

Abstract
Glucagon is a potent glucose‐elevating hormone that is secreted by pancreatic α‐cells. While well‐controlled glucagon secretion plays an important role in maintaining systemic glucose homeostasis and preventing hypoglycaemia, it is increasingly apparent that defects in the regulation of glucagon secretion contribute to impaired counter‐regulation and hyperglycaemia in diabetes. It has therefore been proposed that pharmacological interventions targeting glucagon secretion/signalling can have great potential in improving glycaemic control of patients with diabetes. However, despite decades of research, a consensus on the precise mechanisms of glucose regulation of glucagon secretion is yet to be reached. Second messengers are a group of small intracellular molecules that relay extracellular signals to the intracellular signalling cascade, modulating cellular functions. There is a growing body of evidence that second messengers, such as cAMP and Ca2+, play critical roles in α‐cell glucose‐sensing and glucagon secretion. In this review, we discuss the impact of second messengers on α‐cell electrical activity, intracellular Ca2+ dynamics and cell exocytosis. We highlight the possibility that the interaction between different second messengers may play a key role in the glucose‐regulation of glucagon secretion.
Keywords: glucagon, pancreatic islet, second messenger
In this review, we discuss the impact of second messengers on α‐cell electrical activity, tracellular Ca2+ dynamics and cell exocytosis. We highlight the possibility that the interaction between different second messengers may play a key role in the glucose‐regulation of glucagon secretion.
1. INTRODUCTION
Diabetes is a metabolic disorder stemming from a loss of proper blood glucose regulation by the islets of Langerhans. Located within the pancreas, these micro‐organs are small cell clusters predominantly composed of insulin‐secreting β‐cells, glucagon‐secreting α‐cells, and somatostatin‐secreting δ‐cells. 1 , 2 , 3 The fact that glucose‐lowering insulin secretion and/or signalling is lost in diabetes has led it to be considered as an insulin‐related mono‐hormonal disease. 4 However, over recent years, this β‐cell centric view has been challenged and it has been proposed that diabetes, in fact, has a multi‐hormonal aetiology, consisting of dysfunction in the secretion of both insulin and glucagon. 5 , 6 , 7
Glucagon is a potent glucose‐elevating hormone and normally it is released by α‐cells in response to a fall in plasma glucose. It constitutes part of the ‘counter‐regulatory' mechanism of the body by rapidly releasing glucose stored in the liver and stimulating gluconeogenesis, efficiently restoring normal blood glucose levels. The glucose‐sensing function of α‐cells and the consequent release of glucagon has been demonstrated to become dysregulated in diabetes. 8 As a consequence, hyperglycaemia becomes exacerbated and the risk of severe, life‐threatening hypoglycaemia increases dramatically. The importance of the role that glucagon plays in the pathophysiology of diabetes is further supported by studies in which application of glucagon receptor antagonists, thereby blocking glucagon function, was capable of restoring normoglycaemia in β‐cell ablated animal models. 9 , 10 , 11 , 12 Whereas efforts have already been made to apply glucagon receptor for the treatment of diabetes, 13 , 14 , 15 it is undoubtedly clear that therapeutic approaches aiming to restore normal α‐cell function could provide more optimal treatment options for diabetes, given its role in the prevention of hypoglycaemia.
Unlike the well‐established β‐cell stimulation‐secretion coupling model, the precise mechanisms that regulate α‐cell glucagon secretion have yet to reach a consensus. This knowledge gap hampers efforts to fully understand how dysfunction occurs in diabetes. The electrical excitability of α‐cells naturally led to the hypotheses that high glucose inhibits glucagon secretion by modulating α‐cell electrical activity and downstream Ca2+‐dependent exocytosis. To date, proposed mechanisms can be categorised as intrinsic and paracrine: intrinsic mechanisms suggest that glucose metabolism directly modulate activity of α‐cells; while paracrine mechanisms involve factors released from neighbouring β‐ and δ‐cells that mediate the inhibitory effects of glucose. These two types of mechanisms are not mutually exclusive and are likely complementary to each other; many of the effects have been proposed to be mediated through second messengers.
Second messengers are a group of small intracellular molecules (e.g., cAMP, Ca2+, IP3) that amplify/relay extracellular signals (or the ‘first messengers’) to execute biological functions. The effects of glucose on intracellular concentrations of second messengers in β‐cells have been well documented, 16 , 17 and recent reports indicate metabolic regulation of the same group of molecules also control non‐β‐cell activity and hormone secretion. 18 , 19 , 20 , 21 In this review, we summarise the current research on glucose regulation of glucagon secretion, with a special focus on the involvement of the ubiquitous second messengers cAMP and Ca2+.
2. α‐CELL ELECTRICAL ACTIVITY
Different from β‐cells, α‐cells are electrically active and exhibit Ca2+ oscillations at low glucose concentrations (<3 mmol/L). Therefore, for a long time, Ca2+ activity at low glucose was used as the identifying signature of α‐cells in islets. 22 Interestingly, many of the proteins involved in glucose sensing are similar to that of β‐cells, for example, the ATP‐sensitive potassium (KATP) channels are identical in both islet cell types. 23 Considering this, it is rather intriguing that the basal activity of the KATP channels is approximately ten‐fold lower than that recorded in β‐cells under the same conditions. 24 As a result, the high input resistance of α‐cells enables the firing of action potentials even when the ambient glucose concentration is low (Figure 1A). The action potentials of α‐cells are often overshooting (>+10 mV), suggesting the rapid activating and inactivating voltage‐gated Na+ (Nav) channels are involved in the upstroke of the action potentials. Indeed, Nav1.3 (encoded by Scn3a) and 1.7 (encoded by Scn9a) are expressed in the α‐cells with Nav1.3 playing an important role in shaping the action potentials. 25 It is intriguing though that Nav1.7, although contributing significantly to the Na+ current in α‐cells, provides little input to the action potential upstroke – attributable to its negative voltage‐dependent inactivation.
Figure 1.
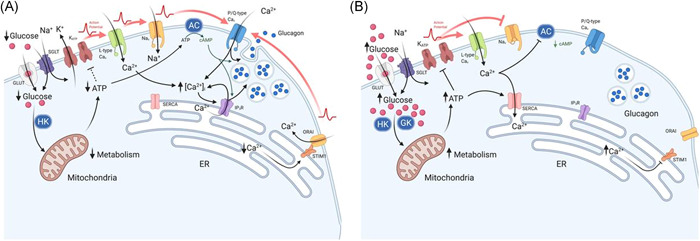
Effect of glucose on α‐cell Ca2+ and cAMP at (A) low glucose and (B) high glucose concentrations, which result in changes to glucagon secretion. (A) At low glucose, there is a low rate of glucose transport across the membrane via GLUT and SGLT proteins. Glucose is then converted to glucose‐6‐phosphate, by hexokinase 1 (HK), to generate ATP via glycolysis. A modest increase in the ATP:ADP ratio, leads to partial closure of KATP‐channels, maintaining the membrane potential at the point of allowing action potential (AP) generation: this enables the activation of L‐type Cav and Nav channels. The resulting high‐amplitude action potentials activate P/Q‐type channels, triggering exocytosis and glucagon release. In addition, adenylyl cyclase (AC) production of cAMP primes vesicles for exocytosis and potentiates Ca2+‐induced Ca2+ release from the endoplasmic reticulum (ER). Consequently, exocytosis of glucagon‐containing vesicles is high. The firing of action potentials can also be attributable to the activation of store‐operated Ca2+ entry (SOCE) via ORAI and STIM1, which is triggered by ER Ca2+ depletion due to low SERCA activity. (B) At high glucose concentrations, there is a high rate of glucose transport into the cytosol, leading to membrane depolarisation. This occurs as a result of increased ATP production via glycolysis, due to substrate abundance: high Km glucokinase (GCK) phosphorylates glucose to produce glucose‐6‐phosphate. High intracellular ATP results in complete closure of KATP‐channels and, consequently, membrane depolarisation. Membrane depolarisation can also be induced by the process of glucose transport through sodium/glucose co‐transporters (SGLTs): Na+ is co‐transported with glucose and thus provides a depolarising membrane conductance. Nav channels, at this membrane potential, become inactivated, whereas L‐type Cav channels remain activatable and contribute to action potential generation. However, action potential amplitude is reduced at high glucose concentrations (despite firing at a higher frequency) and are no longer able to activate P/Q‐type Cav channels, the exocytosis‐related Cav channel in α‐cells. The effect of membrane depolarisation and higher action potential firing frequency may also lead to increased cytosolic Ca2+ within domains adjacent to Ca2+‐sensitive adenylyl cyclase, inhibiting cAMP production and vesicle priming. However, higher ATP may also result in α‐cell membrane repolarisation. High ATP‐activated SERCA uptake of Ca2+ replenish ER Ca2+, deactivating ORAI‐mediated Ca2+ entry, resulting in suppression of α‐cell excitability and glucagon secretion. Red arrows indicate action potential propagation. Figure created using BioRender.com
Although most electrophysiological evidence showed that α‐cells are electrically active at low glucose, there are conflicting reports regarding their electrical activity when challenged by high glucose (Figure 1B). Using the patch‐clamp technique with intact islets, we demonstrated that glucose metabolism induced α‐cell membrane depolarisation and increased firing frequency of action potentials. This coincides with a 20 mV reduction in the action potential amplitude. 24 We attributed this to reduction in α‐cell KATP channel activity. Once transported into α‐cells, glucose is phosphorylated by high affinity hexokinase 1 (HK1) and low affinity glucokinase (GCK) before being broken down to produce pyruvate, and entering the main ATP‐generating citric acid cycle. 26 ADP is then converted to ATP, increasing the ATP:ADP ratio and closing the KATP channels. The consequent membrane depolarisation partially inactivates Nav channels, thereby reducing action potential amplitude. 24 , 27 , 28 , 29 While HK1 may control the basal glucose metabolic rate, metabolism of elevated glucose concentrations (≥5 mmol/L) is controlled by GCK. Conditional tissue specific ablation of GCK expression in α‐cells resulted in a loss of glucose induced membrane depolarisation and inhibition of glucagon secretion in a mouse model. 30
It has also been proposed that high glucose induces membrane depolarisation through the process of glucose entry into the cell. Besides glucose transporters (GLUTs), glucose entry can also be facilitated via the activity of sodium‐glucose co‐transporters (SGLTs), utilising the Na+ concentration gradient. Both SGLT 1 and 2 are expressed in α‐cells, with respective Kms of 2 and 5 mmol/L. 31 , 32 , 33 These differing affinities between subtypes allows each to play a specific role in the cell. The low Km SGLT1 transports glucose at low extracellular concentrations, whereas the high Km SGLT2, functions at glucose concentrations that are maximally inhibitory of glucagon secretion. Mathematic modelling suggested the additional Na+ conductance from the activation of SGLT2 contributes to α‐cell membrane depolarisation at high glucose. 34 However, this cannot be the sole determinant of glucose‐induced membrane potential change. If that were the case, glucose‐induced depolarisation should occur almost immediately subsequent to elevation of extracellular glucose as it would bypasses glucose metabolism. This differs from experimental evidence demonstrating that membrane depolarisation often develops a few minutes after high glucose was applied. 24 It is therefore more likely that, rather than delivering a depolarising ion conductance, SGLT2 predominantly provides an entry route for glucose (which subsequently goes under metabolism, leading to the closure of KATP channels) parallel to GLUTs.
