

Conspectus:
Enolate alkylation and conjugate addition into an α,β-unsaturated system have served as longstanding strategic disconnections for the installation of α or β-substituents on carbonyl-containing compounds. At the onset of our efforts to develop C–H activation reactions for organic synthesis, we set our eye towards developing asymmetric β-C–H activation reactions of aliphatic acids, with the perspective that this bond-forming event could serve as a more flexible retrosynthetic surrogate for both canonical carbonyl-related asymmetric transformations. In this review, we describe our early efforts using strong-coordinating chiral oxazolines to probe reaction mechanism and the stereochemical nature of the C–H cleavage transition state. The characterization of key reactive intermediates through X-ray crystallography and computational studies suggested a transition state with C–H and Pd–OAc bonds being approximately coplanar for optimum interaction. We then moved forward to develop more practical, weakly-coordinating monodentate amide directing groups; a necessary advance towards achieving the β-C–H activation of weakly-coordinating native carboxylic acids. Throughout this journey, gradual deconvolution between a substrate’s directing effect and its intimate interplay with ligand properties has culminated in the design of new ligand classes that ultimately allowed the competency of native carboxylic acids in β-C–H activation. These efforts established the importance of ligand acceleration in Pd-catalyzed C–H activation, where the substrate’s weak-coordination is responsible for positioning the catalyst for C–H cleavage, while the direct participation from the bifunctional ligand is responsible for enthalpically stabilizing the C–H cleavage transition state. Building upon these principles, we developed five classes of chiral ligands (MPAA, MPAQ, MPAO, MPAThio, MPAAM) to enable enantioselective β-C–H activation reactions, including carbon–carbon and carbon–heteroatom bond formation. The accumulated data from our developed enantioselective C–H activation reactions indicate that ligands possessing point chirality are most effective for imparting stereoinduction in the C–H activation step; the application of which enabled the desymmetrization and subsequent C–H functionalization of enantiotopic carbon and protons across a range of weakly-coordinating arylamides, and more recently, with free carboxylic acids. Progress in ligand design, in conjunction with the enabling nature of alkali metal countercations, led to the realization of a suite of β-methyl, and now methylene, C(sp3)–H activation reactions. These advancements also enabled the use of economical oxidants, such as peroxides and molecular oxygen, to facilitate catalyst turnover. In the future, continued progression in designing more efficient bifunctional chiral ligands is likely to provide a myriad of enantioselective β-C–H activation reactions of readily-available native substrates.
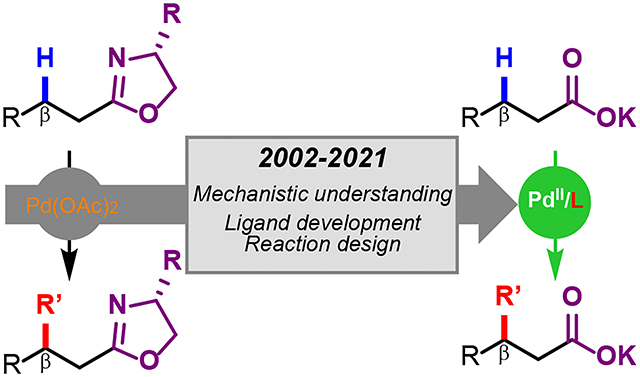
Graphical Abstract
1. Introduction
Since its advent in the early 1800s, synthetic organic chemistry has enabled breakthroughs in medicine, agriculture, and materials science.5,6 Although modern chemists are well-equipped to construct an immense range of complex molecules, organic synthesis still contains an abundance of unsolved problems. One of the most fundamental limitations of organic synthesis is its dependence on reactive functional groups to forge new chemical bonds. Because organic molecules are composed almost entirely of unreactive C–C and C–H bonds, the transformation of simple starting materials into intricate target molecules can often be circuitous to install or manage reactive functional handles that will ultimately be replaced or removed.
The field of C–H activation has accrued tremendous interest over the past several decades due to its potential to overcome the inefficiencies by redefining the inert C–H bond as a latent source of functionality.7 However, the ubiquity of C–H bonds in organic molecules poses one of the most formidable regiochemical challenges in transition metal-catalyzed C–H functionalization. To this end, leveraging Lewis basic groups to direct a metal catalyst is a commonly employed strategy to impart regioselection, precisely positioning the catalyst close to a target C–H bond and minimizing the entropic cost of activation required to select for one of many C–H bonds.8 Over the years, advancements in directing group, catalyst and ligand design have enabled C(sp2)–H functionalization to become a well-established area of research, and also enabled the previously-elusive aliphatic C–H activation a possibility despite the distinct challenges posed by these systems.9
At the outset of our research program, we were particularly interested in developing robust strategies in the β-functionalization of aliphatic free acids, generating products analogous to enolate alkylation in β-methyl functionalization or conjugate addition for β-methylene functionalization (Scheme 1). This was motivated by their ubiquitous presence in chemical feedstocks and natural products, facile diversification, as well as their relative stability compared with carbonyl compounds of lower oxidation states. Such a β-C–H activation approach can widen the versatility of carbonyl functionalization reactions by circumventing the need for: (1) prefunctionalization, and (2) polarity-matching resultant from electronic properties (e.g. innately-nucleophilic enolates, or innately-electrophilic α,β-unsaturated systems). In effect, this reconceptualizes canonical d2/a3 synthons by avoiding the need for/generation of reactive nucleophilic reagents, while enabling broad compatibility with a range of mild nucleophiles and electrophiles.10
Scheme 1.
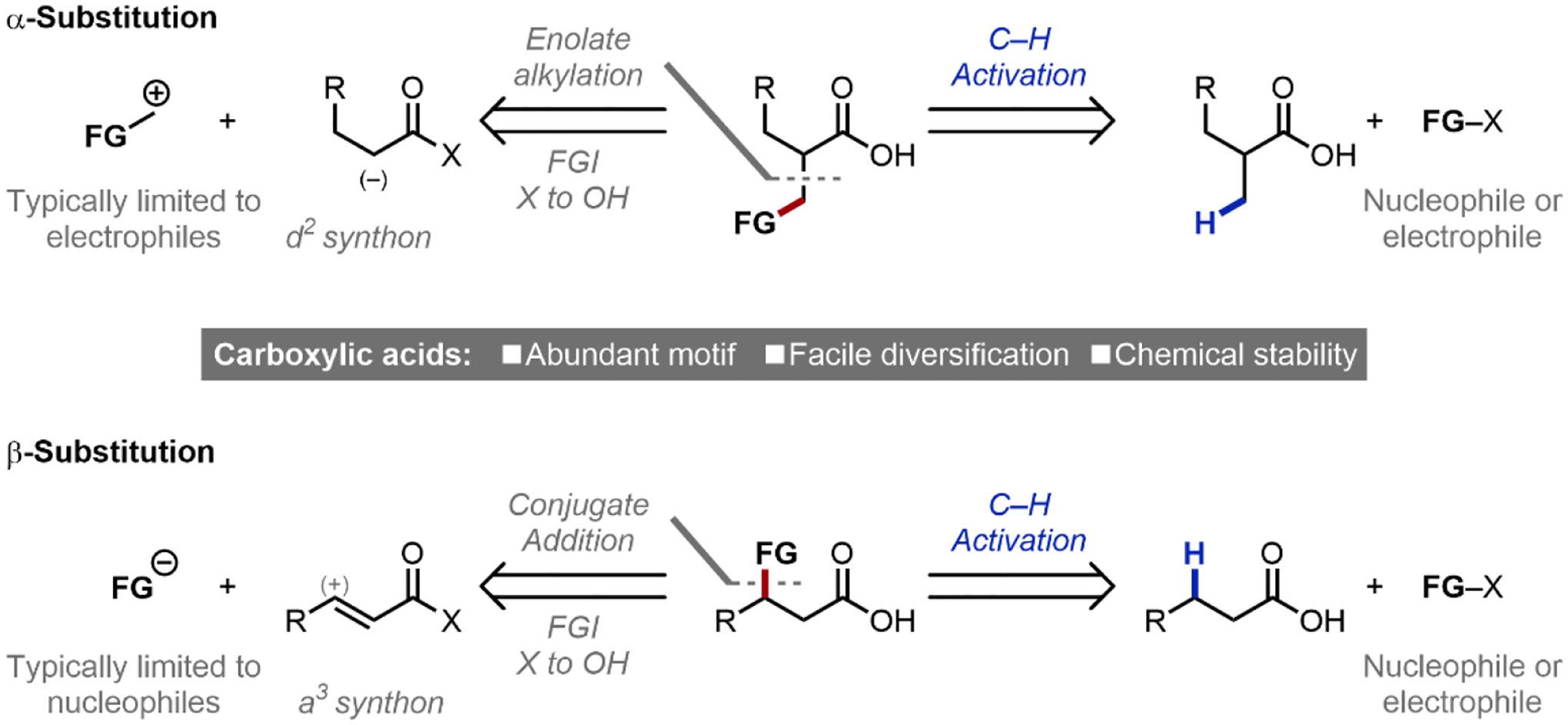
β-C–H functionalization provides an alternative disconnection for the synthesis of functionalized carbonyl derivatives that avoids reactive reagents. FG: Functional group; FGI: functional group interconversion.
Over the decades, fervent research efforts have been devoted toward controlling the π-facial selectivity of enolate or conjugate additions, leading to many established asymmetric variants for these processes. Despite these advancements, free acid asymmetric α-alkylation and conjugate addition into their α,β-unsaturated derivatives represent still to this day, an outstanding methodological gap in the synthetic canon. To this end, β-C–H functionalization provides a unique opportunity in addressing this synthetic shortfall. Contrasting the community’s extensive history in controlling π-facial selectivity, enantioselective C–H functionalization processes—where a chiral transition metal complex stereoselectively reacts with a prochiral C–H bond to generate a chiral organometallic intermediate—remained unknown when we initiated this program.11 These topological differences necessitate the establishment of new stereomodels, given that previously-privileged scaffolds for controlling π-facial selectivity (e.g. C2-symmetic motifs) may not be cross-transferable for the desymmetrization of tetrahedral sp3 centers. Specifically, β-C–H functionalization could be rendered enantioselective through two main categories: (1) desymmetrization of enantiotopic carbons (Scheme 2a), and (2) desymmetrization of enantiotopic protons (Scheme 2b). Given the synthetic importance of these carbonyl-related reactions, and in particular their asymmetric variants, we envisioned that the rigorous development of an alternative retrosynthetic disconnection can potentially reconceptualize how carbonyl substitutions are asymmetrically constructed, providing a unified aliphatic retron to access these diverse products.
Scheme 2.
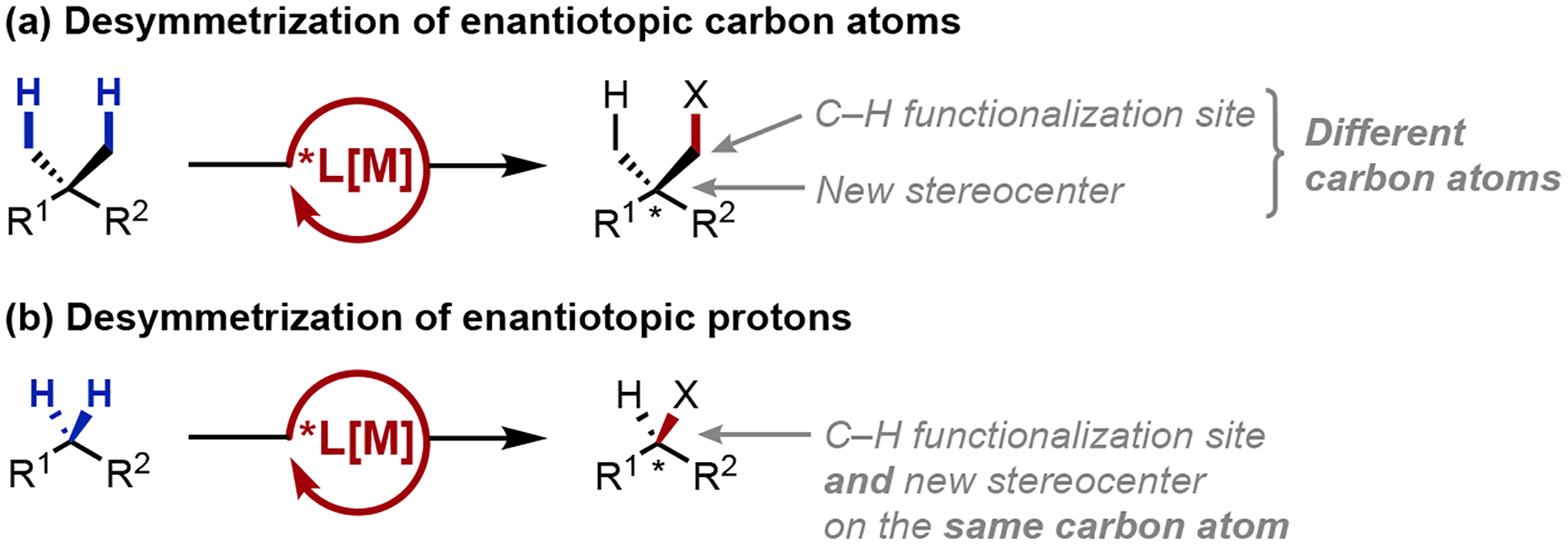
Desymmetrization of (a) enantiotopic carbon atoms vs. (b) enantiotopic protons
However, these ambitions were notably challenging at the outset as the reactivity of Pd(OAc)2 for cleaving C(sp3)–H is poor and, at the time, was limited to cyclopalladation directed by nitrogen or sulfur atoms. Native functional groups are often ineffective directing groups for C–H activation owing to their suboptimal catalyst binding abilities, and therefore require the installation of exogenous directing groups to obtain the desired reactivity; the weak coordinative ability of aliphatic acids is no exception to this. Finally, we were also drawn to the potential of using the cyclopalladated intermediate as a linchpin towards its conversion into a range of C–C or C–X bonds, but recognized that to do so required the development of new ligand scaffolds to enhance the reactivity of the Pd(II) species in order to enable native substrates such as aliphatic acids to direct C–H activation.
In this account, we chronicle the development of palladium-catalyzed β-C(sp3)–H activation reactions of aliphatic acid derivatives and its diverse reactivity evolution through the lens of ligand development. We begin our discussion with our group’s early examples employing chiral oxazolines, highlighting the mechanistic insight into both the redox catalytic cycle and the C–H cleavage transition states established from these initial studies. Next, we describe the progress made towards allowing the use of weakly-coordinating groups in β-C–H activation; a particularly important development towards our ultimate goal: functionalization of free acids. Finally, we cover emerging examples that successfully utilize free acids without requiring exogenous directing group installation—a culmination of a journey since 2002 in ligand design and reaction development. Interwoven through this account, we illustrate how changes in a substrate’s directing effect required synergistic modulations in ligand design, and the crucial developments that enable these scaffolds to achieve the required ligand acceleration required for enantioselective aliphatic β-C(sp3)–H functionalization.12 We note that this review is not designed to be comprehensive, but rather to summarize key developments and to emphasize ongoing challenges and opportunities for the field at-large.13
2. Oxazoline Directing Groups (2002–2007): Mechanistic Insights in Diastereoselective C–H Activation
In our early efforts to develop asymmetric β-C–H activation reactions (supported by a Royal Society Research grant in 2002), we envisioned that a σ-chelating acid-derived chiral auxiliary such as an oxazoline could promote the assembly of a square planar palladium complex poised for cyclometallation. In 2005, our laboratory reported early examples of β-C(sp3)–H functionalization employing oxazolines as auxiliaries for carboxylic acids.14 In this report, the iodination of unactivated methyl and cyclopropane C–H bonds was effected under mild conditions, with the pivotal C–H cleavage step mediated by palladium-bound acetates (Scheme 3).15,16 The C–H functionalization was selective for primary over secondary C–H bonds, and the use of a chiral oxazoline resulted in diastereoselective methyl C(sp3)–H iodination for substrates where R1≠R2. For diastereoselective variants, the oxazoline auxiliary could be cleaved to unveil chiral β-functionalized carboxylic acids in >99% ee. In the same year, we disclosed a related diastereoselective C(sp3)–H acetoxylation reaction (Scheme 4a).17 A trinuclear palladium complex bearing an anti-configured arrangement was isolated and characterized by X-ray crystallography, an observation that eventually formed the basis of future ligand design for enantioselective β-functionalization (Scheme 4b). We also found that the addition of Ac2O was vital to promote catalyst turnover, regenerating the active Pd(OAc)2 catalyst presumably via ligand exchange to afford Pd(IV) diacetate prior to the reductive elimination (Scheme 4c). Subsequent studies on the C–H cleavage step shed light that the Pd–OAc lay approximately coplanar with the target C–H bond, selecting for the C–H bond that minimized substrate steric interactions.18 These insights gathered from these early examples of aliphatic acid β-C–H activation established important foundations in ligand and reaction development that guided our eventual successes in the free acid-directed lactonization and acetoxylation processes.
