

Summary
Carnation (Dianthus caryophyllus) is one of the most popular ornamental flowers in the world. Although numerous studies on carnations exist, the underlying mechanisms of flower color, fragrance, and the formation of double flowers remain unknown. Here, we employed an integrated multi‐omics approach to elucidate the genetic and biochemical pathways underlying the most important ornamental features of carnation flowers. First, we assembled a high‐quality chromosome‐scale genome (636 Mb with contig N50 as 14.67 Mb) of D. caryophyllus, the ‘Scarlet Queen’. Next, a series of metabolomic datasets was generated with a variety of instrumentation types from different parts of the flower at multiple stages of development to assess spatial and temporal differences in the accumulation of pigment and volatile compounds. Finally, transcriptomic data were generated to link genomic, biochemical, and morphological patterns to propose a set of pathways by which ornamental traits such as petal coloration, double flowers, and fragrance production are formed. Among them, the transcription factors bHLHs, MYBs, and a WRKY44 homolog are proposed to be important in controlling petal color patterning and genes such as coniferyl alcohol acetyltransferase and eugenol synthase are involved in the synthesis of eugenol. The integrated dataset of genomics, transcriptomics, and metabolomics presented herein provides an important foundation for understanding the underlying pathways of flower development and coloration, which in turn can be used for selective breeding and gene editing for the development of novel carnation cultivars.
Keywords: Dianthus caryophyllus, petal pigments, eugenol, petaloid stamens, genome
Introduction
Carnation (Dianthus caryophyllus L., 2n = 2x = 30) is one of the most important ornamental flowers worldwide, second only to rose in terms of popularity. Carnation has high economic and cultural value and is sold for multiple ornamental uses, including fresh‐cut flowers, potted plants, and landscaping. Carnation cultivation has a history of more than 2000 years. Carnation flowers are used to commemorate numerous varieties of ceremonies, events, and holidays such as in celebration of weddings in China, banquets in France, and ‘prom’ dances by students in the United States. Plant breeders have used interspecific hybridization to develop cultivars with novel floral and growth characteristics (Nimura et al., 2006; Sparnaaij et al., 1990).
The diversity of floral forms and fragrances has attracted humans for millennia, as evidenced by the myriad aesthetic uses of flowers in daily life. Color is among the most important characteristics that humans perceive when considering the aesthetic quality of a flower. Flower colors among carnation cultivars are very diverse and include red, pink, white, magenta, and yellow, as well as various color patterns consisting of multiple colors on the same petal. Since novel colors and/or patterns are highly sought after and can result in an increase in sales, researchers are constantly trying to develop new and interesting cultivars. Previous studies have reported the molecular mechanisms of carnations with pure color series, including white (Onozaki et al., 1999), pink (Mato et al., 2001), and yellow (Itoh et al., 2002; Yoshida et al., 2004). Based on these studies, a novel blue flower carnation was developed using genetic engineering techniques (Fukui et al., 2003; Matsuba et al., 2010). Breeding novel carnation cultivars will attract more attention and yield high economic value. However, the underlying molecular mechanisms of multicolor flowers must be understood urgently.
Floral morphology, one of the most attractive features among consumers, has also been an important topic for carnation researchers. The well‐known ABC model (Coen and Meyerowitz, 1991) and ‘Floral quartets’ model (Pelaz et al., 2000; Theißen and Saedler, 2001) in Arabidopsis showed that genes of classes A, A + B, B + C, and C are involved in sepal, petal, stamen, and carpel development. Because the double‐flower phenotype is the most popular, genetic linkage maps were constructed using F2 progenies derived from a cross between a simple‐flower parent and a double‐flower parent, which resulted in the identification of key regulatory factors (such as APETALA2‐like (AP2L) gene) contributing to these phenotypes (Wang et al., 2020a; Yagi et al., 2013). The spicy scent of carnations is also considered a desirable trait, with eugenol identified as its main component (Clery et al., 1999; Kishimoto et al., 2011; Onozaki, 2018). Due to the extraordinary aroma, carnation obtained its Chinese name ‘Xiāng shízhú’, which means fragrance, and ‘Shízhú’ refers to the genus name, Dianthus. Previous studies have vastly improved our knowledge of key ornamental traits in carnations (Yagi et al., 2013, 2014, 2017; Zhang et al., 2018), but the currently available genome is a draft assembly containing 45,088 scaffolds, which needs to be improved. It is important to understand the underlying molecular mechanisms behind key traits and apply this knowledge to develop improved cultivars through marker‐assisted breeding and gene editing.
Here, we present an assembled and annotated high‐quality reference genome for the D. caryophyllus cultivar, popularly referred to as the ‘Scarlet Queen’, based on sequence data generated from the PromethION platform from Oxford Nanopore Technologies (ONT), and constructing pseudo‐chromosomes using high‐throughput chromosome conformation capture (Hi‐C) techniques. Based on this reference genome, genetic regulators of important ornamental traits for carnation, including coloration of petal limbs and margins (technically the margin of the apex; hereafter referred to as the ‘margin’ for simplicity), eugenol biosynthesis, and formation of petaloid stamens were identified by means of comprehensive homology search, gene family analyses, integration of metabolomic data, and co‐expression analyses from RNA‐seq datasets. This work provides high‐quality genomic resources needed to develop improved and novel ornamental traits in carnations.
Results
Assembly and annotation of a high‐quality, chromosome‐scale D. caryophyllus genome
To fully facilitate the analysis of the molecular mechanism of ornamental traits in D. caryophyllus, we generated a high‐quality genome for carnations. The previously published carnation genome (‘Francesco’_r1.0, National Center for Biotechnology Information (NCBI) Accession GCA_000512335.1) released in 2014 was a draft assembly and it was generated using only Illumina and GS FLX+ short‐read technology, which resulted in 45,088 scaffolds spanning 568.9 Mb and a scaffold N50 of 60.73 kb (Yagi et al., 2014). In this study, we generated 95× coverage of ONT 60.65 Gb pass reads and assembled the genome of D. caryophyllus ‘Scarlet Queen’ (a well‐known cultivar with petal margin coloration) (Figure 1a). Illumina short reads (45.41 Gb ~62.60× coverage, after filtering) were used to further polish ONT data (Table S1 and Figure S1). Because of its 1.2% heterozygosity rate (Figures S2 and S3 and Table S2), redundant sequences were removed from the initially assembled genome to obtain a de‐redundant genome. The final 636.30 Mb genome assembly (D. caryophyllus v1.0) contained only 75 contigs with a contig N50 of 14.67 Mb, which is close to the estimated genome size of ~640 Mb obtained by K‐mer survey and flow cytometry (Table S2 and Figure S4). The paired‐end reads from Hi‐C were used to cluster, order, and orient (Table S3), then 580.05 Mb (91.16%) was anchored onto 15 pseudochromosomes (2n = 30) using the LACHESIS package with manual adjustments. The final assembly was achieved to N50 with 38.55 Mb and the pseudochromosome lengths ranged from 30.95 to 53.54 Mb (Table 1, Figure 1b and Figure S5). The Benchmarking Universal Single‐Copy Orthologs (BUSCO) analysis results show the completeness reached 97.15% (Table S4). The long terminal repeat (LTR)‐assembly index was 12.11. We remapped and obtained high mapping rates for Illumina (99.77%) and ONT (99.77%) data (Tables S5–S7). The genetic maps of D. caryophyllus ‘72L’ (Yagi et al., 2017) were used to assess the quality of the assembled genome. Results showed higher collinearity between the assembled genome of ‘Scarlet Queen’ and the genetic map of ‘72L’ (average 91.47%) (Figure S6). All the above‐mentioned genome indices indicate that the newly assembled genome for carnation was of high quality.
Figure 1.
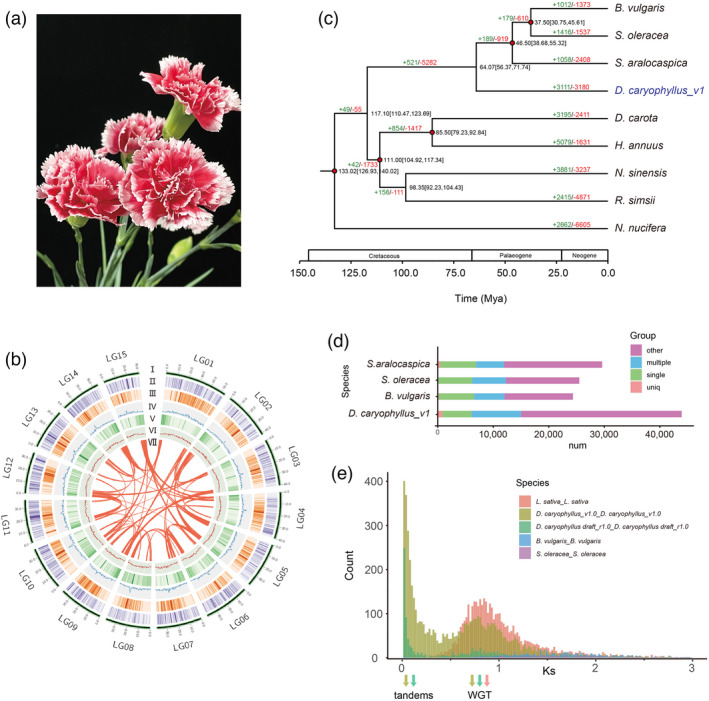
Phylogenetic analysis of the D. caryophyllus genome. (a) Flower of D. caryophyllus ‘Scarlet Queen’. (b) Distribution of carnation genomic features. Circular representation of the 15 pseudochromosomes (I) chromosome length in Mb, (II) gene density, (III) the density of all LTRs, (IV) the density of all Gypsy‐LTRs, (V) the density of all Copia‐LTRs, (VI) GC content in 500 kb windows, and (Ⅶ) each linking line in the center of the circle connects a pair of homologous genes. (c) Phylogenetic tree for D. caryophyllus and eight other eudicot species. Gene family expansions are indicated in green, and contractions in red. The estimated divergence time (million years ago, MYA) is indicated at each node; numbers in brackets are the 95% confidence intervals (each center is defined as mean value). The red dot represents a calibration point. (d) Genes in different groups of D. caryophyllus_v1 and other sequenced genomes were shown and the values are shown with bar charts. (e) Ks values reveal a polyploidy event in carnation compared with S. oleracea and B. vulgaris which have not experienced recent polyploidy events and L. sativa which experienced a recent WGT event.
