

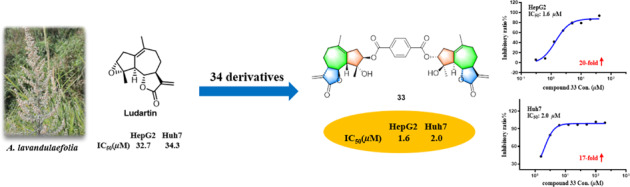
Abstract
Our previous study demonstrated that guaiane-type sesquiterpenoid ludartin showed potent antihepatoma activity against two human hepatocellular carcinoma cell lines, HepG2 and Huh7, with IC50 values of 32.7 and 34.3 μM, respectively. In this study, 34 ludartin derivatives were designed, synthesized and evaluated for their cytotoxic activities against HepG2 and Huh7 cell lines using an MTT assay in vitro. As a result, 17 compounds increased the activity against HepG2 cells, and 20 compounds enhanced the activity against Huh7 cells; 14 derivatives 2, 4-7, 9, 11, 17, 24, 28-30 and 32-33 were superior to ludartin on both HepG2 and Huh7 cells. In particular, dimeric derivative 33 as the most active compound showed 20-fold and 17-fold enhancement of cytotoxicity against HepG2 and Huh7 cells compared to that of ludartin. These results suggested that compound 33 could serve as a promising lead compound against liver cancer.
Graphical abstract
Keywords: Ludartin derivatives, Antihepatoma activity, HepG2 cells, Huh7 cells
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths in the world, with an annual mortality over 4.2 million [1]. HCC can be caused by a variety of factors, including fibrosis and cirrhosis, chronic hepatitis B and C virus infection, aflatoxin infection and alcoholism [2]. The main drugs currently available for the treatment of HCC include the multikinase inhibitors sorafenib, regorafenib, lenvatinib, cabozantinib, and ramucirumab and the immune checkpoint inhibitors nivolumab and pembrolizumab. Recently, combination strategies have become a novel and effective strategy for the treatment of liver cancer (i.e., atezolizumab and bevacizumab) [3]. Icaritin, a prenylated flavonol found in the Epimedium genus, has been approved by the National Medical Products Administration for the treatment of HCC in China [4]. Despite the huge advances have been made in targeted therapy and immunotherapy, the 5-year relative survival rate of patients with HCC for all stages is still lower than 15% [5]. In this context, developing new antihepatoma active molecules with novel structures and different mechanisms of action is highly desirable.
Natural products play a very important role in the discovery of antitumor drugs [6]. Guaianolides, one of the largest subgroups of naturally occurring sesquiterpenoids consisting of a tricyclic 5,7,5-ring system, exhibit significant antitumor activities and have been widely investigated for their potential in the treatment of cancers [7]. Among them, the hydrochloride salt of dimethylamino-arglabin has been developed as an antitumor agent for the treatment of breast, colon, ovarian and lung cancer in Kazakhstan [8]. The fumaric acid salt of dimethylaminomicheliolide [9] and thapsigargin prodrug G-202 [10] are undergoing clinical evaluation for the treatment of malignant tumors.
As one of our ongoing programs to discover antiheptoma sesquiterpenoids from Artemisia species, our previous bioactivity-guided fractionation of A. atrovirens and A. myriantha led to the isolation of 96 antihepatoma sesquiterpenoids, including 67 guaianolides, 15 germacranolides, eight eudesmanolides and two ent-longipinane-type sesquiterpenoids, one xanthanolide and three other sesquiterpenoids [11–17]. Cytotoxicity studies indicated that guaianolide dimers and mono sesquiterpenoids containing α-methylene-γ-lactones displayed cytotoxicity against HepG2, Huh7, and SMMC-7721 cell lines. Seven compounds exhibited strong cytotoxicity against all three tested cancer cell lines with IC50 values ranging from 3.8 to 9.6 μM being more potent than sorafenib. Especially, lavandiolide H could induce G2/M cell cycle arrest and cause HepG2 cell apoptosis by regulating the expression of Bcl-2 and PARP-1 [11]. Artematrolide A exhibited significant anti-cervical cancer effects via the ROS/ERK/mTOR pathway and a metabolic shift on HeLa S3 and SiHa cells [18]. The synthesis of lavandiolides H, I, and K and artematrolide F via a biomimetic Diels−Alder reaction was accomplished in our laboratory [19]. Ludartin, a 6,12-guaianolide isolated from A. lavandulaefolia [20], exhibited moderate cytotoxicity against HepG2 and Huh7 cell lines with IC50 values of 32.7 and 34.3 μM, respectively. Ludartin was first isolated from A. carruthii [21], which was also found in Stevia yaconensi [22] and other Artemisia species [23–25]. The reported investigations have demonstrated that ludartin possesses promising bioactivities, including gastric cytoprotective effects [26], inhibitory action on aromatase [27], activity on TRP ion channels [28], and antiproliferative activity against human tumor cell lines. For instance, ludartin was found to exhibit significant cytotoxicity against T98G, A-549, THP-1, PC-3, HCT-116 and MCF-7 cells in vitro with IC50 values in the range of 0.5–7.5 μM [28, 29]. The anticancer effects of ludartin were associated with the induction of DNA damage and a reduction of mitochondrial membrane potential in MCF-7 cells [30]. Administration of ludartin separately or coadministration with capecitabine effectively inhibited colon tumor growth and angiogenesis in mice [31]. Reported structure–activity relationship (SAR) studies on ludartin involved the synthesis of amino [29] and triazolyl [32] analogs, which indicated that the α, β-unsaturated ketone moiety was a key pharmacophore. Although various pharmacological properties of ludartin have been reported, no investigation of chemically modified ludartin derivatives has been conducted for their antihepatoma effects. In this research, 34 ludartin derivatives were synthesized and evaluated for their cytotoxicity against HepG2 and Huh7 cell lines. The preliminary structure–activity relationships of the synthetic derivatives were also discussed.
Results and discussion
Chemistry
Structurally, ludartin bears a 5,7,5-tricyclic ring system and is characterized by an epoxide on the cyclopentane as well as an exocyclic methylene group conjugated with the carbonyl of the lactone. The synthesis of ludartin derivatives is described in Schemes 1–7. A series of derivatives 1–6 were obtained by epoxide ring opening with different nucleophiles including hydroxide ion, methoxide, formate, fluoride and chloride anions. The hydrolysis of ludartin using a 6% solution of HClO4 in 1,2-dimethoxyethane delivered diol 1 in 87% yield, and switching the solvent to DMF predominantly formed formylated product 2. Treatment of ludartin with 0.1 M H2SO4 in MeOH at room temperature gave methylated compound 3, the addition of methanol selectively took place at the C-4 position. Chlorinated compounds 4-5 were obtained by the reaction of ludartin with TMSCl, while the treatment of ludartin with HF·Py in CH2Cl2 gave fluorinated derivative 6. (Scheme 1)
Scheme 1.
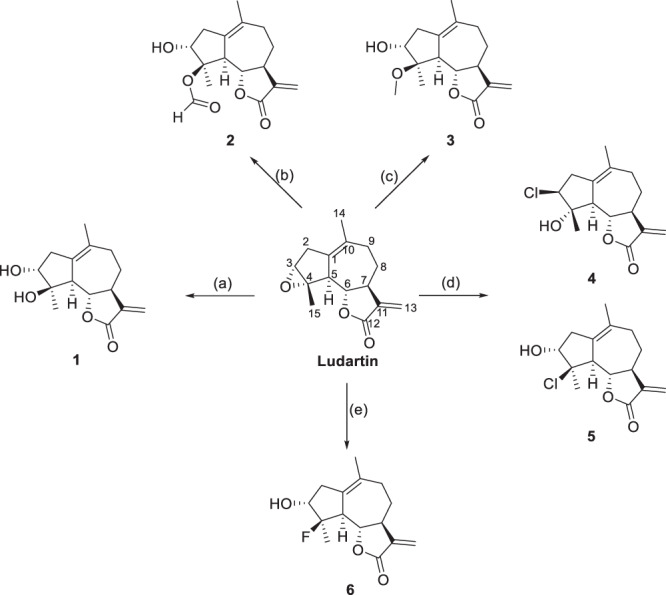
Reagents and conditions: a 6% HClO4, DME, r.t., 87%, b HClO4, DMF, r.t., 76%, c H2SO4, MeOH, r.t., 67%, d TMSCl, CH2Cl2, r.t., 4 (21%), 5 (42%), e HF·Py, CH2Cl2, r.t., 78%
Scheme 7.

Reagents and conditions: EDC, DMAP, appropriate carboxylic acid, CH2Cl2, r.t., 33 (15%), 34 (48%)
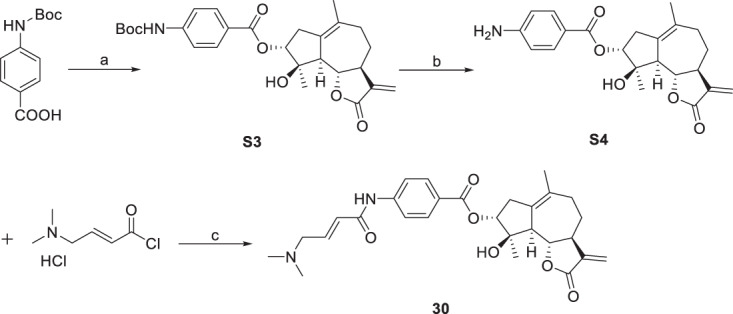
The esterification of sesquiterpenoids with carboxylic acid was proven to be an effective way to enhance their anticancer activity [33]. Thus, compounds 1–3 and 5, 6 were esterified with benzoic acid to obtain derivatives 7–11 (Scheme 2). Since the benzoylation product derived from compound 1 showed the most promising activity against HepG2 cells among compounds 7–11, compound 1 was selected for further 3-O-derivatization studies. The treatment of compound 1 with different carboxylic acids in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and 4-dimethylaminopyridine (DMAP) gave compounds 14–27 (Scheme 3). Alkyl-, aryl-, and heterocycle substituted acyl groups were introduced into the C3-position of compound 1, which were evaluated for their cytotoxicity to explore the influence of the ester side chain. It has been demonstrated that the incorporation of an additional Michael receptor into a certain molecule is an effective strategy for enhancing its biological activities [34]. Thus, compounds 28 and 29 containing acrylamide moieties were prepared from compound 1, acryloyl chloride and 4-aminobenzoic acid or 3-aminobenzoic acid in two steps (Scheme 4). To clarify the impact of the substituent at the acrylamide moieties on the activity, compound 30 with β-dimethylaminomethyl substituents was then prepared from 4-(Boc-amino) benzoic acid and (2E)-4-(dimethylamino)but-2-enoyl chloride hydrochloride in three steps (Scheme 5).
Scheme 2.
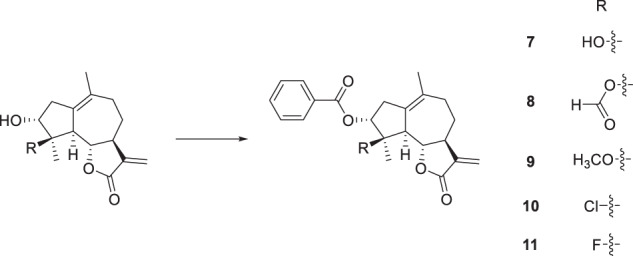
Reagents and conditions: EDC, DMAP, benzoic acid, appropriate ludartin derivative, CH2Cl2, r.t. or reflux, 7 (83%), 8 (76%), 9 (71%), 10 (55%), 11 (83%)
Scheme 3.

Reagents and conditions: HCOOH, r.t. 12 (64%); Ac2O, DMAP, CH2Cl2, r.t., 13 (82%); EDC, DMAP, CH2Cl2, r.t., 14 (24%), 15 (77%), 16 (87%), 17 (52%), 18 (38%), 19 (42%), 20 (41%), 21 (32%), 22 (90%), 23 (71%), 24 (51%), 25 (95%), 26 (87%), 27 (89%)
Scheme 4.

Reagents and conditions: a K2CO3, CH2Cl2, r.t., S1 (67%), S2 (68%), b compound 1, DCC, DMAP, CH2Cl2, r.t., 28 (56%), 29 (88%)
Scheme 5.
Reagents and conditions: a compound 1, DCC, DMAP, CH2Cl2, r.t., 54%, b CF3COOH, CH2Cl2, r.t., 92% (c) Et3N, CH2Cl2, r.t., 51%
Hybridization of natural products with known anticancer agents is an efficient strategy to achieve enhanced activities due to multitargeting features and synergistic effects between the prototype molecules and other anticancer pharmacophores [35, 36]. Thus, a hybrid of ludartin derivative and 5-fluorouracil was synthesized by reaction of compound 1 with 5-fluorouracil-1-yl acetic acid. Alkylation of 31 with benzyl bromide delivered conjugate 32 (Scheme 6). Inspired by the discovery that dimers of sesquiterpenoids such as artemisinin [36] and parthenolide [37] showed elevated anticancer activity, we designed and synthesized two dimers linked by ester bonds. Coupling of 1 with terephthalic acid or succinic acid afforded dimers 33, 34 (Scheme 7).