Scheme 3.
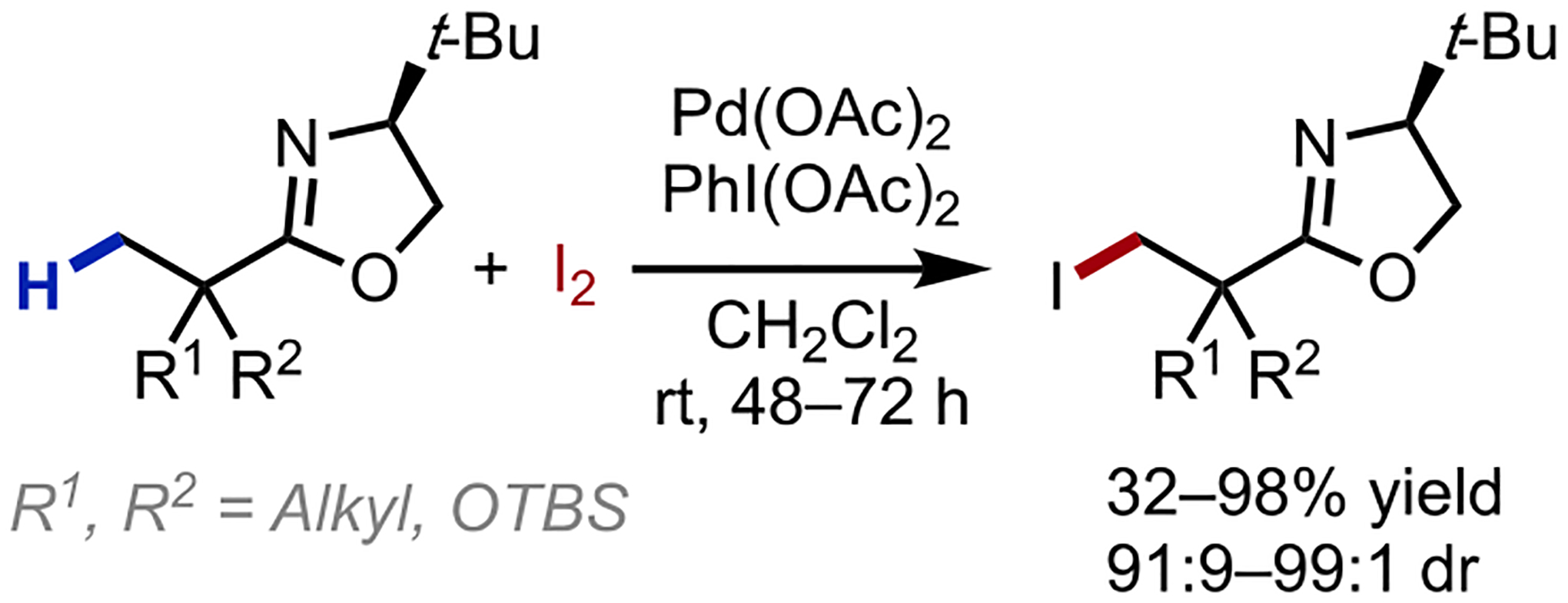
Oxazoline-directed diastereoselective β-C(sp3)–H iodination reaction
Scheme 4.
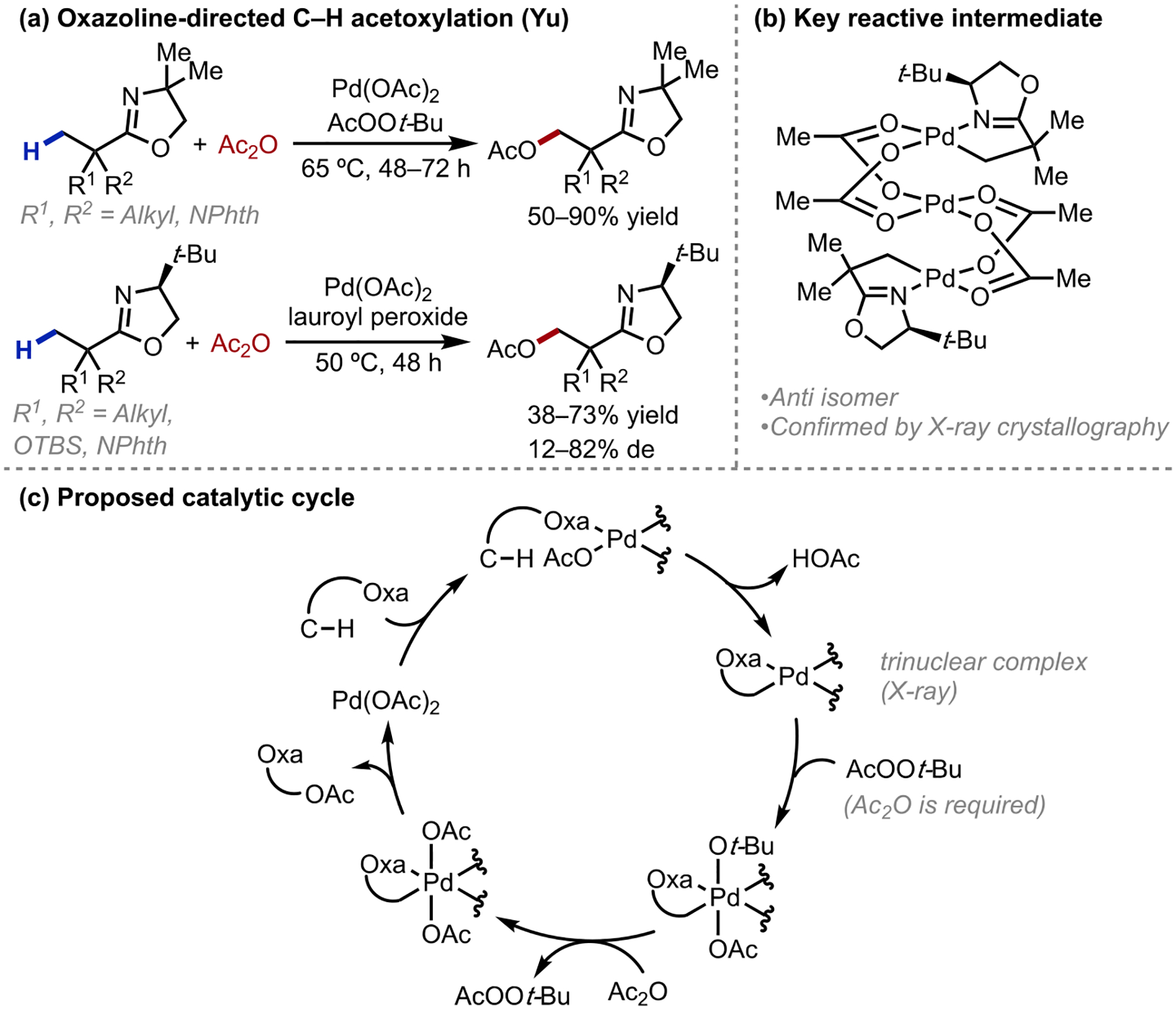
Oxazoline-directed diastereoselective β-C(sp3)–H acetoxylation reaction
3. Weakly-Coordinating Amide Directing Groups as Acid Surrogates (2007–2020)
3.1. Overview and reactivity development
Amides are commonly employed as exogenous directing groups for the β-C(sp3)–H functionalization of carboxylic acids, and can be roughly divided into two categories: 1) strongly-coordinating, bidentate amides (Figure 1),19,20 and 2) weakly-coordinating, monodentate amides.21 While widely employed in transition metal-catalyzed C–H functionalization reactions, strongly-coordinating bidentate amide directing groups are burdened with disadvantages.22 First, the thermodynamic stability of cyclometallated intermediates is a double-edged sword; although C–H activation is more favored due to the thermodynamic stability of the resulting cyclometallated intermediates, subsequent functionalization of these metallacycles can be challenging due to the a) lack of reactivity of stable strongly-bound intermediates, and b) lack of vacant coordination sites limiting the scope of coupling partners.22 The latter prohibits downstream transformations such as cross-coupling from being realized, as these intrinsically require two vacant coordination sites on the catalyst to allow transmetallation to occur. Second, employment of external chiral ligands for catalyst-controlled stereoinduction is often not feasible because the strong directing group coordination outcompetes catalyst binding. This critical limitation renders ligand acceleration—necessary for enantioselective catalytic functionalization—particularly challenging.
Figure 1.
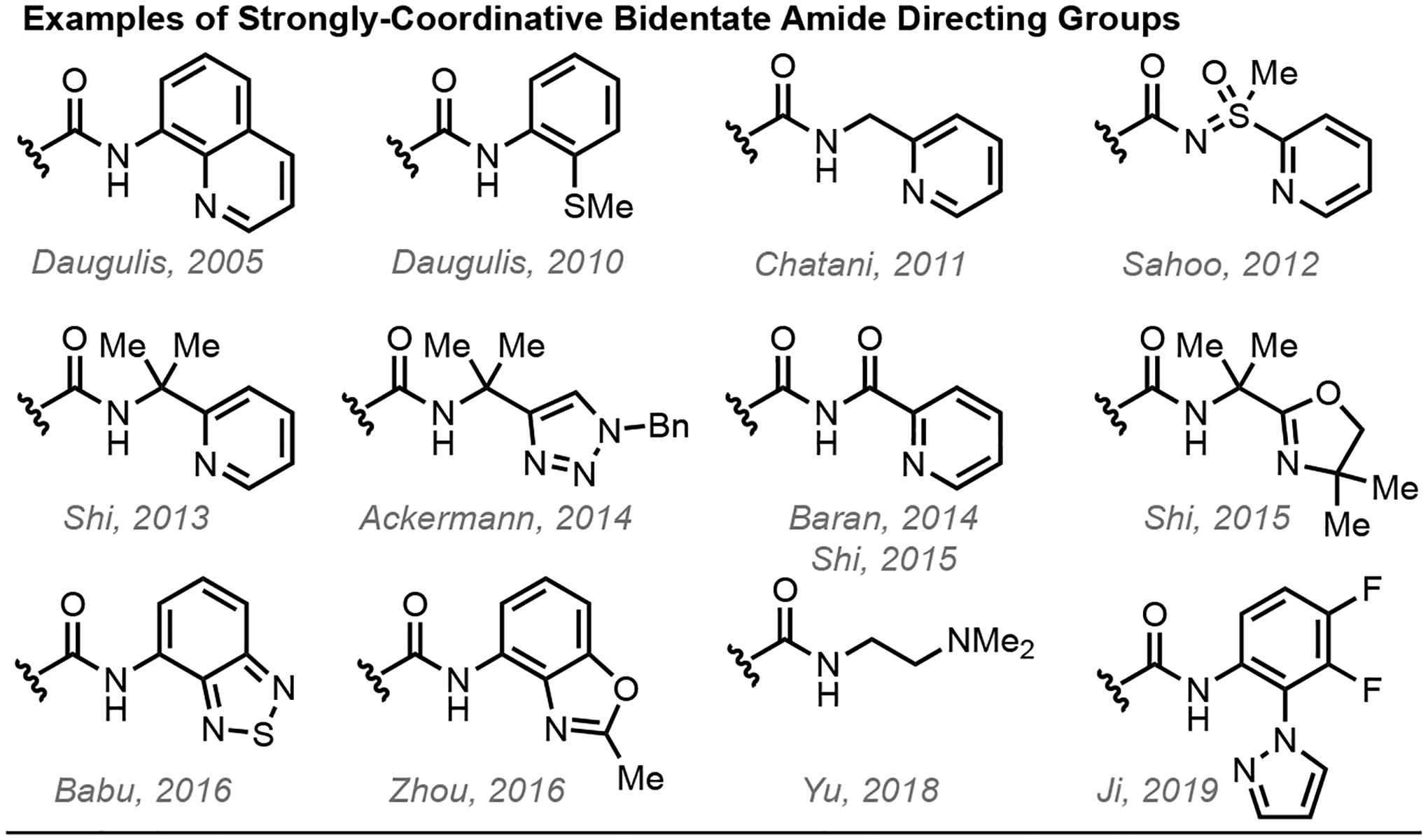
Examples of strongly-coordinative bidentate amide directing groups commonly used for β-C(sp3)–H functionalization
With our vision towards achieving the enantioselective β-C(sp3)–H functionalization of weakly-coordinating free acids, our laboratory moved away from the established reactivity of strongly-coordinating amides. As a steppingstone, we targeted the development of weakly-coordinating monodentate amide directing groups to facilitate C–H functionalization. We envisaged that the extra vacant coordinating site on the catalyst might enable the use of chiral bidentate ligand scaffolds to impart enantioinduction while broadening transformation scope, thus directly addressing the shortcomings imposed by strongly-coordinating bidentate directing groups. The departure from established directing groups posed energetic challenges we needed to overcome, as the enthalpic contribution from the strong coordination, as well as the conformational restriction from these often-rigid directing groups, all assist in favoring the challenging process of C–H metallation. With only weak-coordination available from the directing motif, the requisite catalytically-relevant substrate-Pd(II) complex poised for cyclometallation is more transient or elusive. This means that a suitably-designed ligand is required to accelerate the C–H cleavage process via reducing the energy barrier (Figure 2). Such ligand acceleration is crucial for achieving asymmetric induction, as any competitive racemic pathway arising from a ligand-free process will erode enantioselectivity.
Figure 2.
