

Abstract
Murine double minute 2 (MDM2) and X-linked inhibitor of apoptosis protein (XIAP) are important cell survival proteins in tumor cells. As a dual MDM2/XIAP inhibitor reported previously, compound MX69 has low potency with an IC50 value of 7.5 μM against an acute lymphoblastic leukemia cell line EU-1. Herein, we report the structural optimization based on the MX69 scaffold, leading to the discovery of a 25-fold more potent analogue 14 (IC50 = 0.3 μM against EU-1). We demonstrate that 14 maintains its mode of action by dual targeting of MDM2 and XIAP through inducing MDM2 protein degradation and inhibiting XIAP mRNA translation, respectively, which resulted in cancer cell growth inhibition and cell death. The results strongly suggest that the scaffold based on 14 is promising for further optimization to develop a new therapeutic agent for leukemia and possibly other cancers where MDM2 and XIAP are dysregulated.
Graphical Abstract
INTRODUCTION
The human murine double minute 2 (MDM2) is amplified/overexpressed in various human cancers, including childhood acute leukemia lymphoma (ALL) and neuroblastoma (NB).1–4 In cancer patients, MDM2 overexpression is associated with disease progression and poor treatment outcomes.5–8 The main oncogenic function of MDM2 is to inhibit the tumor suppressor p53;9,10 thus, the p53 function becomes inactivated in MDM2-overexpressing cells, resulting in cancer cell proliferation and growth. In addition, MDM2 plays p53-independent roles in oncogenesis, as reported in p53-deficient cancer patients, in which MDM2 overexpression is also involved in cancer progression and treatment resistance.11–13 This is because in addition to interacting with and regulating p53, MDM2 also interacts with other molecules involved in oncogenesis. For instance, MDM2 can bind to nucleic acids and specific small RNA molecules,14–17 playing many p53-independent roles in cancer pathogenesis. Changes in MDM2 expression level occur in multiple ways, including via gene amplification, transcriptional induction by p53, and regulation at the post-translational level by a self-ubiquitination mechanism. MDM2, a member of the RING (really interesting new gene)-finger-type family of E3 ubiquitin ligases, is also a substrate of its own RING domain E3 ligase.18,19 Under certain conditions, such as upon binding nucleic acids or RNA, the capacity of the MDM2 RING domain E3 ligase to target itself for ubiquitination becomes inhibited.20,21
X-linked inhibitor of apoptosis protein (XIAP) is an important member of the inhibitors of apoptosis protein (IAP) family. It specifically binds to and inhibits the activated forms of caspases 3, 7, and 9; these enzymes induce the intrinsic (mitochondrial) apoptotic pathway, the major cell death mechanism activated by many chemotherapy drugs.22 Upregulated XIAP is detected in tumor cells from a number of cancer types, and a high level of XIAP expression is associated with treatment resistance and a poor prognosis.23–25 XIAP expression is mainly regulated by an internal ribosome entry site (IRES)-mediated mechanism at the translational level.26 The IRES-mediated translation of XIAP is specifically activated when cells undergo cellular stress, such as exposure to radiation or treatment with chemotherapeutic drugs.27
Our recently published studies revealed that expression of XIAP and MDM2 in tumors is mutually regulated.15,21 We found that the C-terminal RING domain of MDM2 binds to the XIAP IRES, increasing the IRES-mediated XIAP translation; this in turn results in increased expression of XIAP and drug resistance.15 Conversely, binding of XIAP IRES to the MDM2 RING domain decreases its E3 ubiquitin-ligase activity for MDM2 self-ubiquitination, which leads to MDM2 stabilization, resulting in p53 inhibition and cancer progression.21 Thus, the interaction between the MDM2 RING protein and the XIAP IRES causes simultaneously increased expression of both MDM2 and XIAP, contributing to cancer progression and drug resistance. These mechanistic studies suggest that simultaneous inhibition of MDM2 and XIAP could be a promising strategy in developing targeted agents for cancer treatment.
Toward this direction, we have recently published the discovery of MX69 as a dual MDM2/XIAP inhibitor by high-throughput screening compound libraries available at Emory University using fluorescence polarization assays, followed by extensive target validation studies.28 MX69 binds to the C-terminal RING domain of MDM2, which is critical for its E3-ligase function, and is capable of blocking or disrupting the interaction between the MDM2 RING domain and the XIAP IRES, leading to simultaneous inhibition of both MDM2 and XIAP. This dual inhibition results in the activation of p53 in wild-type p53 tumor cells, similar to that of nutlin-3, a known MDM2 inhibitor that works by disrupting the binding of MDM2 and p53,29 but has the added advantage of inducing caspases 3, 7, and 9 that is independent of the p53 status.28 Such an agent should be capable of inducing apoptosis in p53-deficient cancer cells that are dependent on the expression of high levels of both MDM2 and XIAP.
While MX69 is well tolerated in mice, its potency is low (IC50 = 7.5 μM against cancer cells) and thus requires a high dose (up to 100 mg/kg) in order to achieve good in vivo efficacy.28 We report here our medicinal chemistry efforts to increase the potency of MX69, which led to the discovery of its more potent analogue 14, N-(3,4-dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]-quinoline-8-sulfonamide. We demonstrate that 14 maintains on-target inhibition for MDM2 and XIAP and shows more than 25-fold improvements in potency compared with MX69. Although its unfavorable pharmacokinetic (PK) properties prevented its further development, the current study paves a foundation to further optimize this scaffold to ultimately develop a more metabolically stable analogue for future potential clinical development.
RESULTS AND DISCUSSION
Structural Modification of the A-ring.
For convenient discussion on the synthesis of MX69 analogues and the subsequent structure–activity relationship (SAR) analysis, we designated the four rings in MX69 as rings A to D as shown in Figure 1. The MX69 scaffold is generally assembled through the Povarov reaction, a three-component Aza–Diels–Alder reaction via in situ imine formation and subsequent formal [4 + 2]-cycloaddition to form the tetrahydroquinoline scaffold by the coupling of amines, aldehydes, and alkenes. The amines could be obtained by palladium-catalyzed hydrogenation of the corresponding nitro compounds, which were synthesized by the reaction of substituted anilines with 4-nitrobenzenesulfonyl chlorides in the presence of pyridine. All the new MX69 analogues were tested for their antiproliferative activities, as stereoisomeric mixtures as synthesized, against the human leukemia EU-1 cell line, and MX69 was used as the reference for comparison (Table 1).
Figure 1.
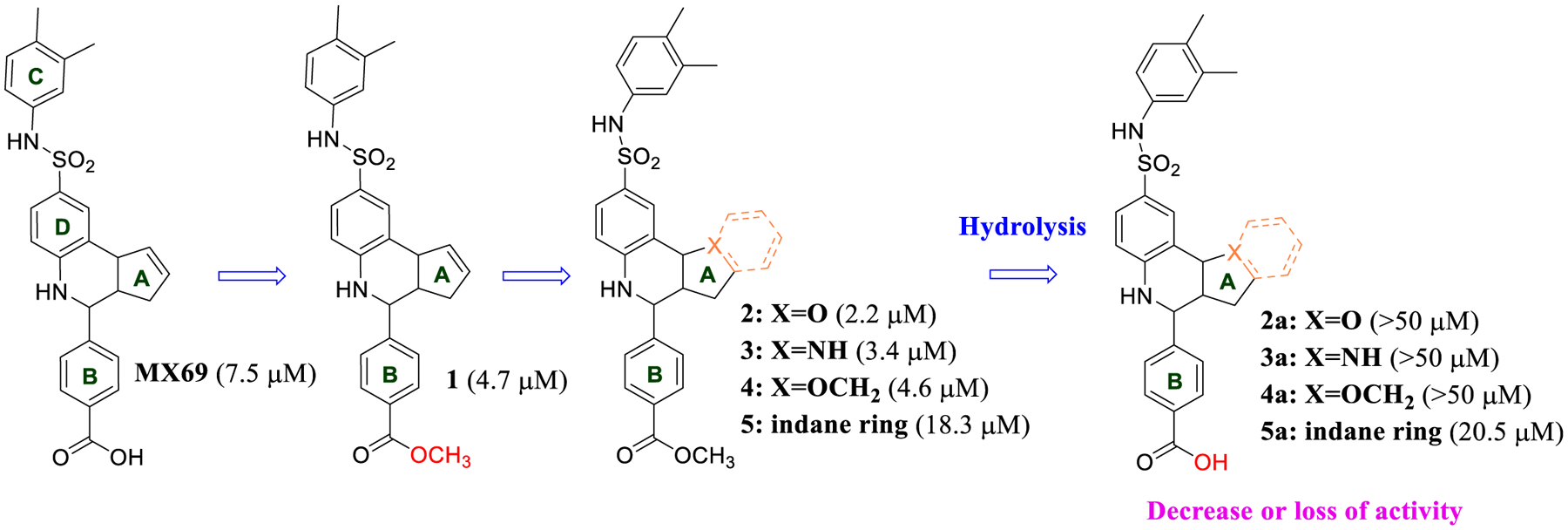
Structural modification of the A-ring in the MX69 scaffold.
Table 1.
Antiproliferative Activities for New MX69 Analogues against the Leukemia EU-1 Cell Line (Data Represent the Mean ± SD of Three Independent Experiments)
| compound | IC50 (μM) | compound | IC50 (μM) |
|---|---|---|---|
| 1 | 4.7 ± 0.6 | 26 | 3.2 ± 0.3 |
| 2 | 2.2 ± 0.3 | 27 | 2.7 ± 0.2 |
| 2a | >50 | 28 | 7.0 ± 0.9 |
| 3 | 3.5 ± 0.2 | 30 | 3.8 ± 0.4 |
| 3a | >50 | 31 | 3.3 ± 0.2 |
| 4 | 4.6 ± 0.3 | 32 | 1.4 ± 0.2 |
| 4a | >50 | 33 | 3.2 ± 0.4 |
| 5 | 18.3 ± 2.6 | 34 | 1.6 ± 0.2 |
| 5a | 20.5 ± 2.9 | 35 | 6.3 ± 0.8 |
| 10 | 4.2 ± 0.4 | 36 | 5.6 ± 0.5 |
| 11 | 3.6 ± 0.3 | 37 | 5.2 ± 0.7 |
| 12 | 4.1 ± 0.4 | 38 | 2.9 ± 0.3 |
| 13 | 0.5 ± 0.1 | 39 | 7.2 ± 0.8 |
| 14 | 0.3 ± 0.1 | 40 | >50 |
| 15 | 2.0 ± 0.2 | 41 | >50 |
| 16 | 2.0 ± 0.2 | 42 | 6.2 ± 0.7 |
| 17 | 3.6 ± 0.3 | 43 | >50 |
| 18 | 6.1 ± 0.5 | 44 | 23.4 ± 3.0 |
| 22 | 5.2 ± 0.5 | 45 | 1.2 ± 0.2 |
| 23 | 30 ± 3.3 | 46 | 1.6 ± 0.2 |
| 24 | 2.9 ± 0.3 | MX69 | 7.5 ± 0.5 |
| 25 | 3.1 ± 0.4 |
Since there are no crystal structures of the MDM2 RING domain available to guide our structural optimization, to get an initial idea about the key functional groups in the MX69 scaffold for the antiproliferative activity, we first probed whether the p-carboxylic acid in MX69 is required by converting it to a more hydrophobic moiety. Compound 1 (IC50 = 4.7 μM, Figure 1) formed by a simple esterification of the acid group shows moderately improved potency, suggesting that the acid moiety is not absolutely required. Next, we bioisosterically replaced the double bond in the A-ring with an oxygen atom to form 2 (IC50 = 2.2 μM) or a nitrogen atom to form 3 (IC50 = 3.5 μM), with the goal of adding polarity to the molecule and thus providing enhanced aqueous solubility from 1. Both analogues also showed slightly increased potency, with the oxygen analogue 2 being the more potent one. Further expansion of the A-ring from a five- to six-membered ring to form 4 (IC50 = 4.6 μM) resulted in a decrease of antiproliferative potency. Interestingly, hydrolysis of the ester substitution in the B-ring back to the corresponding acids 2a, 3a, or 4a (IC50 > 50 μM) all resulted in a significant loss of activity. Attempts to add another ring to the A-ring for either the ester 5 (IC50 = 18.3 μM) or the acid 5a (IC50 = 20.5 μM) also resulted in significant loss of potency. Collectively, these results are consistent with the trend in the transformation from the original MX69 to compound 1, further suggesting that a hydrophobic substitution in this B-ring is beneficial. The activities for these compounds are summarized in Table 1 and their syntheses are shown in Scheme 1.
Scheme 1. Synthesis of 1–5 and 2a–5a through Modification of the A-ringa.
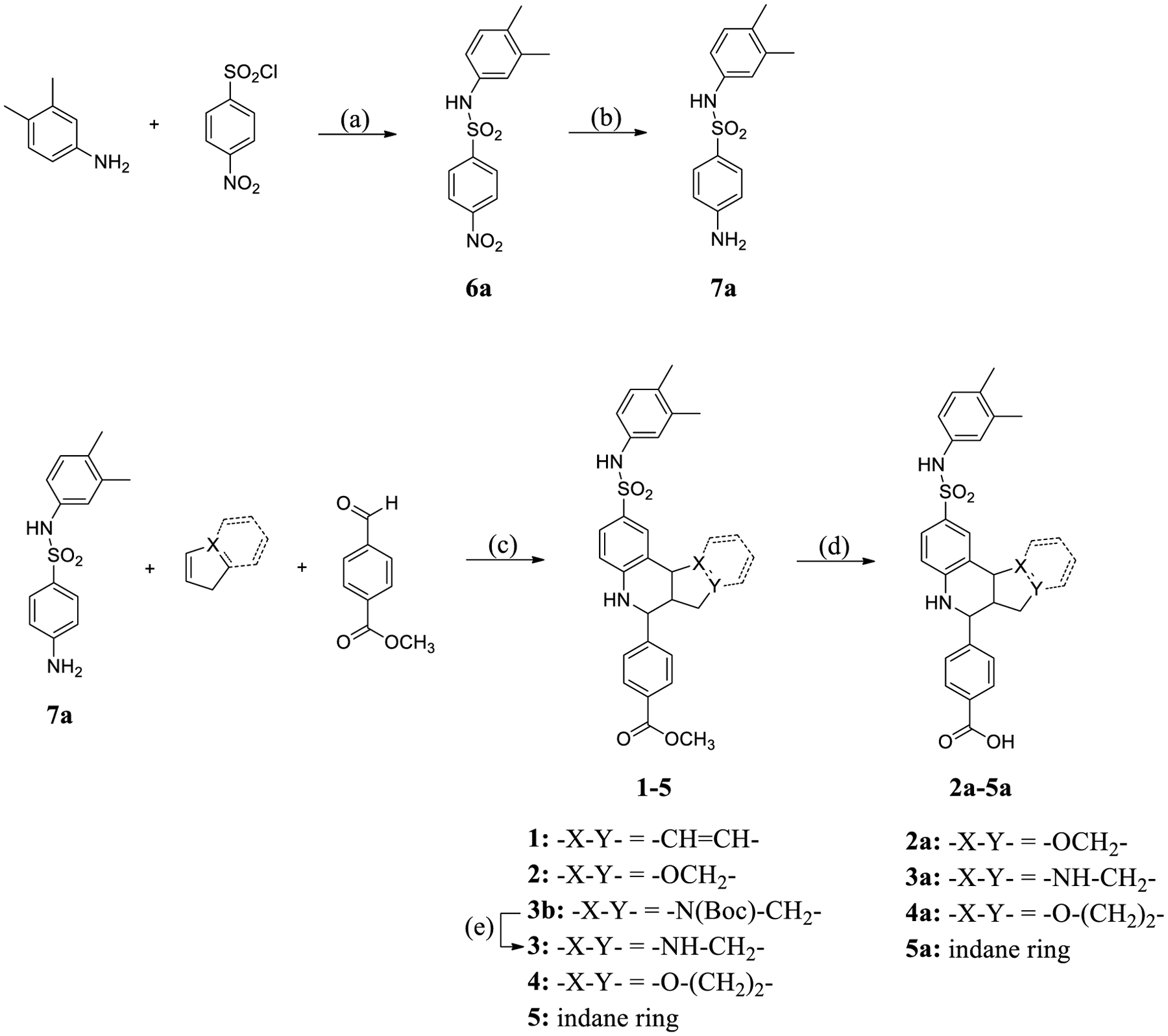
a(a) Pyridine, CH2Cl2, and r.t.; (b) Pd/C, H2, MeOH, and r.t.; (c) Sc(OTf)3, 4 Å molecular sieves, CH3CN, and r.t.; (d) KOH, MeOH/H2O, and r.t.; and (e) TFA, CH2Cl2, and r.t.
Structural Modification of the B-ring Substitution.
Since compound 2 bearing the oxygen atom in the A-ring displayed the highest potency, this scaffold was used for subsequent SAR exploration. The ester substitution on the B-ring is not metabolically stable in vivo where esterase is abundant. In order to increase the metabolic stability of compound 2, analogues were designed with other more stable substitutions in the B-ring (Scheme 2B). Unfortunately, replacement of the ester group by isopropyl (10, IC50 = 4.2 μM), bromo (11, IC50 = 3.6 μM), or methanesulfonyl moieties (12, IC50 = 4.1 μM) did not exhibit improved potency compared to 2. Replacing the substituted phenyl group with a naphthyl group (18, IC50 = 6.1 μM) did not help either. However, compounds 13 and 14 displayed an approximately 4-fold increased potency after replacing the para-ester moiety by acetyl or isobutyryl groups, with IC50 values of 0.5 and 0.3 μM, respectively. Moving the acetyl group and isobutyryl group from para to meta position afforded 16 (IC50 = 2.0 μM) and 17 (IC50 = 3.6 μM), leading to 4-fold and 7-fold drops in potency, respectively. Reduction of the carbonyl group of isobutyryl moiety to the hydroxyl group to form 15 (IC50 = 2.0 μM) caused an approximately 4-fold potency loss compared to 14. Analogues with different ketone moieties were further synthesized (Scheme 2C) based on the results that substitution of B-ring with the ketone group demonstrated a significant improvement in potency. However, cyclization of the isopropyl group to form 22 (IC50 = 5.2 μM) showed a 10-fold reduced potency compared to 14. Replacement of the isopropyl group with linear chains such as ethylene (23, IC50 = 30.0 μM) and n-propyl (24, IC50 = 2.9 μM) failed to further increase the anticancer activity either. Compounds 25 (IC50 = 3.1 μM) and 26 (IC50 = 3.2 μM) with a larger group, cyclohexyl and phenyl, respectively, were approximately 6-fold less potent than 14. Since compound 13 does not bind to the MDM2 target based on subsequent mechanistic studies, the isobutyryl group was considered the optimal substituent at the para position of the B-ring.
Scheme 2.
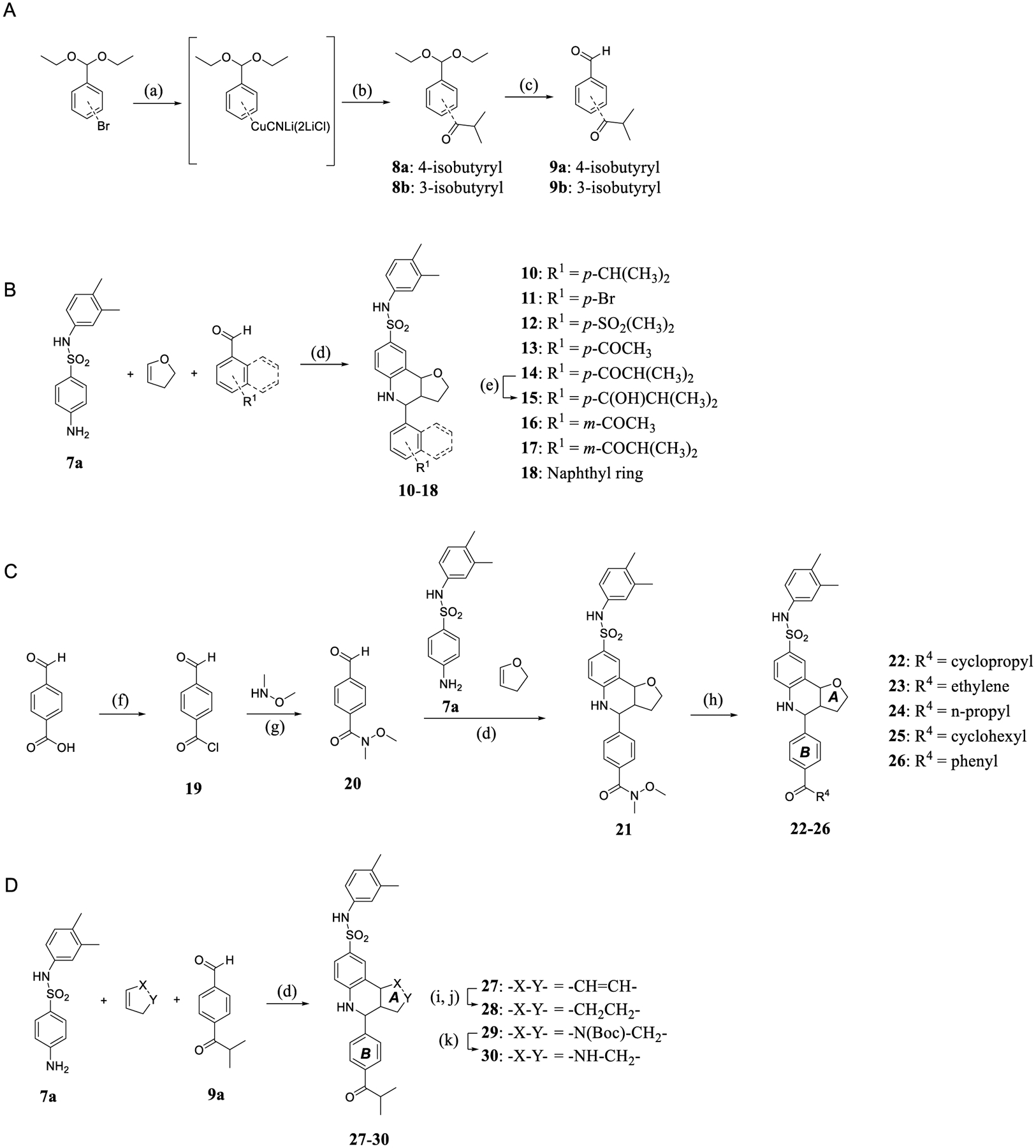
(A) Synthesis of Intermediates 9a and 9b; Reagents and Conditions: (a) N-Butyllithium, CuCN, LiCl, THF, and −78 °C; (b) Isobutyryl Chloride, THF, and −78 °C; (c) HCl (aq, 3 M), CH2Cl2/H2O, and r.t; (B) Synthesis of 10–18 through Modification of the B-ring; Reagents and Conditions: (d) Sc(OTf)3, 4 Å Molecular Sieves, CH3CN, and r.t.; (e) Pd/C, H2, MeOH, and r.t.; (C) Synthesis of 22–26 with Different Ketone Moieties in the B-ring; Reagents and Conditions: (f) Oxalyl Chloride, DMF, CH2Cl2, and 0 °C–r.t; (g) N,O-Dimethylhydroxylamine Hydrochloride, NEt3, CH2Cl2, and 0 °C–r.t; (h) THF, Corresponding Lithium Reagents, −78 °C or Grignard Reagents, and r.t.; (D) Synthesis of 27–30 through the A-ring Modification on Parent Compound 14; Reagents and Conditions: (i) Pd/C, H2, MeOH, and r.t.; (j) Dess–Martin Reagent, CH2Cl2, and r.t.; and (k) TFA, CH2Cl2, and r.t.