Scheme 6.

Reagents and conditions: a EDC, DMAP, CH2Cl2, r.t., 77%; b BnBr, K2CO3, DMF, 73%
Biology
The structure and purity of the synthetic compounds (higher than 95%) were demonstrated by 1H and 13C NMR spectroscopic data, and HRESIMS. All the derivatives were assessed for their cytotoxicity against the proliferation of the human hepatocellular carcinoma cell lines HepG2 and Huh7 using the MTT method with sorafenib as a positive control. As shown in Table 1, 17 compounds (2, 4–7, 9, 11–12, 16–17, 22, 24, 28–30, and 32–33) increased the activity against HepG2 cells, and 20 compounds (2, 4–7, 9, 11, 14, 17, 20, 21, 24–30 and 32, 33) enhanced the activity against Huh7 cells; 14 derivatives were superior to ludartin on both HepG2 and Huh7 cells. Fluorine, chlorine, and formate ester substituted derivatives 2, 4–6 displayed higher activity than ludartin with IC50 values ranging from 10.9 to 26.0 μM, and methoxylation product 3 showed similar cytotoxicity with ludartin. Compounds 7 and 9 enhanced the activity after esterification (1 vs 7, 3 vs 9); among them, compound 7 was 5-fold and 2-fold more potent than ludartin against HepG2 and Huh7 cells with IC50 values of 5.9 μM and 16.2 μM, respectively.
Table 1.
Cytotoxicity of compounds 1–34 against HepG2 and Huh7 cells
| compound | IC50 ± SD (µM)a | compound | IC50 ± SD (µM)a | ||
|---|---|---|---|---|---|
| HepG2 | Huh7 | HepG2 | Huh7 | ||
| ludartin | 32.7 ± 2.4 | 34.3 ± 2.1 | 18 | 43.1 ± 1.8 | 38.0 ± 3.4 |
| 1 | 53.7 ± 0.9 | 49.6 ± 1.6 | 19 | 49.5 ± 0.1 | 30.2 ± 0.5 |
| 2 | 19.9 ± 3.2 | 26.0 ± 0.4 | 20 | 32.7 ± 1.9 | 17.3 ± 0.4 |
| 3 | 29.7 ± 0.8 | 29.1 ± 2.3 | 21 | 26.9 ± 1.6 | 8.1 ± 0.3 |
| 4 | 14.9 ± 1.5 | 16.8 ± 1.9 | 22 | 10.3 ± 1.1 | 28.3 ± 1.5 |
| 5 | 13.1 ± 0.7 | 20.0 ± 1.0 | 23 | 25.0 ± 3.2 | 36.9 ± 0.8 |
| 6 | 14.2 ± 0.8 | 10.9 ± 0.8 | 24 | 10.3 ± 0.4 | 14.5 ± 0.3 |
| 7 | 5.9 ± 0.4 | 16.2 ± 0.2 | 25 | 58.7 ± 0.6 | 22.9 ± 0.8 |
| 8 | 25.5 ± 0.5 | 29.6 ± 1.9 | 26 | 115.2 ± 7.1 | 23.1 ± 0.5 |
| 9 | 11.1 ± 0.5 | 23.3 ± 0.9 | 27 | 32.9 ± 1.6 | 21.4 ± 0.9 |
| 10 | 27.4 ± 2.5 | 31.2 ± 0.3 | 28 | 7.9 ± 0.3 | 8.5 ± 0.2 |
| 11 | 10.0 ± 0.5 | 20.2 ± 0.4 | 29 | 9.6 ± 0.4 | 16.6 ± 0.2 |
| 12 | 23.3 ± 1.5 | 27.9 ± 0.5 | 30 | 5.3 ± 0.1 | 16.0 ± 0.3 |
| 13 | 61.4 ± 2.8 | 61.7 ± 2.7 | 31 | >100 | 78.9 ± 1.9 |
| 14 | 32.1 ± 0.2 | 22.6 ± 1.4 | 32 | 11.6 ± 1.0 | 7.9 ± 0.1 |
| 15 | 33.6 ± 1.2 | 37.3 ± 0.8 | 33 | 1.6 ± 0.6 | 2.0 ± 0.4 |
| 16 | 15.5 ± 1.1 | 33.3 ± 4.4 | 34 | 30.1 ± 0.6 | 35.0 ± 1.3 |
| 17 | 21.6 ± 1.4 | 25.1 ± 0.3 | sorafenibb | 8.2 ± 0.9 | 10.4 ± 0.7 |
aData are expressed as the means ± SD (n = 3)
bSorafenib was used as the positive control
Among aliphatic derivatives, compound 16 with a cyclopentyl moiety exhibited the most potent cytotoxicity against HepG2 cells with an IC50 value of 15.5 μM. For HepG2 cells, the introduction of substituents on the phenyl ring of the ester moiety was detrimental to activity as compared to compound 7. For the trifluoromethyl-substituted compounds, para-substituted derivative 20 was more active than ortho- and meta-substituted derivatives (18 and 19). Pentafluorobenzoyl analog 21 showed a 4-fold increase in cytotoxicity against Huh7 with an IC50 value of 8.1 μM, indicating that a polyfluorinated substituent was favorable. When the benzoyl group (7) was replaced by a cinnamoyl group, the anticancer activity decreased. Further replacing the phenyl moiety of compound 7 with a different heterocyclic ring (24–26) led to a decrease in the antiproliferative efficacy. Compounds 28 and 29 bearing acrylamide structural moieties were 2 to 4 fold more active than ludartin with IC50 values of 7.9 and 9.6 μM (HepG2), 8.5 and 16.6 μM (Huh7), suggesting that the incorporation of an additional unsaturated carbonyl was favorable.
Further modifications based on 28 was performed by introducing an aminomethyl substituent at the β–position of acrylamide afford compound 30. For HepG2 cells, compound 30 exhibited 1.5-fold more potent cytotoxicity than compound 28 with an IC50 value of 5.3 μM, indicating that a β-dimethylaminomethyl (DMAM) substituent on acrylamide was beneficial for promoting cytotoxicity. Compound 27, also containing a dimethylamino group with a fumaric acid linker only showed moderate cytotoxicity. The ludartin-5-fluorouracil hybrids 31 and 32 demonstrated different anticancer activities, and the activity of hybrid 32 (11.6 and 7.9 μM) was more potent than that of 31 (>100 and 78.9 μM). These results suggest that the substituent on the 3-N-5-FU moiety was important for anti-proliferative activity.
For dimeric products, compound 33 with a terephthalic acid linker displayed the most potent activity against both HepG2 and Huh7 cells with IC50 values of 1.6 and 2.0 μM, which was 20-fold and 17-fold more potent than ludartin and was 5-fold and 5-fold higher than the positive control sorafenib. Compound 34 with a succinic acid linker was significantly less potent (30.1 and 35.0 μM), suggesting that the linkers were critical for the cytotoxicity of the dimers.
From the above structure and activity results, preliminary SARs can be drawn: the acyloxys at the C-3 position had a different effect on the activity, and dimeric derivative with a terephthalic acid linker could dramatically increase the activity.
Conclusions
In summary, 34 derivatives of ludartin were synthesized and evaluated for their anti-HCC activity against HepG2 and Huh7 cells. Seventeen derivatives showed higher activities against HepG2 cells with IC50 values superior to ludartin, and 20 compounds increased cytotoxicity against Huh7 cells. The most active compound 33 demonstrated 20-fold and 17-fold improvement compared to ludartin, and was 5-fold and 5-fold more potent than the clinically used anticancer drug sorafenib. These results provide a new insight for the design of ludartin derivatives to enhance the efficacy of candidates.
Experimental
Chemistry
General
All reagents and solvents were obtained from commercial suppliers and used without further purification. All compounds were purified by silica gel (200–300 mesh, Qingdao Makall Group Co., Ltd., Qingdao, China). 1H NMR and 13C NMR spectra were obtained on a 400 or 600 MHz spectrometer (Bruker, Bremerhaven, Germany) with tetramethylsilane as an internal standard. HRMS was measured by a Shimadzu LC/MS-IT-TOF (Shimadzu, Kyoto, Japan). Melting points were obtained on an SGW® X-4B microscopic melting point apparatus (Shanghai Precision & Scientific Instrument Co., Ltd., Shanghai, China). Optical rotations were measured in MeOH, CDCl3 or acetone with an Autopol VI (Serial #91058) polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). The human hepatocellular carcinoma cell lines HepG2 and Huh7 were purchased from Shanghai Jining Biotechnology Co., Ltd. (Shanghai, China) and were cultured in Dulbecco′s Modified Eagle′s Medium (DMEM; Gibco, Thermo Fisher Scientific Co., Ltd., Suzhou, China) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Life Technologies, NY, USA).
Synthesis
3α,4β-dihydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (1) To a stirred solution of ludartin (1 g, 4.07 mmol) in 1,2-dimethoxyethane (DME, 50 mL) was added perchloric acid (25 mL, 6% in water) at room temperature. The reaction mixture was stirred for 3 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) to yield compound 1 (934 mg, 87% yield) as a white powder. mp 140~142 °C; − 0.4 (c 0.093, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.28-2.14 (2H, m, H-2), 3.84 (1H, d, J = 3.8 Hz, H-3), 2.83-2.79 (1H, m, H-5), 3.94 (1H, t, J = 10.2 Hz, H-6), 2.35 (1H, d, J = 17.0 Hz, H-7), 1.87 (1H, br s, H-8), 1.38-1.29 (1H, m, H-8), 2.10-2.15 (2H, m, H-9), 6.16 (1H, d, J = 3.1 Hz, H-13), 5.44 (1H, d, J = 2.8 Hz, H-13), 1.73 (3H, s, H-14), 1.57 (3H, s, H-15); 13C NMR (100 MHz, CDCl3) δ 132.2 (C, C-1), 39.3 (CH2, C-2), 82.9 (CH, C-3), 83.1 (C, C-4), 54.1 (CH, C-5), 79.0 (CH, C-6), 51.5 (CH, C-7), 25.8 (CH2, C-8), 34.4 (CH2, C-9), 134.5 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 118.8 (CH2, C-13), 24.1 (CH3, C-14), 23.9 (CH3, C-15). HRESIMS calcd for C15H21O4 [M + H]+ 265.1434, found 265.1445.
3α-hydroxy-4β-O-formyl-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (2) To a stirred solution of ludartin (25 mg, 0.1 mmol) in DMF (1 mL) two drops of HClO4 (70%) was added at room temperature. The reaction mixture was stirred for 30 min. Then, it was quenched with saturated NaHCO3 aqueous solution and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) compound 2 (24 mg, 76% yield) as a white powder. mp 199~201 °C; + 25.2 (c 0.104, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.97-2.93 (1H, m, H-2), 2.75-2.67 (1H, m, H-2), 4.64 (1H, d, J = 4.4 Hz, H-3), 2.39 (1H, d, J = 17.0 Hz, H-5), 4.04 (1H, t, J = 10.1 Hz, H-6), 2.11-2.06 (1H, m, H-7), 2.34-2.30 (1H, m, H-8), 1.41-1.31 (1H, m, H-8), 2.27-2.20 (2H, m, H-9), 6.17 (1H, d, J = 3.2 Hz, H-13), 5.46 (1H, d, J = 2.9 Hz, H-13), 1.74 (3H, s, H-14), 1.90 (3H, s, H-15), 8.00 (1H, s, H-16); 13C NMR (100 MHz, CDCl3) δ 131.6 (C, C-1), 38.9 (CH2, C-2), 74.0 (CH, C-3), 93.2 (C, C-4), 55.4 (CH, C-5), 81.7 (CH, C-6), 51.0 (CH, C-7), 25.7 (CH2, C-8), 34.8 (CH2, C-9), 132.7 (C, C-10), 139.2 (C, C-11), 170.1 (C, C-12), 118.8 (CH2, C-13), 24.7 (CH3, C-14), 19.4 (CH3, C-15), 160.0 (CH, C-16). HRESIMS calcd for C16H20O5Na [M + Na]+ 315.1203, found 315.1224.
