

Abstract
Antibody therapies represent a valuable tool to reduce COVID-19 deaths and hospitalizations. Multiple antibody candidates have been granted emergency use authorization by the Food and Drug Administration and many more are in clinical trials. Most antibody therapies for COVID-19 are engineered to bind to the receptor-binding domain (RBD) of the SARS-CoV-2 Spike protein and disrupt its interaction with angiotensin-converting enzyme 2 (ACE2). Notably, several SARS-CoV-2 strains have accrued mutations throughout the RBD that improve ACE2 binding affinity, enhance viral transmission and escape some existing antibody therapies. Here, we measure the binding affinity of 33 therapeutic antibodies against a large panel of SARS-CoV-2 variants and related strains of clinical significance using AlphaSeq, a high-throughput yeast mating-based assay to determine epitopic residues, determine which mutations result in loss of binding and predict how future RBD variants may impact antibody efficacy.
Keywords: CoV-2 clinical therapeutics, yeast display, yeast mating, epitope, RBD variants
One-Sentence Summary: By measuring protein binding in vitro, we identify which clinical antibodies retain binding to various mutant SARS-CoV-2 strains.
Statement of Significance: This work represents the first published demonstration of AlphaSeq, a synthetic yeast mating assay, as a tool for highly multiplexed characterization of clinical antibody candidates. We focused on the epitope and cross-reactivity characterization of 33 clinically relevant CoV-2 antibody therapeutics and found very good concordance with literature results, when available.
Antibody therapies represent a valuable tool to reduce COVID-19 deaths and alleviate the burden on healthcare systems. During the vaccine rollout, antibody therapies to treat COVID-19 have served as a stopgap to save lives. With vaccines widely available, antibody therapies continue to play an essential role in treating patients who are unvaccinated, such as the immunocompromised, and those infected with viral variants that escape vaccine protection. Antibody therapies can be developed quickly, making them well suited for rapid response to newly emerging strain variants, and have proven to reduce both viral loads and hospitalizations [1, 2]. Multiple antibody candidates have been granted emergency use authorization by the Food and Drug Administration (FDA) and many more are in phase 2 and phase 3 clinical trials [3, 4].
Most antibody therapies for COVID-19 are engineered to bind to the receptor-binding domain (RBD) of the SARS-CoV-2 Spike protein and disrupt its interaction with angiotensin-converting enzyme 2 (ACE2) [5–7]. Since the first human transmission of COVID-19 over two years ago, SARS-CoV-2 has undergone significant antigenic drift arising from mutations throughout the RBD that improve ACE2 binding affinity, enhance viral transmission and generate resistance to existing antibody therapies [8–11]. Understanding the impact of observed and likely RBD variants on the effectiveness of antibody therapies is of critical importance.
A handful of antibody therapies have been granted emergency use authorization by the FDA, over a dozen are in late-stage clinical development, and many more are in preclinical development [4]. However, previous efforts to measure the effect of RBD variants on the efficacy of antibody candidates for COVID-19 have mostly been limited to characterizing individual SARS-CoV-2 RBD variants on a small number of antibody candidates [6, 12–16]. A notable exception is the method introduced by Starr et al. which involves the construction of a yeast surface display library of RBD variants and enables the complete mapping of binding between a single antibody and all single mutations of the RBD [10, 11, 17, 18]. The low-antibody throughput of this method, however, precludes characterization of a wide variety of clinically relevant antibodies.
The AlphaSeq assay, previously described as yeast synthetic agglutination, is presented here as a method for overcoming existing throughput challenges for characterizing the binding profiles of tens of antibodies against thousands of RBD variants [19]. Here, we leveraged the AlphaSeq assay to measure ~176 760 protein–protein interactions (PPIs) between 33 therapeutically relevant antibody candidates and most single-amino-acid mutations to SARS-CoV-2 RBD, along with selected widely circulating RBD variants containing multiple mutations. The PPI measurements are analyzed to derive key epitope residues for each antibody, determine individual or multiple mutations that result in loss of binding for each antibody and predict how future RBD variants may impact antibody efficacy. Figure 1 presents the AlphaSeq assay and its application to antibody/RBD binding in schematic form.
Figure 1.

