

Summary
Genome phasing is a recently developed assembly method that separates heterozygous eukaryotic genomic regions and builds haplotype‐resolved assemblies. Because differences between haplotypes are ignored in most published de novo genomes, assemblies are available as consensus genomes consisting of haplotype mixtures, thus increasing the need for genome phasing. Here, we review the operating principles and characteristics of several freely available and widely used phasing tools (TrioCanu, FALCON‐Phase, and ALLHiC). An examination of downstream analyses using haplotype‐resolved genome assemblies in plants indicated significant differences among haplotypes regarding chromosomal rearrangements, sequence insertions, and expression of specific alleles that contribute to the acquisition of the biological characteristics of plant species. Finally, we suggest directions to solve addressing limitations of current genome‐phasing methods. This review provides insights into the current progress, limitations, and future directions of de novo genome phasing, which will enable researchers to easily access and utilize genome‐phasing in studies involving highly heterozygous complex plant genomes.
Keywords: de novo phasing, haplotype‐resolved assembly, autopolyploid, chromosomal rearrangement, haplotype‐specific sequence insertion, allele‐specific expression, plant genome
Introduction
The construction of high‐quality genome assemblies is now possible due to recent advances in sequencing technologies that enable the generation of long‐read sequences (LRS), including single‐molecule real‐time sequencing (SMRT), and Oxford Nanopore Technologies (ONT), with chromosome‐scale mate‐pair reads such as high‐throughput chromatin conformation capture (Hi‐C). However, most genome assemblies currently available for highly heterozygous diploid or polyploid species contain crucial errors because haplotype differences were ignored during the de novo assembly process that generates consensus chimeric sequences without separating haplotype alleles (Church et al., 2011, 2015; Korlach et al., 2017). Several reports have described phasing approaches for constructing haplotype‐resolved genome assemblies by distinguishing sequences of haplotype alleles inherited through maternal and paternal genetic materials (Edge et al., 2017; Koren et al., 2018; Kronenberg et al., 2021; Patterson et al., 2015; Shi et al., 2019; Zhang et al., 2019b). Since the launching of the international HapMap project in 2002 (International HapMap et al., 2007), many genomes, particularly those of animals, have been assembled at the haplotype level and deposited in public databases (Bredemeyer et al., 2021; Cao et al., 2015; Mott, 2007). However, haplotype‐resolved genome assembly is not commonly used in plant research due to the highly non‐inbred character and complex genomic structures of plants (Kyriakidou et al., 2018; Van de Peer et al., 2017; Wu et al., 2018).
In plants, haplotype‐resolved genome assembly has been primarily implemented for diploid genomes using reference‐guided or de novo assembly based approaches. If a reference genome is available, haplotype structural variants can be identified by aligning reads with the reference genome. Each haplotype is grouped and assembled separately based on heterozygous variant sites. The generation of haplotype assemblies using the reference‐guided method demands less computational work; however, the results obtained depend on the quality and structural complexity of the reference genome. De novo assembly based phasing generates contigs from two or more parental haplotypes by unzipping heterozygous regions and grouping them into individual haplotypes using Hi‐C reads. If short‐read sequences (SRS) obtained from Illumina platform analyses of parents with LRS from SMRT or ONT of their offspring are available, accurate offspring phasing is possible by identifying sequences specific to each parent and classifying the contigs of the offspring from the maternal and paternal genomes. In addition to these approaches, gamete‐cell sequencing can be used to generate raw sequences of each haplotype; although this is an ideal method for constructing haplotype‐level assemblies, it has technical limitations (Iqbal et al., 2020; Jia et al., 2016; Shi et al., 2019).
Previous studies reported plant genomes assembled as chromosome‐scale haplotypes and demonstrated the importance of phasing by comparing each haplotype allele with the consensus assembly. Specifically, structural variations (SVs) between haplotype alleles such as chromosomal rearrangements and single‐nucleotide polymorphisms (SNPs) that might have been disregarded during consensus genome assembly have been identified. Based on the variation in gene level, a number of presence‐absence variations (PAVs), allele‐specific expression (ASE) patterns, and dominant‐recessive alleles that contribute to phenotype alternation also have been identified (Hasing et al., 2020; Seo et al., 2016; Zhou et al., 2020). Here, we review various de novo assembly based phasing methods, focusing primarily on the most popular and freely available pipelines: TrioCanu (Koren et al., 2018), FALCON‐Phase (Kronenberg et al., 2021), and ALLHiC (Zhang et al., 2019b). Input data, operating processes, output characteristics, and limitations are discussed to provide a guide for novice researchers engaged in plant genome phasing. Finally, we present cases that illustrate significant differences between haplotypes for genomic structures, gene repertoires, and gene expression, demonstrating the fundamental importance of genome phasing in plants.
Data preparation for de novo genome phasing pipelines for haplotype‐resolved assembly
The basic workflow for constructing a de novo genome assembly for each haplotype allele generally consists of (1) sequencing read data, (2) assembly and phasing, (3) scaffolding, and (4) post‐processing (Figure 1). This workflow is similar to the general de novo assembly process but implements an additional process to assign contigs to the appropriate haplotypes to achieve the correct genome constitution. Although a variety of de novo phasing tools are available, we focused on illustrating the operation and characteristics of three major and freely available tools, the TrioCanu (Koren et al., 2018), FALCON‐Phase (Kronenberg et al., 2021), and ALLHiC (Zhang et al., 2019b) pipelines.
Figure 1.
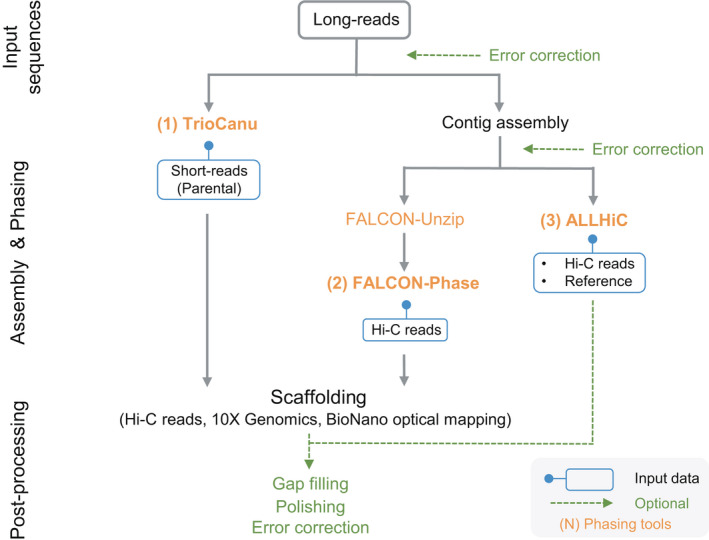
De novo genome phasing using TrioCanu, FALCON‐Phase, and ALLHiC pipelines. Diagram depicts the simplified genome phasing process using TrioCanu, FALCON‐Phase, and ALLHiC. The three major phasing tools are highlighted in orange. All phasing tools use long‐read sequences (LRS) or assembled contigs as basic input data. Additional input data for each tool is indicated in the blue box below the tool name. TrioCanu uses LRS of the target genome and short‐read sequences (SRS) of the parental genomes to construct haplotype‐resolved genome assemblies. FALCON‐Phase requires Hi‐C reads and the results from FALCON and FALCON‐Unzip. Because TrioCanu and FALCON‐Phase results are generated at the contig level, scaffolding with Hi‐C reads, 10× Genomics, and/or BioNano optical mapping are options for constructing chromosome‐scale assemblies. ALLHiC uses assembled contigs, Hi‐C reads, and genomic data of closely related species to construct haplotype‐resolved genome assemblies at the chromosome level without an additional scaffolding step. Optional steps to improve the quality of haplotype‐resolved genomes are indicated by green dashed lines.
