels in the TME. Indeed, emergent work demonstrates the antitumour activity of various exercise paradigms in a variety of preclinical cancer models, 25,26 with related correlative studies indicating that TME-specific reprogramming of metabolic and immune networks plays a major role in orchestrating the exercise – cancer pathogenesis link (Fig. 2a).
Figure 2:
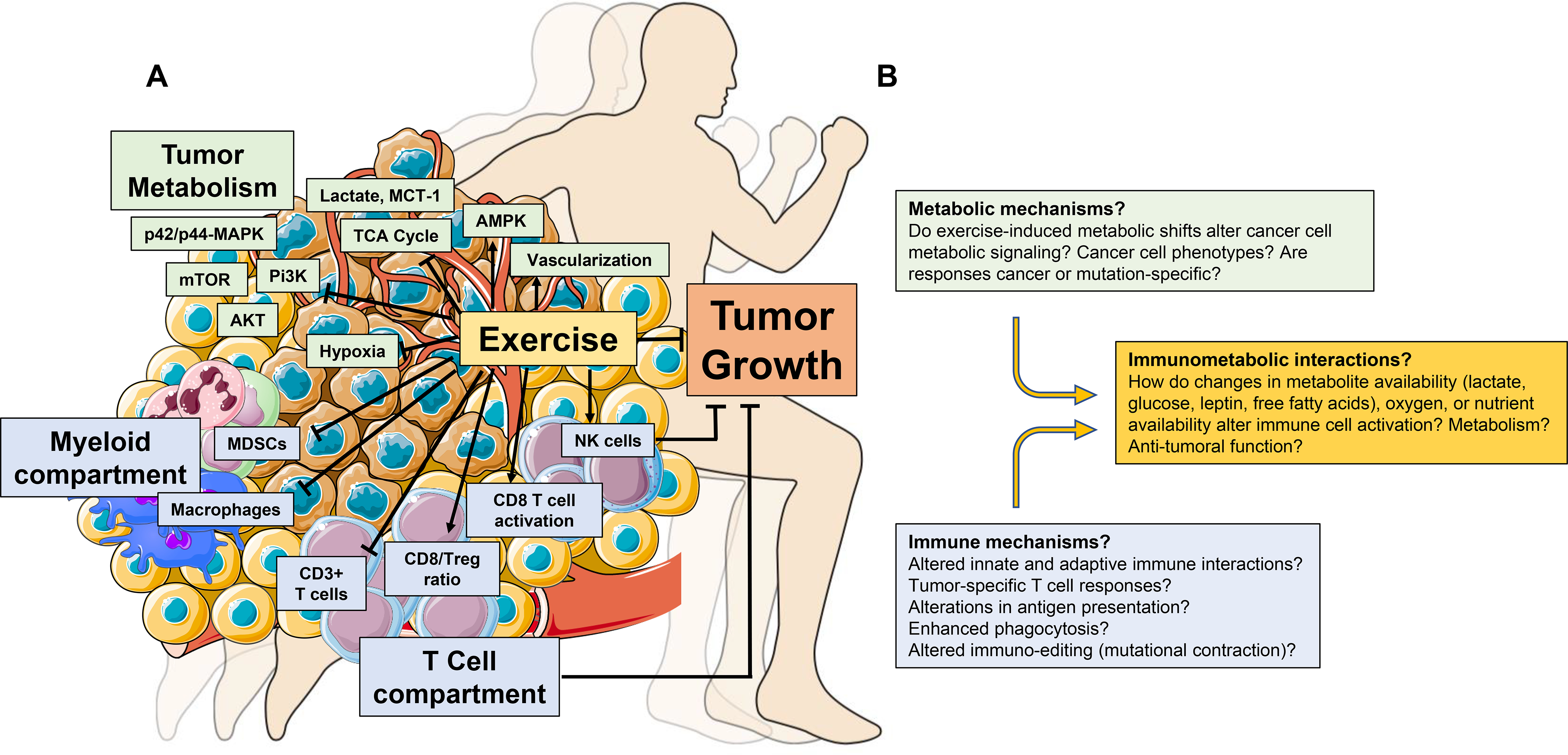
Exercise-induced regulation of immune and metabolic function in the TME. (A) Exercise alters the immune composition of the TME (blue boxes), decreasing the proportion of innate immune cell populations (macrophages and myeloid derived suppressor cells (MDSCs) and increasing CD3+ T cells and NK cells. Furthermore, the ratio of CD8+ T cells versus regulatory T cells (Treg), as well as the activation of CD8+ T cells (CD69+) is increased with exercise. Exercise also alters TME metabolism (green boxes). Decreased hypoxia and increased vascularization occur alongside decreased levels of lactate and MCT1, and the relative concentration of metabolites that comprise the TCA cycle are also reduced. Increased intratumoural AMPK activity and reduced AKT, mTOR, Pi3K, and p42/p44-MAPK have all been reported with exercise. T cells and NK cells are required for exercise-induced tumour inhibition in mouse models of cancer. (B) Proposed immunometabolic mechanisms that may drive exercise-induced inhibition or delay of tumourigenesis. NK: natural killer; MCT-1: Monocarboxylate transporter 1; TCA: tricarboxylic acid cycle; AMPK: adenosine monophosphate kinase; mTOR: mammalian target of rapamycin; Pi3K; Phosphoinositide 3-kinase; MAPK: mitogen-activated protein kinase.