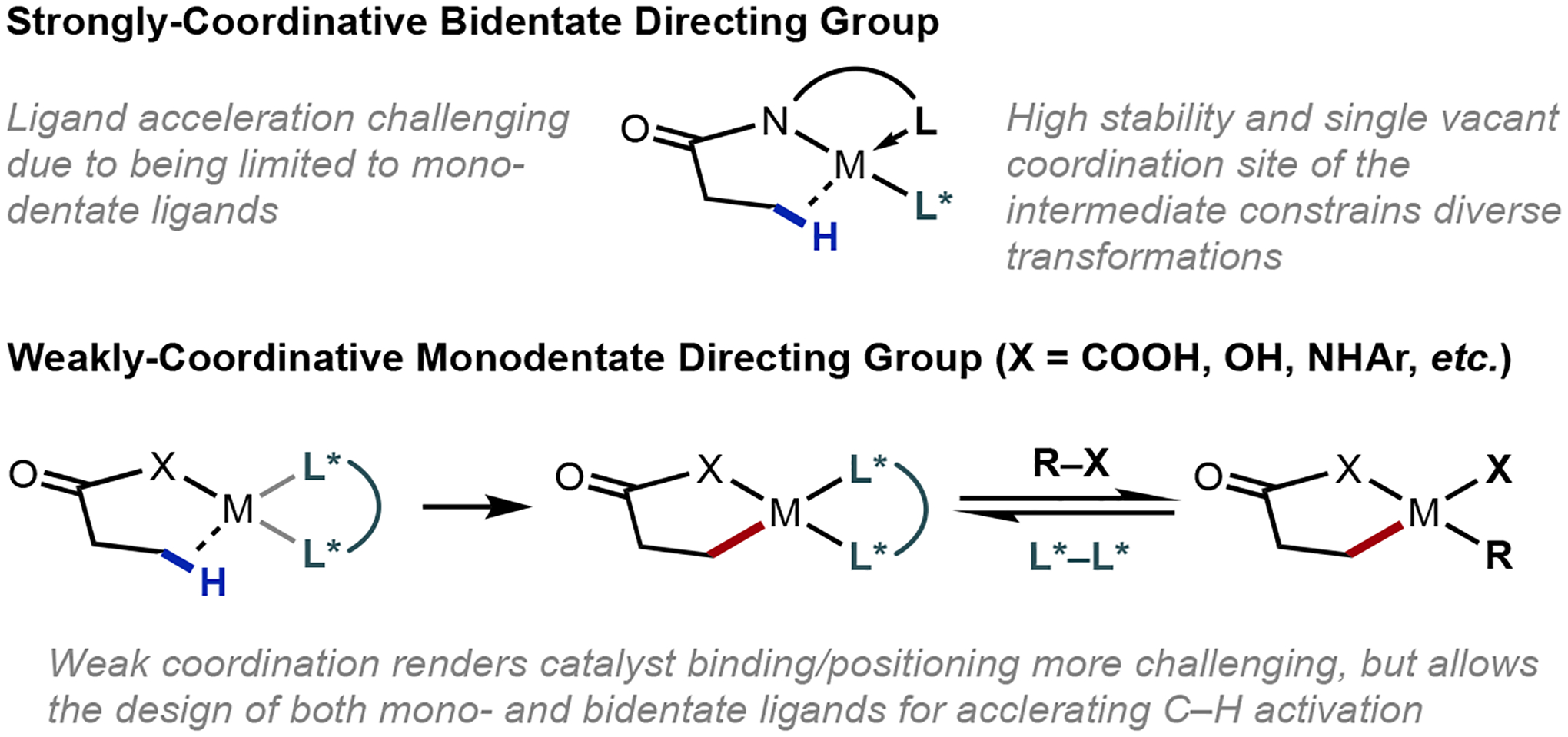
Challenges associated with strongly-coordinating bidentate directing groups and rationale for moving towards weakly-coordinative monodentate directing groups
To this end, we discovered that N-methoxyamides were competent weak-coordinating groups, enabling the first example of β-cross-coupling of aliphatic acids with aryl and alkyl boronic acids (2008, Scheme 5a).21a Critically, the shift to monodenticity facilitated cross-coupling, a previously inaccessible transformation, to proceed efficiently. Later, we identified that electron-deficient monodentate N-arylamides are privileged weak-coordinating motifs surrogating free acids; particularly noting that the acidic amide proton behaves analogously to the acidic carboxyl proton. The innovations developed to meet the challenges imposed by weakly-directing motifs facilitated the broad discovery of novel transformations across diverse catalytic cycles. One such innovation was reported in 2009, where our laboratory reported the first example of a palladium-catalyzed β-C(sp3)–H arylation directed by weakly-coordinating N-arylamides (Scheme 5b).21b The use of CyJohnPhos (L-1) in conjunction with CsF enabled the first example of β-methyl C–H functionalization proceeding via a Pd(0)/Pd(II) catalytic cycle. Shortly thereafter, we designed a modified N-arylamide directing group that proved enabling in the discovery of diverse reactivities. Beyond enabling cross-coupling processes, the weakly-coordinating arylamide directing group proved instrumental in the pioneering discovery of a range of aliphatic β-functionalizations, reporting unprecedented examples in C(sp3)–H arylation,1,2,23 alkylation,24 olefination,2,23b,e,25 alkynylation,2,26 carbonylation,27 borylation,28 amination,29 and halogenation30 (Scheme 5c).
Scheme 5.
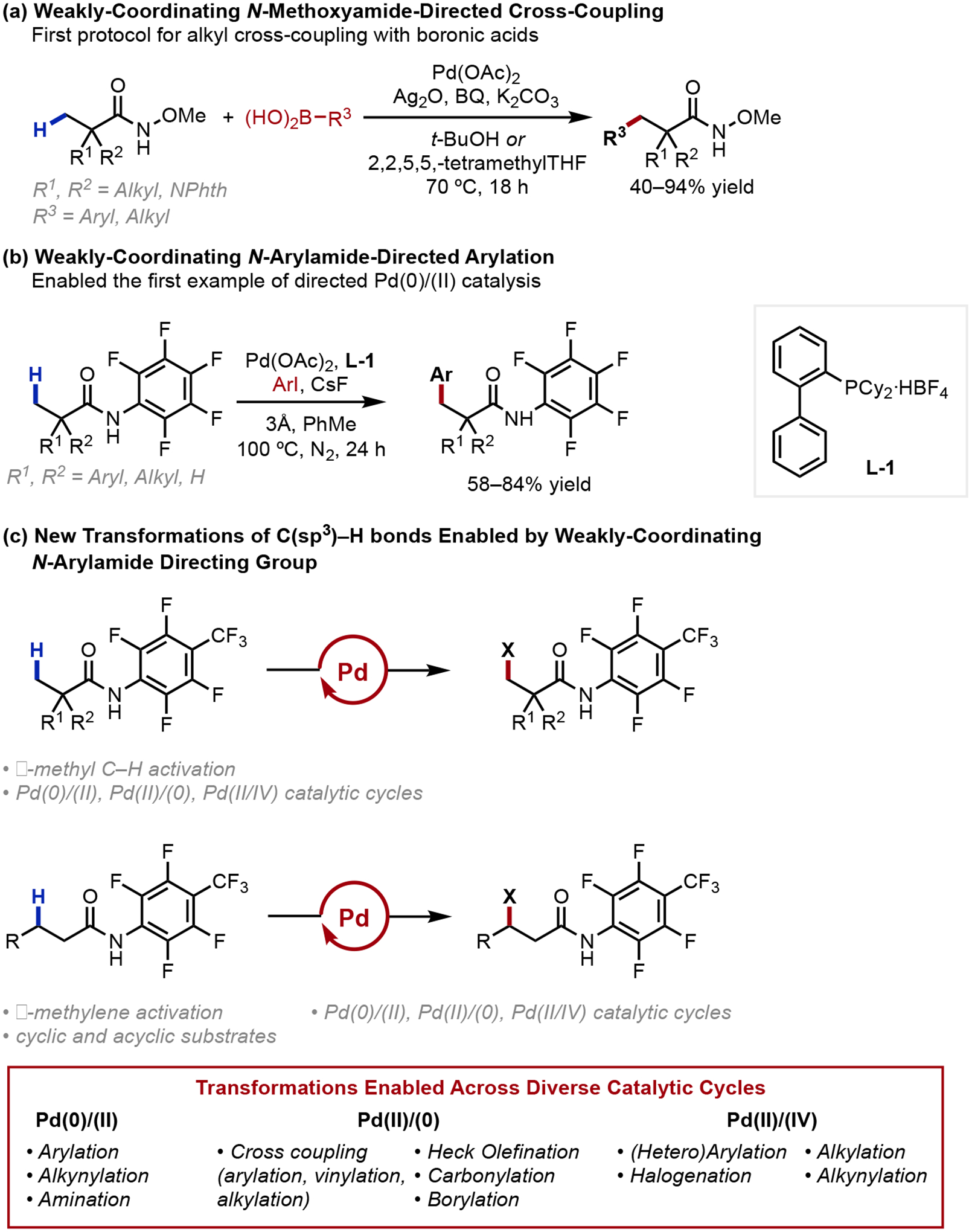
Overview of pioneering transformations facilitated by weak coordination-enabled C–H functionalization
Realizing such a range of transformations validated that diverse functionalization can be enabled by weak substrate coordination. In addition, the shift to a monodentate directing group provided the crucial opportunity for the use of previously inaccessible chiral bidentate ligand scaffolds bearing internal base motifs.31 These internal base motifs participate in and accelerate C–H cleavage, enabling the catalyst to outcompete against potential background reactivity in the enantiodetermining step.1,2 The ability for bifunctional chiral bidentate ligands to directly participate in C–H cleavage transition state then allowed us to achieve a range of enantioselective β-C(sp3)–H activation reactions, with the bidentate binding providing the required spatial organization to enable facile C–H cleavage and the requisite stereoenvironment for enantioinduction despite direction by a weakly-coordinating motif.
In the following section, we highlight the number of enantioselective transformations directed by weakly-coordinative amide directing groups, categorizing by the nature of desymmetrization (i.e. desymmetrization of enantiotopic carbons or enantiotopic protons on the same carbon) during the C–H activation process. These representative examples demonstrate the broad scope of transformations achieved, and highlight key ligand developments that guided our eventual success in achieving free acid functionalization.
3.2. Desymmetrization of Enantiotopic Carbon Atoms
The discovery of bifunctional MPAA ligands—which not only modulate the stereo and electronic environments of the Pd(II) center,32 but also participate in the C–H cleavage step directly —paved the way for subsequent bidentate ligand design.33 Beginning in 2011, our group developed several examples of amide-directed asymmetric C–H functionalization via desymmetrization of enantiotopic carbons. First, we established the enantioselective C–H arylation, alkylation, and alkenylation of cyclopropanes through cross-coupling with a variety of boronates directed by a weakly-coordinating N-arylamide (Scheme 6).34 Systematic tuning of chiral ligands, especially using the N-TcBoc-based protecting group that was likely involved in the C–H cleavage step, led to the development of a new bidentate MPAA ligand (L-2) crucial for high enantioselectivity. Later, our group reported an enantioselective β-C(sp3)–H arylation of cyclobutanes (Scheme 7),35 utilizing the same weakly-coordinating N-arylamide directing group but employs a chiral N-protected bidentate hydroxamic acid ligand (L-3).36 In addition, a small number of acyclic amides were also competent to afford chiral β-aryl amides in moderate yields and ee’s.
Scheme 6.
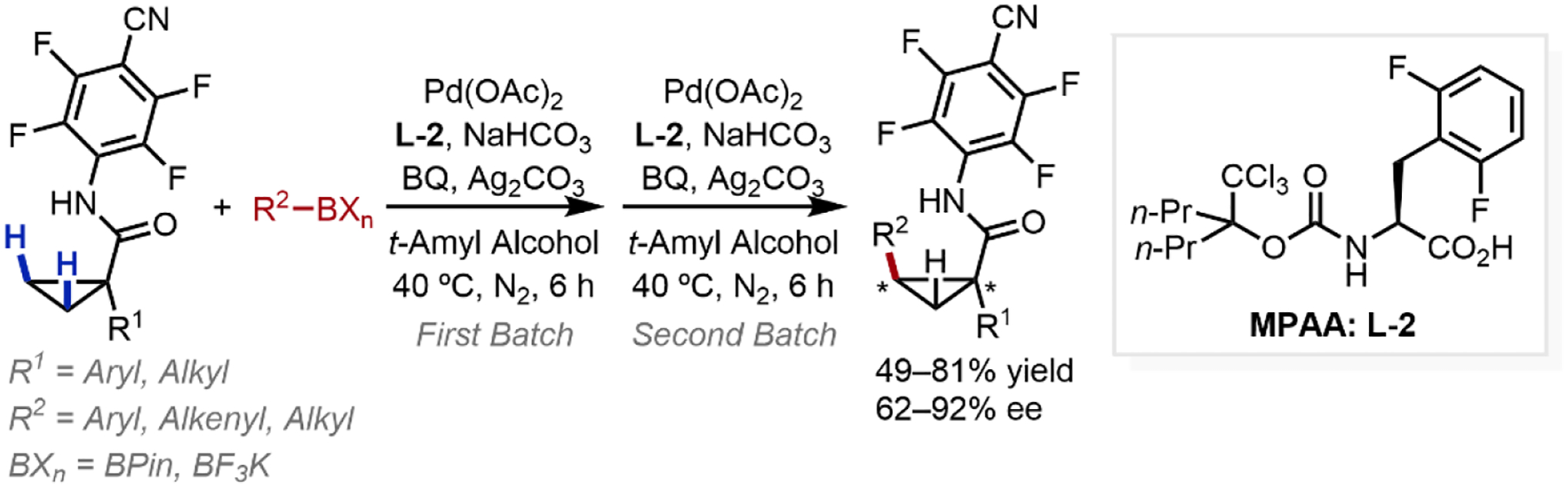
Enantioselective C–H Arylation, Alkenylation, and Alkylation of Cyclopropanes
Scheme 7.
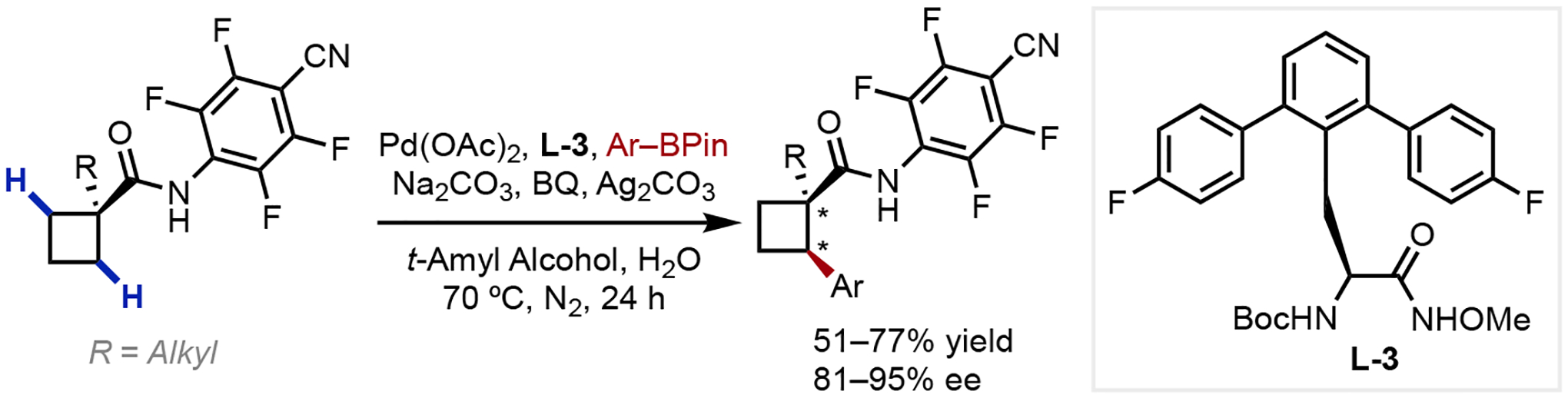
Enantioselective C–H Arylation of Cyclobutanes
The desymmetrization of isopropyl groups represents a far more formidable challenge than our previously reported desymmetrative activated β-methylene C–H functionalization due to the more flexible nature of the substrate (Scheme 8a). In 2017, our laboratory established the enantioselective β-C–H arylation, alkenylation, and alkynylation of isobutyramide substrates, directed by a weakly-coordinating perfluorotolyl N-arylamide group (Scheme 8b).2 Inspired by our early studies in oxazoline directing groups, we found success using chiral bidentate mono-protected aminomethyl oxazoline (MPAO) ligands. The rigidity and previously-established strong-binding conferred by the oxazoline unit was important to achieve high reactivity and enantioselectivity for this process, with the (S,S)-configuration in L-4 proving optimal for enantioinduction. Importantly, the proposed stereomodel was enabled by our early observations with chiral oxazoline-directed C–H activation, leveraging the anti-disposed nature of the substrate and the oxazoline unit to impart enantioinduction. By instead employing a chiral benzoyl-protected MPAO ligand, in conjunction with a weak-coordinating N-methoxyamides directing group, 2-aminoisobutyric acid derivatives also underwent enantioselective C–H arylation to afford synthetically valuable chiral α,α-dialkyl α-amino acids. Notably, the prevailing stereomodel proposed for the transformation was enabled by our early observations with chiral oxazolines, noting that sterically-large groups are preferentially anti-disposed across the metal catalyst. The remaining α-methyl group in the products could undergo an additional C–H arylation, alkynylation, alkylation, bromination, or borylation to afford diverse α-chiral carboxylic acids.
