

Abstract
Residual pulmonary vascular obstruction (RPVO) and chronic thromboembolic pulmonary hypertension (CTEPH) are both long-term complications of acute pulmonary embolism (PE), but it is unknown whether RPVO can be predicted by variants of fibrinogen associated with CTEPH.
We use Akaike Information Criterion to select the best predictive models for RPVO in two prospectively followed cohorts of acute PE patients, using as candidate variables the extent of the initial obstruction, clinical characteristics and fibrinogen-related data. We measured the selected models’ goodness of fit by analysis of deviance and compared models by χ2 test.
RPVO occurred in 29/102 (28.4%) subjects in the first cohort and 46/182 (25.3%) subjects in the second. The best-fit predictive model derived in the first cohort (p = 0.0002) and validated in the second cohort (p = 0.0005) implicated fibrinogen Bβ-chain monosialylation in the development of RPVO. When the derivation procedure excluded clinical characteristics, fibrinogen Bβ-chain monosialylation remained a predictor of RPVO in the best-fit predictive model (p = 0.00003). Excluding fibrinogen characteristics worsened the predictive model (p = 0.03).
Fibrinogen Bβ-chain monosialylation, a common structural attribute of fibrin, helped predict RPVO after acute PE. Fibrin structure may contribute to the risk of developing RPVO.
Keywords: pulmonary embolism, residual vascular obstruction, fibrinogen, V/Q lung scan
Introduction
After acute pulmonary embolism (PE), thrombotic material in the pulmonary arteries typically disintegrates, although in some cases it transforms into permanent obstructing scars.[1] Residual pulmonary vascular obstruction of 10% or more (RPVO) is common after PE and can diminish physical activity, reduce quality of life and worsen overall prognosis. [2–7] RPVO is associated with dyspnea,[8] hypoxemia,[9] gas exchange deficits,[9] exercise intolerance[2] and other serious clinical impairments.[9–11] RPVO incurs the risk of recurrence[12] and of chronic thromboembolic pulmonary hypertension (CTEPH),[2, 13] an uncommon[14, 15] but severe complication of PE[16, 17] in which obstruction is extensive and pulmonary arterial pressure rises.[18]
The mechanisms by which fibrin-rich thromboemboli transform into permanent obstructions characteristic of RPVO and of CTEPH are incompletely understood. RPVO is not associated with “thrombophilia” or with impaired fibrinolytic enzymes.[2] We observed that fibrin itself derived from some patients with CTEPH forms clots resistant to lysis[19, 20] with aberrations in molecular configuration[21] and clot structure[22] that reflect otherwise rare fibrinogen gene mutations.[20] CTEPH-associated dysfibrinogenemia may be a model for more common fibrin characteristics that might tip the balance from clot resolution towards RPVO.
To gain insights into the development of RPVO, the Prediction of Residual Obstruction Manifested after Pulmonary Embolism Treatment (PROMPT) study followed two cohorts of patients after acute PE to determine the best predictors of RPVO. We investigated whether fibrinogen post-translational modifications and other common fibrin structural attributes might help predict the development of RPVO after acute PE. If fibrin properties are consistently associated with RPVO, then they might be implicated as a mechanism of its development.
Methods
Study design and study population
We used clinical data and biological material from two single-center, prospective, observational cohort studies conducted on separate groups of acute PE patients in an academic teaching hospital (Hôpital Européen Georges Pompidou, Paris, France). The two studies were approved by the local ethics committee. All patients provided written informed consent before enrollment.
For cohort 1, we enrolled consecutive patients with PE from our center who had been included in the FARIVE study, a multicentre case-control study that evaluated the interactions of environmental, genetic and biological risk factors on the risk of a first venous thromboembolism (VTE). The study included consecutive subjects who were above 18 years and had their first symptomatic occurrence of pulmonary embolism. Patients were excluded if they had active cancer unless they had been treated successfully and had no evidence of recurrence for the previous 5 years. Patients were also excluded if they had short life expectancies secondary to other associated pathologies, anticipated impossibility of follow up or a previously diagnosed thrombophilia.
For cohort 2, we enrolled consecutive patients with PE followed-up at our center between February 1999 and May 2006 who had not been enrolled in the FARIVE study.[2] This study was designed to assess the clinical significance of residual pulmonary vascular obstruction after an episode of acute PE. Patients were included if they were above 18 years of age and had completed at least 3 months of oral anticoagulant therapy without having recurrent PE.
In both cohorts, the diagnosis of PE was established through high-probability lung scintigraphy, thoracic CT scan, pulmonary angiography, sonography showing the existence of a proximal venous thrombosis in the absence of a differential diagnosis for the reported pulmonary symptoms, or nondiagnostic pulmonary scintigraphy associated with lower extremity ultrasound disclosing proximal DVT. The type and duration of long-term therapy was left to the discretion of physician in charge of the patients. The standard practice was to treat provoked PE for at least 3 months and unprovoked PE for at least 6 months.
Residual Pulmonary Vascular Obstruction
All patients underwent a standardized ventilation/perfusion (V/Q) lung scan at least six months after the acute PE.[23] A trained investigator (BP) blinded to all other study data identified and scored mismatched perfusion defects according to a pre-specified protocol.[24] We defined RPVO as a residual vascular obstruction of 10% or more, which corresponds to an amputation of at least two pulmonary segments (the minimal obstruction defining a high-probability result on diagnostic V/Q lung scans[25]) and is associated with dyspnea and exercise intolerance after acute PE.[2]
Demographic and clinical data
A standardized tool was used upon study enrollment to record clinical data (including the type of acute therapy and the duration of long-term therapy) and conduct a structured interview. Except for the fibrinogen-related laboratory data described in the following section, all laboratory values were measured in the clinical laboratory of Hôpital Européen Georges Pompidou. Physicians blinded to the other data of each patient used a validated score to calculate the initial pulmonary vascular obstruction on the original diagnostic CT scan[26] or V/Q scan.[24] None of the subjects had pulmonary angiography as the initial diagnostic test.