Metabolism.
Initial studies in this area focus on metabolic effectors and/or pathways known to regulate tumourigenesis. For instance, Zhu et al. found that exercise (voluntary wheel running) in a rat model of chemically-induced breast cancer reduced tumour incidence and multiplicity (number of tumours per animal), which occurred in conjunction with reduced systemic levels of insulin, IGF1 and leptin, increased intratumoural activation of AMPK, and inhibition of activated AKT and mTOR.67 Extending these observations, Xie et al. identified similar metabolic alterations (i.e., reduced AKT, PI3K and p42/p44-MAPK) in skin tissues following 10 weeks of forced treadmill running in the TPA-induced model of skin cancer.68 Furthermore, Aveseh and colleagues69 found exercise inhibition of the mammary MC4-L2 cancer model coupled with a shift in the tumour lactate dehydrogenase (LDH) isozyme profile towards LDH-1, together with a reduction in LDHA expression, increase in LDHB expression, and concomitant decrease in tumour lactate and the lactate transporter MCT-1. Since excess lactate is a common byproduct of reprogrammed cancer cell metabolism, lower TME lactate levels suggests exercise either shifted metabolism to produce less lactate, increased lactate utilization, and/or increased lactate clearance.
More recent work has focused on exercise regulation of tumour metabolism. Lu et al. utilized six patient-derived xenografts (PDXs) of colorectal cancer to test how voluntary wheel running altered tumour metabolite composition.70 Exercise inhibition of tumour growth was observed in three models although accompanying metabolomic analysis of tumour lysates signified metabolic alteration in all PDXs compared to control.
Specifically, tumours from exercised mice showed alterations in 47 metabolites with pathway enrichment in nucleotide, vitamin B6, and amino acid metabolism, as well as the tricarboxylic acid cycle (TCA). Interestingly, the TCA cycle activity was downregulated by exercise, as the majority of TCA cycle intermediates (with the exception of succinate and glutamate) were lower compared to control. Comparison of tumour metabolic profiles from exercise-responsive and non-responsive tumours revealed that the differences in TCA cycle metabolites, however, no longer persisted yet changes in nucleotide metabolism remained. Thus, cancer cell-autonomous intrinsic variations in nucleotide metabolism, and not the TCA cycle, may explain differential sensitivity of tumours to exercise, at least in PDX models of colorectal cancer.
Supporting the notion that heterogeneity in cancer cell autonomous metabolic programming may mediate tumour response to exercise, Glass et al.71 found that differential sensitivity of two murine breast cancer cell lines, E0771 and C3(1)SV40Tag-p16-luc, to exercise was paralleled by activation or lack of activation of intratumoural hypoxia-inducible factor 1 (HIF-1α) and its downstream targets PDK-1 and GLUT-1. Specifically, activation of HIF-1α and associated shifts towards glycolysis occurred in exercise-induced acceleration (C3(1)SV40Tag-p16-luc), but not inhibition (E0771), of tumour growth. Blocking of HIF-1α activation using digoxin abrogated exercise-induced acceleration of C3(1)SV40Tag-p16-luc growth rates.
Overall, exercise appears to be a regulator of TME-specific metabolites (e.g. lactate), pathway and transcription factor signaling (e.g. PI3K, HIF1α) and central carbon metabolism (e.g. TCA cycle). Further, exercise-induced metabolic alterations, as well as the pattern of metabolic alterations, does not appear to be uniform across cancer models or tumour types, suggesting cancer-cell intrinsic effects regulate the exercise response. Further exploration of how metabolic alterations mechanistically underpin the antitumour activity of exercise is an exciting area of future research.
Immunity.
In early work, Zielinski et al. reported that two weeks of treadmill running delayed tumour growth in mice, which occurred in conjunction with decreased intratumoural accumulation of macrophages and neutrophils in the EL4 lymphoma model.72 Similar findings were observed in the syngeneic ehrlich tumour model following six weeks of forced swimming.73
Pedersen and colleagues74 significantly extended this work by showing immune activation was required for exercise-induced tumour inhibition. Specifically, voluntary wheel running prior to implantation of B16 melanoma cells inhibited tumour growth, with tumours displaying higher numbers of cytotoxic NK cell and CD3+ T cells compared to control.74 The tumour inhibitory effect of exercise persisted in athymic nude mice, yet was abrogated with NK cell depletion, suggesting that NK cells, and not T cells, were responsible for exercise growth inhibition. Further, blockade of either β-adrenergic or IL-6 signaling prevented NK cell tumour infiltration, abrogating exercise growth inhibition.74
A subsequent study by Hagar et al. reported eight weeks of treadmill running before tumour inoculation inhibited tumour growth and extended survival, alongside a two-fold increase in the intratumoural cytotoxic T cell (CD8+)/regulatory T cell (Foxp3+) ratio in the syngeneic 4T1 breast cancer model.75 Exercise-induced antitumour activity was mitigated in athymic nude mice in this model, suggesting that exercise induced effects on T cells were required for growth inhibition.