Using AlphaSeq to characterize interactions between an antibody library and a library of coronavirus RBD variants in high throughput. (A) Two DNA fragment libraries are constructed with homology to the MATa or MATα genome for integration into the chromosome. Each library contains a diversity of proteins of interest, either antibodies or RBD variants, for display on the yeast cell surface, a diversity of randomized 25 nucleotide DNA barcodes and a lox recombination site. MATa and MATα yeast strains lacking expression of native sexual agglutination proteins are transformed with their respective fragment library and subsequently mixed in liquid culture. (B) In liquid culture, MATa-MATα agglutination is facilitated by interactions between surface displayed antibodies and RBD variants. Agglutination leads to mating between MATa and MATα haploid cells to produce a diploid cell. (C) The number of diploids formed by a haploid pair is dependent on the interaction strength between the antibody and RBD variant expressed on their surfaces. (D) Diploid cells are cultured with β-estradiol to induce for CRE recombinase expression and recombine the engineered chromosome to pair DNA barcodes. Diploids are then lysed and sequenced to count the abundance of each barcode pair and determine the relative interaction strength between each antibody and coronavirus RBD variant.
First, to determine the epitope for each antibody, an AlphaSeq experiment was performed where 33 antibodies were screened against a site-saturation mutagenesis (SSM) library comprised of (after library preparation and sequencing) ~75% of all single-residue mutants of 165 sites within the SARS-CoV-2 RBD (Figs 2A,S1,S2). While many RBD sites were intolerant to mutations (i.e. observed binding was poor across all antibodies and all mutants at that site), other sites revealed differential patterns of mutation-sensitivity among antibodies indicating epitopic diversity among the tested antibodies (Fig. 2B). We carried out a pairwise comparison of binding among antibodies to distinguish epitope residues from intolerant sites where substitutions resulted in poor functional expression of the RBD (Fig. 2C). For each site in the RBD included in the SSM library, all mutant affinities (scaled to each antibody’s affinity to Wild type (WT) RBD) were compared between each pair of antibodies with a Mann–Whitney U test. A putative epitope residue was called if binding was found to be significantly impacted by substitutions relative to at least four other antibodies in the dataset (Bonferroni-corrected P-values ≤ .05). Sites without a putative epitope residue call are either non-epitope residues or lie beyond the limit of detection of the assay. Overall, we found putative epitope residues for 22 antibodies (Fig. 2D, Tables S1,S2). For antibodies with known RBD–Ab structures and with multiple putative epitope residues, we found that our results correctly place key epitopic residues at the interface between the antibody and RBD (Fig. 2E). However, for antibody CR3022, our method only found a single putative epitope residue (residue 455) and this residue is not present at the interface between the antibody and RBD (PDB ID 6ZLR). Overall, our results agree with previously published epitope data for COR-101, imdevimab, casirivimab, bamlavimab, CC12.1 [13, 20, 21] while providing new insight into the epitopes targeted by the remaining antibodies and therefore allowing us to prospectively identify which RBD sites should be of particular concern for antibody binding in new strains of COVID-19.
Figure 2.

(A) Schematic of epitope mapping AlphaSeq experiment, comparing antibodies against a CoV-2 RBD SSM library. (B) Heatmap of AlphaSeq binding data showing all interactions measured in the assay in log10 KD (nM). RBD–CoV-2 intolerant substitutions results in loss binding for all antibodies in the set and appear as vertical dark blue streaks. (C) Method summary for determination of epitope residues; each antibody was compared against all others, and at each RBD site a Mann–Whitney U test was performed to determine if binding was more impacted by a RBD mutations at that site in one antibody; results were filtered by U test significance and for significance in four or more pairwise comparisons. (D) Summary of epitope determination results. A putative epitope residue is represented as a navy color in the figure. An RBD residue is determined to be a putative epitope residue if binding was found to be significantly impacted by substitutions in that residue relative to at least four other antibodies in the dataset. This comparison is necessary to differentiate between substitutions that ablate binding to all antibodies, likely due to misfolding or low expression, versus substitutions that decrease binding to only a fraction of antibodies and therefore are more likely to be at the binding interface. The differential residue counts indicate the number of comparison antibodies for which a relative difference was found for a given antibody and RBD residue (summing the results from light-heavy and heavy-light orientations when both were tested). (E) Representative antibodies (in blue) with known structure binding to RBD (in red), with residues called as epitope locations marked in white or yellow.