Fibrin structural attributes
Upon enrollment, blood samples were collected by venipuncture into anticoagulant-containing tubes. Plasma was separated by two consecutive centrifugations and stored at −70°C for analysis. Plasma fibrinogen levels were measured in a clinical laboratory at the time of blood collection.
Research assays were performed on fibrinogen that had been purified from citrated plasma by ethanol precipitation and exposure to Gelatin Sepharose (GE Healthcare) to remove residual fibronectin, followed by dilution to 4mg/mL.[20] Appendix 1 includes the detailed assay methodologies, which have been previously reported.[19, 20, 22] They are briefly described below.
Fibrinogen, thrombin, and calcium chloride were combined to form fibrin clots in the amounts appropriate for each structural assay. We determined the turbidity of the fibrin clots, which corresponds to fibrin network fibril dispersal and branching as validated by microscopy.[20, 22] We measured fibrin clot susceptibility to lysis through the rates of turbidity decrease, measured consecutively after clots were exposed to a mixture of plasminogen and tPA.[20] We measured fibrin clot permeability, which reflects the organization with which fibrin polymers are formed,[22] by the rate of passage of permeability buffer through fibrin clots. More organized fibrin networks with large pores have higher flow compared to disorganized fibrin networks with small pores. We measured by SDS-PAGE the degree that fibrin crosslinks its α-chains as a reflection of the potential to form stabilized clots during thrombosis.[19] We used I125-labeled antibodies specific for the β15–42 peptide sequence chain (anti-β15–42) to measure the accessibility within the fibrin clots of the fibrin β -chain amino termini (residues 15–42), which are implicated in a variety of physiological events that affect thrombus remodeling into intravascular scars.[27–29]
We measured precise fibrinogen masses with liquid chromatography, followed by mass spectrometry (LC/MS) to identify post-translational modifications.[20] We analyzed the LC/MS spectra representing the fibrinogen Aα-chains, Bβ-chains, and γ-chains and measured the proportional heights of mass peaks corresponding to variants we had observed in CTEPH-associated dysfibrinogenemias and any other mass peaks that represented 10% or more of the associated fibrinogen chains.
Statistical analysis
Categorical variables are presented as numbers and percentages, and continuous variables are presented as means ± SD. All available demographic, clinical and fibrinogen data were compared between the two cohorts, using t-tests, Wilcoxon-Mann-Whitney tests, Fisher’s exact tests or Chi-square tests, as appropriate. Univariate assessment of predictors of RPVO was performed for each cohort, using the same tests, as appropriate.
Because the interaction of lysis, inflammation and cellular remodeling that leads to RPVO likely develops through a complex interaction among clinical factors and biological factors, the careful selection of a predictive model was critical to our goal. A joint predictive effect would be obscured if simplistic univariate analyses are used.[30] For this reason, we used a step-wise multivariate logistic regression procedure based on Akaike Information Criterion (AIC)[30, 31] to select the most parsimonious and accurate multivariable prediction model for RPVO in each cohort, using as candidate predictors both clinical risk factors and fibrin structural properties. If the best predictive models selected in this fashion depended on fibrin structural attributes, then the analysis would support the role of fibrin properties in the development of RPVO after acute PE. The AIC-based approach avoids selecting models solely on the basis of apparent significance of the individual terms or global fit, both of which are likely to be over-stated; overfitting is penalized.[31] We used deviance-based goodness of fit, a standard logistic regression metric, to assess the quality of the selected models in each cohort.
Multivariable modeling was performed on information from subjects who had complete data (without missing values), to enable model comparisons. For cohort 1, AIC-based stepwise logistic regression was used to identify the best clinical and demographic predictors of RPVO. Then, AIC-based stepwise logistic regression was used to select the best model to predict RPVO, using as candidate predictors each patient’s previously identified relevant demographic and clinical measures, as well as the results of the fibrinogen experiments we described. We used the Receiver Operating Characteristic (ROC) curve [32] to find the best probability cutoff for predicting RPVO and assessed the model’s sensitivity and specificity. We then validated the model derived in cohort 1 by using it to predict RPVO in cohort 2.
To confirm the role of specific fibrin structural attributes in RPVO, we performed the prediction model selection process again in cohort 1 and included in the candidate predictor pool only the fibrinogen properties and the initial degree of obstruction (excluding the clinical data). The initial degree of obstruction is such a strong predictor of residual obstruction[2] that all predictive models would include it. We were interested in finding additional, independently predictive measures. Finally, to further verify the role of fibrinogen, we used a χ2 test of residual deviances to determine whether the model that included the initial obstruction as well as fibrinogen properties were significantly better predictors of RPVO than the model that excluded fibrinogen properties.
To confirm the generalizability of individual RPVO predictors identified in cohort 1, we repeated the multivariate modeling process in cohort 2, but limited the candidate variables to those that were disclosed by the cohort 1 analysis. We reasoned that variables predictive of RPVO in both cohorts might be implicated in RPVO development in the general population of patients with acute PE. If a variable derived from cohort 1 was not included in the optimal prediction equation generated for cohort 2, or if its coefficient was in the opposite direction, we reasoned that it would be less likely to be predictive of RPVO in other PE populations.
Results
Subject populations
From January 2003 to January 2009, 137 patients were recruited into cohort 1 (Figure 1A). Two patients (1.4%) were not assessed for RPVO because they were lost to follow up, in six (4.4%), the follow-up V/Q lung scans were missing. Plasma samples for fibrin(ogen) laboratory analysis were unavailable from 27 subjects (20.9%). There were no significant differences between the 35 patients excluded from cohort 1 and the included patients from cohort 1 on age, sex, time from symptoms to diagnosis or residual obstruction. Among the 102 subjects included in the analysis, 29 (28.4%) had RPVO greater than or equal to 10% (Table 1). The mean (± SD) time from PE diagnosis to V/Q Lung scan was 8.3 (± 2.5) months and mean degree of persistent perfusion defect was 8.1% (± 11.7%).