Exercise may regulate additional systemic and intratumoural immune responses. 4T1 breast tumours induce a marked increase in extramedullary hematopoiesis, splenic accumulation of myeloid-derived suppressor cells (MDSCs), and associated splenomegaly.76 Forced treadmill running in the 4T1 model inhibited tumour growth, reduced splenic weight, and splenic and intratumoural MDSCs.77 Consistent with the immunosuppressive effects of MDSCs, exercise-induced effects were associated with increased intratumoural activation of CD8+ T cells (CD69+), although CD8+ T cell function was not assessed. Of interest, exercise potentiated growth inhibition of local radiation plus PD-1 blockade, occurring alongside reduced intratumoural MDSC accumulation compared to control.
These collective findings indicate that modulation of both innate and adaptive immunity is complicit in exercise antitumour activity. Future studies to address how these changes mechanistically regulate tumourigenesis are discussed below.
Future Directions
A sufficient evidence base now exists to launch the next generation of studies leveraging advances in mainstream immuno-metabolism oncology research to comprehensively interrogate exercise efficacy. Herein, we discuss some of the key knowledge gaps in regulation of metabolism and immunity, and perhaps more importantly, elucidation of exercise action at the nexus of these regulatory networks (Fig. 2b).
Metabolism.
Exercise appears to regulate metabolite availability at host and tissue levels, occurring in conjunction with regulation of tumour metabolic pathways, including a shift in central carbon metabolism. The mechanistic drivers of these alterations, or whether such changes are necessary for exercise antitumour activity, however, are unknown. Furthermore, how exercise regulates other aspects of cellular metabolism known to play key roles in the TME (e.g. glycogen metabolism)78,79 are an important area of future study.
Utilization of unbiased metabolomic platforms may reveal global overarching patterns correlating with exercise sensitivity and/or resistance, facilitating in-depth mechanistic interrogation. It is unlikely that shifts in single metabolites and/or resulting pathways will be responsible for exercise phenotypes. As such, experimental approaches with the capacity to capture broad metabolic shifts will be required. Illustration of one such elegant approach was reported by Vande Voorde et al. and Cantor et al, wherein in vitro recapitulation of physiologic (systemic) levels of a multitude of extracellular nutrient and metabolite levels induced profound effects on tumour cell behavior, which better recapitulated in vivo tumour metabolic profiles compared to standard cell culture media.80,81 Similar strategies could be applied to understand how exercise-induced alterations in metabolite/nutrient availability in blood regulates metabolic signaling and cancer cell response in vitro as well as in vivo. Such work could be followed by in vitro screening studies across different oncogenic / mutational profiles, with in vivo validation to determine how tumour-intrinsic features mediate the metabolic response to exercise.
Immunity.
How TME immune cell composition and phenotypes shift in exercise states is poorly characterized. In particular, the interaction between innate and adaptive immune populations, the specific cell types involved, and the phenotypic and functional states are open areas of investigation. This is mainly due to the fact that exercise research is typically restricted to orthotopic models that do not recapitulate the complex immune responses unique to in situ cancer initiation and development.82 Use of genetically engineered models with established characterization of immune populations and phenotypes, neo-antigen-specificity, and/or capacity to evaluate early tumourigenesis or metastatic seeding more closely recapitulate human cancer pathogenesis and hence offer superior model systems for such investigations.82–84 Such models could be exploited to investigate the fundamental questions such as whether the exercise-conditioned host more effectively orchestrates innate and/or adaptive antitumour immune responses, including tumour-specific T cell responses; enhances antigen presentation; or eradicates damaged or mutated cells more effectively.
In parallel, elucidation of how exercise-induced changes in TME immune milieu alters cancer cell phenotypes (and possibly genomic landscape) is also currently unexplored. For example, does exercise induce immunoediting; that is, mutational contraction through depletion of neoantigens and clones harboring them to alter tumour genomic landscapes? Are certain genomic signatures in tumours more sensitive to exercise-induced immune alterations?
Exercise regulation of immune-metabolic interaction.
To our knowledge, how exercise modulates immune and metabolic interaction to alter tumourigenesis has not been investigated. It is plausible that exercise-induced reductions in key metabolic growth factors such as leptin, free-fatty acids, and lactate in the TME (which in excess drive tumour immune suppression6,7,36,37) may, in turn, facilitate broad, sustained immune activation.