Table 1.
Statistics for carnation ‘Scarlet Queen’_v1.0 genome and ‘Francesco’_r1.0 draft genome
| ‘Scarlet Queen’ | ‘Francesco’ | |
|---|---|---|
| Sequencing | ||
| ONT | ~60.65G | * |
| Illumina‐Short | ~48.86G | * |
| Illumina‐HIC | ~81.70G | * |
| Assembly | ||
| Contig N50 | 14.67 Mb | 17.55 kb |
| Contig N90 | 4.53 Mb | 2.68 kb |
| scaffolds N50 | 38.55 M | 62.62 kb |
| scaffolds N90 | 30.95 M | 8.0 kb |
| Longest contig | 34.83 Mb | 1.29 kb |
| Number of contigs | 75 | 88,654 |
| Number of scaffolds | 61 | 45088 |
| De‐redundancy total | 636.30 Mb | 568.89 Mb |
| BUSCO | 97.15% | – |
| Total GC content (%) | 38% | 36% |
| TE | 70.62% | 53.96% |
| Heterozygotic | 1.2% | 0.2% |
| Annotation | ||
| Number of predicted genes | 43925 | 43266 |
| Swiss‐prot | 21700 (49.4%) | * |
| Interproscan | 37656 (85.73%) | * |
| GO | * | 16423 (37.8%) |
| NR | 21133 (48.11) | * |
| KOG | 13896 (31.64%) | 19005 (43.7%) |
| Annotated | 40332 (91.82%) | * |
| Unannotated | 3593 (8.18%) | * |
| BUSCO | 95% | 82.1% |
A total of 43,925 protein‐coding genes were predicted (Tables S8 and S9 and Figure S7). Finally, 91.82% of the predicted genes were annotated using different databases (Table S10). The number of genes annotated from the carnation genome was significantly larger than that in Spinacia oleracea, Suaeda aralocaspica, and Beta vulgaris (Figure 1d). In addition, 1453 transfer RNA, 594 ribosomal RNA (rRNA), 2433 small nuclear RNA, and 95 microRNA (mRNA) genes were predicted using Infernal (Table S11). BUSCO analysis of the newly assembled genome was 95% complete, whereas the D. caryophyllus_r1 draft genome (Yagi et al., 2014) was only 82.1% complete using the same pipeline (Embryophyta_odb10 database) (Table 1), which implied that our genome had higher integrity. In our carnation genome, the repeat sequences accounted for 70.62%, with LTR retrotransposons accounting for 36.14% of the genome, of which 9.03% were of the copia type and the remaining 27.11% of the gypsy type (Figure 1b, Tables S12 and S13 and Figure S8). The genome of the carnation referred to as ‘Scarlet Queen’ was highly heterozygous. Therefore, we examined sequence polymorphisms within our newly assembled genome using a previously published method (Zhong et al., 2021). We found 1.79 million single nucleotide polymorphisms (SNPs), of which 1.15 million were transitions and 0.74 million were transversions (Tables S14 and S15, Figures S9–S11). A quantity of 36.96% of the SNPs was located in intergenic regions. SNPs resulted in the loss/gain of their start or stop codons, affecting 2044 genes. We further identified 0.53 million indels, of which 598 led to the loss or gain of start or stop codons (Tables S14 and S15).
Evolution and whole‐genome duplication
We constructed a phylogenetic tree and estimated the divergence times of the nine plant species using 1433 shared single‐copy genes identified by OrthoFinder (Figure 1c). Results showed that D. caryophyllus was most closely related to the Chenopodiaceae species, S. oleracea, B. vulgaris, and S. aralocaspica. The divergence time of carnation and Chenopodiaceae was estimated at ~64.07 million years ago (MYA) using different calibration points (Figures 1c and S12). In the genome of D. caryophyllus, 3111 and 3180 gene families underwent expansion and contraction, respectively (Figures 1c and S13). By the Gene Ontology (GO) enrichment analysis, the expansion gene families were enriched in RNA glycosylase activity, rRNA N‐glycosylase activity, ADP binding, hydrolase activity, hydrolysing N‐glycosyl compounds, transition metal‐ion binding, catalytic activity, acting on an rRNA, zinc ion binding, and nucleic acid binding (Table S16). In addition, synonymous substitutions per synonymous site (Ks) for the paralogs of S. oleracea and B. vulgaris, which the two no experienced recent polyploidy events (Xu et al., 2017) and lettuce (Lactuca sativa) which experienced a recent WGT event (Badouin et al., 2017), and D. caryophyllus and 4dTv values indicate that a whole‐genome polyploidy event occurred (Figures 1e and S14). The collinearity patterns were 3:1 between D. caryophyllus_v1 and Vitis vinifera, B. vulgaris, and S. oleracea (Figures S15–S21). Results of the Ks, 4dTv value, and collinearity pattern suggested that the carnation may have had a whole‐genome triplication event after the divergence of Caryophyllaceae from Chenopodiaceae.
Identification and quantification of the main pigments in petals
In this work, we sampled tissue from ‘Lorca’ which has a red margin and yellow limb flower petals to elucidate spatial and temporal differences in petal pigments (Figure 2a). Petals at three different stages were selected for metabolite detection: stage 3 (entirely closed flowers, petals without red color), stage 4 (petals begin red coloring), and stage 6 (open flowers, petals with red coloration).
Figure 2.
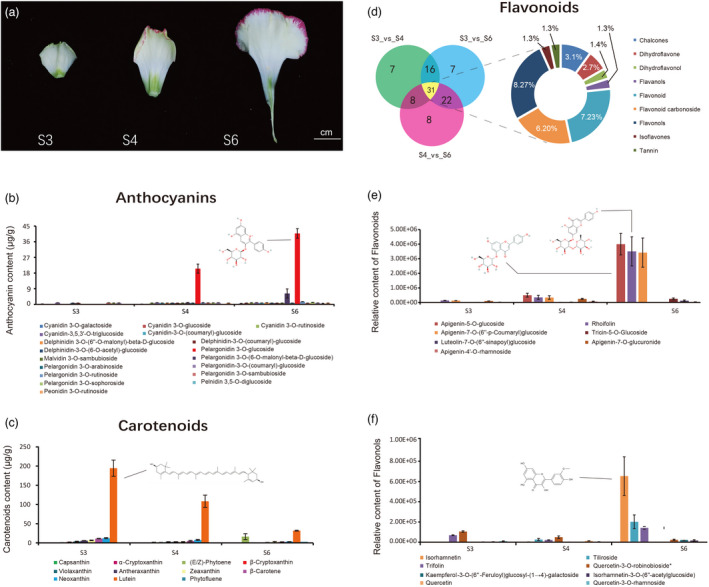
Identification and quantification of the pigments in carnation. (a) Three stages, S3: petals without color and S4 and S6: petals coloration. Petals were used for metabolite detection. (b) Eighteen anthocyanins were identified and quantified from petals of S3, S4, and S6. (c) Eleven carotenoids were identified and quantified from petals of S3, S4, and S6. (d–f) Flavonoids were identified and quantified from petals at S3, S4, and S6. (d) Venn diagram of differential accumulated metabolites between S3 vs. S4, S3 vs. S6, and S4 vs. S6. (e) The quantity of seven high abundance flavonoids at different stages. (f) The quantity of eight high‐abundance flavanols at different stages.
Using liquid chromatography–electrospray ionization tandem mass spectrometry (LC–ESI–MS/MS), 30 anthocyanin compounds were identified and quantified (against 99 anthocyanin standards) in petals (Figure S22 and Table S17). By comparing these anthocyanins (top 18 are shown in Figure 2b) across different stages of development, we found that pelargonidin 3‐O‐glucoside content was the highest in the S6 petals, followed by pelargonidin 3‐O‐(6‐O‐malonyl‐beta‐d‐glucoside) (Figure 2b). Given that the increases in these two compounds were associated with petal development and deeper red margins, we propose that these anthocyanins were the main compounds responsible for the red coloration of petal margins. The carotenoid content in flowers at different stages was quantified via ultra‐performance liquid chromatography (UPLC) separation (Supplementary Notes and Figure S23). Among the carotenoids detected, lutein content was the highest across the three stages for which comparisons were made (Figure 2c and Table S18). Although lutein was the most abundant carotenoid, it decreased with flower development. Phytoene increased in S6 but remained at a lower concentration than lutein. In addition, we also detected and quantified 184 flavonoid compounds (Table S19 and Figure S24) in the three stages of petals using UPLC (Supplementary Methods Notes). From our comparative analyses (S3 vs. S4, S4 vs. S6, and S3 vs. S6), the difference in flavonoid content (fold change ≥2, fold change ≤0.5, and variable importance in projection score ≥1) varied between each stage of floral development; however, 31 flavonoids were shared across all comparison groups with the main shared compounds being dihydroflavones (numbers = 8, quantity 27%), flavonoids (7, 23%), and dihydroflavonols (6, 20%) (Figure 2d and Table S20). Among the flavonoids, apigenin‐5‐O‐glucoside, rhoifolin, and apigenin‐7‐O‐(6″‐p‐coumarin)‐glucoside contents at S6 were significantly higher than those at S3 and S4 (Figure 2e). Among the flavanols, isorhamnetin was the most abundant at S6. These contents probably contribute to the coloration of ‘Lorca’ petals ranging from white‐green to yellow with development (Figure 2f).
Characterization of pigment synthesis pathway genes involved with petal color
To gain further insights into pigment biosynthesis, especially the anthocyanin biogenesis pathway, seven different stages (S1–S3 samples exhibit colorless petals, and S4–S7 petals exhibit red margins, and among them, ‘Lorca’ petals form red margins gradually during S4) were sampled to extract and sequence gene transcripts.