Figure 1. Study outline for both cohorts.
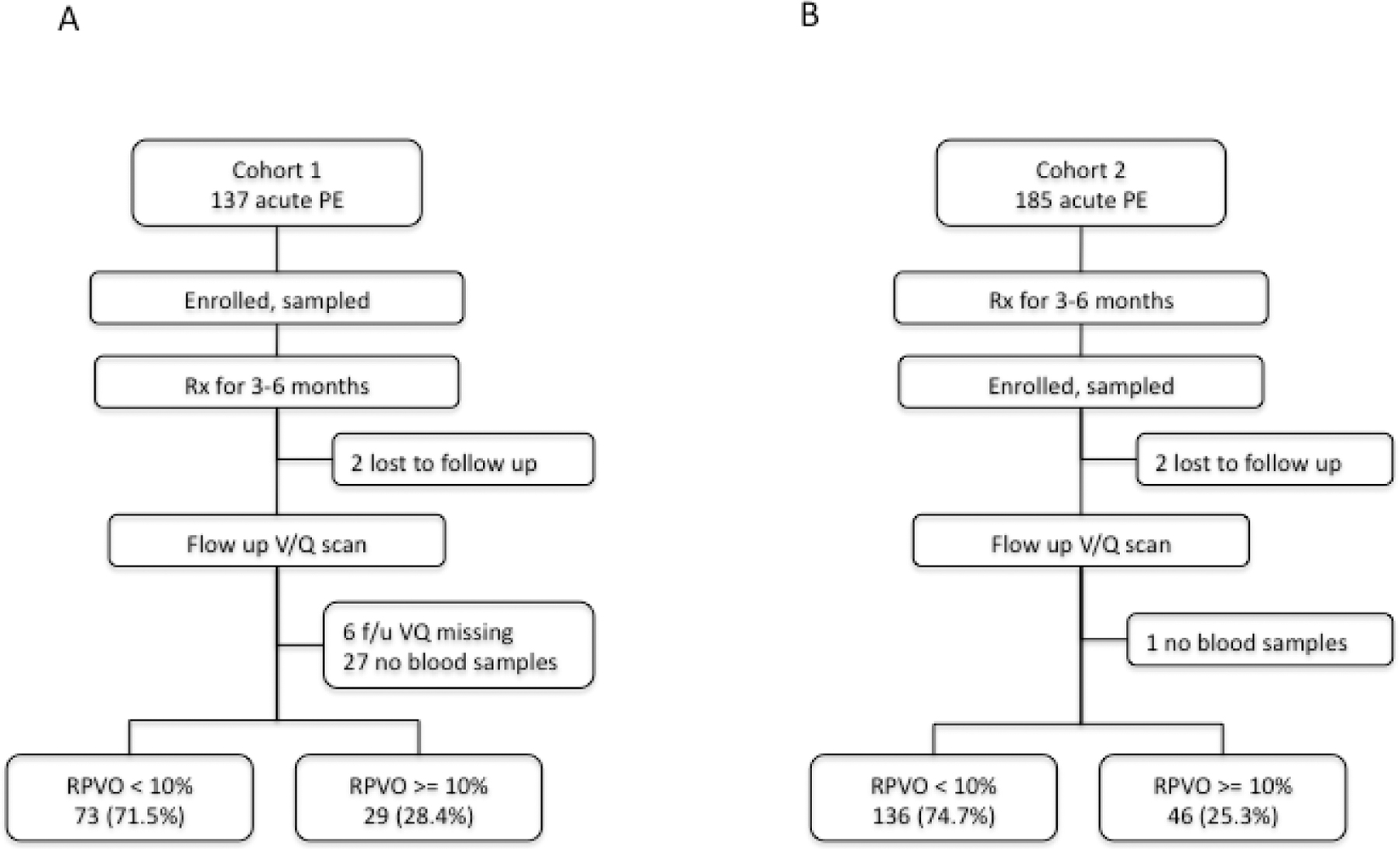
A. In cohort 1, of 137 subjects recruited at the time of acute PE diagnosis, 102 subjects could be evaluated, 29 (28.4%) of whom had RPVO of at least 10%. B. In cohort 2, of the 185 subjects recruited after long-term treatment for acute PE, 182 subjects could be evaluated, 46 (25.3%) of whom developed RPVO of at least 10%.
Table 1. Comparison of characteristics between cohort 1 and cohort 2.
Wilcoxon-Mann-Whitney was used to calculate p-values for continuous variables. Fisher’s exact test was used for binary variables.
