

Dear editor,
In the past decade, genome‐wide characterization of the three‐dimensional chromatin structure using high‐throughput methods has greatly advanced our knowledge in plant genome architecture (Liu and Weigel, 2015; Ouyang et al., 2020). However, due to the limitation of Illumina paired‐end short read, most contacts obtained by Hi‐C/ChIA‐PET are pairwise, and interactions among three or more sites of chromatin (multi‐way) can only be inferred from the two‐way data. To directly capture multi‐way interaction and associated methylation modification in Arabidopsis, we applied a long‐read‐based method called Pore‐C that directly sequences the DNA multivalent fragments joined by proximity‐based ligation (Ulahannan et al., 2019).
Compared with Hi‐C, Pore‐C replaced the Illumina sequencing with nanopore long‐read sequencing, which drastically simplified the library construction procedure by removing the biotin labelling, fragmentation, and several purification steps (Figure 1a). Ligation within the nucleus makes Pore‐C reads to retain the interaction information in different cells. We generated two Pore‐C libraries using 12‐day‐old Arabidopsis seedlings of wildtype Col‐0 and analyzed them with the Pore‐C pipeline (Ulahannan et al., 2019). We obtained 6.5 million (11.7 Gb) and 3.5 million (7.9 Gb) high‐quality reads (Q7 filter) with N50 up to 2654 and 3006 bp, respectively (Table S1). Over 96% of the reads can be mapped to the Arabidopsis TAIR10 reference genome in both libraries. From ~10 million Pore‐C reads in total, we obtained 50.7 million pairwise interactions, a similar amount but with fewer reads compared to Hi‐C data (59.1 million pairwise contacts from 239.5 million Illumina reads) (Wang et al., 2015), demonstrating the efficiency of long‐read sequencing in harvesting chromosomal interaction, although with higher cost (Table S1). The two Pore‐C libraries showed high consistency with each other (Figure 1b, Figures S2 and S3) and with the Hi‐C data in their contact feature (Figure 1c), suggesting high reproducibility and accuracy of Pore‐C.
Figure 1.
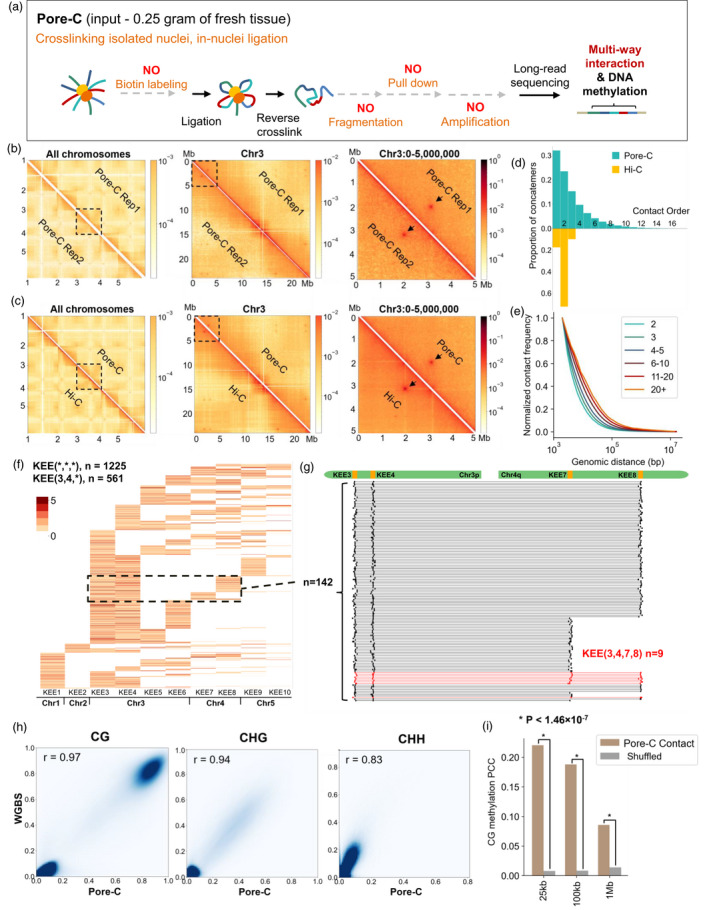
Pore‐C captures multi‐way contacts and DNA methylation in Arabidopsis. (a) Schematic of the Pore‐C protocol. (b, c) The consistency between two Pore‐C libraries (b), between Hi‐C and Pore‐C (c), respectively. Contact maps for all chromosomes (left), chromosome 3 (middle), and chr3: 0–5 Mb (right) are shown. The interactions between KEEs are marked by black arrows. (d) Comparison of Contact Order distribution between previous Hi‐C data and Pore‐C data in Arabidopsis. (e) Contact frequency between different genomic distances of various order groups of Pore‐C. (f) Combination information of reads related to at least three different KEEs. Each row of the heatmap represents one read. The color intensities in the heatmap indicate the counts of interaction capture within one KEE on one read. KEE(*,*,*) means the interaction between at least three individual KEEs, ‘*’ indicates at least one KEE, and ‘n’ shows the read counts. (g) Alignment locations of individual reads related to KEE3, KEE 4, KEE7, and KEE8. The red reads (n = 9) show the direct interactions between all the four KEEs. (h) The consistency of CG, CHG, and CHH DNA methylation between Pore‐C and WGBS data. (i) Pearson correlation coefficient (PCC) of CG methylation level between Pore‐C contacts with a genomic distance larger than 25 kb, 100 kb, and 1 Mb, respectively.