The first step in the haplotype‐resolved assembly process is preparing the read data. The three pipelines basically require LRS, such as SMRT or ONT, which are more suitable than SRS given the complex structure of the plant genome, which includes a large number of repeat or heterozygous regions (Van de Peer et al., 2017). An advanced option that can be used for high‐quality genome phasing is PacBio High‐Fidelity (HiFi) reads, which is the latest sequencing technology and exhibits high accuracy (up to 99.8%; Wenger et al., 2019). A recent study reported an improved version of the haplotype‐resolved human genome using HiFi reads (Nurk et al., 2020). In addition to the LRS, the SRS of each parent species are essential when using the TrioCanu pipeline, and FALCON‐Phase and ALLHiC require input sequences of Hi‐C reads. When preparing the raw sequences for TrioCanu, preparation of parental SRS is recommended with >30× coverage for each parent and >40× LRS coverage for the target offspring (Koren et al., 2018). When using FALCON‐Phase, PacBio LRS or HiFi reads are needed with >60× or >30× coverage, respectively (Kronenberg et al., 2021).
Phasing process using the TrioCanu, FALCON‐Phase, and ALLHiC pipelines
The LRS of a diploid target genome can be classified into maternal and paternal haplotypes based on the SRS of the target’s parents using the TrioCanu module in the Canu assembler. Prior to contig assembly, TrioCanu constructs a list of k‐mer subsequences from the two parental SRS datasets and identifies parent‐specific k‐mers as distinctive markers to infer which offspring LRS are derived from which parental haplotype. The LRS of the offspring are then assigned to the maternal and paternal haplotype groups based on parent‐unique k‐mers (Figure 2a). Finally, the offspring LRS in individual haplotype groups are assembled using Canu. Because the offspring LRS in each haplotype are provided, users can assemble haplotype groups using other long‐read assemblers such as FALCON (Chin et al., 2016) or Flye (Kolmogorov et al., 2019). Although highly accurate assemblies are possible with TrioCanu, the pipeline is difficult to use if the raw data of the parents are unavailable. To assemble without parental sequence information, FALCON‐Phase and ALLHiC were developed to generate haplotype‐resolved genome assemblies using Hi‐C reads instead.
Figure 2.
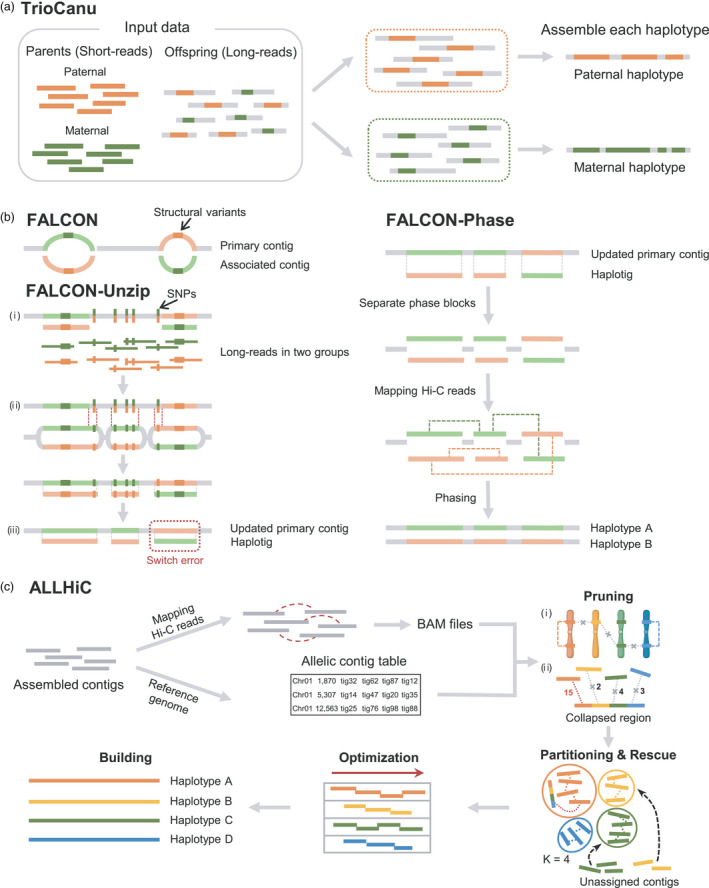
Detailed phasing pipelines of TrioCanu, FALCON‐Phase, and ALLHiC. (a) In TrioCanu, target offspring LRS and parental SRS are used as input data. Paternal and maternal SRS are represented in orange and green, respectively. The offspring LRS are partitioned into paternal and maternal groups based on parental‐specific k‐mers. Partitioned offspring LRS in each group are assembled into each respective haplotype. (b) The FALCON‐Phase pipeline proceeds according to the order FALCON, FALCON‐Unzip, and FALCON‐Phase. In each contig, the heterozygous regions are shown in light green and orange, and structural variants and SNPs in heterozygous regions are shown in the same dark colors. FALCON generates two types of contigs: primary and associated contigs. Associated contigs are separated from primary contigs during FALCON assembly because of structural variants between haplotypes. FALCON‐Unzip consists of three steps: (i) heterozygous SNPs are detected by mapping LRS to the primary and associated contigs obtained from FALCON, (ii) heterozygous regions containing SNPs (indicated by red dashed lines) are unzipped to updated primary contigs and haplotigs, and (iii) switch error is calculated between updated primary contigs and haplotigs as depicted in the dashed red box. In FALCON‐Phase, updated primary contigs and haplotigs are separated into phase blocks and a collapsed region (grey). Hi‐C reads, which are marked with dashed green and orange lines, are mapped to the phase blocks. (c) ALLHiC uses assembled contigs, Hi‐C reads, and the reference genome of the target species as input data. By mapping Hi‐C reads to assembled contigs, linkage information between contigs can be saved as BAM files. An allelic contig table is generated using assembled contigs and the reference genome. ALLHiC resolves polyploid genomes into each haplotype via five steps (pruning, partitioning, rescue, optimization, and building). Different haplotype chromosomes and contigs are depicted in different colors. Hi‐C linkages between contigs are depicted with dashed lines. Hi‐C signals of Hi‐C linkages are shown as numbers next to the Hi‐C linkages. In the pruning step: (i) Hi‐C linkages between contigs in different haplotypes are removed and (ii) collapsed regions in which some of all the haplotype regions are fused have Hi‐C linkages with contigs in all haplotypes. The Hi‐C linkage with the strongest Hi‐C signal is marked by a red dashed line. Pruned contigs are partitioned in each haplotype group based on the K number (e.g., K = 4) in the partitioning step, and unassigned contigs are assigned to appropriate groups in the rescue step. The order and orientation of partitioned contigs are optimized in the optimization step. Finally, a haplotype‐resolved genome at the chromosome level can be achieved. The figure is adapted from previous studies (Koren et al., 2018; Kronenberg et al., 2021; and Zhang et al., 2019b) and created with BioRender.com.