| Cohort 1 (n=102) | Cohort 2 (n=182) | p* | |
|---|---|---|---|
| Age (years) Mean ± SD | 52.2 ± 19.6 | 61.3 ± 18.5 | 0.0002 |
| Sex (% male) | 45.1 | 36.3 | 0.1644 |
| VTE provoked (%) | 52.9 | 54.4 | 0.9013 |
| Recent travel for > 3 hours (%) | 12.7 | 12.1 | 0.8532 |
| Trauma or LE fracture within 3 months (%) | 10.8 | 9.3 | 0.6838 |
| Bed rest (%) | 8.8 | 20.3 | 0.0118 |
| Surgery or anesthesia within 3 months (%) | 9.9 | 17.6 | 0.1153 |
| Family history of VTE (%) | 31.4 | 18.0 | 0.0121 |
| Active cancer (%) | 2.0 | 12.1 | 0.003 |
| Concomitant DVT (%) | 34.3 | 54.9 | 0.0009 |
| Time from first symptom to diagnosis (days) Mean ± SD | 11.2 ± 17.1 | 14.3 ± 28.6 | 0.0409 |
| Diagnosis by CTPA, rather than VQ (%) | 74.5 | 71.4 | 0.6778 |
| Initial pulmonary vascular obstruction, (%) mean ± SD | 38.8 ± 25.9 | 34.1 ± 22.9 | 0.1383 |
| Thrombolysis (%) | 0 | 5.5 | 0.0157 |
| Inferior vena caval filter (%) | 0 | 4.9 | 0.0288 |
| Days of anticoagulation Mean ± SD | 212.5 ± 82.80 | 231.8 ± 171.42 | 0.91 |
| Extended duration anticoagulation (%) | 16.7 | 24.2 | 0.1752 |
| Interval from diagnosis to study VQ scan (days), mean ± SD | 258 ± 75 | 330 ± 111 | <0.0001 |
| Factor VLeiden (%) | 15.9 | 6.2 | 0.025 |
| Prothrombin 20210a (%) | 13.0 | 5.6 | 0.0637 |
| Fibrinogen Aα T312A (%) | 57.6 | 37.3 | 0.0019 |
| Antithrombin (IU/dL) Mean ± SD** | 97.54 ± 12.95 | 96.50 ± 11.13 | 0.5982 |
| Protein C value (IU/dL) Mean ± SD** | 98.45 ± 24.81 | 104.10 ± 19.69 | 0.0749 |
| Protein S value (IU/dL) Mean ± SD** | 89.18 ± 20.76 | 89.60 ± 21.53 | 0.9643 |
| Fibrinogen (g/L) Mean ± SD | 4.28 ± 1.13 | 3.52 ± 0.89 | <0.0001 |
| Platelet (G/l) Mean ± SD | 258.8 ± 86.43 | 258.9 ± 68.48 | 0.6496 |
| Prothrombin ratio (% control) Mean ± SD | 78.97 ± 18.52 | 83.75 ± 20.38 | 0.001 |
| Thrombin (%) Mean ± SD | 83.35 ± 30.13 | 93.76 ± 27.03 | 0.0811 |
| FV (%) Mean ± SD | 106.2 ± 26.58 | 103.8 ± 25.32 | 0.9201 |
| FVII (%) Mean ± SD | 86.67 ± 40.75 | 100.30 ± 34.10 | 0.1526 |
p value for comparison between cohort 1 and cohort 2.
Antithrombin 3 levels were not measured in 23/102 subjects in cohort 1 and 19/182 subjects in cohort 2.
Protein C values were not measured in 38/102 subjects in cohort 1and 27/182 subjects in cohort 2. Protein S values were not measured from 42/102 subjects in cohort 1 and 35/182 subjects in cohort 2.
From February 1999 to May 2006, 185 patients were recruited into cohort 2 (Figure 1B). Two patients (1.0%) were lost to follow up. Plasma samples for fibrin(ogen) laboratory analysis were unavailable from one subject (0.5%). The excluded patients were not outliers on any of the studied characteristics. Among the remaining 182 subjects, 46 (25.3%) had RPVO greater than 10%. The mean (± SD) time from PE diagnosis to V/Q Lung scan was 10.9 (± 3.7) months and mean RPVO was 6.8 % (± 12.7 %).
The subject characteristics are compared between the two cohorts in Table 1 (a comprehensive list is included in appendix 2). Cohort 1 was significantly younger than Cohort 2 (52.2 ± 19.6 years vs 61.3 ± 18.5 years, p = 0.0002). Cohort 1 had a lower prevalence of cancer (2.0 % vs 12.1 %, p = 0.003), a lower prevalence of bed rest (8.8 % vs 20.3 %, p = 0.01), a lower use of thrombolytics (0 % vs 5.5 %, p = 0.02) and a lower use of IVC filters (0 % vs 4.5 %, p = 0.03) than Cohort 2. However, family histories of VTE were more common in Cohort 1 than in Cohort 2 (31.4 % vs 18.0 %, p = 0.01). PE was diagnosed somewhat earlier after the onset of symptoms in Cohort 1 than in Cohort 2 (11.2 ± 17.1 days vs 14.3 ± 28.6 days, p = 0.04).
A univariate analysis of the factors associated with RPVO is displayed in Table 2. In both cohorts, the initial pulmonary vascular obstructions were smaller among those who recovered without RPVO, than in those with RPVO. Notably, a CTEPH-associated mutation (fibrinogenSan Diego III)[20] was found in one subject from Cohort 1, who had RPVO of 29.5 %.
Table 2. Univariate comparison of characteristics between patients with RPVO and without.
Wilcoxon-Mann-Whitney was used to calculate p-values for continuous variables. Fisher’s exact test was used for binary variables.