3α-hydroxy-4β-methoxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (3) To a stirred solution of 0.1 M H2SO4 in MeOH (2 mL) was added ludartin (100 mg, 0.4 mmol) at room temperature. The reaction mixture was stirred for 10 min. Then, it was quenched with saturated NaHCO3 aqueous solution and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 15:85) to yield compound 3 (74 mg, 67% yield) as a white powder. mp 162~164 °C; − 4.5 (c 0.112, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.84-2.74 (2H, m, H-2), 4.12 (1H, d, J = 4.0 Hz, H-3), 2.30 (1H, d, J = 16.3 Hz, H-5), 4.04 (1H, t, J = 10.1 Hz, H-6), 2.08-2.02 (1H, m, H-7), 2.71-2.64 (1H, m, H-8), 1.41-1.31 (1H, m, H-8), 2.25-2.18 (2H, m, H-9), 6.15 (1H, d, J = 3.2 Hz, H-13), 5.42 (1H, d, J = 2.9 Hz, H-13), 1.73 (3H, s, H-14), 1.58 (3H, s, H-15), 3.26 (3H, s, H-16); 13C NMR (100 MHz, CDCl3) δ 131.6 (C, C-1), 40.0 (CH2, C-2), 82.3 (CH, C-3), 86.7 (C, C-4), 55.5 (CH, C-5), 73.9 (CH, C-6), 50.9 (CH, C-7), 25.8 (CH2, C-8), 35.0 (CH2, C-9), 133.0 (C, C-10), 139.9 (C, C-11), 170.8 (C, C-12), 118.3 (CH2, C-13), 24.8 (CH3, C-14), 17.1 (CH3, C-15), 50.3 (CH3, C-16). HRESIMS calcd for C16H23O4 [M + H]+ 279.1591, found 279.1596.
3β-chloro-4α-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (4) and 3α-hydroxy-4β-chloro-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (5) To a stirred solution of ludartin (200 mg, 0.8 mmol) in CH2Cl2 (5 mL) was added trimethylchlorosilane (TMSCl, 510 µL, 4 mmol) in portions at 0 °C under argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred overnight before quenching with saturated NaHCO3 aqueous solution. Then, it was extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 5:95) to yield compounds 4 (47 mg, 21% yield) and 5 (95 mg, 42% yield) as a white powder.
Compound 4: mp 147~149 °C; + 41.1 (c 0.107, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.92-2.86 (1H, m, H-2), 2.42-2.33 (1H, m, H-2), 4.16 (1H, dd, J = 11.6 Hz, J = 8.2 Hz, H-3), 2.79-2.77 (1H, m, H-5), 3.86 (1H, t, J = 10.2 Hz, H-6), 2.13-2.09 (1H, m, H-7), 2.30-2.26 (2H, m, H-8), 2.70-2.64 (2H, m, H-9), 6.24 (1H, d, J = 3.3 Hz, H-13), 5.53 (1H, d, J = 2.9 Hz, H-13), 1.70 (3H, s, H-14), 1.37 (3H, s, H-15); 13C NMR (100 MHz, CDCl3) δ 133.0 (C, C-1), 39.6 (CH2, C-2), 63.9 (CH, C-3), 80.7 (C, C-4), 57.0 (CH, C-5), 83.7 (CH, C-6), 49.7 (CH, C-7), 25.6 (CH2, C-8), 35.0 (CH2, C-9), 125.8 (C, C-10), 138.3 (C, C-11), 169.4 (C, C-12), 120.0 (CH2, C-13), 24.1 (CH3, C-14), 17.5 (CH3, C-15). HRESIMS calcd for C15H19O3ClNa [M + Na]+ 305.0915, found 305.0923;
Compound 5: mp 189~191 °C; + 8.1 (c 0.111, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.05-3.02 (2H, m, H-2), 4.16 (1H, d, J = 4.0 Hz, H-3), 2.38 (1H, d, J = 17.8 Hz, H-5), 4.14-4.09 (1H, m, H-6), 2.77-2.71 (1H, m, H-7), 2.30-2.27 (1H, m, H-8), 1.46-1.37 (1H, m, H-8), 2.13-2.07 (2H, m, H-9), 6.20 (1H, d, J = 3.3 Hz, H-13), 5.48 (1H, d, J = 3.0 Hz, H-13), 1.76 (3H, s, H-14), 1.92 (3H, s, H-15); 13C NMR (100 MHz, CDCl3) δ 132.8 (C, C-1), 39.9 (CH2, C-2), 80.1 (CH, C-3), 80.5 (C, C-4), 50.0 (CH, C-5), 83.6 (CH, C-6), 55.5 (CH, C-7), 25.7 (CH2, C-8), 35.0 (CH2, C-9), 131.1 (C, C-10), 139.2 (C, C-11), 170.1 (C, C-12), 119.0 (CH2, C-13), 25.2 (CH3, C-14), 25.0 (CH3, C-15). HRESIMS calcd for C15H20O3Cl [M + H]+ 283.1095, found 283.1113.
3α-hydroxy-4β-fluoro-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (6) To a stirred solution of ludartin (50 mg, 0.2 mmol) in CH2Cl2 (2 mL) was added HF·Py (27 μL, 0.3 mmol) at room temperature. The reaction mixture was stirred for 1 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 15:85) to yield compound 6 (42 mg, 78% yield) as a white powder. mp 190~192 °C; − 14.7 (c 0.109, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.79-2.70 (2H, m, H-2), 4.01-3.98 (1H, m, H-3), 2.39 (1H, d, J = 16.9 Hz, H-5), 3.92 (1H, t, J = 10.2 Hz, H-6), 2.24-2.18 (1H, m, H-7), 2.32-2.25 (1H, m, H-8), 1.40-1.30 (1H, m, H-8), 2.95-2.86 (1H, m, H-9), 2.10-2.05 (1H, m, H-9), 6.15 (1H, d, J = 3.1 Hz, H-13), 5.43 (1H, d, J = 2.8 Hz, H-13), 1.75 (6H, s, H-14, H-15); 13C NMR (100 MHz, CDCl3) δ 132.6 (C, C-1), 39.3 (CH2, C-2), 81.4 (CH, C-3), 81.5 (C, C-4), 51.7 (CH, C-5), 77.0 (CH, C-6), 53.3 (CH, C-7), 25.7 (CH2, C-8), 34.8 (CH2, C-9), 133.1 (C, C-10), 139.5 (C, C-11), 170.2 (C, C-12), 118.6 (CH2, C-13), 24.6 (CH3, C-14), 20.2 (CH3, C-15). HRESIMS calcd for C15H20O3F [M + H]+ 267.1391, found 267.1408.
3α-O-benzoyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (7) To a stirred solution of benzoic acid (73 mg, 0.6 mmol) in CH2Cl2 (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 105 μL, 0.6 mmol), compound 1 (53 mg, 0.2 mmol) and 4-dimethylaminopyridine (DMAP, 5 mg, 0.04 mmol) at room temperature. Then the reaction mixture was equipped with a reflux condenser, and heated to reflux for 10 h. After cooling to room temperature, the mixture was diluted with water and extracted with ethyl acetate (3 × 5 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 7 (61 mg, 83% yield) as a white powder. mp 88~90 °C; + 98.8 (c 0.016, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.01-2.91 (2H, m, H-2), 5.14 (1H, d, J = 4.6 Hz, H-3), 2.53 (1H, d, J = 17.2 Hz, H-5), 4.06 (1H, t, J = 10.1 Hz, H-6), 2.12-2.06 (1H, m, H-7), 2.34-2.26 (1H, m, H-8), 1.43-1.33 (1H, m, H-8), 2.84-2.77 (1H, m, H-9), 2.24-2.17 (1H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.47 (1H, d, J = 2.9 Hz, H-13), 1.70 (3H, s, H-14), 1.63 (3H, s, H-15), 8.00-7.97 (2H, m, H-3ʹ, H-7ʹ), 7.45-7.42 (2H, m, H-4ʹ, H-6ʹ), 7.57-7.54 (1H, m, H-5ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.4 (CH2, C-2), 81.6 (CH, C-3), 82.7 (C, C-4), 55.3 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.2 (C, C-10), 139.3 (C, C-11), 170.1 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 24.3 (CH3, C-15), 165.8 (C, C-1ʹ), 130.4 (C, C-2ʹ), 129.7 (CH, C-3ʹ, C-7ʹ), 128.6 (CH, C-4ʹ, C-6ʹ), 133.4 (CH, C-5ʹ). HRESIMS calcd for C22H25O5 [M + H]+ 369.1697, found 369.1701.
3α-O-benzoyl-4β-O-formyl-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (8) To a stirred solution of benzoic acid (24 mg, 0.2 mmol) in CH2Cl2 (1 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 35 μL, 0.2 mmol), compound 2 (29 mg, 0.1 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred for 4 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 8 (30 mg, 76% yield) as a white powder. mp 50~52 °C; − 50.7 (c 0.108, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.86-2.76 (2H, m, H-2), 5.88 (1H, d, J = 4.7 Hz, H-3), 2.61 (1H, d, J = 18.0 Hz, H-5), 4.13 (1H, t, J = 10.1 Hz, H-6), 3.07-3.04 (1H, m, H-7), 2.17-2.11 (1H, m, H-8), 1.46-1.36 (1H, m, H-8), 2.35-2.24 (2H, m, H-9), 6.21 (1H, d, J = 3.2 Hz, H-13), 5.50 (1H, d, J = 2.9 Hz, H-13), 1.71 (3H, s, H-14), 1.97 (3H, s, H-15), 7.99-7.97 (2H, m, H-3ʹ, H-7ʹ), 7.48-7.43 (2H, m, H-4ʹ, H-6ʹ), 7.59-7.55 (1H, m, H-5ʹ), 8.07 (1H, s, -CHO); 13C NMR (100 MHz, CDCl3) δ 130.6 (C, C-1), 37.2 (CH2, C-2), 76.8 (CH, C-3), 91.9 (C, C-4), 56.7 (CH, C-5), 81.6 (CH, C-6), 50.7 (CH, C-7), 25.8 (CH2, C-8), 35.0 (CH2, C-9), 133.1 (C, C-10), 139.1 (C, C-11), 170.0 (C, C-12), 119.2 (CH2, C-13), 24.8 (CH3, C-14), 20.0 (CH3, C-15), 165.5 (C, C-1ʹ), 130.0 (C, C-2ʹ), 129.7 (CH, C-3ʹ, C-7ʹ), 128.6 (CH, C-4ʹ, C-6ʹ), 133.4 (CH, C-5ʹ), 159.6 (CH, -CHO). HRESIMS calcd for C23H24O6Na [M + Na]+ 419.1465, found 419.1472.
3α-O-benzoyl-4β-methoxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (9) To a stirred solution of benzoic acid (12 mg, 0.1 mmol) in CH2Cl2 (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 18 μL, 0.1 mmol), compound 3 (15 mg, 0.05 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred for 10 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 9 (13 mg, 71% yield) as a white powder. mp 138~140 °C; − 85.1 (c 0.079, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2,95-2.89 (2H, m, H-2), 5.42 (1H, d, J = 4.9 Hz, H-3), 2.48 (1H, d, J = 17.6 Hz, H-5), 4.13 (1H, t, J = 10.1 Hz, H-6), 2.11-2.05 (1H, m, H-7), 2.76-2.70 (1H, m, H-8), 1.45-1.35 (1H, m, H-8), 2.34-2.22 (2H, m, H-9), 6.19 (1H, d, J = 3.2 Hz, H-13), 5.46(1H, d, J = 2.9 Hz, H-13), 1.70 (3H, s, H-14), 1.59 (3H, s, H-15), 8.01-7.99 (2H, m, H-3ʹ, H-7ʹ), 7.47-7.43 (2H, m, H-4ʹ, H-6ʹ), 7.59-7.56 (1H, m, H-5ʹ), 3.34 (3H, -OMe); 13C NMR (100 MHz, CDCl3) δ 131.7 (C, C-1), 38.0 (CH2, C-2), 82.1 (CH, C-3), 86.2 (C, C-4), 56.9 (CH, C-5), 76.7 (CH, C-6), 50.5 (CH, C-7), 25.9 (CH2, C-8), 35.2 (CH2, C-9), 132.0 (C, C-10), 139.8 (C, C-11), 170.6 (C, C-12), 118.6 (CH2, C-13), 24.9 (CH3, C-14), 17.7 (CH3, C-15), 166.2 (C, C-1ʹ), 130.3 (C, C-2ʹ), 129.7 (CH, C-3ʹ, C-7ʹ), 128.6 (CH, C-4ʹ, C-6ʹ), 133.3 (CH, C-5ʹ), 50.6 (CH3, -OMe). HRESIMS calcd for C23H26O5Na [M + Na]+ 405.1672, found 405.1679.