Having the currently most potent compound 14 in hand, we went back to investigate the effect of A-ring on the antiproliferative activity (Scheme 2D). Similarly, incorporation of a carbon double bond (27, IC50 = 2.7 μM) or nitrogen (30, IC50 = 3.8 μM) into the A-ring decreased the potencies by 5- or 6-fold, respectively, compared to 14. Further saturation of the double bond of the cyclopentene ring of 27 to form 28 (IC50 = 7.0 μM) also exhibited an approximately 3-fold drop in potency. All these results are consistent with the previous finding that a tetrahydrofuran moiety displayed higher activity than other modifications on the A-ring.
Optimization of the C-ring Moiety.
Having optimized the A- and B-rings in the MX69 scaffold, next we investigated the substitutions on the C-ring by removing either of the two methyl substituents in parent compound 14 or by bioisosterically replacing the methyl group(s) with halogens (Scheme 3). Removing either (31, IC50 = 3.3 μM; 32, IC50 = 1.4 μM) or both of the methyl substituents (33, IC50 = 3.2 μM) in 14 all led to reduced antiproliferative potency. Replacing one (34, IC50 = 1.6 μM; 35, IC50 = 6.3 μM) or both methyl substitutions (36, IC50 = 5.6 μM) with a chlorine atom also resulted in lower potency. Substitutions using other halogen atoms that are larger (37, IC50 = 5.2 μM) or smaller (38, IC50 = 2.9 μM) than chlorine did not help, nor did replace a methyl with a trifluoromethyl group (39, IC50 = 7.2 μM). Further analyses of this SAR indicated that when keeping the meta-methyl substitution intact, increasing the size of the para substitution decreases the antiproliferative potency with the order H (32) > F (38) > Cl (35) > CF3 (39). Thus, bulkier substituents at either position seem to reduce the activity. However, since compound 14 is more potent than compounds 32, 35, and 38 despite the former’s larger substituents, these results suggest that the activity also correlates with the C-ring substituents’ electronic effects, with the two electron-donating methyl groups of 14 striking the best balance for antiproliferative activity at this stage of the study.
Scheme 3. Synthesis of 31–39 through Modification of the C-ringa.
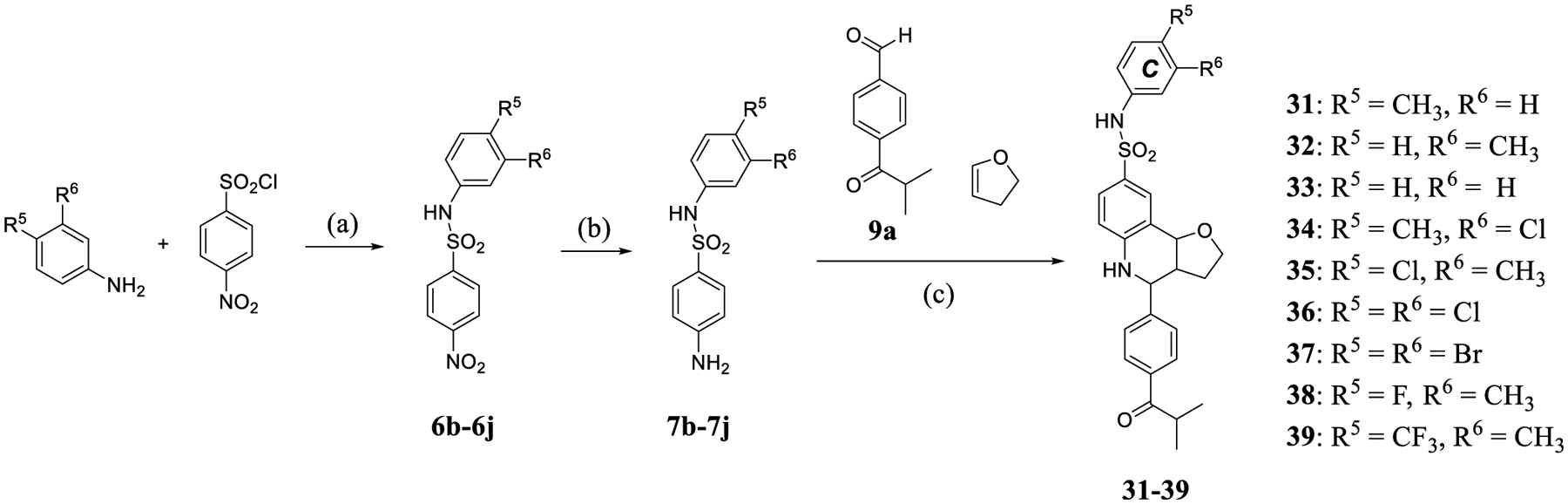
a(a) Pyridine, CH2Cl2, r.t. (b) Pd/C, H2, MeOH, r.t. (c) Sc(OTf)3, 4 Å molecular sieves, CH3CN, r.t.
Effects of Substitutions on the D-ring and on the NH Moieties.
In the next phase of our SAR analysis, modifications in the D-ring were designed by a simple introduction of a chlorine substituent to different positions. Compound 2 was selected as a parent scaffold, and incorporation of chlorine into the 6- or 7-position afforded compounds 40 and 41 (Scheme 4A). An alternative pathway yielded 7-chlorinated compound 43 and 9-chlorinated compound 44 based on the scaffold bearing a methanesulfonyl group in the B-ring and a carbon double bond in the A-ring (Scheme 4B). These compounds all exhibited abolishment or major reduction in anticancer activity. The best, 44 (IC50 = 23.4 μM), is approximately 6-fold less potent than its parent compound 42 (IC50 = 6.2 μM), and the others all have IC50 values larger than 50 μM. These outcomes suggest that it is not appropriate to occupy the position of the D-ring with substituents. The incorporation of methyl group(s) on the nitrogen(s) of 14 was investigated as well. The resulting 45 (IC50 = 1.2 μM) and 46 (IC50 = 1.6 μM) have 2-fold and 3-fold decreased potency, respectively, so the NH groups are important for antiproliferative activity (Scheme 4C).
Scheme 4. (A,B) Synthesis of 40–44 through Modification of the D-ring and (C) Synthesis of 45 and 46 by Methylation of the NH Group.a.
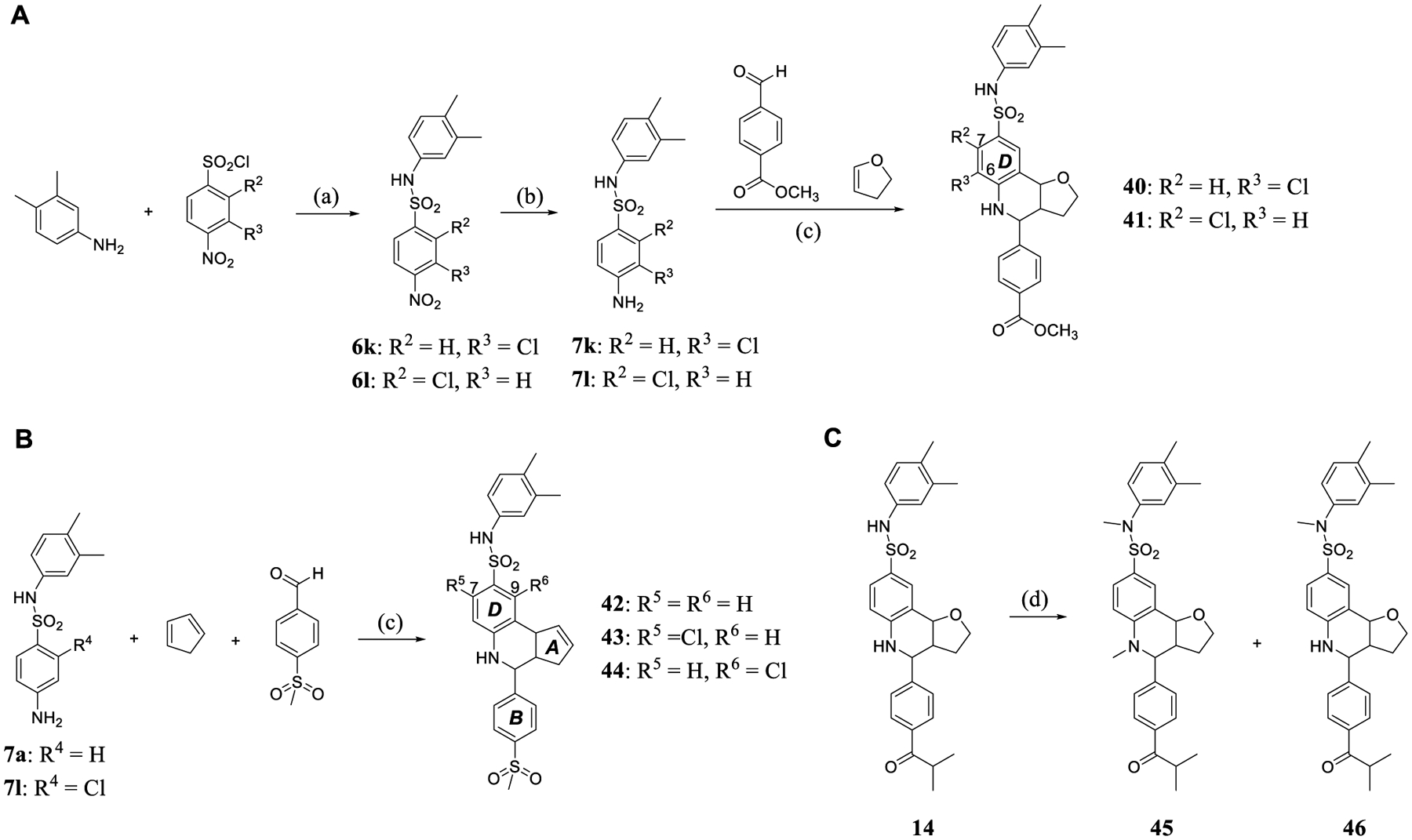
aReagents and conditions: (a) pyridine, CH2Cl2, and r.t.; (b) Pd/C, H2, MeOH, and r.t.; (c) Sc(OTf)3, 4 Å molecular sieves, CH3CN, and r.t; (d) NaH, MeI, THF, and r.t.
Biological Characterization of Compound 14.
Compound 14 Attenuates the Proliferation in Cancer Cells but Shows a Negligible Inhibitory Effect on Normal Hematopoiesis.
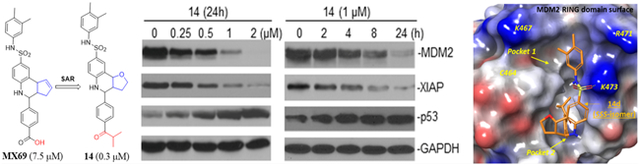
Based on the screening on the 40 new MX69 analogues using the water-soluble tetrazolium salt (WST) assay in the EU-1 cell line, compound 14 exhibited significantly enhanced anticancer activity with an IC50 value of 0.3 μM, representing a 25-fold increase in activity compared to MX69 (IC50 = 7.5 μM). Furthermore, we tested four additional cancer cell lines including one ALL (EU-3) and three NB (NB-1643, SHEP1, and LA1–55N). WST cytotoxicity assays showed that 14 consistently exhibited potent cytotoxicity against these tested cell lines with more sensitivity of ALL (IC50 = 0.3–0.4 μM) than NB (IC50 = 0.5–1.2 μM) to 14 (Figure 2A). Furthermore, the results of colony formation assays showed that 14 potently inhibits cell growth in all the three NB cell lines. We observed a significant reduction in both colony number and size in 14-treated cells as compared with controls (Figure 2B).
Figure 2.
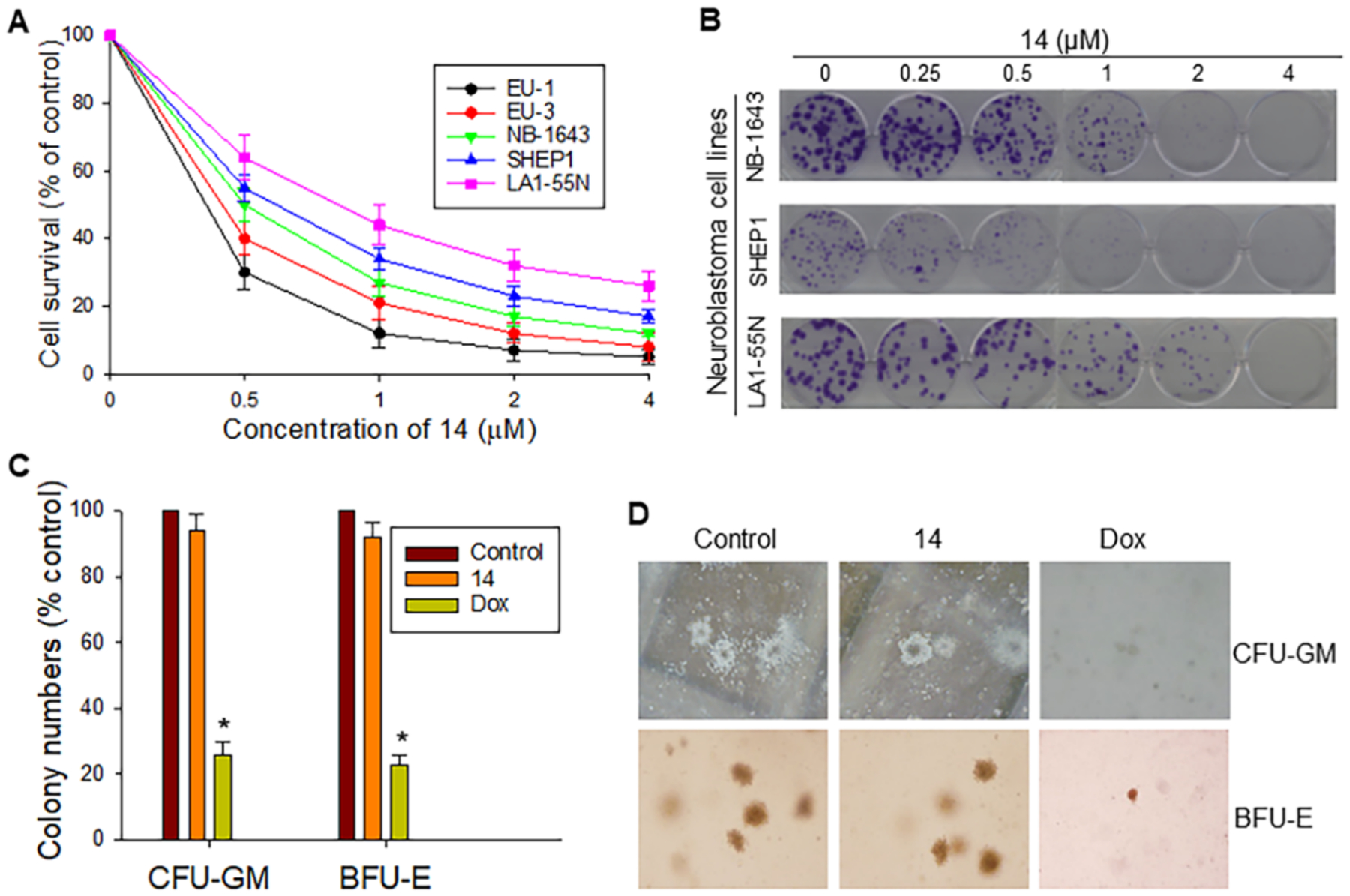
Effects of 14 on cancer cell viability and growth as well as on normal human hematopoiesis. (A) WST assay for cytotoxic effects of 14 on two ALL cell lines (EU-1 and EU-3) and three NB cell lines (NB-1643, SHEP1, and LA1–55N). The cells were treated at the doses indicated for 48 h. Data represent the mean ± SD of three independent experiments. (B) Representative colony formation of NB cell lines as indicated treated with or without 14 for 2 weeks. (C,D) Comparison of inhibitory effects of 14 and Dox on CFU-GM and BFU-E in NBMM cells using in vitro colony formation analysis. NBMM cells (1 × 105) were incubated with GM-CSF or Epo in the absence or presence of 1 μM either 14 or Dox. The colonies were counted after 14 days of culture. Comparison of colony numbers, *p < 0.01.
To evaluate the possible inhibitory effect of 14 on normal hematopoiesis, we performed Clonogenic assays for CFU-GM and BFU-E using human normal bone marrow mononuclear (NBMM) cells, with doxorubicin (Dox) as the reference control. The CFU-GM and BFU-E colony numbers and size in 14-treated samples were similar to the control, whereas both the colony number and size were significantly reduced in Dox-treated samples (Figure 2C,D).
Compound 14 Inhibits Expression of MDM2 through Post-translational Modification.
To determine whether the more potent compound 14 maintains its mechanism of action as a dual MDM2/XIAP inhibitor, similar to its parental compound MX69, we performed Western blot assays for the effects of 14 on MDM2 and XIAP expression. Results showed that 14 induced a remarkable downregulation of MDM2 and XIAP in a dose-dependent manner and occurred at approximately 2–4 h after treatment, followed by steady-state suppression (Figure 3A). Downregulation of MDM2 was accompanied by increased expression of p53. To further evaluate the mechanism by which 14 inhibits MDM2, we performed MG132 (protein degradation inhibitor) treatment and cycloheximide (CHX, protein synthesis inhibitor) chase assays in EU-1 cells treated with 14; results showed that 14 induced increased MDM2 protein degradation. As can be seen in Figure 3B, the observed downregulation of MDM2 by 14 was blocked by MG132. The CHX chase assay results showed that the half-life of MDM2 in untreated EU-1 cells was >120 min, whereas 14 treatment decreased the MDM2 half-life to <60 min (Figure 3C). In contrast, the half-life of p53 in untreated cells was <30 min, and the time was increased to >120 min by treatment with 14. Since the MDM2 protein stability is regulated by a self-ubiquitination mechanism, we tested whether 14-induced MDM2 protein degradation is mediated through this mechanism. Immunoprecipitation–Western blot assay results showed that 14 induced ubiquitination of endogenous MDM2 in EU-1 cells (Figure 3D). These results suggest that 14 downregulates MDM2 through induction of MDM2 self-ubiquitination and degradation. Furthermore, activation of p53 following 14-mediated MDM2 ubiquitination and degradation led to activation of the p53 downstream targets p21 and PUMA that induce cell cycle arrest and apoptosis (Figure 3E).
Figure 3.
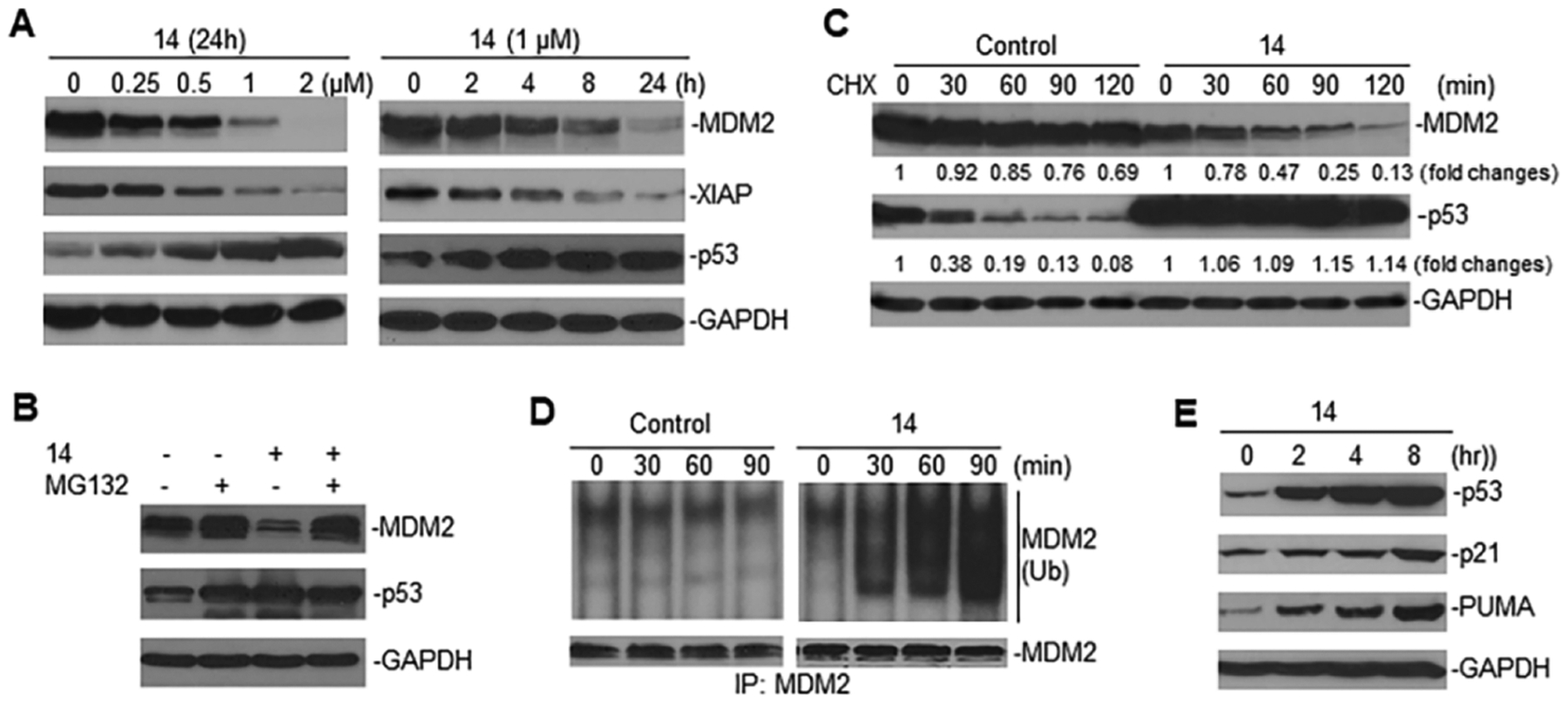
Effects of 14 on the expression of MDM2 and XIAP and activation of p53. (A) Western blot assays showed the dose–response and time course of MDM2 and XIAP inhibition as well as p53 induction by 14 in the EU-1 cell line treated with doses and times as indicated. (B) The EU-1 cells with or without 14 treatment (1 μM for 8 h) were treated with 10 μM of MG132 for an additional 4 h and then Western blot assays were performed for the expression of proteins as indicated. (C) CHX chase assay for the detection of protein turnover in EU-1 cells treated with or without (control) 1 μM of 14 for 4 h. Numerical labels under each band of Western blots represent the expression levels after normalization for GAPDH, compared with untreated samples (defined as 1 unit). (D) IP and Western blot assay using anti-MDM2 and anti-ubiquitin antibodies, respectively, to detect the effects of 14 on the ubiquitination of endogenous MDM2 in EU-1 cells. (E) Western blot for the expression of p53 and its targets p21 and PUMA in EU-1 cells treated with 14.