Scheme 8.
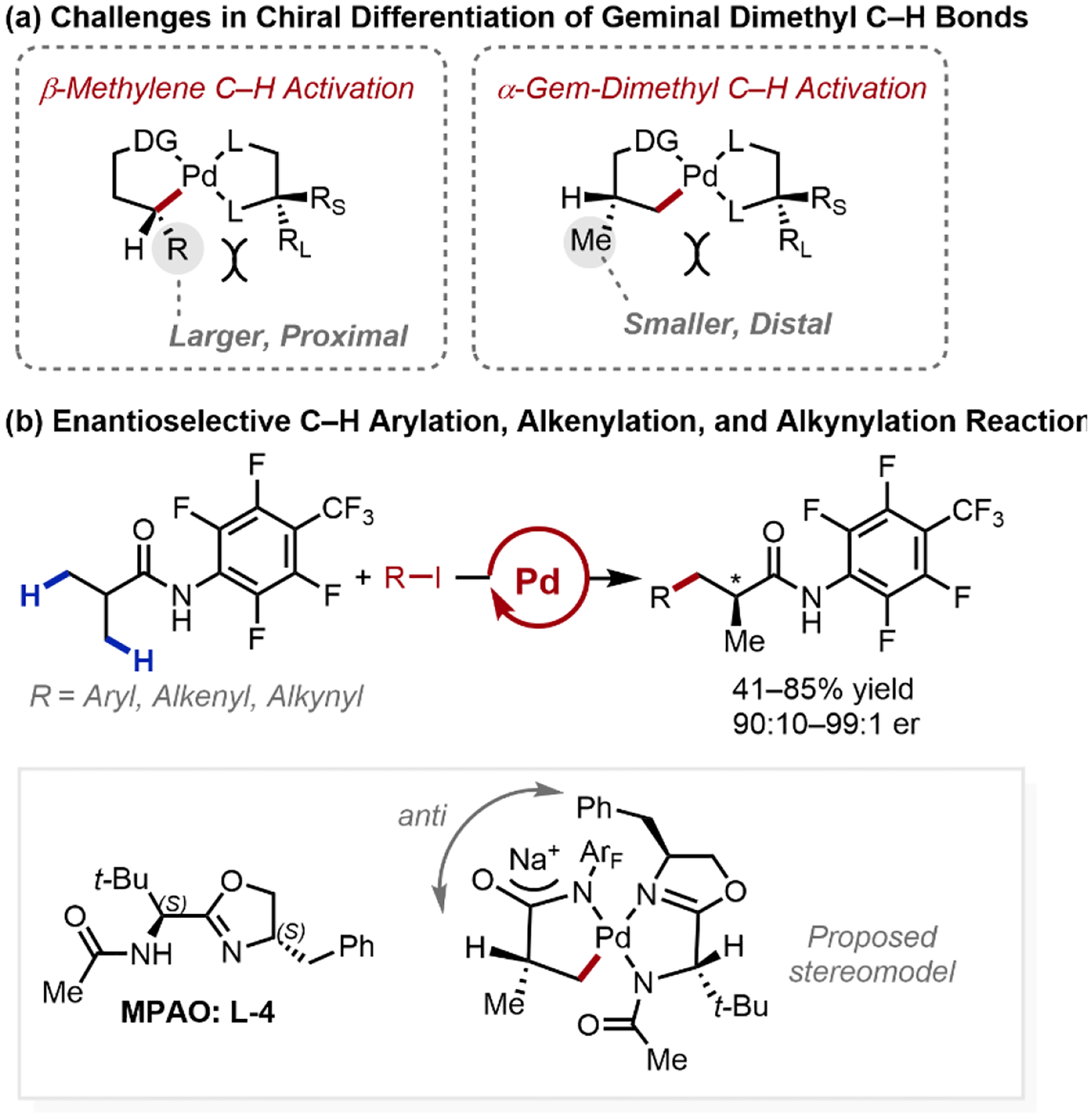
Enantioselective C–H Arylation, Alkenylation, and Alkynylation of Isobutyric Acid Derivatives
Taking the success of the MPAO ligand scaffold, our group developed the first enantioselective C(sp3)–H borylation platform, capable of desymmetrizing methylene C–H bonds in achiral cyclobutylamide substrates using Chiral (S,R)-configured bidentate MPAO ligand L-5 and O2 as the sole oxidant (Scheme 9a).28b Enantioinduction was proposed to occur through minimizing steric repulsion between the cyclobutyl ring and the ligand substituents on both MPAO stereocentres (Scheme 9b). Notably, cyclic amides of various ring sizes engaged in the C–H borylation and substrates containing α-tertiary and quaternary carbon centers were tolerated. This study highlights the enabling nature of MPAO ligands in discriminating prochiral carbons for this substrate class given the structural similarity between the isopropyl and cyclobutyl motifs (which can be seen as a constrained isopropyl unit). At the same time, these results illustrated the profound effects on enantioinduction imparted by subtle differences in ligand configuration; the (S,R)-MPAO diastereomer effective for cyclobutyl substrates was found to be ineffective for isopropyl desymmetrization.
Scheme 9.
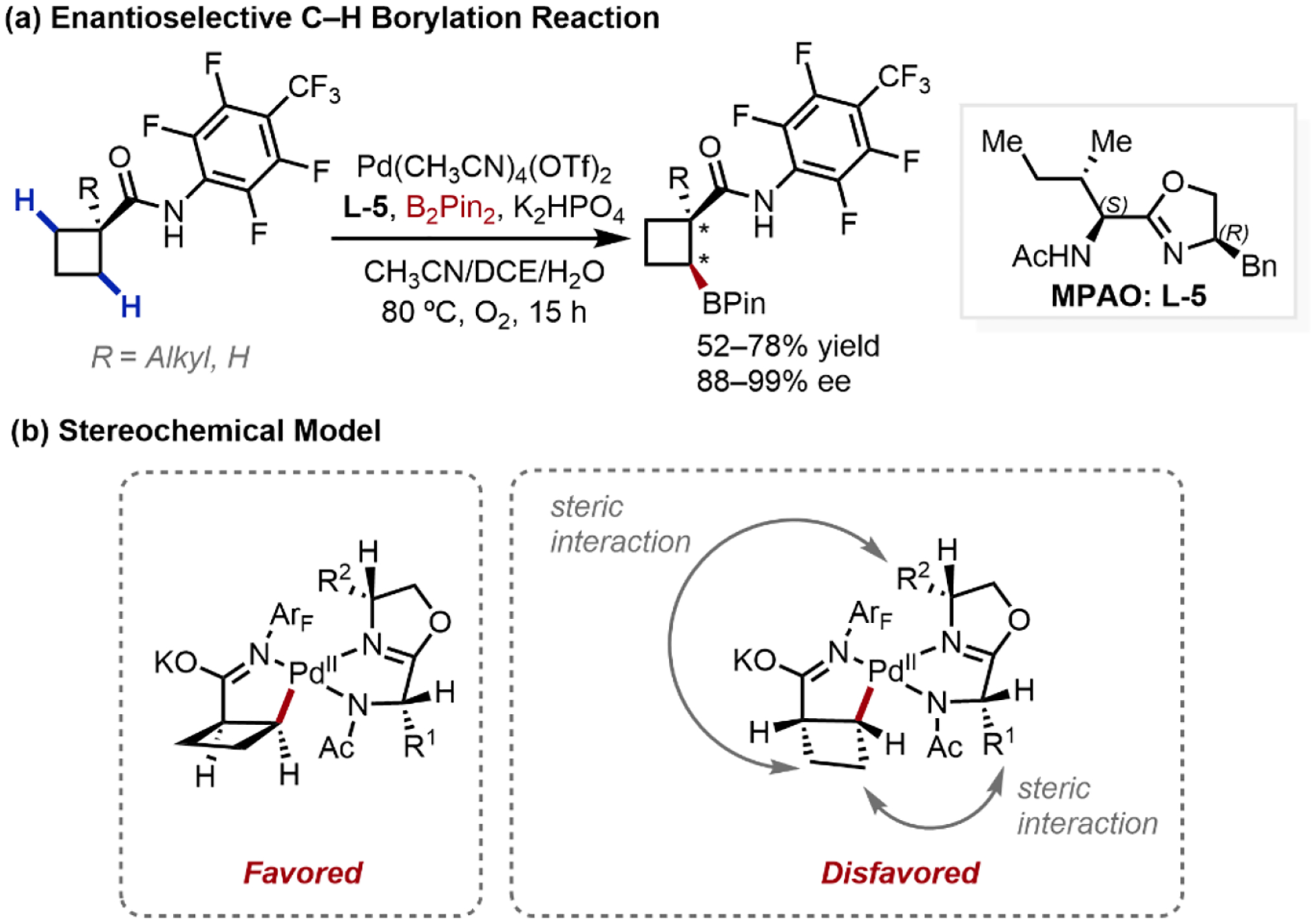
Enantioselective C–H Borylation of Cyclobutanylamides
In 2018, our group extended the applicability of MPAO ligands to enantioselective β-C(sp3)–H arylation and vinylation of cyclobutylamides using L-6 (Scheme 10).23e This new transformation overcame several of the key limitations associated with our earlier work (Scheme 7, vide supra). First, cyclobutane substrates possessing α-hydrogen atoms were now compatible, and vinylation reactions were performed in addition to arylation. To demonstrate the utility of the transformation, sequential C–H arylation and vinylation were carried out to rapidly construct substituted chiral cyclobutanes bearing three contiguous stereocenters challenging to construct using traditional means.
Scheme 10.
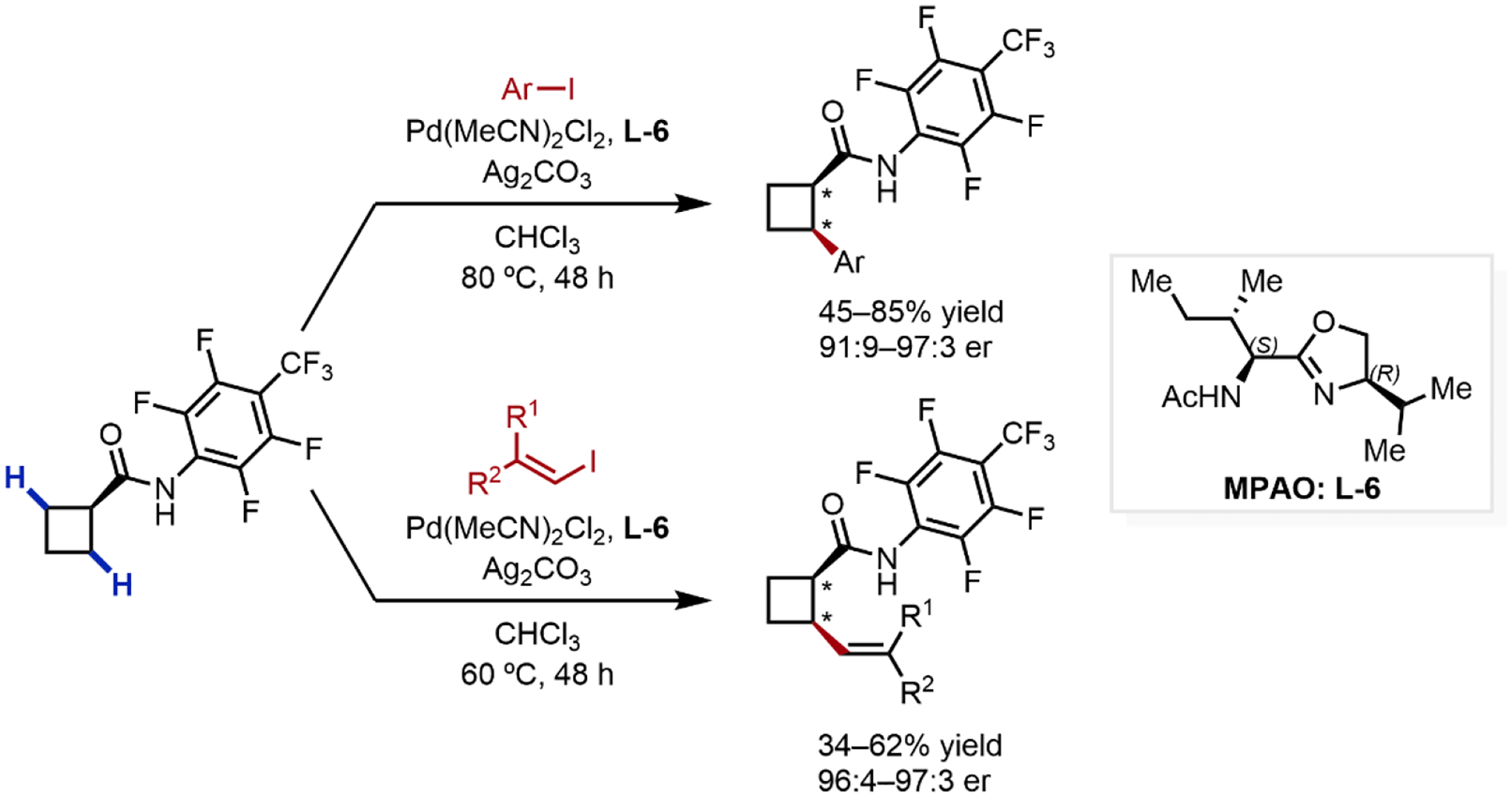
Enantioselective C–H Arylation and Vinylation of Cyclobutanes
In addition to examples developed by our laboratory, the Colobert group has also reported amide-directed enantioselective β-C(sp3)–H activation reactions. Inspired by their previous work utilizing chiral sulfoxides as auxiliary directing groups,37 the Colobert group recently designed a new family of bidentate chiral N-protected aminosulfoxide ligands (L-7) for enantioselective β-C(sp3)–H activation (Scheme 11).38 N-Arylamide derivatives of cyclopropanecarboxylic acid underwent enantioselective C–H arylation in good yields and enantioselectivities utilizing a broad scope of aryl iodides. The proposed stereochemical model invokes key π-stacking interactions between the substrate and ligand, as well as minimized steric interactions between the aryl substituent and N-Ac moiety on the ligand.
Scheme 11.