3α-O-benzoyl-4β-chloro-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (10) To a stirred solution of benzoic acid (49 mg, 0.4 mmol) in CH2Cl2 (5 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 71 μL, 0.4 mmol), compound 5 (42 mg, 0.15 mmol) and 4-dimethylaminopyridine (DMAP, 5 mg, 0.04 mmol) at room temperature. The reaction mixture was stirred for 9 h. The mixture was diluted with water and extracted with ethyl acetate (3 × 5 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) to yield compound 10 (32 mg, 55% yield) as a white powder. mp 72~74 °C; − 44.2 (c 0.110, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.22-3.11 (2H, m, H-2), 5.44 (1H, d, J = 4.8 Hz, H-3), 2.54 (1H, d, J = 17.7 Hz, H-5), 4.18 (1H, t, J = 9.8 Hz, H-6), 2.83-2.76 (1H, m, H-7), 2.17-2.10 (1H, m, H-8), 1.51-1.40 (1H, m, H-8), 2.38-2.26 (2H, m, H-9), 6.23 (1H, d, J = 3.2 Hz, H-13), 5.51 (1H, d, J = 2.9 Hz, H-13), 1.94 (3H, s, H-14), 1.73 (3H, s, H-15), 8.01-7.98 (2H, m, H-3ʹ, H-7ʹ), 7.48-7.44 (2H, m, H-4ʹ, H-6ʹ), 7.60-7.57 (1H, m, H-5ʹ); 13C NMR (100 MHz, CDCl3) δ 130.2 (C, C-1), 38.1 (CH2, C-2), 81.9 (CH, C-3), 79.4 (C, C-4), 49.6 (CH, C-5), 83.6 (CH, C-6), 57.2 (CH, C-7), 25.9 (CH2, C-8), 35.3 (CH2, C-9), 133.1 (C, C-10), 139.1 (C, C-11), 169.9 (C, C-12), 119.3 (CH2, C-13), 25.7 (CH3, C-14), 25.1 (CH3, C-15), 165.5 (C, C-1ʹ), 130.0 (C, C-2ʹ), 129.8 (CH, C-3ʹ, C-7ʹ), 128.7 (CH, C-4ʹ, C-6ʹ), 133.5 (CH, C-5ʹ). HRESIMS calcd for C22H24O4Cl [M + H]+ 387.1358, found 387.1353.
3α-O-benzoyl-4β-fluoro-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (11) To a stirred solution of benzoic acid (20 mg, 0.16 mmol) in CH2Cl2 (1 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 28 μL, 0.16 mmol), compound 6 (21 mg, 0.08 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred for 4 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 15:85) to yield compound 11 (25 mg, 83% yield) as a white powder. mp 53~55 °C; − 92.4 (c 0.097, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.07-2.93 (2H, m, H-2), 5.29-5.25 (1H, m, H-3), 2.59 (1H, d, J = 17.7 Hz, H-5), 4.01 (1H, t, J = 10.1 Hz, H-6), 2.14-2.09 (1H, m, H-7), 2.82-2.75 (1H, m, H-8), 1.45-1.35 (1H, m, H-8), 2.35-2.22 (2H, m, H-9), 6.20 (1H, d, J = 3.2 Hz, H-13), 5.48 (1H, d, J = 2.9 Hz, H-13), 1.80-1.73 (6H, m, H-14, H-15), 8.00-7.98 (2H, m, H-3ʹ, H-7ʹ), 7.47-7.44 (2H, m, H-4ʹ, H-6ʹ), 7.60-7.57 (1H, m, H-5ʹ); 13C NMR (100 MHz, CDCl3) δ 131.4 (C, C-1), 37.4 (CH2, C-2), 78.9 (CH, C-3), 81.2 (C, C-4), 51.2 (CH, C-5), 81.3 (CH, C-6), 54.5 (CH, C-7), 25.7 (CH2, C-8), 34.9 (CH2, C-9), 133.2 (C, C-10), 139.3 (C, C-11), 169.9 (C, C-12), 118.9 (CH2, C-13), 24.7 (CH3, C-14), 20.6 (CH3, C-15), 165.5 (C, C-1ʹ), 129.9 (C, C-2ʹ), 129.8 (CH, C-3ʹ, C-7ʹ), 128.6 (CH, C-4ʹ, C-6ʹ), 133.5 (CH, C-5ʹ). HRESIMS calcd for C22H24O4F [M + H]+ 371.1653, found 371.1676.
3α-O-formyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (12) Compound 1 (21 mg, 0.08 mmol) was added to formic acid (4 mL) at room temperature. The reaction mixture was stirred for 9 h. Then, it was quenched with saturated NaHCO3 aqueous solution and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 15:85) to yield compound 12 (15 mg, 64% yield) as a white powder. mp 180~182 °C; − 61.4 (c 0.130, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.91-2.87 (1H, m, H-2), 2.23-2.17 (1H, m, H-2), 5.04 (1H, d, J = 4.5 Hz, H-3), 2.43 (1H, d, J = 17.2 Hz, H-5), 3.97 (1H, t, J = 10.2 Hz, H-6), 2.12-2.07 (1H, m, H-7), 2.33-2.25 (1H, m, H-8), 1.40-1.31 (1H, m, H-8), 2.82-2.74 (2H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.46 (1H, d, J = 3.0 Hz, H-13), 1.72 (3H, s, H-14), 1.54 (3H, s, H-15), 7.99 (1H, s, H-16); 13C NMR (100 MHz, CDCl3) δ 132.4 (C, C-1), 37.1 (CH2, C-2), 80.6 (CH, C-3), 82.2 (C, C-4), 54.9 (CH, C-5), 82.5 (CH, C-6), 51.2 (CH, C-7), 25.7 (CH2, C-8), 34.5 (CH2, C-9), 133.2 (C, C-10), 139.1 (C, C-11), 169.8 (C, C-12), 119.0 (CH2, C-13), 24.2 (CH3, C-14), 24.0 (CH3, C-15), 160.2 (CH, C-16). HRESIMS calcd for C16H21O5 [M + H]+ 293.1384, found 293.1410.
3α-O-acetyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (13) To a stirred solution of compound 1 (21 mg, 0.08 mmol) in CH2Cl2 (1 mL) was added acetic anhydride (27 μL, 0.24 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred for 3 h. Then, it was quenched with saturated NaHCO3 aqueous solution and extracted with ethyl acetate (3×3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 13 (20 mg, 82% yield) as a white powder. mp 129~131 °C; − 62.6 (c 0.109, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.88-2.83 (1H, m, H-2), 2.28-2.25 (1H, m, H-2), 4.92 (1H, d, J = 4.7 Hz, H-3), 2.37 (1H, d, J = 17.2 Hz, H-5), 3.98 (1H, t, J = 10.2 Hz, H-6), 2.22-2.16 (1H, m, H-7), 2.11-2.09 (1H, m, H-8), 1.40-1.30 (1H, m, H-8), 2.79-2.73 (2H, m, H-9), 6.17 (1H, d, J = 3.2 Hz, H-13), 5.46 (1H, d, J = 3.0 Hz, H-13), 1.71 (3H, s, H-14), 1.51 (3H, s, H-15), 2.05 (3H, s, H-2ʹ); 13C NMR (100 MHz, CDCl3) δ 132.1 (C, C-1), 37.3 (CH2, C-2), 80.9 (CH, C-3), 82.5 (C, C-4), 55.1 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.6 (CH2, C-9), 133.6 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 24.0 (CH3, C-15), 170.4 (C, C-1ʹ), 21.4 (CH3, C-2ʹ). HRESIMS calcd for C17H23O5 [M + H]+ 307.1540, found 307.1555.
General procedure for the synthesis of compounds 14–26
To a stirred solution of acid (0.16 mmol) in CH2Cl2 (1 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 28 μL, 0.16 mmol), compound 1 (21 mg, 0.08 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred for 10 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum.
3α-O-acryloyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (14) White powder, 24% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 105~107 °C; − 51.2 (c 0.050, acetone); 1H NMR (400 MHz, CDCl3) δ 2.92-2.88 (1H, m, H-2), 2.12-2.07 (1H, m, H-2), 5.00 (1H, d, J = 4.7 Hz, H-3), 2.43 (1H, d, J = 17.2 Hz, H-5), 4.00 (1H, t, J = 10.2 Hz, H-6), 2.24-2.22 (1H, m, H-7), 2.33-2.26 (1H, m, H-8), 1.42-1.32 (1H, m, H-8), 2.84-2.75 (2H, m, H-9), 6.19 (1H, d, J = 3.2 Hz, H-13), 5.47 (1H, d, J = 3.0 Hz, H-13), 1.72 (3H, s, H-14), 1.54 (3H, s, H-15), 6.11 (1H, dd, J = 17.3 Hz, J = 10.4 Hz, H-2ʹ), 5.84 (1H, d, J = 10.4 Hz, H-3ʹ), 6.40 (1H, d, J = 17.3 Hz, H-3ʹ); 13C NMR (100 MHz, CDCl3) δ 132.2 (C, C-1), 37.3 (CH2, C-2), 81.1 (CH, C-3), 82.6 (C, C-4), 55.2 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.5 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 24.1 (CH3, C-15), 165.5 (C, C-1ʹ), 128.6 (CH, C-2ʹ), 131.2 (CH2, C-3ʹ). HRESIMS calcd for C18H23O5 [M + H]+ 319.1540, found 319.1556.
3α-O-octanoyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (15) White powder, 77% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 103~105 °C; − 50.0 (c 0.130, acetone); 1H NMR (400 MHz, CDCl3) δ 2.87-2.83 (1H, m, H-2), 2.22-2.16 (1H, m, H-2), 4.91 (1H, d, J = 4.6 Hz, H-3), 2.36 (1H, d, J = 17.2 Hz, H-5), 3.98 (1H, t, J = 10.2 Hz, H-6), 2.10-2.07 (1H, m, H-7), 1.40-1.32 (2H, m, H-8), 2.79-2.73 (2H, m, H-9), 6.17 (1H, d, J = 3.2 Hz, H-13), 5.45 (1H, d, J = 3.0 Hz, H-13), 1.70 (3H, s, H-14), 1.51 (3H, s, H-15), 6.11 (2H, t, J = 7.4 Hz, H-2ʹ), 1.61-1.57 (2H, m, H-3ʹ), 1.30-1.24 (8H, m, H-4ʹ~7ʹ), 0.86 (3H, t, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3) δ 132.0 (C, C-1), 37.4 (CH2, C-2), 80.7 (CH, C-3), 82.5 (C, C-4), 55.1 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.6 (CH2, C-9), 133.6 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.3 (CH3, C-14), 24.0 (CH3, C-15), 173.1 (C, C-1ʹ), 34.7 (CH2, C-2ʹ), 25.1 (CH2, C-3ʹ), 29.0 (CH2, C-4ʹ), 29.1 (CH2, C-5ʹ), 31.8 (CH2, C-6ʹ), 22.7 (CH2, C-7ʹ), 14.2 (CH3, C-8ʹ). HRESIMS calcd for C23H35O5 [M + H]+ 391.2479, found 391.2495.
3α-O-cyclopentane formyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (16) White powder, 87% yield after chromatography on silica gel (acetone-petroleum ether, 20:80). mp 120~122 °C; −72.8 (c 0.081, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.88-2.82 (1H, m, H-2), 2.28-2.25 (1H, m, H-2), 4.90 (1H, d, J = 4.6 Hz, H-3), 2.34 (1H, d, J = 17.3 Hz, H-5), 3.98 (1H, t, J = 10.1 Hz, H-6), 2.22-2.16 (1H, m, H-7), 2.72-2.66 (1H, m, H-8), 1.40-1.30 (1H, m, H-8), 2.80-2.73 (2H, m, H-9), 6.17 (1H, d, J = 3.1 Hz, H-13), 5.45 (1H, d, J = 2.8 Hz, H-13), 1.70 (3H, s, H-14), 1.51 (3H, s, H-15), 2.11-2.06 (1H, m, H-2ʹ), 1.90-1.74 (4H, m, H-3ʹ, H-6ʹ), 1.68-1.54 (4H, m, H-4ʹ, H-5ʹ); 13C NMR (100 MHz, CDCl3) δ 131.9 (C, C-1), 37.4 (CH2, C-2), 80.5 (CH, C-3), 82.5 (C, C-4), 55.1 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.6 (CH2, C-9), 133.6 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.3 (CH3, C-14), 24.0 (CH3, C-15), 175.9 (C, C-1ʹ), 44.1 (CH, C-2ʹ), 30.3 (CH2, C-3ʹ), 25.8 (CH2, C-4ʹ, C-5ʹ), 29.8 (CH, C-6ʹ). HRESIMS calcd for C21H29O5 [M + H]+ 361.2010, found 361.2013.