Exercise also modulates TME cellular architecture, characterized by increased angiogenesis and vascular function, leading to a decrease in hypoxia.26,85 Such changes likely further alter metabolite availability, as well as the infiltration, spatial composition and function of intratumoural immune cells. Experimental approaches with the capacity to evaluate broad metabolic and immune alterations, including cancer cell responses to such alterations, are needed. In an exemplar of one such approach, Leone and colleagues found that glutamine blockade reduced intratumoural hypoxia, acidosis, and nutrient depletion in the TME. These alterations occurred alongside T cell activation and concurrent suppression of cancer cell metabolism (e.g. reductions in glycolytic and oxidative metabolism), leading to decreased tumour growth.86 It is intriguing to speculate that exercise could similarly restore (immunometabolic) function in the TME.
Clinical Translation
At least two, multicenter phase 3 randomized control trials (RCTs) are investigating the efficacy of structured exercise therapy on cancer outcomes in individuals with primary colon cancer (disease-free survival)87 and metastatic prostate cancer (overall survival, progression-free survival), alongside several other ongoing clinical trials of structured exercise therapy in cancer prevention and prognosis (Table).88 A critical corollary to these ongoing trials are correlative science studies to interrogate whether exercise-induced alterations in systemic (host) immunometabolic factors link with alterations in “normal” tissue microenvironments among individuals at high-risk of cancer (i.e., exercise to prevent primary cancer incidence) as well as the TME in patients with cancer (i.e., exercise to prevent recurrence or progression of cancer).89
Table:
Ongoing clinical trials of structured exercise therapy in cancer prevention and prognosis
| Trial | Design | NCT# | Sample size / Population | Exercise Intervention | Cancer-specific Endpoints |
|---|---|---|---|---|---|
|
| |||||
|
Post Diagnosis
Phase 3 Trials |
|||||
| Intense Exercise for Survival Among Men With Metastatic Castrate-Resistant Prostate Cancer (INTERVAL)88 | Phase 3 RCT | NCT02730338 | N=866; advanced prostate cancer | 2 years – individualized, progressive moderate to high intensity aerobic and resistance exercise. Supervised (Year 1) and home-based (Year 2) | Primary: Overall survival Secondary: Disease progression, program safety |
| The Colon Health and Life-Long Exercise Change Trial (CHALLENGE)87 | Phase 3 RCT | NCT00819208 | N=962; stage II/III colon cancer patients | 3 years – combined behavior support with supervised / unsupervised activity sessions with goal to increase recreational physical activity up to 27 MET-hrs/wk | Primary: Disease-free survival Secondary: Overall survival |
| Other Trials | |||||
| Effect of Physical Exercise on Tumour Proliferation of Luminal B Breast Cancer Patients | Case Control | NCT03860740 | N=60; operable and untreated HR+ HER2− breast cancer patients | 2–3 wks prior to surgery - 60% to 100% VO2peak, 10 sessions minimum | Primary: Tumour Proliferation (Ki67), Proliferation score (PAM50) Secondary: change from baseline molecular subtypes (PAM50), intratumoural VEGF, HIF-1, cleaved caspase-3 |
| Exercise Treatment with Standard Therapy for Metastatic Breast Cancer | Phase 1a/b | NCT03988595 | N=60; HR+ metastatic breast cancer | 24 wks - 3 to 5x/wk - 4 escalated doses: 90 mins/wk, 150 mins/wk, 225 mins/wk, or 300 mins/wk | Phase 1a (dose escalation): Primary: Maximum feasible dose Secondary: Change in circulating tumour DNA, tumour proliferation (Ki67), HR signaling Phase 1b (dose expansion): Primary: Change in circulating tumour DNA Secondary: tumour proliferation (Ki67), HR signaling |
| Exercise Interventions for Breast Cancer Patients Undergoing Neoadjuvant Chemotherapy (BENEFIT) | 3-arm RCT | NCT02999074 | N=240; breast cancer patients scheduled for neoadjuvant chemotherapy | 18 wks, 2 interventions: Resistance exercise (2 x wk) - 8 machine-based exercises, each performed in 3 sets, 12 repetitions at 60–80% of one repetition maximum (1-RM); or aerobic exercise (2 x wk) – up to 60–70% VO2peak |
Primary: Tumour Size - change from baseline (before start of neoadjuvant chemotherapy) to breast surgery Secondary: CPS-EG score, pathological complete response |
| Combination of Exercise and a Plant-Based Diet in Overweight Postmenopausal Women with Breast Cancer | 2-arm RCT | NCT04298086 | N=62; HR+ HER2− stage I-III breast cancer patients receiving aromatase inhibitor | 24 wks; 7 x wk to achieve the patient-specific goal energy expenditure in conjunction with a plant-based diet | Primary: Change in breast aromatase levels Secondary: Change in breast tissue gene expression |
| Effects of Pre-Surgical Aerobic Training in Patients with Solid Tumours | Phase 0 / 1a/b | NCT03813615 | N=78; Phase 0: early stage breast, endometrial and prostate cancer Phase 1a/b: operable, untreated prostate cancer |
Phase 0: > 2 wks aerobic training - 150 mins/wk, 5 sessions/wk Phase 1a.: > 2 wks aerobic training - 150 mins/wk, 225 mins/wk, 300 mins/wk, or 375 mins/wk; 3 to 6 sessions/wk Phase 1b: Dose expansion |
Phase 1a. Maximal feasible dose with biological activity Phase 1b. Further examination of tolerability and activity |
| Prevention | |||||
| Physical Activity, Proliferation and Immune Markers in Benign Breast Tissue | Single arm | NCT03657628 | N=60; premenopausal women with high breast density | 12 wks - supervised, moderate-intensity aerobic exercise | Primary: Proliferation (Ki67) |