Compound 14 Inhibits XIAP Translation and Activity.
We investigated whether compound 14-mediated inhibition of XIAP, such as MX69-mediated, occurs at the translational level. We performed linear sucrose gradient fractionation to assess the state of polyribosome association of XIAP mRNA in EU-1 cells subjected to 14 treatment. We found that 14 induced a downregulation in polyribosome association. This was shown by a shift in XIAP mRNA from fractions containing enriched translating polyribosomes to fractions containing translation-inactive complexes, monoribosomes (Figure 4A). We also tested the effects of 14-mediated inhibition of XIAP on the activation of caspases 3 and 9, as well as the cleavage of the death substrate PARP. As shown in Figure 4B, the cleavage of caspases 3 and 9 and PARP can be detected 8 h after 14 treatment in EU-1 cells. Simultaneously, we treated EU-1 cells with MX69 for comparison, and results showed that 14 induced a stronger cleavage of caspases 3 and 9 and PARP at a much lower concentration (1 μM) than MX69 (5 μM). These results suggest that the increased potency of 14 relative to MX69 in inhibiting EU-1 cells is closely associated with the enhanced inhibition of XIAP function as well as the activation of p53.
Figure 4.
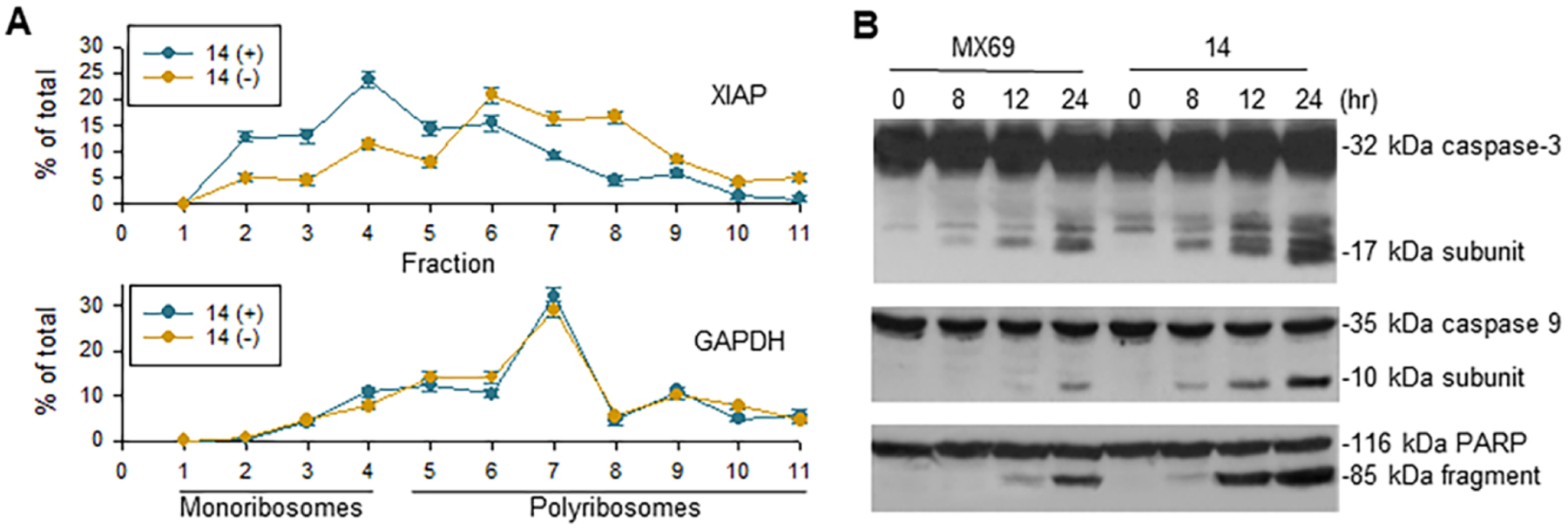
(A) EU-1 cells were treated with or without 1 μM of 14 for 4 h and their cytoplasmic lysates were fractionated on a sucrose gradient. RNA was extracted from each of the fractions and subjected to qRT-PCR for the analysis of the distribution of XIAP and GAPDH mRNAs. Data show the percentage of the total amount of the corresponding mRNA in each fraction and represent the mean ± SD of three independent experiments. (B) Western blot showed the activation of caspases 3 and 9 as well as the cleavage of the death substrate PARP in EU-1 cells following treatment with 5 μM of MX69 and 1 μM of 14 for times indicated.
PK Study Exhibits Limited In Vivo Metabolic Stability of 14.
Prior to embarking on the efficacy studies in murine cancer models to further assess the potential in vivo efficacy of compound 14, we investigated its PK behavior after intravenous (IV) and oral administration in rats to ensure that potentially efficacious drug exposures could be reached and maintained in vivo. The resulting average concentration–time profiles are shown in Figure S1. After an IV administration of 10 mg/kg, plasma concentrations followed a reproducible biexponential profile with peak concentrations of 8.20 ± 2.47 μg/mL (mean ± SD) and an area under the concentration–time curve (AUC) of 4.75 ± 0.51 μg h/mL. The clearance was with 2.12 ± 0.21 L/h/kg relatively fast, indicating limited in vivo metabolic stability. Consequently, despite a relatively large volume of distribution of 2.88 ± 0.51 L/kg as expected for a lipophilic compound such as 14, the elimination half-life remained very short at 0.94 ± 0.11 h. After oral administration of 25 mg/kg of 14, the exposure remained relatively limited with an AUC of 0.71 ± 0.31 μg h/mL. The resulting low oral bioavailability of 5.9% was likely the consequence of the limited in vivo stability and the related high hepatic first-pass inactivation after oral absorption. These unfavorable PK properties prevented a further comprehensive in vivo evaluation of compound 14, and efforts are underway to create more viable analogues with increased metabolic stability.
Separation of Stereoisomers 14 and Identification of Absolute Configurations for Individual Isomers.
All the current MX69 analogues contain three chiral centers, with two chiral centers in the A-ring restricted to the same geometry (either both α- or both β-configurations) due to the structural constriction during the synthesis. Thus, the most potent compound 14 is a mixture of four stereoisomers. Since different stereoisomers often have different biological activities, the separation was undertaken by chiral HPLC and several analytical conditions were tested. Utilization of a cellulose-4 column with methanol as the mobile phase was found to provide the best separation for the four isomers (14a–d shown in Figure 5A). These conditions were applied to a preparative method to give the individual isomers with good purities (>98%). Preliminary identification by 1H NMR indicated that 14a/14c and 14b/14d are two pairs of racemates (trans- and cis-isomers). A comparative depiction of the 1H NMR spectra of the stereoisomers of 14 is presented in Figure S2, panel A. To elucidate the absolute configuration of these stereoisomers, single-crystal X-ray diffraction (SCXRD) studies were undertaken. Isomer 14a provided crystals suitable for SCXRD analysis, and its absolute configuration was determined as 3aR,4S,9bR (Figure 5C). Multiple attempts to crystallize 14b and 14d under various conditions failed to yield crystals of sufficient quality for X-ray analysis. Several alternatives were attempted until ultimately, Boc-14b and Boc-14d, Boc derivatives prepared by N-protection of 14b and 14d, respectively, yielded suitable crystals. As shown in Figure 5D,E, the absolute configurations 3aR,4R,9bR and 3aS,4S,9bS were established for 14b and 14d, respectively, based on the SCXRD of the corresponding Boc derivatives. By elimination, the absolute configuration of the remaining isomer, 14c, is unambiguously established as 3aS,4R,9bS. The absolute structures of 14a–d are shown in Figure 5B. The antiproliferative activity of the four isomers was subsequently tested by the WST cytotoxicity assays. Compound 14d (IC50 = 0.3 μM) exhibited an outstanding antiproliferative activity, with 5- to 8-fold higher potency compared to the other three isomers (IC50 values of 14a–c are 1.9, 1.6, and 2.4 μM, respectively). It is interesting that 14d exhibited the same activity as 14, implying a complicated interaction among the isomers or between the isomers with a site of action.
Figure 5.
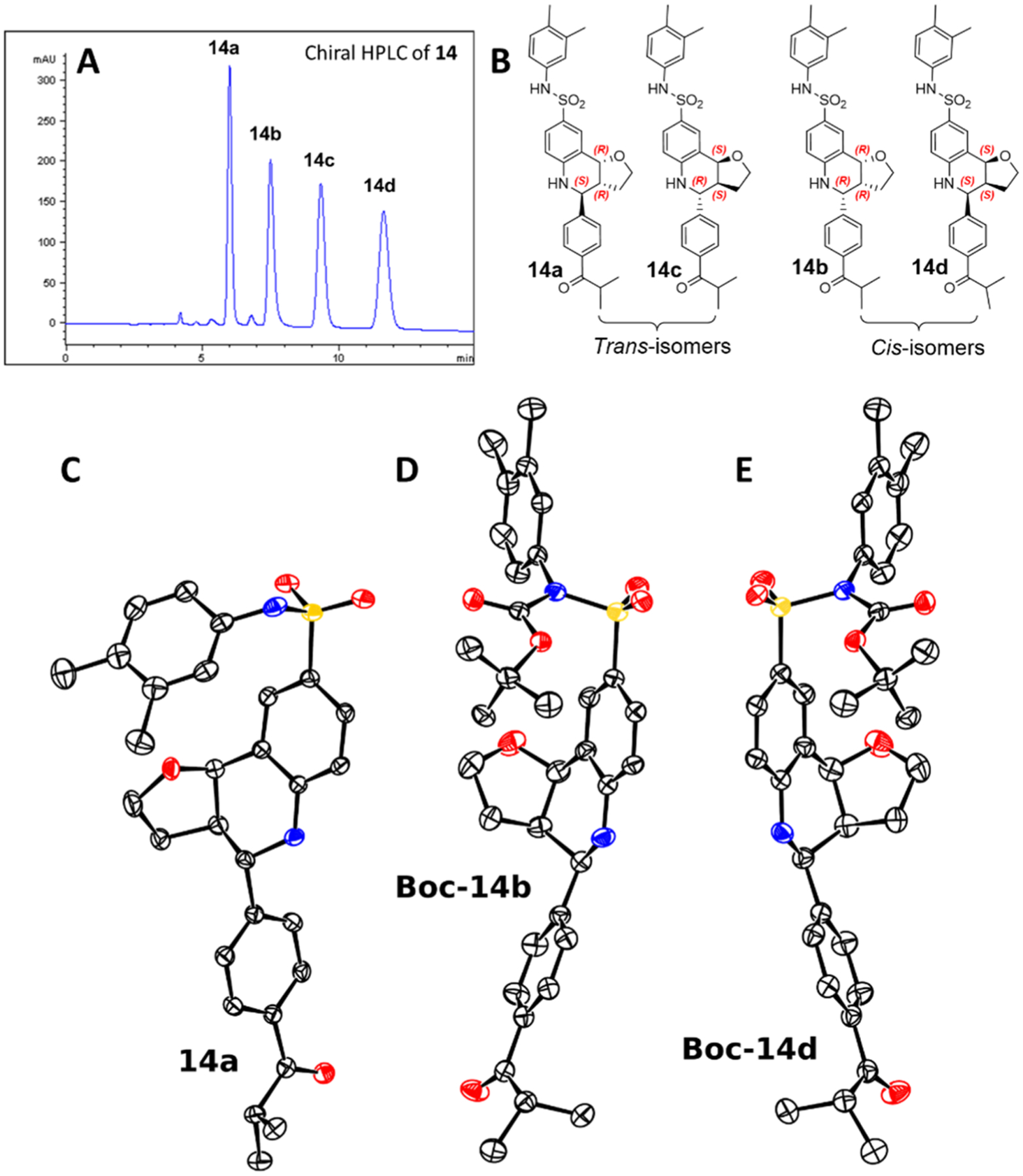
(A) Chiral HPLC separation of the stereoisomeric mixture 14. (B) Stereostructure of isomers 14a–d. ORTEP plot (thermal ellipsoids set at 50% probability) of the structures obtained from the SCXRD analyses of (C) 14a, (D) Boc-14b, and (E) Boc-14d. Hydrogen atoms are removed for clarity. The reliability of the absolute stereochemistry assignments is established by Flack parameters of −0.017(9) for 14a, −0.020(11) for Boc-14b, and −0.005(5) for Boc-14d.
Molecular Modeling Studies to Preliminarily Understand the SAR of the MX69 Scaffold.
Since no crystal structure of the MDM2 RING domain is presently available and we are actively working to generate this protein and to determine its complexes with potent MX69 analogues, as a preliminary step to understand the observed SAR described above, we employed the published NMR structure of the MDM2 RING domain protein (PDB: 2HDP) for modeling.30 We examined the MDM2 RING domain and identified the most likely binding site for MX69 (Figure 6, top panels) that also fulfilled two important requirements: (1) the size of the site must be large enough to accommodate MX69 or its larger analogues such as compound 14 and (2) the site must be close to the critical residue Cys464 because it is well known that the point mutation C464A will abolish the E3-ligase activity of MDM2.31 Consistent with these requirements, this site has a “7” shape, which is highly compatible with the overall geometry of MX69 and its analogues. Subsequent docking studies generated reasonable binding conformations for MX69 analogues, as exemplified for MX69 and 14d shown in Figure 6, since isomer 14d with absolute configuration 3aS,4S,9bS exhibited the highest potency.
Figure 6.
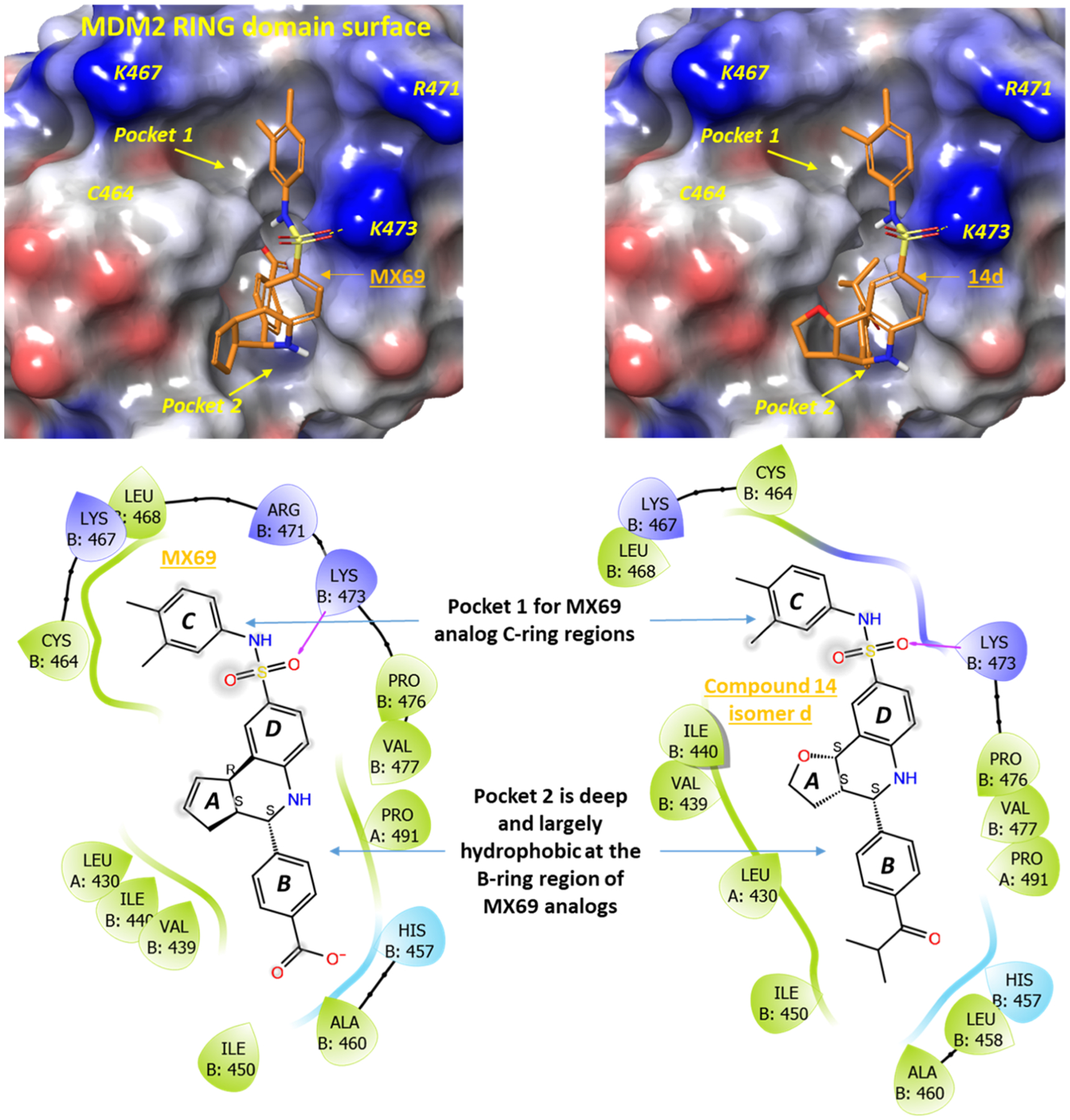
A molecular model of MX69 and compound 14d binding the MDM2 RING domain (PDB: 2HDP). The binding site is identified using SiteMap in Schrodinger suites. Top panels show best docking poses on the MDM2 electrostatic surface (blue: positive; red: negative). The deep, hydrophobic pocket 2 may explain the improved binding of compound 14d over MX69 at the B-ring substitution and the untolerated substitutions at the sulfonamide. Bottom panels show 2D ligand interactions (green: hydrophobic residues).
This model revealed several interesting features that are consistent with the SAR results described above for the MX69 scaffold. First, the B-ring moiety of MX69 occupies the deeper pocket 2. Serendipitously, this pocket is large and is formed mostly by the nearby hydrophobic residues. This may explain why compound 14 with a less polar but a larger B-ring substitution is significantly better than MX69 for MDM2 binding. Second, the sulfonamide NH in the head moiety is very close to the MDM2 surface due to the strong interactions between K473 and both the SO2 moiety (hydrogen bond) and the dimethylphenyl ring (cation–π interaction). Thus, the sulfonamide NH between the C/D rings has limited tolerance for large substitutions, as we found with reduced activities in compounds 45 and 46. The other NH in the central moiety of MX69 also has limited surrounding space. This is consistent with our SAR data that a substitution at either NH is highly detrimental to MDM2 binding and thus the antiproliferative potency. Third, substitutions on the D-ring, especially to the K473 side of the MDM2 RING domain surface, are predicted to reduce the potency because of the tight space between the D-ring and the MDM2 RING domain. This is consistent with the significant loss of potency for compounds 40, 41, 43, and, to a lesser extent, 44. Fourth, the five-member A-ring in the central body of MX69 is partially exposed to solvents and does not seem to significantly contribute to MDM2 binding except for maintaining the proper geometry for the B-ring moiety to enter pocket 2. This is consistent with the marginal improvement observed when we bioisosterically replaced the C=C double bond of this ring with O or NH and the detrimental effects when we added a hydrophobic fused ring. Last, both methyl substitutions in the C-ring fit very well to a hydrophobic region in pocket 1, consistent with the optimal potency observed for compound 14 over compounds 31–39. Although very preliminary and hypothetical at this stage, this binding model does explain many of our observed SAR. When an X-ray structure of the MDM2 RING domain becomes available, we will revisit this model and refine it, so as to better guide our ongoing efforts to optimize the MX69 scaffold.
CONCLUSIONS
In this report, we performed a preliminary, systemic SAR investigation based on the MX69 scaffold as a dual MDM2/XIAP inhibitor and discovered compound 14 that achieves a 25-fold increase in potency over the original MX69 hit. We also reported a preliminary molecular binding model that could explain most of the observed SAR for the MX69 scaffold. We demonstrated that 14 sustains the on-target of MDM2 and XIAP activity by inducing MDM2 protein degradation and inhibiting XIAP mRNA translation, which leads to cancer inhibition and death.
However, PK studies revealed that the in vivo stability of 14 is not ideal. We are currently working to design new analogues with improved PK properties. Nevertheless, the presented data strongly suggest that this scaffold is promising for further optimization to develop a new therapeutic agent for leukemia and possibly other cancers where MDM2 and XIAP are dysregulated.
EXPERIMENTAL SECTION
General Chemistry.
All solvents and chemical reagents were purchased from commercial sources and used directly without further purification. Glassware was oven-dried before use. All reactions were performed under an argon atmosphere. Analytical thin-layer chromatography was performed on silica gel 60 GF254 and monitored under UV light. Flash chromatography was performed on 230–400 mesh silica gel (Fisher Scientific). NMR spectra were obtained on a Bruker Ascend 400 (Billerica, MA) spectrometer. The purity of all final compounds (as stereoisomers not separable on the UPLC column used) was ≥95% as determined by UPLC and further confirmed by proton NMR (details given in the Supporting Information). High-resolution mass spectrometry data (HRMS) were obtained on a Waters Acquity UPLC linked to a Waters Acquity Photodiode Array Detector and a Waters qTof mass detector. UPLC analyses were performed using a BEH C18 (2.1 × 50 mm, 1.7 μm) column and a mixture of solvent methanol/water at a flow rate of 0.3 mL/min, monitored by UV absorption at the appropriate wavelength. Chemical shifts are given in ppm. Tetramethylsilane is used as an internal reference for NMR spectra taken in chloroform-d. All coupling constants (J) are given in hertz (Hz).
General Procedure for the Synthesis of Sulfonamides 6a–6l.
Substituted benzenesulfonyl chloride (26.58 mmol) and 3,4-dimethylaniline (29.23 mmol) were dissolved in anhydrous methylene chloride (100 mL) at room temperature under argon. Pyridine (2.31 g, 29.23 mmol) was added dropwise via a syringe at room temperature. The reaction mixture was stirred at room temperature overnight under an argon atmosphere. Then, the volatile was removed under reduced pressure. The yellow solid residues were subjected to flash column chromatography (silica gel, CH2Cl2) to afford compounds 6a–6l.