3α-O-(4-bromobenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (17) White powder, 52% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 51~53 °C; −62.7 (c 0.118, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.99-2.89 (2H, m, H-2), 5.12 (1H, d, J = 4.6 Hz, H-3), 2.51 (1H, d, J = 17.3 Hz, H-5), 4.04 (1H, t, J = 10.1 Hz, H-6), 2.84-2.78 (1H, m, H-7), 2.31-2.29 (1H, m, H-8), 1.42-1.33 (1H, m, H-8), 2.23-2.08 (2H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.47 (1H, d, J = 2.9 Hz, H-13), 1.70 (3H, s, H-14), 1.60 (3H, s, H-15), 7.83 (2H, d, J = 8.4 Hz, H-3ʹ, H-7ʹ), 7.56 (2H, d, J = 8.4 Hz, H-4ʹ, H-6ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.4 (CH2, C-2), 81.9 (CH, C-3), 82.5 (C, C-4), 55.3 (CH, C-5), 82.7 (CH, C-6), 51.1 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.2 (C, C-10), 139.2 (C, C-11), 170.0 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 24.2 (CH3, C-15), 165.1 (C, C-1ʹ), 129.2 (C, C-2ʹ), 131.9 (CH, C-3ʹ, C-7ʹ), 131.2 (CH, C-4ʹ, C-6ʹ), 128.3 (C, C-5ʹ). HRESIMS calcd for C22H23O5BrNa [M + Na]+ 469.0621, found 469.0628.
3α-O-(2-trifluoromethylbenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (18) White powder, 38% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 154~156 °C; −47.1 (c 0.079, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.82-2.73 (2H, m, H-2), 5.14 (1H, d, J = 4.5 Hz, H-3), 2.61 (1H, d, J = 17.2 Hz, H-5), 4.00 (1H, t, J = 10.2 Hz, H-6), 2.98-2.92 (1H, m, H-7), 2.31-2.25 (1H, m, H-8), 1.42-1.32 (1H, m, H-8), 2.23-2.10 (2H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.46 (1H, d, J = 3.0 Hz, H-13), 1.74 (3H, s, H-14), 1.59 (3H, s, H-15), 7.75-7.72 (2H, m, H-3ʹ, H-6ʹ), 7.65-7.58 (2H, m, H-4ʹ, H-5ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 36.9 (CH2, C-2), 82.7 (CH, C-3), 82.5 (C, C-4), 55.0 (CH, C-5), 83.0 (CH, C-6), 51.5 (CH, C-7), 25.9 (CH2, C-8), 34.5 (CH2, C-9), 133.4 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.2 (CH3, C-14), 24.1 (CH3, C-15), 166.3 (C, C-1ʹ), 126.9 (C, C-2ʹ), 130.3 (CH, C-3ʹ), 131.3 (C, C-4ʹ), 132.0 (CH, C-5ʹ), 126.8 (CH, C-6ʹ), 131.5 (CH, C-7ʹ), 124.8 (C, CF3). HRESIMS calcd for C23H23O5F3Na [M + Na]+ 459.1390, found 459.1396.
3α-O-(3-trifluoromethylbenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (19) White powder, 42% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 65~67 °C; −66.8 (c 0.136, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.02-2.92 (2H, m, H-2), 5.17 (1H, d, J = 4.6 Hz, H-3), 2.55 (1H, d, J = 17.4 Hz, H-5), 4.05 (1H, t, J = 10.1 Hz, H-6), 2.87-2.81 (1H, m, H-7), 2.36-2.29 (1H, m, H-8), 1.43-1.34 (1H, m, H-8), 2.21-2.09 (2H, m, H-9), 6.20 (1H, d, J = 3.2 Hz, H-13), 5.49 (1H, d, J = 2.9 Hz, H-13), 1.72 (3H, s, H-14), 1.63 (3H, s, H-15), 8.23 (1H, s, H-3ʹ), 8.17 (1H, d, J = 7.8 Hz, H-5ʹ), 7.59 (1H, t, J = 7.8 Hz, H-6ʹ), 7.82 (1H, d, J = 7.7 Hz, H-7ʹ); 13C NMR (100 MHz, CDCl3) δ 132.5 (C, C-1), 37.4 (CH2, C-2), 82.3 (CH, C-3), 82.6 (C, C-4), 55.3 (CH, C-5), 82.7 (CH, C-6), 51.1 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.2 (C, C-10), 139.2 (C, C-11), 169.9 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 24.3 (CH3, C-15), 164.6 (C, C-1ʹ), 131.1 (C, C-2ʹ), 126.6 (CH, C-3ʹ), 131.5 (C, C-4ʹ), 129.7 (CH, C-5ʹ), 129.3 (CH, C-6ʹ), 132.9 (CH, C-7ʹ), 131.1 (C, CF3) HRESIMS calcd for C23H24O5F3 [M + H]+ 437.1570, found 437.1585.
3α-O-(4-trifluoromethylbenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (20) White powder, 41% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 82~84 °C; −63.8 (c 0.079, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.03-2.91 (2H, m, H-2), 5.16 (1H, d, J = 4.6 Hz, H-3), 2.54 (1H, d, J = 17.3 Hz, H-5), 4.05 (1H, t, J = 10.1 Hz, H-6), 2.85-2.79 (1H, m, H-7), 2.31-2.27 (1H, m, H-8), 1.44-1.34 (1H, m, H-8), 2.25-2.08 (2H, m, H-9), 6.19 (1H, d, J = 3.2 Hz, H-13), 5.48 (1H, d, J = 2.9 Hz, H-13), 1.71 (3H, s, H-14), 1.62 (3H, s, H-15), 8.09 (2H, d, J = 8.1 Hz, H-3ʹ, H-7ʹ), 7.70 (2H, d, J = 8.1 Hz, H-4ʹ, H-6ʹ); 13C NMR (100 MHz, CDCl3) δ 132.5 (C, C-1), 37.4 (CH2, C-2), 82.3 (CH, C-3), 82.5 (C, C-4), 55.3 (CH, C-5), 82.7 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.1 (C, C-10), 139.2 (C, C-11), 170.0 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 24.2 (CH3, C-15), 164.6 (C, C-1ʹ), 134.5 (C, C-2ʹ), 130.1 (CH, C-3ʹ, C-7ʹ), 125.6 (CH, C-4ʹ, C-6ʹ), 134.9 (C, C-5ʹ), 133.6 (C, CF3). HRESIMS calcd for C23H24O5F3 [M + H]+ 437.1570, found 437.1595.
3α-O-(2,3,4,5,6-pentafluorobenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (21) White powder, 32% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 40~42 °C; −44.1 (c 0.160, CH3OH); 1H NMR (600 MHz, CDCl3) δ 2.84-2.76 (2H, m, H-2), 5.20 (1H, d, J = 4.6 Hz, H-3), 2.54 (1H, d, J = 17.4 Hz, H-5), 3.99 (1H, t, J = 10.2 Hz, H-6), 2.99-2.95 (1H, m, H-7), 2.30-2.25 (1H, m, H-8), 1.40-1.34 (1H, m, H-8), 2.23-2.19 (1H, m, H-9), 2.12-2.08 (1H, m, H-9), 6.19 (1H, d, J = 3.3 Hz, H-13), 5.47 (1H, d, J = 3.1 Hz, H-13), 1.73 (3H, s, H-14), 1.62 (3H, s, H-15); 13C NMR (150 MHz, CDCl3) δ 132.7 (C, C-1), 37.2 (CH2, C-2), 82.6 (CH, C-3), 82.3 (C, C-4), 55.0 (CH, C-5), 83.7 (CH, C-6), 51.3 (CH, C-7), 25.8 (CH2, C-8), 34.6 (CH2, C-9), 132.9 (C, C-10), 139.1 (C, C-11), 169.9 (C, C-12), 119.2 (CH2, C-13), 24.3 (CH3, C-14), 24.1 (CH3, C-15), 158.3 (C, C-1ʹ). HRESIMS calcd for C22H20O5F5 [M + H]+ 459.1225, found 459.1235.
3α-O-nicotinoyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (22) White powder, 90% yield after chromatography on silica gel (acetone-petroleum ether, 20:80). mp 167~169 °C; −82.1 (c 0.085, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.01-2.96 (1H, m, H-2), 2.24-2.18 (1H, m, H-2), 5.17 (1H, d, J = 4.3 Hz, H-3), 2.53 (1H, d, J = 17.2 Hz, H-5), 4.04 (1H, t, J = 10.1 Hz, H-6), 2.14-2.08 (1H, m, H-7), 2.36-2.29 (1H, m, H-8), 1.43-1.33 (1H, m, H-8), 2.94-2.91 (1H, m, H-9), 2.86-2.80 (1H, m, H-9), 6.18 (1H, d, J = 2.9 Hz, H-13), 5.48 (1H, d, J = 2.5 Hz, H-13), 1.71 (3H, s, H-14), 1.61 (3H, s, H-15), 8.31-8.29 (1H, m, H-3ʹ), 7.47-7.43 (1H, m, H-4ʹ), 8.85-8.78 (1H, m, H-5ʹ), 9.24-9.18 (1H, m, H-6ʹ); 13C NMR (100 MHz, CDCl3) δ 132.6 (C, C-1), 37.3 (CH2, C-2), 82.3 (CH, C-3), 82.4 (C, C-4), 55.3 (CH, C-5), 82.7 (CH, C-6), 51.1 (CH, C-7), 25.8 (CH2, C-8), 34.7 (CH2, C-9), 133.0 (C, C-10), 139.2 (C, C-11), 170.0 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 24.2 (CH3, C-15), 164.2 (C, C-1ʹ), 123.8 (C, C-2ʹ), 137.8 (CH, C-3ʹ, C-4ʹ), 153.4 (CH, C-5ʹ), 150.8 (CH, C-6ʹ). HRESIMS calcd for C21H24NO5 [M + H]+ 370.1649, found 370.1650.
3α-O-cinnamoyl-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (23) White powder, 71% yield after chromatography on silica gel (acetone-petroleum ether, 20:80). mp 87~89 °C; −63.1 (c 0.084, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.96-2.86 (2H, m, H-2), 5.06 (1H, d, J = 4.6 Hz, H-3), 2.47 (1H, d, J = 17.2 Hz, H-5), 4.02 (1H, t, J = 10.1 Hz, H-6), 2.82-2.76 (1H, m, H-7), 2.35-2.27 (1H, m, H-8), 1.42-1.32 (1H, m, H-8), 2.24-2.19 (1H, m, H-9), 2.13-2.08 (1H, m, H-9), 6.19 (1H, d, J = 3.2 Hz, H-13), 5.47 (1H, d, J = 3.0 Hz, H-13), 1.73 (3H, s, H-14), 1.58 (3H, s, H-15), 6.42 (1H, d, J = 16.0 Hz, H-2ʹ), 7.67 (1H, d, J = 16.0 Hz, H-3ʹ), 7.53-7.51 (2H, m, H-5ʹ, H-9ʹ), 7.38-7.36 (3H, m, H-6ʹ, H-7ʹ, H-8ʹ); 13C NMR (100 MHz, CDCl3) δ 132.1 (C, C-1), 37.5 (CH2, C-2), 81.0 (CH, C-3), 82.6 (C, C-4), 55.2 (CH, C-5), 82.8 (CH, C-6), 51.2 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.6 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.4 (CH3, C-14), 24.1 (CH3, C-15), 166.3 (C, C-1ʹ), 118.2 (CH, C-2ʹ), 145.3 (CH, C-3ʹ), 134.4 (C, C-4ʹ), 128.2 (CH, C-5ʹ, C-9ʹ), 129.0 (CH, C-6ʹ, C-8ʹ), 130.5(CH, C-7ʹ). HRESIMS calcd for C24H27O5 [M + H]+ 395.1853, found 395.1859.
3α-O-(pyridine-4-acryloyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (24) White powder, 51% yield after chromatography on silica gel (methanol- dichloromethane, 2:98). mp 123~125 °C; −58.0 (c 0.100, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.98-2.91 (1H, m, H-2), 2.32-2.27 (1H, m, H-2), 5.07 (1H, d, J = 4.6 Hz, H-3), 2.47 (1H, d, J = 17.3 Hz, H-5), 4.02 (1H, t, J = 10.2 Hz, H-6), 2.14-2.08 (1H, m, H-7), 2.25-2.19 (1H, m, H-8), 1.43-1.33 (1H, m, H-8), 2.88-2.76 (2H, m, H-9), 6.19 (1H, d, J = 3.2 Hz, H-13), 5.48 (1H, d, J = 2.9 Hz, H-13), 1.73 (3H, s, H-14), 1.58 (3H, s, H-15), 6.59 (1H, d, J = 16.0 Hz, H-2ʹ), 7.58 (1H, d, J = 16.0 Hz, H-3ʹ), 7.40-7.37 (2H, m, H-5ʹ, H-8ʹ), 8.68-8.65 (2H, m, H-6ʹ, H-7ʹ); 13C NMR (100 MHz, CDCl3) δ 132.4 (C, C-1), 37.4 (CH2, C-2), 81.6 (CH, C-3), 82.5 (C, C-4), 55.2 (CH, C-5), 82.7 (CH, C-6), 51.3 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.4 (C, C-10), 139.2 (C, C-11), 170.0 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 24.1 (CH3, C-15), 165.3 (C, C-1ʹ), 122.1 (CH, C-2ʹ), 142.3 (CH, C-3ʹ), 141.9 (C, C-4ʹ), 123.1 (CH, C-5ʹ, C-8ʹ), 150.6 (CH, C-6ʹ, C-7ʹ). HRESIMS calcd for C23H26NO5 [M + H]+ 396.1805, found 396.1814.