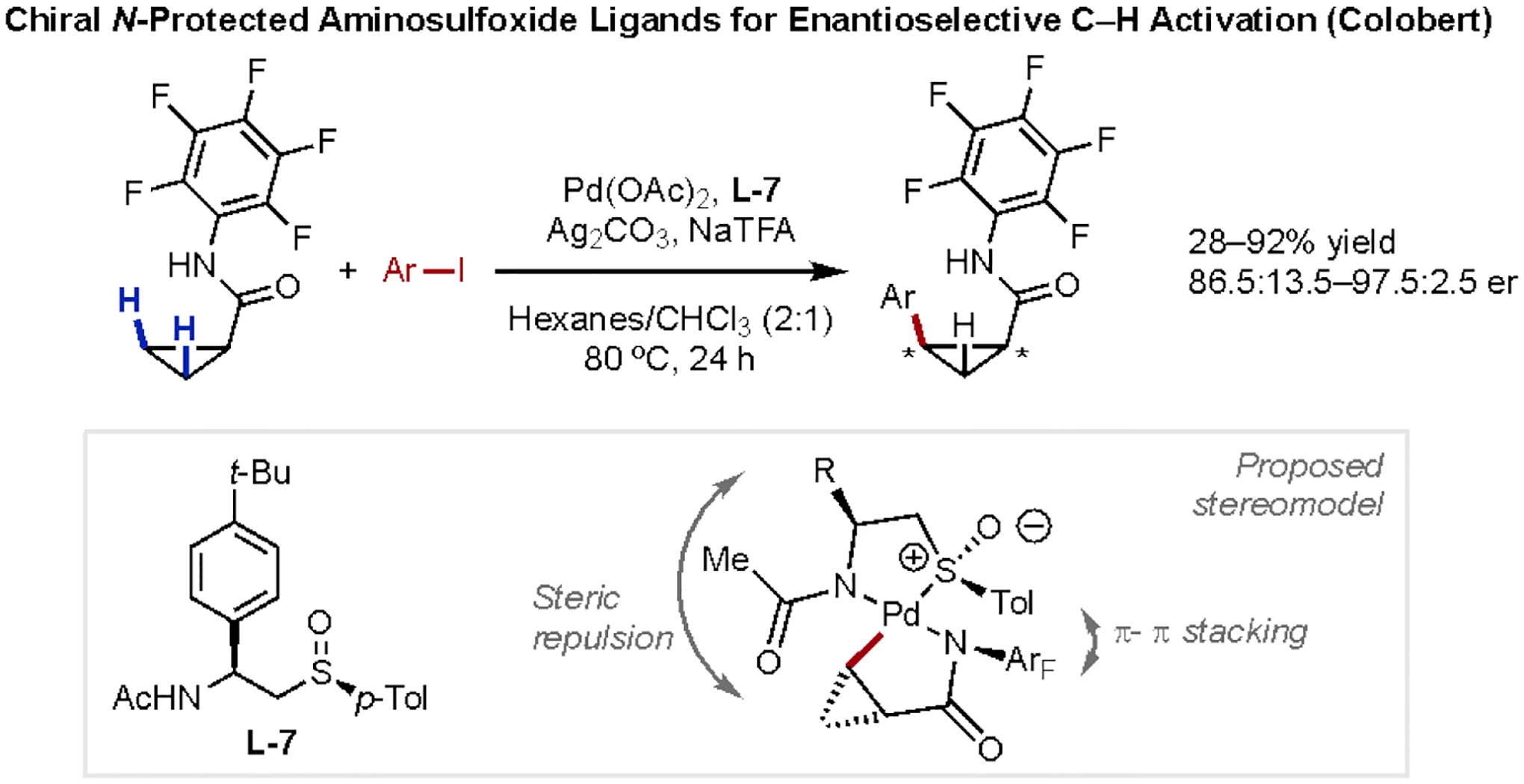
Weakly-coordinative Amide-Directed Enantioselective β-C(sp3)–H Arylation Reaction Developed by the Colobert Group
3.3. Desymmetrization of Enantiotopic Protons
Compared to the desymmetrization of enantiotopic carbons, the enantioselective activation of enantiotopic protons represents a far greater challenge, owing both to the precision required to select for only one of two target protons (rather than differentiating sets of several protons), as well as the higher energetic barrier of methylene C–H activation especially for unactivated aliphatic substrates. In 2016, our group developed the enantioselective β-C(sp3)–H arylation of aliphatic amides, enabled by the discovery of chiral bidentate acetyl-protected aminoethyl quinoline (MPAQ) ligands (L-8, Scheme 12).1,39 The design of MPAQ ligands was inspired by combining features from quinoline ligands, previously employed by our group to stabilize and reliably speciate the active catalyst,23a,33 and MPAA ligands, previously utilized to accelerate C(sp3)–H activation and imbue enantioselection.31,32 In a similar manner, the quinolyl motif was incorporated to provide strong σ-coordination to the metal catalyst, positioning the internal base to favorably cleave the target C–H bond. Extensive ligand evaluation revealed that scaffolds that generated a six-membered Pd chelate was crucial for reactivity. This was later rationalized computationally noting that the five-membered chelate favors the formation of a catalytically incompetent dimeric Pd species, as well as accruing disfavored steric interactions between the arylamide unit with the quinoline ligand due to restricted conformational freedom of the smaller chelate.40 With the optimized MPAQ ligand (L-8), this transformation tolerated a broad scope of aryl iodides and aliphatic amides, proceeding in excellent yields and enantioselectivities.
Scheme 12.
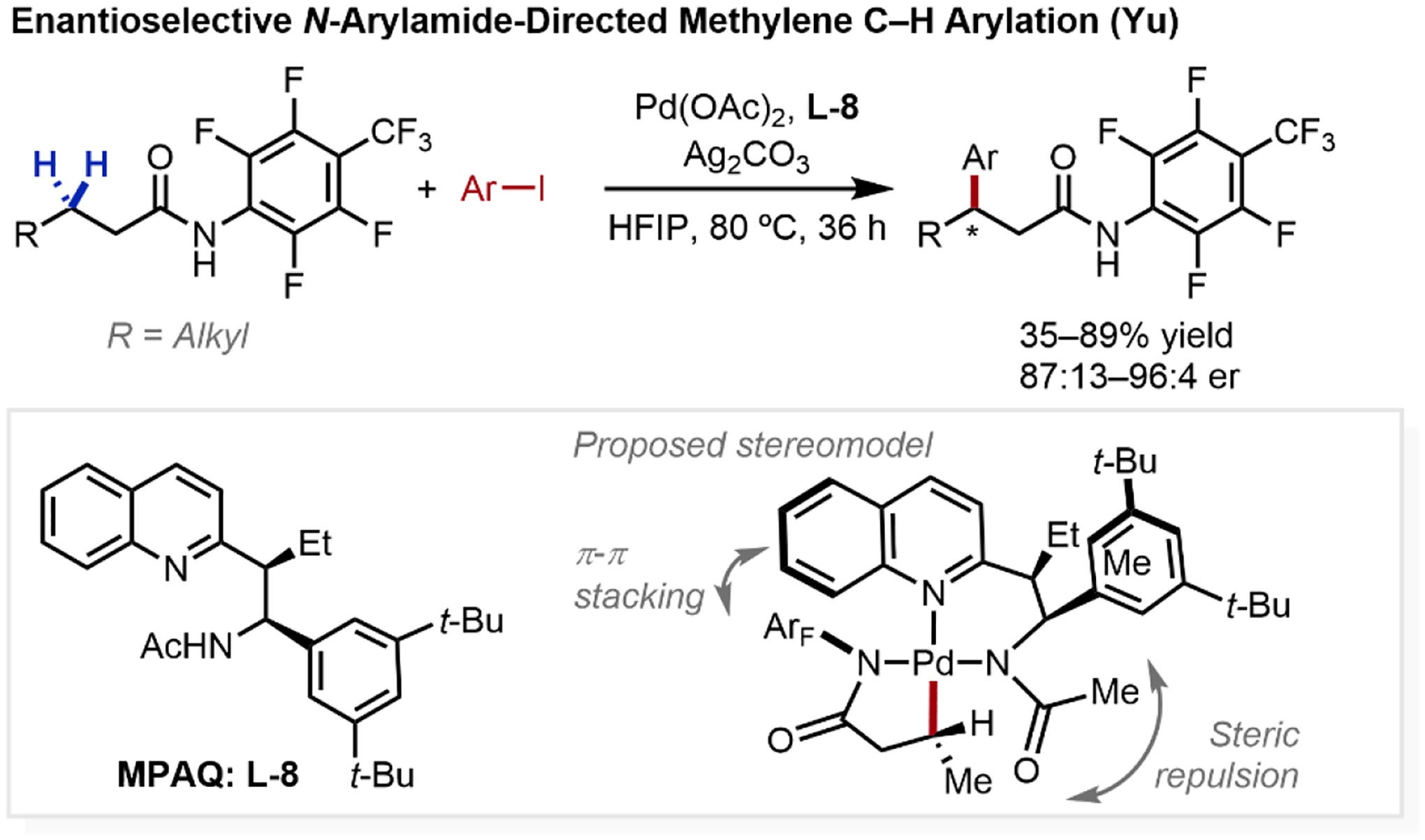
Intermolecular Amide-Directed Enantioselective β-C(sp3)–H Activation Reactions
Extending this finding, we developed an enantio- and diastereoselective methylene C–H arylation of unstrained cycloalkylcarboxamides enabled using analogous bidentate MPAQ ligands (Scheme 13),23f which represents the first example of diastereoselective C–H activation through catalyst rather than substrate control. By selecting the optimal N,N-ligand (L-9, L-10) differing in its stereochemical nature, the reaction enabled the selective C–H cleavage of any of the four possible β-C–H bonds in the cycloalkylcarboxamides substrate, selectively generating any of the four possible stereoisomers of the 2-arylated products.
Scheme 13.
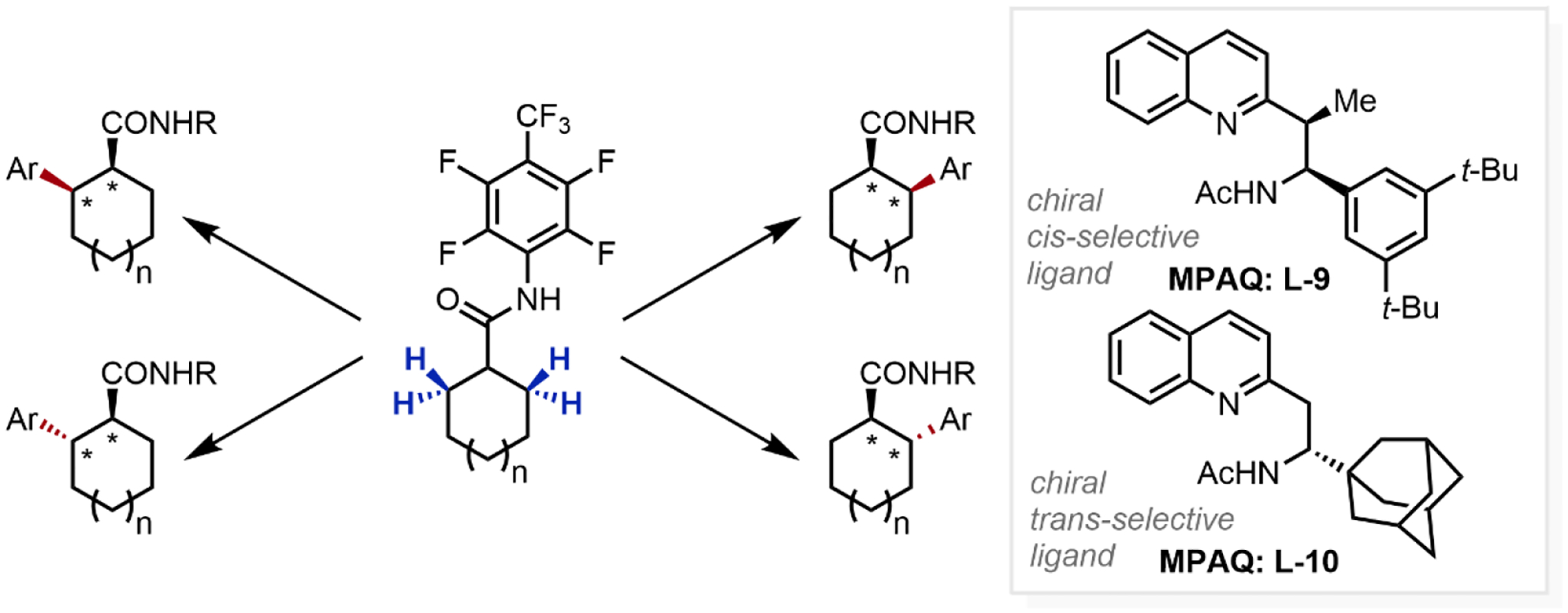
Enantio- and Diastereoselective C–H Arylation of Cycloalkylcarboxamides
4. β-C(sp3)–H Activation Reactions of Free Carboxylic Acids
4.1. Overview
So far, bespoke directing groups previously discussed have been crucial for the advancement of aliphatic C–H activation, though separate installation and often-adverse removal conditions hamper their tractability. While the direct functionalization of free acid is a desirable venture, their weak coordination diminishes the amount of catalyst-bound substrate present, thus abating reactivity.41 Additionally, competition between κ1 and κ2 coordination modes is problematic, as the κ2-bound complex does not possess adequate geometry required for cyclometallation. Taking lessons learned from studies incorporating weak-coordinating amides, we discuss the developments that has led to the successful β-C(sp3)–H functionalization of native carboxylic acids without exogenous directing groups.
The first example of native acid-directed aliphatic C–H activation was reported by the Sen group in 1991 (Scheme 14a).42 This C–H oxidation reaction was stoichiometric in platinum and required superstoichiometric amounts of the substrate. Despite these shortcomings, this work served as an early proof-of-concept that free carboxylic acids could direct a transition metal-catalyzed C–H functionalization reaction at the β-position of an aliphatic substrate. In 2007, our laboratory reported the first catalytic carboxylic acid-directed β-C(sp3)–H activation reaction under ligand free conditions (Scheme 14b).43 A key finding we discovered was the dramatic acceleration in reactivity through the use of alkali metal carboxylate-derivatives of free acids; noteworthily, the use of a preformed palladium carboxylate substrate in the absence of an alkali metal cation was not reactive.43,44 This remarkable effect in reactivity was later ascribed to the alkali metal preferentially binding to the carboxylate in a κ2 manner, facilitating carboxylate-catalyst binding in a productive κ1 fashion.45 This finding was a major enabler for subsequent reaction development, with nearly all future discoveries requiring a source of alkali metal cation to promote reactivity. Simple α-quaternized aliphatic acids were functionalized with arylboron reagents with good mono selectivity under ligand-free conditions, albeit in low yields. β-Functionalization was also achieved with aryl iodides as coupling partners in good yields and with moderate selectivity for monoarylation. In addition, one example was demonstrated for the arylation of a cyclopropyl C–H bond, notable due to the rarity of methylene C–H activation reactions at the time.
Scheme 14.