| Cohort 1 (n=102) | Cohort 2 (n=182) | |||||
|---|---|---|---|---|---|---|
| Patients with RPVO ≤ 10% (n=73) | Patients with RPVO >= 10% (n=29) | p* | Patients with RPVO ≤ 10% (n=136) | Patients with RPVO >= 10% (n=47) | p* | |
| Presentation and treatment | ||||||
| Age (years) Mean ± SD | 49.3 ± 18.1 | 59.4 ± 21.6 | 0.023 | 59.2 ± 19.3 | 67.4 ± 14.4 | 0.019 |
| Provoked VTE | 60.3 % | 34.5 % | 0.027 | 55.1 % | 52.2 % | 0.735 |
| Interval from symptoms to diagnosis (days) Mean ± SD | 8.7 ± 15.5 | 17.1 ± 19.4 | 0.001 | 12.5 ± 25.4 | 19.8 ± 36.3 | 0.336 |
| Initial pulmonary vascular obstruction (%) Mean ± SD | 32.4 ± 23.2 | 54.9 ± 25.9 | 0.0001 | 30.7 ± 22.1 | 44.1 ± 22.4 | 0.001 |
| Extended anticoagulation | 9.6 % | 34.5 % | 0.006 | 20.6 % | 34.8 % | 0.072 |
| Interval from diagnosis to VQ scan (days) Mean ± SD | 266.7 ± 78.0 | 234.5 ± 64.5 | 0.047 | 337.6 ± 113.1 | 305.6 ± 100.1 | 0.088 |
| Clinical laboratory | ||||||
| Fibrinogen (g/L) Mean ± SD | 4.3 ± 1.1 | 4.2 ± 1.1 | 0.504 | 3.4 ± 0.8 | 3.8 ± 1.1 | 0.019 |
| Platelets amount/nL) Mean ± SD | 245.2 ± 87.2 | 293.8 ± 75.6 | 0.008 | 258.3 ± 70.1 | 260.3 ± 64.4 | 0.341 |
| Prothrombin ratio (% of control) Mean ± SD | 80.1 ± 17.6 | 76.3 ± 20.7 | 0.570 | 86.3 ± 18.5 | 77.0 ± 23.7 | 0.013 |
| Fibrin property | ||||||
| des-gln461 Bβ-chains (%) Mean ± SD | 6.03 ± 1.81 | 5.74 ± 1.34 | 0.678 | 15.53 ± 11.49 | 12.11 ± 8.52 | 0.042 |
p value for comparison between patients with and without RPVO.
Derivation of RPVO predictive model in cohort 1
Prediction model creation and selection was performed on 94 subjects in cohort 1 who had complete clinical and fibrin-related data sets. When all candidate variables were considered, the optimal model to predict the probability of RPVO included the initial extent of vascular obstruction, fibrinogen Bβ-chain monosialylation, the days between symptoms and diagnosis and fibrinogenγ’(Table 3). The goodness of fit of the model was highly significant (difference between null and residual deviances = 21.8, χ2 p = 0.0002). The area under the receiver operator characteristic (ROC) curve was 0.762 (p = 0.0001). The sensitivity was 0.679 (95% CI 0.476 0.841) and the specificity was 0.727 (95% CI 0.604, 0.830).
Table 3. Predictors of RPVO in Cohort 1.
Candidate predictors with p values listed in the table were incorporated in the corresponding models that had been selected by the Akaike Information Criterion to be the optimally predictive of RPVO after acute pulmonary embolism for the cohort.
| Candidate predictive variables | OR (95% CI) |
p* |
|---|---|---|
| Presentation and treatment | ||
| Initial obstruction | 1.041 (1.018, 1.064) | 0.000369 |
| Interval symptoms to diagnosis | 1.026 (0.997, 1.056) | 0.078758 |
| Fibrin property | ||
| Bβ-chain mono sialylation | 1.074 (0.986, 1.168) | 0.100657 |
| Fibrinogen γ’ | 0.641 (0.419, 0.980) | 0.040079 |
| Model goodness of fit** | 0.00022 | |
p value from multivariate analysis.
Deviance-based goodness of fit, comparing the fit of each model with the fit of the null model.
Validation of RPVO predictive model in cohort 2
The optimal predictive model derived from all candidate variables in cohort 1 was applied to the 141 subjects in cohort 2 who had complete data sets. The area under the receiver operator characteristic (ROC) curve was 0.693 (p = 0.0005). The sensitivity was 0.622 (95% CI 0.448, 0.775) and the specificity was 0.692 (95% CI 0.595, 0.777).
Derivation of RPVO predictive model in cohort 1 using fibrinogen properties
When the candidate predictor pool excluded the clinical and demographic data, and encompassed only the initial extent of obstruction and fibrinogen data, the best prediction model’s goodness of fit remained highly significant (difference between null and residual deviances = 26.2, χ2 p = 0.00003). Initial obstruction and fibrinogen Bβ-chain monosialylation remained as predictors of RPVO in that model, which also included fibrinogen γ’and the fibrin lysis rate (Table 4). The area under the receiver operator characteristic (ROC) curve was 0.802. However, when the candidate predictor pool also excluded the fibrinogen data, χ2 tests of residual deviances disclosed that the best-fit models did not predict RPVO as well as when fibrinogen data were included (difference between the two models’ deviances = 9.0, χ2 p = 0.03).
Table 4. Predictors of RPVO in Cohort 1, considering only the initial obstruction and fibrin properties.
Candidate predictors with p values listed in the table were incorporated in the corresponding models that had been selected by the Akaike Information Criterion to be the optimally predictive of RPVO after acute pulmonary embolism for the cohort.
| Candidate predictive variables | OR (95% CI) | p* |
|---|---|---|
| Presentation and treatment | ||
| Initial obstruction | 1.054 (1.028, 1.080) | 0.00003 |
| Fibrin property | ||
| Bβ-chain mono sialylation | 1.095 (0.994, 1.206) | 0.0655 |
| Fibrin clot lysis rate | 10.872 (0.349, 338) | 0.1737 |
| Fibrinogen γ’ | 0.614 (0.386, 0.975) | 0.0388 |
| Model goodness of fit** | 0.00003 | |
p value from multivariate analysis.
Deviance-based goodness of fit, comparing the fit of each model with the fit of the null model.
Derivation of RPVO predictive model in cohort 2 using predictor pool from cohort 1
A prediction model was created for cohort 2, with the pool of candidate predictors restricted to only the four predictors identified by the model creation and predictor selection process performed for cohort 1. When only those candidate variables were considered, the optimal model to predict the probability of RPVO included the initial extent of vascular obstruction, fibrinogen Bβ-chain monosialylation and the days between symptoms and diagnosis. Fibrinogenγ’was no longer included as a significant predictor. The goodness of fit of the model remained highly significant (difference between null and residual deviances = 18.8, χ2 p = 0.0009). The area under the receiver operator characteristic (ROC) curve was 0.702.