| Dose-Response of Aerobic Training in Women at High-Risk for Development of Breast Cancer | 3-arm RCT | NCT02494869 | N=75; women at high risk for breast cancer (family history, atypical hyperplasia) | 24 wks, 2 exercise doses: 150 minutes/wk aerobic training (3 sessions of 50 minutes) at 55–100% VO2peak; or 300 minutes/wk aerobic training (5 sessions of 60 minutes) at 55–100% VO2peak |
Primary: Changes in gene expression patterns of non-neoplastic breast epithelial cells Secondary: Changes in (epi)-genomic profile |
Abbreviations: RCT: randomized control trial; HR: hormone receptor; HER2: human epidermal growth factor receptor 2; Met: metabolic equivalent of task; VO2peak: peak rate of oxygen consumption; CPS-EG: Clinical-Pathologic Stage score
To our knowledge, only one trial to date has directly investigated the effects of exercise on biological end points in a tissue / organ other than the skeletal muscle or adipose tissue. McTiernan et al. performed a two-arm RCT to examine the effects of structured exercise (60 minutes/d, 6 d/wk) on change in number of Ki67-stained cells, a marker of cellular proliferation, in colon mucosal crypts among women and men undergoing routine screening. At 12 months, men exercising at least 250 min/wk had significant reductions in colon crypt cell proliferation.90 Data from the same trial indicated that exercise training increased expression of the proapoptotic protein (Bax) in the bottom of the colon crypts among men, whereas it decreased expression in the middle of colon crypts among women.91
Similarly, investigation of exercise regulation of the TME is limited to one trial. Taking advantage of the pre-operative “window of opportunity”, which permits testing of candidate strategies on tumour biology without the confounding impact of other anticancer therapies, Ligibel et al.92 studied the effects of a combined aerobic and resistance exercise regimen (planned dose, ~200 mins/wk) compared to usual care in 49 operable breast cancer patients. No differences were observed in tumour cell proliferation, apoptosis or insulin receptor expression in response to exercise. However, exploratory whole transcriptome sequencing before and after exercise revealed enrichment of immune- and inflammation-associated pathways including NFκB signaling, NK cell mediated cytotoxicity, and T cell receptor signaling. Given inherent limitations of whole-tumour transcriptome analyses, an independent team re-analyzed this data set with deconvolution analyses, permitting evaluation of changes in tumour immune cell composition, and revealed exercise-associated changes, albeit non-significant, in macrophages and B cells.93
These preliminary findings suggest that both short-term and longer-term treatment with exercise regulates the microenvironments of tissues / organs harboring cells potentially primed for malignant transformation as well as the TME. Furthermore, they also demonstrate conduct of exercise trials with correlative science tissue biology end points are feasible in both the prevention and post-diagnosis settings. These vanguard efforts together with advancements in molecular and computational biology now provide an unprecedented opportunity to interrogate exercise reprogramming of tissue-specific immunometabolic pathways in normal tissue as well as the TME. For instance, exercise trials could incorporate deep, dynamic phenotyping of exercise response at the level of the whole-organism via longitudinal profiling at the level of the microbiome, metabolome, or immunome. If such studies are conducted in high-risk patients or the pre-operative window, there is also the exciting opportunity to combine phenotyping of host response with profiling of the tissue or tumour landscape at the cellular, genomic and epigenomic level using tissue obtained from routine and/or research-directed biopsies.
Recent advancements in single cell and spatiotranscriptomic technologies may be particularly relevant in the exercise context.94 The pleiotropic nature of exercise action suggests that approaches with the capacity to uncover dynamics of cellular phenotypes including compositional changes in tissue and TME architecture are likely required for comprehensive molecular interrogation of the exercise – cancer link not possible with current bulk tissue sequencing approaches. The large datasets generated by digital medicine approaches will require advanced computational methods, including use of machine and deep learning artificial intelligence tools, to fully comprehend the abundance of data generated.95 Although daunting, such efforts would provide remarkable insights into exercise reprogramming of the complex, dynamic interplay between organismal physiology, immunometabolic network signaling, and tumourigenesis at an unprecedented level of resolution.
Conclusion
Unhealthy lifestyle factors accelerate tumourigenesis, in part via dysregulation of the immune-metabolic axis at the organismal, tissue, cellular, and molecular levels. As reviewed here, exercise is one strategy with the capacity to regulate immune and metabolic networks, and potentially their interaction. Further, modulation of these processes may underpin the exercise – tumourigenesis inhibition link. Over the next decade we anticipate preclinical and correlative clinical studies will provide exciting new insights on how exercise regulates immunometabolic phenotypes in the host and distant tissues. Such work will bridge the gap between the long-standing epidemiological findings to direct mechanistic evidence on how exercise may inhibit cancer pathogenesis.