Differing from our observations, other groups observed α‐cell membrane repolarisation in the presence of high glucose concentration. 35 , 36 One of the ion channels suggested to mediate this response is the two‐pore domain channel TASK‐1. 36 TASK‐1 channels are small conductance, outwardly rectifying K+ channels active at physiological voltages. 37 , 38 Inhibition of TASK‐1 channels increases α‐cell electrical activity and, intriguingly, glucose‐inhibition of glucagon secretion. Therefore, these channels may function to limit excitability of α‐cells at high glucose concentrations. 36 However, it remains to be established if/how the TASK‐1 channel activity changes in response to glucose metabolism.
In addition to background leak K+ channels, voltage‐dependant K+ (Kv) channels are involved in the excitability of α‐cells. α‐cell Kv channels open at the peak of the action potential, repolarising the membrane and shaping the downstroke of action potentials. 39 This hyperpolarising current, activated via Kv2 channels, enables reactivation of voltage‐gated Ca2+ (Cav) and Nav channels, preventing depolarisation‐induced inhibition of α‐cell action potential firing. 40
K+ and Na+ are not the only cations involved in the electrical activity of the α‐cell. Ca2+ currents, through Cav channels, also play an important role in the upstroke of the action potential. Blockade of L‐type channels, which carry the bulk of the overall Ca2+ current in mice, eliminates action potential generation. 41 It is possible that L‐type Ca2+ channels function as the pace making channels in α‐cells, initiating the action potential firing. The same Cav channel may play a similar role in human α‐cell electrical activity. 42 However, it is rather counterintuitive that dihydropyridines (e.g., nifedipine or isradipine), potent L‐type Cav channel blockers, exert no apparent inhibitory effect on glucagon secretion at low glucose.
2.1. Intracellular Ca2+ store in α‐cell electrical activity
Transmembrane Ca2+ currents are not the sole determinants of cellular Ca2+ dynamics and electrical activity. The Ca2+ stored within the endoplasmic reticulum (ER) has also been proposed to be part of the α‐cell metabolic sensing machinery, regulating α‐cell electrical activity (Figure 1). It was suggested that the low [ATP]i at low glucose restricts the sarco/endoplasmic Ca2+ ATPase (SERCA) activity, depleting the ER Ca2+ store. This, in turn, activates ER‐bound Ca2+ sensing stromal interaction molecules (STIM), which undergo conformational changes, activating Orai1 Ca2+ channel on the plasma membrane. The Ca2+ influx through the Orai1 channels induces membrane depolarisation and α‐cell electrical activity. 43 This is reversed in response to an increase in extracellular glucose, whereupon higher [ATP]i replenishes ER Ca2+ by the activation of SERCA and subsequently inactivates the store‐operated channels (SOCs) to repolarise the membrane. 44 , 45 However, we note here that expression of both Stim and Orai1 is fairly low in the α‐cells and direct measurements of Orai1 current is technically challenging. Future studies using newly developed ER Ca2+ probes in conjunction with membrane potential recordings, would provide enlightenment to this intriguing hypothesis.
2.2. Ca2+ in exocytosis
Electrical activity, or action potentials, provide signals for the release of hormones through exocytosis, a process where hormone containing granules fuse with the plasma membrane to release their cargo. As with most neural and exocrine cells, α‐cell exocytosis is Ca2+‐dependent. 46 , 47 , 48 A‐cells are equipped with SNARE proteins (a group of proteins that form a complex required for exocytosis): VAMP2; SNAP‐25; and Syntaxin‐1. 49 , 50 Furthermore, α‐cells also express synaptotagmin VII (encoded by Syt7) a SNARE‐related Ca2+ sensing protein. 51 , 52 Ablating Syt7 reduced glucose‐ and depolarisation‐triggered cell exocytosis. It is interesting however, that residual exocytosis of Syt7‐/‐ α‐cells is sensitive to the N‐type Ca2+ channel blocker, ω‐conotoxin. This suggests that: (1) α‐cells express Ca2+‐sensing proteins other than Syt7 and (2) α‐cell granules are tightly coupled to a subtype of Cav (N‐type) channel. These, together with observations that N‐ or P/Q‐type Ca2+ channels are the exocytosis‐relevant Cav channels, 24 , 28 despite their relatively low contribution in transmembrane Ca2+ currents, raises the possibility that α‐cells exhibit exocytosis ‘hotspots’. It is possible that in these areas N‐ and/or P/Q‐type Ca2+ channels, SNARE complexes, and secretory granules are tightly coupled to form releasing sites for rapid secretion of glucagon through interaction with lipid rafts. 53 This is similar to that has been describe in L‐type Ca2+ channels and insulin‐containing granules in β‐cells. 54 Interestingly, L‐type Ca2+ channels, although conducting >50% of transmembrane Ca2+ currents, play an insignificant role in α‐cell exocytosis and glucagon secretion further supporting the existence of specialised release ‘hotspots’ in α‐cells. 41
As discussed above, increase in intracellular Ca2+ triggers α‐cell exocytosis and glucagon secretion. However, interestingly, removal or lowering extracellular Ca2+ below physiological range paradoxically stimulates glucagon secretion. 55 , 56 , 57 Perfusion of rat pancreas with solutions containing low Ca2+ (170 µmol/L), transiently stimulated glucagon secretion at both low and high concentrations of glucose, an effect reversed by re‐introduction of physiological concentrations of Ca2+ (1.94 mmol/L). 46 Furthermore, chelating extracellular Ca2+ by EGTA exerted similar effect on glucagon secretion. 55 However, with prolonged exposure to low Ca2+, the initially high secretion of glucagon begins to decline, presumably due to depletion of intracellular Ca2+ stores. 46
Mediation of this response to extracellular Ca2+ can be attributed to the Ca2+‐sensing receptor (CaSR), a Gq‐coupled GPCR, expressed in both α‐ and β‐cells. 58 , 59 CaSR may also be involved in the glucose‐regulation of glucagon secretion and hyperactivity of these receptors (gain‐of‐function mutations) is correlated to impaired glucose‐suppression of glucagon secretion and glucose tolerance. 60 However, it is interesting to note that loss‐of‐function CaSR mutations do not appear to influence glucose tolerance or insulin secretion. 61
3. PARACRINE REGULATION OF GLUCAGON
In addition to its intrinsic mechanisms, it has also been proposed that α‐cell glucose sensing is, at least in part, mediated by the factors released by neighbouring β‐ and/or δ‐cells, as well as incretins released from the gut. 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 Because this has been discussed in a number of excellent reviews in the recent years, here we will only briefly review the literature relevant to the topic of this review. We categorise these external regulatory factors as inhibitory or stimulatory (Table 1).
Table 1.