In a post-hoc analysis, the median extent of Bβ-chain monosialylation among the entire (two cohort) group was 59.2%. Among the half of patients with Bβ-chain monosialylation below the median level, 19.4% had RPVO, whereas 31.9% of those with higher levels of Bβ-chain monosialylation had RPVO (p = 0.0169).
Discussion
We followed two cohorts of patients after acute PE to determine the factors that were most predictive of RPVO. High proportions of monosialylation of the fibrinogen Bβ-chain were consistently predictive of RPVO across the two cohorts. Among the other candidate variables, only the initial degree of vascular obstruction was more predictive of RPVO, which is in agreement with our previous observations.[2]
We defined RPVO as persistent lung perfusion defects in 10% or more of the lungs (i.e. in at least two lung segments). Although we did not objectively measure quality of life or other symptomatic outcomes in the current study, we previously reported that our definition of RPVO, which occurred in 29% of acute PE patients, was significantly associated with dyspnea, elevated pulmonary artery pressures and exercise intolerance.[2] Among PE patients with this degree of RPVO, 16% were subsequently diagnosed with chronic thromboembolic pulmonary hypertension (CTEPH).[2]
The predictors selected by the model were highly associated with the subsequent development of RPVO (p = 0.0001 in the derivation cohort and p = 0.0005 in the validation cohort). The diagnostic accuracies of the model were not sufficiently high to exclude of predict RPVO for clinical purposes (AUC 0.762 and 0.693 in the derivation and validation cohorts, respectively). However, the association between RPVO and fibrinogen properties provides insights into the mechanism whereby some patients develop RPVO or even CTEPH after acute PE. No association with genetic thrombophilia has been identified,[2] which raises the possibility that impairments in the process of fibrinolysis are responsible.[33] We focused our investigation on fibrin itself, since the resistance of purified fibrin clots to lysis has been observed in some patients with CTEPH[19, 20] and, to a lesser extent, in some PE patients.[33, 34] We used as our model the properties of fibrin[21, 22] that we had observed in CTEPH-associated dysfibrinogenemias.[20] We reasoned that variations in post-translational modification patterns might influence fibrin formation and lysis[35, 36] less severely, but more commonly, than CTEPH-associated dysfibrinogenemias and that they may have a role in persistence of thromboemboli after acute PE.
Our results support the hypothesis that attenuation of the body’s normal mechanisms for resolution of fibrin-rich thrombi is an important pathophysiological step in the etiology of chronic syndromes after venous thromboembolism.[5, 18] One possible mechanism is that properties of the fibrin clot would delay or limit plasmin-mediated fibrinolysis or modify other clot resolution mechanisms such as angiogenic and inflammation responses.[18] Our observations suggest that fibrin structural attributes may influence fibrin clot structure sufficiently to contribute to poor clot resolution and chronic disease after acute PE.
High proportions of monosialiated Bβ-chains were predictive of RPVO in both PE cohorts that we studied. Sialylation occurs during fibrinogen formation and has various effects on fibrin polymer organization and resolution due to the electrostatic effect of the negatively charged sialic acid. Experimental sialic acid removal results in disordered fibrin clot formation,[35, 37, 38] while excessive sialylation of Bβ-chains and γ-chains also results in abnormal clots.[22, 36] Monosialylation is increased when clotting might be beneficial, such as during pregnancy[39] and acute infection.[40]
The results for the two cohorts we studied differed somewhat in the predictive power of low fibrinogen γ’, which has been implicated in the regulation of fibrin crosslinking. The differences might be due to chance. Alternatively, they might reflect subtle differences between the two cohorts or even different implications of fibrin(ogen) characteristics at the time of the acute PE (when blood samples were collected from the first cohort) and several months later (when the second cohort was sampled).
Our study is limited by the fact that we did not independently confirm the clinical significance of RPVO in our patients by objective measurements of dyspnea, hemodynamics or subsequent clinical outcomes after completion of the study protocol. We have, however, validated the association between RPVO and those findings in a previous series of similar PE patients.[2] We also acknowledge that 20% of patients in the first cohort were excluded because they did not have adequate plasma samples for fibrinogen analysis, which could have introduced a potential for bias in that cohort’s analysis. While we did not disclose an association between the type of acute or long-term anticoagulation therapy and RPVO, the study was performed prior to the widespread use of direct oral anticoagulants, so we cannot comment on the influence of those therapies. Finally, despite the consistency of our findings in both cohorts who had similar rates of RPVO, they should be confirmed in subsequent prospective studies.
Conclusion
The extent of pulmonary vascular obstruction at the onset of PE, as well as fibrinogen Bβ-chain monosialylation, were predictive of RPVO after acute PE in the two cohorts we studied. Our results confirm our previous observations and support the role of fibrin properties in the resolution or persistence of thrombi after acute PE, which is similar to the role played in the development of CTEPH. It is possible that abnormal fibrin properties could help identify acute PE patients at high risk for RPVO for whom adjuvant therapy to modify disease progression might be evaluated. For example, since fibrinogen properties such as fibrinogen Bβsialylation may be influenced pharmacologically,[39] the effect on RPVO of such interventions to alter fibrinogen or the body’s response to fibrin may have clinical implications.
Supplementary Material
Bullet point:
Fibrinogen helps predict RPVO after PE: results from the PROMPT study
Acknowledgements
The authors would like to thank Dr. Russell Doolittle for his helpful advice and suggestions on the experimental methods and manuscript. Dr. Jess Mandel also provided helpful comments on the manuscript. The study was funded by research grants from Bayer Pharmaceuticals, Fonds de Recherche en Santé Respiratoire, France, R01HL095089 and through collaboration with the UCSD DNA Sequencing Facility (supported, in part, by grant 2 P30 CA023100-23 from the NIH) for performing DNA sequence analysis; and the City of Hope Mass Spectrometry and Proteomics Core Facility (supported, in part, by grant CA033572 from the NIH) for performing fibrinogen mass spectrometry analysis. The authors had full discretion over the study design, performance and creation of the manuscript.