FALCON‐Phase was designed to classify primary and associated contigs from the FALCON pipeline and generate individual haplotype assemblies for diploid genomes. FALCON basically assembles LRS into consensus sequences (referred to as primary contigs) based on a string graph regardless of haplotype variations. During graph construction, the large heterozygous variations, such as structural variants, are detected and separated into primary and associated contigs (Figure 2b). Because haplotype‐fused contigs will remain as a result of ignoring small heterozygous variations such as SNPs, the FALCON module FALCON‐Unzip can be used to identify haplotype‐fused regions by mapping LRS to the assembly, including both primary and associated contigs. In addition, FALCON‐Unzip divides mapped LRS into two groups for each haplotype‐fused region, given the small heterozygous variations (Figure 2b). FALCON‐Unzip then reassembles the LRS in the two groups for each haplotype‐fused region and generates additional primary and associated contigs, which are fused in the same contigs (Figure 2b). Finally, updated primary contigs and haplotigs (updated associated contigs) are constructed. In the updated contigs, it is not determined which haplotype generates which primary contigs or haplotigs, and this generates switch errors because FALCON‐Unzip divided the fused regions into two groups without assignments in specific haplotypes (Figure 2b). FALCON‐Phase corrects these switch errors and generates two complete haplotype assemblies via Hi‐C reads mapping to the updated primary contigs and haplotigs generated by FALCON‐Unzip. Specifically, the assembled contigs obtained from FALCON‐Unzip are separated and classified as phase blocks and collapsed regions (homozygous contigs; Figure 2b). The phase blocks are assigned to each haplotype group based on the results of Hi‐C reads mapping (Figure 2b). By merging the collapsed regions (shown in grey in Figure 2b) into two sets of resolved haplotype phase blocks, FALCON‐Phase generates two haplotype‐resolved contig sets (Figure 2b). FALCON‐Phase requires FALCON‐Unzip result files, so users should assemble the LRS using FALCON and FALCON‐Unzip before genome phasing using FALCON‐Phase. Both TrioCanu and FALCON‐Phase generate haplotype‐resolved assemblies at the contig level. To construct chromosome‐level genome assemblies, contigs should be ordered and oriented using additional data for scaffolding, such as 10× Genomics Linked‐Reads and/or BioNano optical mapping (Figure 1).
ALLHiC is a de novo phasing tool that generates haplotype‐resolved assemblies for complex polyploid species beyond diploid. ALLHiC was developed to enable the construction of chromosome‐level assemblies of individual haplotypes using Hi‐C reads without any additional scaffolding step. Unlike TrioCanu and FALCON‐Phase, which utilize raw sequences of the target genome, ALLHiC requires previously assembled contigs, Hi‐C reads of the target species, and protein‐coding gene sequences of a close relative with their genomic positions as input data (Figure 2c). Before performing ALLHiC phasing, users prepare the input assembly that contains as many of the heterozygous contigs as possible to obtain more accurate individual assemblies, including specific sequences in individual haplotypes. Then, Canu assembler with the polyploid parameter can be one option to secure possible heterozygous contigs. ALLHiC cannot handle erratic sequences in the input assembly, such as haplotype‐fused contigs; therefore, it is recommended that those erratic regions are corrected in contigs via Hi‐C read mapping using juicer, juicebox, or 3D‐DNA pipelines. A recent study showed that using error‐corrected contigs from the 3D‐DNA pipeline as input data could improve the quality of the haplotype‐resolved tea plant genome (Zhang et al., 2021). After preparing the input data, ALLHiC first performs Hi‐C reads mapping to assembled contigs and then saves linkage information for the contigs in the same and different haplotypes in BAM file format (Figure 2c). Allelic contig information is then generated, providing the names of contigs located at allelic positions by comparing the input assembly to the coding sequences of a closely related species. Based on the Hi‐C mapping data in BAM files and the allelic contig information, the pruning step, which involves resolving haplotypes in the ALLHiC pipeline, is implemented to retain Hi‐C linkages between the same haplotype contigs after removal of linkages between different haplotypes. Specifically, ALLHiC first removes the Hi‐C linkages between allelic contigs based on the allelic contig information (Figure 2c). The Hi‐C linkages between contigs in different haplotypes are primarily removed based on the Hi‐C signal score (the number of read pairs per length of the contiguous contig) of the linkages to prevent the generation of haplotype‐fused chromosomes (Figure 2c). The pruned contigs are partitioned into each haplotype set according to the user‐defined group number (K), generally specified as the number of chromosomes (Figure 2c). Because contigs in the same haplotype but with low Hi‐C signals are unassigned into haplotype groups, the rescue process assigns unassigned contigs to the partitioned haplotype groups based on the Hi‐C signal in the original BAM file data (Figure 2c). After determining the order and orientation of the contigs partitioned in each haplotype group, a chromosome‐level haplotype‐resolved assembly is generated and saved in a single FASTA file (Figure 2c). If the number of haplotype sequences in the FASTA file does not match the number of chromosomes, users need to perform additional curation to correct the separation of haplotypes. For example, the haplotype‐resolved genome of autotetraploid sugarcane (Saccharum spontaneum) was assembled into 48 super‐scaffolds containing 32 chromosomes with 16 super‐scaffolds after running ALLHiC (Zhang et al., 2018). The authors manually assigned unanchored super‐scaffolds into 32 chromosomes when the specific scaffold(s) had the best Hi‐C signal score with a specific chromosome (Zhang et al., 2018). Finally, users can determine which haplotypes are grouped into the homologous chromosomes in the FASTA file using a dot plot.
Evaluating the accuracy of the phasing tools
Previous studies generally validated individual haplotype assemblies using the three tools examined in this study via comparison to the parental genomes as a means of evaluating phasing accuracy. Koren et al. (2018) validated the F1 haplotypes from the two accessions of Arabidopsis thaliana (Col‐0 and Cvi‐0) via comparison with the parental data using TrioCanu. Haplotype A was covered with 99.5% of Col‐0, and haplotype B was covered with 99.0% of Cvi‐0. Kronenberg et al. (2021) also evaluated the accuracy of the haplotype‐resolved F1 genomes using FALCON‐Phase via comparison with the parental genomes. Phasing accuracy was examined using zebra finch, cow, and human samples (HG00733 and mHomSap3). The authors reported phasing accuracies of 91%, 96%, 80%, and 91% for zebra finch, cow, HG00733, and mHomSap3, respectively. To evaluate the accuracy of phasing using ALLHiC, Zhang et al. (2019b) combined previously assembled Oryza sativa spp. Japonica and O. sativa spp. Indica genomes as a synthetic genome for both rice subspecies. They tested the efficiency of the pruning process and reported a reduced ratio of Hi‐C linkages between different subspecies, suggesting that the pruning step improved the phasing accuracy of ALLHiC. Based on the evaluations of several cases, these results suggest that the three tools achieve highly accurate genome phasing. However, these results are not sufficient to validate that the three tools enable accurate genome phasing for complex genomes because the evaluation cases are limited to relatively small genomes of specific species that exhibit low complexity compared with polyploid species.