Next, a second AlphaSeq experiment was performed to assess binding of the same 33 clinically relevant antibodies against a curated panel of coronavirus RBD variants. Included were 34 unique SARS-CoV-2 RBD variants with single, double or triple mutations, including five CDC-defined variants of concern, B.1.1.7, B.1.351, P.1, B.1.427 and B.1.429, and four related coronavirus RBDs (Fig. 3A). From this single experiment, we quantitatively measured the binding of each antibody to each SARS-CoV-2 variant and characterized the cross-reactivity profile of each antibody to related coronavirus strains (Fig. S3, Table S3).
Figure 3.
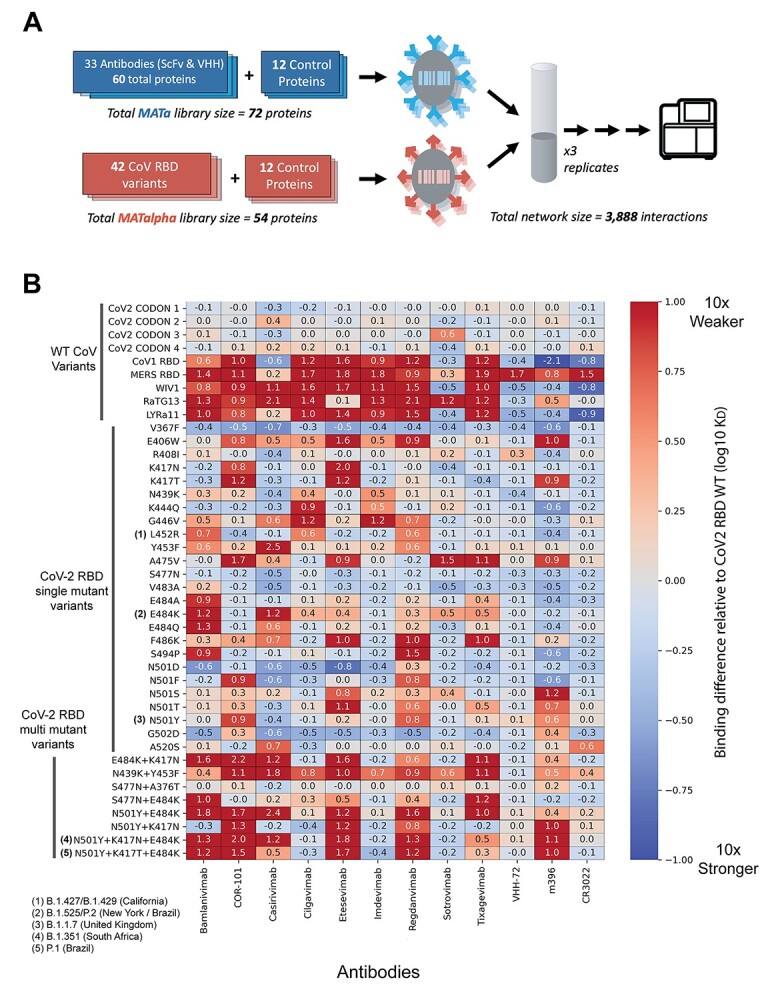
(A) Schematic of AlphaSeq experiment, comparing antibodies against a curated panel of coronavirus RBD variants. (B) Binding affinity of selected SARS-CoV-2 RBD variants to selected antibodies. Antibodies are on the x-axis, SARS-CoV-2 RBD variants are on the y-axis; in each column, values are mean difference in predicted binding affinity (measured on a log10 scale) between a given CoV variant and WT SARS-CoV-2 RBD for each antibody. Values below 0 (blue) represent improved binding relative to WT SARS-CoV-2 RBD and values above 0 (red) represent reduced binding affinity. In these data, cilgavimab/AZD1061, imdevimab/REGN10987 and sotrovimab/GSK4182136 retain high affinity for the widest variety of clinically relevant RBD variants.