References
- 1.Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest 1993: 103(3): 685–692. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez O, Helley D, Couchon S, Roux A, Delaval A, Trinquart L, Collignon MA, Fischer AM, Meyer G. Perfusion defects after pulmonary embolism: risk factors and clinical significance. J Thromb Haemost 2010: 8(6): 1248–1255. [DOI] [PubMed] [Google Scholar]
- 3.Klok FA, van Kralingen KW, van Dijk AP, Heyning FH, Vliegen HW, Kaptein AA, Huisman MV. Quality of life in long-term survivors of acute pulmonary embolism. Chest 2010: 138(6): 1432–1440. [DOI] [PubMed] [Google Scholar]
- 4.Wartski M, Collignon MA. Incomplete recovery of lung perfusion after 3 months in patients with acute pulmonary embolism treated with antithrombotic agents. THESEE Study Group. Tinzaparin ou Heparin Standard: Evaluation dans l’Embolie Pulmonaire Study. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2000: 41(6): 1043–1048. [PubMed] [Google Scholar]
- 5.Klok FA, van der Hulle T, den Exter PL, Lankeit M, Huisman MV, Konstantinides S. The post-PE syndrome: a new concept for chronic complications of pulmonary embolism. Blood reviews 2014: 28(6): 221–226. [DOI] [PubMed] [Google Scholar]
- 6.Planquette B, Ferre A, Peron J, Vial-Dupuy A, Pastre J, Mourin G, Emmerich J, Collignon MA, Meyer G, Sanchez O. Residual pulmonary vascular obstruction and recurrence after acute pulmonary embolism. A single center cohort study. Thrombosis research 2016: 148: 70–75. [DOI] [PubMed] [Google Scholar]
- 7.Meneveau N, Ider O, Seronde MF, Chopard R, Davani S, Bernard Y, Schiele F. Long-term prognostic value of residual pulmonary vascular obstruction at discharge in patients with intermediate- to high-risk pulmonary embolism. Eur Heart J 2013: 34(9): 693–701. [DOI] [PubMed] [Google Scholar]
- 8.Phear D Pulmonary embolism. A study of late prognosis. Lancet 1960: 2: 832–835. [DOI] [PubMed] [Google Scholar]
- 9.Prediletto R, Paoletti P, Fornai E, Perissinotto A, Petruzzelli S, Formichi B, Ruschi S, Palla A, Giannella-Neto A, Giuntini C. Natural course of treated pulmonary embolism. Evaluation by perfusion lung scintigraphy, gas exchange, and chest roentgenogram. Chest 1990: 97(3): 554–561. [DOI] [PubMed] [Google Scholar]
- 10.Paraskos JA, Adelstein SJ, Smith RE, Rickman FD, Grossman W, Dexter L, Dalen JE. Late prognosis of acute pulmonary embolism. N Engl J Med 1973: 289(2): 55–58. [DOI] [PubMed] [Google Scholar]
- 11.Donnamaria V, Palla A, Petruzzelli S, Carrozzi L, Pugliesi O, Giuntini C. Early and late follow-up of pulmonary embolism. Respiration; international review of thoracic diseases 1993: 60(1): 15–20. [DOI] [PubMed] [Google Scholar]
- 12.Wan T, Rodger M, Zeng W, Robin P, Righini M, Kovacs MJ, Tan M, Carrier M, Kahn SR, Wells PS, Anderson DR, Chagnon I, Solymoss S, Crowther M, White RH, Vickars L, Bazarjani S, Le Gal G. Residual pulmonary embolism as a predictor for recurrence after a first unprovoked episode: Results from the REVERSE cohort study. Thrombosis research 2018: 162: 104–109. [DOI] [PubMed] [Google Scholar]
- 13.Pesavento R, Filippi L, Palla A, Visona A, Bova C, Marzolo M, Porro F, Villalta S, Ciammaichella M, Bucherini E, Nante G, Battistelli S, Muiesan ML, Beltramello G, Prisco D, Casazza F, Ageno W, Palareti G, Quintavalla R, Monti S, Mumoli N, Zanatta N, Cappelli R, Cattaneo M, Moretti V, Cora F, Bazzan M, Ghirarduzzi A, Frigo AC, Miniati M, Prandoni P, Investigators S. Impact of residual pulmonary obstruction on the long-term outcome of patients with pulmonary embolism. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology 2017: 49(5). [DOI] [PubMed] [Google Scholar]
- 14.Pengo V, Lensing AW, Prins MH, Marchiori A, Davidson BL, Tiozzo F, Albanese P, Biasiolo A, Pegoraro C, Iliceto S, Prandoni P. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004: 350(22): 2257–2264. [DOI] [PubMed] [Google Scholar]
- 15.Guerin L, Couturaud F, Parent F, Revel MP, Gillaizeau F, Planquette B, Pontal D, Guegan M, Simonneau G, Meyer G, Sanchez O. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thrombosis and haemostasis 2014: 112(3): 598–605. [DOI] [PubMed] [Google Scholar]
- 16.Piazza G, Goldhaber SZ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2011: 364(4): 351–360. [DOI] [PubMed] [Google Scholar]
- 17.Lang IM, Madani M. Update on chronic thromboembolic pulmonary hypertension. Circulation 2014: 130(6): 508–518. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes T, Planquette B, Sanchez O, Morris T. From Acute to Chronic Thromboembolic Disease. Ann Am Thorac Soc 2016: 13 Suppl 3: S207–214. [DOI] [PubMed] [Google Scholar]