Sources of Funding.
GJK is supported by the Louis and Rachel Rudin Foundation. XZ and TT are supported in part by Josie Robertson, Rita Allen and V Foundation Scholarships and the Stanley and Fiona Druckenmiller Center for Lung Cancer Research at MSK. AS is supported in part by funding from the National Cancer Institute (DP2 CA225212, U54 CA209975), the Josie Robertson Foundation, and the Cancer Research Institute. LWJ is supported in part by funding from the National Cancer Institute and AKTIV Against Cancer. This work is supported by the Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748).
Footnotes
Competing interests: L.W.J. owns stock in Pacylex, Inc. G.J.K., X.Z., T.T., A.S. declare no competing interests.
Disclosures. LWJ – stock ownership in Pacylex, Inc.
References
- 1.Song M & Giovannucci E Preventable Incidence and Mortality of Carcinoma Associated With Lifestyle Factors Among White Adults in the United States. JAMA Oncol 2, 1154–1161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song M, Vogelstein B, Giovannucci EL, Willett WC & Tomasetti C Cancer prevention: Molecular and epidemiologic consensus. Science 361, 1317–1318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotas ME & Medzhitov R Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chovatiya R & Medzhitov R Stress, inflammation, and defense of homeostasis. Mol Cell 54, 281–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schworer S, Vardhana SA & Thompson CB Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab 29, 576–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka AK & Coussens LM The Basis of Oncoimmunology. Cell 164, 1233–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen DS & Mellman I Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Quail DF & Joyce JA Microenvironmental regulation of tumour progression and metastasis. Nat Med 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engblom C, Pfirschke C & Pittet MJ The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447–462 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Wang A, Luan HH & Medzhitov R An evolutionary perspective on immunometabolism. Science 363(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehla K & Singh PK Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 5, 822–834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitale I, Manic G, Coussens LM, Kroemer G & Galluzzi L Macrophages and Metabolism in the Tumour Microenvironment. Cell Metab 30, 36–50 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Dyck L & Lynch L Cancer, obesity and immunometabolism - Connecting the dots. Cancer Lett 417, 11–20 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Turbitt WJ, Buchta Rosean C, Weber KS & Norian LA Obesity and CD8 T cell metabolism: Implications for anti-tumour immunity and cancer immunotherapy outcomes. Immunol Rev (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drew DA, Cao Y & Chan AT Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer 16, 173–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demierre MF, Higgins PD, Gruber SB, Hawk E & Lippman SM Statins and cancer prevention. Nat Rev Cancer 5, 930–942 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Zaleska M, Mozenska O & Bil J Statins use and cancer: an update. Future Oncol 14, 1497–1509 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Barron TI, Connolly RM, Sharp L, Bennett K & Visvanathan K Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol 29, 2635–2644 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Ganz PA, Habel LA, Weltzien EK, Caan BJ & Cole SW Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort. Breast Cancer Res Treat 129, 549–556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pernicova I & Korbonits M Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 10, 143–156 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Moore SC, et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern Med 176, 816–825 (2016). Meta-analysis showing that high vs low levels of leisure-time physical activity is associated with lower risk of 13 cancers.
- 24. Friedenreich CM, Neilson HK, Farris MS & Courneya KS Physical Activity and Cancer Outcomes: A Precision Medicine Approach. Clin Cancer Res 22, 4766–4775 (2016). Systematic review showing postdiagnosis physical activity is associated with lower risk of cancer recurrence or progression across breast, prostate, and colorectal cancer.