The Pore‐C pipeline defined the number of genome‐matched fragments on a single Pore‐C read as the ‘Contact Order’ for that read (Ulahannan et al., 2019) (e.g., the Contact Order of the nanopore read shown in Figure 1a is 5). Our results show that 44% reads contain multi‐way interactions (Contact Order ≥3), similar to Pore‐C in human cells (47.56%) (Ulahannan et al., 2019). Whereas the Contact Orders are mostly at two in Hi‐C (Wang et al., 2015), consistent with its design (Figure 1d). Pore‐C results in Arabidopsis and animals show a higher efficiency in capturing multi‐way interaction compared to SPRITE (12%) (Quinodoz et al., 2018), demonstrating that the long‐read approach overmatches the labeling approach in capturing multi‐way interaction. Reads with higher contact order detected interactions across longer genomic regions in Pore‐C (Figure 1e), which helps genome assembly.
To validate the effectiveness of Pore‐C in multi‐way interaction in Arabidopsis, we examined the interactions among KNOT ENGAGED ELEMENT (KEE) (Grob et al., 2014) with our Pore‐C data. In Arabidopsis, the pairwise interactions of ten KEEs (named KEE1 to KEE10) can be visualized as discrete dots on the contact matrix of Hi‐C and were confirmed by light microscopy‐based FISH assay (Grob et al., 2014). However, it remains unclear if multiple KEEs simultaneously interact in the same nucleus. In our Pore‐C data, 1145 reads detect contacts among three KEEs, higher than that of the 100 randomly selected control regions (mean 279.29 reads, P‐value = 0.04), and 78 reads capture the contacts among four KEEs and more (Figure 1f). Interestingly, nearly half of these high‐order interactions involve both KEE3 and KEE4 (561 reads), suggesting that these two KEEs serve as key hubs in the KEE interacting network (Figure 1f). Direct interaction among four KEEs was also detected, including 9 reads for KEE3, KEE4, KEE7, and KEE8 (referred to as KEE(3,4,7,8), Figure 1g), 4 reads for KEE(1,3,4,6), and 11 reads for KEE(3,4,5,6). These results demonstrate the existence of multi‐way contacts of KEEs. Additional analyses on KEEs and telomeres are in the Supplementary material.
The PCR‐free strategy of Pore‐C enables to directly detect DNA methylation and higher‐order chromatin interaction by long‐read sequencing and thus helps to reveal their coordination on the same read, without bisulfite conversion required in Methyl‐HiC (Li et al., 2019). The CG, CHG, and CHH methylation level we called from Pore‐C reads are highly consistent with whole‐genome bisulfite sequencing (WGBS), the gold standard for DNA methylation (Figure 1h) (Ni et al., 2021; Stroud et al., 2012), and between the two Pore‐C replicates (Figure S5). We used nanopolish (Simpson et al., 2017) to detect 5mCG related to each Pore‐C read, and found a higher correlation of CG methylation level among interacting fragments in Arabidopsis (Figure 1i), consistent with the previous report in mouse ESCs (Li et al., 2019).
In summary, Pore‐C efficiently captures the genome‐wide multi‐way chromatin interaction landscape at a single‐molecular level, and reveals the epigenetic modification of interacting DNA fragments, which may help to explore the heterogeneity of the chromatin interaction and DNA methylation in nuclei of different cell types. With this method, we validated the multi‐way contacts among KEEs and telomeres, and found CG methylated regions on Arabidopsis genome tend to contact. Taken together, our results demonstrate that Pore‐C is a simple, accurate, and effective method for exploring multi‐way chromatin interaction in the plant which might help to explore the interactions among several homologous chromosomes in polyploid plants. More bioinformatics tools developed for Nanopore will facilitate further analysis of Pore‐C multidimensional information.
Materials and methods
Please refer to Supplementary material.
Conflict of interests
The authors declare no competing interests.