- 19.Morris TA, Marsh JJ, Chiles PG, Auger WR, Fedullo PF, Woods VL Jr. Fibrin derived from patients with chronic thromboembolic pulmonary hypertension is resistant to lysis. Am J Respir Crit Care Med 2006: 173(11): 1270–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris TA, Marsh JJ, Chiles PG, Magana MM, Liang NC, Soler X, Desantis DJ, Ngo D, Woods VL Jr. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood 2009: 114(9): 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsh JJ, Guan HS, Li S, Chiles PG, Tran D, Morris TA. Structural insights into fibrinogen dynamics using amide hydrogen/deuterium exchange mass spectrometry. Biochemistry 2013: 52(32): 5491–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsh JJ, Chiles PG, Liang NC, Morris TA. Chronic thromboembolic pulmonary hypertension-associated dysfibrinogenemias exhibit disorganized fibrin structure. Thrombosis research 2013: 132(6): 729–734. [DOI] [PubMed] [Google Scholar]
- 23.Miller RF, O’Doherty MJ. Pulmonary nuclear medicine. Eur J Nucl Med 1992: 19(5): 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer G, Collignon MA, Guinet F, Jeffrey AA, Barritault L, Sors H. Comparison of perfusion lung scanning and angiography in the estimation of vascular obstruction in acute pulmonary embolism. Eur J Nucl Med 1990: 17(6–8): 315–319. [DOI] [PubMed] [Google Scholar]
- 25.Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). The PIOPED Investigators. JAMA : the journal of the American Medical Association 1990: 263(20): 2753–2759. [DOI] [PubMed] [Google Scholar]
- 26.Qanadli SD, El Hajjam M, Vieillard-Baron A, Joseph T, Mesurolle B, Oliva VL, Barre O, Bruckert F, Dubourg O, Lacombe P. New CT index to quantify arterial obstruction in pulmonary embolism: comparison with angiographic index and echocardiography. AJR Am J Roentgenol 2001: 176(6): 1415–1420. [DOI] [PubMed] [Google Scholar]
- 27.Qi J, Kreutzer DL. Fibrin activation of vascular endothelial cells. Induction of IL-8 expression. J Immunol JID - 2985117R 1995: 155(2): 867–876. [PubMed] [Google Scholar]
- 28.Martinez J, Ferber A, Bach TL, Yaen CH. Interaction of fibrin with VE-cadherin. Annals of the New York Academy of Sciences 2001: 936: 386–405. [DOI] [PubMed] [Google Scholar]
- 29.Bunce LA, Sporn LA, Francis CW. Endothelial cell spreading on fibrin requires fibrinopeptide B cleavage and amino acid residues 15–42 of the beta chain. J Clin Invest JID - 7802877 1992: 89(3): 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burnham KP, Anderson DR. Information and Likelihood Theory: A Basis for Model Selection and Inference. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach. Springer-Verlag, New York, 2002; pp. 49–97. [Google Scholar]
- 31.Akaike H Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F, eds. International Symposium on Information Theory. Akademia Kiado, Budapest, 1973; pp. 267–281. [Google Scholar]
- 32.Hosmer DW, Lemeshow S. Assessing the Fit of the Model. Applied Logistic Regression, Second Edition John Wiley & Sons, Inc., New York, 2000; pp. 143–202. [Google Scholar]
- 33.Lami D, Cellai AP, Antonucci E, Fiorillo C, Becatti M, Grifoni E, Cenci C, Marcucci R, Mannini L, Miniati M, Abbate R, Prisco D. Residual perfusion defects in patients with pulmonary embolism are related to impaired fibrinolytic capacity. Thrombosis research 2014: 134(3): 737–741. [DOI] [PubMed] [Google Scholar]
- 34.Miniati M, Fiorillo C, Becatti M, Monti S, Bottai M, Marini C, Grifoni E, Formichi B, Bauleo C, Arcangeli C, Poli D, Nassi PA, Abbate R, Prisco D. Fibrin resistance to lysis in patients with pulmonary hypertension other than thromboembolic. Am J Respir Crit Care Med 2010: 181(9): 992–996. [DOI] [PubMed] [Google Scholar]
- 35.Okude M, Yamanaka A, Morimoto Y, Akihama S. Sialic acid in fibrinogen: effects of sialic acid on fibrinogen-fibrin conversion by thrombin and properties of asialofibrin clot. Biological & pharmaceutical bulletin 1993: 16(5): 448–452. [DOI] [PubMed] [Google Scholar]
- 36.Morris TA, Marsh JJ, Chiles PG, Kim NH, Noskovack KJ, Magana MM, Gruppo RA, Woods VL Jr. Abnormally sialylated fibrinogen gamma-chains in a patient with chronic thromboembolic pulmonary hypertension. Thrombosis research 2007: 119(2): 257–259. [DOI] [PubMed] [Google Scholar]
- 37.Dang CV, Shin CK, Bell WR, Nagaswami C, Weisel JW. Fibrinogen sialic acid residues are low affinity calcium-binding sites that influence fibrin assembly. The Journal of biological chemistry 1989: 264(25): 15104–15108. [PubMed] [Google Scholar]
- 38.Martinez J, Palascak J, Peters C. Functional and metabolic properties of human asialofibrinogen. J Lab Clin Med 1977: 89(2): 367–377. [PubMed] [Google Scholar]
- 39.Maghzal GJ, Brennan SO, George PM. The sialic acid content of fibrinogen decreases during pregnancy and increases in response to fibrate therapy. Thrombosis research 2005: 115(4): 293–299. [DOI] [PubMed] [Google Scholar]
- 40.Brennan SO. Variation of fibrinogen oligosaccharide structure in the acute phase response: Possible haemorrhagic implications. BBA Clin 2015: 3: 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.