- 25.Ashcraft KA, Peace RM, Betof AS, Dewhirst MW & Jones LW Efficacy and Mechanisms of Aerobic Exercise on Cancer Initiation, Progression, and Metastasis: A Critical Systematic Review of In Vivo Preclinical Data. Cancer Res 76, 4032–4050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koelwyn GJ, Quail DF, Zhang X, White RM & Jones LW Exercise-dependent regulation of the tumour microenvironment. Nat Rev Cancer 17, 620–632 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Golemis EA, et al. Molecular mechanisms of the preventable causes of cancer in the United States. Genes Dev 32, 868–902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quail DF & Dannenberg AJ The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol 15, 139–154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hopkins BD, Goncalves MD & Cantley LC Obesity and Cancer Mechanisms: Cancer Metabolism. J Clin Oncol 34, 4277–4283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Font-Burgada J, Sun B & Karin M Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab 23, 48–62 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Iyengar NM, Gucalp A, Dannenberg AJ & Hudis CA Obesity and Cancer Mechanisms: Tumour Microenvironment and Inflammation. J Clin Oncol 34, 4270–4276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howe LR, Subbaramaiah K, Hudis CA & Dannenberg AJ Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res 19, 6074–6083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clements VK, et al. Frontline Science: High fat diet and leptin promote tumour progression by inducing myeloid-derived suppressor cells. J Leukoc Biol 103, 395–407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, et al. Paradoxical effects of obesity on T cell function during tumour progression and PD-1 checkpoint blockade. Nat Med 25, 141–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baek AE, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8, 864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumour Growth. Cell Metab 31, 148–161 e145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michelet X, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumour responses. Nat Immunol 19, 1330–1340 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Krall JA, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumours in mouse models of dormancy. Sci Transl Med 10(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koelwyn GJ, et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat Med (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meijers WC, et al. Heart Failure Stimulates Tumour Growth by Circulating Factors. Circulation 138, 678–691 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Hasin T, et al. Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer. J Am Coll Cardiol 68, 265–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasin T, et al. Patients with heart failure have an increased risk of incident cancer. J Am Coll Cardiol 62, 881–886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peake JM, et al. Modulating exercise-induced hormesis: Does less equal more? J Appl Physiol (1985) 119, 172–189 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Hawley JA, Hargreaves M, Joyner MJ & Zierath JR Integrative biology of exercise. Cell 159, 738–749 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Egan B, Hawley JA & Zierath JR SnapShot: Exercise Metabolism. Cell Metab 24, 342–342 e341 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Koelwyn GJ, Wennerberg E, Demaria S & Jones LW Exercise in Regulation of Inflammation-Immune Axis Function in Cancer Initiation and Progression. Oncology (Williston Park) 29, 908–920, 922 (2015). [PMC free article] [PubMed] [Google Scholar]
- 47.Joyner MJ & Dempsey JA Physiological Redundancy and the Integrative Responses to Exercise. Cold Spring Harb Perspect Med 8(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones LW, Eves ND, Haykowsky M, Freedland SJ & Mackey JR Exercise intolerance in cancer and the role of exercise therapy to reverse dysfunction. Lancet Oncol 10, 598–605 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Neufer PD, et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab 22, 4–11 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Severinsen MCK & Pedersen BK Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr Rev 41(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedersen BK & Febbraio MA Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8, 457–465 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Michelotti GA, Machado MV & Diehl AM NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 10, 656–665 (2013). [DOI] [PubMed] [Google Scholar]
- 53. Gehrke N, et al. Voluntary exercise in mice fed an obesogenic diet alters the hepatic immune phenotype and improves metabolic parameters - an animal model of life style intervention in NAFLD. Sci Rep 9, 4007 (2019). Preclinical study showing exercise protects against diet-induced non-alcoholic fatty liver disease (NAFLD) in mice, which coincided with improvements in liver-specific metabolic and inflammatory alterations.
- 54.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olen O, et al. Colorectal cancer in ulcerative colitis: a Scandinavian population-based cohort study. Lancet 395, 123–131 (2020). [DOI] [PubMed] [Google Scholar]
- 56. Qin L, et al. Swimming attenuates inflammation, oxidative stress, and apoptosis in a rat model of dextran sulfate sodium-induced chronic colitis. Oncotarget 8, 7391–7404 (2017). Preclinical study showing exercise protection from chronic colitis alongside reductions in colon-specific inflammation.
- 57. Frodermann V, et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat Med 25, 1761–1771 (2019). Study showing that exercise in mice reduces bone marrow hematopoiesis via leptin signaling and epigenetic alterations in bone marrow hematopoietic progenitors
- 58.Wu WC, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci U S A 111, 4221–4226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shipp C, Speigl L, Janssen N, Martens A & Pawelec G A clinical and biological perspective of human myeloid-derived suppressor cells in cancer. Cell Mol Life Sci 73, 4043–4061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stanford KI & Goodyear LJ Exercise and type 2 diabetes: molecular mechanisms regulating glucose uptake in skeletal muscle. Adv Physiol Educ 38, 308–314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bigley AB & Simpson RJ NK cells and exercise: implications for cancer immunotherapy and survivorship. Discov Med 19, 433–445 (2015). [PubMed] [Google Scholar]
- 62.Timmerman KL, Flynn MG, Coen PM, Markofski MM & Pence BD Exercise training-induced lowering of inflammatory (CD14+CD16+) monocytes: a role in the anti-inflammatory influence of exercise? J Leukoc Biol 84, 1271–1278 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Peters C, Lotzerich H, Niemeir B, Schule K & Uhlenbruck G Exercise, cancer and the immune response of monocytes. Anticancer Res 15, 175–179 (1995). [PubMed] [Google Scholar]
- 64.Fairey AS, et al. Randomized controlled trial of exercise and blood immune function in postmenopausal breast cancer survivors. J Appl Physiol (1985) 98, 1534–1540 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Irwin ML, et al. Randomized controlled trial of aerobic exercise on insulin and insulin-like growth factors in breast cancer survivors: the Yale Exercise and Survivorship study. Cancer Epidemiol Biomarkers Prev 18, 306–313 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fairey AS, et al. Effects of exercise training on fasting insulin, insulin resistance, insulin-like growth factors, and insulin-like growth factor binding proteins in postmenopausal breast cancer survivors: a randomized controlled trial. Cancer Epidemiol Biomarkers Prev 12, 721–727 (2003). [PubMed] [Google Scholar]
- 67.Zhu Z, et al. Effect of nonmotorized wheel running on mammary carcinogenesis: circulating biomarkers, cellular processes, and molecular mechanisms in rats. Cancer Epidemiol Biomarkers Prev 17, 1920–1929 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie L, et al. Effects of dietary calorie restriction or exercise on the PI3K and Ras signaling pathways in the skin of mice. J Biol Chem 282, 28025–28035 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Aveseh M, Nikooie R & Aminaie M Exercise-induced changes in tumour LDH-B and MCT1 expression are modulated by oestrogen-related receptor alpha in breast cancer-bearing BALB/c mice. J Physiol 593, 2635–2648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lu M, et al. Exercise inhibits tumour growth and central carbon metabolism in patient-derived xenograft models of colorectal cancer. Cancer Metab 6, 14 (2018). Study showing differential sensitivity to exercise across patient-derived xenograft models, occurring in conjunction with whole tumour metabolism alterations.