Based on homology search and genome functional annotation, we identified several genes involved in pigment biosynthesis, including PAL, C4H, 4CL, CHS, CHI, F3H, FLS, DFR, ANS, and AT/GT. We outlined the putative pigment biosynthetic pathway (Figure 3) based on the Kyoto Encyclopedia of Genes and Genomes database and those previously published for other higher plants (Belwal et al., 2020; Falcone Ferreyra et al., 2012). We found that DFR and ANS mRNA levels were higher during the S4 to S7 stages than during the first three flower developmental stages (Figure 3a). A similar expression pattern was observed for Dca_v1_10 g.0063700_FLS. The occurrence of carotenoid biosynthesis has been described in numerous plant species (Sun et al., 2018). Based on the functional annotation, we identified two PSY, three PDS, five ZDS/CRTISO, two LCYB/LCYE, one CHYB, eight ZEP, and two VDE carotenoid biosynthesis genes in carnations (Figure 3b). As petals develop, the expression of Dca_v1_ 13g.0238600_PSY, which is thought to be the rate‐limiting enzyme in carotenoid skeleton production, decreases. Other downstream genes, such as homologous genes of Dca_v1_07g.0027500_PDS, Dca_v1_08g.0280000_LCYE, Dca_v1_01g.0015600_ZEP, Dca_v1_10 g.0104200_ZEP, and Dca_v1_15g.0018200_ZEP, also showed the same expression pattern, resulting in a decrease in carotenoid accumulation. These results are of particular interest to understand pigment accumulation and coloration.
Figure 3.
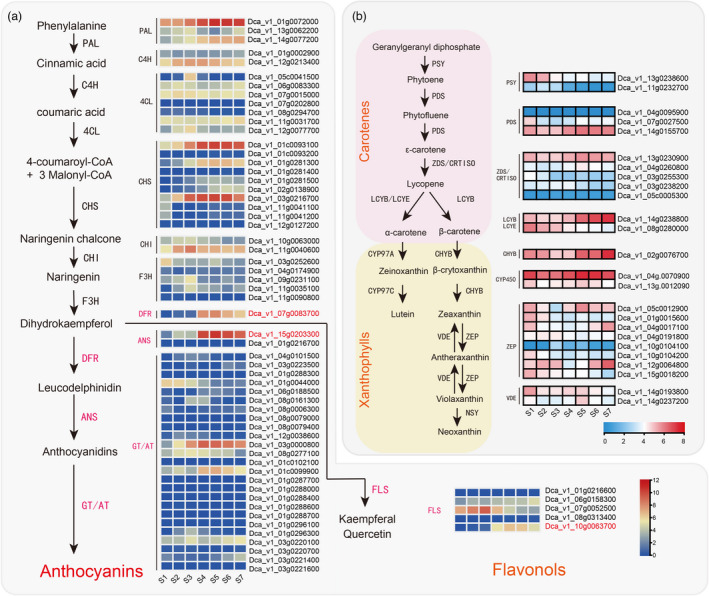
The biosynthesis pathways of pigments. (a) Proposed synthesis pathway of anthocyanins and flavonols. (b) Proposed synthesis pathway of xanthophylls and carotenes. Phenylalanine ammonia‐lyase (PAL), cinnamate‐4‐hydroxylase (C4H), 4‐coumarate CoA ligase 4 (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3‐hydroxylase (F3H), dihydroflavonol 4‐reductase (DFR), anthocyanidin synthase (ANS), acyltransferase (AT), glucosyltransferase (GT), flavonol synthase (FLS). phytoene synthase (PSY), phytoene desaturase (PDS), carotene isomerase (CRTISO), lycopene β‐cyclase (LCYB), lycopene ε‐cyclase (LCYE), cytochrome P450 carotene β‐hydroxylase (CYP97A), cytochrome P450 carotene ε‐hydroxylase (CYP97C), zeaxanthin epoxidase (ZEP), violaxanthin de‐epoxidase (VDE), and neoxanthin synthase (NXS). Gene expression profile (FPKM) at different stages. Low to high expression is indicated by a change in color from blue (low accumulation) to red (high accumulation).
MYBs, bHLHs, and WRKYs were involved in petal margin coloration
To further clarify genes involved in the red coloration of petal margins, we conducted a comparative analysis of differentially expressed genes (DEGs) with two groups of combined samples representing major changes in petal coloration (margin vs. limb and S3 vs. S4) (Figure 4a). There were 4140 (S3 vs. S4) and 626 (margin vs. limb) DEGs specific to the two comparisons (Figure 4b and Tables S21 and 22). We identified 275 DEGs that overlapped between the two groups during carnation flower development. GO enrichment analysis of DEGs indicated that these genes were enriched for oxidoreductase, hydrolase (acting on glycosyl bonds, hydrolysing O‐glycosyl compounds), and transcription regulator activities, and other metabolic and biological processes (Figure 4c). Previous studies have shown that MYBs and bHLHs are involved in anthocyanin biosynthesis (Xu et al., 2015). We identified the MYB family by analysing their phylogenetic and gene structure (Figure S25) and location (Figure S26), and analysed expressions of MYBs and bHLHs from 275 DEGs (Figures S27 and S28) in carnations. Combining characteristic traits, nine candidate genes related to red margin coloration were identified (Figures 4d and S29). Among them, we found that FLS, DFR, ANS, two MYBs, and WRKY44 had similar expression patterns to flower development. The expression of these genes increased with reddening and then decreased; the expression of these genes at the margin of the petal was higher than that in the limb of the petal. By examining the co‐expression network inferred from WGCNA (an R package), we constructed weighted gene co‐expression networks and obtained 30 clusters (Figures S30 and S31). In addition, a regulatory network was constructed based on genes related to anthocyanin synthesis by cystoscopy (Figure S32). The regulatory network of the nine candidate genes mentioned above was also proposed (Figure 4e), which suggested that the MYBs, bHLHs, and WRKY44 TFs were involved in anthocyanin synthesis. To comprehensively examine the putative gene regulation involved in the anthocyanin/flavonol biosynthesis pathways, a regulatory network for the formation of red margins on carnation petals was proposed (Figure 4f); when the expression of the Dca_v1_10g0111900_MYB gene is specifically upregulated in petal margins together with other MYBs, bHLHs, and WRKY44, it promotes the increase of downstream DFR and ANS expressions contributing to red margins.
Figure 4.
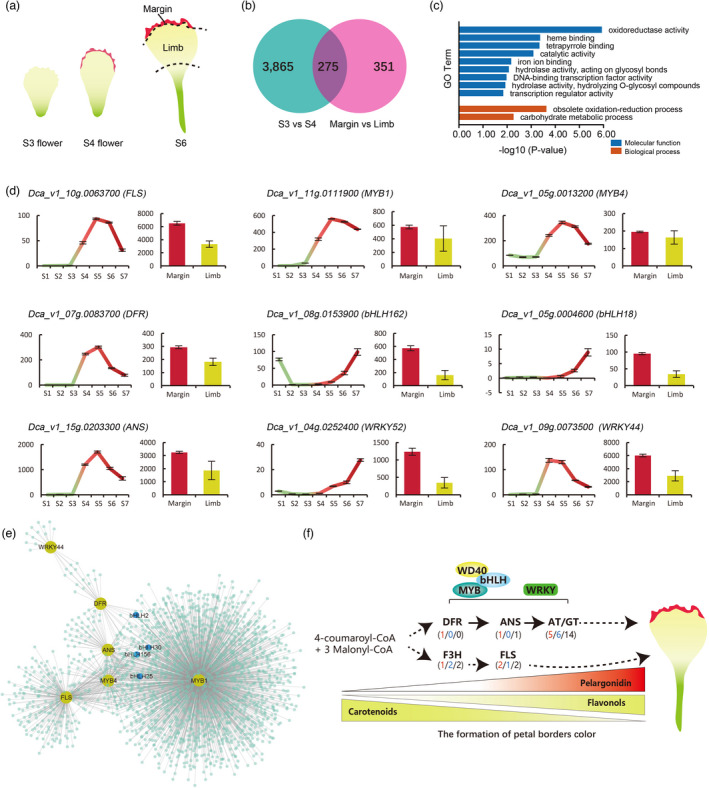
The formation of petal margin coloration. (a) Flower petal (corolla lobe) morphology and coloration at different stages of development (b) Venn diagram indicates the number of DEGs in two comparisons. (c) Gene Ontology (GO) enrichment analysis of DEGs in two comparisons. (d) The expression of anthocyanin synthesis‐related genes in 7 stages and two tissues. (e) Sub‐network for Dca_v1_10g0111900_MYB, low values to small size circle. Low to high expression is indicated by a change in color from blue to red. (f) Proposed regulatory pathways for petals coloration.
Proposed biosynthetic pathway of eugenol synthesis in carnation
We found that three main volatiles, cis‐3‐hexenyl benzoate, β‐caryophyllene, and eugenol, were the primary components of the carnation flower fragrance from S6 flowers by GC–MS (Figure 5a). Eugenol is often referred to as a characteristic aroma of carnations. To further assess eugenol biosynthesis, steady‐state levels of eugenol were quantified in flowers at various stages (S2–S6) of floral development. Results showed that the eugenol content changed with flower development. Specifically, eugenol increased across the stages of flower development from 0.00708 mmol/g at S4 to 0.02596 mmol/g at S5, and 0.04247 mmol/g at S6 (Figure 5b). Of particular interest was the finding that both the accumulation of eugenol and anthocyanins began at S4 with similar accumulation patterns thereafter.
Figure 5.