Uncovering genomic information for individual haplotypes
General de novo genome‐assembly methods are designed to generate collapsed monoploid assemblies, ignoring differences between haplotypes (Michael and VanBuren, 2020). Because the majority of published genomes were assembled ignoring differences between haplotypes, resulting in chimeric monoploid genome assemblies, these genomes could contain critical errors, such as (i) incorrect chromosome structures that result from ignoring rearrangements between individual haplotypes, especially in autopolyploid species (Tang, 2017); (ii) distorted gene repertoires resulting from the omission of genes in unassembled haplotype regions; and (iii) the presence of chimeric sequences generated by ignoring variations between haplotypes, thus hindering the identification of important haplotype characteristics such as PAV and ASE (Seo et al., 2016). In this review, we examined 9 published plant haplotype‐resolved de novo assemblies and found significant differences between haplotypes (Table 1). The difference in total length between haplotypes ranged from 1 Mb (Medicago sativa L.) to 130 Mb (Camellia sinensis; Table 1). The copy number variations of annotated genes between haplotypes were ranged from a minimum of 13 in Vanilla planifolia to a maximum of 6,964 in C. sinensis (Table 1). Genomic variations between haplotypes such as SNPs, indels, SVs, and PAVs also have been reported (Zhou et al., 2020). To demonstrate the importance of constructing haplotype‐resolved assemblies, we investigated and discussed significant differences between haplotypes such as chromosomal structures, haplotype‐specific insertions, PAVs, and ASEs through specific examples.
Table 1.
Summary of available haplotype‐resolved plant genome assemblies
| Scientific name | Ploidy | Phased in chromosome‐level | Genome size (Mb) | Number of genes | Phasing pipeline | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HA | HB | HC | HD | HA | HB | HC | HD | |||||
| Vanilla planifolia | Diploid | O | 737 | 744 | – | – | 29 167 | 29 180 | – | – | FALCON‐Phase | Hasing et al. (2020) |
| Hydrangea macrophylla | Diploid | X | 2256 | 2227 | – | – | 32 205 | 32 222 | – | – | FALCON‐Phase | Nashima et al. (2021) |
| Cerasus × yedoensis | Diploid | X | 350 | 340 | – | – | 48 280 | 46 796 | – | – | TrioCanu | Shirasawa et al. (2019) |
| Malus domestica cv. Gala a | Diploid | O | 658 | 577 | – | – | 46 165 | 40 018 | – | – | DeNovo‐MAGIC3 | Zhang et al. (2019a) |
| Solanum tuberosum L. a | Diploid | O | 810 | 800 | – | – | 37 115 | 37 094 | – | – | ALLHiC | Zhou et al. (2020) |
| Saccharum spontaneum L. a | Tetraploid | O | 734 | 744 | 723 | 698 | 21 829 | 21 182 | 20 079 | 20 736 | ALLHiC | Zhang et al. (2018) |
| Medicago sativa L. a | Tetraploid | O | 679 | 700 | 682 | 676 | 39 532 | 40 180 | 39 982 | 39 200 | ALLHiC | Chen et al. (2020) |
| Camellia sinensis a | Diploid | O | 3058 | 2928 | – | – | 29 792 | 22 828 | – | – | ALLHiC | Zhang et al. (2021) |
| Dendrocalamus latiflorus Munro b | Allohexaploid | O | 1374 | 1363 | – | – | 67 646 | 67 585 | – | – | ALLHiC | Zheng et al. (2022) |
Unanchored scaffolds and genes were excluded in statistics.
Statistics for allohexaploid bamboo (A1A2B1B2C1C2) were subdivided into diploid subgenomes (HA; A1B1C1 and HB; A2B2C2).
Chromosomal rearrangements
Genome phasing enables the construction of precise chromosome structures for individual haplotypes, particularly for autopolyploid species. Plant genomes often undergo basic chromosome fission and fusion during evolution (Jones, 1998). These phenomena contribute to the wide range of genomic diversity observed in plants, including basal chromosome number variation and polyploidization, and serve as important mechanisms of interspecies hybridization and family ploidy speciation (Keeler and Cheplick, 1998; Svacina et al., 2020). Although autopolyploid species contain three or more chromosome pairs, current assembly methods only provide single representative chromosomal structures that include haplotype chimeras but ignore rearrangements (Tang, 2017). However, haplotype‐resolved assemblies could be used to distinguish chromosomal rearrangements between haplotypes in sugarcane, a complex autotetraploid species (Zhang et al., 2018).
Zhang et al. (2018) utilized the ALLHiC pipeline to assemble the sugarcane genome using a combination of SRS and LRS phased into eight chromosome sets, each consisting of four homologous alleles. Specifically, they identified inversions among homologous alleles on chromosomes 2, 6, and 7 during two rounds of whole‐genome duplication (WGD; Figure 3). Based on genomic evidence obtained by comparing individual haplotype structures, they clearly demonstrated that chromosomal inversions had occurred in the lower and upper regions of chromosomes 2 and 7 (Ss2 and Ss7), respectively, during the first WGD, which was followed by the second WGD that resulted in two inversions among a total of four haplotypes (Figure 3a). An additional inversion had occurred during the second WGD in the upper region of chromosome 6 (Ss6); consequently, one of the four haplotypes of chromosome 6 (Ss6_C) contained an inverted chromosome region (Figure 3b). These results demonstrate that genome phasing provides unprecedented genomic resources and enables the detection of comprehensive genome rearrangements in complex autotetraploid species by investigating chromosomal structures of individual haplotypes in detail.
Figure 3.
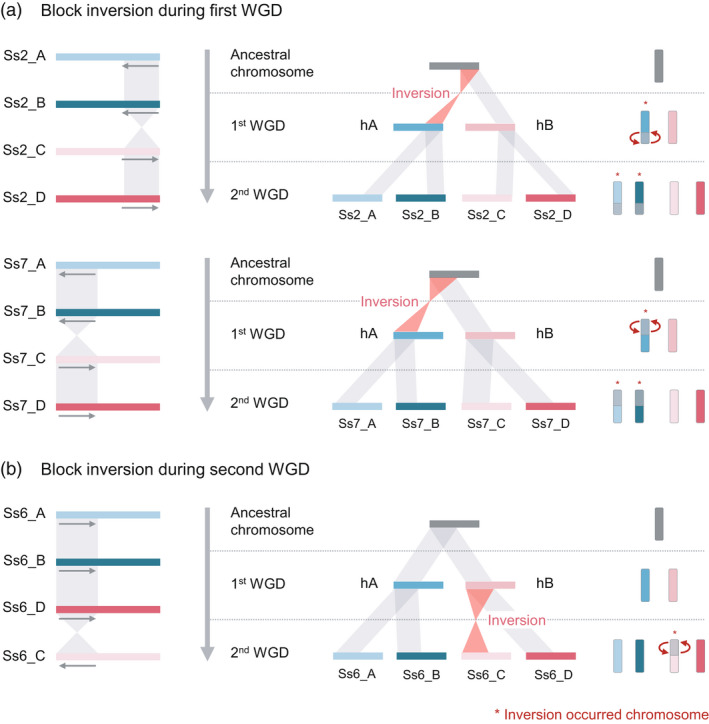
Chromosome rearrangements between sugarcane haplotypes. (a, b) Colored bars represent each of four haplotype chromosomes of sugarcane (Ss: Saccharum spontaneum). Chromosome numbers and haplotypes are presented next to Ss and underlined, respectively. Left and middle alignments show the synteny block between the four haplotypes and the predicted ancestral chromosomes. The arrow on the left under the haplotype bars indicates the direction of the alignment. The grey‐shaded area shows the synteny region, and the red‐shaded area indicates the inverted region. (a) Scheme of chromosomal inversion during the first whole‐genome duplication (WGD) of sugarcane. The diagram shows that the long arm (short arm) in the ancestral haplotype for Ss2_A‐B (Ss7_A‐B) was inverted during the first WGD; the subsequent second WGD caused the duplication of the ancestral haplotypes hA to Ss2_A‐B (Ss7_A‐B) and hB to Ss2_C‐D (Ss7_C‐D), resulting in chromosome rearrangement among the haplotypes. (b) Chromosomal inversion during the second WGD of sugarcane. The Ss6 haplotypes on the left side show the reverse synteny in the short arm between Ss6_A‐B‐D and Ss6_C. The diagram on the right side depicts chromosome inversion specifically occurring in Ss6_C during the second WGD. The figure is adapted from a previous study (Zhang et al., 2018).