Non‐nutrient factors regulating α‐cell glucagon secretion
| Factors | Origin | Effects on α‐cells | Effect on glucagon secretion | References | ||
|---|---|---|---|---|---|---|
| Somatostatin | Islet δ‐cells | Electrical activity/cAMP | Inhibitory | 47 , 62 , 63 , 76 | ||
| γ‐Aminobutyric acid | Islet β‐cells | Electrical activity | Inhibitory | 66 , 67 , 77 , 81 | ||
| Insulin | Islet β‐cells | Electrical activity/cAMP | Inhibitory/no effect | 76 , 77 , 78 | ||
| Zn2+ | Islet β‐cells | Electrical activity | Inhibitory/no effect | 64 , 78 | ||
| GLP‐1 | Intestinal L‐cells, islet α‐cells | Electrical activity, cAMP | Inhibitory | 68 , 82 , 83 , 84 , 85 | ||
| Adrenaline | Adrenal glands | Electrical activity, cAMP | Stimulatory | 85 , 89 , 90 | ||
| Glucagon | Islet α‐cells | cAMP | Stimulatory | 94 | ||
| GIP | Intestinal L‐ or L/K‐cells, islet α‐cells | cAMP | Stimulatory | 95 , 97 , 98 | ||
GLP‐1: Glucagon‐like peptide‐1; GIP: glucose‐dependent insulinotropic polypeptide
3.1. Inhibitory factors
Somatostatin (SST), produced by δ‐cells, is secreted in response to high glucose and potentiated by both insulin and glucagon. 21 , 70 , 71 , 72 It is a potent inhibitor of both glucagon and insulin, activating Gαi‐coupled somatostatin receptor (SSTR) 2 and 5, on α‐ and β‐ cells respectively. 63 , 70 , 73 , 74 , 75 The effect of SSTRs activation is two‐fold. On one hand, it inhibits adenylyl cyclases, lowering intracellular cAMP, a second messenger that stimulates glucagon secretion. 76 On the other hand, it also activates the G‐protein‐coupled inwardly‐rectifying K+ (GIRK) channels. 62 , 73 Opening of the GIRK channels rapidly repolarises α‐cells, abolishing α‐cells electrical activity and subsequent glucagon secretion.
Beta cells may exert their effects on α‐cells through insulin and co‐released factors, such as Zn2+ and γ‐Aminobutyric acid (GABA). Insulin has long been proposed as a negative regulator of glucagon secretion. 77 However, with conflicting effects of insulin on α‐cell electrical activity, 41 , 78 doubt has been cast on the idea that insulin acts as the primary factor in the suppression of glucagon secretion at high glucose. More recent evidence has demonstrated that the glucagonostatic effect of insulin is exerted by through stimulation of δ‐cell SST secretion via SGLTs. 71 Zn2+ is packed into insulin‐containing granules through Zn2+ transporters and is required to stabilise the hexameric insulin crystals within. 79 Consequently, glucose‐stimulated insulin secretion will greatly increase intra‐islet Zn2+ concentration. It has been proposed that Zn2+ may activate α‐cell KATP channels to inhibit α‐cell electrical activity. 78 However, its impact on glucagon release from either islets or αTC1‐9 cells was unclear. 64 GABA, meanwhile, is an inhibitory neurotransmitter released by β‐cells in response to glucose. 80 It activates the A‐type GABA receptor (GABAAR), a Cl‐ channel present on α‐cells. 66 , 67 This induces membrane hyperpolarization, inhibiting electrical activity and lowering of [Ca2+]i. 77 Indeed, inhibition of GABAAR increased glucagon secretion at low glucose and reduced the inhibitory effect of glucose on glucagon secretion. 67 It is interesting to note that the GABAAR mediated effect can be enhanced in the presence of high glucose, where intra‐islet qinsulin induces higher membrane insertion of the Cl‐ channel. 81
Glucagon‐like peptide‐1 (GLP‐1), an incretin produced by intestinal L‐cells, was shown to modulate glucagon secretion from α‐cells. 68 Although, the low level of GLP‐1 receptor expression in α‐cells 69 , 78 and the observation that GLP‐1 inhibited glucagon secretion can be restored by inhibiting SSTR2, led to the hypothesis that GLP‐1 effect is mediated by the stimulation of SST secretion. 82 , 83 However, we showed that GLP‐1 receptors are expressed in α‐cells, albeit at low level, 84 and mediate the SST‐independent effects of GLP‐1 via the activation of PKA. 85
3.2. Stimulatory factors
Adrenaline's stimulatory effect on glucagon secretion has been well established. Released in response to various physiological stresses, such as fight‐or‐flight, exercise, infection and hypoglycaemia. 86 Adrenaline rapidly elevates blood glucose by (1) direct stimulation of hepatic glucose production, (2) stimulation of glucagon and (3) inhibition of insulin secretion. 87 , 88 It stimulates α‐cells through the activation of α1‐ and β‐adrenoceptors, which are Gq/11 and Gs coupled receptors, respectively. 85 , 89
While activation of Gs‐proteins exerts a strong direct stimulatory effect on α‐cell exocytosis via increasing intracellular cAMP concentration, 90 the effect mediated by Gq/11‐protein is through PKC, and, potentially, the release of ER Ca2+. The Gq/11‐protein activates the Phospholipase C (PLC), which cleaves phosphatidylinositol 4, 5 bisphosphate (PIP2) into 1,4,5‐inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 subsequently activates the ligand‐gated IP3 receptor, a Ca2+‐releasing channel located on the ER membrane, increasing cytosolic Ca2+ concentration. 91 DAG binds to and activates protein kinase C (PKC), a serine/threonine kinase, on the plasma membrane. This process can be further potentiated by Ca2+‐induced translocation of PKC to cell membrane 92 It has been shown that PKC activation by phorbol 12‐myristate 12‐acetate (PMA) stimulates glucagon secretion, without direct effect on α‐cell electrical activity. 85 , 93 It is therefore possible that PKC is able to potentiate IP3R release ER Ca2+ and stimulate glucagon release.
Interestingly, glucagon itself can serve as a positive autocrine signal for α‐cell glucagon secretion. α‐cells are equipped with glucagon receptors (a Gs‐coupled receptor) and glucagon was shown to elevate intracellular cAMP concentration, promoting exocytosis. 94 This mechanism can play an important role in glucagon secretion in response to hypoglycaemia, whereupon a large amount of glucagon is required within a short period.
Unlike GLP‐1, another incretin produced by gut cells (K or L/K‐cells), glucose‐dependent insulinotropic polypeptide (GIP) potentiates glucagon secretion. Upon binding to its receptor, GIP stimulates α‐cell cAMP production via GαS‐dependent pathway, 95 , 96 , 97 enhancing glucagon secretion at high glucose levels. 98 It is interesting to note that GIP is also produced by α‐cells, albeit relatively low in quantity, possibly contributing to intra‐islet paracrine and autocrine regulation. 99
4. cAMP
As discussed above, much of the physiological regulation of glucagon secretion is exerted via intracellular changes in α‐cell cAMP concentrations. Downstream to the activation of Gs‐coupled receptors, the predominant effect of cAMP is to potentiate glucagon secretion, and this can be replicated with the uncaging of cAMP or application of forskolin. 37 , 100 Increases in cAMP concentration stimulate granules docking to the membrane, priming them for exocytosis, and forming the ready releasable pool of granules that can be rapidly released when proximal Ca2+ channels activated, thereby enhancing glucagon secretion. 41 , 85 , 90 Therefore, it is possible that cAMP represents the key molecule for the glucose sensing in α‐cells, as proposed by a recent study. 18 Cyclic AMP is catalysed by adenylyl cyclases (ACs) and hydrolysed by phosphodiesterases (PDEs). Intracellular cAMP concentration can fluctuate rapidly depending on the activities of these two enzymes with opposing actions, resulting in oscillations similar to that seen with Ca2+. 19 Furthermore, the concentration of cAMP is not homogenous throughout the cytoplasm. It is likely to be compartmentalised, constrained by the localised distribution and activities of ACs and PDEs. 101
4.1. Production and degradation of cAMP
There are a 10 different AC subtypes, 102 with AC3, AC5, AC6, and AC9 expressed in α‐cells, AC6 being the most highly expressed. 49 Of these 4 subtypes, AC5 and AC6 have had their presence demonstrated at the protein level by immunohistochemistry. 103