The dataset includes nine antibodies in clinical trials with publicly available sequences [22]. We observed a wide range of binding to the panel of SARS-CoV-2 RBD variants (Fig. 3B). Notably, we found good concordance with previous literature reports for most antibody-variant interactions (Table S1). Of the nine clinical antibodies, three antibodies have been extensively studied in the literature: imdevimab, casirivimab and bamlanivimab [10, 12, 15, 16, 18, 23, 24]. We recapitulated the binding profiles for the B.1.1.7, B.1.351, and P.1 variants to imdevimab, casirivimab and bamlanivimab [16, 23, 24]. To summarize: we observed no significant changes in binding to the B.1.1.7 variant for imdevimab, casirivimab or bamlanivimab. We observed a decrease in binding affinity for casirivimab and bamlanivimab to B.1.351 and P.1 variants, whereas imdevimab retained WT binding. Our findings with these four antibodies are highly consistent with previous literature reports. To date, there have been minimal or no reports on sotrovimab, regdanvimab, tixagevimab, cilgavimab and COR-101 antibodies [13, 17]. We found that regdanvimab and COR-101 display reduced binding affinity to B.1.1.7, B.1.351 and P.1 variants. Tixagevimab had reduced binding affinity to B.1.351 and P.1 variants but maintained WT binding affinity with the B.1.1.7 variant, whereas sotrovimab and COR-101 showed no changes in binding affinity to the B.1.1.7, B.1.351 and P.1 variants.
As part of the same experiment, cross-reactivity was evaluated by measuring antibody binding to four additional RBDs from related coronaviruses: SARS-CoV-1, LYRa11, WIV1 and RaTG13. Some antibodies were found to be highly specific to SARS-CoV-2, including imdevimab, bamlanivimab, regdanvimab, tixagevimab, cilgavimab and COR-101. In contrast, casirivimab, sotrovimab and etesevimab demonstrated varying degrees of cross-reactivity.
From two AlphaSeq assays, we mapped binding of 33 anti-CoV antibodies against a panel of SARS-CoV-2 RBD variants, including B.1.1.7, B.1.351 and P.1, and identified key epitope residues for each antibody. Binding measurements for both AlphaSeq assays are included as supplementary files along with the naming convention used for each antibody included in the dataset (Tables S4–S6). This experiment has provided, for the first time, a comprehensive view of the impact of potential escape mutants (including several strains of substantial clinical concern) on the binding of RBD-targeted antibody therapeutics. For the small fraction of the dataset for which binding has been assayed previously, or for which the epitope is known via crystal structure, good agreement with previous methods establishes the reliability of AlphaSeq as a method. Specifically, 59 out 68 of the binding measurements in this work are in directional agreement with previous results that examined the effect of RBD mutations on the binding affinity of clinical antibodies (Table S7). However, we did not attempt to derive a correlation between our results and affinity values reported in the literature since reported values are dependent on the choice of antibody format, spike protein format and binding method, all of which vary among published datasets.
One of the limitations of this work is the expression of clinical antibodies as single-chain variable fragments (scFv) since we expect that this antibody format may not capture the viral neutralization capacity of an immunoglobulin G (IgG) or the native antibody conformation. A second limitation of this work is that our binding measurements were carried out with an RBD truncation (amino acids 319–527) expressed on the surface of yeast. Yeast primarily uses high-mannose N-glycosylation that can have implications for binding and structure. We also expect that differences in antibody and target expression, steric hindrance in binding due to the display orientation on the yeast surface and the type of the assay used for comparison may explain some of the differences between AlphaSeq binding measurements and those reported in the literature. Finally, it is important to note that relative changes in antibody binding to yeast surface displayed RBD do not necessarily translate directly into changes of neutralization potency and require neutralization assays with fully assembled and human glycosylated virus for confirmation.
The results presented here begin to provide a deeper understanding of the impact of RBD mutations on the binding of therapeutic antibodies. Further validation of these results with additional orthogonal binding assays or viral neutralization assays is essential to prioritize the development of therapies effective against the widest possible variety of circulating and newly emerging viral variants and to enable the eventual possibility of regional or even personal prioritization of the most effective therapies for circulating SARS-CoV-2 variants.