N-(3,4-Dimethylphenyl)-4-nitrobenzenesulfonamide (6a).
87% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.37 (s, 1H); 8.36 (d, J = 8.0 Hz, 2H), 7.96 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 1H), 6.88 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 2.11 (s, 3H), 2.09 (s, 3H).
N-(3-Bromo-4-methylphenyl)-4-nitrobenzenesulfonamide (6b).
94.3% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.75 (s, 1H), 8.39 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 8.0 Hz, 2H), 7.30 (s, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 2.23 (s, 3H).
4-Nitro-N-(m-tolyl)benzenesulfonamide (6c).
99% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.56 (s, 1H), 8.38 (d, J = 8.0 Hz, 2H), 8.00 (d, J = 8.0 Hz, 2H), 7.15−7.11 (m, 1H), 6.93−6.88 (m, 3H), 2.20 (s, 3H).
4-Nitro-N-phenylbenzenesulfonamide (7d).
92.7% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.63 (s, 1H), 8.37 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.28−7.24 (m, 1H), 7.11−7.06 (m, 1H).
N-(3-Chloro-4-methylphenyl)-4-nitrobenzenesulfonamide (6e).
86.1% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H), 8.38 (d, J = 8.8 Hz, 2H), 8.00 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 2.0 Hz, 1H), 6.99 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H).
N-(4-Chloro-3-methylphenyl)-4-nitrobenzenesulfonamide (6f).
97.5% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.00 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 2.0 Hz, 1H), 6.99 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 2.21 (s, 3H).
N-(3,4-Dichlorophenyl)-4-nitrobenzenesulfonamide (6g).
99.6% yield; 1H NMR (400 MHz, DMSO-d6): δ 11.03 (s, 1H), 8.40 (d, J = 8.4 Hz, 2H), 8.03 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.8 Hz, 1H), 7.32 (d, J = 2.4 Hz, 1H), 7.11 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H).
N-(3,4-Dibromophenyl)-4-nitrobenzenesulfonamide (6h).
94.9% yield; 1H NMR (400 MHz, DMSO-d6): δ 11.03 (s, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.03 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 8.8 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.08 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H).
N-(4-Fluoro-3-methylphenyl)-4-nitrobenzenesulfonamide (6i).
96.6% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.51 (s, 1H), 8.38 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.8 Hz, 2H), 7.06−6.99 (m, 2H), 6.91−6.87 (m, 1H), 2.14 (s, 3H).
N-(3-Methyl-4-(trifluoromethyl)phenyl)-4-nitrobenzenesulfonamide (6j).
89.8% yield; 1H NMR (400 MHz, DMSO-d6): δ 11.15 (s, 1H), 8.40 (d, J = 8.8 Hz, 2H), 8.09 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 1H), 7.14 (s, 1H), 7.12 (d, J = 8.0 Hz, 1H), 2.24 (s, 3H).
3-Chloro-N-(3,4-dimethylphenyl)-4-nitrobenzenesulfonamide (6k).
87.5% yield; 1H NMR (400 MHz, CDCl3): δ 7.88−7.86 (m, 2H), 7.65 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.21 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.98 (1, 1H), 6.93 (d, J = 8.0 Hz, 1H), 2.23 (s, 6H).
2-Chloro-N-(3,4-dimethylphenyl)-4-nitrobenzenesulfonamide (6l).
84% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H), 8.43 (d, J = 2.0 Hz, 1H), 8.30−8.22 (m, 2H), 6.98 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 1.6 Hz, 1H), 6.82 (dd, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 2.10 (s, 3H), 2.08 (s, 3H).
General Procedure for Hydrogenation to form Anilines 7a–7l.
Sulfonamide (16.32 mmol) and Pd/C (10% Pd base) were mixed in MeOH (50 mL) at room temperature. Hydrogen gas was introduced via a H2 balloon. The reaction mixture was stirred under a H2 atmosphere overnight at room temperature. The reaction mixture was filtered to remove the solid. The solution was concentrated under reduced pressure and the resulting residues were purified by column chromatography (silica gel, CH2Cl2/MeOH = 9/1 v/v) to afford compounds 7a–7l.
4-Amino-N-(3,4-dimethylphenyl)benzenesulfonamide (7a).
87.6% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, 1H); 7.37 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 8.0 Hz, 1H), 6.84 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.52 (d, J = 8.0 Hz, 2H), 5.91 (s, 2H), 2.09 (s, 3H), 2.08 (s, 3H).
4-Amino-N-(p-tolyl)benzenesulfonamide (7b).
Yield 59.4%. 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.50 (d, J = 8.8 Hz, 2H), 5.95 (s, 2H), 2.17 (s, 3H).
4-Amino-N-(m-tolyl)benzenesulfonamide (7c).
87.2% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.80 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.09−7.05 (m, 1H), 6.86 (s, 2H), 6.77 (d, J = 7.2 Hz, 1H), 6.52 (d, J = 8.4 Hz, 2H), 5.97 (s, 2H), 2.18 (s, 3H).
4-Amino-N-phenylbenzenesulfonamide (7d).
96.3% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.86 (s, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.21−7.17 (m, 2H), 7.05 (d, J = 8.0 Hz, 2H), 6.98−6.94 (m, 1H), 6.52 (s, 2H), 5.97 (s, 2H).
4-Amino-N-(3-chloro-4-methylphenyl)benzenesulfonamide (7e).
90.3% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.98 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.0 Hz, 1H), 7.07 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 8.4 Hz, 2H), 6.01 (s, 2H), 2.19 (s, 3H).
4-Amino-N-(4-chloro-3-methylphenyl)benzenesulfonamide (7f).
94.1% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.98 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.0 Hz, 1H), 7.07 (s, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 8.4 Hz, 2H), 6.01 (s, 2H), 2.19 (s, 3H).
4-Amino-N-(3,4-dichlorophenyl)benzenesulfonamide (7g).
99.2% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.28 (s, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 2.0 Hz, 1H), 7.05 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 6.55 (d, J = 8.4 Hz, 2H), 6.07 (s, 2H).
4-Amino-N-(3,4-dibromophenyl)benzenesulfonamide (7h).
60.3% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.98 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.0 Hz, 1H), 7.07 (s, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 8.4 Hz, 2H), 6.01 (s, 2H), 2.19 (s, 3H).
4-Amino-N-(4-fluoro-3-methylphenyl)benzenesulfonamide (7i).
97.5% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.74 (s, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.00−6.92 (m, 2H), 6.88−6.86 (m, 1H), 6.52 (d, J = 8.0 Hz, 2H), 5.98 (s, 2H), 2.12 (s, 3H).
4-Amino-N-(3-methyl-4-(trifluoromethyl)phenyl)-benzenesulfonamide (7j).
93.8% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.42 (s, 1H), 7.51−7.46 (m, 3H), 7.07−7.04 (m, 2H), 6.55 (d, J = 8.8 Hz, 2H), 6.07 (s, 2H), 2.32 (s, 3H).
4-Amino-3-chloro-N-(3,4-dimethylphenyl)benzenesulfonamide (7k).
84.2% yield; 1H NMR (400 MHz, DMSO-d6): δ 7.44 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1H), 7.20−7.16 (m, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.80 (s, 1H), 6.76 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 6.53−6.49 (m, 1H), 5.98 (s, 2H), 2.07 (s, 3H), 2.06 (s, 3H).
4-Amino-2-chloro-N-(3,4-dimethylphenyl)benzenesulfonamide (7l).
87% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.90 (s, 1H); 7.61 (d, J = 8.8 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 2.0 Hz, 1H), 6.77 (dd, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 6.58 (d, J = 2.0 Hz, 1H), 6.45 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 6.23 (s, 2H), 2.08 (s, 3H), 2.07 (s, 3H).
General Procedure for the Synthesis of Aldehydes 9a and 9b.
To a solution of 1-bromo-4-(diethoxymethyl)benzene or 1-bromo-3-(diethoxymethyl)benzene (45.30 g, 174.81 mmol) in 200 mL of anhydrous tetrahydrofuran under an argon atmosphere was added dropwise n-butyllithium (76.88 mL of 2.5 N hexanes solution, 192.2 mmol) at −78 °C in a dry ice/acetone bath. After stirring at −78 °C for 2 h, a solution of CuCN (15.66 g, 184.81 mmol) and LiCl (14.82 g, 349.62 mmol) in 200 mL of anhydrous tetrahydrofuran was added dropwise with stirring at −78 °C under argon. The resulting solution was stirred at −78 °C for 30 min, then slowly warmed to room temperature, and stirred for another hour. Isobutyryl chloride (18.08 mL, 174.81 mmol) was added dropwise into the solution at −78 °C within 1 h. The reaction mixture was allowed to warm up to room temperature and stirred for another 4 h. The reaction was quenched by the addition of 100 mL of water at room temperature with vigorous stirring. THF solvent was removed under reduced pressure. The residue 8a or 8b was extracted with methylene chloride (3 × 100 mL). HCl (100 mL, aq, 3 M) was then added to the solution and the mixture was stirred for 3 h. The organic layer was separated and dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude liquid was purified by column chromatography (silica gel, CH2Cl2) to afford pure compound 9a or 9b.
4-Isobutyrylbenzaldehyde (9a).
91.5% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.11 (s, 1H); 8.14 (d, J = 8.0 Hz, 2H), 8.05 (d, J = 8.0 Hz, 2H), 3.70 (m, 1H), 1.12 (d, J = 6.8 Hz, 6H).
3-Isobutyrylbenzaldehyde (9b).
94.1% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.12 (s, 1H); 8.48 (s, 1H), 8.29 (d, J = 7.6 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 3.77−3.70 (m, 1H), 1.14 (d, J = 6.8 Hz, 6H).
Synthesis of 4-Formyl-N-methoxy-N-methylbenzamide (20).
4-Formylbenzoic acid (10.00 g, 66.61 mmol) was dissolved in anhydrous methylene chloride (100 mL) at room temperature under argon. Oxalyl chloride (10.15 g, 79.93 mmol) was added dropwise via a syringe at room temperature. The reaction mixture was cooled to 0 °C and 0.5 mL of anhydrous DMF was added. The resulted mixture was stirred at room temperature for 4 h. Then, N,O-dimethylhydroxylamine hydrochloride (9.75 g, 99.92 mmol) was added at room temperature. Triethylamine (20.22 g, 199.83 mmol) was added to the reaction mixture at 0 °C. After stirring at room temperature overnight, the reaction was quenched by adding water. The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The yellow oil residue was subjected to flash column chromatography (silica gel, CH2Cl2/MeOH = 19/1 v/v) to afford a pale-yellow oil product (10.58 g, 82.2% yield). 1H NMR (400 MHz, CDCl3): δ 10.07 (s, 1H), 7.94 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 8.0 Hz, 2H), 3.54 (s, 3H), 3.39 (s, 3H).
General Procedure for the Synthesis of MX69 Analogues.
Anilines (0.27 g, 0.87 mmol), aldehydes (0.87 mmol), and Sc(OTf)3 (0.17 mmol) were mixed together and dissolved in anhydrous CH3CN (10 mL) at room temperature under argon. The reaction mixture was stirred for 1 h at room temperature. Alkene (1.74 mmol) was then added via a syringe. The resulting mixture was stirred at room temperature under argon overnight. The reaction was quenched by adding 50 mL of water at room temperature and neutralized by adding NaHCO3. The solution was extracted with ethyl acetate (3 × 50 mL). The extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to dryness. The solid residues were purified by column chromatography to afford the desired compounds.
Methyl 4-(8-(N-(3,4-Dimethylphenyl)sulfamoyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-4-yl)benzoate (1).
79.2% yield; 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.40 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.36 (s, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.81 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.28 (s, 1H), 5.64−5.62 (m, 2H), 4.75 (d, J = 3.2 Hz, 1H), 4.20 (s, 1H), 4.05 (d, J = 8.8 Hz, 1H), 3.95 (s, 3H), 3.05−2.98 (m, 1H), 2.54−2.47 (m, 1H), 2.21 (s, 6H), 1.80−1.74 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 166.8, 149.2, 146.8, 137.6, 134.4, 133.8, 133.4, 130.4, 130.2, 130.0, 129.5, 129.0, 128.2, 126.3, 125.9, 125.5, 123.6, 119.7, 115.5, 57.2, 52.2, 45.6, 45.3, 31.4, 19.8, 19.2. HRMS (ESI): calcd for C28H28N2NaO4S, 511.1662 [M + Na]+; found, 511.1650. Purity: 97.6% by UPLC (Rt = 3.44 min).
Methyl 4-(8-(N-(3,4-Dimethylphenyl)sulfomoyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-4-yl)benzoate (2).
68.5% yield; 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 2.0 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.45 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 2 Hz, 1H), 6.81 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 1H), 6.56 (d, J = 8.4 Hz, 1H), 6.27 (s, 1H), 5.20 (d, J = 7.6 Hz, 1H), 4.88 (d, J = 2.8 Hz, 1H), 4.30 (s, 1H), 3.95 (s, 3H), 3.77−3.72 (m, 1H), 3.69−3.67 (m, 1H), 2.80−2.74 (m, 1H), 2.21 (s, 3H), 2.20 (s, 3H), 2.09−1.97 (m, 1H), 1.51−1.47 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 166.7, 148.7, 148.0, 146.3, 145.9, 137.4, 134.6, 134.5, 133.3, 133.2, 131.3, 130.1, 129.9, 129.6, 128.6, 128.2, 128.0, 127.9, 127.2, 126.4, 123.2, 123.0, 121.4, 119.1, 119.0, 118.7, 114.5, 114.3, 75.2, 74.8, 66.3, 65.0, 56.6, 56.0, 52.3, 52.2, 44.4, 42.4, 28.3, 24.1, 19.8, 19.1. HRMS (ESI): calcd for C27H28N2NaO5S, 515.1611 [M + Na]+; found, 515.1586. Purity: 98.9% by UPLC (Rt = 3.28 min).
tert-Butyl 8-(N-(3,4-Dimethylphenyl)sulfamoyl)-4-(4-(methoxycarbonyl)phenyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo-[3,2-c]quinoline-1-carboxylate (3b).
The crude product was used directly for the next step without further purification.
Methyl 4-(9-(N-(3,4-Dimethylphenyl)sulfamoyl)-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)benzoate (4).
1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 2.0 Hz, 0.3H), 7.72 (d, J = 2.0 Hz, 0.7H), 7.53 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.3H), 7.44 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 0.7H), 7.01−6.98 (m, 1H), 6.90 (s, 1H), 6.84−6.80 (m, 1H), 6.59 (d, J = 8.8 Hz, 0.3H), 6.48 (d, J = 8.8 Hz, 0.7H), 6.41 (s, 0.3H), 6.35 (s, 0.7H), 5.23 (d, J = 5.6 Hz, 0.3H), 4.80 (s, 0.7H), 4.78 (s, 0.3H), 4.57 (s, 0.7H), 4.37−4.36 (m, 1H), 4.04−4.02 (m, 0.7H), 3.95 (s, 3H), 3.71−3.69 (m, 1H), 3.45−3.40 (m, 0.7H), 3.07−3.02 (m, 0.3H), 2.20 (s, 3H), 2.19 (s, 3H), 2.08−2.05 (m, 0.7H), 1.85−1.82 (m, 1H), 1.74−1.62 (m, 1H), 1.57−1.41 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 166.7, 148.0, 146.5, 137.5, 134.5, 133.5, 130.9, 130.2, 128.9, 127.6, 126.5, 123.3, 119.5, 119.3, 113.6, 54.7, 52.3, 38.3, 34.7, 31.6, 25.3, 23.7, 22.7, 22.0, 19.8, 19.2, 14.1. HRMS (ESI): calcd for C28H31N2O5S, 507.1948 [M + H]+; found, 507.1957. Purity: 95.4% by UPLC (Rt = 1.19 min).
Methyl 4-(2-(N-(3,4-Dimethylphenyl)sulfamoyl)-6,6a,7,11b-tetrahydro-5H-indeno[2,1-c]quinolin-6-yl)benzoate (5).
1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 7.6 Hz, 2H), 7.74 (s, 1H), 7.53 (d, J = 7.6 Hz, 2H), 7.42 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.15−7.14 (m, 2H), 7.05 (s, 1H), 6.91 (d, J = 7.2 Hz, 1H), 6.77 (s, 1H), 6.72 (d, J = 8.0 Hz, 1H), 6.58 (d, J = 8.4 Hz, 1H), 6.21 (s, 1H), 4.87 (s, 1H), 4.50 (d, J = 4.8 Hz, 1H), 4.36 (s, 1H), 3.97 (s, 3H), 3.16−3.11 (m, 2H), 2.38−2.30 (m, 1H), 2.19 (s, 3H), 2.13 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 166.8, 157.9, 151.0, 149.2, 146.4, 144.3, 140.6, 137.85, 137.81, 136.9, 134.2, 134.1, 131.6, 130.6, 130.4, 130.0, 129.4, 128.7, 127.8, 126.4, 125.6, 124.64, 124.57, 124.2, 123.6, 122.7, 119.7, 52.4, 42.3, 34.6, 31.6, 29.7, 25.2, 22.6, 19.8, 19.1, 14.1. HRMS (ESI): calcd for C32H31N2O4S, 539.1999 [M + H]+; found, 539.1990. Purity: 98.4% by UPLC (Rt = 3.46 min).
N-(3,4-Dimethylphenyl)-4-(4-isopropylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (10).
75.6% yield; 1H NMR (400 MHz, CDCl3): δ 7.89 (s, 0.6H), 7.80 (s, 0.4H), 7.44−7.39 (m, 1H), 7.36−7.32 (m, 2H), 7.28−7.26 (m, 3H), 7.01−6.97 (m, 1H), 6.99 (d, J = 1.6 Hz, 1H), 6.80 (d, J = 7.6 Hz, 1H), 6.50 (d, J = 8.8 Hz, 1H), 6.48 (d, J = 4.4 Hz, 0.4H), 6.42 (s, 0.6H), 5.19 (d, J = 7.2 Hz, 0.4H), 4.78 (d, J = 2.4 Hz, 0.4H), 4.62 (s, 0.6H), 4.54 (d, J = 4.8 Hz, 0.6H), 4.31 (s, 0.4H), 4.06−4.02 (m, 0.6H), 3.89−3.87 (m, 0.4H), 3.82 (d, J = 11.2 Hz, 0.6H), 3.76−3.69 (m, 1H), 2.97−2.92 (m, 1H), 2.76−2.71 (m, 0.4H), 2.41−2.35 (m, 0.6H), 2.19 (s, 6H), 2.09−2.04 (m, 1H), 1.78−1.72 (m, 0.6H), 1.65−1.57 (m, 0.4H), 1.29−1.27 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 149.3, 148.9, 148.8, 148.2, 138.3, 137.9, 137.5, 134.6, 134.5, 133.4, 133.3, 131.4, 130.12, 130.11, 130.09, 128.6, 128.1, 127.8, 127.7, 127.0, 126.9, 126.8, 126.4, 123.2, 123.1, 121.7, 119.2, 119.1, 118.9, 114.2, 114.0, 75.5, 75.0, 66.5, 65.0, 56.6, 56.1, 44.7, 42.3, 33.83, 33.78, 28.6, 24.3, 24.0, 19.8, 19.8, 19.2. HRMS (ESI): calcd for C28H32N2NaO3S, 499.2026 [M + Na]+; found, 499.1997. Purity: 99.5% by UPLC (Rt = 3.49 min).
4-(4-Bromophenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (11).
79.2%; 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.44 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 7.31 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.56 (d, J = 2.0 Hz, 1H), 5.18 (d, J = 7.6 Hz, 1H), 4.77 (d, J = 3.2 Hz, 1H), 4.31 (s, 1H), 3.95 (s, 3H), 3.75−3.71 (m, 1H), 3.69−3.65 (m, 1H), 2.72−2.70 (m, 1H), 2.20 (s, 3H), 2.19 (s, 3H), 2.03−1.97 (m, 1H), 1.55−1.51 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 148.7, 147.8, 140.1, 139.6, 137.6, 134.4, 134.3, 133.6, 133.5, 132.0, 131.9, 131.7, 131.4, 130.5, 130.2, 130.14, 130.06, 129.8, 128.6, 128.2, 128.0, 127.9, 127.5, 123.3, 123.2, 122.4, 121.8, 121.7, 119.24, 119.15, 119.0, 114.4, 114.2, 75.3, 74.8, 66.4, 65.0, 56.4, 55.8, 44.6, 42.5, 29.7, 28.4, 24.2, 19.8, 19.2. HRMS (ESI): calcd for C25H25BrN2NaO3S, 535.0661 [M + Na]+; found, 535.0659. Purity: 95.3% by UPLC (Rt = 3.41 min).
Methyl N-(3,4-Dimethylphenyl)-4-(4-(methylsulfonyl)phenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (12).
73% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.75 (s, 0.5H), 9.71 (s, 0.5H), 7.96−7.94 (m, 2H), 7.75−7.70 (m, 2H), 7.60 (s, 0.5H), 7.55 (s, 0.5H), 7.40 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.5H), 7.33 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H), 7.19 (s, 0.5H), 6.96 (d, J = 8.0 Hz, 1H), 6.89−6.80 (m 3H), 6.73−6.70 (m, 1H), 5.12 (d, J = 7.2 Hz, 0.5H), 4.89 (d, J = 2.8 Hz, 0.5H), 4.44 (d, J = 4.4 Hz, 0.5H), 3.92−3.84 (m, 1H), 3.75−3.50 (m, 2.5H), 2.72−2.67 (m, 0.5H), 2.33−2.29 (m, 0.5H), 2.11 (s, 3H), 2.10 (s, 3H), 1.99−1.92 (m, 0.5H), 1.81−1.76 (m, 1H), 1.56−1.53 (m, 0.5H), 1.48−1.28 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 148.9, 148.1, 140.2, 137.1, 136.3, 131.9, 130.3, 129.3, 128.0, 127.6, 127.3, 127.3, 121.7, 121.0, 118.0, 114.6, 74.5, 66.0, 54.9, 44.0, 43.8, 24.4, 20.0, 19.1. HRMS (ESI): calcd for C26H28N2NaO5S2, 535.1332 [M + Na]+; found, 535.1329. Purity: 99.5% by UPLC (Rt = 3.03 min).
4-(4-Acetylphenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (13).