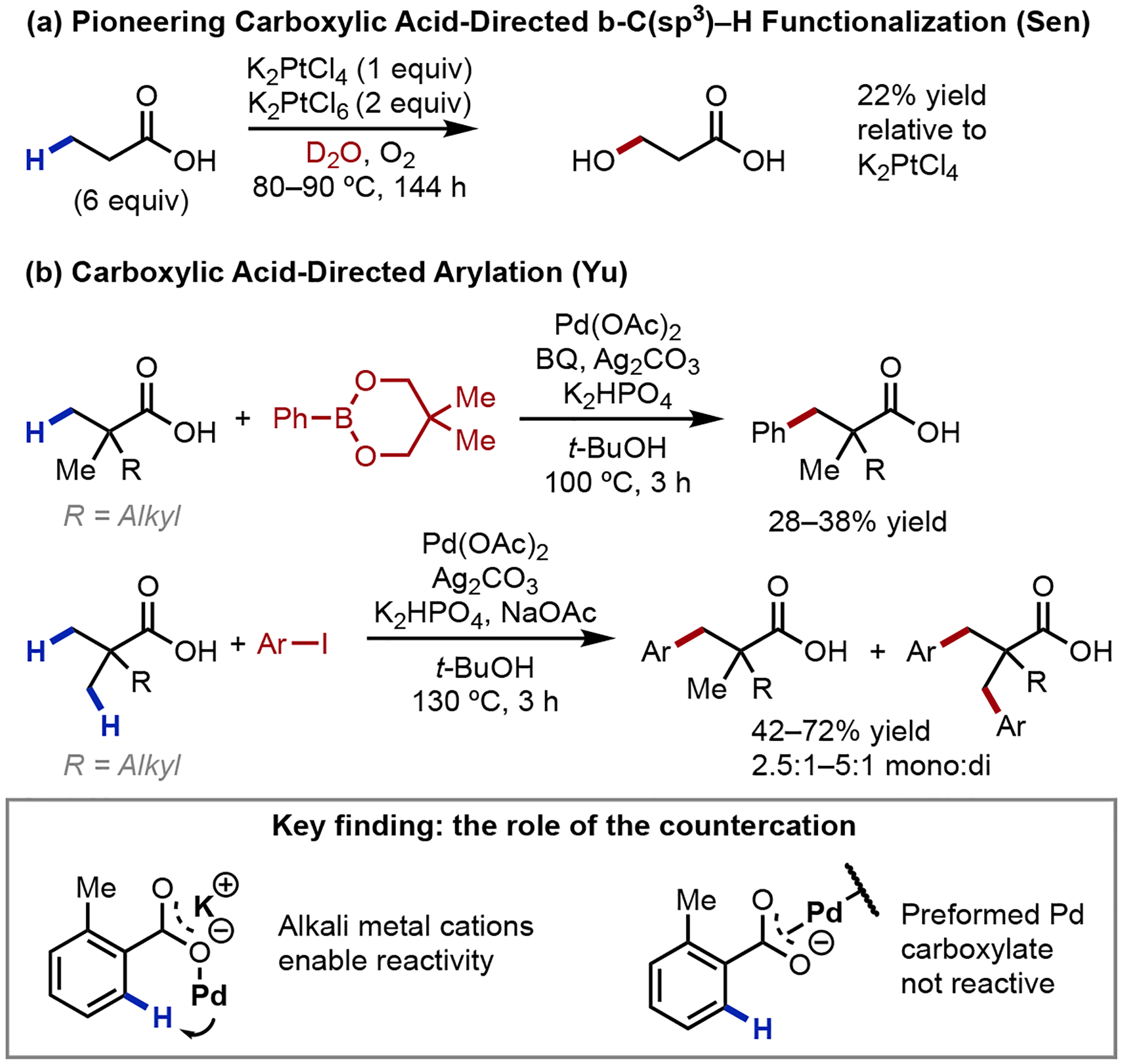
Pioneering Examples of Carboxylic Acid-Directed β-C(sp3)–H Activation Reactions
4.2. Carboxylic acid directed methyl functionalization (2017–present)
Carboxylic acid-directed aliphatic β-C–H activation remained dormant until a decade later, when several examples of β-C(sp3)–H arylation of substrates without α-quaternary centers were developed. In 2017, our laboratory disclosed the β-C–H arylation of α-amino acids employing aryl iodides as coupling partners (Scheme 15a and b),46 where a strongly-coordinating pyridine-type ligand L-11 was crucial for the success of our C–H arylation reaction. Zhao group found that our bidentate MPAA ligand could also effect this arylation reaction.47 In the same year, the van Gemmeren group reported the β-C(sp3)–H arylation of simple, unfunctionalized carboxylic acids with aryl iodides to afford hydrocinnamic acid derivatives (Scheme 15c).48 This transformation again employed a bidentate β-MPAA ligand, tolerated a wide variety of aryl iodides, and performed well with carboxylic acids bearing α-secondary, tertiary, and quaternary centers.
Scheme 15.
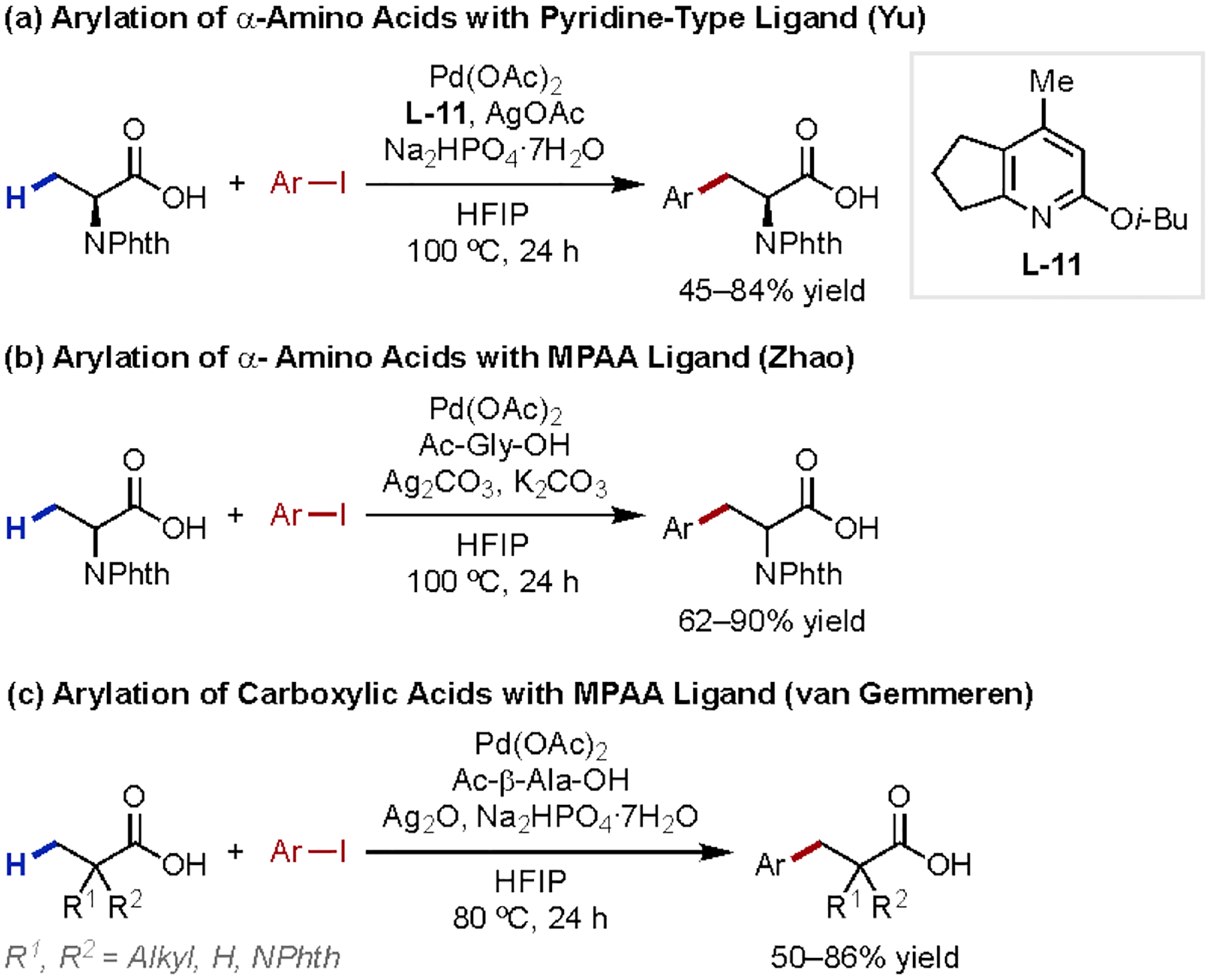
β-C(sp3)–H Arylation of Carboxylic Acids Without α-Quaternary Centers
Over the years, cumulative advances have broadened the scope of free acid functionalization to include olefination, carbonylation, alkynylation, acetoxylation, lactonization, and acyloxylation reactions, with these reactions uniformly enabled by bidentate MPAA and related ligands.3,49–54 In 2018, our laboratory reported the β-C(sp3)–H olefination, and subsequent lactonization, of aliphatic carboxylic acids to afford γ-lactones (Scheme 16a).3 This transformation was enabled by the development of a new class of bifunctional acetyl-protected aminoethyl phenyl thioether ligand (L-12), with the thioether motif anchoring the catalyst through strong σ-coordination to favorably position the NHAc internal base for C–H cleavage. The use of this novel ligand later also permitted the carboxylic acid-directed β-C(sp3)–H carbonylation to afford acid anhydrides, giving a rapid entry point to a range of β-carboxylated products (Scheme 16b).49 Alongside our initial disclosure in 2018, the van Gemmeren group developed the β-C(sp3)–H acetoxylation of aliphatic carboxylic acids (Scheme 16b).50 This transformation, though scalable to 5 mmol, was mostly limited to carboxylic acids bearing α-quaternary centers, although one example with an α-tertiary center was demonstrated in low yield.
Scheme 16.
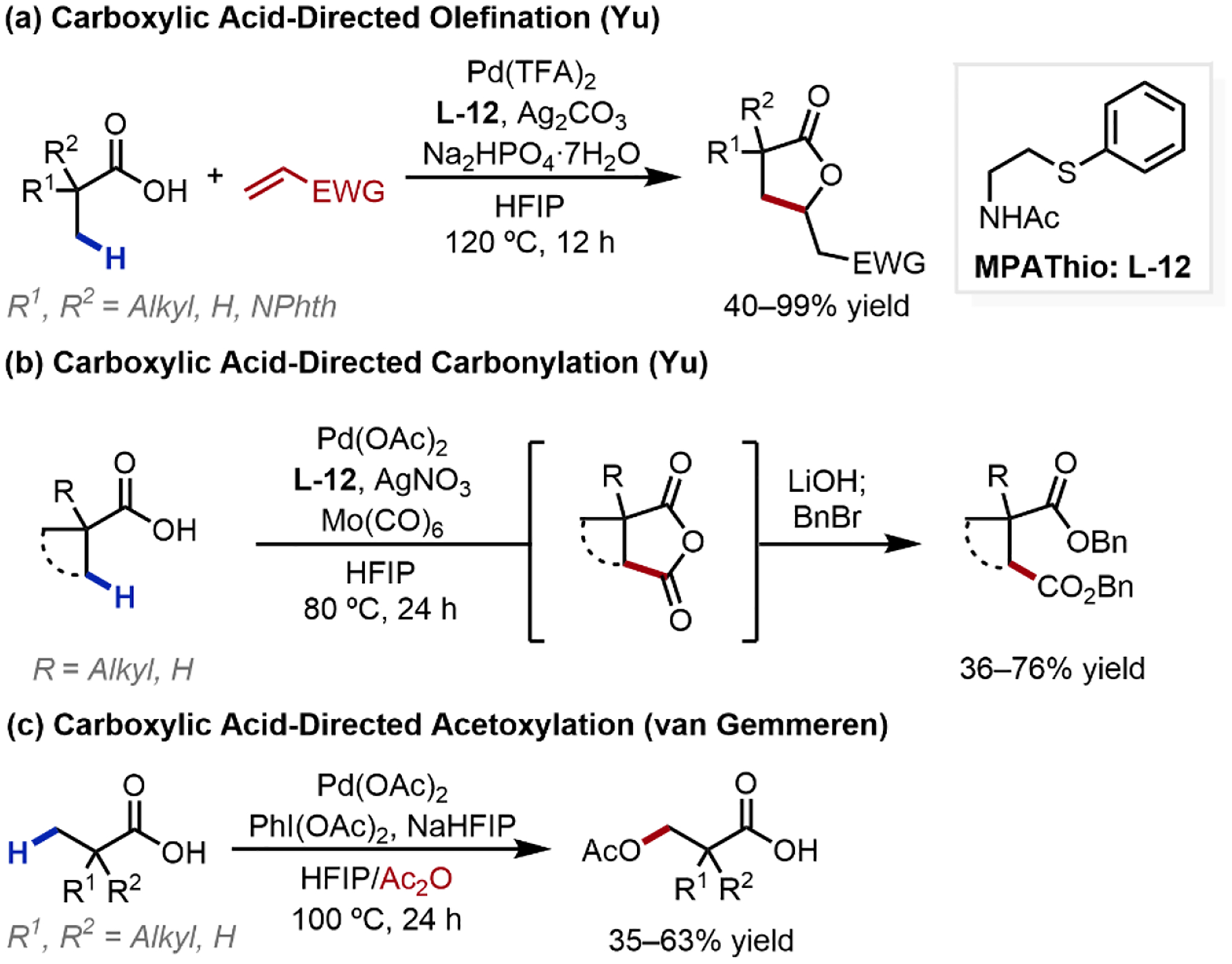
Diverse Carboxylic Acid-Directed β-C(sp3)–H Activation Transformations
Recently, our laboratory discovered a carboxylic acid-directed β-C(sp3)–H lactonization to afford β-lactones using an inexpensive tert-butyl hydroperoxide (TBHP) oxidant enabled by β-MPAA ligand L-13 (Scheme 17a).51 This provided a modular synthetic route toward β-substituted carboxylic acids by nucleophilic ring-opening of β-lactones to construct diverse C–C and C–X bonds. This C–H lactonization could be carried out on gram scale, catalyst loading could be lowered to 1% and the β-lactone product could be isolated with a simple aqueous wash without chromatography. Modification of these conditions next enabled the discovery of carboxylic acid-directed β-C(sp3)–H acyloxylation and intramolecular lactonization (to generate γ-, δ-, and ε-lactones), enabled by a new cyclopentane-based β-MPAA ligand (L-14).52 Such conditions also facilitated a free acid-directed dehydrogenative cross-coupling reaction, whereby an initial methyl β-C(sp3)–H activation generates a weakly bound alkylpalladium species that could then activate and cross-couple with a second aryl C–H bond (Scheme 17b).53 This discovery enabled a range of tetralin, chromane and indane motifs to be generated, and facilitated a four-step total synthesis of (±)-russujaponol F. Coming full circle, the use of inexpensive TBHP oxidant in conjunction with Ac2O was inspired by our early mechanistic studies in oxazoline-directed C–H acetoxylation; both requiring a similar combination of peroxide oxidant with Ac2O for their success.
Scheme 17.