MATERIALS AND METHODS
Materials
Antibody candidates
Clinically relevant antibodies were identified based on those designated to be in clinical trials in the COVID-19 Antibody Therapeutics tracker on 9 January 2021 [22] Nine antibody candidates were found to have amino acid sequences publicly available at the time after a literature search and were included in this work: REGN10933 (casirivimab), REGN10987 (imdevimab), bamlanivimab (LY3819253, LY-CoV555), regdanvimab (CT-P59), sotrovimab (VIR-7831/GSK4182136), AZD8895/tixagevimab/COV2–2196, AZD1061/cilgavimab, etesevimab, COR-101/STE90-C11. Additional antibodies were identified from the literature and their sequences were obtained from the Coronavirus-Binding Antibody Sequences & Structures (CoV-AbDab) [25] Overall, a total of 33 distinct antibodies were selected for this work (27 IgG’s and 6 VHH formats). IgG format antibodies were built as scFvs in both heavy-light and light-heavy orientations. We selected the antibody orientation with strongest binding against WT CoV-2 RBD when we found drastic differences in binding between the scFv orientations (Table S8). More detailed information for each antibody can be found in Table S9.
Coronavirus variants
SARS-CoV-2 (isolate Wuhan-Hu-1, Genbank accession number MN908947, residues 319–527) and additional sarbecovirus homologs (RaTG13, Genbank MN996532; SARS-CoV-1 Urbani, Genbank AY278741; WIV1, Genbank KF367457; LYRa11, Genbank KF569996) CoV RBD sequences can be found in Table S10.
Yeast media
Yeast peptone dextrose, yeast peptone galactose and synthetic drop out (SDO) media supplemented with 80 mg/l adenine were made according to standard protocols. Suppliers used for our yeast media are as follows: Bacto Yeast Extract (Life Technologies), Bacto Tryptone (Fisher BioReagents), Dextrose (Fisher Chemical), Galactose (Millipore Sigma), Adenine (ACROS Organics), Yeast Nitrogen Base w/o Amino Acids (Thermo Scientific), SC-His-Leu-Lys-Trp-Ura Powder (Sunrise Science Products), L-Histidine (Fisher BioReagents), L-Tryptophan (Fisher BioReagents), Uracil (ACROS Organics) and Bacto Agar (Fisher BioReagents).
Methods
Isogenic yeast plasmid transformation
AlphaSeq compatible plasmids encoding yeast surface display cassettes were constructed by Twist Bioscience and resuspended at 100 ng/μl. A total of 100 ng of plasmid was digested with PmeI enzyme for 1 h at 37°C to linearize, leaving chromosomal homology for integration into the ARS314 locus at both the 5′ and 3′ ends as described in [19]. Yeast transformations were performed with Frozen-EZ Yeast Transformation Kit II (Zymo Research) according to manufacturer’s instructions. Yeast were plated on SDO–Trp plates and grown at 30°C for 2–3 days. Successful transformants were struck out onto yeast peptone adenine dextrose (YPAD) plates and grown overnight at 30°C.
Isogenic yeast fragment transformation
AlphaSeq compatible fragments encoding yeast surface display cassettes were constructed by Twist Bioscience and resuspended at 100 ng/μl. A three-piece yeast transformation was performed with upstream and downstream fragments that contained chromosomal homology for integration into the ARS314 locus at both the 5′ and 3′ ends as described in [19]. Yeast transformations were performed with Frozen-EZ Yeast Transformation Kit II (Zymo Research) according to manufacturer’s instructions. Yeast were plated on SDO–Trp plates and grown at 30°C for 2–3 days. Successful transformants were struck out onto YPAD plates and grown overnight at 30°C.
Protein expression validation—flow cytometry
Yeast was inoculated in YPAD and grown overnight at 30°C. Yeast were labelled with FITC-anti-C-myc antibody (Immunology Consultants Laboratory, Inc.) in phosphate-buffered saline (PBS; Gibco) + 0.2% bovine serum albumin (BSA; Thermo) for 30 min at room temperature. Yeast were pelleted and resuspended in PBS + 0.2% BSA and read on a LSRII cytometer.
DNA library construction
AlphaSeq compatible fragments were synthesized by Twist Bioscience and were resuspended at 1 ng/μl in molecular grade water and pooled together. Fragment libraries were polymerase chain reaction (PCR) amplified using KAPA DNA polymerase (Roche). A second DNA fragment with a randomized DNA barcode was PCR amplified. Fragments were run on a 0.8% agarose gel and extracted using Monarch Gel Purification kit (NEB).
SSM library construction
SSM library of SARS-CoV-2 RBD was synthesized by Twist Bioscience and resuspended at 1 ng/μl in molecular grade water. The SSM library fragments were PCR amplified using KAPA DNA polymerase (Roche). Quatitative PCR (qPCR) was terminated before saturation to minimize PCR bias, generally between 12 and 15 cycles. A second DNA fragment with a randomized DNA barcode was PCR amplified. Fragments were run on a 0.8% agarose gel and extracted using Monarch Gel Purification kit (NEB).