Haplotype‐specific sequence insertions
Haplotype‐specific insertions, which are a type of SV, induce sequence dissimilarities between haplotypes; these dissimilarities, in turn, affect a variety of biological characteristics, including plant phenotype (Stancu et al., 2017). Because consensus genome assemblies include one of the heterozygous regions in one of the haplotypes, haplotype‐specific insertions may go undetected, ultimately leading to the potential omission of genomic regions related to important traits. However, haplotype‐resolved genome assemblies reflect both regions, thereby enabling the identification of trait‐related genomic regions in a specific haplotype. Two examples of haplotype‐specific insertion in allele‐associated genomic regions that affect phenotypic alterations are described below (Figure 4).
Figure 4.
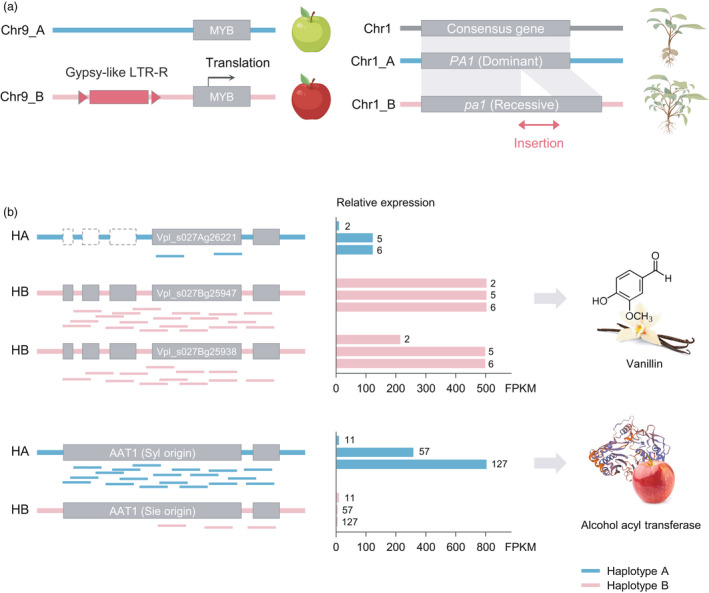
Haplotype‐specific sequence insertion and allele‐specific expression. (a) Schematic diagram illustrating phenotypic changes mediated by haplotype‐specific sequence insertion. The left scheme shows that the Gypsy‐like long terminal repeat retrotransposon (LTR‐R) is inserted upstream of the MYB gene in haplotype B of chromosome 9 in the Gala apple genome. The recessive allele pa1 (57‐bp long) in potato resulted in the insertion of a sequence encoding an additional 19 amino acids compared with the dominant allele PA1 (right). (b) Examples of allele‐specific expression between haplotypes. The scheme on the left side shows differentially mapped RNA‐seq data between haplotypes. Grey boxes represent exons, and dotted boxes indicate the absence of exons in the corresponding allelic position. In graphs on the right side, the numbers at the end of each relative expression bar indicate seed tissue 2, 5, and 6 months after pollination in vanilla (top) and Gala fruit (bottom) 11, 57, and 127 days after full bloom, respectively. The figure is adapted from previous studies (Sun et al., 2020; Hasing et al., 2020; and Zhou et al., 2020) and created with BioRender.com.
Sun et al. (2020) reported the haplotype‐resolved genome of the Gala variety of apple, which was assembled using DeNovoMAGIC3 (NRGene), a commercial phasing method based on Illumina SRS, 10× Genomics reads, and PacBio HiFi reads. They observed a specific long terminal repeat retrotransposon (LTR‐R), denoted as redTE insertion, upstream of the MYB gene in haplotype B of chromosome 9, which is related to color determination in fruit (Figure 4a). This was consistent with the results of a previous study reporting that the yellowish‐red color of Gala apples derives from the insertion of a single redTE LTR‐R on a single side of the haplotype upstream of the MYB gene (Mdg_09g022880), which has a crucial role in determining red skin color in fruit (Zhang et al., 2019a). They found that redTE was present only in Malus sieversii and Malus domestica, suggesting that the haplotype B‐specific redTE insertion region originated from the M. sieversii haplotype, as demonstrated by comparing the wild‐type progenitor (M. sieversii and Malus sylvestris) haplotypes and the Gala apple haplotypes (Sun et al., 2020). These results demonstrate that haplotype‐resolved assembly can provide accurate information regarding variations between alleles that have vital roles in the emergence of functional genes related to important agricultural traits.
The sequence diversity between dominant and recessive genes associated with growth vigour in potatoes has been characterized (Zhou et al., 2020). Diploid potato (RH89‐039‐16) was sequenced using a combination of strategies, including Illumina whole‐genome sequencing, 10× Genomics linked‐reads, ONT, and circular consensus sequencing (CCS), and haplotypes were determined using the ALLHiC pipeline. The authors identified a 57‐bp nucleotide insertion in pa1 (RHC01H2G0765.2, recessive allele) that resulted in the inclusion of 19 extra amino acids in the protein encoded by the recessive allele compared with PA1 (RHC01H1G0699.2, dominant allele; Figure 4a). This result was obtained via haplotype‐resolved assembly of the potato genome and provided accurate information regarding the PAV of specific sequences in a trait‐related gene.
Allele‐specific expression
Not all alleles are expressed, even for the same allele in each haplotype (Springer and Stupar, 2007). Thus, genes that are actually expressed in specific haplotype(s) may not be identified in the consensus genome assembly containing a bisected gene annotation. However, haplotype‐resolved genome assembly enables the entire gene repertoire of an individual haplotype to be determined, thereby enhancing our understanding of accurate allele expression.
The CPLP gene in the vanilla genome is an intuitive example to verify ASE in a specific haplotype. Hasing et al. (2020) reported a draft haplotype‐resolved vanilla genome assembled via ONT LRS and Illumina SRS and phased via FALCON‐Phase. They found that CPLP alleles (vanillin biosynthesis‐related alleles) were differentially expressed in each haplotype (Figure 4b). Vpl_s027Ag26221, a candidate CPLP allele in haplotype A, exhibited relatively low transcript abundance compared with candidate CPLP alleles Vpl_s027Bg25947 and Vpl_s027Bg25938 in haplotype B. Vpl_s027Bg25947 was highly expressed under all conditions examined, whereas Vpl_s027Bg25938 was highly expressed in seed tissues 5–6 months after pollination. These results illustrate the contribution of ASE to vanillin biosynthesis in haplotype B. Another example of ASE is the gene encoding alcohol acyltransferase in Gala apple (AAT1) (Sun et al., 2020). Although the sequences of both AAT1 alleles (control ester production in Gala apple) are similar, the haplotype A AAT1 (derived from M. sylvestris) is expressed at a higher level than haplotype B AAT1 (derived from M. sieversii; Figure 4b). The ASE of AAT1 alleles was due to the insertion of a specific sequence in the upstream region of haplotype A AAT1 derived from M. sylvestris. These examples highlight the importance of understanding ASE as it relates to trait regulation and using haplotype‐resolved assembly to accurately estimate the expression levels of each allele in individual haplotypes.