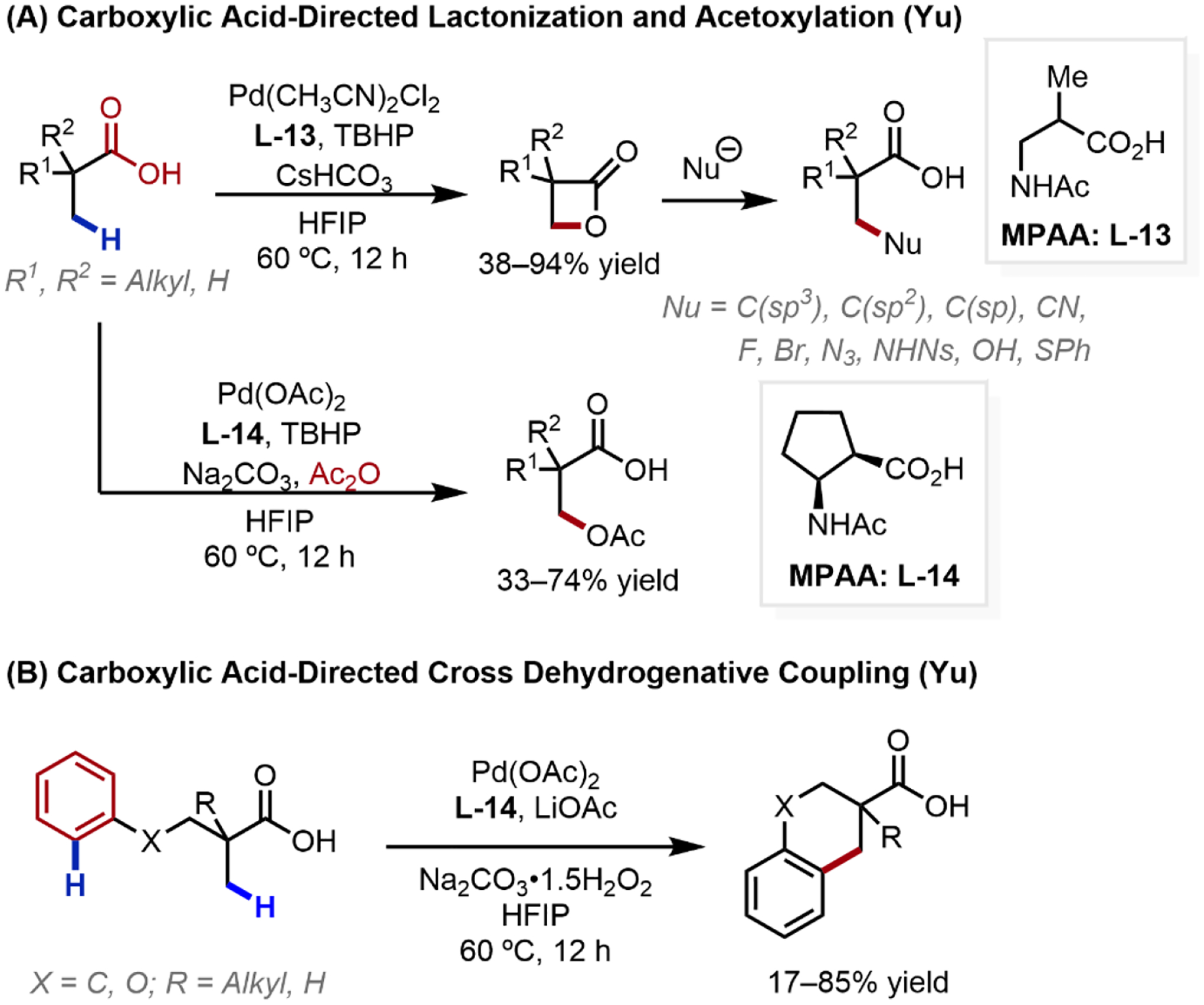
Carboxylic Acid-Directed β-C(sp3)–H Activation Transformations Using Inexpensive Peroxide Oxidants
4.2. Current Challenges: Enantioselective and β-Methylene Functionalization Functionalization of Carboxylic Acids
Beyond its weak directing ability, the utilization of free carboxylic acids as directing groups for enantioselective C–H activation is especially challenging due to the flexibility of the metal-carboxylate complex, demanding the design of new chiral ligands to promote the desired reactivity and stereoselectivity. The first examples of carboxylic acid-directed enantioselective β-C(sp3)–H activation reactions via differentiation of enantiotopic carbon have been reported by our laboratory. In 2018, we reported the carboxylic acid-directed C(sp3)–H arylation of cyclopropanes, enabled by a new chiral mono-protected aminoethyl amine (MPAAm) ligand L-15 (Scheme 18a);55 the replacement of the planar azines (e.g. L-8) with the sterically more hindered dialkylamino group was crucial for achieving enantioselectivity. More recently, we reported the carboxylic acid-directed cross-coupling of cyclopropanes and cyclobutanes to introduce β-aryl or vinyl groups, enabled by chiral MPAAm (L-16) and MPAA ligands (L-17, Scheme 18b).56 In light of these promising results and the challenges highlighted above, the development of diverse enantioselective carboxylic acid-directed functionalizations of unbiased methylene C(sp3)–H bonds forms an ongoing ambition within our laboratory.
Scheme 18.
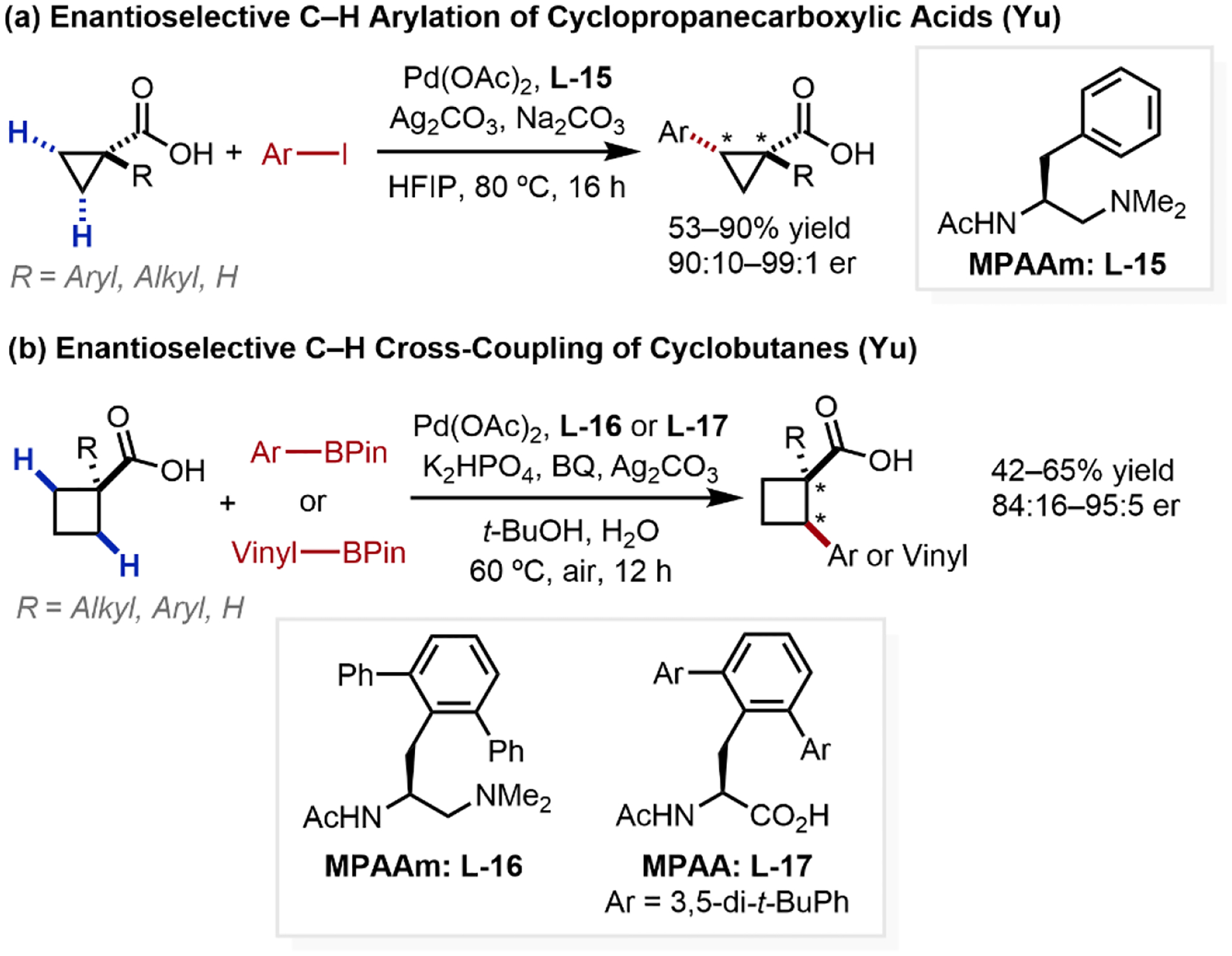
Carboxylic Acid-Directed Enantioselective β-C(sp3)–H Activation Reactions
Advancing bifunctional ligand design and incorporating peroxide-mediated reaction conditions inspired from our early stoichiometric studies enabled a growing number of functionalizations directed by free acids. However, these successes tend to be limited to substrates bearing β-methyl or highly activated β-methylene groups, even though deuteration of β-methylene C–H bonds has been observed using MPAAm type ligands.57 As unactivated methylene C–H bonds are more recalcitrant towards cyclopalladation, an enduring challenge relates to their effective β-methylene C–H activation and downstream functionalization. Building on its enabling ability in free acid-directed C(sp2)–H oxygenations,58 we discovered that a new class of pyridine-pyridone (PyriPyri) ligand scaffold (L-18 and L-19) uniquely enabled the β-methylene dehydrogenation of free acids in the presence of other enolizable motifs (Scheme 19).4 Through this discovery, we further demonstrated that this dehydrogenated intermediate can be intercepted with alkynyl bromides as a second coupling partner, generating diverse butenolide scaffolds. These emerging results augur hope that diverse β-methylene C(sp3)–H functionalization of native carboxylic acids is indeed possible, and we project that further development along this enabling class of ligand could unveil new reactivities, and achieve enantioselection, for free acid-directed unactivated methylene C(sp3)–H activation.
Scheme 19.
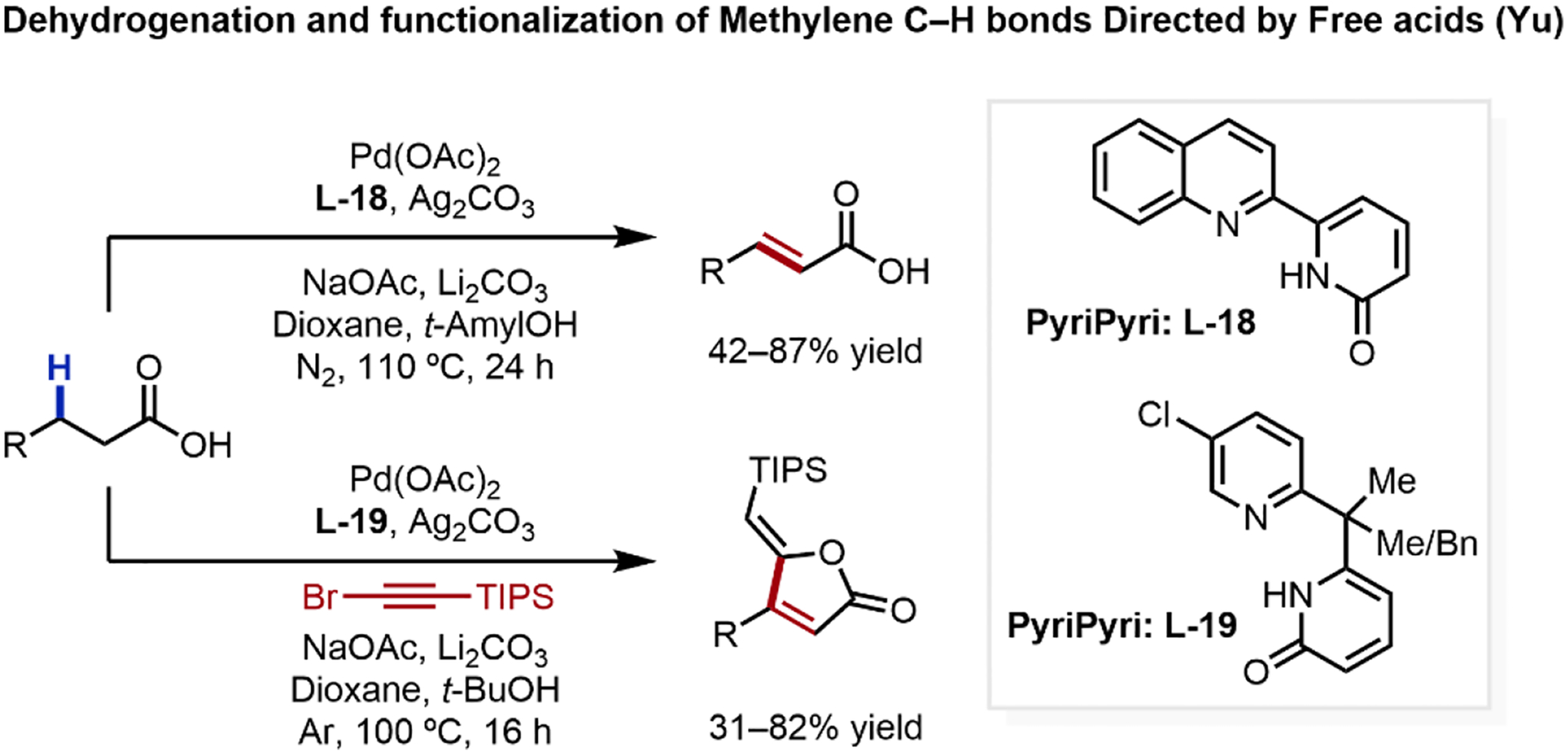
Carboxylic Acid-Directed β-Methylene C(sp3)–H Activation Enables Dehydrogenation and Further Functionalization
6. Summary and Outlook
Over the past two decades, significant advances have been made towards the diverse β-functionalization of aliphatic acids and their derivatives. These transformations typically circumvent the need for prefunctionalization, generation of reactive intermediates and polarity-matching constraints that characterize traditional enolate or conjugate addition transforms, highlighting the potential flexibility of C–H activation as an alternative retrosynthetic strategy. The early oxazoline β-C(sp3)–H activation reaction reported by our laboratory in 2005 provided a valuable framework for subsequent reaction and chiral ligand design. To overcome the limitations posed by the well-established reactivity of strongly-coordinating bidentate directing groups (e.g. unreactive cyclometallated intermediates and limited use of external ligands), our group first developed β-C–H activation reactions that employ weakly-coordinating N-arylamide directing groups as free acid surrogates. These studies establish that the combination of weak substrate coordination and bifunctional ligand acceleration is a promising approach for advancing C(sp3)–H activation, which is essential for realizing the β-C–H activation/functionalization of aliphatic acid substrates.
Central to these advances are the concomitant development of new ligand topologies, enabled both by increased mechanistic understanding and a shift to substrate monodenticity. Through this, specific ligand motifs were engineered to enable productive ligand participation at various steps within the catalytic cycle. Through these developments, we broadened the range of coupling partners possible through diverse catalytic cycles, and established the ability to impart ligand acceleration in these processes. The latter providing the framework necessary for the development of enantioselective variants, where we designed chiral ligands to facilitate enantioselective β-C(sp3)–H activation reactions. Transformations which desymmetrize both enantiotopic carbon atoms and protons have been established, though the latter remaining less developed. To date, emerging examples show that challenging free acid-directed methylene and enantioselective transformations are possible, though significant research effort is necessary to broaden substrate and transformation scopes for these reactions. Through gaining insight into their reactivity, continued advances will be reliant on the development of new ligand scaffolds to address current shortfalls, which we project can improve the practicality, imbue stereoselectivity, surpass the current site-selectivity and enable further iterative functionalizations for diverse free acid-directed transformations (Scheme 20). These achievements can allow C(sp3)–H functionalization of free acids to firmly stand beside or even exceed enolate and conjugate addition as a venerable strategy for the facile synthesis of diversely β-substituted carbonyl compounds.
Scheme 20.