Yeast library transformation
MATa or MATalpha AlphaSeq yeast were grown for 6 h in yeast peptone adenine galactose (YPAG) media to induce SceI expression, as described in [19]. All spin steps were performed at 3000 RPM for 5 min. Yeast was spun down and washed once in 50 ml 1 M Sorbitol (Teknova) + 1 mM CaCl2 solution. Washed yeast were resuspended in a solution of 0.1 M LiOAc/1 mM DTT and incubated shaking at 30°C for 30 min. After 30 min, yeast was spun down and washed once in 50 ml 1 M Sorbitol +1 mM CaCl2 solution. Yeast was resuspended to a final volume of 400 μl in 1 M Sorbitol +1 mM CaCl2 solution and incubated with DNA for at least 5 min on ice. Yeast were electroporated at 2.5 kV and 25 μF (BioRad). Immediately following electroporation, yeast were resuspended in 5 ml of 1:1 solution of 1 M Sorbitol:YPAD and incubated shaking at 30°C for 30 min. Recovered yeast cells were spun down and resuspend in 50 ml of SDO-Trp media and transferred to a 250 ml baffled flask. A total of 20 μl of resuspended cells were plated on SDO–Trp to determine transformation efficiency. Both the flask and plate were incubated at 30°C for 2–3 days. After 2–3 days, transformation efficiency was determined by counting colonies on the SDO–Trp plate.
Nanopore barcode mapping
Genomic DNA from yeast libraries was extracted using Yeast DNA Extraction Kit (Thermo Scientific) following the manufacturer’s instructions. A single round of qPCR was performed to amplify a fragment pool from the genomic DNA containing the gene through the associated DNA barcode. qPCR was terminated before saturation to minimize PCR bias, generally between 15 and 20 cycles. The final amplified fragment was concentrated with KAPA beads, quantified with a Quantus (Promega), prepped with a SQK-LSK-110 ligation kit (Oxford Nanopore) and sequenced with a Minion R10 flow cell (Oxford Nanopore) following the manufacturer’s instructions.
Library-on-library AlphaSeq assays
A total 2 ml of saturated MATa and MATalpha library were combined in 800 ml of YPAD media and incubated at 30°C in a shaking incubator. Three technical replicates were performed for each assay. After 16 h, 100 ml of yeast culture was washed once in 50 ml of sterile water and transferred to 600 ml of SDO–lys–leu with 100 nM ß-estradiol (Sigma) for 24 h. After 24 h, 100 ml of yeast was transferred to fresh SDO–lys–leu with 100 nM ß-estradiol for an additional 24 h.
Library preparation for next-generation sequencing
Genomic DNA was extracted using Yeast DNA Extraction Kit (Thermo Scientific) following manufacturer’s instructions. qPCR was performed to amplify a fragment pool from the genomic DNA and to add standard Illumina sequencing adaptors and assay specific index barcodes. qPCR was terminated before saturation to minimize PCR bias, generally between 23 and 27 cycles. The final amplified fragment was concentrated with KAPA beads, quantified with a Quantus (Promega) and sequenced with an NextSeq 500 sequencer (Illumina).