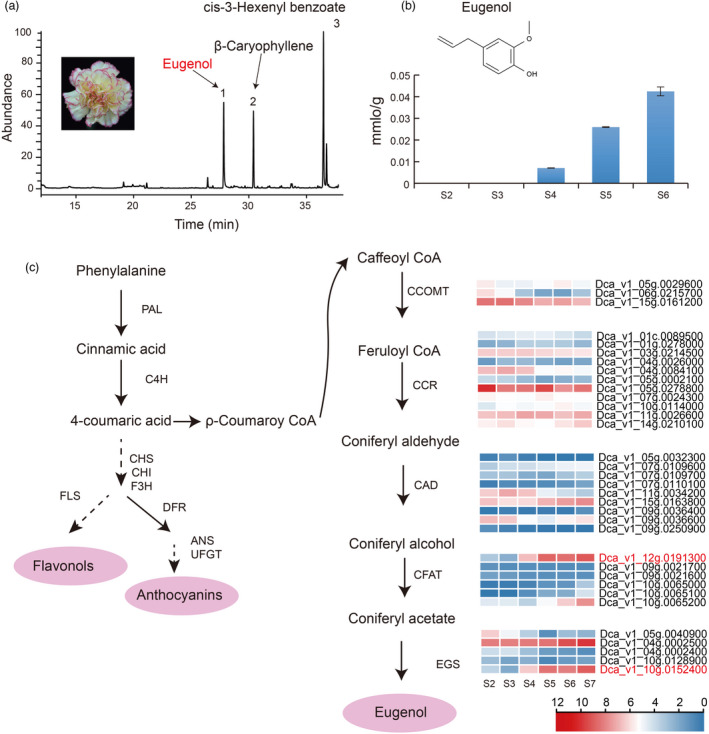
Proposed biosynthetic pathway of eugenol synthesis in carnation. (a) Three volatiles were identified by SPME of GC_MS. (b) The content of eugenol at different stages of floral development. (c) Proposed biosynthetic pathway of eugenol synthesis in carnation. Phenylalanine ammonia‐lyase (PAL), cinnamate‐4‐hydroxylase (C4H), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3‐hydroxylase (F3H), dihydroflavonol 4‐reductase (DFR), anthocyanidin synthase (ANS), UDP‐flavonoid glucosyltransferase (UFGT), caffeoyl‐CoA O‐methyl transferase (CCOMT), cinnamoyl‐CoA reductase (CCR), cinnamyl alcohol dehydrogenase (CAD), coniferyl alcohol acetatyltransferase (CFAT), eugenol synthase (EGS).
To resolve the pathway of eugenol synthesis, we identified 34 genes related to eugenol synthesis, including three CCOMT genes, eleven CCR, nine CAD, six coniferyl alcohol acetyltransferase (CFAT), and five eugenol synthase (EGS) genes in carnation by combining data from the carnation genome with transcriptome data. Among these, the expansion genes mentioned above contained CFAT genes, which belong to GO terms of zinc ion binding, involved in the pathway of eugenol synthesis. We also found that CFAT Dca_v1_12 g.0191300 and the EGS homolog Dca_v1_10g.0152400 increased from S4, which corresponds to the time when eugenol began to accumulate (Figure 5c). Several important differences were observed from phylogenetic analysis, sequence alignment, and promoter element comparison of EGS homologs in the ‘Lorca’ carnation cultivar (rich in eugenol) and ‘Francesco’ cultivar (not rich in eugenol) (Kishimoto, 2020) (Figure S33). A premature stop codon in Dca27403 from ‘Francesco’ was found in the last exon, shortening the translated protein by 15 amino acids. The promoter of Dca_v1_10g.0152400 in ‘Lorca’ containing fewer regulatory elements compared to Dca36983 and Dca27403 genes in ‘Francesco’ was found.
Expression patterns of ABC genes in double‐flower carnation
To characterize the genetic patterns of flower initiation and development, seven stages, including S1: active axillary buds, stem apical meristem transformation to flower primordium, S2: floral meristem and early floral organ development, S3: entirely closed flower, S4: flower coloration begins, S5: buds before flowering and coloration, S6: open flower, and S7: senescing flowers, were selected for analysis (Figure 6a).
Figure 6.
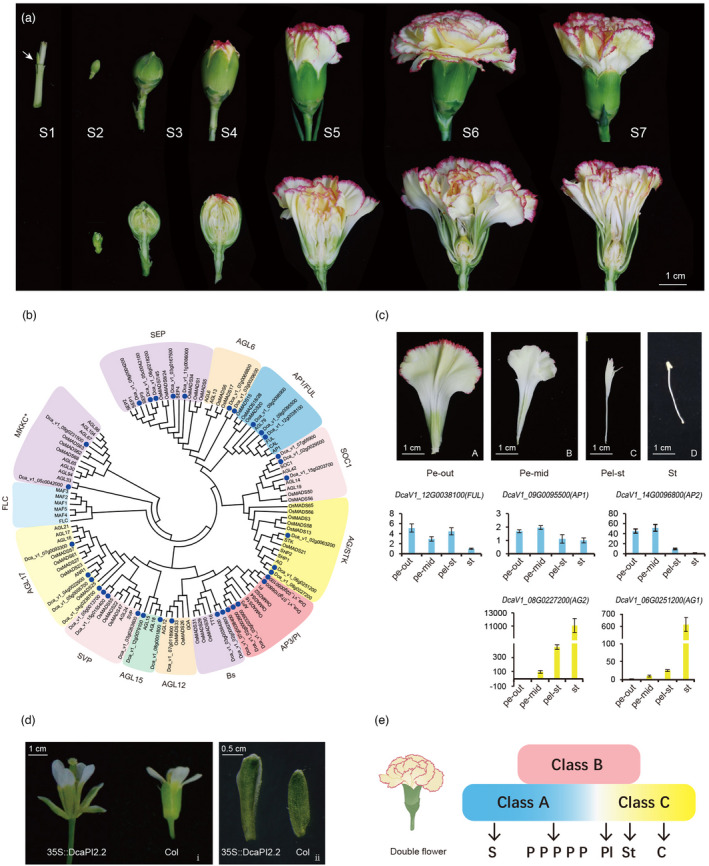
Genetic basis of flower development. (a) Flower developmental stages (S1–S7). (b) Phylogenetic tree of type Ⅱ MADS‐box family, carnation genes denoted by blue dots. (c) The gene expression patterns of A‐/C‐function genes from various floral organs. Pe‐out (A): outer petals; Pe‐mid (B): inner petals; Pel‐st (C): petaloid stamens; and St (D): stamens. (d) The phenotype of 35S::DcaPI2.2 in Arabidopsis. The flower of 35S::DcPI2.2 transgenic are spread out (ⅰ) and contained petaloid sepals (ⅱ), Col means Colombia Arabidopsis. (e) ABC model in the double flower phenotype in carnation where in class A gene expression is maintained across floral organs resulting in the petalization of the inner whorls. S: sepal; P: petal; PI: petaloid stamen; St: stamen; C: carpel.
By examining the co‐expression network mentioned above and combining annotation information from the genome, we found that the dark‐grey and floral‐white modules were related to the initiation of flowering, and the antique‐white, pale‐turquoise, and brown modules were related to the development of floral organs, while dark‐olive green modules were related to senescence (Figure S34). And hence, the identification of these modules facilitates the analysis of these biological processes. Based on the reported flower development genes in Arabidopsis, homologous genes were identified in the carnation genome, and a phylogenetic tree was constructed using genes from both species. From these, 35 type II MADS genes were identified in carnation, including five SEP genes, two AGL6, three AP1/FUL, three SOC1, three AG/STK, five AP3/PI (two PI, two AP3, and one TM6), two Bs, one AGL12, two AGL15, three SVP, four AGL17, and two MIKC* (Figure 6b).
To assess the double flowers in carnations, flower petals from outside to inside were categorized into four whorls: outer petals, middle petals, petaloid stamens, and sterile stamens (Figure 6c). Scanning electron microscopy was performed on the abaxial surface of the petals to assess epidermal changes from the outer to inner whorls. The shapes of cells in proximal epidermal surfaces from petal to stamen changed from an irregular circle to an oblong circle, and that in proximal epidermal surfaces from petals to stamens from a long shuttle type to a shortened type (Figure S35). The distal abaxial epidermal cell of the petaloid stamen showed a gradient shape between the petal and stamen (Figure S35). Patterns of gene expression were compared to assess the types and levels of expressed genes to determine the effects of shifts in gene expression on double flowers. We interpreted our results in reference to the ABC model for flower development, where A genes determine sepals, A + B genes determine petals, B + C genes determine stamens, and C genes determine carpel formation (Coen and Meyerowitz, 1991; Theißen and Saedler, 2001). We found that AP1 and FUL of class A were expressed in all tissues, but their expression in petals was higher than that in stamens, as measured by qRT‐PCR. AP2L was mainly expressed in both the outer and middle petals and it was expressed at low levels in petaloid stamens. The expression of class C genes was detected not only in stamens but also in petaloid stamens and middle petals, with the highest levels of expression in the stamens, followed by petaloid stamens (Figure 6c). In addition, we have analysed the functions of class C genes of carnations (Wang et al., 2020b). In our results, five B genes were expressed in all detected flower organs of the outer petals, middle petals, petaloid stamens, and stamens, and the PI homolog expression was higher than those of AP3 and TM6 homologs (Figure S36).
To further investigate the function of class B genes in petal identity, DcaPI2 with higher expression levels among class B genes was selected for further experiments. The DcaPI2 gene in the two carnation cultivars ‘Lorca’ and ‘L’ was amplified, and there was a one‐amino acid difference between them (Figure S37). We chose the longer sequence of ‘L’ named ‘DcaPI2.2’, for further study. In 35S: DcaPI2.2 transgenic plants, the sepals were converted to organs classified as petaloid sepals, based on shape and color (Figure 6d). Using a scanning electron microscope, we observed sepal marginal cells of the transgenic line specialized in petal‐specific cells (Figure S38E). The shape of these cells was between the epidermal cells of the sepals (Figure S38F) and the epidermal cells of the petals (Figure S38G) in wild‐type Arabidopsis thaliana, showing an irregular square shape. The expression of DcaPI2.2, AtPI, and AtAP3 genes in the transgenic lines of Arabidopsis were detected and showed similar expression patterns (Figure S39). These results indicate that PI2 plays an important role in petal identification in carnations.
Within the context of the ABC model, our results indicated that in the model of carnation double flower formation, the boundary of the class A/C gene expression slides (the A gene slides inward and the C gene slides outward) and fades, resulting in the boundary between petals and stamens intergrading, as indicated by the progression of petal forms from the outside (most petal‐like) to the inside (most stamen‐like) (Figure 6e).
