

Clinical Presentation
A 34-year-old woman with a medical history of migraines and bipolar disorder is referred to nephrology clinic for elevated serum creatinine and polyuria. In clinic, she states she has had “constant” urination for the past year, noting that she urinates almost 15 times daily, including 3 episodes of nocturia. She drinks nearly 100 ounces of water daily, which is driven by her perpetual thirst. She otherwise describes her diet as “typical,” and does not consume abnormal amounts of sodium or protein. Her medications include as-needed NSAIDs for migraines and lithium carbonate for bipolar disorder, which she has taken since age 17. Laboratory data are listed in Table 1. She has no family history of kidney disease, calcium disorders, or parathyroid disease. She does not endorse musculoskeletal or abdominal pain.
Table 1.
Laboratory values
| Test | Lab Value | Reference Range |
|---|---|---|
| Serum creatinine | ||
| At time of presentation, mg/dL | 1.34 | 0.6–1.2 |
| Four months prior, mg/dL | 1.40 | |
| eGFR, mL/min/1.73m2 | 51–53 | >60 |
| Serum Bicarbonate, mmol/L | 22 | 22–26 |
| Phosphorus, mg/dL | 4.0 | 2.4–4.7 |
| Total calcium, mg/dL | 11.1 | 8.9–10.3 |
| Albumin, g/dL | 4.2 | 3.5–5.1 |
| Intact PTH, pmol/L | 13.4 | 1.6–6.9 |
| Thyroid Stimulating Hormone, μIU/mL | 2.4 | 0.3–4.2 |
| 25-hydroxyvitamin D, ng/mL | 34 | 25–80 |
| 24-hour urine calcium, mg/d | 88 | >200* |
| Urine Specific Gravity | 1.005 | 1.010 – 1.030 |
| Urinalysis | No blood, no protein | -- |
Abbreviations: PTH, parathyroid hormone; eGFR, estimated glomerular filtration rate.
Typical value; varies with intake.
What are likely differential diagnostic possibilities for hypercalcemia in this patient, and what diagnosis is likely?
What is the pathophysiology of this patient’s hypercalcemia?
What is the cause of her polyuria?
What treatment options are possible for this patient?
Discussion
What are the differential diagnostic possibilities of hypercalcemia in this patient, and what diagnosis is likely?
Hypercalcemia suppresses parathyroid hormone (PTH), and workup is predicated on knowing if PTH is appropriately suppressed. She demonstrates PTH-dependent hypercalcemia. The differential for PTH-dependent hypercalcemia includes primary hyperparathyroidism, tertiary hyperparathyroidism, familial hypocalciuric hypercalcemia (FHH), and lithium therapy.
Measurement of 24-hour urine calcium is an important next step. Elevated urine calcium (> 200 mg/d) is consistent with either primary or tertiary hyperparathyroidism. She exhibits hypocalciuria, which is consistent with either FHH, primary hyperparathyroidism with low vitamin D stores, or lithium therapy. Without a family history of hypercalcemia or low vitamin D levels, FHH and primary hyperparathyroidism are unlikely. She most likely has lithium-associated hypercalcemia, which can be seen in up to 25–30% of individuals receiving lithium therapy.1
What is the pathophysiology of this patient’s hypercalcemia?
Lithium increases the set point of serum calcium for PTH inhibition by partially agonizing the calcium-sensing receptor (CaSR). Through a series of intracellular signals in parathyroid chief cells, lithium reduces activation of protein kinase C, which would normally reduce PTH release. Inability to reduce PTH release, and shifting of the set point for serum calcium leads to the clinical scenario of hypercalcemia with elevated PTH (figure 1).2, 3
Figure 1.
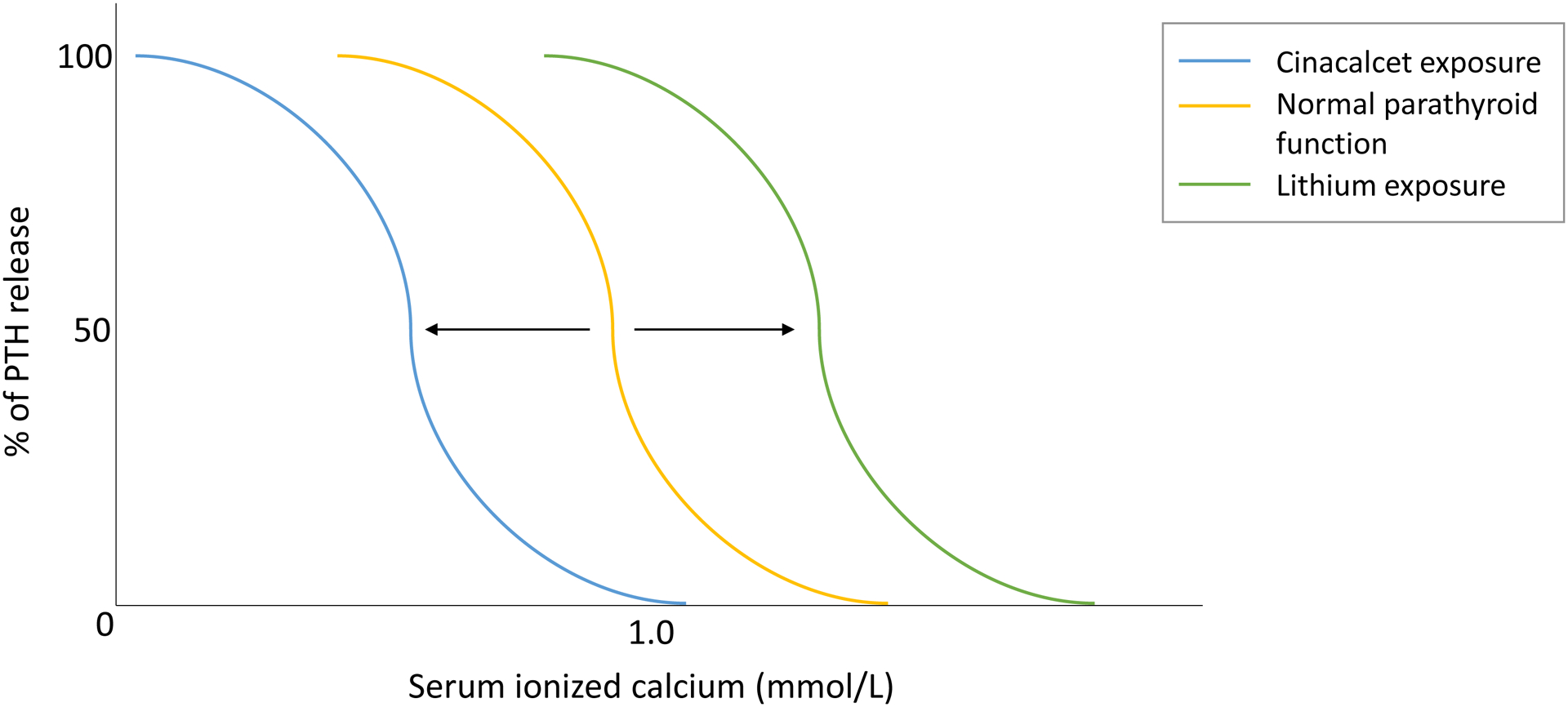
Changes in PTH release and serum ionized calcium level with exposure to lithium and cinacalcet. Lithium exposure shifts setpoint higher at any given ionized calcium level by partial agonism of CaSR. Cinacalcet activates the CaSR to reduce PTH release at lower calcium levels.