1H NMR (400 MHz, CDCl3): δ 8.02−7.99 (m, 2H), 7.89 (d, J = 2.0 Hz, 0.5H), 7.81 (d, J = 2.0 Hz, 0.5H), 7.54−7.51 (m, 2H), 7.47−7.43 (m, 1H), 7.00 (s, 0.5H), 6.98 (s, 0.5H), 6.90 (s, 1H), 6.82−6.80 (m, 1H), 6.57 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 6.39 (s, 0.5H), 6.35 (s, 0.5H), 5.20 (d, J = 7.2 Hz, 0.5H), 4.88 (d, J = 3.2 Hz, 0.5H), 4.61 (s, 0.5H), 4.54 (d, J = 4.8 Hz, 0.5H), 4.34 (s, 0.5H), 4.17−4.01 (m, 1H), 3.93−3.85 (m, 1H), 3.77−3.63 (m, 1H), 2.78−2.76 (m, 0.5H), 2.64 (s, 3H), 2.45−2.39 (m, 0.5H), 2.19 (s, 6H), 2.08−2.00 (m, 0.5H),1.75−1.70 (m, 0.5H), 1.54−1.47 (m, 0.5H). 13C NMR (100 MHz, CDCl3): δ 197.87, 197.85, 148.7, 148.0, 146.5, 146.1, 137.5, 137.4, 137.0, 136.6, 134.6, 134.5, 133.33, 133.26, 131.3, 130.1, 129.9, 128.9, 128.5, 128.4, 128.0, 127.8, 127.3, 126.6, 123.2, 123.0, 119.1, 119.0, 118.7, 114.6, 114.3, 75.2, 74.7, 66.3, 65.0, 56.5, 56.0, 44.4, 42.4, 28.3, 26.71, 26.67, 24.1, 19.8, 19.1. HRMS (ESI): calcd for C27H28N2NaO4S, 499.1662 [M + Na]+; found, 499.1631. Purity: 95.3% by UPLC (Rt = 3.19 min).
N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (14).
41.7% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.78,9.75 (s, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.62−7.55 (m, 3H), 7.39 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.55H), 7.33 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.45H), 7.18 (s, 0.55H), 6.96 (d, J = 8.4 Hz, 1H), 6.89−6.86 (m, 1.45H), 6.84−6.80 (m, 1H), 6.74−6.70 (m, 1H), 5.11 (d, J = 7.2 Hz, 0.45 H), 4.84 (d, J = 2.8 Hz, 0.45H), 4.43 (d, J = 4.8 Hz, 0.55H), 3.90−3.88 (m, 0.55H), 3.79 (d, J = 10.8 Hz, 0.55H), 3.72−3.59 (m, 2H), 3.51−3.49 (m, 0.55H), 2.69−2.67 (m, 0.45H), 2.30−2.27 (m, 0.55H), 2.11 (s, 3H), 2.09 (s, 3H), 1.96−1.93 (m, 0.55H), 1.83−1.72 (m, 0.55H), 1.56−1.52 (m, 0.55H), 1.32−1.30 (m, 0.45H), 1.11 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 204.0, 203.9, 149.6, 149.1, 147.3, 137.2, 137.1, 136.4, 135.7, 135.3, 131.9, 131.8, 131.0, 130.32, 130.28, 129.3, 129.2, 128.9, 128.8, 128.0, 127.4, 127.3, 127.1, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.0, 74.5, 66.1, 64.6, 55.8, 55.0, 44.0, 42.0, 35.0, 28.5, 24.5, 20.0, 19.5, 19.4, 19.1. HRMS (ESI): calcd for C29H33N2O4S, 505.2161 [M + H]+; found, 505.2151. Purity: 99.3% by UPLC (Rt = 3.29 min).
4-(3-Acetylphenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (16).
67.4% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.77 (s, 0.5H), 9.73 (s, 0.5H), 8.00 (d, J = 3.6 Hz, 1H), 7.96−7.91 (m, 1H), 7.73−7.70 (m, 1H), 7.60−7.55 (m, 2H), 7.39 (d, J = 8.8 Hz, 0.5H), 7.33 (d, J = 8.4 Hz, 0.5H), 7.16 (s, 0.5H), 6.96 (d, J = 8.0 Hz, 1H), 6.89−6.80 (m, 2.5H), 6.74−6.71 (m, 1H), 5.11 (d, J = 7.2 Hz, 0.5H), 4.86 (s, 0.5H), 4.43 (d, J = 4.8 Hz, 0.5H), 3.94−3.88 (m, 0.5H), 3.80 (d, J = 10.8 Hz, 0.5H), 3.74−3.68 (m, 0.5H), 3.63−3.57 (m, 0.5H), 3.52−2.50 (m, 0.5H), 2.70−2.65 (m, 0.5H), 2.59 (s, 3H), 2.34−2.29 (m, 0.5H), 2.11 (s, 3H), 2.09 (s, 3H), 1.98−1.92 (m, 0.5H),1.81−1.76 (m, 0.5H), 1.56−1.50 (m, 0.5H), 1.31−1.29 (m, 0.5H). 13C NMR (100 MHz, DMSO-d6): δ 198.4, 149.7, 149.2, 142.7, 142.5, 137.5, 137.3, 137.2, 137.1, 136.39, 136.36, 133.7, 131.89, 131.86, 131.8, 131.0, 130.32, 130.28, 129.4, 129.3, 128.3, 128.03, 127.96, 127.3, 127.1, 126.5, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.1, 74.6, 66.0, 64.6, 55.8, 55.0, 44.2, 42.0, 28.6, 27.30, 27.27, 24.4, 20.03, 20.02, 19.1. HRMS (ESI): calcd for C27H28N2NaO4, 499.1662 [M + Na]+; found, 499.1648. Purity: 98.7% by UPLC (Rt = 3.19 min).
N-(3,4-Dimethylphenyl)-4-(3-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (17).
59.4% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.77 (s, 0.6H), 9.74 (s, 0.4H), 8.03 (d, J = 3.6 Hz, 1H), 7.96−7.91 (m, 1H), 7.73−7.71 (m, 1H), 7.68−7.55 (m, 2H), 7.39 (d, J = 8.8 Hz, 0.6H), 7.18 (s, 0.6H), 6.96 (d, J = 8.0 Hz, 1H), 6.89−6.80 (m, 2.4H), 6.74−6.71 (m, 1H), 5.10 (d, J = 7.2 Hz, 0.4H), 4.86 (s, 0.4H), 4.43 (d, J = 4.8 Hz, 0.6H), 3.94−3.88 (m, 0.6H), 3.80 (d, J = 10.8 Hz, 0.6H), 3.71−3.68 (m, 2H), 3.63−3.57 (m, 0.8H), 3.52−2.50 (m, 0.6H), 2.70−2.65 (m, 0.4H), 2.34−2.29 (m, 0.6H), 2.09−2.11 (m, 9H), 1.98−1.92 (m, 1H),1.87−1.76 (m, 1H), 1.51−1.50 (m, 1H), 1.31−1.29 (m, 0.4H), 1.19−1.05 (m, 6.6H). 13C NMR (100 MHz, DMSO-d6): δ 204.25, 204.22, 149.7, 149.2, 142.8, 142.6, 137.2, 137.1, 136.40, 136.36, 136.3, 136.2, 133.5, 131.9, 131.8, 131.0, 130.32, 130.27, 129.5, 129.4, 128.4, 128.3, 128.0, 127.8, 127.3, 127.1, 126.6, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.1, 74.6, 66.1, 64.6, 55.8, 55.0, 44.2, 42.0, 35.11, 35.08, 30.0, 28.5, 24.4, 20.0, 19.6, 19.5, 19.3, 19.1. HRMS (ESI): calcd for C29H32N2NaO4S, 527.1975 [M + Na]+; found, 527.1965. Purity: 95.5% by UPLC (Rt = 3.33 min).
N-(3,4-Dimethylphenyl)-4-(naphthalene-1-yl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (18).
76.4% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.76 (s, 0.6H), 9.73 (s, 0.4H), 8.27−8.25 (m, 1H), 8.00−7.88 (m, 2H), 7.78 (d, J = 7.2 Hz, 0.4H), 7.66−7.64 (m, 1H), 7.58−7.53 (m, 3.6H), 7.41−7.29 (m, 1.6H), 6.99−6.96 (m, 1H), 6.89−6.72 (m, 3.4H), 5.62 (d, J = 2.0 Hz, 0.4H), 5.29 (d, J = 7.2 Hz, 0.4H), 4.50 (d, J = 4.8 Hz, 0.6H), 4.48 (s, 0.4H), 3.92−3.88 (m, 0.6H), 3.74−3.69 (m, 0.6H), 3.56−3.51 (m, 1H), 2.85−2.67 (m, 0.4H), 2.70−2.65 (m, 0.6H), 2.11−2.10 (m, 6H), 2.02−1.92 (m, 0.6H), 1.84−1.78 (m, 0.4H), 1.48−1.43 (m, 0.6H), 1.19−1.10 (m, 0.6H). 13C NMR (100 MHz, DMSO-d6): δ 149.8, 137.4, 137.2, 137.1, 136.5, 134.1, 133.8, 132.0, 131.84, 131.77, 131.1, 130.4, 130.3, 129.3, 128.8, 128.2, 128.0, 127.2, 127.0, 126.84, 126.76, 126.2, 126.0, 125.9, 123.7, 123.1, 121.6, 121.4, 118.2, 117.8, 117.5, 114.8, 114.2, 75.3, 74.5, 66.0, 64.8, 60.2, 51.0, 42.5, 31.2, 29.0, 25.0, 20.0, 19.1. HRMS (ESI): calcd for C29H29N2O3S, 485.1893 [M + H]+; found, 485.1888. Purity: 97.3% by UPLC (Rt = 1.29 min).
N-((3,4-Dimethylphenyl)-sulfonyl-2,3,3a,4,5,9b-hexahydrofuran-[3,2-c]quinolin-4-yl)-N-methoxy-N-methylbenzamide (21).
87.3% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.75, 9.71 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.55−7.50 (m, 3H), 7.38 (dd, J1 = 8.4 Hz, J2 = 4.4 Hz, 0.38 H), 7.32 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H), 7.17 (s, 0.38 H), 6.96 (d, J = 8.0 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.84−6.80 (m, 1H), 6.75−6.70 (m, 1H), 5.11 (d, J = 7.6 Hz, 0.67 H), 4.81 (d, J = 2.8 Hz, 0.66H), 4.33 (d, J = 2.8 Hz, 0.43H), 3.90−3.88 (m, 0.5H), 3.77−3.70 (m, 1H), 3.52 (s, 3H), 3.26 (s, 3H), 2.69−2.67 (s, 0.76H), 2.32−2.26 (m, 0.65H), 2.11−2.09 (m, 6H), 1.99−1.93 (m, 0.85H), 1.82−1.75 (m, 1H), 1.57−1.55 (m, 0.67H), 1.37−1.33 (m, 0.72H).
N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide (27).
72.6% yield; 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.40−7.37 (m, 2H), 6.98 (d, J = 8.0 Hz, 1H), 6.85 (s, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.76−6.71 (m, 1H), 6.60 (d, J = 8.4 Hz, 1H), 5.60 (d, J = 10.4 Hz, 2H), 4.71 (s, 1H), 4.25 (s, 1H), 4.02 (d, J = 8.4 Hz, 1H), 3.55 (sep, J = 6.8 Hz, 1H), 3.02−2.96 (m, 1H), 2.51−2.45 (m, 1H), 2.17 (s, 3H), 1.78−1.72 (m, 2H), 1.22 (d, J = 6.4 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 204.1, 149.2, 146.7, 137.6, 135.5, 134.4, 133.8, 133.4, 130.4, 130.2, 129.0, 128.8, 128.2, 126.5, 125.9, 125.5, 123.6, 119.7, 115.5, 57.2, 45.6, 45.3, 35.4, 31.5, 19.8, 19.2, 19.1. HRMS (ESI): calcd for C30H32N2NaO3S, 523.2026 [M + Na]+; found, 523.2003. Purity: 97.9% by UPLC (Rt = 3.47 min).
tert-Butyl 8-(N-(3,4-Dimethylphenyl)sulfamoyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]quinoline-1-carboxylate (29).
The crude product was used directly for the next step without further purification.
4-(4-Isobutyrylphenyl)-N-(p-tolyl)-2,3,3a,4,5,9b-hexahydrofuro-[3,2-c]quinoline-8-sulfonamide (31).
72.2% yield; 1H NMR (400 MHz, CDCl3): δ 7.98−7.95 (m, 2H), 7.86 (d, J = 2.0 Hz, 0.6H), 7.76 (d, J = 1.6 Hz, 0.4H), 7.52−7.48 (m, 2H), 7.42−7.38 (m, 1H), 7.03−6.93 (m, 4H), 6.84 (s, 0.4H), 6.78 (s, 0.6H), 6.56−6.53 (m, 1H), 5.18 (d, J = 7.2 Hz, 0.4H), 4.84 (d, J = 2.8 Hz, 0.4H), 4.74 (s, 0.6H), 4.50 (d, J = 4.8 Hz, 0.6H), 4.44 (s, 0.4H), 4.05−4.01 (m, 0.6H), 3.90 (d, J = 10.8 Hz, 0.6H), 3.87−3.83 (m, 0.6H), 3.72−3.64 (m, 0.8H), 2.56−2.51 (m, 1H), 2.76−2.72 (m, 0.4H), 2.42−2.38 (m, 0.6H), 2.26 (s, 3H), 2.05−1.99 (m, 1H), 1.71−1.68 (m, 0.6H), 1.52−1.43 (m, 0.4H), 1.22 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 204.0, 148.7, 147.9, 146.0, 145.6, 136.3, 134.9, 134.2, 131.4, 130.0, 129.8, 128.9, 128.6, 128.4, 127.9, 126.6, 122.1, 122.0, 114.4, 114.2, 75.2, 74.8, 66.4, 65.1, 56.7, 56.1, 44.4, 42.5, 35.5, 35.4, 28.4, 24.2, 20.8, 19.14, 19.11. HRMS (ESI): calcd for C28H30N2NaO4S, 513.1818 [M + Na]+; found, 513.1788. Purity: 96.3% by UPLC (Rt = 3.29 min). HRMS (ESI): calcd for C28H30N2NaO4S, 513.1818 [M + Na]+; found, 513.1788. Purity: 96.3% by UPLC (Rt = 3.29 min).
4-(4-Isobutyrylphenyl)-N-(m-tolyl)-2,3,3a,4,5,9b-hexahydrofuro-[3,2-c]quinoline-8-sulfonamide (32).
58.7% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.93 (s, 0.5H), 9.90 (s, 0.5H), 7.99 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.6 Hz, 2H), 7.57 (s, 1H), 7.42 (d, J = 8.4 Hz, 0.5H), 7.35 (d, J = 8.8 Hz, 0.5H), 7.21 (s, 0.5H), 7.11−7.07 (m, 1H), 6.91−6.88 (m, 2.5H), 6.80 (d, J = 7.2 Hz, 1H), 6.75−6.71 (m, 1H), 5.11 (d, J = 7.2 Hz, 0.5H), 4.85 (s, 0.5H), 4.43 (d, J = 4.4 Hz, 0.5H), 3.91−3.87 (m, 0.5H), 3.80 (d, J = 10.8 Hz, 0.5H), 3.74−3.45 (m, 3H), 2.71−2.65 (m, 0.5H), 2.31−2.24 (m, 0.5H), 2.20 (s, 3H), 1.97−1.92 (m, 0.5H), 1.80−1.75 (m, 0.5H), 1.58−1.53 (m, 0.5H), 1.32−1.30 (m, 0.5H), 1.10 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 203.95, 203.92, 149.7, 149.2, 147.3, 147.0, 138.8, 138.73, 138.69, 135.7, 135.3, 131.0, 129.4, 129.3, 129.2, 128.9, 128.8, 128.0, 127.4, 127.3, 127.0, 126.1, 124.6, 124.5, 121.0, 120.4, 120.2, 118.4, 117.0, 116.7, 114.6, 114.4, 75.0, 74.5, 66.1, 64.6, 55.8, 55.0, 44.0, 42.0, 35.1, 35.0, 28.5, 24.5, 21.54, 21.53, 19.5, 19.45, 19.43. HRMS (ESI): calcd for C28H30N2NaO4S, 513.1818 [M + Na]+; found, 513.1788. Purity: 98.3% by UPLC (Rt = 3.28 min).
4-(4-Isobutyrylphenyl)-N-phenyl-2,3,3a,4,5,9b-hexahydrofuro-[3,2-c]quinoline-8-sulfonamide (33).
56.6% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.99, 9.97 (s, 1H), 8.00 (d, J = 8.0 Hz, 2H), 7.62−7.56 (m, 3H), 7.41 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.5H), 7.35 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.5H), 7.24−7.21 (m, 2.5H), 7.11−7.08 (m, 2H), 7.01−6.98 (m, 1H), 6.92 (s, 0.5H), 6.73 (t, J = 8.4 Hz, 1H), 5.11 (d, J = 7.6 Hz, 0.5H), 4.85 (d, J = 2.8 Hz, 0.5H), 4.43 (d, J = 4.8 Hz, 0.5H), 3.91−3.88 (m, 0.5H), 3.80 (d, J = 10.4 Hz, 0.5H), 3.72−3.59 (m, 2H), 3.52−3.49 (m, 0.5H), 2.72−2.67 (m, 0.5H), 2.33−2.27 (m, 0.5H),1.99−1.92 (m, 0.5H), 1.82−1.72 (m, 0.5H), 1.57−1.52 (m, 0.5H), 1.34−1.29 (m, 0.5H), 1.11 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 204.0, 203.9, 149.7, 149.2, 147.3, 147.0, 138.8, 135.7, 135.3, 131.0, 129.50, 129.45, 129.3, 129.2, 128.9, 128.8, 128.0, 127.5, 127.4, 127.0, 126.0, 123.9, 123.8, 121.1, 120.0, 119.8, 118.4, 114.6, 114.4, 75.0, 74.5, 66.1, 64.6, 55.8, 55.0, 43.9, 41.9, 35.1, 35.0, 28.5, 24.5, 19.5, 19.45, 19.43. HRMS (ESI): calcd for C27H28N2NaO4S, 499.1662 [M + Na]+; found, 499.1629. Purity: 99.2% by UPLC (Rt = 3.23 min).
N-(3-Chloro-4-methylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (34).
56.6% yield; 1H NMR (400 MHz,, DMSO-d6): δ 10.13 (s, 0.6H), 10.08 (s, 0.4H), 8.00 (d, J = 8.0 Hz, 2H), 7.62−7.58 (m, 3H), 7.41 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 0.6H), 7.35 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 0.4H), 7.26 (s, 0.6H), 7.21 (d, J = 8.4 Hz, 1H), 7.10 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1H), 7.01−6.99 (m, 1.4H), 6.77−6.72 (m, 1H), 5.12 (d, J = 7.2 Hz, 0.4H), 4.87 (d, J = 3.2 Hz, 0.4H), 4.44 (d, J = 4.8 Hz, 0.6H), 3.92−3.88 (m, 0.6H), 3.81 (d, J = 10.8 Hz, 0.6H), 3.74−3.60 (m, 2H), 3.52−3.49 (m, 0.4H), 2.72−2.67 (m, 0.4H), 2.33−2.27 (m, 0.6H), 2.21 (s, 3H), 1.99−1.91 (m, 0.6H), 1.82−1.72 (m, 0.6H), 1.57−1.53 (m, 0.4H), 1.34−1.31 (m, 0.4H), 1.11 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 203.9, 149.9, 149.3, 147.2, 147.0, 138.0, 135.7, 135.3, 133.7, 133.6, 132.0, 132.0, 131.0, 130.7, 130.6, 129.3, 129.2, 128.9, 128.8, 128.0, 127.4, 127.3, 126.9, 126.4, 125.5, 121.1, 120.0, 119.7, 118.8, 118.5, 114.7, 114.4, 75.0, 74.4, 66.1, 64.6, 55.7, 55.0, 43.9, 41.9, 35.05, 35.02, 28.5, 24.5, 19.5, 19.4, 19.2. HRMS (ESI): calcd for C28H29ClN2NaO4S, 547.1429 [M + Na]+; found, 547.1453. Purity: 96.0% by UPLC (Rt = 3.39 min).
N-(4-Chloro-3-methylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (35).
67.5% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.10 (s, 0.6H), 10.06 (s, 0.4H), 8.00 (d, J = 7.6 Hz, 2H), 7.62−7.55 (m, 3H), 7.42 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 0.6H), 7.36 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 0.4H), 7.28−7.25 (m, 1.6H), 7.04 (s, 1H), 6.96−6.92 (m, 1.4H), 6.76−6.72 (m, 1H), 5.12 (d, J = 7.2 Hz, 0.4H), 4.86 (d, J = 2.8 Hz, 0.4H), 4.44 (d, J = 4.8 Hz, 0.6H), 3.92−3.89 (m, 0.6H), 3.81 (d, J = 10.8 Hz, 0.6H), 3.74−3.58 (m, 2H), 3.52−3.49 (m, 0.4H), 2.71−2.65 (m, 0.4H), 2.33−2.26 (m, 0.6H), 2.21 (s, 3H), 1.98−1.93 (m, 0.6H), 1.83−1.75 (m, 0.6H), 1.57−1.53 (m, 0.4H), 1.35−1.29 (m, 0.4H), 1.15 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 203.97, 149.8, 149.3, 147.2, 147.0, 137.7, 136.5, 135.7, 135.3, 131.0, 129.8, 129.2, 128.9, 128.1, 127.5, 126.5, 125.6, 122.3, 122.1, 121.0, 119.0, 118.7, 118.4, 114.4, 105.8, 75.0, 74.5, 66.1, 66.0, 64.6, 55.7, 55.0, 43.9, 41.9, 35.05, 35.02, 28.5, 24.5, 20.2, 19.4. HRMS (ESI): calcd for C28H29ClN2NaO4S, 547.1429 [M + Na]+; found, 547.1440. Purity: 96.9% by UPLC (Rt = 3.41 min).
N-(3,4-Dichlorophenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (36).
69.8% yield; 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.2 Hz, 2H), 7.84−7.76 (m, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.49−7.43 (m, 1H), 7.32 (d, J = 8.7 Hz, 0.7H), 7.18 (d, J = 3.6 Hz, 1H), 7.10−7.06 (m, 0.3H), 7.06−7.02 (m, 0.3H), 7.00 (dd, J1 = 8.7, J2 = 2.6 Hz, 1H), 6.64−6.60 (m, 0.7H), 6.59−6.55 (m, 1H), 5.21 (d, J = 7.5 Hz, 1H), 4.89 (d, J = 3.0 Hz, 1H), 4.38 (s, 0.7H), 4.36 (s, 0.3H), 3.80−3.64 (m, 2H), 3.60−3.50 (m, 1H), 2.82−2.72 (m, 1H), 2.18 (s, 1H), 2.09−1.94 (m, 1H), 1.56−1.46 (m, 1H), 1.24 (s, 3H), 1.22 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 204.0, 149.1, 148.2, 145.8, 145.4, 136.8, 136.7, 136.4, 135.9, 132.9, 131.5, 130.8, 130.2, 130.1, 128.9, 128.6, 128.4, 128.3, 127.9, 127.1, 126.6, 126.4, 122.3, 121.9, 120.2, 120.0, 119.1, 114.6, 114.4, 75.2, 74.7, 66.5, 65.1, 56.6, 56.1, 44.3, 42.4, 35.51, 35.46, 28.4, 24.2, 19.14, 19.10. HRMS (ESI): calcd for C27H26Cl2N2NaO4S, 567.0883 [M + Na]+; found, 567.0944. Purity: 96.8% by UPLC (Rt = 3.43 min).