A comprehensive genome‐editing protocol for complex plant genomes
The speed of genome editing can be increased using haplotype‐resolved assembly based on precise genome sequence information for individual haplotypes, and this is particularly useful for polyploid species with a complex genome structure (Chen et al., 2020). For accurate CRISPR/Cas9‐based genome editing, identifying a specific genome target site is an essential prerequisite. However, because polyploid or highly heterozygous diploid plants generally harbor numerous dissimilar alleles, the target region may not cover all these alleles, which can ultimately decrease the accuracy of editing when target sequences are determined using only a consensus reference genome. Therefore, Chen et al. (2020) suggested that target regions in dissimilar alleles should be identified using the haplotype‐resolved assembly of autotetraploid alfalfa (Medicago sativa L.; Figure 5).
Figure 5.
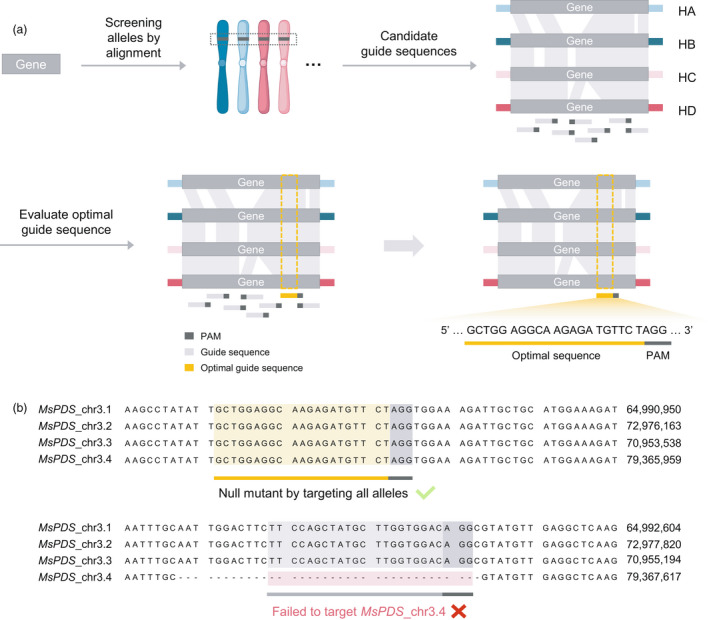
Genome‐editing protocol using haplotype‐resolved assembly of autotetraploid Medicago sativa L. (a) Protocol for obtaining optimal guide sequences for CRISPR/Cas9. Chromosome colors represent different haplotypes: HA, HB, HC, and HD. Dark‐grey bars on each haplotype indicate alleles. Grey‐shaded areas show corresponding blocks between alleles. (b) Validation of the optimal guide sequence targeting for all alleles. The top alignment is an optimal case in which sequences contain all alleles, and the bottom alignment is a suboptimal case in which sequences contain unmatched regions. Sequences following the suboptimal candidate are artificially generated for intuitive understanding. Numbers to the right of the sequence represent the chromosomal position. The figure is adapted from a previous study (Chen et al., 2020) and created with BioRender.com.
The cultivated alfalfa genome was assembled using PacBio CCS LRS and Illumina HiSeq2000 SRS, and constructed as a haplotype‐level assembly using ALLHiC (Chen et al., 2020). The authors established an efficient CRISPR/Cas9‐based genome‐editing protocol to accurately identify target regions covering four dissimilar alleles using the haplotype‐resolved genome assembly of alfalfa. Allele‐aware optimal guide sequences are designed as follows using this protocol (Figure 5): (i) homologous alleles of a target gene are screened based on alignments among allele sequences in each haplotype; (ii) multiple guide sequence candidates are extracted from these homologous alleles using Perl scripts (https://github.com/stanleyouth/‐/blob/master/crispr.sgRNA.finder.pl); and (iii) optimal guide sequence(s) are selected from among the multiple guide candidates based on specific evaluation criteria, such as covering all alleles, clear off‐target sites, guide sequence position, and conservation. Chen et al. (2020) verified the feasibility of this protocol for a clearly mutated gene in the alfalfa genome, MsPDS, which mediates the formation of pentafoliate leaves in legume plants (Chen et al., 2010). These results suggest that allele‐aware genome editing is an accurate and rapid alternative solution for molecular breeding of complex polyploid or highly heterozygous diploid plant genomes to speed up the breeding procedure by exactly targeting all alleles in the haplotype.
Conclusions and perspectives
Current advanced sequencing technologies generate considerably longer sequences and chromosome‐scale mate‐pairs than was possible with previously described methods. Current de novo assembly methods are designed to generate consensus monoploid assemblies by integrating genomic sequences without considering differences between haplotypes. This results in the generation of incorrect assemblies, especially for complex plant genomes. Genome phasing is a recently proposed alternative approach for constructing haplotype‐resolved assemblies for highly heterozygous genomes.
Our investigation revealed that the representative freely available and widely used phasing tools TrioCanu, FALCON‐Phase, and ALLHiC are suitable for assembling genomes of individual haplotypes, even in complex autopolyploid plant species beyond diploid plants. TrioCanu and FALCON‐Phase are designed for haplotype‐resolved assemblies only in diploid genomes. In TrioCanu, haplotype information from parental sequence data makes it possible to partition reads into haplotypes with high accuracy. FALCON‐Phase is developed to create a haplotype‐resolved genome by utilizing information on large SVs detected in FALCON and heterozygous SNPs detected in FALCON‐Unzip without parental data. Unlike the above two tools, ALLHiC is specially designed for polyploid genome phasing, which can be useful in phasing polyploid plant genomes. The results of downstream analyses using haplotype‐resolved assemblies indicated the importance of haplotype‐resolved assemblies in plants as tools for enhancing our understanding of the fundamental characteristics of species by identifying crucial variations among haplotypes. The results of haplotype sequence‐based genome‐editing studies suggest that haplotype‐resolved assemblies that generate optimal target sequences might be valuable tools for efficient breeding research for complex autopolyploid species.
Despite the necessity of haplotype‐resolved assemblies in plant research and breeding, there are two major limitations to consider for future studies: (i) the dependency of these tools in the phasing process and (ii) the lack of standard validation methods of phasing accuracy. Because TrioCanu relies on raw sequences of the parental species as input data for accurate phasing, the tool is difficult to use in cases where parental materials are not available. Unlike TrioCanu and ALLHiC, which can use any contig assemblers, FALCON‐Phase is compatible only with contig assemblies from FALCON, although more suitable tools can be used for contig assembly (Murigneux et al., 2020). ALLHiC overcomes the obstacle of TrioCanu and FALCON‐Phase, which can only be applied to the diploid genome; however, its application is restricted because of the requirement for whole‐gene information of closely related species. These dependency problems suggest a future direction for a phasing tool update based on the classification of parental‐specific haplotypes only using offspring sequences and the operation of phasing tools with any assemblers and without genomic information of closely related species.