Normally, elevated serum calcium binds to and activates the CaSR on the basolateral membrane of the thick ascending limb (TAL). Here, CaSR activation causes 1) decreased TAL paracellular permeability to calcium through alteration of claudins, and 2) inhibition of ROMK (renal outer medullary potassium) channels, which dissolves the transcellular voltage gradient that drives calcium reabsorption.4 Hypercalcemia thus leads to hypercalciuria. On account of abrogating this response by changing the setpoint of the CaSR, lithium leads to hypocalciuria, often mimicking the physiology of FHH.2, 5 Lithium may directly stimulate parathyroid gland hyperplasia or adenoma growth, but it is unclear if this is a primary process or an unmasking and new discovery of an existing parathyroid hyperplasia or adenomatous change.2, 3
What is the cause of her polyuria?
Hypercalcemia itself can cause reversible nephrogenic diabetes insipidus (NDI) via induction of autophagic degradation of aquaporin 2 channels.6 Additionally, lithium decreases insertion of aquaporin 2 channels into the distal nephron, leading to NDI and polyuria. When therapy is prolonged, lithium can lead to medullary interstitial fibrosis and irreversible NDI.1
What treatment options are possible for her hypercalcemia?
Treatment options for lithium-associated hypercalcemia include drug cessation, cinacalcet, and surgical management. Drug cessation may reverse hypercalcemia, although sustained hypercalcemia may persist.3 Cinacalcet, a calcimimetic that reduces PTH secretion at a lower serum calcium level (figure 1), has demonstrated effectiveness in case reports.7 Surgical management may be preferred in cases where a parathyroid adenoma is present in conjunction with hypercalcemia.3
Final Diagnosis: Lithium-associated hypercalcemia.
Support:
Dr. Spiardi is supported by National Institutes of Health grant 5T32DK007006-48.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure: The authors declare that they have no other relevant financial interests.
Patient Protections: The authors declare that the patient described in this Quiz is a fictionalized case, and thus no patient protection measures were implemented.
References
- 1.Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. May 2009;5(5):270–6. doi: 10.1038/nrneph.2009.43 [DOI] [PubMed] [Google Scholar]
- 2.Szalat A, Mazeh H, Freund HR. Lithium-associated hyperparathyroidism: report of four cases and review of the literature. Eur J Endocrinol. Feb 2009;160(2):317–23. doi: 10.1530/eje-08-0620 [DOI] [PubMed] [Google Scholar]
- 3.Meehan AD, Udumyan R, Kardell M, Landén M, Järhult J, Wallin G. Lithium-Associated Hypercalcemia: Pathophysiology, Prevalence, Management. World J Surg. Feb 2018;42(2):415–424. doi: 10.1007/s00268-017-4328-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. Jul 7 2015;10(7):1257–72. doi: 10.2215/cjn.09750913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McHenry CR, Lee K. Lithium therapy and disorders of the parathyroid glands. Endocr Pract. Mar-Apr 1996;2(2):103–9. doi: 10.4158/ep.2.2.103 [DOI] [PubMed] [Google Scholar]
- 6.Khositseth S, Charngkaew K, Boonkrai C, et al. Hypercalcemia induces targeted autophagic degradation of aquaporin-2 at the onset of nephrogenic diabetes insipidus. Kidney Int. May 2017;91(5):1070–1087. doi: 10.1016/j.kint.2016.12.005 [DOI] [PubMed] [Google Scholar]
- 7.Sloand JA, Shelly MA. Normalization of lithium-induced hypercalcemia and hyperparathyroidism with cinacalcet hydrochloride. Am J Kidney Dis. Nov 2006;48(5):832–7. doi: 10.1053/j.ajkd.2006.07.019 [DOI] [PubMed] [Google Scholar]