N-(3,4-Dibromophenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (37).
56.6%; 1H NMR (400 MHz, DMSO-d6): δ 10.41 (s, 0.6H), 10.36 (s, 0.4H), 8.00 (d, J = 7.6 Hz, 2H), 7.63−7.55 (m, 4H), 7.46−7.36 (m, 2H), 7.32 (s, 0.4H), 7.09−7.04 (m, 1.6H), 6.79−6.75 (m, 1H), 5.13 (d, J = 7.6 Hz, 0.4H), 4.88 (d, J = 2.8 Hz, 0.4H), 4.45 (d, J = 4.8 Hz, 0.6H), 3.92−3.88 (m, 0.6H), 3.83 (d, J = 10.8 Hz, 0.6H), 3.75−3.59 (m, 2H), 3.52−3.49 (m, 0.4H), 2.72−2.67 (m, 0.4H), 2.33−2.27 (m, 0.6H), 1.99−1.91 (m, 0.4H), 1.80−1.72 (m, 0.6H), 1.59−1.52 (m, 0.6H), 1.36−1.32 (m, 0.4H), 1.11 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 204.0, 203.9, 150.1, 149.5, 147.1, 146.9, 139.6, 135.8, 135.3, 134.61, 134.57, 131.0, 129.3, 129.2, 128.9, 128.8, 128.1, 127.5, 127.4, 125.9, 125.0, 124.51, 124.46, 123.8, 123.6, 121.1, 120.3, 119.9, 118.5, 117.9, 117.7, 114.8, 114.5, 74.9, 74.4, 66.1, 64.6, 55.7, 54.9, 43.8, 41.8, 35.1, 35.0, 28.5, 24.5, 19.5, 19.45, 19.43. HRMS (ESI): calcd for C27H26Br2N2NaO4S, 654.9872 [M + Na]+; found, 654.9943. Purity: 95.3% by UPLC (Rt = 3.46 min).
N-(4-Fluoro-3-methylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (38).
68.4% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.88 (s, 0.4H), 9.83 (s, 0.6H), 8.00 (d, J = 8.0 Hz, 2H), 7.62−7.58 (m, 2.4H), 7.51 (s, 0.6H), 7.37 (d, J = 8.8 Hz, 0.4H), 7.32 (d, J = 8.4 Hz, 0.6H), 7.22 (s, 0.4H), 7.03−6.89 (m, 3.6H), 6.72 (t, J = 8.4 Hz, 1H), 5.11 (d, J = 7.6 Hz, 0.6H), 4.86 (d, J = 2.8 Hz, 0.6H), 4.43 (d, J = 4.8 Hz, 0.4H), 3.92−3.88 (m, 0.4H), 3.81 (d, J = 10.4 Hz, 0.4H), 3.72−3.57 (m, 2H), 3.52−3.46 (m, 0.6H), 2.71−2.65 (m, 0.6H), 2.33−2.27 (m, 0.4H), 2.14 (s, 3H), 1.99−1.90 (m, 0.4H), 1.80−1.75 (m, 0.4H), 1.56−1.53 (m, 0.6H), 1.34−1.29 (m, 0.6H), 1.11 (d, J = 6.4 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 203.9, 149.7, 149.2, 147.2, 147.0, 135.7, 135.3, 134.7, 131.0, 129.3, 129.2, 128.9, 128.8, 128.0, 127.5, 127.3, 126.7, 125.8, 125.2, 125.0, 123.8, 123.6, 121.0, 120.04, 119.96, 119.6, 118.4, 115.8, 115.6, 114.6, 114.4, 75.0, 74.5, 66.0, 64.6, 55.8, 55.0, 43.9, 41.9, 35.05, 35.02, 28.5, 24.5, 19.5, 19.4, 14.8, 14.8. HRMS (ESI): calcd for C28H29FN2NaO4S, 531.1724 [M + Na]+; found, 531.1708. Purity: 99.0% by UPLC (Rt = 3.31 min).
4-(4-Isobutyrylphenyl)-N-(3-methyl-4-(trifluoromethyl)phenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (39).
77.1% yield; 1H NMR (400 MHz, CDCl3): δ 7.97−7.94 (m, 2.5H), 7.88 (s, 0.5H), 7.70−7.67 (m, 1H), 7.57−7.47 (m, 3H), 7.43−7.38 (m, 1H), 7.01−6.99 (m, 2H), 6.64−6.60 (m, 1H), 5.20 (d, J = 7.2 Hz, 0.5H), 4.89−4.85 (m, 1H), 4.58 (s, 0.5H), 4.52 (d, J = 4.4 Hz, 0.5H), 4.02−3.98 (m, 0.5H), 3.91−3.84 (m, 1H), 2.74−2.68 (m, 0.5H), 3.63−3.51 (m, 1.5H), 2.76−2.74 (m, 0.5H), 2.37 (s, 3H), 2.07−1.98 (m, 1H), 1.72−1.69 (m, 0.5H), 1.50−1.48 (m, 0.5H), 1.21 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 204.2, 149.1, 148.3, 145.9, 145.5, 140.3, 140.2, 138.2, 136.3, 135.8, 131.6, 130.2, 128.9, 128.6, 128.4, 127.9, 127.3, 127.0, 126.6, 125.8, 124.4, 124.1, 123.1, 122.1, 121.7, 118.9, 115.9, 115.8, 114.8, 114.5, 75.2, 74.7, 66.4, 65.0, 56.5, 56.0, 44.3, 42.3, 35.5, 35.4, 28.4, 24.2, 19.4, 19.13, 19.10, 19.07. HRMS (ESI): calcd for C29H29F3N2NaO4S, 581.1692 [M + Na]+; found, 581.1707. Purity: 99.5% by UPLC (Rt = 3.42 min).
Methyl 4-(6-Chloro-8-(N-(3,4-dimethylphenyl)sulfamoyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-4-yl)benzoate (40).
83% yield; 1H NMR (400 MHz, CDCl3): δ 8.03−8.01 (m, 2H), 7.64 (s, 1H), 7.47 (m, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.02−6.96 (m, 1H), 6.90−6.72 (m, 3H), 6.13,5.99 (s, 1H), 5.15 (d, J = 7.6 Hz, 0.37 H), 4.49 (d, J = 4.8 Hz, 1H), 4.00−3.97 (m, 1H), 3.95 (s, 3H), 3.86−3.84 (m, 0.7H), 3.71−3.65 (m, 1.3H), 2.64−2.72 (m, 0.3H), 2.31−2.27 (m, 0.7H), 2.23−2.09 (m, 6H), 2.07−1.97 (m, 0.7H), 1.74−1.70 (m, 1.7H), 1.45−1.36 (m, 0.3H). 13C NMR (100 MHz, CDCl3): δ 166.6, 145.7, 145.5, 142.0, 141.8, 138.03, 137.95, 136.3, 135.8, 135.24, 135.18, 133.0, 132.9, 130.4, 130.2, 130.14, 130.09, 129.7, 129.5, 128.8, 127.8, 126.2, 126.0, 125.3, 123.4, 122.33, 122.29, 122.0, 121.8, 121.5, 121.2, 74.9, 66.7, 65.1, 56.3, 55.7, 52.31, 52.28, 43.9, 42.2, 28.4, 24.0, 19.72, 19.69, 19.34, 19.32. HRMS (ESI): calcd for C27H27ClN2NaO5S, 549.1221 [M + Na]+; found, 549.1230. Purity: 96.4% by UPLC (Rt = 3.50 min).
Methyl 4-(7-Chloro-8-(N-(3,4-dimethylphenyl)sulfamoyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-4-yl)benzoate (41).
66.7% yield; 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.0 Hz, 2H), 8.00 (s, 1H), 7.47 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.93 (s, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.97 (s, 1H), 6.66 (s, 1H), 5.14 (d, J = 7.2 Hz, 1H), 4.86 (d, J = 2.8 Hz, 1H), 4.43 (s, 1H), 3.95 (s, 3H), 3.76−3.70 (m, 1H), 3.64−3.59 (m, 1H), 2.77−2.71 (m, 1H), 2.19 (s, 3H), 2.17 (s, 3H), 1.99−1.89 (m, 1H), 1.52−1.45 (m, 1H), 0.90−0.85 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 166.6, 149.1, 145.2, 137.7, 135.9, 133.8, 133.8, 131.8, 130.5, 130.3, 130.2, 128.1, 124.4, 123.0, 118.8, 117.3, 116.1, 74.6, 65.0, 56.5, 52.3, 42.3, 28.3, 19.9, 19.2. HRMS (ESI): calcd for C27H27ClN2NaO5S, 549.1221 [M + Na]+; found, 549.1241. Purity: 96.8% by UPLC (Rt = 3.32 min).
N-(3,4-Dimethylphenyl)-4-(4-(methylsulfonyl)phenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (42).
73% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.75 (s, 0.5H), 9.71 (s, 0.5H), 7.96−7.94 (m, 2H), 7.75−7.70 (m, 2H), 7.60 (s, 0.5H), 7.55 (s, 0.5H), 7.40 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.5H), 7.33 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H), 7.19 (s, 0.5H), 6.96 (d, J = 8.0 Hz, 1H), 6.89−6.80 (m 3H), 6.73−6.70 (m, 1H), 5.12 (d, J = 7.2 Hz, 0.5H), 4.89 (d, J = 2.8 Hz, 0.5H), 4.44 (d, J = 4.4 Hz, 0.5H), 3.92−3.84 (m, 1H), 3.75−3.50 (m, 2.5H), 2.72−2.67 (m, 0.5H), 2.33−2.29 (m, 0.5H), 2.11 (s, 3H), 2.10 (s, 3H), 1.99−1.92 (m, 0.5H), 1.81−1.76 (m, 1H), 1.56−1.53 (m, 0.5H), 1.48−1.28 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 150.2, 148.4, 140.0, 137.1, 136.4, 134.5, 131.9, 130.3, 128.5, 128.0, 127.7, 127.4, 125.6, 124.5, 121.7, 118.0, 115.6, 56.0, 45.3, 45.0, 44.0, 31.6, 20.0, 19.1. HRMS (ESI): calcd for C27H28N2NaO4S2, 531.1383 [M + Na]+; found, 531.1387. Purity: 97.5% by UPLC (Rt = 3.21 min).
7-Chloro-N-(3,4-dimethylphenyl)-4-(4-(methylsulfonyl)phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone-8-sulfonamide (43).
28% yield; 1H NMR (400 MHz, DMSO-d6): δ 10.02 (s, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 8.0 Hz, 2H), 7.64 (s, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.87 (s, 1H), 6.83−6.82 (m, 3H), 5.90 (s, 1H), 5.59 (d, J = 5.2 Hz, 1H), 4.78 (d, J = 2.8 Hz, 1H), 4.05 (d, J = 8.4 Hz, 1H), 3.23 (s, 3H), 3.01−2.95 (m, 1H), 2.30−2.24 (m, 1H), 2.10 (s, 3H), 2.08 (s, 3H), 1.62−1.56 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 151.2, 148.0, 140.1, 137.2, 135.8, 134.4, 133.4, 131.4, 130.3, 128.9, 128.0, 127.5, 123.8, 123.0, 120.1, 117.1, 116.5, 55.6, 44.8, 44.7, 44.0, 31.6, 20.0, 19.0. HRMS (ESI): calcd for C27H28ClN2O4S2, 543.1174 [M + H]+; found, 543.1434. Purity: 97.1% by UPLC (Rt = 3.16 min).
9-Chloro-N-(3,4-dimethylphenyl)-4-(4-(methylsulfonyl)phenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolone-8-sulfonamide (44).
34%; 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 8.0 Hz, 1H), 6.92 (s, 1H), 6.87−6.85 (m, 2H), 6.55 (d, J = 8.8 Hz, 1H), 6.10 (s, 1H), 5.70 (d, J = 3.6 Hz, 1H), 4.68 (d, J = 3.2 Hz, 1H), 4.42 (d, J = 9.2 Hz, 1H), 4.24 (s, 1H), 3.25−3.21 (m, 1H), 3.10 (s, 3H), 2.64−2.58 (m, 1H), 2.18 (s, 3H), 2.17 (s, 3H), 1.90−1.84 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 151.6, 147.5, 139.5, 137.6, 133.9, 133.8, 131.9, 131.2, 130.4, 130.2, 127.7, 127.4, 125.7, 125.6, 123.1, 123.0, 119.0, 113.9, 113.8, 57.2, 45.6, 45.6, 45.4, 44.5, 32.2, 19.82, 19.79, 19.17, 19.15. HRMS (ESI): calcd for C26H28N2NaO5S2, 565.0993 [M + Na]+; found, 565.0996. Purity: 99.4% by UPLC (Rt = 3.21 min).
Hydrolysis of Methyl Ester 2–5 to Afford 2a–5a.
To a solution of 2–5 (0.3 mmol) in MeOH (3 mL) was added 1 mL of NaOH (aq, 3 M). The solution was stirred overnight, and then the solvent was removed under reduced pressure. Water was added, and the reaction mixture was extracted with ethyl acetate (3 × 10 mL). Combined solvent was evaporated under reduced pressure to provide a solid residue, which was purified via flash chromatography on silica. Elution with DCM/MeOH (10:1−3:1) afforded 2a–5a as off-white to yellowish solids.
4-(8-(N-(3,4-Dimethylphenyl)sulfamoyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-4-yl)benzoic Acid (2a).
92.1%; 1H NMR (400 MHz, DMSO-d6): δ 12.96 (s, 1H), 9.74 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.58 (t, J = 5.8 Hz, 3H), 7.38 (dd, J1 = 8.4 Hz, J2 = 2.2 Hz, 1H), 7.16 (s, 1H), 6.96 (d, J = 8.1 Hz, 1H), 6.87 (s, 1H), 6.82 (dd, J1 = 8.4 Hz, J2 = 2.2 Hz, 1H), 6.73 (d, J = 8.7 Hz, 1H), 4.43 (d, J = 4.8 Hz, 1H), 3.89 (q, J = 8.3 Hz, 1H), 3.79 (d, J = 10.6 Hz, 1H), 3.71 (dt, J1 = 14.3 Hz, J2 = 7.3 Hz, 1H), 2.33−2.24 (m, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 2.02−1.87 (m, 1H), 1.61−1.48 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 167.7, 149.6, 149.1, 147.0, 146.7, 137.2, 137.1, 136.4, 131.9, 131.8, 131.2, 131.0, 130.32, 130.28, 129.9, 129.3, 129.0, 128.0, 127.3, 127.2, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.0, 74.5, 66.1, 64.6, 55.8, 55.1, 44.0, 42.0, 28.5, 24.5, 20.0, 19.1. HRMS (ESI): calcd for C26H26N2NaO5S, 501.1455 [M + Na]+; found, 501.1410. Purity: 99.8% by UPLC (Rt = 3.18 min).
4-(8-(N-(3,4-Dimethylphenyl)sulfamoyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]quinolin-4-yl)benzoic Acid (3a).
88.3%; 1H NMR (400 MHz, DMSO-d6): δ 9.77 (s, 1H), 7.96 (d, J = 7.8 Hz, 2H), 7.75 (s, 1H), 7.46 (d, J = 7.7 Hz, 2H), 7.36 (d, J = 8.4 Hz, 1H), 7.12 (s, 1H), 6.95 (d, J = 8.1 Hz, 1H), 6.89 (s, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.73 (d, J = 8.6 Hz, 1H), 3.96 (s, 1H), 3.90 (d, J = 10.4 Hz, 1H), 3.10−3.00 (s, 1H), 2.95−2.85 (m, 1H), 2.30−2.20 (s, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 1.86−1.76 (s, 1H), 1.50−1.40 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 169.4, 149.9, 143.9, 137.2, 136.4, 136.2, 131.6, 131.1, 130.3, 129.9, 128.2, 127.9, 126.2, 121.3, 117.4, 116.3, 114.5, 57.2, 55.2, 42.1, 41.5, 27.4, 20.0, 19.1. HRMS (ESI): calcd for C26H28N3O4S2, 478.1795 [M + H]+; found, 478.1746. Purity: 97.7% by UPLC (Rt = 2.72 min).
4-(9-(N-(3,4-Dimethylphenyl)sulfamoyl)-3,4,4a,5,6,10b-hexahydro-2H-pyrano[3,2-c]quinolin-5-yl)benzoic Acid (4a).
91.7%; 1H NMR (400 MHz, DMSO-d6): δ 9.67−9.64 (m, 1H), 7.96−7.93 (m, 2H), 7.52−7.46 (m, 3H), 7.37−7.34 (m, 1H), 7.12 (s, 0.7H), 6.97−6.94 (m, 1H), 6.91 (s, 0.3H), 6.86−6.81 (m, 2H), 6.71 (d, J = 8.4 Hz, 0.3H), 6.62 (d, J = 8.4 Hz, 0.7H), 5.18 (d, J = 5.6 Hz, 0.3H), 4.81 (d, J = 2.4 Hz, 0.3H), 4.61 (d, J = 9.6 Hz, 0.7H), 4.31 (d, J = 2.8 Hz, 0.7H), 3.78−3.76 (m, 1H), 3.62−3.57 (m, 1H), 2.95−2.89 (m, 0.3H), 2.11 (s, 3H), 2.09 (s, 3H), 1.97−1.94 (m, 1H), 1.78−1.62 (m, 2H), 1.35−1.32 (m, 1H), 1.21−1.17 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 167.6, 149.8, 149.0, 147.5, 145.8, 137.1, 137.0, 136.4, 132.0, 131.8, 130.7, 130.3, 130.2, 130.0, 129.6, 128.3, 128.2, 127.5, 127.4, 126.9, 126.3, 125.2, 122.0, 121.6, 118.8, 118.3, 118.1, 117.7, 114.0, 113.4, 71.3, 59.9, 57.6, 54.1, 37.7, 37.5, 30.0, 25.4, 23.7, 22.2, 20.02, 19.98, 19.1, 18.4. HRMS (ESI): calcd for C27H29N2O5S, 493.1792 [M + H]+; found, 493.1798. Purity: 95.0% by UPLC (Rt = 3.11 min).
4-(2-(N-(3,4-Dimethylphenyl)sulfamoyl)-6,6a,7,11b-tetrahydro-5H-indeno[2,1-c]quinolin-6-yl)benzoic Acid (5a).
90.5%; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (s, 1H), 9.77 (s, 1H), 7.97 (d, J = 7.6 Hz, 2H), 7.65 (s, 1H), 7.58 (d, J = 7.2 Hz, 2H), 7.50 (d, J = 7.2 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.19−7.15 (m, 1H), 7.13−7.09 (m, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.81 (s, 3H), 4.81 (s, 1H), 4.50 (d, J = 8.0 Hz, 1H), 3.12−3.10 (m, 1H), 2.95−2.88 (m, 1H), 2.20−2.14 (m, 1H), 2.07 (s, 3H), 2.05 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 167.7, 150.1, 147.2, 146.0, 142.2, 137.1, 136.4, 131.7, 130.3, 130.2, 129.8, 128.9, 127.41, 127.38, 127.3, 127.0, 126.0, 125.3, 125.1, 122.5, 121.2, 117.4, 115.2, 55.8, 46.9, 45.2, 31.3, 20.0, 19.1. HRMS (ESI): calcd for C31H29N2O4S, 525.1843 [M + H]+; found, 525.1835. Purity: 99.6% by UPLC (Rt = 3.32 min).
Preparation of N-(3,4-Dimethylphenyl)-4-(4-(1-hydroxy-2-methylpropyl)phenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]-quinoline-8-sulfonamide (15).
Compound 14 (0.20 g, 0.40 mmol) and Pd/C (10% Pd base, 0.10 g) were mixed in MeOH (50 mL) and ethyl acetate (50 mL) at room temperature. Hydrogen gas was introduced via a H2 balloon. The reaction mixture was stirred under a H2 atmosphere overnight at room temperature and then filtered to remove the precipitate. The solution was concentrated under reduced pressure to afford a pale-yellow solid, which was purified by column chromatography (silica gel, CH2Cl2/MeOH = 9/1 v/v) to afford a white solid product (90% yield). 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 2.0 Hz, 0.5H), 7.78 (d, J = 2.0 Hz, 0.5H), 7.43−7.32 (m, 4H), 6.97 (d, J = 8.0 Hz, 1H), 6.88 (s, 1H), 6.80−6.77 (m, 1H), 6.53−6.50 (m, 1H), 6.35 (s, 0.5H), 6.30 (s, 0.5H), 5.17 (d, J = 7.6 Hz, 0.5H), 4.78 (d, J = 2.8 Hz, 0.5H), 4.59 (s, 0.5H), 4.51 (d, J = 4.8 Hz, 0.5H), 4.42−4.39 (m, 1H), 4.29 (s, 0.5H), 4.03−3.98 (m, 0.5H), 3.86−3.82 (m, 1H), 3.73−3.65 (m, 1H), 2.75−2.70 (m, 0.5H), 2.40−2.36 (m, 0.5H), 2.17 (s, 6H), 2.02−1.94 (m, 2H), 1.88 (s, 1H), 1.74−1.68 (m, 0.5H), 1.55−1.50 (m, 0.5H), 1.01−0.99 (m, 3H), 0.82 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 148.9, 148.1, 144.0, 143.5, 140.1, 139.6, 137.5, 134.5, 134.4, 133.5, 133.4, 131.4, 130.13, 130.12, 130.07, 128.6, 127.93, 127.88, 127.8, 127.1, 127.0, 126.2, 123.3, 123.1, 121.6, 119.2, 119.1, 118.9, 114.2, 114.0, 79.59, 79.56, 75.4, 74.9, 66.4, 65.0, 56.6, 56.1, 56.0, 53.7, 44.7, 42.4, 35.3, 29.2, 28.5, 24.3, 19.8, 19.2, 19.0, 18.2. HRMS (ESI): calcd for C29H33N2O4S, 529.2131 [M + Na]+; found, 529.2148. Purity: 95.7% by UPLC (Rt = 3.31 min).
General Coupling between Weinreb Amide 21 and Lithium Reagents or Grignard Reagents to Form 22–26.