Discussion
In the present study, we describe the first chromosome‐scale genome assembly for a carnation. This high‐quality reference genome for carnations is an essential resource for carnation cultivar developers as well as for researchers studying the molecular mechanisms of flower development and evolution.
Carnation is one of the most popular ornamental flowers in the world, partly because of the diversity of flower colors, fragrance, and phenotype, making it attractive to consumers. Previous studies have reported functions of genes encoding relevant enzymes and transcription factors in the carnation pigment pathway (Abe et al., 2008; Fukui et al., 2003; Momose et al., 2013; Morimoto et al., 2019; Yagi et al., 2013). However, these studies focused on the accumulation of metabolites in different varieties and lacked complementary gene expression data across developmental stages, which is necessary for the complete elucidation of the underlying pathways. Here, using a newly assembled genome, along with transcriptomic and metabolomic data across a developmental series of flowering stages and different tissues, we propose a co‐expression network for petal coloration and infer the contribution of key transcription factors. Our study suggests that bHLHs and MYBs together regulate anthocyanin pigmentation, which is similar to the findings in other species (Huang et al., 2018; Wang et al., 2021; Xu et al., 2015).
In recent years, more studies have found the conservation of WRKY‐based regulatory mechanisms in the anthocyanin pathway, such as in Arabidopsis, where WRKY factors combined with the MYB‐bHLH‐WD40 (MBW) complex to regulate the anthocyanin pathways (Verweij et al., 2016). Another study also reported that WRKY proteins can control flavonoid pigment pathways through its relationship with the MBW complex (Lloyd et al., 2017). In addition, WRKY was found to be involved in recently published research on rhododendron color (Yang et al., 2020). These studies illustrate the important role of the WRKY factor in the pigment pathway. Interestingly, our results also showed that the WRKY44 transcription factor was specifically expressed in petal margins, and it was predicted to interact with the DFR gene, suggesting that it is involved in petal margin coloration in carnations. WRKY transcription factors participate in the synthesis of anthocyanins across a diversity of different species; therefore, more research is needed to understand how WRKY has evolved throughout angiosperms and what function it serves in other lineages. Previous attempts to create novel blue pigment pathways in carnations with genetic engineering have been successful (Matsuba et al., 2010), but efforts were slow for multicolor petals given the lack of complete genomic resources. It is well known that complete high‐quality genomes and the resolution of gene regulatory networks improve the efficiency and reduce off‐target effects of gene editing techniques (Manghwar et al., 2020; Scheben et al., 2017). As such, the high‐quality genome and proposed gene regulatory networks presented herein provide a blueprint for the development of carnation cultivars with novel multi‐coloration, both through conventional marker‐assisted breeding and various gene‐editing techniques.
To clarify the diversity of carnation scents, Clery et al. (1999) studied the volatile organic compounds of carnations and found that modern cultivars have lost the spicy fragrance of eugenol. The diversity of cultivated carnation scents was much lower than that of wild Dianthus species. In a review of floral fragrance, the study suggested that in modern flower cultivars fragrance was unintentionally selected against other traits (Schade et al., 2001; Vainstein et al., 2001). In a study of emitted volatiles in 25 carnation cultivars, it was found that many commonly grown cultivars also lost the ability to produce fragrance (Kishimoto, 2020). In this study, we identified candidate genes, CFAT and EGS, that contributed to eugenol synthesis. The structural changes of the EGS gene may explain the lack of eugenol in cultivars like ‘Francesco’. This finding provides a basis for testing cultivars that lack fragrance for these structural gene differences and potential return of functional genes to newly developed cultivars, such that, fragrant carnation can once again be grown while preserving important morphological and color traits. Lastly, we propose that together with class B genes, the expression of class A genes across floral whorls may contribute to the petal formation of all whorls and in turn be a part of producing the double‐flower form that is most sought after in carnations (Figure 6d). Overall, the high‐quality carnation genome reported in this study provides new insights into the ornamental traits of carnations and provides a basis for further development of research questions and techniques in plant biotechnology.
Methods
Plant material and tissue collection
The carnation (D. caryophyllus L.) cultivars, ‘Scarlet Queen’ and ‘Lorca’ (well‐known cultivars possessing petals with marginal coloration), were grown in a greenhouse at the experimental growth facility of Huazhong Agricultural University (located at 30°28′36.5 N and 114°21′59.4 E) in Wuhan, Hubei Province, China. Young leaves of ‘Scarlet Queen’ were collected to extract high‐quality DNA for Illumina and ONT sequencing. RNA‐seq data from stems, leaves, and roots of ‘Scarlet Queen’ and published data from the NCBI Sequence Read Archive database (BioProject: PRJDB5916) were used for genome annotation.
DNA extraction, library construction, and genome sequencing
For the ONT library preparation, high‐quality genomic DNA was extracted using the Qiagen Genomic DNA extraction kit (Cat. No.10,243 and 19,060, Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Quality control of the extracted DNA was carried out using a NanoDrop One UV–Vis spectrophotometer (Thermo Fisher Scientific, USA) to assess DNA purity (OD260/280 ranging from 1.8 to 2.0, and OD260/230 between 2.0 and 2.2). Following the Nanopore library construction protocol, a library was constructed for use on the PromethION sequencer platform (Oxford Nanopore Technologies, UK). For Illumina sequencing, we generated ~50× Illumina short reads on the HiSeq 2000 platform (Illumina, San Diego, CA) with insert sizes of 400 bp to polish the ONT genome assembly. Raw sequencing data were processed using FastQ (v0.20.0) (Chen et al., 2018) and FastQC (Brown et al., 2017) to filter and detect poor‐quality reads and over‐represented sequences. The Hi‐C library (PE 150bp) was constructed by chromatin extraction and digestion using a standard procedure. Details of the Hi‐C library procedure, including DNA ligation, purification, and fragmentation, are described in Supplementary Notes. The HiC library was sequenced on an Illumina NovaSeq platform and resulting data were used to construct the chromosome‐level genome assembly.
Genome assembly and pseudochromosome scaffolding
Illumina clean reads were analysed using kmerfreq to generate the k‐mer depth distribution with a k‐mer size of 17 bp. These data were then used to estimate the genome size. Correction of ONT long reads and de novo assembly were performed using NextDenovo (reads_cutoff: 1k and seed_cutoff: 32k). For genome polishing, ONT reads and Illumina sequencing reads were separately subjected to three and four rounds of genome correction, respectively, using NextPolish v1.2.4 (https://github.com/Nextomics/NextPolish.git). A subprogram of Purge_Haplotigs (Roach et al., 2018) was used to generate the final contig assembly to retain only one copy of each contig from the heterozygous regions. For Hi‐C sequence data, we also initially filtered out low‐quality reads using fastp and validated paired‐end reads using Bowtie2 v2.3.2 (Langmead and Salzberg, 2012). Then we employed LACHESIS (Burton et al., 2013) software (cluster_min_re_sites = 100; cluster_max_link_density = 2.5; cluster_noninformative_ratio = 1.4; order_min_n_res_in_trunk = 60; order_min_n_res_in_ shreds = 60) to cluster, reorder, and orient the contig‐scale genome assembly, and manually checked the placement and orientation errors apparent in chromosomes using the Hi‐C heatmap. To evaluate the newly assembled genome, the completeness of the genome assembly was assessed using BUSCO (Simao et al., 2015) against the Embryophyta_odb10 database. Quality was assessed using ‘72L’ genetic maps of carnation published (Yagi et al., 2017) using ALLMAPS (Tang et al., 2015). Finally, we judged the assembly quality by mapping our Illumina and ONT reads back to the genome.
Annotation of repeat elements and genes
Before gene prediction, repetitive sequences were masked by an extensive de novo TE annotator pipeline (Ou et al., 2019). Transcriptome‐based, homology‐based, and de novo approaches were used to predict high‐quality protein‐coding genes (Supplementary Notes). For homology‐based prediction, we selected transcript protein sequences from eight species, including A. thaliana, Oryza sativa, Rosa chinensis, V. vinifera, Carica papaya, D. caryophyllus_draft _r1.0, Solanum lycopersicum, and B. vulgaris. For ab initio annotation, Augustus (Stanke et al., 2004), SNAP (https://github.com/KorfLab/SNAP), and GlimmerHMM (Majoros et al., 2004) were employed. For transcriptome‐based prediction, the transcriptome data were produced by Illumina sequencing of materials including leaves, shoots, and roots of ‘Scarlet Queen’ and RNA‐seq data of flowers from NCBI. RNA‐seq alignment files were generated using HISAT2 (Kim et al., 2015) and the PASA program was used to align spliced transcripts and annotate candidate genes. Evidence Modeler v1.1.1 (Haas et al., 2008) was used to generate the final consensus set of gene models obtained using the three approaches and results were updated using PASA. Functional annotation of protein‐coding genes was performed by BLASTp (e‐value 1e‐5 cutoff) using the SwissProt, NR, and KOG databases. InterProScan (Jones et al., 2014) was used to annotate the protein domains by searching the InterPro database. GO terms for each gene were obtained from the corresponding InterPro entries. Non‐coding RNAs were identified using Rfam and Infernal software.
Phylogenetic analyses
To investigate the evolutionary history of D. caryophyllus, eight other species with complete genomes, B. vulgaris, S. oleracea, S. aralocaspica, Daucus carota, Helianthus annuus, Nesolagus sinensis, Rhododendron simsii, and Nelumbo nucifera were selected for using OrthoFinder (Emms and Kelly, 2015) with default parameters to generate a matrix for phylogenetic analysis. Single‐copy orthologs were identified from this dataset and used to construct a phylogenetic tree. Protein sequences were aligned using MAFFT (Katoh and Standley, 2013). We further estimated the divergence times between species using the MCMCTree in the PAML package (Yang, 2007). For the estimation of divergence time, we calibrated the model using the divergence time between N. nucifera and H. annuus (121.6–134.9 MYA), B. vulgaris and S. aralocaspica (40–62 MYA), B. vulgaris and S. oleracea (22–61 MYA), D. carota and H. annuus (77.3–91.7 MYA), and H. annuus and R. simsii (99–114 MYA) obtained from the TimeTree database (http://www.timetree.org/) and the crown age of 89.8 MYA (~90 MYA) of either the Cornales or Ericales (Li et al., 2019).