Dissociation constant estimation
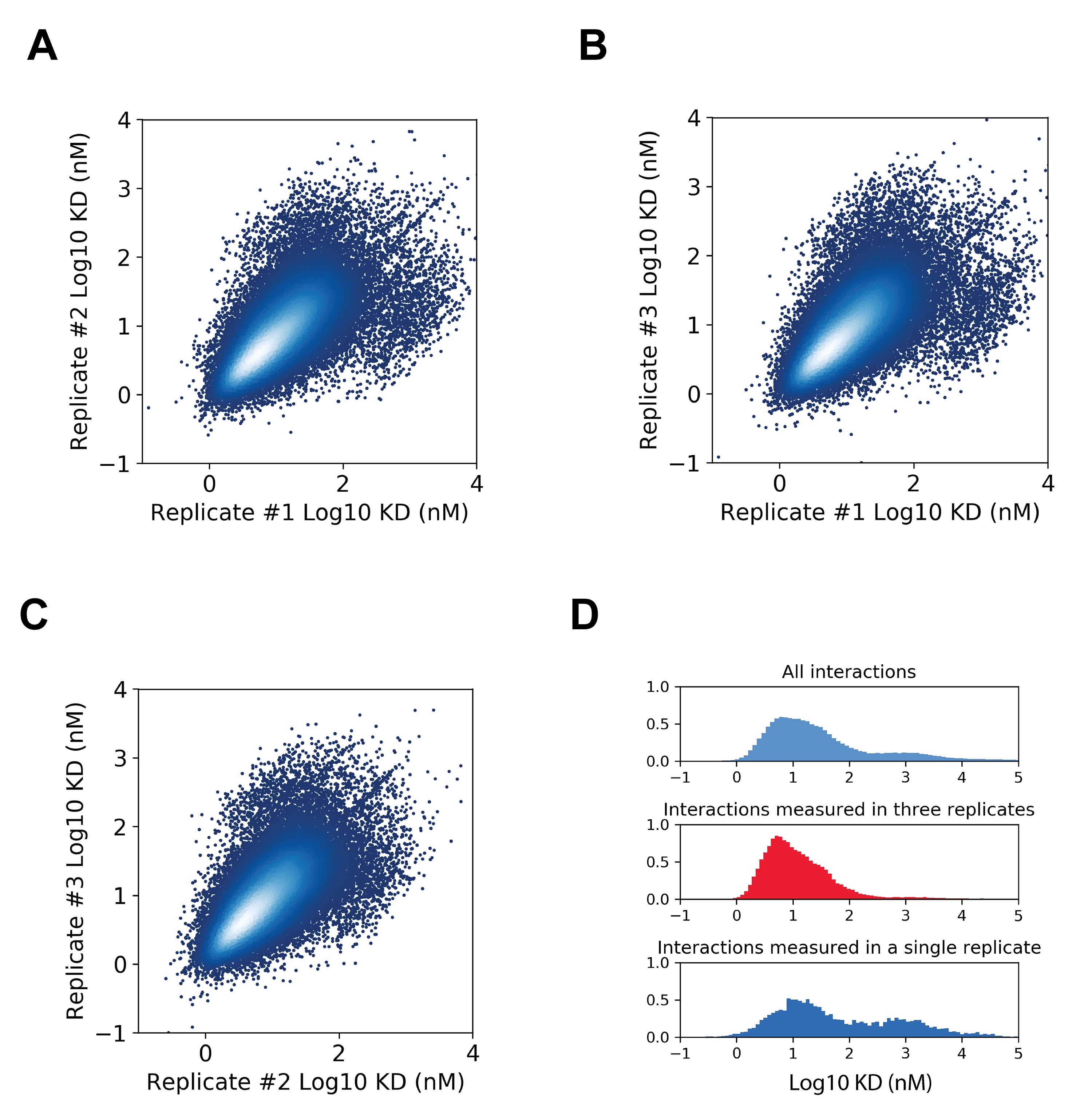
Both diploid barcode pairs counts and haploid barcode counts were generated using Illumina sequencing as described above. Counts of diploid barcode pairs were normalized by dividing them by the product of each corresponding haploid barcode counts to account for differences in library representation. A standard curve of proteins with known interacting partners and dissociation constant values was included in the assay and used to extrapolate dissociation constant values for the experimental protein interactions. Additional details on the computational approach to estimate dissociation constant values can be found in [19].
DATA AND MATERIALS AVAILABILITY
Code is available at: https://github.com/A-AlphaBio/cov2_antibodies_variants. Protein sequences are included in Tables S9 and S10. Raw affinity datasets and processed epitope datasets are included in supplementary Tables S1,S4 and S5. These tables are stored the GitHub repository. Heatmap figures were generated using Seaborn, a Python data visualization library. Protein structure figures were generated using software Pymol version
AUTHOR CONTRIBUTIONS
Conceptualization: D.Y., R.L., E.E., R.E. Formal analysis: R.L., E.E., R.E. Funding acquisition: D.Y., R.L. Investigation: E.E., C.L., M.K., D.G., C.S. Methodology: R.L. Project administration: R.L., E.E., M.K. Supervision: R.L. Visualization: R.L., E.E., R.E. Writing—original draft: D.Y., R.L., E.E., R.E. Writing—review and editing: D.Y., R.L., E.E., R.E., C.L.