The specific expression of PDEs, meanwhile, is yet to be fully elucidated in α‐cells, in contrast to β‐cells where their roles have been extensively studied. 104 Transcriptomic studies have revealed the expression of a large number of isoforms in α‐cells. 49 , 105 Inhibition of PDE3B and PDE4, with cilostamide and rolipram respectively, increases secretion of glucagon at high glucose, while blocking PDE4 also increased glucagon secretion at low glucose. 76 Both PDEs are activated by PKA‐dependent phosphorylation and inhibited by cGMP and have been proposed to play a key role in cAMP regulation in α‐cells. 76 PDE1C, however, an isozyme expressed at a similar level in both α‐ and β‐cells, may be of particular importance to α‐cell function. 106 In β‐cells, glucose enhances PDE1C responsiveness through the activity of Ca2+/calmodulin. The higher responsiveness of the isozyme breaks down cAMP faster at high glucose to form a negative feedback pathway for insulin secretion. 107 It can be postulated that the same PDE may play a similar role in the downregulation of glucagon secretion in α‐cells, preventing over‐secretion of glucagon.Effectors of cAMP
4.2. Effectors of cAMP
The cellular response to cAMP is mediated by two effectors: the serine/threonine kinase Protein kinase A (PKA) and exchange protein directly activated by cAMP 2 (EPAC2). PKA binds to four cAMP molecules on the two R subunits of the inactive holoenzyme. This induces a conformational change, releasing the two catalytic subunits, 108 which are then able to phosphorylate PDEs, 109 ACs, glucose transporters, 110 and ion channels. 111
PKA activation potentiates glucagon secretion at high glucose in murine and human islets, however, this did not occur at low glucose concentrations. 76 It was suggested that PKA is maximally activated at low glucose, but activity at high glucose is curtailed by lower concentrations of cAMP. The main effect can be through the phosphorylation of SNARE proteins, particularly SNAP‐25, which primes vesicles for exocytosis. 112 PKA is also able to phosphorylate the IP3R, potentiating Ca2+‐induced Ca2+ release from the ER. 113 Finally, PKA is able to self‐regulate the levels of cAMP, providing a negative feedback loop, preventing overstimulation. This is performed by phosphorylation of several amino acid sites on ACs, particularly AC5 and AC6, reducing their enzymic activity, converting ATP to cAMP at a lower rate. 114 They also are able to phosphorylate, and consequently activate, PDEs leading to enhanced degradation of cAMP. 109
The second, and less well studied, of the two major enzymes activated by cAMP binding is EPAC2. Upon binding of cAMP, EPAC2 undergoes a conformational change, freeing the catalytic lobe from the regulatory region. 115 This allows it to act as a guanine‐nucleotide‐exchange factor (GEF) for a number of targets involved in exocytosis, such as Rap2B, RAP1, RAB3a, and RIM2. 116 , 117 , 118 , 119
EPAC2 is key to cAMP‐stimulated glucagon secretion. Mice lacking EPAC2 expression have reduced, though not completely abolished, glucagon secretion in response to adrenaline. 85 This is similar to that seen in β‐cells. 117 Unlike PKA, EPAC2 agonists augment glucagon secretion independent of glucose concentration. 76 This is possibly because EPAC2 contains two different cAMP binding domains with different affinities. 120 The effect of EPAC2 on glucagon secretion may be exerted at the level of gene expression and protein production, 121 as well as via modulation of α‐cell exocytosis. For example, SNAP‐25 is a target for EPAC2, 122 although these effects may be mediated indirectly via the scaffold‐associated protein Rab‐interacting molecule 2 (RIM2). 119 , 123 RIM2 is key in the interactions within the SNARE complex, interacting with synaptotagmin, RAB3, Cav channels, munc13, and SNAP‐25. 124 In β‐cells, separation of RIM2 from the exocytotic machinery through deletion of the RAB3 binding domain, impairs exocytosis. 125 RIM2 is highly expressed in α‐cells and glucagon secretion in response to insulin increased in Rim2α −/− mice. 49 , 126
Cyclic AMP can therefore function as a molecular switch for α‐cell exocytosis, the final step of glucagon secretion. Indeed, capacitance measurements with intracellular application of cAMP demonstrated a steep dependence of α‐cell exocytosis to cAMP. 90 Given that α‐cell intracellular cAMP concentration is glucose‐dependent, it is highly likely to constitute part of the mechanism by which glucose regulates glucagon secretion.
4.3. Effect of cAMP on electrical activity
In addition to their direct effect on the exocytotic machinery, it is possible that cAMP effectors are able to modulate α‐cell electrical activity too. Hyperpolarisation‐activated cyclic‐nucleotide‐gated (HCN) channels are members of a superfamily of voltage‐gated cation channels that are permeable to both Na+ and K+ ions and produce a slowly activating inward current. The presence of HCN channels has been demonstrated in both primary islet and cells lines. 127 Unlike most voltage‐dependent channels, HCN channels are activated by membrane hyperpolarisation as well as activation of GPCRs that elevate cAMP levels. 128 These greatly facilitate the activation of the channels by shifting their gating voltage by 10 mV or more to more positive potentials. Similarly, Nav channels may be phosphorylated by PKA. 129 This acts to reduce the peak Na+ current through the channel. 130 , 131
5. INTERACTION BETWEEN Ca2+ AND cAMP
5.1. cAMP effect on Ca2+
As discussed above, both Ca2+ and cAMP play an important role in α‐cell glucagon secretion. Their effects can be exerted on electrical activity and/or cell exocytosis. Furthermore, the interaction between the two molecules, is a key element of the regulatory mechanism.
The effect of cAMP on intracellular Ca2+ has been well documented. Cyclic AMP‐dependent PKA phosphorylation of L‐type Ca2+ channels increases channel activity. 132 , 133 The increased conductance and role in secretion after GPCR stimulation has led to the assertion that L‐type Cav channels become coupled to α‐cells exocytotic machinery and influx through these channels directly triggers exocytosis. 41 , 125 Furthermore, α‐cell Ca2+ conductance is also increased through phosphorylation of N‐type or P/Q‐type Cavs, though via EPAC2‐dependant mechanisms, 85 and additionally enhanced by RIM2‐meidated co‐localisation of these with secretory granules. 134
In addition to the effects on transmembrane Ca2+ influx, α‐cell cAMP is able to directly potentiate Ca2+ release from intracellular stores. 135 It was later identified that cAMP‐induced ER Ca2+ release is mediated by the PKA/EPAC2‐dependent activation of Tpc2 channels located at the lysosomal acidic stores. 90
5.2. Ca2+effect on cAMP
When compared with the well‐established effect elicited by cAMP on intracellular Ca2+, little is known about how Ca2+ may regulate intracellular cAMP in α‐cells. However, all AC isoforms expressed in α‐cells can be modulated by factors within Ca2+‐signalling pathway, such as PKA, Ca2+/calmodulin‐dependent protein kinase (CaMK), or Ca2+ itself. AC3 and AC9 are inhibited indirectly, with CaMKII inhibiting AC3, though only when previously stimulated by Gs, 136 , 137 while AC9 is inhibited by phosphorylation by calcineurin. 138 By comparison, AC5 and AC6 are directly inhibited by free Ca2+ through the competitive displacement of free Mg2+ ions key to the catalytic activity of the enzymes. 139 , 140 It is therefore possible that Ca2+ plays a regulatory role in the α‐cell intracellular cAMP concentration too. As an increase in glucose metabolism is stimulatory to α‐cell electrical activity, 24 it is reasonable to postulate that this may lead to an increase in the cytosolic Ca2+ concentration (though the changes may be subtle and restricted within a certain subcellular compartment). This could, in principle, inhibit AC activity and lower the intracellular cAMP concentration, reducing α‐cell exocytosis (Figure 1B). This idea was partially tested in a recent study 18 using simultaneous imaging of cAMP and Ca2+. While the authors concluded that Ca2+ had little effect on cAMP oscillations in α‐cell, it should be noted the Ca2+ probe used in the study is a strong chelator of Ca2+ and therefore the experimental condition may not have been ideal to address this question. Future studies using less invasive methods may bring novel insights to this interesting interaction of the two second messengers.