Gene families that underwent expansion or contraction were identified in the eight sequenced species using Computational Analysis of Gene Family Evolution (Han et al., 2013). Homologous pairs of D. caryophyllus proteins were identified using an all‐to‐all search in BLASTp with an e‐value cutoff of 1e−5. MCScanX (Wang et al., 2012) with default parameters to identify collinear blocks, each containing at least 15 collinear gene pairs. To look for polyploidy events, add_ka_and_ks_to_collinearity. pl of the downstream MCScanX script was used to calculate Ks between collinear genes in each pair of D. caryophyllus_v1, D. caryophyllus r1_draft, B. vulgaris, S. oleracea, and L. sativa within each species. 4DTv values for these species were obtained using the PAML package.
RNA sequencing, gene expression analysis, and co‐expression analyses
Samples at each of the different stages of flower development of ‘Lorca’ (i.e., S1: active axillary buds, stem apical meristem transforms to flower primordium, S2: floral meristem and early floral organs development, S3: entirely closed flowers, S4: coloration of flowers begins, S5: buds before flowering and coloration, S6: open flowers, and S7: senescing flowers), as well as red margin tissue and yellow limb tissue, were collected for RNA‐seq. We used HISAT2 (Kim et al., 2015) to align RNA‐seq short reads from 27 different tissues to our chromosome‐scale genome. Read counts of these samples were calculated using HTSeq, and differential gene expression analysis was conducted using the R package DESeq2 (|log2FC| >1 and padj <0.05). Other samples of different floral organs (outer petals, middle petals, petaloid stamens, and stamens) were collected for qRT‐PCR. We followed the qRT‐PCR procedures and analyses described in a previous paper (Zhang et al., 2018) to quantify the gene expression among the different floral whorls. To identify relationships between DEGs, co‐expression networks were constructed using the WGCNA package (Langfelder and Horvath, 2008). The parameters used in the construction of WGCNA were weighted network, unsigned, hierarchal clustering tree, dynamic hybrid tree cut algorithm, power 8, and minModuleSize = 30. We then used a Cytoscape to display the network. Network statistics were calculated using NetworkAnalyzer in Cytoscape (Shannon et al., 2003). Expression heatmaps were generated using TBtools software (Chen et al., 2020) with log and row scales and online heatmap software v0.2.2 (https://hiplot.com.cn/basic/heatmap) with default parameters.
Metabolite profiling
The petals of ‘Lorca’ at S3, S4, and S6 were collected and immediately frozen in liquid nitrogen. anthocyanin, carotenoid, and flavonoid compounds were identified and quantified using a UPLC–ESI–MS/MS system (HPLC, UFLC Shimadzu Nexera X2, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q Trap, www.appliedbiosystems.com.cn/) and a UPLC–APCI–MS/MS system (UPLC, ExionLC™ AD, https://sciex.com.cn/; MS, Applied Biosystems 6500 Triple Quadrupole, https://sciex.com.cn/). For each developmental stage, three biological replicates were evaluated for each part of the flower (i.e., samples of the same segment from four individuals were mixed to form one sample) for a total of nine samples. The freeze‐dried samples were extracted, identified, and quantified using the methods detailed in Supplementary Notes.
Volatile compounds of ‘Lorca’ emitted from flowers were collected by solid‐phase microextraction. These samples were added to a 15‐mL headspace bottle, weighed, and balanced. After extraction, the extracted fiber was immediately inserted into the GC injection port for thermal analysis. Three independent replicates were performed for each stage for each sample. Analysis was performed using a GCMS‐QP2010 system (Shimadzu, Japan). The ion source and interface temperatures were 200 and 250 °C, respectively. After obtaining the chromatograms and mass spectrograms of the floral compounds, these data were assessed by qualitative analysis using the NIST2011 library, and the identified compounds were checked against published plant volatiles in The Pherobase Database (www.pherobase.com).
Plant transformation
Full‐length coding sequences of the PI2 gene were amplified from the floral cDNA of two cultivated carnations D. caryophyllus ‘Lorca’ and D. chinensis ‘L’ using gene‐specific primers and cloned into pRI101 vectors. The primers used were those described by Zhang et al. (2018). All the reconstructed vectors were confirmed by sequencing. For genetic transformation, we used A. thaliana (Columbia) plants transformed using the floral dip method (Logemann et al., 2006). After RNA extraction, qRT‐PCR was conducted using SYBR® Premix Ex TaqTM II (Takara, Japan) in an Applied Biosystems Real‐Time PCR System (Life Technologies), with each gene analysed in three biological replicates and three technical replicates. The relative expression values were calculated using the comparative 2−▵▵CT method. The primers used for qRT‐PCR were those published by Zhang et al. (2018) and Wang et al. (2020).
These and other methods are further detailed in Supplementary Notes.
Conflicts of interest
The authors have no conflicts of interest to declare.
Author contributions
X.F., Zh.W., Ma.B., and X.Z. conceived the ideas of this paper. X.Z., S.L., and D.P. performed the bioinformatic analysis. K.X. and D.P. contributed evolutionary analysis. Q.W. and Ze.W prepared the sequencing samples and the qRT‐PCR. X.Z. and X.L. participated in metabolite profiling and RNA‐seq. X.Z. drafted the manuscript with the help of L.R.T., X.F., Zh.W., and Mo.B. All authors read and approved the final manuscript.
Supporting information
Figure S1 The length distribution of filtered Oxford Nanopore reads from the D. caryophyllus cv ‘Scarlet Queen’ sample.
Figure S2 K‐mer analysis for estimating the genome size of D. caryophyllus indicating high heterozygosity.
Figure S3 Sequence depth and GC content for the carnation genome after redundant sequences.
Figure S4 Estimate of the genome size of carnation ‘Scarlet Queen’ on a BC 524C CytoFLEX flow cytometry.
Figure S5 Hi‐C interaction heatmap of the D. caryophyllus genome.
Figure S6 Collinearity between the ‘806‐46b’ x ‘Mizuki’ (named ‘72L’) carnation genetic map and D. caryophyllus ‘Scarlet Queen’ pseudomolecules.
Figure S7 Characteristics of predicted D. caryophyllus protein‐coding genes.
Figure S8 The proportions of different classes of repetitive elements in the D. caryophyllus genome.
Figure S9 Distribution of SNP polymorphisms along each carnation chromosome.
Figure S10 SNP substitution types in carnation genome.
Figure S11 Indel polymorphisms in carnation genome.
Figure S12 Phylogenetic tree for D. caryophyllus and six other eudicot species.
Figure S13 The expansion genes associated with each category are indicated in brackets after the term description.
Figure S14 The rate of transversions on fourfold degenerate synonymous sites (4DTv) among paralogs for D. caryophyllus, B. vulgaris, and S. oleracea.
Figure S15 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Vitis vinifera genomes.
Figure S16 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Vitis vinifera genomes.
Figure S17 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Beta vulgaris genomes.
Figure S18 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Beta vulgaris genomes.
Figure S19 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Spinacia oleracea genomes.
Figure S20 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Spinacia oleracea.
Figure S21 Dot plot showing the gene sequence alignment of 15 chromosomes in Dianthus caryophyllus_v1.
Figure S22 Histogram of 30 contents of anthocyanin levels in different stages of carnation petals.
Figure S23 Eleven carotenoids were identified and relatively quantified from three stages of petals using an LC–ESI–MS/MS system.
Figure S24 Heat map visualization of flavonoids from three stages of petals.
Figure S25 Phylogenetic tree (a) and the conserved motifs (b) of the candidate MYBs identified in D. caryophyllus genome.
Figure S26 MYB genes position on chromosomes.
Figure S27 Expression patterns of MYB genes of DEGs (S3_vs_S4) in different stages of flowers of D. caryophyllus.
Figure S28 Expression patterns of bHLH genes of DEGs (S3_vs_S4) in different stages of flowers of D. caryophyllus.
Figure S29 Expression patterns of 16 candidate genes in different stages of flowers of D. caryophyllus.
Figure S30 Sample clustering for different tissues from WGCNA.
Figure S31 Weighted gene co‐expression network analysis for carnation RNA‐seq data.
Figure S32 The WGCNA related to anthocyanin biosynthesis is represented by a node and edge graph.
Figure S33 Gene structure of the eugenol synthase (EGS).
Figure S34 The genes involved in flower development.
Figure S35 SEM images of the abaxial epidermal cells of width upper and narrow lower flower organs.
Figure S36 Relative expression levels of five class B genes from various floral organs.
Figure S37 The carnation PI2 amino acid sequence alignment.
Figure S38 The analysis of DcPI2.2 overexpression in A. thaliana.
Figure S39 Relative expression levels of petal development‐related genes in transgenic lines.
Table S1 Summary of genome sequencing and read‐out for D. caryophyllus.
Table S2 Survey statistic results of D. caryophyllus.
Table S3 Summary of HIC reads mapping to the D. caryophyllus.
Table S4 Result of the evaluation of the D. caryophyllus genome by BUSCO.
Table S5 Genome Illumina data comparison statistics.
Table S6 Genome ONT data comparison statistics.
Table S7 Genomic single‐base accuracy statistics.
Table S8 Gene numbers and features of D. caryophyllus.
Table S9 Basic statistical results of gene structure prediction of D. caryophyllus.
Table S10 The statistical results of gene function annotation of D. caryophyllus.
Table S11 The statistical results of non‐coding RNA of D. caryophyllus.
Table S12 Summary of Repeat contents in D. caryophyllus genome (EDTA software).
Table S13 Summary of Repeat contents in D. caryophyllus genome (RepeatMasker software).
Table S14 SNP s and indels count s upon mapping the Illumina reads to the carnation genome.
Table S15 SNP and indel variants distribution among carnation chromosomes.