3α-O-(quinoline-7-formyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (25) White powder, 95 % yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 98~100 °C; −43.8 (c 0.101, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.05-2.97 (2H, m, H-2), 5.21 (1H, d, J = 4.5 Hz, H-3), 2.57 (1H, d, J = 17.3 Hz, H-5), 4.10 (1H, t, J = 10.1 Hz, H-6), 2.87-2.83 (1H, m, H-7), 2.35-2.29 (1H, m, H-8), 1.43-1.33 (1H, m, H-8), 2.24-2.18 (1H, m, H-9), 2.12-2.07 (1H, m, H-9), 6.17 (1H, d, J = 3.1 Hz, H-13), 5.46 (1H, d, J = 2.8 Hz, H-13), 1.71 (3H, s, H-14), 1.67 (3H, s, H-15), 8.52 (1H, s, H-3ʹ), 8.28-8.21 (2H, m, H-5ʹ, H-10ʹ), 7.48-7.45 (1H, m, H-6ʹ), 8.99-8.98 (1H, m, H-7ʹ), 8.15-8.13 (1H, m, H-9ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.5 (CH2, C-2), 82.1 (CH, C-3), 82.4 (C, C-4), 55.4 (CH, C-5), 82.7 (CH, C-6), 51.0 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.1 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.4 (CH3, C-14), 24.1 (CH3, C-15), 165.3 (C, C-1ʹ), 128.3 (C, C-2ʹ), 131.1 (CH, C-3ʹ), 127.5 (C, C-4ʹ), 137.5 (CH, C-5ʹ), 122.0 (CH, C-6ʹ), 152.6 (CH, C-7ʹ), 150.1 (C, C-8ʹ), 128.9 (CH, C-9ʹ), 129.9 (CH, C-10ʹ). HRESIMS calcd for C25H26NO5 [M + H]+ 420.1805, found 420.1809.
3α-O-(quinoline-2-formyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (26) White powder, 87% yield after chromatography on silica gel (acetone-petroleum ether, 15:85). mp 90~92 °C; −57.1 (c 0.165, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.07-2.99 (2H, m, H-2), 5.26 (1H, d, J = 4.5 Hz, H-3), 2.64 (1H, d, J = 17.4 Hz, H-5), 4.06 (1H, t, J = 10.2 Hz, H-6), 2.84-2.78 (1H, m, H-7), 2.32-2.26 (1H, m, H-8), 1.42-1.32 (1H, m, H-8), 2.22-2.17 (1H, m, H-9), 2.12-2.06 (1H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.46 (1H, d, J = 2.9 Hz, H-13), 1.70 (6H, s, H-14, H-15), 8.04-8.02 (1H, m, H-4ʹ), 7.79-7.74 (1H, m, H-5ʹ), 7.87-7.85 (1H, m, H-6ʹ), 8.29-8.26 (2H, m, H-7ʹ, H-9ʹ), 7.64-7.61 (1H, m, H-10ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.4 (CH2, C-2), 82.6 (CH, C-3), 82.7 (C, C-4), 55.3 (CH, C-5), 82.7 (CH, C-6), 51.0 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.2 (C, C-10), 139.3 (C, C-11), 170.0 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 24.2 (CH3, C-15), 164.4 (C, C-1ʹ), 148.0 (C, C-2ʹ), 147.9 (C, C-3ʹ), 130.4 (CH, C-4ʹ), 131.0 (CH, C-5ʹ), 128.7 (CH, C-6ʹ), 127.5 (CH, C-7ʹ), 129.3 (C, C-8ʹ), 137.3 (CH, C-9ʹ), 120.9 (CH, C-10ʹ). HRESIMS calcd for C25H25NO5Na [M + Na]+ 442.1625, found 442.1629.
3α-O-(4-dimethylamino-2-butenoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (27) To a stirred solution of maleic anhydride (49 mg, 0.5 mmol) in CH2Cl2 (2 mL), dimethylamine (250 μL, 0.5 mmol, 2.0 M in THF) and 4-dimethylaminopyridine (DMAP, 7 mg, 0.06 mmol) was added at room temperature. The reaction mixture was stirred for 1 h and the solvent was evaporated under vacuum. To a stirred solution of crude residue in CH2Cl2 (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 86 μL, 0.5 mmol) and compound 1 (26 mg, 0.1 mmol) at room temperature. The reaction mixture was stirred overnight. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 25:75) to yield compound 27 (35 mg, 89 % yield) as a white powder. mp 92~94 °C; [α]20 D−76.0 (c 0.080, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.81-2.74 (2H, m, H-2), 4.98 (1H, d, J = 4.5 Hz, H-3), 2.40 (1H, d, J = 17.2 Hz, H-5), 3.97 (1H, t, J = 10.1 Hz, H-6), 2.92-2.85 (1H, m, H-7), 2.10-2.05 (1H, m, H-8), 1.38-1.29 (1H, m, H-8), 2.27-2.19 (2H, m, H-9), 6.15 (1H, d, J = 3.2 Hz, H-13), 5.45 (1H, d, J = 3.0 Hz, H-13), 1.69 (3H, s, H-14), 1.51 (3H, s, H-15), 6.74 (2H, d, J = 15.3 Hz, H-2ʹ), 7.39 (2H, d, J = 15.3 Hz, H-3ʹ), 3.01 (1H, s, H-5ʹ), 3.11 (1H, s, H-6ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.2 (CH2, C-2), 81.7 (CH, C-3), 82.3 (C, C-4), 55.0 (CH, C-5), 82.7 (CH, C-6), 51.2 (CH, C-7), 25.8 (CH2, C-8), 34.6 (CH2, C-9), 133.2 (C, C-10), 139.2 (C, C-11), 170.0 (C, C-12), 119.0 (CH2, C-13), 24.3 (CH3, C-14), 24.0 (CH3, C-15), 165.1 (C, C-1ʹ), 131.1 (CH, C-2ʹ), 134.5 (CH, C-3ʹ), 164.7 (CH, C-4ʹ), 35.9 (CH, C-5ʹ), 37.7 (CH, C-6ʹ). HRESIMS calcd for C21H28NO6 [M + H]+ 390.1911, found 390.1922.
3α-O-(4-acrylamidebenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (28) To a stirred solution of 4-aminobenzoic acid (680 mg, 5 mmol) in CH2Cl2 (5 mL) was added K2CO3 (1.73 g, 10 mmol) and acryloyl chloride (490 µL, 6 mmol) at 0 °C under an argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred overnight before it was quenched with water. Then, it was extracted with ethyl acetate (3 × 20 mL). The combined aqueous phases were acidified by 5% HCl aqueous solution and filtered to provide 4-acrylamidobenzoic acid (640 mg, 67% yield) as yellow powder. To a stirred solution of 4-acrylamidobenzoic acid (38 mg, 0.2 mmol) in CH2Cl2 (1 mL) was added dicyclohexylcarbodiimide (DCC, 41 mg, 0.2 mmol), compound 1 (26 mg, 0.1 mmol) and 4-dimethylaminopyridine (DMAP, 2 mg, 0.02 mmol) at room temperature. The reaction mixture was stirred overnight. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 28 (13 mg, 56% yield) as a white powder. mp 120~122 °C; −62.4 (c 0.092, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.99-2.92 (2H, m, H-2), 5.11 (1H, d, J = 4.4 Hz, H-3), 2.53 (1H, d, J = 17.2 Hz, H-5), 4.05 (1H, t, J = 10.1 Hz, H-6), 2.86-2.81 (1H, m, H-7), 2.13-2.09 (1H, m, H-8), 1.42-1.37 (1H, m, H-8), 2.31-2.23 (2H, m, H-9), 6.19 (1H, d, J = 3.1 Hz, H-13), 5.49 (1H, d, J = 2.8 Hz, H-13), 1.71 (3H, s, H-14), 1.60 (3H, s, H-15), 7.96-7.94 (2H, m, H-3ʹ, H-7ʹ), 7.71-7.69 (2H, m, H-4ʹ, H-6ʹ), 6.31 (1H, dd, J = 16.8, 10.2 Hz H-9ʹ), 6.47 (1H, d, J = 16.8 Hz, H-10ʹ), 5.80 (1H, d, J = 10.2 Hz, H-10ʹ); 13C NMR (100 MHz, CDCl3) δ 132.3 (C, C-1), 37.5 (CH2, C-2), 81.5 (CH, C-3), 82.7 (C, C-4), 55.3 (CH, C-5), 82.9 (CH, C-6), 51.1 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.3 (C, C-10), 139.3 (C, C-11), 170.2 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 24.3 (CH3, C-15), 163.9 (C, C-1ʹ), 125.9 (C, C-2ʹ), 131.0 (CH, C-3ʹ, C-4ʹ, C-6ʹ, C-7ʹ), 142.5 (C, C-5ʹ), 165.3 (C, C-8ʹ), 119.3 (CH, C-9ʹ), 128.9 (CH2, C-10ʹ). HRESIMS calcd for C25H28NO6 [M + H]+ 438.1911, found 438.1918.
3α-O-(3-acrylamidebenzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (29) To a stirred solution of 3-aminobenzoic acid (680 mg, 5 mmol) in CH2Cl2 (5 mL) was added K2CO3 (1.73 g, 10 mmol) and acryloyl chloride (490 µL, 6 mmol) at 0 °C under an argon atmosphere. The reaction mixture was allowed to warm to room temperature and stirred overnight before it was quenched with water. Then, it was extracted with ethyl acetate (3×20 mL). The combined aqueous phases were acidified by 5% HCl aqueous solution and filtered to provide 4-acrylamidobenzoic acid (650 mg, 68% yield) as a yellow powder. In a sealed tube, dicyclohexylcarbodiimide (DCC, 82 mg, 0.4 mmol), compound 1 (53 mg, 0.4 mmol) and 4-dimethylaminopyridine (DMAP, 5 mg, 0.04 mmol) was added to a stirred solution of 3-acrylamidobenzoic acid (76 mg, 0.4 mmol) in CH2Cl2 (2 mL) at room temperature. The reaction mixture was heated to 50 °C and stirred for 7 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 25:75) to yield compound 29 (76 mg, 88% yield) as a white powder. mp 111~113 °C; −58.6 (c 0.103, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.97-2.86 (2H, m, H-2), 5.06 (1H, d, J = 4.5 Hz, H-3), 2.49 (1H, d, J = 17.2 Hz, H-5), 4.05 (1H, t, J = 10.1 Hz, H-6), 2.74-2.71 (1H, m, H-7), 2.07-2.03 (1H, m, H-8), 1.39-1.29 (1H, m, H-8), 2.29-2.14 (2H, m, H-9), 6.15 (1H, d, J = 2.9 Hz, H-13), 5.45 (1H, d, J = 2.5 Hz, H-13), 1.67 (3H, s, H-14), 1.55 (3H, s, H-15), 8.04 (1H, s, H-3ʹ), 7.68-7.66 (1H, m, H-5ʹ), 7.38-7.34 (1H, m, H-6ʹ), 8.12-8.10 (1H, m, H-7ʹ), 5.73-5.70 (1H, m, H-9ʹ), 6.43-6.30 (1H, m, H-10ʹ); 13C NMR (100 MHz, CDCl3) δ 132.2 (C, C-1), 37.3 (CH2, C-2), 81.9 (CH, C-3), 82.2 (C, C-4), 55.1 (CH, C-5), 82.8 (CH, C-6), 50.7 (CH, C-7), 25.7 (CH2, C-8), 34.6 (CH2, C-9), 132.7 (C, C-10), 139.2 (C, C-11), 170.5 (C, C-12), 119.1 (CH2, C-13), 24.4 (CH3, C-14), 23.8 (CH3, C-15), 164.2 (C, C-1ʹ), 130.8 (C, C-2ʹ), 120.9 (CH, C-3ʹ), 138.5 (C, C-4ʹ), 125.2 (CH, C-5ʹ), 129.2 (CH, C-6ʹ), 124.8 (CH, C-7ʹ), 165.6 (C, C-8ʹ), 131.1 (CH, C-9ʹ), 128.1 (CH2, C-10ʹ). HRESIMS calcd for C25H28NO6 [M + H]+ 438.1911, found 438.1917.