Author contributions
Y.L. and Z.L. developed the method and performed the experiments. Z.L., Y.Y., F.Z., and Y.L. analyzed the data, J.Z. conceived and oversaw the study. Z.L., Y.L., and J.Z. wrote the manuscript. All authors revised the manuscript.
Supporting information
Figure S1 Description of Pore‐C pipeline.
Figure S2 Size and order distribution of the two Pore‐C replicates.
Figure S3 Pore‐C identifies the multi‐way interactions between KEEs.
Figure S4 Pore‐C reveals multi‐way interactions of telomeres in Arabidopsis.
Figure S5 The consistency of CG, CHG and CHH methylation level from WGBS and the two replicates of Pore‐C.
Table S1 The sequencing statistics of Pore‐C libraries.
Table S2 Commands and parameters used in this study.
Acknowledgements
Group of J.Z. is supported by the National Key R&D Program of China Grant (2019YFA0903903), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2016ZT06S172), the Shenzhen Sci‐Tech Fund (KYTDPT20181011104005), a Stable Support Plan Program of Shenzhen Natural Science Fund Grant (20200925153345004), and Key Laboratory of Molecular Design for Plant Cell Factory of Guangdong Higher Education Institutes (2019KSYS006).
Li, Z. , Long, Y. , Yu, Y. , Zhang, F. , Zhang, H. , Liu, Z. , Jia, J. , Mo, W. , Tian, S. Z. , Zheng, M. and Zhai, J. (2022) Pore‐C simultaneously captures genome‐wide multi‐way chromatin interaction and associated DNA methylation status in Arabidopsis. Plant Biotechnol. J., 10.1111/pbi.13811
Data availability statement
The Pore‐C data are available in the Genome Sequence Archive in National Genomics Data Center under accession number CRA005105 (https://ngdc.cncb.ac.cn/gsa/s/INkjuqHu).
References
- Grob, S. , Schmid, M.W. and Grossniklaus, U. (2014) Hi‐C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell, 55, 678–693. [DOI] [PubMed] [Google Scholar]
- Li, G. , Liu, Y. , Zhang, Y. , Kubo, N. , Yu, M. , Fang, R. et al. (2019) Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods, 16, 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. and Weigel, D. (2015) Chromatin in 3D: progress and prospects for plants. Genome Biol. 16, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, P. , Huang, N. , Nie, F. , Zhang, J. , Zhang, Z. & Wu, B. et al. (2021) Genome‐wide detection of cytosine methylations in plant from Nanopore sequencing data using deep learning. bioRxiv, 2021.2002.2007.430077. [DOI] [PMC free article] [PubMed]
- Ouyang, W. , Xiong, D. , Li, G. and Li, X. (2020) Unraveling the 3D genome architecture in plants: present and future. Mol. Plant, 13, 1676–1693. [DOI] [PubMed] [Google Scholar]
- Quinodoz, S.A. , Ollikainen, N. , Tabak, B. , Palla, A. , Schmidt, J.M. , Detmar, E. et al. (2018) Higher‐order inter‐chromosomal hubs shape 3D genome organization in the nucleus. Cell, 174, 744–757.e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, J.T. , Workman, R.E. , Zuzarte, P.C. , David, M. , Dursi, L.J. and Timp, W. (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods, 14, 407–410. [DOI] [PubMed] [Google Scholar]
- Stroud, H. , Hale, C.J. , Feng, S. , Caro, E. , Jacob, Y. , Michaels, S.D. et al. (2012) DNA methyltransferases are required to induce heterochromatic re‐replication in Arabidopsis. PLoS Genet. 8, e1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulahannan, N. , Pendleton, M. , Deshpande, A. , Schwenk, S. , Behr, J.M. & Dai, X. et al. (2019) Nanopore sequencing of DNA concatemers reveals higher‐order features of chromatin structure. bioRxiv, 833590.
- Wang, C. , Liu, C. , Roqueiro, D. , Grimm, D. , Schwab, R. , Becker, C. et al.(2015) Genome‐wide analysis of local chromatin packing in Arabidopsis thaliana . Genome Res. 25, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Description of Pore‐C pipeline.
Figure S2 Size and order distribution of the two Pore‐C replicates.
Figure S3 Pore‐C identifies the multi‐way interactions between KEEs.
Figure S4 Pore‐C reveals multi‐way interactions of telomeres in Arabidopsis.
Figure S5 The consistency of CG, CHG and CHH methylation level from WGBS and the two replicates of Pore‐C.
Table S1 The sequencing statistics of Pore‐C libraries.
Table S2 Commands and parameters used in this study.
Data Availability Statement
The Pore‐C data are available in the Genome Sequence Archive in National Genomics Data Center under accession number CRA005105 (https://ngdc.cncb.ac.cn/gsa/s/INkjuqHu).