Table S16 GO enrichment information for D. caryophyllus expanded gene families.
Table S17 Summary of anthocyanins contents.
Table S18 Summary of carotenoids contents.
Table S19 Summary of flavonoids contents.
Table S20 31 contents were identified in all comparison groups (S3_vs_S4, S4_vs_S6, and S4_vs_S6) with differences
Table S21 DEGs of S3_vs_S4.
Table S22 DEGs of margin_vs_limb.
Note S1 Plant materials and genome analyses.
Note S2 Genome assembly and annotation.
Note S3 Phylogenetic tree.
Note S4 Metabolite profiling.
Note S5 Plant transformation.
Acknowledgements
This work was supported by funding from the National Natural Science Foundation of China (32072607, 32002074, and 31872135). This work was funded by the Science, Technology and Innovation Commission of Shenzhen Municipality (STIC: RCYX20200714114538196). Finally, this work was also supported in part by the Chinese Academy of Agricultural Sciences Elite Youth Program to Zhiqiang Wu.
Zhang, X. , Lin, S. , Peng, D. , Wu, Q. , Liao, X. , Xiang, K. , Wang, Z. , Tembrock, L. R. , Bendahmane, M. , Bao, M. , Wu, Z. and Fu, X. (2022) Integrated multi‐omic data and analyses reveal the pathways underlying key ornamental traits in carnation flowers. Plant Biotechnol. J., 10.1111/pbi.13801
Contributor Information
Zhiqiang Wu, Email: wuzhiqiang@caas.cn.
Xiaopeng Fu, Email: fuxiaopeng@mail.hzau.edu.cn.
Data availability statement
All data supporting the results of this study are included in the manuscript and its additional files. Genome assembly and annotations were deposited in the NCBI BioProject under accessions PRJNA796118.
References
- Abe, Y. , Tera, M. , Sasaki, N. , Okamura, M. , Umemoto, N. , Momose, M. , Kawahara, N. et al. (2008) Detection of 1‐O‐malylglucose: pelargonidin 3‐O‐glucose‐6''‐O‐malyltransferase activity in carnation (Dianthus caryophyllus). Biochem. Biophys. Res. Commun. 373, 473–477. [DOI] [PubMed] [Google Scholar]
- Badouin, H. , Gouzy, J. , Grassa, C.J. , Murat, F. , Staton, S.E. , Cottret, L. , Lelandais‐Briere, C. et al. (2017) The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature, 546, 148–152. [DOI] [PubMed] [Google Scholar]
- Belwal, T. , Singh, G. , Jeandet, P. , Pandey, A. , Giri, L. , Ramola, S. , Bhatt, I.D. et al. (2020) Anthocyanins, multi‐functional natural products of industrial relevance: recent biotechnological advances. Biotechnol. Adv. 43, 107600. [DOI] [PubMed] [Google Scholar]
- Brown, J. , Pirrung, M. and McCue, L.A. (2017) FQC Dashboard: integrates FastQC results into a web‐based, interactive, and extensible FASTQ quality control tool. Bioinformatics, 33, 3137–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R. , Kitzman, J.O. and Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Chen, H. , Zhang, Y. , Thomas, H.R. , Frank, M.H. , He, Y. and Xia, R. (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant, 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. and Gu, J. (2018) fastp: An ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clery, R.A. , Owen, N.E. , Chambers, S.F. and Thornton‐Wood, S.P. (1999) An investigation into the scent of Carnations. J. Essent. Oil Res. 11, 355–359. [Google Scholar]
- Coen, E.S. and Meyerowitz, E.M. (1991) The war of the whorls: genetic interactions controlling flower development. Nature, 353, 31–37. [DOI] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone Ferreyra, M.L. , Rius, S.P. and Casati, P. (2012) Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui, Y. , Tanaka, Y. , Kusumi, T. , Iwashita, T. and Nomoto, K. (2003) A rationale for the shift in colour towards blue in transgenic carnation flowers expressing the flavonoid 3',5'‐hydroxylase gene. Phytochemistry, 63, 15–23. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, M.V. , Thomas, G.W. , Lugo‐Martinez, J. and Hahn, M.W. (2013) Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. [DOI] [PubMed] [Google Scholar]
- Huang, D. , Wang, X. , Tang, Z. , Yuan, Y. , Xu, Y. , He, J. , Jiang, X. et al. (2018) Subfunctionalization of the Ruby2‐Ruby1 gene cluster during the domestication of citrus. Nat. Plants, 4, 930–941. [DOI] [PubMed] [Google Scholar]
- Itoh, Y. , Higeta, D. , Suzuki, A. , Yoshida, H. and Ozeki, Y. (2002) Excision of transposable elements from the chalcone isomerase and dihydroflavonol 4‐reductase genes may contribute to the variegation of the yellow‐flowered carnation (Dianthus caryophyllus). Plant Cell Physiol. 43, 578–585. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.Y. , Fraser, M. , Li, W. , McAnulla, C. , McWilliam, H. et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto, K. (2020) The characteristics of flower scents in carnations. In The Carnation Genome ( Onozaki, T. and Yagi, M. , eds), pp. 147–157. Singapore: Springer Singapore. [Google Scholar]
- Kishimoto, K. , Nakayama, M. , Yagi, M. , Onozaki, T. and Oyama‐Okubo, N. (2011) Evaluation of wild Dianthus species as genetic resources for fragrant carnation breeding based on their floral scent composition. J. Jpn Soc. Hortic. Sci. 80, 175–181. [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–U354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H.T. , Yi, T.S. , Gao, L.M. , Ma, P.F. , Zhang, T. , Yang, J.B. , Gitzendanner, M.A. et al. (2019) Origin of angiosperms and the puzzle of the Jurassic gap. Nature Plants, 5, 461–470. [DOI] [PubMed] [Google Scholar]
- Lloyd, A. , Brockman, A. , Aguirre, L. , Campbell, A. , Bean, A. , Cantero, A. and Gonzalez, A. (2017) Advances in the MYB‐bHLH‐WD repeat (MBW) pigment regulatory model: Addition of a WRKY factor and co‐option of an anthocyanin MYB for betalain regulation. Plant Cell Physiol. 58, 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logemann, E. , Birkenbihl, R.P. , Ulker, B. and Somssich, I.E. (2006) An improved method for preparing Agrobacterium cells that simplifies the Arabidopsis transformation protocol. Plant Methods, 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Manghwar, H. , Li, B. , Ding, X. , Hussain, A. , Lindsey, K. , Zhang, X. and Jin, S. (2020) CRISPR/Cas Systems in genome editing: Methodologies and tools for sgRNA design, off‐Target evaluation, and strategies to mitigate off‐target effects. Adv. Sci. 7, 1902312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato, M. , Onozaki, T. , Ozeki, Y. , Higeta, D. , Itoh, Y. , Hisamatsu, T. , Yoshida, H. et al. (2001) Flavonoid biosynthesis in pink‐flowered cultivars derived from 'William Sim' carnation (Dianthus caryophyllus). J. Jpn Soc. Hortic. Sci. 70, 315–319. [Google Scholar]
- Matsuba, Y. , Sasaki, N. , Tera, M. , Okamura, M. , Abe, Y. , Okamoto, E. , Nakamura, H. et al. (2010) A novel glucosylation reaction on anthocyanins catalyzed by acyl‐glucose‐dependent glucosyltransferase in the petals of carnation and delphinium. Plant Cell, 22, 3374–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose, M. , Nakayama, M. , Itoh, Y. , Umemoto, N. , Toguri, T. and Ozeki, Y. (2013) An active hAT transposable element causing bud mutation of carnation by insertion into the flavonoid 3'‐hydroxylase gene. Mol. Genet. Genomics, 288, 175–184. [DOI] [PubMed] [Google Scholar]
- Morimoto, H. , Narumi‐Kawasaki, T. , Takamura, T. and Fukai, S. (2019) Analysis of flower color variation in carnation (Dianthus caryophyllus L.) cultivars derived from continuous bud mutations. Horticult. J. 88, 116–128. [Google Scholar]
- Nimura, M. , Kato, J. and Mii, M. (2006) Interspecific hybrid production by reciprocal crosses between Dianthus caryophyllus L. and Dianthus X isensis Hirahata et Kitamura. J. Hortic. Sci. Biotechnol. 81, 995–1001. [Google Scholar]
- Onozaki, T. (2018) Dianthus. In Ornamental Crops ( Van Huylenbroeck, J. , ed), pp 349–381. Cham: Springer International Publishing. [Google Scholar]
- Onozaki, T. , Mato, M. , Shibata, M. and Ikeda, H. (1999) Differences in flower color and pigment composition among white carnation (Dianthus caryophyllus L.) cultivars. Sci. Hortic. 82, 103–111. [Google Scholar]
- Ou, S.J. , Su, W.J. , Liao, Y. , Chougule, K. , Agda, J.R.A. , Hellinga, A.J. , Lugo, C.S.B. et al. (2019) Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz, S. , Ditta, G.S. , Baumann, E. , Wisman, E. and Yanofsky, M.F. (2000) B and C floral organ identity functions require SEPALLATA MADS‐box genes. Nature, 405, 200. [DOI] [PubMed] [Google Scholar]
- Roach, M.J. , Schmidt, S.A. and Borneman, A.R. (2018) Purge Haplotigs: allelic contig reassignment for third‐gen diploid genome assemblies. BMC Bioinformatics, 19, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade, F. , Legge, R.L. and Thompson, J.E. (2001) Fragrance volatiles of developing and senescing carnation flowers. Phytochemistry, 56, 703–710. [DOI] [PubMed] [Google Scholar]
- Scheben, A. , Wolter, F. , Batley, J. , Puchta, H. and Edwards, D. (2017) Towards CRISPR/Cas crops ‐ bringing together genomics and genome editing. New Phytol. 216, 682–698. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , Amin, N. et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Sparnaaij, L.D. , Koehorst‐van Putten, H.J.J. and Demmink, J.F. (1990) Variation between genotypes of carnation (Dianthus caryophyllus cultivars and interspecific hybrids) in time of flowering and response to long days. II. Variation in shoot development. Euphytica, 50, 43–50. [Google Scholar]