3α-O-(4-(1-dimethylamino-2-butenamide)benzoyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (30) To a stirred solution of 4-(tert-butoxycarbonylamino) benzoic acid (355 mg, 1.5 mmol) in CH2Cl2 (5 mL) was added dicyclohexylcarbodiimide (DCC, 309 mg, 1.5 mmol), compound 1 (264 mg, 1 mmol) and 4-dimethylaminopyridine (DMAP, 25 mg, 0. 2 mmol) at room temperature. The reaction mixture was stirred overnight. Then, it was quenched with water and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) provided intermediate S3 (263 mg, 54% yield) as a white powder. Trifluoroacetic acid was added to a stirred solution of intermediate S3 (240 mg, 0.7 mmol) in CH2Cl2 (3 mL) (1 mL) at room temperature. The reaction mixture was stirred 4 h. Then, it was quenched with saturated sodium bicarbonate solution and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) provided intermediate S4 (247 mg, 92% yield) as a white powder. To a stirred solution of acyl chloride in CH2Cl2 (3 mL), intermediate S4 (23 mg, 0.06 mmol) and triethylamine (138 µL, 1.0 mmol) were added at room temperature. The reaction mixture was stirred 4 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) to provide 30 (15 mg, 51% yield) as a white powder. mp 130~132 °C; −59.0 (c 0.097, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.98-2.90 (2H, m, H-2), 5.09 (1H, d, J = 4.4 Hz, H-3), 2.50 (1H, d, J = 17.2 Hz, H-5), 4.06 (1H, t, J = 10.1 Hz, H-6), 2.84-2.79 (1H, m, H-7), 1.42-1.33 (2H, m, H-8), 2.19-2.07 (2H, m, H-9), 6.20-6.15 (2H, m, H-13, H-9ʹ), 5.45 (1H, d, J = 2.7 Hz, H-13), 1.70 (3H, s, H-14), 1.58 (3H, s, H-15), 7.92 (2H, d, J = 8.3 Hz, H-3ʹ, H-7ʹ), 7.69 (2H, d, J = 8.4 Hz, H-4ʹ, H-6ʹ), 7.01-6.94 (1H, m, H-10ʹ), 3.09-3.07 (2H, m, H-11ʹ), 2.25 (6H, s, H-12ʹ, H-13ʹ); 13C NMR (100 MHz, CDCl3) δ 132.2 (C, C-1), 37.5 (CH2, C-2), 81.6 (CH, C-3), 82.5 (C, C-4), 55.3 (CH, C-5), 82.9 (CH, C-6), 51.1 (CH, C-7), 25.9 (CH2, C-8), 34.7 (CH2, C-9), 133.3 (C, C-10), 139.3 (C, C-11), 170.4 (C, C-12), 119.2 (CH2, C-13), 24.5 (CH3, C-14), 24.1 (CH3, C-15), 164.0 (C, C-1ʹ), 125.6 (C, C-2ʹ), 130.9 (CH, C-3ʹ, C-4ʹ, C-6ʹ, C-7ʹ), 142.8 (C, C-5ʹ), 165.5 (C, C-8ʹ), 125.8 (CH, C-9ʹ), 143.2 (CH, C-10ʹ), 60.3 (CH2, C-11ʹ), 45.6 (CH3, C-12ʹ, C-13ʹ). HRESIMS calcd for C28H35N2O6 [M + H]+ 495.2490, found 495.2494.
3α-O-(2,4-dioxo-5-fluoro-3,4-dihydro-1(2H)-pyrimidineacetyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (31) To a stirred solution of 5-fluoro-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidineacetic acid (75 mg, 0.4 mmol) in CH2Cl2 (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 70 μL, 0.4 mmol), compound 1 (53 mg, 0.2 mmol) and 4-dimethylaminopyridine (DMAP, 5 mg, 0.04 mmol) at room temperature. The reaction mixture was stirred for 22 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 25:75) to provide compound 31 (66 mg, 77% yield) as a white powder. mp 121~123 °C; −21.5 (c 0.094, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.29-2.16 (2H, m, H-2), 5.00 (1H, br.s, H-3), 2.36 (1H, d, J = 17.2 Hz, H-5), 3.97 (1H, t, J = 10.1 Hz, H-6), 2.89-2.85 (1H, m, H-7), 2.08-2.05 (1H, m, H-8), 1.37-1.28 (1H, m, H-8), 2.77-2.68 (2H, m, H-9), 6.14 (1H, s, H-13), 5.45 (1H, s, H-13), 1.69 (3H, s, H-14), 1.46 (3H, s, H-15), 4.51-4.39 (2H, m, H-2ʹ), 7.35-7.33 (1H, m, H-6ʹ); 13C NMR (100 MHz, CDCl3) δ 132.6 (C, C-1), 37.1 (CH2, C-2), 82.1 (CH, C-3), 82.6 (C, C-4), 54.9 (CH, C-5), 82.9 (CH, C-6), 51.1 (CH, C-7), 25.7 (CH2, C-8), 34.6 (CH2, C-9), 132.7 (C, C-10), 139.2 (C, C-11), 170.4 (C, C-12), 119.2 (CH2, C-13), 24.4 (CH3, C-14), 23.7 (CH3, C-15), 166.6 (C, C-1ʹ), 49.3 (CH2, C-2ʹ), 149.9 (C, C-3ʹ), 157.5 (C, C-4ʹ), 141.7 (C, C-5ʹ), 129.3 (CH, C-6ʹ). HRESIMS calcd for C21H24N2O7F [M + H]+ 435.1562, found 435.1564.
3α-O-(3-benzyl-2,4-dioxo-5-fluoro-3,4-dihydro-1(2H)-pyrimidineacetyl)-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide (32) To a stirred solution of compound 31 (22 mg, 0.05 mmol) in DMF (1 mL) was added benzyl bromide (12 μL, 0.1 mmol) and K2CO3 (28 mg, 0.2 mmol) at room temperature. The reaction mixture was stirred for 10 h. Then, it was quenched with 5% HCl aqueous solution and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 25:75) to yield compound 32 (19 mg, 73% yield) as a white powder. mp 93~95 °C; −13.5 (c 0.110, CH3OH); 1H NMR (400 MHz, CDCl3) δ 2.91-2.85 (1H, m, H-2), 2.62-2.59 (1H, m, H-2), 5.01 (1H, d, J = 4.4 Hz, H-3), 2.38 (1H, d, J = 17.2 Hz, H-5), 3.92 (1H, t, J = 10.1 Hz, H-6), 2.10-2.04 (1H, m, H-7), 2.68-2.63 (1H, m, H-8), 1.38-1.30 (1H, m, H-8), 2.21-2.19 (2H, m, H-9), 6.18 (1H, d, J = 3.2 Hz, H-13), 5.47 (1H, d, J = 3.0 Hz, H-13), 1.72 (3H, s, H-14), 1.46 (3H, s, H-15), 5.16-5.08 (2H, m, H-2ʹ), 7.32-7.26 (3H, m, H-6ʹ, H-9ʹ, H-13ʹ), 4.53 (1H, d, J = 17.4 Hz, H-7ʹ), 4.34 (1H, d, J = 17.4 Hz, H-7ʹ), 7.48-7.46 (2H, m, H-10ʹ, H-12ʹ), 7.20-7.18 (1H, m, H-11ʹ); 13C NMR (100 MHz, CDCl3) δ 132.8 (C, C-1), 37.1 (CH2, C-2), 82.4 (CH, C-3), 82.2 (C, C-4), 54.9 (CH, C-5), 82.8 (CH, C-6), 51.3 (CH, C-7), 25.7 (CH2, C-8), 34.5 (CH2, C-9), 132.9 (C, C-10), 139.1 (C, C-11), 169.9 (C, C-12), 119.2 (CH2, C-13), 24.3 (CH3, C-14), 24.0 (CH3, C-15), 166.4 (C, C-1ʹ), 50.1 (CH2, C-2ʹ), 157.1 (C, C-3ʹ), 157.4 (C, C-4ʹ), 150.1 (C, C-5ʹ), 127.0 (CH, C-6ʹ), 45.4 (CH2, C-7ʹ), 135.9 (C, C-8ʹ), 128.7 (CH, C-9ʹ, C-13ʹ), 129.4 (CH, C-10ʹ, C-12ʹ). HRESIMS calcd for C28H30N2O7F [M + H]+ 525.2032, found 525.2038.
1,4-bis(3α-hydroxy-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide)phthalate (33) To a stirred solution of terephthalic acid (17 mg, 0.1 mmol) in CH2Cl2 (2 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 70 μL, 0.4 mmol), compound 1 (53 mg, 0.2 mmol) and 4-dimethylaminopyridine (DMAP, 5 mg, 0.04 mmol) at room temperature. The reaction mixture was stirred for 10 h. Then, it was quenched with water and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 10:90) to yield compound 33 (10 mg, 15% yield) as a white powder. mp 138~140 °C; −89.7 (c 0.117, CH3OH); 1H NMR (400 MHz, CDCl3) δ 3.01-2.97 (2H, m, H-2, H-2ʹ), 2.24-2.20 (2H, m, H-2, H-2ʹ), 5.16 (2H, d, J = 4.5 Hz, H-3, H-3ʹ), 2.55 (2H, d, J = 17.2 Hz, H-5, H-5ʹ), 4.04 (2H, t, J = 10.1 Hz, H-6, H-6ʹ), 2.93-2.90 (2H, m, H-7, H-7ʹ), 2.34-2.27 (2H, m, H-8, H-8ʹ), 1.44-1.34 (2H, m, H-8, H-8ʹ), 2.83-2.78 (2H, m, H-9, H-9ʹ), 2.13-2.11 (2H, m, H-9, H-9ʹ), 6.21 (2H, d, J = 3.2 Hz, H-13, H-13ʹ), 5.49 (2H, d, J = 2.9 Hz, H-13, H-13ʹ), 1.72 (6H, s, H-14, H-14ʹ), 1.62 (6H, s, H-15, H-15ʹ), 8.06 (4H, s, H-18, H-18ʹ, H-19, H-19ʹ); 13C NMR (100 MHz, CDCl3) δ 132.5 (C, C-1, C-1ʹ), 37.4 (CH2, C-2, C-2ʹ), 82.1 (CH, C-3, C-3ʹ), 82.6 (C, C-4, C-4ʹ), 55.4 (CH, C-5, C-5ʹ), 82.7 (CH, C-6, C-6ʹ), 51.3 (CH, C-7, C-7ʹ), 25.9 (CH2, C-8, C-8ʹ), 34.7 (CH2, C-9, C-9ʹ), 133.2 (C, C-10, C-10ʹ), 139.2 (C, C-11, C-11ʹ), 170.0 (C, C-12, C-12ʹ), 119.3 (CH2, C-13, C-13ʹ), 24.4 (CH3, C-14, C-14ʹ), 24.3 (CH3, C-15, C-15ʹ), 164.9 (C, C-16, C-16ʹ), 134.3 (C, C-17, C-17ʹ), 129.8 (CH, C-18, C-18ʹ, C-19, C-19ʹ). HRESIMS calcd for C38H43O10 [M + H]+ 659.2851, found 659.2861.
1,4-bis(3α-hydroxy-4β-hydroxy-5,7α,6β(H)-guaia-1(10),11(13)-dien-12,6-olide)succinate (34) To a stirred solution of succinic acid (73 mg, 0.6 mmol) in CH2Cl2 (1 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 21 μL, 0.12 mmol), compound 1 (32 mg, 0.12 mmol) and 4-dimethylaminopyridine (DMAP, 3 mg, 0.025 mmol) at room temperature. Then the reaction mixture was equipped with a reflux condenser, and heated to 50 °C for 24 h. After cooling to room temperature, the mixture was diluted with saturated NaHCO3 aqueous solution and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under vacuum, the crude residue was purified by silica gel column chromatography (acetone-petroleum ether, 20:80) to yield compound 34 (15 mg, 48% yield) as a white powder. mp 97~99 °C; + 48.3 (c 0.012, CHCl3); 1H NMR (400 MHz, CDCl3) δ 2.88-2.84 (2H, m, H-2, H-2ʹ), 2.22-2.17 (2H, m, H-2, H-2ʹ), 4.94 (2H, d, J = 4.6 Hz, H-3, H-3ʹ), 2.37 (2H, d, J = 17.2 Hz, H-5, H-5ʹ), 3.97 (2H, t, J = 10.1 Hz, H-6, H-6ʹ), 2.11-2.09 (2H, m, H-7, H-7ʹ), 2.53-2.26 (2H, m, H-8, H-8ʹ), 1.40-1.31 (2H, m, H-8, H-8ʹ), 2.80-2.74 (4H, m, H-9, H-9ʹ), 6.18 (2H, d, J = 3.2 Hz, H-13, H-13ʹ), 5.47 (2H, d, J = 2.9 Hz, H-13, H-13ʹ), 1.71 (6H, s, H-14, H-14ʹ), 1.51 (6H, s, H-15, H-15ʹ), 2.61 (4H, s, H-17, H-17ʹ); 13C NMR (100 MHz, CDCl3) δ 132.2 (C, C-1, C-1ʹ), 37.3 (CH2, C-2, C-2ʹ), 81.2 (CH, C-3, C-3ʹ), 82.5 (C, C-4, C-4ʹ), 55.1 (CH, C-5, C-5ʹ), 82.7 (CH, C-6, C-6ʹ), 51.2 (CH, C-7, C-7ʹ), 25.9 (CH2, C-8, C-8ʹ), 34.7 (CH2, C-9, C-9ʹ), 133.4 (C, C-10, C-10ʹ), 139.2 (C, C-11, C-11ʹ), 170.0 (C, C-12, C-12ʹ), 119.2 (CH2, C-13, C-13ʹ), 24.4 (CH3, C-14, C-14ʹ), 24.1 (CH3, C-15, C-15ʹ), 171.5 (C, C-16, C-16ʹ), 29.5 (C, C-17, C-17ʹ). HRESIMS calcd for C34H42O10Na [M + Na]+ 633.2670, found 633.2678.