The accuracy and quality of haplotype‐resolved assemblies were assessed as described above (the ‘Evaluating the accuracy of the phasing tools’ section). However, it is difficult to generalize and conclusively state that those phasing tools enable the construction of accurate haplotype‐resolved assemblies in most cases. Therefore, it is necessary to develop more robust approaches to estimate the accuracy level of haplotype‐resolved assemblies, suggesting the need for a qualified standard of phasing accuracy. A recent evaluation of phasing accuracy offers an alternative solution. Zhang et al. (2021) used a new tool called calc_switchErr to assess the phasing accuracy of the tea plant (C. sinensis) genome, which was resolved into haplotypes using ALLHiC. The calc_switchErr tool was designed to identify the oppositely located SNPs (designated as switched SNPs) and common SNPs (designated as truly phased SNPs). For example, if haplotype A and B after genome phasing have C and T in a specific site, but calc_switchErr detects T and C in the same position of haplotype A and B, respectively, this switched SNP is classified as an error. The calc_switchErr tool estimates the error rate by calculating the ratio of switched SNPs to truly phased SNPs [(the number of switched SNPs) × 100 ÷ (the total number of truly phased SNPs)]. The accuracy of the haplotype‐resolved tea plant genome was evaluated with a switch error rate of 5.9% (8473 of 144 868) which was much lower than the switch error rate of the monoploid genome (23.6%, 94 273 of 399 821) (Zhang et al., 2021). To estimate the phasing accuracy using the method for haplotype‐resolved plant genomes reviewed in this study, we used calc_switchErr to validate four plant genomes with raw sequences available in public databases (Table 2). Our results were slightly different from the results in a recent study due to differences in the input data used by the two studies (Zhang et al., 2021; Zheng et al., 2022). For example, we were unable to use 10× Genomics reads data, which were not available in public databases (Table 2). As a result, our data revealed specific scores for phasing errors of haplotype‐resolved assemblies, providing standards for researchers to evaluate the phasing quality of those assemblies. The calc_switchErr tool is open‐source and, thus, applicable in most cases to estimate the accuracy of haplotype‐resolved assemblies by calculating the error score.
Table 2.
Estimation of switch errors in published haplotype‐resolved genome assemblies using the calc_switchErr pipeline
| Scientific name | Phasing pipeline | Generated results in this study | Percentage of switch error in other studies (%) | ||
|---|---|---|---|---|---|
| Total number of SNPs | Total number of switched SNPs | Percentage of switch error (%) | |||
| Vanilla planifolia | FALCON‐Phase | 565 976 | 248 779 | 44.0 | NA |
| Malus domestica cv. Gala | DeNovoMAGIC3 | 639 051 | 141 656 | 22.2 | 29.6 (Zheng et al., 2022) |
| Solanum tuberosum L. | ALLHiC | 209 522 | 35 823 | 17.1 | 15.4 (Zheng et al., 2022) |
| Camellia sinensis | ALLHiC | 371 067 | 27 775 | 7.5 | 5.9 (Zhang et al., 2021) |
The evaluation of switch errors was only performed when both raw short‐ and long‐read sequencing data were available and assembly phased at the chromosome level.
10× Genomics‐linked raw reads data were excluded from input data during evaluation for the criteria consistency between the various phasing pipelines.
Our review provides new insights into the details of phasing principles to enhance the practical understanding of genomics researchers. We also provide valuable information regarding biological trait‐related sequences in haplotype‐resolved assemblies in plants. Research in plant genome phasing and the development of phasing pipelines are still in the early stages. We suggest that genome phasing will become indispensable for understanding biological phenomena and accelerating breeding research for complex plant species by overcoming current limitations.
Conflicts of interest
All authors declare they have no conflicts of interest.
Funding
This work was supported by a grant from the Korea Forest Service of the Korean Government through the R&D Program for Forestry Technology (grant number 2014071H10‐2022‐AA04 to S.K.), and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) (grant number NRF‐2017R1A6A3A04004014 to S.K.).
Author contributions
S.K. designed and conceptualized the review. J.‐Y.G., Y.M.L, and S.K. reviewed phasing operation principles, and M.‐J.J., J.‐W.C., and S.K. reviewed examples of downstream phasing analyses. J.‐Y.G and M.‐J.J wrote the initial manuscript, and all authors edited and reviewed the final version.
Acknowledgment
We thank the NBIT program at Kangwon National University for motivating this review.
Guk, J.‐Y. , Jang, M.‐J. , Choi, J.‐W. , Lee, Y. M. and Kim, S. (2022) De novo phasing resolves haplotype sequences in complex plant genomes. Plant Biotechnol. J., 10.1111/pbi.13815
Contributor Information
Ji‐Yoon Guk, Email: mentorlg195@gmail.com.
Min‐Jeong Jang, Email: minj.9411@gmail.com.
Jin‐Wook Choi, Email: argon502@gmail.com.
Yeon Mi Lee, Email: codsuak@gmail.com.
Seungill Kim, Email: ksi2204@uos.ac.kr.
References
- Bredemeyer, K.R. , Harris, A.J. , Li, G. , Zhao, L. , Foley, N.M. , Roelke‐Parker, M. , O'Brien, S.J. et al. (2021) Ultracontinuous single haplotype genome assemblies for the domestic cat (Felis catus) and Asian leopard cat (Prionailurus bengalensis). J. Hered. 112, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H.Z. , Wu, H.L. , Luo, R.B. , Huang, S.J. , Sun, Y.H. , Tong, X. , Xie, Y.L. et al. (2015) De novo assembly of a haplotype‐resolved human genome. Nat. Biotechnol. 33, 617–622. [DOI] [PubMed] [Google Scholar]
- Chen, H.T. , Zeng, Y. , Yang, Y.Z. , Huang, L.L. , Tang, B.L. , Zhang, H. , Hao, F. et al. (2020) Allele‐aware chromosome‐level genome assembly and efficient transgene‐free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.H. , Yu, J.B. , Ge, L.F. , Wang, H.L. , Berbel, A. , Liu, Y. , Chen, Y.H. et al. (2010) Control of dissected leaf morphology by a Cys(2)His(2) zinc finger transcription factor in the model legume Medicago truncatula . Proc. Natl Acad. Sci. 107, 10754–10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.S. , Peluso, P. , Sedlazeck, F.J. , Nattestad, M. , Concepcion, G.T. , Clum, A. , Dunn, C. et al. (2016) Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church, D.M. , Schneider, V.A. , Graves, T. , Auger, K. , Cunningham, F. , Bouk, N. , Chen, H.C. et al. (2011) Modernizing reference genome assemblies. PLoS Biol. 9, e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church, D.M. , Schneider, V.A. , Steinberg, K.M. , Schatz, M.C. , Quinlan, A.R. , Chin, C.S. , Kitts, P.A. et al. (2015) Extending reference assembly models. Genome Biol. 16, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge, P. , Bafna, V. and Bansal, V. (2017) HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 27, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasing, T. , Tang, H. , Brym, M. , Khazi, F. , Huang, T. and Chambers, A.H. (2020) A phased Vanilla planifolia genome enables genetic improvement of flavour and production. Nature Food, 1, 811–819. [DOI] [PubMed] [Google Scholar]