6. CODA
Considered to be an “emergency hormone”, glucagon prevents life‐threatening hypoglycaemia by raising blood glucose levels when they begin to fall. Furthermore, glucagon is key to ensuring normal glucose tolerance. 141 , 142 This is not only due to the potent glucose‐elevating capacity of glucagon, but may also be attributed to other beneficial effects exerted on the β‐cells (Reviewed by Stanojevic and Habener 143 ). In diabetes, α‐cell glucose sensing is often impaired, glucagon secretion is no longer stimulated by hypoglycaemia and often cannot be inhibited by hyperglycaemia. Not only does this lead to recurrent hypoglycaemia, a potentially life‐threatening medical condition that is currently the limiting factor in glycaemic management of the disease, 144 but also exacerbates hyperglycaemia. 145 Therefore, pharmacological interventions targeting α‐cell function will offer safer and better treatment for diabetes. However, despite extensive research on α‐cell physiology, the exact mechanism by which glucose regulates glucagon secretion remains obscure. As discussed above, the two ubiquitous second messengers, Ca2+ and cAMP, are implicated in glucagon secretion, and importantly, can be effectively regulated by glucose metabolism. Therefore, it is highly likely that the synergistic action of these two signalling molecules is at the core of glucose regulation of glucagon secretion. The interaction of intracellular Ca2+ and cAMP is complex. Although there is currently no direct experimental evidence, the possible dual role of Ca2+ as both a triggering signal and limiting factor (through the inhibition of adenylyl cyclases, reducing cAMP) for cell exocytosis could potentially explain the inhibitory action of glucose (and sulfonylurea) on glucagon secretion. With the improvement of cAMP and Ca2+ sensors, future studies focusing on their interaction will undoubtedly shed light on α‐cell physiology and pathophysiology in diabetes, opening novel avenues for developing better and safer treatment of the disease.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
Acreman S, Zhang Q. Regulation of α‐cell Glucagon Secretion: The Role of Second Messengers. Chronic Dis Transl Med. 2022;8:7‐18. 10.1016/j.cdtm.2021.06.001
Edited by Yi Cui
REFERENCES
- 1. Brissova M, Fowler MJ, Nicholson WE, et al. Assessment of Human Pancreatic Islet Architecture and Composition by Laser Scanning Confocal Microscopy. J Histochem Cytochem. 2005;53:1087‐1097. 10.1369/jhc.5C6684.2005 [DOI] [PubMed] [Google Scholar]
- 2. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A. 2006;103:2334‐2339. 10.1073/pnas.0510790103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steiner DJ, Kim A, Miller K, Hara M. Pancreatic islet plasticity: Interspecies comparison of islet architecture and composition. Islets. 2010;2:135‐145. 10.4161/isl.2.3.11815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weir GC, Bonner‐Weir S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci. 2013;1281:92‐105. 10.1111/nyas.12031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetees mellitus. J Clin Endocrinol Metab. 2000;85:4053‐4059. 10.1210/jc.85.11.4053 [DOI] [PubMed] [Google Scholar]
- 6. Greenbaum CJ, Prigeon RL, D'Alessio DA. Impaired β‐cell function, incretin effect, and glucagon suppression in patients with type 1 diabetes who have normal fasting glucose. Diabetes. 2002;51:951‐957. 10.2337/diabetes.51.4.951 [DOI] [PubMed] [Google Scholar]
- 7. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4‐12. 10.1172/JCI60016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Müller WA, Faloona GR, Unger RH. Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med. 1973;54:52‐57. 10.1016/0002-9343(73)90083-1 [DOI] [PubMed] [Google Scholar]
- 9. Wang M‐Y, Yan H, Shi Z, et al. Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proc Natl Acad Sci. 2015;112:2503‐2508. 10.1073/pnas.1424934112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Damond N, Thorel F, Moyers JS, et al. Blockade of glucagon signaling prevents or reverses diabetes onset only if residual β‐cells persist. eLife. 2016;5:1‐18. 10.7554/eLife.13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei R, Gu L, Yang J, et al. Antagonistic Glucagon Receptor Antibody Promotes α‐Cell Proliferation and Increases β‐Cell Mass in Diabetic Mice. iScience. 2019;16:326‐339. 10.1016/j.isci.2019.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang MY, Dean ED, Quittner‐Strom E, et al. Glucagon blockade restores functional β‐cell mass in type 1 diabetic mice and enhances function of human islets. Proc Natl Acad Sci U S A. 2021;118:1‐8. 10.1073/pnas.2022142118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheen AJ, Paquot N, Lefèbvre PJ. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin Investig Drugs. 2017;26:1373‐1389. 10.1080/13543784.2017.1395020 [DOI] [PubMed] [Google Scholar]
- 14. Guan HP, Yang X, Lu K, et al. Glucagon receptor antagonism induces increased cholesterol absorption. J Lipid Res. 2015;56:2183‐2195. 10.1194/jlr.M060897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kazda CM, Ding Y, Kelly RP, et al. Evaluation of efficacy and safety of the glucagon receptor antagonist LY2409021 in patients with type 2 diabetes: 12‐and 24‐week phase 2 studies. Diabetes Care. 2016;39:1241‐1249. 10.2337/dc15-1643 [DOI] [PubMed] [Google Scholar]
- 16. Howell SL, Jones PM, Persaud SJ. Regulation of insulin secretion: the role of second messengers. Diabetologia. 1994;37:S30‐S35. 10.1007/BF00400823 [DOI] [PubMed] [Google Scholar]
- 17. Tengholm A. Cyclic AMP dynamics in the pancreatic β‐cell. Ups J Med Sci. 2012;117:355‐369. 10.3109/03009734.2012.724732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia. 2019;62:1212‐1224. 10.1007/s00125-019-4857-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tian G, Sandler S, Gylfe E, Tengholm A. Glucose‐ and hormone‐induced cAMP oscillations in α‐ and β‐cells within intact pancreatic islets. Diabetes. 2011;60:1535‐1543. 10.2337/db10-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Marinis YZ, Zhang E, Amisten S, et al. Enhancement of glucagon secretion in mouse and human pancreatic alpha cells by protein kinase C (PKC) involves intracellular trafficking of PKCalpha and PKCdelta. Diabetologia. 2010;53:717‐729. 10.1007/s00125-009-1635-x [DOI] [PubMed] [Google Scholar]
- 21. Denwood G, Tarasov A, Salehi A, et al. Glucose stimulates somatostatin secretion in pancreatic δ‐cells by cAMP‐dependent intracellular Ca2+ release. J Gen Physiol. 2019;151:1094‐1115. 10.1085/jgp.201912351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rorsman P, Hellman B. Voltage‐activated currents in guinea pig pancreatic α2 Cells: Evidence for Ca2+‐dependent action potentials. J Gen Physiol. 1988;91:223‐242. 10.1085/jgp.91.2.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bokvist K, Olsen HL, Høy M, et al. Characterisation of sulphonylurea and ATP‐regulated K+ channels in rat pancreatic A‐cells. Pflugers Arch. 1999;438:428‐436. 10.1007/s004249900076 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Q, Ramracheya R, Lahmann C, et al. Role of KATP channels in glucose‐regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013;18:871‐882. 10.1016/j.cmet.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Q, Chibalina MV, Bengtsson M, et al. Na+ current properties in islet α‐ and β‐cells reflect cell‐specific Scn3a and Scn9a expression. J Physiol. 2014;592:4677‐4696. 10.1113/jphysiol.2014.274209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heimberg H, De Vos A, Moens K, et al. The glucose sensor protein glucokinase is expressed in glucagon‐producing α‐cells. Proc Natl Acad Sci U S A. 1996;93:7036‐7041. 10.1073/pnas.93.14.7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP‐sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179‐183. 10.1038/387179a0 [DOI] [PubMed] [Google Scholar]
- 28. MacDonald PE, De Marinis YZ, Ramracheya R, et al. A KATP channel‐dependent pathway within α cells regulates glucagon release from both rodent and human islets of langerhans. PLoS Biol. 2007;5:1236‐1247. 10.1371/journal.pbio.0050143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gopel SO, Kanno T, Barg S, Rorsman P. Patch‐clamp characterisation of somatostatin‐secreting δ‐cells in intact mouse pancreatic islets. J Physiol. 2000;528:497‐507. 10.1111/j.1469-7793.2000.00497.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basco D, Zhang Q, Salehi A, et al. Α‐Cell Glucokinase Suppresses Glucose‐Regulated Glucagon Secretion. Nat Commun. 2018;9:9. 10.1038/s41467-018-03034-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hummel CS, Lu C, Loo DDF, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D‐glucose cotransporters SGLT1 and SGLT2. Am J Physiol ‐ Cell Physiol. 2011;300:14‐21. 10.1152/ajpcell.00388.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonner C, Kerr‐Conte J, Gmyr V, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med. 2015;21:512‐517. 10.1038/nm.3828 [DOI] [PubMed] [Google Scholar]