- Stanke, M. , Steinkamp, R. , Waack, S. and Morgenstern, B. (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, T.H. , Yuan, H. , Cao, H.B. , Yazdani, M. , Tadmor, Y. and Li, L. (2018) Carotenoid metabolism in plants: the role of plastids. Mol. Plant, 11, 58–74. [DOI] [PubMed] [Google Scholar]
- Tang, H. , Zhang, X. , Miao, C. , Zhang, J. , Ming, R. , Schnable, J.C. , Schnable, P.S. et al. (2015) ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theißen, G. and Saedler, H. (2001) Floral quartets. Nature, 409, 469–471. [DOI] [PubMed] [Google Scholar]
- Vainstein, A. , Lewinsohn, E. , Pichersky, E. and Weiss, D. (2001) Floral fragrance. New inroads into an old commodity. Plant Physiol. 127, 1383–1389. [PMC free article] [PubMed] [Google Scholar]
- Verweij, W. , Spelt, C.E. , Bliek, M. , de Vries, M. , Wit, N. , Faraco, M. , Koes, R. et al. (2016) Functionally similar WRKY proteins regulate vacuolar acidification in Petunia and Hair development in Arabidopsis. Plant Cell, 28, 786–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Ji, W. , Liu, Y. , Zhou, P. , Meng, Y. , Zhang, P. , Wen, J. et al. (2021) The antagonistic MYB paralogs RH1 and RH2 govern anthocyanin leaf markings in Medicago truncatula. New Phytol, 229, 3330–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q.J. , Dan, N.Z. , Zhang, X.N. , Lin, S.N. , Bao, M.Z. and Fu, X.P. (2020b) Identification, characterization and functional analysis of C‐Class genes associated with double flower trait in carnation (Dianthus caryphyllus L.). Plants, 9, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhang, X. , Lin, S. , Yang, S. , Yan, X. , Bendahmane, M. , Bao, M. et al. (2020a) Mapping a double flower phenotype‐associated gene DcAP2L in Dianthus chinensis . J. Exp. Bot. 71, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T.H. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Jiao, C. , Sun, H.H. , Cai, X.F. , Wang, X.L. , Ge, C.H. , Zheng, Y. et al. (2017) Draft genome of spinach and transcriptome diversity of 120 Spinacia accessions. Nat Commun. 8, 15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Dubos, C. and Lepiniec, L. (2015) Transcriptional control of flavonoid biosynthesis by MYB‐bHLH‐WDR complexes. Trends Plant Sci. 20, 176–185. [DOI] [PubMed] [Google Scholar]
- Yagi, M. , Kosugi, S. , Hirakawa, H. , Ohmiya, A. , Tanase, K. , Harada, T. , Kishimoto, K. et al. (2014) Sequence analysis of the genome of carnation (Dianthus caryophyllus L.). DNA Res. 21, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi, M. , Shirasawa, K. , Waki, T. , Kume, T. , Isobe, S. , Tanase, K. and Yamaguchi, H. (2017) Construction of an SSR and RAD marker‐based genetic linkage map for carnation (Dianthus caryophyllus L.). Plant Mol. Biol. Rep. 35, 110–117. [Google Scholar]
- Yagi, M. , Yamamoto, T. , Isobe, S. , Hirakawa, H. , Tabata, S. , Tanase, K. , Yamaguchi, H. et al. (2013) Construction of a reference genetic linkage map for carnation (Dianthus caryophyllus L.). BMC Genom. 14, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F.S. , Nie, S. , Liu, H. , Shi, T.L. , Tian, X.C. , Zhou, S.S. , Bao, Y.T. et al. (2020) Chromosome‐level genome assembly of a parent species of widely cultivated azaleas. Nat. Commun. 11, 5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yoshida, H. , Itoh, Y. , Ozeki, Y. , Iwashina, T. and Yamaguchi, M. (2004) Variation in chalcononaringenin 2 '‐O‐glucoside content in the petals of carnations (Dianthus caryophyllus) bearing yellow flowers. Sci. Hortic. 99, 175–186. [Google Scholar]
- Zhang, X. , Wang, Q. , Yang, S. , Lin, S. , Bao, M. , Bendahmane, M. , Wu, Q. et al. (2018) Identification and characterization of the MADS‐Box genes and their contribution to flower organ in carnation (Dianthus caryophyllus L.). Genes, 9, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, M.‐C. , Jiang, X.‐D. , Yang, G.‐Q. , Cui, W.‐H. , Suo, Z.‐Q. , Wang, W.‐J. , Sun, Y.‐B. et al. (2021) Rose without prickle: genomic insights linked to moisture adaptation. Nat. Sci. Rev. 8, nwab092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The length distribution of filtered Oxford Nanopore reads from the D. caryophyllus cv ‘Scarlet Queen’ sample.
Figure S2 K‐mer analysis for estimating the genome size of D. caryophyllus indicating high heterozygosity.
Figure S3 Sequence depth and GC content for the carnation genome after redundant sequences.
Figure S4 Estimate of the genome size of carnation ‘Scarlet Queen’ on a BC 524C CytoFLEX flow cytometry.
Figure S5 Hi‐C interaction heatmap of the D. caryophyllus genome.
Figure S6 Collinearity between the ‘806‐46b’ x ‘Mizuki’ (named ‘72L’) carnation genetic map and D. caryophyllus ‘Scarlet Queen’ pseudomolecules.
Figure S7 Characteristics of predicted D. caryophyllus protein‐coding genes.
Figure S8 The proportions of different classes of repetitive elements in the D. caryophyllus genome.
Figure S9 Distribution of SNP polymorphisms along each carnation chromosome.
Figure S10 SNP substitution types in carnation genome.
Figure S11 Indel polymorphisms in carnation genome.
Figure S12 Phylogenetic tree for D. caryophyllus and six other eudicot species.
Figure S13 The expansion genes associated with each category are indicated in brackets after the term description.
Figure S14 The rate of transversions on fourfold degenerate synonymous sites (4DTv) among paralogs for D. caryophyllus, B. vulgaris, and S. oleracea.
Figure S15 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Vitis vinifera genomes.
Figure S16 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Vitis vinifera genomes.
Figure S17 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Beta vulgaris genomes.
Figure S18 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Beta vulgaris genomes.
Figure S19 Syntenic dotplot illustrating the comparative analysis of the Dianthus caryophyllus_v1 and Spinacia oleracea genomes.
Figure S20 The syntenic depth ratio between the Dianthus caryophyllus_v1 and Spinacia oleracea.
Figure S21 Dot plot showing the gene sequence alignment of 15 chromosomes in Dianthus caryophyllus_v1.
Figure S22 Histogram of 30 contents of anthocyanin levels in different stages of carnation petals.
Figure S23 Eleven carotenoids were identified and relatively quantified from three stages of petals using an LC–ESI–MS/MS system.
Figure S24 Heat map visualization of flavonoids from three stages of petals.
Figure S25 Phylogenetic tree (a) and the conserved motifs (b) of the candidate MYBs identified in D. caryophyllus genome.
Figure S26 MYB genes position on chromosomes.
Figure S27 Expression patterns of MYB genes of DEGs (S3_vs_S4) in different stages of flowers of D. caryophyllus.
Figure S28 Expression patterns of bHLH genes of DEGs (S3_vs_S4) in different stages of flowers of D. caryophyllus.
Figure S29 Expression patterns of 16 candidate genes in different stages of flowers of D. caryophyllus.
Figure S30 Sample clustering for different tissues from WGCNA.
Figure S31 Weighted gene co‐expression network analysis for carnation RNA‐seq data.
Figure S32 The WGCNA related to anthocyanin biosynthesis is represented by a node and edge graph.
Figure S33 Gene structure of the eugenol synthase (EGS).
Figure S34 The genes involved in flower development.
Figure S35 SEM images of the abaxial epidermal cells of width upper and narrow lower flower organs.
Figure S36 Relative expression levels of five class B genes from various floral organs.
Figure S37 The carnation PI2 amino acid sequence alignment.
Figure S38 The analysis of DcPI2.2 overexpression in A. thaliana.
Figure S39 Relative expression levels of petal development‐related genes in transgenic lines.
Table S1 Summary of genome sequencing and read‐out for D. caryophyllus.
Table S2 Survey statistic results of D. caryophyllus.
Table S3 Summary of HIC reads mapping to the D. caryophyllus.
Table S4 Result of the evaluation of the D. caryophyllus genome by BUSCO.
Table S5 Genome Illumina data comparison statistics.
Table S6 Genome ONT data comparison statistics.
Table S7 Genomic single‐base accuracy statistics.
Table S8 Gene numbers and features of D. caryophyllus.
Table S9 Basic statistical results of gene structure prediction of D. caryophyllus.
Table S10 The statistical results of gene function annotation of D. caryophyllus.
Table S11 The statistical results of non‐coding RNA of D. caryophyllus.
Table S12 Summary of Repeat contents in D. caryophyllus genome (EDTA software).
Table S13 Summary of Repeat contents in D. caryophyllus genome (RepeatMasker software).
Table S14 SNP s and indels count s upon mapping the Illumina reads to the carnation genome.
Table S15 SNP and indel variants distribution among carnation chromosomes.
Table S16 GO enrichment information for D. caryophyllus expanded gene families.
Table S17 Summary of anthocyanins contents.
Table S18 Summary of carotenoids contents.
Table S19 Summary of flavonoids contents.
Table S20 31 contents were identified in all comparison groups (S3_vs_S4, S4_vs_S6, and S4_vs_S6) with differences
Table S21 DEGs of S3_vs_S4.
Table S22 DEGs of margin_vs_limb.
Note S1 Plant materials and genome analyses.
Note S2 Genome assembly and annotation.
Note S3 Phylogenetic tree.
Note S4 Metabolite profiling.
Note S5 Plant transformation.
Data Availability Statement
All data supporting the results of this study are included in the manuscript and its additional files. Genome assembly and annotations were deposited in the NCBI BioProject under accessions PRJNA796118.