- InternationalHapMap, C. , Frazer, K.A. , Ballinger, D.G. , Cox, D.R. , Hinds, D.A. , Stuve, L.L. and Gibbs, R.A. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature, 449, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal, M.M. , Hurgobin, B. , Holme, A.L. , Appels, R. and Kaur, P. (2020) Status and potential of single‐cell transcriptomics for understanding plant development and functional biology. Cytometry Part A, 97, 997–1006. [DOI] [PubMed] [Google Scholar]
- Jia, X. , Zhang, X. , Qu, J. and Han, R. (2016) Optimization conditions of wheat mesophyll protoplast isolation. Agric. Sci. 7, 850–858. [Google Scholar]
- Jones, K. (1998) Robertsonian fusion and centric fission in karyotype evolution of higher plants. Bot. Rev. 64, 273–289. [Google Scholar]
- Keeler, K.H. and Cheplick, G.P. (1998) Population Biology of Intraspecific Polyploidy in Grasses. Cambridge: Cambridge University Press Cambridge. [Google Scholar]
- Kolmogorov, M. , Yuan, J. , Lin, Y. and Pevzner, P.A. (2019) Assembly of long, error‐prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. [DOI] [PubMed] [Google Scholar]
- Koren, S. , Rhie, A. , Walenz, B.P. , Dilthey, A.T. , Bickhart, D.M. , Kingan, S.B. , Hiendleder, S. et al. (2018) De novo assembly of haplotype‐resolved genomes with trio binning. Nat. Biotechnol. 36, 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach, J. , Gedman, G. , Kingan, S.B. , Chin, C.‐S. , Howard, J.T. , Audet, J.‐N. , Cantin, L. et al. (2017) De novo PacBio long‐read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. Gigascience, 6, gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg, Z.N. , Rhie, A. , Koren, S. , Concepcion, G.T. , Peluso, P. , Munson, K.M. , Porubsky, D. et al. (2021) Extended haplotype‐phasing of long‐read de novo genome assemblies using Hi‐C. Nat. Commun. 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakidou, M. , Tai, H.H. , Anglin, N.L. , Ellis, D. and Stromvik, M.V. (2018) Current strategies of polyploid plant genome sequence assembly. Front. Plant Sci. 9, 1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, T.P. and VanBuren, R. (2020) Building near‐complete plant genomes. Curr. Opin. Plant Biol. 54, 26–33. [DOI] [PubMed] [Google Scholar]
- Mott, R. (2007) A haplotype map for the laboratory mouse. Nat. Genet. 39, 1054–1056. [DOI] [PubMed] [Google Scholar]
- Murigneux, V. , Rai, S.K. , Furtado, A. , Bruxner, T.J.C. , Tian, W. , Harliwong, I. , Wei, H.M. et al. (2020) Comparison of long‐read methods for sequencing and assembly of a plant genome. Gigascience, 9, giaa146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashima, K. , Shirasawa, K. , Ghelfi, A. , Hirakawa, H. , Isobe, S. , Suyama, T. , Wada, T. et al. (2021) Genome sequence of Hydrangea macrophylla and its application in analysis of the double flower phenotype. DNA Res. 28, dsaa026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Walenz, B.P. , Rhie, A. , Vollger, M.R. , Logsdon, G.A. , Grothe, R. , Miga, K.H. et al. (2020) HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high‐fidelity long reads. Genome Res. 30, 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, M. , Marschall, T. , Pisanti, N. , Van Iersel, L. , Stougie, L. , Klau, G.W. and Schönhuth, A. (2015) WhatsHap: weighted haplotype assembly for future‐generation sequencing reads. J. Comput. Biol. 22, 498–509. [DOI] [PubMed] [Google Scholar]
- Seo, J.S. , Rhie, A. , Kim, J. , Lee, S. , Sohn, M.H. , Kim, C.U. , Hastie, A. et al. (2016) De novo assembly and phasing of a Korean human genome. Nature, 538, 243–247. [DOI] [PubMed] [Google Scholar]
- Shi, D. , Wu, J. , Tang, H. , Yin, H. , Wang, H. , Wang, R. , Wang, R. et al. (2019) Single‐pollen‐cell sequencing for gamete‐based phased diploid genome assembly in plants. Genome Res. 29, 1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasawa, K. , Esumi, T. , Hirakawa, H. , Tanaka, H. , Itai, A. , Ghelfi, A. , Nagasaki, H. et al. (2019) Phased genome sequence of an interspecific hybrid flowering cherry, ‘Somei‐Yoshino’(Cerasus× yedoensis). DNA Res. 26, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, N.M. and Stupar, R.M. (2007) Allelic variation and heterosis in maize: how do two halves make more than a whole? Genome Res. 17, 264–275. [DOI] [PubMed] [Google Scholar]
- Stancu, M.C. , van Roosmalen, M.J. , Renkens, I. , Nieboer, M.M. , Middelkamp, S. , de Ligt, J. , Pregno, G. et al. (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 8, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X. , Jiao, C. , Schwaninger, H. , Chao, C.T. , Ma, Y. , Duan, N. , Khan, A. et al. (2020) Phased diploid genome assemblies and pan‐genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svacina, R. , Sourdille, P. , Kopecky, D. and Bartos, J. (2020) Chromosome pairing in polyploid grasses. Front. Plant Sci. 11, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H.B. (2017) Disentangling a polyploid genome. Nature Plants, 3, 688–689. [DOI] [PubMed] [Google Scholar]
- Van de Peer, Y. , Mizrachi, E. and Marchal, K. (2017) The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. [DOI] [PubMed] [Google Scholar]
- Wenger, A.M. , Peluso, P. , Rowell, W.J. , Chang, P.C. , Hall, R.J. , Concepcion, G.T. , Ebler, J. et al. (2019) Accurate circular consensus long‐read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S. , Lau, K.H. , Cao, Q.H. , Hamilton, J.P. , Sun, H.H. , Zhou, C.X. , Eserman, L. et al. (2018) Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Zhang, X. , Tang, H. , Zhang, Q. , Hua, X. , Ma, X. , Zhu, F. et al. (2018) Allele‐defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Zhang, L.Y. , Hu, J. , Han, X.L. , Li, J.J. , Gao, Y. , Richards, C.M. , Zhang, C.X. et al. (2019a) A high‐quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.T. , Chen, S. , Shi, L.Q. , Gong, D.P. , Zhang, S.C. , Zhao, Q. , Zhan, D.L. et al. (2021) Haplotype‐resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis . Nat. Genet. 53, 1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Zhang, S. , Zhao, Q. , Ming, R. and Tang, H. (2019b) Assembly of allele‐aware, chromosomal‐scale autopolyploid genomes based on Hi‐C data. Nature Plants, 5, 833–845. [DOI] [PubMed] [Google Scholar]
- Zheng, Y. , Yang, D. , Rong, J. , Chen, L. , Zhu, Q. , He, T. , Chen, L. et al. (2022) Allele‐aware chromosome‐scale assembly of the allopolyploid genome of hexaploid Ma Bamboo (Dendrocalamus latiflorus Munro). J. Integr. Plant Biol. 64, 649–670. [DOI] [PubMed] [Google Scholar]
- Zhou, Q. , Tang, D. , Huang, W. , Yang, Z.M. , Zhang, Y. , Hamilton, J.P. , Visser, R.G.F. et al. (2020) Haplotype‐resolved genome analyses of a heterozygous diploid potato. Nat. Genet. 52, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]