- 71.Glass OK, et al. Differential response to exercise in claudin-low breast cancer. Oncotarget 8, 100989–101004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zielinski MR, Muenchow M, Wallig MA, Horn PL & Woods JA Exercise delays allogeneic tumour growth and reduces intratumoural inflammation and vascularization. J Appl Physiol (1985) 96, 2249–2256 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Almeida PW, et al. Swim training suppresses tumour growth in mice. J Appl Physiol (1985) 107, 261–265 (2009). [DOI] [PubMed] [Google Scholar]
- 74. Pedersen L, et al. Voluntary Running Suppresses Tumour Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab 23, 554–562 (2016). Study showing that exercise inhibits tumour growth across multiple preclinical models, including the B16 mouse melanoma model, which was dependent on natural killer cell tumour infiltration.
- 75.Hagar A, et al. Endurance training slows breast tumour growth in mice by suppressing Treg cells recruitment to tumours. BMC Cancer 19, 536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sio A, et al. Dysregulated hematopoiesis caused by mammary cancer is associated with epigenetic changes and hox gene expression in hematopoietic cells. Cancer Res 73, 5892–5904 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Wennerberg E, et al. Exercise reduces immune suppression and breast cancer progression in a preclinical model. Oncotarget 11, 452–461 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zois CE & Harris AL Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J Mol Med (Berl) 94, 137–154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dauer P & Lengyel E New Roles for Glycogen in Tumour Progression. Trends Cancer 5, 396–399 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Vande Voorde J, et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv 5, eaau7314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cantor JR, et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272 e217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Day CP, Merlino G & Van Dyke T Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franklin RA, et al. The cellular and molecular origin of tumour-associated macrophages. Science 344, 921–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Philip M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Betof AS, et al. Modulation of murine breast tumour vascularity, hypoxia and chemotherapeutic response by exercise. J Natl Cancer Inst 107(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leone RD, et al. Glutamine blockade induces divergent metabolic programs to overcome tumour immune evasion. Science 366, 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Courneya KS, et al. The Colon Health and Life-Long Exercise Change trial: a randomized trial of the National Cancer Institute of Canada Clinical Trials Group. Curr Oncol 15, 279–285 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Newton RU, et al. Intense Exercise for Survival among Men with Metastatic Castrate-Resistant Prostate Cancer (INTERVAL-GAP4): a multicentre, randomised, controlled phase III study protocol. BMJ Open 8, e022899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iyengar NM & Jones LW Development of Exercise as Interception Therapy for Cancer: A Review. JAMA Oncol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McTiernan A, et al. Effect of a 12-month exercise intervention on patterns of cellular proliferation in colonic crypts: a randomized controlled trial. Cancer Epidemiol Biomarkers Prev 15, 1588–1597 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Campbell KL, et al. Effect of a 12-month exercise intervention on the apoptotic regulating proteins Bax and Bcl-2 in colon crypts: a randomized controlled trial. Cancer Epidemiol Biomarkers Prev 16, 1767–1774 (2007). [DOI] [PubMed] [Google Scholar]
- 92. Ligibel J, et al. Impact of a pre-operative exercise intervention on breast cancer proliferation and gene expression: Results from the Pre-Operative Health and Body (PreHAB) Study. Clin Cancer Res (2019). “Window of opportunity’ trial showing enrichment of intratumoural immune and inflammation-associated pathways following short-term exercise in treatment-naïve patients with operable breast cancer.
- 93.Sims AH, Leggate M & Campbell A Exercise Window Trial in Newly Diagnosed Breast Cancer-Letter. Clin Cancer Res 25, 7609–7610 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Hu BC The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shilo S, Rossman H & Segal E Axes of a revolution: challenges and promises of big data in healthcare. Nat Med 26, 29–38 (2020). [DOI] [PubMed] [Google Scholar]