FUNDING
National Science Foundation Small Business Innovation Research Program Phase I Award (#2033772 to R.L.).
CONFLICT OF INTEREST STATEMENT
R.L. and D.Y. are the founders and current employees of A-Alpha Bio, Inc. (A-Alpha Bio) and own stock/stock options of A-Alpha Bio. E.E., R.E., C.L, M.K, C.S. and D.G. are employees of A-Alpha Bio; all employees own stock/stock options of A-Alpha Bio. A-Alpha Bio has a patent (US10988759B2) relating to certain research described in this article. All the authors do not have any additional competing interest.
ETHICS AND CONSENT STATEMENT
Not applicable.
ANIMAL RESEARCH STATEMENT
Not applicable.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Emily Engelhart, A-Alpha Bio, Seattle, WA. 98195. USA.
Randolph Lopez, A-Alpha Bio, Seattle, WA. 98195. USA.
Ryan Emerson, A-Alpha Bio, Seattle, WA. 98195. USA.
Charles Lin, A-Alpha Bio, Seattle, WA. 98195. USA.
Colleen Shikany, A-Alpha Bio, Seattle, WA. 98195. USA.
Daniel Guion, A-Alpha Bio, Seattle, WA. 98195. USA.
Mary Kelley, A-Alpha Bio, Seattle, WA. 98195. USA.
David Younger, A-Alpha Bio, Seattle, WA. 98195. USA.
References
- 1. Gottlieb, RL, Nirula, A, Chen, Pet al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA 2021; 325: 632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinreich, DM, Sivapalasingam, S, Norton, Tet al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med 2021; 384: 238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun, Y, Ho, M. Emerging antibody-based therapeutics against SARS-CoV-2 during the global pandemic. Antib Ther 2020; 3: 246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor, PC, Adams, AC, Hufford, MMet al. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat Rev Immunol 2021; 21: 382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahmed, SF, Quadeer, AA, McKay, MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020; 12: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann, M, Kleine-Weber, H, Schroeder, Set al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020; 181: 271, e278–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walls, AC, Park, YJ, Tortorici, MAet al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020; 181: 281, e286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greaney, AJ, Starr, TN, Gilchuk, Pet al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 2021; 29: 44, e49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li, Q, Wu, J, Nie, Jet al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020; 182: 1284, e1289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Starr, TN, Greaney, AJ, Addetia, Aet al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021; 371: 850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Starr, TN, Greaney, AJ, Hilton, SKet al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020; 182: 1295, e1220–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baum, A, Fulton, BO, Wloga, Eet al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020; 369: 1014–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bertoglio, F, Fuhner, V, Ruschig, Met al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep 2021; 36: 109433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong, J, Zost, SJ, Greaney, AJet al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat Microbiol 2021; 6: 1233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomson, EC, Rosen, LE, Shepherd, JGet al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021; 184: 1171, e1120–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, R, Zhang, Q, Zhang R et al. SARS-CoV-2 omicron variants reduce antibody neutralization and acquire usage of mouse ACE2. Front. Immunol 2022; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Starr, TN, Czudnochowski, N, Liu, Zet al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 2021; 597: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Starr, TN, Greaney, AJ, Dingens, ASet al. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med 2021; 2: 100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Younger, D, Berger, S, Baker, Det al. High-throughput characterization of protein-protein interactions by reprogramming yeast mating. Proc Natl Acad Sci U S A 2017; 114: 12166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hansen, J, Baum, A, Pascal, KEet al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020; 369: 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan, M, Liu, H, Wu, NCet al. Structural basis of a shared antibody response to SARS-CoV-2. Science 2020; 369: 1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang, L, Liu, W, Yu, Xet al. COVID-19 antibody therapeutics tracker: a global online database of antibody therapeutics for the prevention and treatment of COVID-19. Antib Ther 2020; 3: 205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoffmann, M, Arora, P, Gross, Ret al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021; 184: 2384, e2312–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu, H, Wei, P, Zhang, Qet al. 501Y.V2 and 501Y.V3 variants of SARS-CoV-2 lose binding to bamlanivimab in vitro. MAbs 2021; 13: 1919285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raybould, MIJ, Kovaltsuk, A, Marks, Cet al. CoV-AbDab: the coronavirus antibody database. Bioinformatics 2021; 37: 734–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.