Compound 21 (1.05 g, 2.01 mmol) was dissolved in 50 mL of anhydrous THF at room temperature under argon. Lithium reagent or Grignard reagent (6.64 mmol) was added via a syringe at room temperature under argon. The reaction solution was stirred at room temperature overnight and then quenched by adding 100 mL of water. The resulting solution was extracted with ethyl acetate (3 × 50 mL). The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude solid was purified by flash column chromatography (silica gel, CH2Cl2/acetone = 19/1 v/v) to afford the desired products 22–26.
4-(4-(Cyclopropanecarbonyl)phenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (22).
41.7% yield; 1H NMR (400 MHz, DMSO-d6): δ 9.74 (s, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.54 (s, 1H), 7.32 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.91 (s, 1H), 6.86 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 5.11 (d, J = 7.6 Hz, 0.45 H), 4.86 (d, J = 2.4 Hz, 1H), 3.67−3.73 (m, 1H), 3.62−3.56 (m, 1H), 3.52−3.49 (m, 1H), 2.95−2.90 (m, 1H), 2.71−2.68 (m, 1H), 2.11 (s, 3H), 2.09 (s, 3H), 1.80−1.73 (m, 1H), 1.33−1.31 (1H), 1.17−1.09 (m, 2H), 0.86−0.76 (2H). 13C NMR (100 MHz, DMSO-d6): δ 199.8, 149.1, 147.4, 137.1, 136.9, 136.3, 131.9, 130.3, 129.3, 128.5, 127.3, 127.1, 121.7, 121.0, 117.9, 114.6, 74.5, 66.0, 55.0, 54.6, 44.0, 30.9, 24.5, 20.0, 19.1, 17.0, 11.8, 11.7. HRMS (ESI): calcd for C29H30N2NaO4S, 525.1818 [M + Na]+; found, 525.1821. Purity: 98.2% by UPLC (Rt = 3.27 min).
4-(4-Acryloylphenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (23).
1H NMR (400 MHz, DMSO-d6): δ 9.78 (s, 0.4H), 9.74 (s, 0,6H), 8.04 (d, J = 8.0 Hz, 2H), 7.65−7.54 (m, 3H), 7.47−7.40 (m, 1.6H), 7.33 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 0.1H), 7.20 (s, 0.4H), 6.96 (d, J = 8.0 Hz, 1H), 6.91 (s, 0.6H), 6.86 (d, J = 6.4 Hz, 1H), 6.82−6.80 (m, 1H), 6.74−6.38 (m, 1H), 6.36 (d, J = 17.2 Hz, 1H), 6.01 (d, J = 2.8 Hz, 1H), 5.12 (d, J = 7.2 Hz, 0.6H), 4.87 (d, J = 2.8 Hz, 0.6H), 4.43 (d, J = 4.8 Hz, 0.4H), 3.92−3.88 (m, 0.6H), 3.82 (d, J = 10.8 Hz, 0.4H), 3.72−3.69 (m, 0.6H), 3.65−3.59 (m, 0.6H), 3.52−3.48 (m, 0.6H), 3.02−2.98 (m, 2H), 2.72−2.68 (m, 0.6H), 2.33−2.25 (m, 0.4H), 2.11 (s, 3H), 2.09 (s, 3H), 1.96−1.91 (m, 0.4H), 1.80−1.72 (m, 0.6H), 1.59−1.52 (m, 0.4H), 1.34−1.28 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 189.9, 189.8, 149.6, 149.0, 147.8, 147.5, 137.2, 137.1, 136.8, 136.34, 136.29, 132.7, 131.9, 131.8, 131.0, 130.9, 130.8, 130.3, 129.3, 129.2, 127.5, 127.3, 127.2, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.0, 74.5, 66.0, 64.6, 55.8, 55.0, 44.0, 42.0, 34.8, 30.9, 24.5, 20.0, 19.1. HRMS (ESI): calcd for C28H28N2NaO4S, 511.1662 [M + Na]+; found, 511.1643. Purity: 96.0% by UPLC (Rt = 1.02 min).
4-(4-Butyrylphenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (24).
1H NMR (400 MHz, DMSO-d6): δ 9.78,9.74 (s, 1H), 7.99 (d, J = 8.0 Hz, 2H), 7.61−7.54 (m, 3H), 7.39 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.4H), 7.32 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 0.6H), 7.17 (s, 0.40H), 6.96 (d, J = 8.4 Hz, 1H), 6.89−6.86 (m, 1.6H), 6.84−6.80 (m, 1H), 6.77−6.70 (m, 1H), 5.11 (d, J = 7.6 Hz, 0.6 H), 4.84 (d, J = 2.8 Hz, 0.6H), 4.43 (d, J = 4.4 Hz, 0.4H)), 3.91−3.88 (m, 0.6H), 3.79 (d, J = 10.4 Hz, 0.4H), 3.72−3.68 (m, 0.6H), 3.63−3.59 (m, 0.6H), 3.51−3.49 (m, 0.6H), 3.02−2.98 (m, 2H), 2.71−2.67 (m, 0.6H), 2.30−2.26 (m, 0.4H), 2.11 (s, 3H), 2.09 (s, 3H), 1.96−1.91 (m, 0.4H), 1.81−1.72 (m, 0.6H), 1.65 (q, J = 7.6 Hz, 2H), 1.59−1.54 (m, 0.4H), 1.34−1.28 (m, 1H), 0.93 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 200.1, 200.0, 149.6, 149.1, 147.3, 147.0, 137.2, 137.1, 136.8, 136.4, 131.9, 131.8, 131.0, 130.32, 130.27, 129.3, 129.1, 128.7, 128.6, 128.5, 128.2, 128.0, 127.3, 127.2, 126.2, 121.7, 121.5, 121.0, 118.4, 117.9, 117.6, 114.6, 114.3, 75.0, 74.5, 66.0, 64.6, 55.8, 55.0, 54.5, 44.0, 42.0, 28.5, 24.5, 20.01, 19.96, 19.1, 17.7, 17.6, 14.1. HRMS (ESI): calcd for C29H32N2NaO4S, 527.1975 [M + Na]+; found, 527.1967. Purity: 99.2% by UPLC (Rt = 3.36 min).
4-(4-(Cyclohexanecarbonyl)phenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (25).
1H NMR (400 MHz, DMSO-d6): δ 9.74 (s, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.54 (s, 1H), 7.32 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.89 (s, 1H), 6.86 (d, J = 6.4 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.71 (d, J = 8.4 Hz, 1H), 5.11 (d, J = 7.6 Hz, 1H), 4.86 (d, J = 2.4 Hz, 1H), 3.63−3.57 (m, 1H), 3.52−3.48 (m, 1H), 2.71−2.68 (m, 1H), 2.11 (s, 3H), 2.09 (s, 3H), 1.76−1.62 (m, 7H), 1.47−1.31 (m, 3H), 1.24−1.09 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 203.3, 149.1, 147.2, 137.1, 136.3, 135.4, 131.9, 130.3, 129.3, 127.4, 127.3, 127.1, 121.7, 121.0, 117.9, 114.6, 74.5, 66.1, 55.0, 44.8, 44.0, 30.9, 29.5, 26.0, 25.6, 24.5, 20.0, 19.1. HRMS (ESI): calcd for C32H36N2NaO4S, 567.2288 [M + Na]+; found, 567.2289. Purity: 96.4% by UPLC (Rt = 3.48 min).
4-(4-Benzoylphenyl)-N-(3,4-dimethylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (26).
41.7% yield; 1H NMR (400 MHz, CDCl3): δ 7.88−7.79 (m, 5H), 7.64−7.60 (m, 1H), 7.55−7.48 (m, 4H), 7.46−7.42 (m, 1H), 6.98 (d, J = 8.0 Hz, 2H), 6.88 (s, 1H), 6.81−6.78 (m, 1H), 6.58−6.55 (m, 1H), 6.25−6.22 (m, 1H), 5.20 (d, J = 7.6 Hz, 0.55H), 4.88 (d, J = 2.8 Hz, 0.55H), 4.59 (s, 0.45H), 4.54 (d, J = 5.2 Hz, 0.45H), 4.31 (s, 0.55H), 4.05−4.03 (m, 0.55H), 3.94 (d, J = 10.8 Hz, 0.45H), 3.90−3.86 (m, 0.55H), 3.76−3.72 (m, 0.55H), 3.70−3.66 (m, 0.55H), 2.80−2.77 (m, 0.55H), 2.45−2.43 (m, 0.45H), 2.18−2.17 (m, 6H), 2.09−2.01 (m, 1H), 1.76−1.73 (m, 0.55H). 13C NMR (100 MHz, CDCl3): δ 196.2, 148.6, 147.8, 145.7, 145.2, 137.7, 137.6, 137.3, 137.2, 134.5, 134.4, 133.5, 132.7, 132.6, 131.4, 130.6, 130.1, 130.04, 130.01, 128.6, 128.4, 128.1, 127.9, 127.5, 126.3, 123.3, 123.2, 121.7, 119.21, 119.15, 119.0, 114.5, 114.2, 75.2, 74.8, 66.4, 65.1, 56.8, 56.2, 44.5, 42.5, 29.2, 28.4, 24.2, 19.8, 19.2. HRMS (ESI): calcd for C32H30N2NaO4S, 561.1818 [M + Na]+; found, 561.1830. Purity: 99.8% by UPLC (Rt = 3.37 min).
Preparation of N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[c]quinoline-8-sulfonamide (28).
Compound 26 (80 mg, 0.16 mmol) and Pd/C (10% Pd base, 0.04 g) were mixed in MeOH (20 mL) and ethyl acetate (20 mL) at room temperature. Hydrogen gas was introduced via a H2 balloon. The reaction mixture was stirred under a H2 atmosphere overnight at room temperature. The reaction mixture was filtered to remove the solid. The solution was concentrated under reduced pressure to afford a pale-yellow solid, which was purified by column chromatography (silica gel, CH2Cl2/MeOH = 9/1 v/v) to afford a white solid product. The resulting product was dissolved in anhydrous methylene chloride (10 mL) at room temperature. To this solution was added 0.10 g (0.24 mmol) of Dess–Martin reagent at room temperature. The reaction solution was stirred at room temperature for 30 min, then diluted with 50 mL of water, and neutralized with NaHCO3. The resulting mixture was extracted with methylene chloride (3 × 50 mL). The extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to dryness. The brown solid residue was purified by column chromatography (silica gel, CH2Cl2/acetone = 19/1 v/v) to afford a white solid product (0.53 mg, 66.3% yield). 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.43−7.39 (m, 2H), 6.97 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.80 (dd, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 6.69 (s, 1H), 6.55 (d, J = 8.4 Hz, 1H), 4.68 (d, J = 3.2 Hz, 1H), 4.35 (s, 1H), 3.56−3.52 (m, 1H), 3.37 (t, J = 6.8 Hz, 1H), 2.45−2.42 (m, 1H), 2.17 (s, 3H), 2.16 (s, 3H), 2.04−1.99 (m, 1H), 1.63−1.52 (m, 2H), 1.40−1.35 (m, 2H), 1.22 (d, J = 6.4 Hz, 6H), 1.17−1.15 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 204.1, 148.6, 147.3, 137.5, 135.4, 134.6, 133.7, 130.2, 129.2, 128.7, 127.3, 126.6, 126.0, 125.2, 123.4, 119.6, 114.5, 56.7, 45.9, 40.4, 35.4, 34.0, 23.7, 22.7, 19.8, 19.2, 19.14, 19.10. HRMS (ESI): calcd for C30H34N2NaO3S, 525.2182 [M + Na]+; found, 525.2169. Purity: 98.2% by UPLC (Rt = 3.51 min).
Removal of Boc Protecting Group of 3b and 29 to Form 3 and 30.
Compound 3b or 29 (0.52 mmol) was dissolved in CH2Cl2 (20 mL) at room temperature. Trifluoroacetic acid (1 mL) was added dropwise with stirring via a syringe. The reaction solution was stirred at room temperature overnight, and then the reaction was quenched by adding 30 mL of saturated NaHCO3 solution. The organic layer was separated, dried over anhydrous MgSO4, filtered, and concentrated to dryness. The crude product was purified by column chromatography (silica gel, CH2Cl2/MeOH = 9/1 v/v) to afford the desired compounds 3 or 30 as white solids.
Methyl 4-(8-(N-(3,4-Dimethylphenyl)sulfamoyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]quinolin-4-yl)benzoate (3).
45.4% yield (two-step); 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 7.6 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 1H), 6.99−6.97 (m, 2H), 6.87 (d, J = 8.0 Hz, 1H), 6.54 (d, J = 8.8 Hz, 1H), 4.96 (br, 1H), 4.86 (s, 1H), 4.42 (s, 1H), 3.94 (s, 3H), 3.02 (s, 2H), 2.87 (s, 1H), 2.19 (s, 6H), 1.96−1.91 (m, 1H), 1.48−1.47 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 166.7, 148.5, 145.9, 137.4, 134.9, 133.2, 130.4, 130.05, 130.01, 128.3, 128.2, 127.3, 123.6, 119.5, 119.3, 114.1, 57.6, 56.4, 52.2, 43.2, 42.1, 29.7, 27.7, 19.8, 19.1. HRMS (ESI): calcd for C27H30N3O4S, 492.1952 [M + H]+; found, 492.1961. Purity: 99.8% by UPLC (Rt = 3.49 min).
N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydro-1H-pyrrolo[3,2-c]quinoline-8-sulfonamide (30).
51.5% yield (two-step); 1H NMR (400 MHz, DMSO-d6): δ 9.78 (s, 1H), 8.00 (d, J = 8.0 Hz, 2H), 7.74 (s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.8 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.90 (s, 1H), 6.87 (s, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 8.8 Hz, 1H), 4.82 (d, J = 2.8 Hz, 1H), 4.59 (d, J = 7.6 Hz, 1H), 3.67 (sep, J = 6.8 Hz, 1H), 2.87−2.81 (m, 1H), 2.71−2.67 (m, 1H), 2.62−2.56 (m, 1H), 2.11 (s, 3H), 2.09 (s, 3H), 1.69−1.58 (m, 1H), 1.24−1.18 (m, 1H), 1.10 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 204.0, 149.7, 147.0, 137.2, 136.4, 135.7, 131.7, 130.7, 130.3, 129.1, 128.9, 127.7, 126.3, 121.4, 117.5, 114.5, 57.7, 55.4, 43.0, 41.7, 35.1, 27.7, 20.0, 19.4, 19.1. HRMS (ESI): calcd for C29H34N3O3S, 504.2315 [M + H]+; found 504.2307. Purity: 97.8% by UPLC (Rt = 2.81 min).
Methylation of 14 to Form 45 and 46.
Compound 14 (0.30 g, 0.59 mmol) was dissolved in 20 mL of anhydrous DMF at room temperature. NaH (26 mg, 60% weight in mineral oil, 0.65 mmol) was added at room temperature. The mixture was stirred at room temperature for 1 h, and then MeI (0.13 g, 0.89 mmol) was added. The reaction was stirred at room temperature overnight and then quenched by adding 100 mL of saturated NH4Cl solution. The white precipitate formed was isolated by filtration and purified by column chromatography (silica gel, CH2Cl2/acetone = 19/1 v/v) to afford 45 and 46 as white solids.
N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-N,5-dimethyl-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (45).
35.5% yield; 1H NMR (400 MHz, DMSO-d6): δ 7.95 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.37−7.30 (m, 3H), 7.25 (d, J = 2.4 Hz, 0.5H), 7.18 (d, J = 2.0 Hz, 0.5H), 7.08−7.04 (m, 1H), 6.87−6.75 (m, 3H), 4.77 (t, J = 6.0 Hz, 1H), 4.51−4.47 (m, 1H), 3.73−3.60 (m, 2H), 3.52−3.48 (m, 0.5H), 3.03 (s, 1.5H), 3.01 (s, 1.5H), 2.94−2.88 (m, 0.5H), 2.84 (s, 1.5H), 2.80−2.77 (m, 0.5H), 2.76 (s, 1.5H), 2.67−2.60 (m, 0.5H), 2.18−2.13 (m, 6H), 1.79−1.72 (m, 1.5H), 1.09−1.07 (m, 6H). 13C NMR (100 MHz, DMSO-d6): δ 203.9, 203.8, 149.4, 149.0, 147.5, 145.8, 139.8, 137.1, 137.0, 135.6, 135.3, 135.1, 130.0, 129.8, 129.4, 129.3, 129.2, 128.7, 128.5, 127.9, 127.5, 124.03, 123.98, 123.0, 122.6, 121.6, 121.1, 112.0, 111.0, 73.8, 72.2, 65.3, 65.1, 63.3, 62.8, 43.2, 41.6, 38.6, 38.4, 38.2, 35.0, 29.5, 27.2, 19.9, 19.8, 19.5, 19.44, 19.38. HRMS (ESI): calcd for C31H37N2O4S, 533.2469 [M + H]+; found, 533.2504. Purity: 98.5% by UPLC (Rt = 3.49 min).
N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-N-methyl-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (46).
19.6% yield; 1H NMR (400 MHz, DMSO-d6): δ 8.00 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 2.0 Hz, 1H), 7.14 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 7.01 (s, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.77−6.74 (m, 2H), 5.09 (d, J = 7.6 Hz, 1H), 4.89 (d, J = 2.8 Hz, 1H), 3.70−3.50 (m, 3H), 3.00 (s, 3H), 2.70−2.66 (m, 1H), 2.18 (s, 3H), 2.16 (s, 3H), 1.87−1.76 (m, 1H), 1.36−1.30 (m, 1H), 1.10 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 204.0, 150.0, 147.0, 139.9, 137.1, 135.7, 135.6, 131.8, 130.0, 129.2, 128.9, 127.9, 123.8, 122.4, 118.4, 114.2, 74.9, 64.6, 55.8, 42.0, 38.3, 35.1, 28.5, 19.9, 19.5, 19.44, 19.39. HRMS (ESI): calcd for C29H33N2O4S, 519.2312 [M + H]+; found, 519.2358. Purity: 100.0% by UPLC (Rt = 3.47 min).
Separation of Stereoisomers 14a–d by Chiral HPLC.
Analytical setting: Analytical HPLC was performed by using an Agilent HPLC 1100 system and a Phenomenex Cellulose-4 column (250 × 4.6 mm, 5 μm) with MeOH as the mobile phase at 25 °C and a 1.0 mL/min flow rate (diode array detector; wavelength, 275 nm; retention times, 6.0 min for 14a, 7.5 min 14b, 9.3 min for 14c and 11.6 min for 14d). These conditions were also used for purity determination after separation.
(3aR,4S,9bR)-N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (14a).
1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 1.8 Hz, 1H), 7.48 (d, J = 8.2 Hz, 2H), 7.40 (dd, J1 = 8.6 Hz, J2 = 2.0 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.83 (d, J = 11.9 Hz, 2H), 6.75 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 1H), 6.54 (d, J = 8.6 Hz, 1H), 4.72 (s, 1H), 4.50 (d, J = 4.7 Hz, 1H), 4.10−3.95 (m, 1H), 3.90−3.80 (m, 2H), 3.60−3.46 (m, 1H), 2.41−2.33 (m, 1H), 2.14 (s, 6H), 2.09−1.97 (m, 1H), 1.74−1.64 (m, 1H), 1.22 (d, J = 6.8 Hz, 6H).
(3aR,4R,9bR)-N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (14b).
1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.0 Hz, 2H), 7.80 (s, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.48−7.36 (m, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.89 (s, 1H), 6.79 (d, J = 7.8 Hz, 1H), 6.55 (d, J = 8.5 Hz, 1H), 6.42 (s, 1H), 5.20 (d, J = 7.4 Hz, 1H), 4.86 (s, 1H), 4.33 (s, 1H), 3.73 (q, J = 8.0 Hz, 1H), 3.65 (td, J1 = 8.8 Hz, J2 = 3.5 Hz, 1H), 3.55 (dt, J1 = 13.7 Hz, J2 = 6.8 Hz, 1H), 2.80−2.76 (m, 1H), 2.19 (s, 3H), 2.18 (s, 3H), 2.02 (dd, J = 21.0, 10.3 Hz, 1H), 1.56−1.40 (m, 1H), 1.23 (d, J = 6.7 Hz, 6H).
(3aS,4R,9bS)-N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (14c).
1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.1 Hz, 2H), 7.88 (s, 1H), 7.50 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.6 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.88 (s, 1H), 6.78 (d, J = 6.7 Hz, 1H), 6.55 (d, J = 8.6 Hz, 1H), 6.50−6.25 (m, 1H), 4.59 (s, 1H), 4.53 (d, J = 4.7 Hz, 1H), 4.03 (dd, J = 14.6, 8.3 Hz, 1H), 3.95−3.81 (m, 2H), 3.55 (dt, J = 13.6, 6.8 Hz, 1H), 2.40 (s, 1H), 2.18 (s, 6H), 2.10−1.99 (m, 1H), 1.84−1.66 (m, 1H), 1.26−1.18 (m, 6H).
(3aS,4S,9bS)-N-(3,4-Dimethylphenyl)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinoline-8-sulfonamide (14d).
1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.2 Hz, 2H), 7.80 (s, 1H), 7.51 (d, J = 8.2 Hz, 2H), 7.43 (dd, J1 = 8.5 Hz, J2 = 2.0 Hz, 1H), 6.96 (d, J = 8.1 Hz, 1H), 6.88 (s, 1H), 6.80 (dd, J1 = 8.0 Hz, J2 = 1.9 Hz, 1H), 6.76 (s, 1H), 6.55 (d, J = 8.6 Hz, 1H), 5.20 (d, J = 7.4 Hz, 1H), 4.85 (d, J = 2.6 Hz, 1H), 4.43 (s, 1H), 3.72 (q, J = 8.0 Hz, 1H), 3.64 (td, J1 = 8.8 Hz, J2 = 3.5 Hz, 1H), 3.55 (dt, J1 = 13.6 Hz, J2 = 6.8 Hz, 1H), 2.75 (d, J = 8.3 Hz, 1H), 2.17 (d, J = 2.6 Hz, 6H), 2.09−1.92 (m, 1H), 1.49 (dtd, J1 = 11.8 Hz, J2 = 7.8 Hz, J3 = 3.7 Hz, 1H), 1.22 (d, J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 204.0, 147.9, 146.0, 137.5, 135.8, 134.5, 133.5, 130.1, 130.0, 128.9, 128.2, 127.9, 126.6, 123.2, 121.6, 119.2, 114.5, 74.8, 66.3, 56.1, 44.4, 35.4, 24.2, 19.8, 19.2, 19.14, 19.11.