- 33. Suga T, Kikuchi O, Kobayashi M, et al. SGLT1 in pancreatic α cells regulates glucagon secretion in mice, possibly explaining the distinct effects of SGLT2 inhibitors on plasma glucagon levels. Mol Metab. 2019;19:1‐12. 10.1016/j.molmet.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pedersen MG, Ahlstedt I, El Hachmane MF, Gopel SO. Dapagliflozin stimulates glucagon secretion at high glucose: Experiments and mathematical simulations of human A‐cells. Sci Rep. 2016;6:1‐9. 10.1038/srep31214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet β‐cells: A comparison of islet‐cell electrical activity in mouse and rat. Endocrinology. 2006;147:4655‐4663. 10.1210/en.2006-0424 [DOI] [PubMed] [Google Scholar]
- 36. Dadi PK, Luo B, Vierra NC, Jacobson DA. TASK‐1 potassium channels limit pancreatic α‐cell calcium influx and glucagon secretion. Mol Endocrinol. 2015;29:777‐787. 10.1210/me.2014-1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gromada J, Ma X, Høy M, et al. ATP‐sensitive K+ channel‐dependent regulation of glucagon release and electrical activity by glucose in wild‐type and SUR1‐/‐ mouse α‐cells. Diabetes. 2004;53:181‐189. 10.2337/diabetes.53.suppl_3.S181 [DOI] [PubMed] [Google Scholar]
- 38. Dadi PK, Vierra NC, Jacobson DA. Pancreatic β‐cell‐specific ablation of TASK‐1 channels augments glucose‐stimulated calcium entry and insulin secretion, improving glucose tolerance. Endocrinology. 2014;155:3757‐3768. 10.1210/en.2013-2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Macdonald PE, Ha XF, Wang J, et al. Members of the Kv1 and Kv2 voltage‐dependent K+ channel families regulate insulin secretion. Mol Endocrinol. 2001;15:1423‐1435. 10.1210/mend.15.8.0685 [DOI] [PubMed] [Google Scholar]
- 40. Spigelman AF, Dai X, MacDonald PE, Voltage‐dependent K. channels are positive regulators of alpha cell action potential generation and glucagon secretion in mice and humans. Diabetologia. 2010;53:1917‐1926. 10.1007/s00125-010-1759-z [DOI] [PubMed] [Google Scholar]
- 41. Gromada J, Bokvist K, Ding WG, et al. Adrenaline stimulates glucagon secretion in pancreatic A‐cells by increasing the Ca2+ current and the number of granules close to the L‐type Ca2+ channels. J Gen Physiol. 1997;110:217‐228. 10.1085/jgp.110.3.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ramracheya R, Ward C, Shigeto M, et al. Membrane potential‐dependent inactivation of voltage‐gated ion channels in α‐cells inhibits glucagon secretion from human islets. Diabetes. 2010;59:2198‐2208. 10.2337/db09-1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Derler I, Jardin I, Romanin C. Molecular mechanisms of STIM/Orai communication. Am J Physiol ‐ Cell Physiol. 2016;310:C643‐C662. 10.1152/ajpcell.00007.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu YJ, Vieira E, Gylfe E. A store‐operated mechanism determines the activity of the electrically excitable glucagon‐secreting pancreatic α‐cell. Cell Calcium. 2004;35:357‐365. 10.1016/j.ceca.2003.10.002 [DOI] [PubMed] [Google Scholar]
- 45. Vieira E, Salehi A, Gylfe E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia. 2007;50:370‐379. 10.1007/s00125-006-0511-1 [DOI] [PubMed] [Google Scholar]
- 46. Leclercq‐Meyer V, Rebelledo O, Marchand J, Malaisse W. Glucagon release: paradoxical stimulation by glucose during calcium deprivation. Science (80‐). 1975;189:897‐899. 10.1126/science.1098149 [DOI] [PubMed] [Google Scholar]
- 47. Gerich JE, Frankel BJ, Fanska R, West L, Forsham PH, Grodsky GM. Calcium dependency of glucagon secretion from the in vitro perfused rat pancreas. Endocrinology. 1974;94:1381‐1385. 10.1210/endo-94-5-1381 [DOI] [PubMed] [Google Scholar]
- 48. Barg S, Galvanovskis J, Gopel SO, Rorsman P, Eliasson L. Tight coupling between electrical activity and exocytosis in mouse glucagon‐secreting α‐cells. Diabetes. 2000;49:1500‐1510. 10.2337/diabetes.49.9.1500 [DOI] [PubMed] [Google Scholar]
- 49. DiGruccio MR, Mawla AM, Donaldson CJ, et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab. 2016;5:449‐458. 10.1016/j.molmet.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andersson SA, Pedersen MG, Vikman J, Eliasson L. Glucose‐dependent docking and SNARE protein‐mediated exocytosis in mouse pancreatic alpha‐cell. Pflugers Arch Eur J Physiol. 2011;462:443‐454. 10.1007/s00424-011-0979-5 [DOI] [PubMed] [Google Scholar]
- 51. Gustavsson N, Wei SH, Hoang DN, et al. Synaptotagmin‐7 is a principal Ca2+ sensor for Ca2+ ‐induced glucagon exocytosis in pancreas. J Physiol. 2009;587:1169‐1178. 10.1113/jphysiol.2008.168005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY. SNAREing voltage‐gated K+ and ATP‐sensitive K+ channels: Tuning β‐cell excitability with syntaxin‐1A and other exocytotic proteins. Endocr Rev. 2007;28:653‐663. 10.1210/er.2007-0010 [DOI] [PubMed] [Google Scholar]
- 53. Xia F, Leung YM, Gaisano G, et al. Targeting of voltage‐gated K+ and Ca2+ channels and soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor proteins to cholesterol‐rich lipid rafts in pancreatic α‐cells: Effects on glucagon stimulus‐secretion coupling. Endocrinology. 2007;148:2157‐2167. 10.1210/en.2006-1296 [DOI] [PubMed] [Google Scholar]
- 54. Barg S, Ma X, Eliasson L, et al. Fast exocytosis with few Ca2+ channels in insulin‐secreting mouse pancreatic B cells. Biophys J. 2001;81:3308‐3323. 10.1016/S0006-3495(01)75964-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leclercq‐Meyer V, Marchand J, Malaisse WJ. The effect of calcium and magnesium on glucagon secretion. Endocrinology. 1973;93:1360‐1370. 10.1210/endo-93-6-1360 [DOI] [PubMed] [Google Scholar]
- 56. Lundquist I, Fanska R, Grodsky GM. Direct calcium‐stimulated release of glucagon from the isolated perfused rat pancreas and the effect of chemical sympathectomy. Endocrinology. 1976;98:815‐818. 10.1210/endo-98-3-815 [DOI] [PubMed] [Google Scholar]
- 57. Lundquist I, Fanska R, Grodsky GM. Interaction of calcium and glucose on glucagon secretion. Endocrinology. 1976;99:1304‐1312. 10.1210/endo-99-5-1304 [DOI] [PubMed] [Google Scholar]
- 58. Squires PE, Harris TE, Persaud SJ, Curtis SB, Buchan AMJ, Jones PM. The extracellular CaSR on human b‐cells negatively modulates insulin secretion. Diabetes. 2000;49:409‐417. [DOI] [PubMed] [Google Scholar]
- 59. Regard JB, Sato IT, Coughlin SR. Anatomical Profiling of G Protein‐Coupled Receptor Expression. Cell. 2008;135:561‐571. 10.1016/j.cell.2008.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Babinsky VN, Hannan FM, Ramracheya RD, et al. Mutant mice with calcium‐sensing receptor activation have hyperglycemia that is rectified by calcilytic therapy. Endocrinology. 2017;158:2486‐2502. 10.1210/en.2017-00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wolf P, Krššák M, Winhofer Y, et al. Cardiometabolic phenotyping of patients with familial hypocalcuric hypercalcemia. J Clin Endocrinol Metab. 2014;99:E1721‐E1726. 10.1210/jc.2014-1541 [DOI] [PubMed] [Google Scholar]
- 62. Yoshimoto Y, Fukuyama Y, Horio Y, Inanobe A, Gotoh M, Kurachi Y. Somatostatin induces hyperpolarization in pancreatic islet α cells by activating a G protein‐gated K+ channel. FEBS Lett. 1999;444:265‐269. 10.1016/S0014-5793(99)00076-9 [DOI] [PubMed] [Google Scholar]
- 63. Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: An in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology. 2000;141:111‐117. 10.1210/endo.141.1.7263 [DOI] [PubMed] [Google Scholar]
- 64. Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic α‐cells. Diabetes. 2005;54:1789‐1797. 10.2337/diabetes.54.6.1789 [DOI] [PubMed] [Google Scholar]
- 65. Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest. 1984;74:2296‐2299. 10.1172/JCI111658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rorsman P, Berggren PO, Bokvist K, et al. Glucose‐inhibition of glucagon secretion involves activation of GABAA‐receptor chloride channels. Nature. 1989;341:233‐236. 10.1038/341233a0 [DOI] [PubMed] [Google Scholar]
- 67. Wendt A, Birnir B, Buschard K, et al. Glucose Inhibition of Glucagon Secretion from Rat α‐Cells Is Mediated by GABA Released from Neighboring β‐Cells. Diabetes. 2004;53:1038‐1045. 10.2337/diabetes.53.4.1038 [DOI] [PubMed] [Google Scholar]