Cytotoxicity assay
The cytotoxicity of the compounds was tested by the MTT assay. Briefly, cells at a density of 3 × 104 cells/well were seeded into 96-well plates and incubated at 37 °C with 5% CO2 for 24 h. The culture medium was replaced with fresh medium containing different concentrations of compound, and the cells were incubated for an additional 48 h. After removal of the medium, 100 μL of MTT reagent (1 mg/mL) was added to each well, and the plates were kept in an incubator for 4 h. After that, 100 μL of dimethyl sulfoxide (DMSO) was added to each well, and the plates were measured at 490 nm using a microplate reader (BIO-RAD, USA). The inhibitory ratio was calculated as [(A490 control − A490 treated)/A490 control] × 100%. The cytotoxicity of compounds was expressed as IC50 values calculated by GraphPad Prism 5 (GraphPad Software, California, USA).
Supplementary Information
Acknowledgements
This work was supported by the Key Program of the National Natural Science Foundation of China (22137008), the Xingdian Rencai Project (YNWR – KJLJ – 2019-002) the Youth Innovation Promotion Association, CAS (2020386), the Reserve Talents of Young and Middle-aged Academic and Technical Leaders in Yunnan Province (T-ZL), and the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2021-ZZ06).
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dong Xue, Email: xuedong_welcome@snnu.edu.cn.
Ji-Jun Chen, Email: chenjj@mail.kib.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1007/s00044-022-02890-2.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73:4–13. doi: 10.1002/hep.31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu HT, Jiang MJ, Deng ZJ, Li L, Huang JL, Liu ZX, et al. Immune checkpoint inhibitors in hepatocellular carcinoma: Current progresses and challenges. Front Oncol. 2021;11:737497. doi: 10.3389/fonc.2021.737497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Liu Y, Jiang W, Xue J, Cheng Y, Wang J, et al. Icaritin promotes apoptosis and inhibits proliferation by down-regulating AFP gene expression in hepatocellular carcinoma. BMC Cancer. 2021;21:318. doi: 10.1186/s12885-021-08043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Zhang W, Jiang L, Chen Y. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark Res. 2022;10:3. doi: 10.1186/s40364-021-00350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- 7.Ma GH, Chen KX, Zhang LQ, Li YM. Advance in biological activities of natural guaiane-type sesquiterpenes. Med Chem Res. 2019;28:1339–1358. doi: 10.1007/s00044-019-02385-7. [DOI] [Google Scholar]
- 8.Lone SH, Bhat KA, Khuroo MA. Arglabin: from isolation to antitumor evaluation. Chem-Biol Interact. 2015;240:180–198. doi: 10.1016/j.cbi.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Lickliter JD, Jennens R, Lemech CR, Su SYC, Chen Y. Phase 1 dose-escalation study of ACT001 in patients with recurrent glioblastoma and other advanced solid tumors. J Clin Oncol. 2018;36:15. doi: 10.1200/JCO.2018.36.15_suppl.e14048. [DOI] [Google Scholar]
- 10.Mahalingam D, Peguero J, Cen P, Arora SP, Sarantopoulos J, Rowe J, et al. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers. 2019;11:833. doi: 10.3390/cancers11060833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su L, Zhang X, Ma Y, Geng C, Huang X, Hu J, et al. New guaiane-type sesquiterpenoid dimers from Artemisia atrovirens and their antihepatoma activity. Acta Pharm Sin B. 2021;11:1648–1666. doi: 10.1016/j.apsb.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su LH, Geng CA, Li TZ, Ma YB, Huang XY, Zhang XM, et al. Artatrovirenols A and B: Two cagelike sesquiterpenoids from Artemisia atrovirens. J Org Chem. 2020;85:13466–13471. doi: 10.1021/acs.joc.0c01491. [DOI] [PubMed] [Google Scholar]
- 13.Su LH, Li TZ, Geng CA, Ma YB, Huang XY, Wang JP, et al. Trimeric and dimeric sesquiterpenoids from Artemisia atrovirens and their cytotoxicities. Org Chem Front. 2021;8:1249–1256. doi: 10.1039/D0QO01615B. [DOI] [Google Scholar]
- 14.Su L-H, Li TZ, Ma YB, Geng CA, Huang XY, Zhang X, et al. Artematrovirenolides A—D and artematrolides S—Z, sesquiterpenoid dimers with cytotoxicity against three hepatoma cell lines from Artemisia atrovirens. Chine J Chem. 2022;40:104–114. doi: 10.1002/cjoc.202100528. [DOI] [Google Scholar]
- 15.Su LH, Ma YB, Geng CA, Huang XY, Zhang X, Gao Z, et al. Artematrovirenins A–P, guaiane-type sesquiterpenoids with cytotoxicities against two hepatoma cell lines from Artemisia atrovirens. Bioorg Chem. 2021;114:105072. doi: 10.1016/j.bioorg.2021.105072. [DOI] [PubMed] [Google Scholar]
- 16.Tang S, Zhang XT, Ma YB, Huang XY, Geng CA, Li TZ, et al. Artemyrianolides A–S, cytotoxic sesquiterpenoids from Artemisia myriantha. J Nat Prod. 2020;83:2618–2630. doi: 10.1021/acs.jnatprod.0c00396. [DOI] [PubMed] [Google Scholar]
- 17.Tang S, Ma YB, Geng CA, Shen C, Li TZ, Zhang XM, et al. Artemyrianins A–G from Artemisia myriantha and their cytotoxicity against HepG2 Cells. Nat Prod Bioprospect. 2020;10:251–260. doi: 10.1007/s13659-020-00255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XT, Hu J, Su LH, Geng CA, Chen JJ. Artematrolide A inhibited cervical cancer cell proliferation via ROS/ERK/mTOR pathway and metabolic shift. Phytomedicine. 2021;91:153707. doi: 10.1016/j.phymed.2021.153707. [DOI] [PubMed] [Google Scholar]
- 19.Li TZ, Yang XT, Wang JP, Geng CA, Ma YB, Su LH, et al. Biomimetic Synthesis of Lavandiolides H, I, and K and Artematrolide F via Diels–Alder Reaction. Org Lett. 2021;23:8380–8384. doi: 10.1021/acs.orglett.1c03120. [DOI] [PubMed] [Google Scholar]
- 20.Shen C, Huang XY, Geng CA, Li TZ, Tang S, Su LH, et al. Artemlavanins A and B from Artemisia lavandulaefolia and their cytotoxicity against hepatic stellate cell line LX2. Nat Prod Bioprospect. 2020;10:243–LX50. doi: 10.1007/s13659-020-00254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geissman TA, Griffin TS. Sesquiterpene lactones of Artemisia carruthii. Phytochemistry. 1972;11:833–835. doi: 10.1016/0031-9422(72)80059-1. [DOI] [Google Scholar]
- 22.Sosa VE, Oberti JC, Gil RR, Ruveda EA, Goedken VL, Gutierrez AB, et al. 10-Epideoxycumambrin B and other constituents of Stevia yaconensis var. subeglandulosa. Phytochemistry. 1989;28:1925–1929. doi: 10.1016/s0031-9422(00)97888-9. [DOI] [Google Scholar]
- 23.Bohlmann F, Ang W, Trinks C, Jakupovic J, Huneck S. Dimeric guaianolides from Artemisia sieversiana. Phytochemistry. 1985;24:1009–1015. doi: 10.1016/s0031-9422(00)83172-6. [DOI] [Google Scholar]
- 24.Rodriguez G, Pestchanker LJ, Pestchanker MJ, Giordano OS. Guaianolides and other constituents from Artemisia douglasiana. Phytochemistry. 1990;29:3028–3029. doi: 10.1016/0031-9422(90)87129-i. [DOI] [Google Scholar]
- 25.Zeng YT, Jiang JM, Lao HY, Guo JW, Lun YN, Yang M. Antitumor and apoptotic activities of the chemical constituents from the ethyl acetate extract of Artemisia indica. Mol Med Rep. 2015;11:2234–2240. doi: 10.3892/mmr.2014.3012. [DOI] [PubMed] [Google Scholar]
- 26.Ando M, Ibayashi K, Minami N, Nakamura T, Isogai K, Yoshimura H. The syntheses of 11β,13-dihydrokauniolide, estafiatin, isodehydrocostuslactone, 2-oxodesoxyligustrin, arborescin, 1,10-epiarborescin, 11β,13-dihydroludartin, 8-deoxy-11β,13-dihydrorupicolin B, 8-deoxyrupicolin B, 3,4-epiludartin, ludartin, kauniolide, dehydroleucodin, and leucodin. J Nat Prod. 1994;57:433–445. doi: 10.1021/np50106a001. [DOI] [Google Scholar]
- 27.Blanco JG, Gil RR, Alvarez CI, Patrito LC, Genti-Raimondi S, Flury A. A novel activity for a group of sesquiterpene lactones: inhibition of aromatase. FEBS Lett. 1997;409:396–400. doi: 10.1016/S0014-5793(97)00560-7. [DOI] [PubMed] [Google Scholar]
- 28.Giorgi A, Bassoli A, Borgonovo G, Panseri S, Manzo A, Pentimalli D, et al. Extracts and compounds active on TRP ion channels from Waldheimia glabra, a ritual medicinal plant from Himalaya. Phytomedicine. 2017;32:80–87. doi: 10.1016/j.phymed.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 29.Lone SH, Bhat KA, Shakeel UR, Majeed R, Hamid A, Khuroo MA. Synthesis and biological evaluation of amino analogs of ludartin: Potent and selective cytotoxic agents. Bioorg Med Chem Lett. 2013;23:4931–4934. doi: 10.1016/j.bmcl.2013.06.068. [DOI] [PubMed] [Google Scholar]
- 30.Lone SH, Bhat KA, Naseer S, Rather RA, Khuroo MA, Tasduq SA. Isolation, cytotoxicity evaluation and HPLC-quantification of the chemical constituents from Artemisia amygdalina Decne. J Chromatogr B Anal Technol Biomed Life Sci. 2013;940:135–141. doi: 10.1016/j.jchromb.2013.09.027. [DOI] [PubMed] [Google Scholar]
- 31.Huang LX, Zhong MY, Dan D, Yang XM, Qiu H, Guo PZ. Combination of capecitabine and ludartin inhibits colon cancer growth in mice. Trop J Pharm Res. 2017;16:2623–2628. doi: 10.4314/tjpr.v16i11.8. [DOI] [Google Scholar]
- 32.Lone SH, Bhat KA, Majeed R, Hamid A, Khuroo MA. Click chemistry inspired facile synthesis and bioevaluation of novel triazolyl analogs of ludartin. Bioorg Med Chem Lett. 2014;24:1047–1051. doi: 10.1016/j.bmcl.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Kuang B, Kang N, Ding Y, Ge W, Lian L, et al. Synthesis and anti-acute myeloid leukemia activity of C-14 modified parthenolide derivatives. Eur J Med Chem. 2017;127:296–304. doi: 10.1016/j.ejmech.2016.12.044. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Li S, Zhao J, Yang H, Yin F, Ding M, et al. Design and SAR of withangulatin A analogues that act as covalent TrxR inhibitors through the Michael addition reaction showing potential in cancer treatment. J Med Chem. 2020;63:11195–11214. doi: 10.1021/acs.jmedchem.0c01128. [DOI] [PubMed] [Google Scholar]
- 35.Ding Y, Li S, Ge W, Liu Z, Zhang X, Wang M, et al. Design and synthesis of parthenolide and 5-fluorouracil conjugates as potential anticancer agents against drug resistant hepatocellular carcinoma. Eur J Med Chem. 2019;183:111706. doi: 10.1016/j.ejmech.2019.111706. [DOI] [PubMed] [Google Scholar]
- 36.Gao F, Sun Z, Kong F, Xiao J. Artemisinin-derived hybrids and their anticancer activity. Eur J Med Chem. 2020;188:112044. doi: 10.1016/j.ejmech.2020.112044. [DOI] [PubMed] [Google Scholar]
- 37.Ding Y, Xue Q, Liu S, Hu K, Wang D, Wang T, et al. Identification of parthenolide dimers as activators of pyruvate kinase M2 in xenografts of glioblastoma multiforme. vivo J Med Chem. 2020;63:1597–1611. doi: 10.1021/acs.jmedchem.9b01328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.