Preparative Setting: Preparative HPLC was performed by using a Phenomenex Cellulose-4 column (250 × 10 mm, 5 μm) with methanol as the mobile phase at 25 °C and a 5 mL/min flow rate. The method was scaled up for separation of 700 mg of 14 to provide 140 mg of 14a, 125 mg of 14b, 120 mg of 14c, and 120 mg of 14d with purity >98% in all cases.
tert-Butyl (3,4-Dimethylphenyl)(((3aS,4S,9bS)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-8-yl)-sulfonyl)carbamate (Boc-14b) and tert-Butyl (3,4-Dimethylphenyl)(((3aR,4R,9bR)-4-(4-isobutyrylphenyl)-2,3,3a,4,5,9b-hexahydrofuro[3,2-c]quinolin-8-yl)sulfonyl)-carbamate (Boc-14d) by N-Boc Protection.
To a solution of compounds 14b or 14d (20.0 mg, 0.04 mmol) in CH2Cl2 (5 mL) were added DMAP (4.9 mg, 0.04 mmol), Boc2O (17.5 mg, 0.08 mmol), and TEA (10.1 mg, 0.1 mmol). The mixture was heated to reflux and stirred overnight. The reaction mixture was diluted with CH2Cl2 (10 mL) and washed with water (20 mL). The phases were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic fractions were dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, CH2Cl2/acetone = 19/1 v/v) to afford a white solid product (19.8 mg, 97.0%).
Single-Crystal X-ray Structure Determination of 14a, Boc-14b, and Boc-14d.
Crystallization and Sample Preparation: Crystals of 14a, Boc-14b, and Boc-14d were obtained by slow evaporation of an ethanol/water (1/1, v/v) solution of the corresponding compounds. The crystals were removed from the mother liquor and deposited on a glass microscope slide with a small amount of Apiezon L hydrocarbon grease (14a) or Parabar oil (Boc-14b, Boc-14d) to serve as a protectant and adhesive. The selected single crystal was affixed to a sample support and flash-cooled to the experimental temperature via the instrument’s cryogenic crystal cooler.
Data Collection:
Diffraction data for 14a were measured at the St. Jude Children’s Research Hospital X-ray Center on a Bruker X8 kappa goniometer equipped with a Photon III CMOS area detector, an IμS microfocus source producing Cu Kα radiation, multilayer mirror optics, and an Oxford Crystream 700 crystal cooler operating at 100 K. A manifold-redundant full sphere of data was collected using φ and ω scans. Programs used: Bruker Apex3,32 Bruker SAINT,33 and SADABS.34
Diffraction data for Boc-14b and Boc-14d were measured at the John Hopkins University X-ray Crystallography Facility on an Agilent SuperNova kappa goniometer equipped with an Atlas CCD area detector, a microfocus source producing Cu Kα radiation, multilayer mirror optics, and an Oxford Instruments CryoJet crystal cooler operating at 110 K. Redundant full spheres of data were collected using ω scans. Programs used: Rigaku CrysalisPro.35 Crystal parameters and data collection statistics are given in Table S1.
Structure Solution and Refinement:
The structure of 14a, including all non-hydrogen atoms, was solved with program SHELXT.36 Full-matrix least-squares refinement on F2 was performed with SHELXL-2018.37 Hydrogen atoms of the secondary amino functions were located in a difference map and freely refined as isotropic contributors, and all other hydrogen atoms were placed in calculated positions and refined according to a riding model. Approximately 17% of the crystal volume is made up of solvent-accessible channels. No satisfactory atomistic model could be constructed for the contents of these channels, so their contents were modeled via the SQUEEZE38 routine of program PLATON.39
The structures of Boc-14b and Boc-14d were solved with program SHELXS-201840 and completed with atomic positions from difference electron density maps. Full-matrix least-squares refinement on F2 was performed with SHELXL-2018.37 Hydrogen atoms of the secondary amino functions were located in a difference map and refined as isotropic contributors with the use of a N–H bond distance restraint. All other hydrogen atoms were placed in calculated positions and refined according to a riding model. Multiple geometric and thermal parameter restraints were employed in modeling the several significant crystallographic disorders in each structure. The final refinement statistics are shown in Table S1.
The crystal data of compounds 14a, Boc-14b, and Boc-14d have been uploaded to the Cambridge Crystallographic Data Centre with CCDC numbers of 2049512, 2049513, and 2049514, respectively. Data can be downloaded free of charge from CCDC via www.ccdc.cam.ac.uk or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK.; fax: (+44) 1223-336-033; or deposit@ccdc.cam.ac.uk.
Molecular Modeling.
Since there is no published MDM2 RING domain protein crystal structure, we built a consensus docking model41,42 using the published NMR structure of the MDM2 RING domain protein (PDB: 2HDP) and the Schrödinger Molecular Modeling Suite 2018 (Schrödinger LLC, New York, NY). We used the SiteMap module to identify the most like binding site for MX69 or its improved analogues. Subsequently, the MDM2 receptor grid was generated for this potential binding site using the Receptor Grid Generation. Both MX69 and compound 14 were built and prepared for docking using the LigPrep module before they were docked into the receptor. Ligand–receptor interactions, binding pose characteristics, and data analyses were carried out using the Maestro interface of Schrödinger software, similar to what we have reported previously in other projects.43–46
Biology.
Cells and Cell Culture.
This study used two ALL cell lines (EU-1 and EU-3) and three NB cell lines (NB-1643, SHEP1, and LA1–55N). All the five cancer cell lines were established from pediatric ALL or NB patients and were well characterized for their expression of MDM2 and p53 status, as reported previously.3,17,47 All cell lines were grown in a standard culture medium (RPMI 1640 containing 10% fetal bovine serum (FBS), 2 mmol/L l-glutamine, 50 U penicillin, and 50 μg/mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2.
Approved by the Emory University Institutional Review Board (IRB), human NBMM cells were obtained from donors after informed consent and were used for assays to measure the effect of compound 14 on normal hematopoiesis.
Immunoprecipitation and Western Blot Assay.
Cells were lysed in a buffer composed of 50 mM Tris, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 10 mM sodium phosphate, 10 mM NaF, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 10 μg/mL pepstatin. After centrifugation, the clarified cell lysate was separated from the pellet of cell debris and then incubated with 15 μL of protein G/protein A-agarose and 1 μg of antibodies overnight at 4 °C. For the Western blot, the resulting cell lysates or immunoprecipitates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. They were then transferred to a nitrocellulose filter and probed with the specific antibodies. Finally, proteins were visualized with a chemiluminescent detection system.
Polysome Preparation and Analysis.
Cells were incubated with 100 μg/mL of CHX for 15 min to arrest polyribosome migration and then lysed (in order to isolate cytoplasmic extracts) in a buffer containing 20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100, 500 U/mL RNAsin, and a cocktail of protease inhibitors. Fractionation was performed on a 15–45% (w/v) sucrose gradient at 39,000 rpm for 1 h (SW 41 Ti rotor). The fractions were collected by upward replacement in a fractionator (Isco, Lincoln, NE). The RNA from each fraction was subjected to qPCR.
Clonogenic Assay.
Clonogenic assays were used to determine the effect of compound 14 on the in vitro growth of NB and normal human hematopoietic cells. For the NB colony formation assay, treated cells were harvested with treatment by trypsinization, producing a single-cell suspension, and then 200 cells were seeded into a six-well plate and cultured for approximately 2 weeks. The colonies were stained with a mixture of 6.0% glutaraldehyde and 0.5% crystal violet for 30 min, then the staining mixture was carefully removed, rinsed with tap water, and the colonies were counted.
For colony formation of the normal human hematopoiesis assay, a bottom layer of low melting point, 0.5% agarose (in RPMI 1640 medium plus 10% FBS) was poured into gridded 35 mm dishes and allowed to gel. The cells were cultured at the top layer of 0.35% agarose/medium at 37 °C in a humidified atmosphere containing 5% CO2. After 2–3 weeks, the cultures were fixed with formalin and the colonies were scored.
Cytotoxicity Assay.
The cytotoxic effect of MX69 analogues including compound 14 on cancer cells was determined using the WST assay. Briefly, the cells cultured in 96-well microtiter plates were treated with different concentrations of compounds for a 44 h period. WST (25 μg/well) was then added and incubation was allowed to continue for an additional 4 h, after which optical density was read with a microplate reader (test wavelength: 450 nm; reference wavelength: 620 nm). Appropriate controls lacking cells were included to determine background absorbance.
PK Assessments.
All animal studies were performed in adherence to the NIH Principles of Laboratory Animal Care and were initiated only after prior approval by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center. Catheterized male Sprague-Dawley rats (225–250 g; Harlan Bioscience, Indianapolis, IN) were kept at a 12 h light/dark cycle with access to food and water ad libitum. Groups of five animals received either a single IV dose of 10 mg/kg of compound 14 by injection via a femoral vein catheter or a single oral dose of 25 mg/kg of compound 14 by oral gavage. The compound was formulated in dimethyl sulfoxide (10%), Tween 80 (10%), and PEG300 (80%). After drug administration, blood samples (200 μL) were collected via a jugular vein catheter at up to 10 predefined time points over 24 h. Plasma was immediately separated by centrifugation (6000g for 10 min at 4 °C) and stored at −70 °C until analysis. For quantification of compound 14 concentrations in plasma, the samples were processed by protein precipitation with four volumes of methanol and analyzed by LC–MS/MS. Chromatographic separations were carried out on a Zorbax SB-C18 3.5 μm, 150 × 4.6 mm column (Agilent Technologies, Santa Clara, CA) using a Nexera XR liquid chromatograph (Shimadzu Corporation, Columbia, MD). The mobile phase consisted of (a) water with 0.1% formic acid and (b) methanol with 0.1% formic acid and was used in the gradient mode at a flow rate of 0.5 mL/min. The eluate was led directly into an API 5500 triple quadruple mass spectrometer (Applied Biosystems, Foster City, CA) equipped with a turbospray ion source operated in the positive ion mode. Multiple reaction monitoring was performed to track the characteristic mass transitions of compound 14 (m/z 505.2/320.2) and the internal standard (2-(1H-indol-3-yl)-1H-imidazole-4-yl) (3,4,5-trimethoxyphenyl)methanone (m/z 378.4/210.0). Data were acquired and processed with Analyst software version 1.6.2 (Applied Biosystems, Foster City, CA). The obtained plasma concentration–time profiles were analyzed by standard noncompartmental PK analysis using the software package WinNonlin 8.0 (Certara, Princeton, NJ).
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by NIH grants R01CA193609, 1S10OD010678-01, and 1S10RR026377-01 to W.L., R01CA180519 to M.Z., 1R43CA246788-01 to Z.W., 1S10OD016226-01A1 to B.M., and R01CA240447 to W.L. and M.Z. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional supports were provided by the University of Tennessee College of Pharmacy Drug Discovery Center to W.L. and research grants from Rally and CURE to M.Z. and L.G. We appreciate Dr. Hao Chen for technical assistance in single-crystal growth. We also thank Dr. Lei Yang and Dr. Yong Li at St. Jude Children’s Research Hospital for the LC-MS analysis for some of the new MX69 analogues and Stacey Barnett of the UTHSC Lab Animal Care Unit for technical assistance in the PK studies.
ABBREVIATIONS
- ALL
acute leukemia lymphoma
- CHX
cycloheximide
- Dox
doxorubicin
- IAP
inhibitors of apoptosis protein
- IRES
internal ribosome entry site
- MDM2
murine double minute 2
- NB
neuroblastoma
- NBMM
normal bone marrow mononuclear cells
- RING
really interesting new gene
- SAR
structure–activity relationship
- SCXRD
single-crystal X-ray diffraction
- WST
water-soluble tetrazolium salt
- XIAP
X-linked inhibitor of apoptosis protein
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00932.
1H NMR for synthetic compounds; 1H NMR comparison between stereoisomers 14 and individual isomers 14a–d; 13C NMR of 14d; HRMS and HPLC purity for MX-69 analogues; crystal data, diffraction data, and structure refinement statistics of 14a, Boc-14b, and Boc-14d; and PK profile for 14 (PDF)
Molecular formula strings and biological data (CSV)
Docked complex formed by MX69 and the MDM2 RING domain (PDB)
Docked complex formed by compound 14d and the MDM2 RING domain (PDB)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00932
The authors declare the following competing financial interest(s): ZW, LG, MZ, and WL are listed as inventors for the new MX69 analogs reported in this paper. WL is the founder and owner of SEAK Therapeutics LLC, who has signed an exclusive optional licensing agreement with interests to license the reported compounds in this report in the future. However, SEAK Therapeutics LLC does not provide any financial support or has any influence on the work reported in this study.
Contributor Information
Zhongzhi Wu, Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Lubing Gu, Department of Pediatrics and Aflac Cancer and Blood Disorders Center, Emory University School of Medicine, Atlanta, Georgia 30322, United States.
Sicheng Zhang, Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Tao Liu, Department of Pediatrics and Aflac Cancer and Blood Disorders Center, Emory University School of Medicine, Atlanta, Georgia 30322, United States.
Pradeep B. Lukka, Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Bernd Meibohm, Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
John C. Bollinger, Department of Structural Biology, Biomolecular X-Ray Crystallography Center, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, United States
Muxiang Zhou, Department of Pediatrics and Aflac Cancer and Blood Disorders Center, Emory University School of Medicine, Atlanta, Georgia 30322, United States.
Wei Li, Department of Pediatrics and Aflac Cancer and Blood Disorders Center, Emory University School of Medicine, Atlanta, Georgia 30322, United States.
REFERENCES
- (1).Momand J; Jung D; Wilczynski S; Niland J The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bueso-Ramos C; Yang Y; deLeon E; McCown P; Stass S; Albitar M The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82, 2617–2623. [PubMed] [Google Scholar]
- (3).Zhou M; Yeager A; Smith S; Findley H Overexpression of the MDM2 gene by childhood acute lymphoblastic leukemia cells expressing the wild-type p53 gene. Blood 1995, 85, 1608–1614. [PubMed] [Google Scholar]
- (4).Carr J; Bell E; Pearson ADJ; Kees UR; Beris H; Lunec J; Tweddle DA Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006, 66, 2138–2145. [DOI] [PubMed] [Google Scholar]
- (5).Nakayama T; Toguchida J; Wadayama B-I; Kanoe H; Kotoura Y; Sasaki MS MDM2 gene amplification in bone and soft-tissue tumors: association with tumor progression in differentiated adipose-tissue tumors. Int. J. Cancer 1995, 64, 342–346. [DOI] [PubMed] [Google Scholar]
- (6).Teoh G; Urashima M; Ogata A; Chauhan D; DeCaprio JA; Treon SP; Schlossman RL; Anderson KC MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood 1997, 90, 1982–1992. [PubMed] [Google Scholar]
- (7).Zhou M; Gu L; Abshire T; Homans A; Billett A; Yeager A; Findley H Incidence and prognostic significance of MDM2 oncoprotein overexpression in relapsed childhood acute lymphoblastic leukemia. Leukemia 2000, 14, 61–67. [DOI] [PubMed] [Google Scholar]
- (8).Polsky D; Melzer K; Hazan C; Panageas KS; Busam K; Drobnjak M; Kamino H; Spira JG; Kopf AW; Houghton A; Cordon-Cardo C; Osman I HDM2 protein overexpression and prognosis in primary malignant melanoma. J. Natl. Cancer Inst 2002, 94, 1803–1806. [DOI] [PubMed] [Google Scholar]
- (9).Momand J; Zambetti GP; Olson DC; George D; Levine AJ The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- (10).Haupt Y; Maya R; Kazaz A; Oren M Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Z; Zhang R p53-independent activities of MDM2 and their relevance to cancer therapy. Curr. Cancer Drug Targets 2005, 5, 9–20. [DOI] [PubMed] [Google Scholar]
- (12).Arena G; Cissé MY; Pyrdziak S; Chatre L; Riscal R; Fuentes M; Arnold JJ; Kastner M; Gayte L; Bertrand-Gaday C; Nay K; Angebault-Prouteau C; Murray K; Chabi B; Koechlin-Ramonatxo C; Orsetti B; Vincent C; Casas F; Marine J-C; Etienne-Manneville S; Bernex F; Lombès A; Cameron CE; Dubouchaud H; Ricchetti M; Linares LK; Le Cam L Mitochondrial MDM2 regulates respiratory complex I activity independently of p53. Mol. Cell 2018, 69, 594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Todoric J; Antonucci L; Di Caro G; Li N; Wu X; Lytle NK; Dhar D; Banerjee S; Fagman JB; Browne CD; Umemura A; Valasek MA; Kessler H; Tarin D; Goggins M; Reya T; Diaz-Meco M; Moscat J; Karin M Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 2017, 32, 824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Elenbaas B; Dobbelstein M; Roth J; Shenk T; Levine AJ The MDM2 oncoprotein binds specifically to RNA through its RING finger domain. Mol. Med 1996, 2, 439–451. [PMC free article] [PubMed] [Google Scholar]
- (15).Gu L; Zhu N; Zhang H; Durden DL; Feng Y; Zhou M Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009, 15, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou S; Gu L; He J; Zhang H; Zhou M MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Mol. Cell. Biol 2011, 31, 4928–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gu L; Zhang H; He J; Li J; Huang M; Zhou M MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 2012, 31, 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fang S; Jensen JP; Ludwig RL; Vousden KH; Weissman AM Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem 2000, 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- (19).Sharp DA; Kratowicz SA; Sank MJ; George DL Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem 1999, 274, 38189–38196. [DOI] [PubMed] [Google Scholar]
- (20).Linares LK; Scheffner M The ubiquitin-protein ligase activity of Hdm2 is inhibited by nucleic acids. FEBS Lett. 2003, 554, 73–76. [DOI] [PubMed] [Google Scholar]
- (21).Liu T; Zhang H; Xiong J; Yi S; Gu L; Zhou M Inhibition of MDM2 homodimerization by XIAP IRES stabilizes MDM2, influencing cancer cell survival. Mol. Cancer 2015, 14, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schimmer AD Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [DOI] [PubMed] [Google Scholar]
- (23).Berezovskaya O; Schimmer AD; Glinskii AB; Pinilla C; Hoffman RM; Reed JC; Glinsky GV Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005, 65, 2378–2386. [DOI] [PubMed] [Google Scholar]
- (24).Tamm I; Richter S; Oltersdorf D; Creutzig U; Harbott J; Scholz F; Karawajew L; Ludwig W-D; Wuchter C High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin. Cancer Res 2004, 10, 3737–3744. [DOI] [PubMed] [Google Scholar]
- (25).Mizutani Y; Nakanishi H; Li YN; Matsubara H; Yamamoto K; Sato N; Shiraishi T; Nakamura T; Mikami K; Okihara K; Takaha N; Ukimura O; Kawauchi A; Nonomura N; Bonavida B; Miki T Overexpression of XIAP expression in renal cell carcinoma predicts a worse prognosis. Int. J. Oncol 2007, 30, 919–925. [PubMed] [Google Scholar]
- (26).Holcik M; Lefebvre C; Yeh C; Chow T; Korneluk RG A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol 1999, 1, 190–192. [DOI] [PubMed] [Google Scholar]
- (27).Lewis SM; Holcik M IRES in distress: translational regulation of the inhibitor of apoptosis proteins XIAP and HIAP2 during cell stress. Cell Death Differ. 2005, 12, 547–553. [DOI] [PubMed] [Google Scholar]
- (28).Gu L; Zhang H; Liu T; Zhou S; Du Y; Xiong J; Yi S; Qu C-K; Fu H; Zhou M Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell 2016, 30, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vassilev LT; Vu BT; Graves B; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; Fotouhi N; Liu EA In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- (30).Kostic M; Matt T; Martinez-Yamout MA; Dyson HJ; Wright PE Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J. Mol. Biol 2006, 363, 433–450. [DOI] [PubMed] [Google Scholar]
- (31).Wawrzynow B; Zylicz A; Wallace M; Hupp T; Zylicz M MDM2 chaperones the p53 tumor suppressor. J. Biol. Chem 2007, 282, 32603–32612. [DOI] [PubMed] [Google Scholar]
- (32).Apex3; Bruker Analytical X-ray Systems, Inc., 2020. [Google Scholar]
- (33).SAINT, version 8.40A; Bruker Analytical X-ray Systems, Inc., 2020. [Google Scholar]
- (34).SADABS; Bruker Analytical X-ray Systems, Inc., 2016. [Google Scholar]
- (35).CrysAlis PRO, version 1.171.39.29c; Rigaku OD, 2017. [Google Scholar]
- (36).Sheldrick GM SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sheldrick GM Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Spek AL PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr., Sect. C: Struct. Chem 2015, 71, 9–18. [DOI] [PubMed] [Google Scholar]
- (39).Spek AL PLATON: A Multipurpose Crystallographic Tool; Utrecht University: The Netherlands, 2020. [Google Scholar]
- (40).Sheldrick GM A short history of SHELX. Acta Crystallogr., Sect. A: Found. Adv 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- (41).Huang S-Y; Zou X Efficient molecular docking of NMR structures: application to HIV-1 protease. Protein Sci. 2007, 16, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Velazquez HA; Riccardi D; Xiao Z; Quarles LD; Yates CR; Baudry J; Smith JC Ensemble docking to difficult targets in early-stage drug discovery: Methodology and application to fibroblast growth factor 23. Chem. Biol. Drug Des 2018, 91, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chen H; Deng S; Wang Y; Albadari N; Kumar G; Ma D; Li W; White SW; Miller DD; Li W Structure-activity relationship study of novel 6-aryl-2-benzoyl-pyridines as tubulin polymerization inhibitors with potent antiproliferative properties. J. Med. Chem 2020, 63, 827–846. [DOI] [PubMed] [Google Scholar]
- (44).Arnst KE; Banerjee S; Wang Y; Chen H; Li Y; Yang L; Li W; Miller DD; Li W X-ray crystal structure guided discovery and antitumor efficacy of dihydroquinoxalinone as potent tubulin polymerization inhibitors. ACS Chem. Biol 2019, 14, 2810–2821. [DOI] [PubMed] [Google Scholar]
- (45).Wang Q; Arnst KE; Wang Y; Kumar G; Ma D; Chen H; Wu Z; Yang J; White SW; Miller DD; Li W Structural modification of the 3,4,5-trimethoxyphenyl moiety in the tubulin inhibitor VERU-111 leads to improved antiproliferative activities. J. Med. Chem 2018, 61, 7877–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Arnst KE; Wang Y; Hwang D-J; Xue Y; Costello T; Hamilton D; Chen Q; Yang J; Park F; Dalton JT; Miller DD; Li W A potent, metabolically stable tubulin inhibitor targets the colchicine binding site and overcomes taxane resistance. Cancer Res. 2018, 78, 265–277. [DOI] [PubMed] [Google Scholar]
- (47).He J; Gu L; Zhang H; Zhou M Crosstalk between MYCN and MDM2-p53 signal pathways regulates tumor cell growth and apoptosis in neuroblastoma. Cell Cycle 2011, 10, 2994–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.