- 68. Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev. 2007;87:1409‐1439. 10.1152/physrev.00034.2006 [DOI] [PubMed] [Google Scholar]
- 69. Moens K, Heimberg H, Flamez D, et al. Expression and functional activity of glucagon, glucagon‐like peptide I, and glucose‐dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes. 1996;45:257‐261. 10.2337/diabetes.45.2.257 [DOI] [PubMed] [Google Scholar]
- 70. Alberti KGMM, Juel Christensen N, Engkjær Christensen S, et al. INHIBITION OF INSULIN SECRETION BY SOMATOSTATIN. Lancet. 1973;302:1299‐1301. 10.1016/S0140-6736(73)92873-0 [DOI] [PubMed] [Google Scholar]
- 71. Vergari E, Knudsen JG, Ramracheya R, et al. Insulin inhibits glucagon release by SGLT2‐induced stimulation of somatostatin secretion. Nat Commun. 2019;10:1‐11. 10.1038/s41467-018-08193-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Weir GC, Samols E, Day JA, Patel YC. Glucose and glucagon stimulate the secretion of somatostatin from the perfused canine pancreas. Metabolism. 1978;27:1223‐1226. 10.1016/0026-0495(78)90047-1 [DOI] [PubMed] [Google Scholar]
- 73. Kailey B, van de Bunt M, Cheley S, et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β‐ and α‐cells. Am J Physiol ‐ Endocrinol Metab. 2012;303:1107‐1116. 10.1152/ajpendo.00207.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gerich JE, Lorenzi M, Schneider V, et al. Inhibition of pancreatic glucagon responses to arginine by somatostatin in normal man and in insulin dependent diabetics. Diabetes. 1974;23:876‐880. 10.2337/diab.23.11.876 [DOI] [PubMed] [Google Scholar]
- 75. Hauge‐Evans AC, King AJ, Carmignac D, et al. Somatostatin secreted by islet δ‐cells fulfills multipleRoles as a paracrine regulator of islet function. Diabetes. 2009;58:403‐411. 10.2337/db08-0792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Elliott AD, Ustione A, Piston DW. Somatostatin and insulin mediate glucose‐inhibited glucagon secretion in the pancreatic α‐cell by lowering cAMP. Am J Physiol ‐ Endocrinol Metab. 2015;308:E130‐E143. 10.1152/ajpendo.00344.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kawamori D, Kurpad AJ, Hu J, et al. Insulin Signaling in α Cells Modulates Glucagon Secretion In Vivo. Cell Metab. 2009;9:350‐361. 10.1016/j.cmet.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB. Beta‐cell secretory products activate alpha‐cell ATP‐dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54:1808‐1815. 10.2337/diabetes.54.6.1808 [DOI] [PubMed] [Google Scholar]
- 79. Hill CP, Dauter Z, Dodson EJ, Dodson GG, Dunn MF. X‐ray structure of an unusual calcium site and the roles of zinc and calcium in the assembly, stability, and storage of the insulin hexamer. Biochemistry. 1991;30:917‐924. 10.1021/bi00218a006 [DOI] [PubMed] [Google Scholar]
- 80. Braun M, Ramracheya R, Bengtsson M, et al. aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic β‐cells. Diabetes. 2010;59:1694‐1701. 10.2337/db09-0797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xu E, Kumar M, Zhang Y, et al. Intra‐islet insulin suppresses glucagon release via GABA‐GABAA receptor system. Cell Metab. 2006;3:47‐58. 10.1016/j.cmet.2005.11.015 [DOI] [PubMed] [Google Scholar]
- 82. Ørgaard A, Holst JJ. The role of somatostatin in GLP‐1‐induced inhibition of glucagon secretion in mice. Diabetologia. 2017;60:1731‐1739. 10.1007/s00125-017-4315-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ding WG, Renström E, Rorsman P, Buschard K, Gromada J. Glucagon‐like peptide I and glucose‐dependent insulinotropic polypeptide stimulate Ca2+‐induced secretion in rat α‐cells by a protein kinase A‐ mediated mechanism. Diabetes. 1997;46:792‐800. 10.2337/diabetes.46.5.792 [DOI] [PubMed] [Google Scholar]
- 84. Ramracheya R, Chapman C, Chibalina M, et al. GLP‐1 suppresses glucagon secretion in human pancreatic alpha‐cells by inhibition of P/Q‐type Ca 2+ channels. Physiol Rep. 2018;6:1‐17. 10.14814/phy2.13852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. de Marinis YZ, Salehi A, Ward CE, et al. GLP‐1 inhibits and adrenaline stimulates glucagon release by differential modulation of N‐ and L‐type Ca2+ channel‐dependent exocytosis. Cell Metab. 2010;11:543‐553. 10.1016/j.cmet.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Verberne AJM, Korim WS, Sabetghadam A, Llewellyn‐Smith IJ. Adrenaline: Insights into its metabolic roles in hypoglycaemia and diabetes. Br J Pharmacol. 2016;173:1425‐1437. 10.1111/bph.13458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000;43:533‐549. 10.1007/s001250051341 [DOI] [PubMed] [Google Scholar]
- 88. Hirose H, Maruyama H, Ito K, Kido K, Koyama K, Saruta T. Effects of α2‐ and β‐adrenergic agonism on glucagon secretion from perfused pancreata of normal and streptozocin‐induced diabetic rats. Metabolism. 1993;42:1072‐1076. 10.1016/0026-0495(93)90025-J [DOI] [PubMed] [Google Scholar]
- 89. Vieira E, Liu YJ, Gylfe E. Involvement of α1 and β‐adrenoceptors in adrenaline stimulation of the glucagon‐secreting mouse α‐cell. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:179‐183. 10.1007/s00210-003-0858-5 [DOI] [PubMed] [Google Scholar]
- 90. Hamilton A, Zhang Q, Salehi A, et al. Adrenaline stimulates glucagon secretion by Tpc2‐Dependent ca2+ mobilization from acidic stores in pancreatic a‐Cells. Diabetes. 2018;67:1128‐1139. 10.2337/db17-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta ‐ Mol Cell Res. 2009;1793:933‐940. 10.1016/j.bbamcr.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 92. Huang KP. The mechanism of protein kinase C activation. Trends Neurosci. 1989;12:425‐432. 10.1016/0166-2236(89)90091-X [DOI] [PubMed] [Google Scholar]
- 93. Hii CST, Stutchfield J, Howell SL. Enhancement of glucagon secretion from isolated rat islets of Langerhans by phorbol 12‐myristate 13‐acetate. Biochem J. 1986;233:287‐289. 10.1042/bj2330287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ma X, Zhang Y, Gromada J, et al. Glucagon stimulates exocytosis in mouse and rat pancreatic α‐cells by binding to glucagon receptors. Mol Endocrinol. 2005;19:198‐212. 10.1210/me.2004-0059 [DOI] [PubMed] [Google Scholar]
- 95. De Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon‐like peptide‐1, but not glucose‐dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. 2008;51:2263‐2270. 10.1007/s00125-008-1149-y [DOI] [PubMed] [Google Scholar]
- 96. Baggio LL, Drucker DJ. Biology of Incretins: GLP‐1 and GIP. Gastroenterology. 2007;132:2131‐2157. 10.1053/j.gastro.2007.03.054 [DOI] [PubMed] [Google Scholar]
- 97. Gremlich S, Porret A, Hani EH, et al. Cloning, Functional Expression, and Chromosomal Localization of the Human Pancreatic Islet Glucose‐Dependent Insulinotropic Polypeptide Receptor. Diabetes. 1995;44:1202‐1208. 10.2337/diab.44.10.1202 [DOI] [PubMed] [Google Scholar]
- 98. Meier JJ, Gallwitz B, Siepmann N, et al. Gastric inhibitory polypeptide (GIP) dose‐dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia. 2003;46:798‐801. 10.1007/s00125-003-1103-y [DOI] [PubMed] [Google Scholar]
- 99. Fujita Y, Wideman RD, Asadi A, et al. Glucose‐Dependent Insulinotropic Polypeptide Is Expressed in Pancreatic Islet α‐Cells and Promotes Insulin Secretion. Gastroenterology. 2010;138:1966‐1975.e1. 10.1053/j.gastro.2010.01.049 [DOI] [PubMed] [Google Scholar]
- 100. Hermansen K. Forskolin, an Activator of Adenylate Cyclase, Stimulates Pancreatic Insulin, Glucagon, and Somatostatin Release in the Dog: Studies in Vitro*. Endocrinology. 1985;116:2251‐2258. 10.1210/endo-116-6-2251 [DOI] [PubMed] [Google Scholar]
- 101. Willoughby D, Cooper DMF. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965‐1010. 10.1152/physrev.00049.2006 [DOI] [PubMed] [Google Scholar]
- 102. Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145‐174. 10.1146/annurev.pharmtox.41.1.145 [DOI] [PubMed] [Google Scholar]
- 103. Hodson DJ, Mitchell RK, Marselli L, et al. ADCY5 couples glucose to insulin secretion in human islets. Diabetes. 2014;63:3009‐3021. 10.2337/db13-1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tengholm A, Gylfe E. cAMP signalling in insulin and glucagon secretion. Diabetes, Obes Metab. 2017;19:42‐53. 10.1111/dom.12993 [DOI] [PubMed] [Google Scholar]
- 105. Bramswig NC, Everett LJ, Schug J, et al. Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J Clin Invest. 2013;123:1275‐1284. 10.1172/JCI66514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mehats C, Andersen CB, Filopanti M, Jin SLC, Conti M. Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling. Trends Endocrinol Metab. 2002;13:29‐35. 10.1016/S1043-2760(01)00523-9 [DOI] [PubMed] [Google Scholar]
- 107. Han P, Werber J, Surana M, Fleischer N, Michaeli T. The calcium/calmodulin‐dependent phosphodiesterase PDE1C down‐regulates glucose‐induced insulin secretion. J Biol Chem. 1999;274:22337‐22344. 10.1074/jbc.274.32.22337 [DOI] [PubMed] [Google Scholar]
- 108. Krebs EG, Beavo JA. Phosphorylation‐Dephosphorylation of Enzymes. Annu Rev Biochem. 1979;48:923‐959. 10.1146/annurev.bi.48.070179.004423 [DOI] [PubMed] [Google Scholar]
- 109. Pyne NJ, Furman BL. Cyclic nucleotide phosphodiesterases in pancreatic islets. Diabetologia. 2003;46:1179‐1189. 10.1007/s00125-003-1176-7 [DOI] [PubMed] [Google Scholar]
- 110. Thorens B, Dériaz N, Bosco D, et al. Protein kinase A‐dependent phosphorylation of GLUT2 in pancreatic β cells. J Biol Chem. 1996;271:8075‐8081. 10.1074/jbc.271.14.8075 [DOI] [PubMed] [Google Scholar]
- 111. Britsch S, Krippeit‐Drews P, Lang F, Gregor M, Drews G. Glucagon‐like peptide‐1 modulates Ca2+ current but not K+ATP current in intact mouse pancreatic B‐cells. Biochem Biophys Res Commun. 1995;207:33‐39. 10.1006/bbrc.1995.1149 [DOI] [PubMed] [Google Scholar]
- 112. Nagy G, Reim K, Matti U, et al. Regulation of Releasable Vesicle Pool Sizes by Protein Kinase A‐Dependent Phosphorylation of SNAP‐25. Neuron. 2004;41:417‐429. 10.1016/S0896-6273(04)00038-8 [DOI] [PubMed] [Google Scholar]
- 113. Dyachok O, Gylfe E. Ca2+‐induced Ca2+ release via inositol 1,4,5‐trisphosphate receptors is amplified by protein kinase A and triggers exocytosis in pancreatic β‐cells. J Biol Chem. 2004;279:45455‐45461. 10.1074/jbc.M407673200 [DOI] [PubMed] [Google Scholar]
- 114. Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol. 2006;535:1‐12. 10.1016/j.ejphar.2006.01.054 [DOI] [PubMed] [Google Scholar]
- 115. Rehmann H, Prakash B, Wolf E, et al. Structure and regulation of the cAMP‐binding domains of Epac2. Nat Struct Biol. 2003;10:26‐32. 10.1038/nsb878 [DOI] [PubMed] [Google Scholar]
- 116. Schmidt M, Evellin S, Weernink PA, et al. A new phospholipase‐C‐calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol. 2001;3:1020‐1024. 10.1038/ncb1101-1020 [DOI] [PubMed] [Google Scholar]
- 117. Shibasaki T, Takahashi H, Miki T, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A. 2007;104:19333‐19338. 10.1073/pnas.0707054104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Park JH, Kim SJ, Park SH, et al. Glucagon‐like peptide‐1 enhances glucokinase activity in pancreatic β‐cells through the association of Epac2 with Rim2 and Rab3A. Endocrinology. 2012;153:574‐582. 10.1210/en.2011-0259 [DOI] [PubMed] [Google Scholar]
- 119. Kashima Y, Miki T, Shibasaki T, et al. Critical Role of cAMP‐GEFII·Rim2 Complex in Incretin‐potentiated Insulin Secretion. J Biol Chem. 2001;276:46046‐46053. 10.1074/jbc.M108378200 [DOI] [PubMed] [Google Scholar]
- 120. De Rooij J, Rehmann H, Van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP‐dependent RapGEFs. J Biol Chem. 2000;275:20829‐20836. 10.1074/jbc.M001113200 [DOI] [PubMed] [Google Scholar]
- 121. Islam D, Zhang N, Wang P, et al. Epac is involved in cAMP‐stimulated proglucagon expression and hormone production but not hormone secretion in pancreatic α‐ And intestinal L‐cell lines. Am J Physiol ‐ Endocrinol Metab. 2009;296:174‐181. 10.1152/ajpendo.90419.2008 [DOI] [PubMed] [Google Scholar]
- 122. Vikman J, Svensson H, Huang YC, et al. Truncation of SNAP‐25 reduces the stimulatory action of cAMP on rapid exocytosis in insulin‐secreting cells. Am J Physiol ‐ Endocrinol Metab. 2009;297:452‐461. 10.1152/ajpendo.90585.2008 [DOI] [PubMed] [Google Scholar]
- 123. Ozaki N, Shibasaki T, Kashima Y, et al. cAMP‐GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805‐811. 10.1038/35041046 [DOI] [PubMed] [Google Scholar]
- 124. Coppola T, Magnin‐Lüthi S, Perret‐Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct Interaction of the Rab3 Effector RIM with Ca2+ Channels, SNAP‐25, and Synaptotagmin. J Biol Chem. 2001;276:32756‐32762. 10.1074/jbc.M100929200 [DOI] [PubMed] [Google Scholar]
- 125. Shibasaki T, Sunaga Y, Fujimoto K, Kashima Y, Seino S. Interaction of ATP Sensor, cAMP Sensor, Ca2+ Sensor, and Voltage‐dependent Ca2+ Channel in Insulin Granule Exocytosis. J Biol Chem. 2004;279:7956‐7961. 10.1074/jbc.M309068200 [DOI] [PubMed] [Google Scholar]
- 126. Yasuda T, Shibasaki T, Minami K, et al. Rim2α determines docking and priming states in insulin granule exocytosis. Cell Metab. 2010;12:117‐129. 10.1016/j.cmet.2010.05.017 [DOI] [PubMed] [Google Scholar]
- 127. Zhang Y, Zhang N, Gyulkhandanyan AV, et al. Presence of functional hyperpolarisation‐activated cyclic nucleotide‐gated channels in clonal alpha cell lines and rat islet alpha cells. Diabetologia. 2008;51:2290‐2298. 10.1007/s00125-008-1166-x [DOI] [PubMed] [Google Scholar]
- 128. Robinson RB, Siegelbaum SA. Hyperpolarization‐Activated Cation Currents: From Molecules to Physiological Function. Annu Rev Physiol. 2003;65:453‐480. 10.1146/annurev.physiol.65.092101.142734 [DOI] [PubMed] [Google Scholar]
- 129. Li M, West JW, Lai Y, Scheuer T, Catterall WA. Functional modulation of brain sodium channels by cAMP‐dependent phosphorylation. Neuron. 1992;8:1151‐1159. 10.1016/0896-6273(92)90135-Z [DOI] [PubMed] [Google Scholar]
- 130. Cantrell AR, Scheuer T, Catterall WA. Voltage‐dependent neuromodulation of Na+ channels by D1‐like dopamine receptors in rat hippocampal neurons. J Neurosci. 1999;19:5301‐5310. 10.1523/jneurosci.19-13-05301.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltage‐gated sodium current in rat striatal neurones through a protein kinase A. J Physiol. 1995;483:95‐107. 10.1113/jphysiol.1995.sp020570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Bünemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L‐type calcium channels via protein kinase A‐ mediated phosphorylation of the β2 subunit. J Biol Chem. 1999;274:33851‐33854. 10.1074/jbc.274.48.33851 [DOI] [PubMed] [Google Scholar]
- 133. Ämmälä C, Ashcroft FM, Rorsman P. Calcium‐independent potentiation of insulin release by cyclic AMP in single β‐cells. Nature. 1993;363:356‐358. 10.1038/363356a0 [DOI] [PubMed] [Google Scholar]
- 134. Kaeser PS, Deng L, Wang Y, et al. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ‐domain interaction. Cell. 2011;144:282‐295. 10.1016/j.cell.2010.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Johansson H, Gylfe E, Hellman B. Cyclic AMP raises cytoplasmic calcium in pancreatic α2‐cells by mobilizing calcium incorporated in response to glucose. Cell Calcium. 1989;10:205‐211. 10.1016/0143-4160(89)90003-1 [DOI] [PubMed] [Google Scholar]
- 136. Wayman GA, Impey S, Storm DR. Ca2+ inhibition of type III adenylyl cyclase in vivo. J Biol Chem. 1995;270:21480‐21486. 10.1074/jbc.270.37.21480 [DOI] [PubMed] [Google Scholar]
- 137. Wang H, Storm DR. Calmodulin‐Regulated Adenylyl Cyclases: Cross‐Talk and Plasticity in the Central Nervous System. Mol Pharmacol. 2003;63:463‐468. 10.1124/mol.63.3.463 [DOI] [PubMed] [Google Scholar]
- 138. Antoni FA, Palkovits M, Simpson J, et al. Ca2+/calcineurin‐lnhibited adenylyl cyclase, highly abundant in forebrain regions, is important for learning and memory. J Neurosci. 1998;18:9650‐9661. 10.1523/jneurosci.18-23-09650.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Cech SY, Broaddus WC, Maguire ME. Adenylate cyclase: The role of magnesium and other divalent cations. Mol Cell Biochem. 1980;33:67‐92. 10.1007/BF00224572 [DOI] [PubMed] [Google Scholar]
- 140. Guillou JL, Nakata H, Cooper DMF. Inhibition by calcium of mammalian adenylyl cyclases. J Biol Chem. 1999;274:35539‐35545. 10.1074/jbc.274.50.35539 [DOI] [PubMed] [Google Scholar]
- 141. Shah P, Basu A, Basu RR, Rizza RA. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol ‐ Endocrinol Metab. 1999;277:283‐290. 10.1152/ajpendo.1999.277.2.e283 [DOI] [PubMed] [Google Scholar]
- 142. Gilon P. The Role of α‐Cells in Islet Function and Glucose Homeostasis in Health and Type 2 Diabetes. J Mol Biol. 2020;432:1367‐1394. 10.1016/j.jmb.2020.01.004 [DOI] [PubMed] [Google Scholar]
- 143. Stanojevic V, Habener JF. Evolving function and potential of pancreatic alpha cells. Best Pract Res Clin Endocrinol Metab. 2015;29:859‐871. 10.1016/j.beem.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cryer PE. Hypoglycaemia: The limiting factor in the glycaemic management of Type I and Type II diabetes. Diabetologia. 2002;45:937‐948. 10.1007/s00125-002-0822-9 [DOI] [PubMed] [Google Scholar]
- 145. Dinneen S, Alzaid A, Turk D, Rizza R. Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM. Diabetologia. 1995;38:337‐343. 10.1007/BF00400639 [DOI] [PubMed] [Google Scholar]