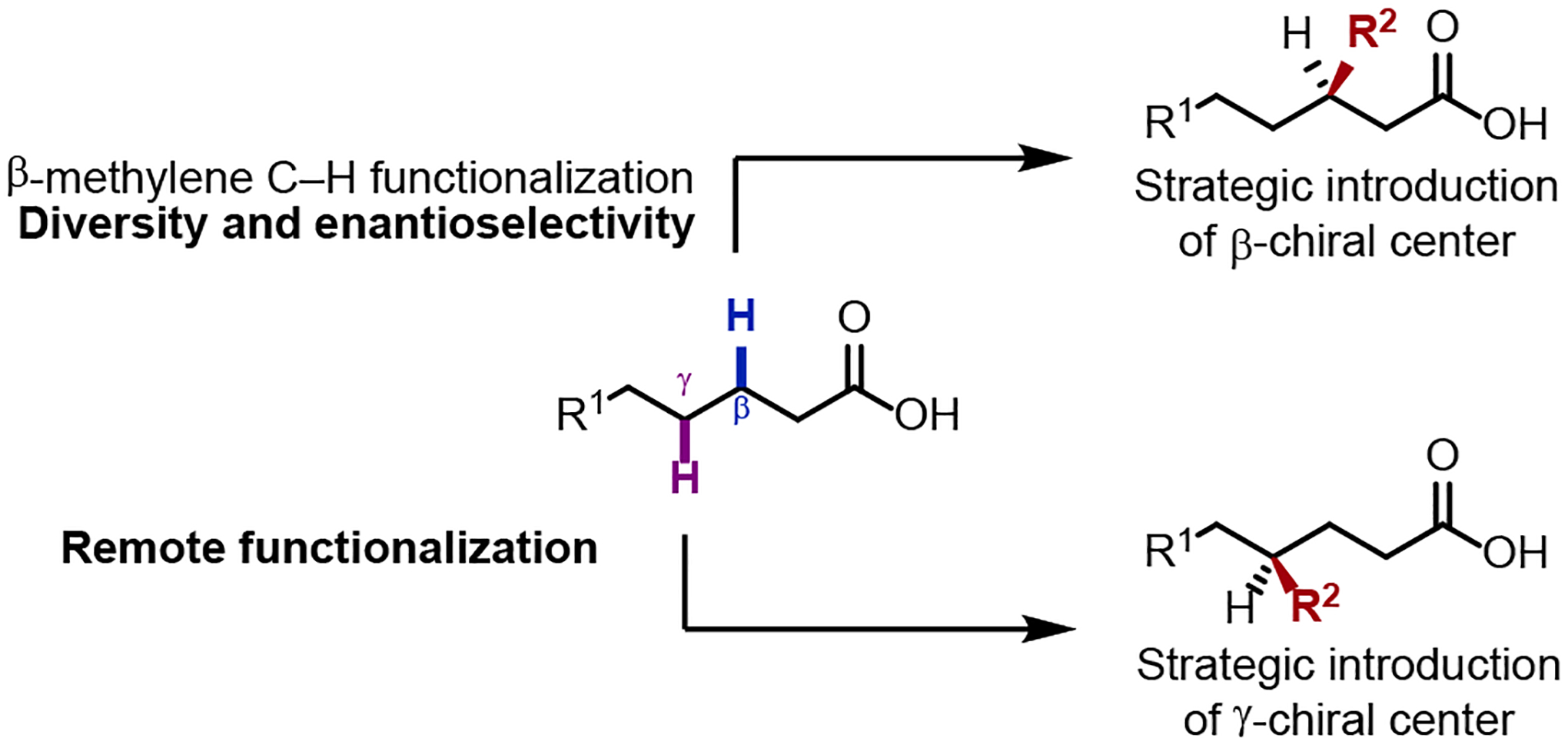
Future ambitions for the development of free acid-directed C(sp3)–H functionalization
Acknowledgement
We gratefully acknowledge the Royal Society for funding our preliminary studies on asymmetric β-C–H functionalization reactions (2002; RSC: 020 7451 2545, RG 36873). We also thank The Scripps Research Institute, the NIH (National Institute of General Medical Sciences grant R01 GM102265), NSF under the CCI Center for Selective C–H Functionalization, CHE-1205646, the Lindemann Trust (fellowship to NYSL) and the Croucher Foundation (fellowship to HSSC) for financial support.
Biographies
Erika L. Lucas received her B.Sc. from the University of California, Santa Cruz (Profs. R. Scott Lokey and Donald R. Smith; 2014). She completed her Ph.D. at the University of California, Irvine, where she studied Ni-catalyzed cross-electrophile coupling reactions (Prof. Elizabeth R. Jarvo; 2019). She worked as a Postdoctoral Associate with Prof. Jin-Quan Yu at the Scripps Research Institute developing new ligands for Pd-catalyzed C–H activation reactions. Erika currently works as a process chemist at Gilead.
Nelson Y. S. Lam received his B.Sc.(Hons) from the University of Auckland (Prof. Christian Hartinger; 2015). He obtained his Ph.D. at the University of Cambridge, where he worked on the total syntheses of polyketide natural products (Prof. Ian Paterson; 2019). He is currently a Lindemann Postdoctoral Fellow with Prof. Jin-Quan Yu at the Scripps Research Institute, developing novel Pd-catalyzed C(sp3)–H functionalization transformations.
Zhe Zhuang received predoctoral training in Prof. Wei-Wei Liao’s lab (B.Sc., 2013, Jilin University) and Prof. Zhi-Xiang Yu’s lab (2014–2015, Peking University). He completed his Ph.D. in Prof. Jin-Quan Yu’s lab, focusing on developing native functional group-directed C–H functionalization transformations using sustainable oxidations. He is currently a postdoctoral scholar at Stanford University with Prof. Nathanael Gray.
Hau Sun Sam Chan received his B.Sc. (Hons) from the University of Hong Kong (Prof. Pauline Chiu; 2015). He completed his Ph.D. at the University of Oxford, where he investigated the organic chemistry of oxonium ions with applications for the total syntheses of Laurencia natural products (Prof. Jonathan Burton; 2019). He is currently a Croucher Postdoctoral Fellow with Prof. Jin-Quan Yu at the Scripps Research Institute, developing novel Pd-catalyzed C(sp3)–H functionalization transformations
Daniel A. Strassfeld received his A. B. from Princeton University (Prof. Erik Sorensen; 2012). He completed his Ph.D. at Harvard University, where he worked on the development and mechanistic study of chiral hydrogen-bond-donor catalyzed reactions (Prof. Eric Jacobsen; 2020). He is currently a Postdoctoral Associate with Prof. Jin-Quan Yu at the Scripps Research Institute developing novel Pd-catalyzed C–H activation reactions.
Jin-Quan Yu received his B.Sc. in Chemistry from East China Normal University and his M.Sc. from the Guangzhou Institute of Chemistry. In 2000, he obtained his Ph.D. at the University of Cambridge with Prof. Jonathan B. Spencer. Following a Junior Research Fellowship at Cambridge, he joined the laboratory of Prof. E. J. Corey at Harvard University as a postdoctoral fellow. He then began his independent career at Cambridge (2003–2004) before moving to Brandeis University (2004–2007) and finally The Scripps Research Institute, where he is currently the Frank and Bertha Hupp Professor of Chemistry.
Footnotes
The authors declare no competing financial interests.
References
- (1).Chen G; Gong W; Zhuang Z; Andrä MS; Chen Y-Q; Hong X; Yang Y-F; Liu T; Houk KN; Yu J-Q Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science 2016, 353, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]; The design of chiral bidentate acetyl-protected aminoethyl quinoline (MPAQ) ligands enabled the desymmetrization of enantiotopic protons via β-C(sp3)–H arylation of aliphatic amides.
- (2).Wu Q-F; Shen P-X; He J; Wang X-B; Zhang F; Shao Q; Zhu R-Y; Mapelli C; Qiao JX; Poss MA; Yu J-Q Formation of α-chiral centers by asymmetric β-C(sp3)–H arylation, alkenylation, and alkynylation. Science 2017, 355, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]; The design of chiral bidentate mono-protected aminomethyl oxazoline (MPAO) ligands enabled the desymmetrization of enantiotopic carbons via β-C(sp3)–H arylation, alkenylation, and alkynylation of isobutyramide substrates.
- (3).Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q Ligand-enabled β-C(sp3)–H olefination of free carboxylic acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]; The design of bidentate acetyl-protected aminoethyl phenyl thioether (MPAThio) ligands enabled the first β-methyl C–H olefination of free carboxylic acids.
- (4).Wang Z; Hu L; Zhuang Z; Qian S; Qiao JX; Yu J-Q Ligand-controlled divergent dehydrogenative reactions of carboxylic acids via C–H activation. Science 2021, 374, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]; The design of bidentate pyridine-pyridone (PyriPyri) ligands enabled the dehydrogenation of free carboxylic acids via β-methylene C–H activation.
- (5).Wöhler F Ueber künstliche Bildung des Harnstoffs. Annalen der Physik und Chemie 1828, 88, 253–256. [Google Scholar]
- (6).Nicolaou KC Catalyst: Synthetic Organic Chemistry as a Force for Good. Chem 2016, 1, 331–334. [Google Scholar]
- (7).For selected reviews on Palladium-catalyzed C-H activation, see:; (a) Chen X; Engle KM; Wang D-H; Yu J-Q Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: versatility and practicality. Angew. Chem. Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lyons TW; Sanford MS Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Daugulis O; Roane J; Tran LD Bidentate, monoanionic auxiliary-directed functionalization of carbon–hydrogen bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For further discussion, see: Meng G; Lam NYS; Lucas EL; Saint-Denis TG; Verma P; Chekshin N; Yu J-Q Achieving site-selectivity for C–H activation processes based on distance and geometry: a carpenter’s approach. J. Am. Chem. Soc 2020, 142, 10571–10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For selected reviews on C(sp3)–H activation, see: He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Serratosa F, Chapter 5. Synthesis of Dissonant Systems, Elsevier, 1996. [Google Scholar]; For further discussion on the role of C–H activation on circumventing the need for ‘polarity matching’ in synthesis, see: Lam NYS; Wu K; Yu J-Q Advancing the logic of chemical synthesis: C–H activation as strategic and tactical disconnections for C–C bond construction. Angew. Chem. Int. Ed 2020, 60, 15767–15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For selected reviews on enantioselective C-H activation, see:; (a) Saint-Denis TG; Zhu R-Y; Chen G; Wu Q-F; Yu J-Q Enantioselective C(sp3)‒H bond activation by chiral transition metal catalysts. Science 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]
- (12). The enantioselective β-C–H activation/functionalizations described in this article pertain only to “point desymmetrization”, or the generation of a new atom-centered stereocenter. For examples of other classes of enantioselective C–H functionalization (e.g. via kinetic resolution or desymmetrization about a plane or axis) see reference 7.
- (13).We limit our discussion in this account to reactions which proceed by metal insertion, rather than alternative mechanisms such as carbene/nitrene insertion or hydrogen atom transfer. For a discussion on other modes of transition metal-catalyzed C–H functionalization via carbene/nitrene transfer, see: Davies HML; Manning JR Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For a review on early σ-chelation-directed C–H functionalization reactions, see: Yu J-Q; Giri R; Chen X σ-Chelation-directed C–H functionalizations using Pd(II) and Cu(II) catalysts: regioselectivity, stereoselectivity and catalytic turnover. Org. Biomol. Chem 2006, 4, 4041–4047. [DOI] [PubMed] [Google Scholar]
- (15).Giri R; Chen X; Yu J-Q Palladium-catalyzed asymmetric iodination of unactivated C–H bonds under mild conditions. Angew. Chem. Int. Ed 2005, 44, 2112–2115. [DOI] [PubMed] [Google Scholar]
- (16).This approach was also employed in a diiodination reaction followed by radical cyclization. See: Giri R; Wasa M; Breazzano SP; Yu J-Q Converting gem-dimethyl groups into cyclopropanes via Pd-catalyzed sequential C–H activation and radical cyclization. Org. Lett 2006, 8, 5685–5688. [DOI] [PubMed] [Google Scholar]
- (17).Giri R; Liang J; Lei J-G; Li J-J; Wang D-H; Chen X; Naggar IC; Guo C; Foxman BM; Yu J-Q Pd-catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: a dual role for carboxylic anhydrides in catalytic C–H bond oxidation. Angew. Chem. Int. Ed 2005, 44, 7420–7424. [DOI] [PubMed] [Google Scholar]
- (18).Giri R; Lan Y; Liu P; Houk N, K.; Yu J-Q Understanding reactivity and stereoselectivity in palladium-catalyzed diastereoselective sp3 C–H bond activation: intermediate characterization and computational studies. J. Am. Chem. Soc 2012, 134, 14118–14126. [DOI] [PubMed] [Google Scholar]
- (19).Zaitsev VG; Shabashov D; Daugulis O Highly regioselective arylation of sp3 C–H bonds catalyzed by palladium acetate. J. Am. Chem. Soc 2005, 127, 13154–13155. [DOI] [PubMed] [Google Scholar]
- (20).For a review on amide-directed C(sp3)–H functionalization, see: Tan PW; Seayad J Advances in amide and thioamide assisted C(sp3)–H functionalization. Tetrahedron Lett 2019, 60, 151338. [Google Scholar]
- (21).(a) Wang D-H; Wasa M; Giri R; Yu J-Q Pd(II)-catalyzed cross-coupling of sp3 C–H bonds with sp2 and sp3 boronic acids using air as the oxidant. J. Am. Chem. Soc 2008, 130, 7190–7191. [DOI] [PubMed] [Google Scholar]; (b) Wasa M; Engle K; Yu J-Q Pd(0)/PR3-catalyzed intermolecular arylation of sp3 C–H bonds. J. Am. Chem. Soc 2009, 131, 9886–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Engle KM; Mei TS; Wasa M; Yu JQ Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Wasa M; Chan KSL; Zhang X-G; He J; Miura M; Yu J-Q Ligand-enabled methylene C(sp3)–H bond activation with a Pd(II) catalyst. J. Am. Chem. Soc 2012, 134, 18570–18572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Li S; Deng Y; Fu H; Laforteza BN; Spangler JE; Homs A; Yu J-Q Ligand-controlled C(sp3)–H arylation and olefination in syntheses of unnatural chiral α-amino acids. Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng Y; Gong W; He J; Yu J-Q Ligand-enabled triple C–H activation reactions: one-pot synthesis of diverse 4-aryl-2-quinolinones from propionamides. Angew. Chem. Int. Ed 2014, 53, 6692–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) He J; Takise R; Fu H; Yu J-Q Ligand-enabled cross-coupling of C(sp3)–H bonds with arylsilanes. J. Am. Chem. Soc 2015, 137, 4618–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wu Q-F; Wang X-B; Shen P-X; Yu J-Q Enantioselective C–H arylation and vinylation of cyclobutyl carboxylic amides. ACS Catal 2018, 8, 2577–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Andrä MS; Schifferer L; Pollok CH; Merten C; Gooßen LJ; Yu J-Q Enantio- and diastereoswitchable C–H arylation of methylene groups in cycloalkanes. Chem. Eur. J 2019, 25, 8503–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhu R-Y; He J; Wang X-C; Yu J-Q Ligand-promoted alkylation of C(sp3)–H and C(sp2)–H bonds. J. Am. Chem. Soc 2014, 136, 13194–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wasa M; Engle KM; Yu J-Q Pd(II)-catalyzed olefination of sp3 C–H bonds. J. Am. Chem. Soc 2010, 132, 3680–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) He J; Wasa M; Chan KSL; Yu J-Q Palladium(0)-catalyzed alkynylation of C(sp3) –H bonds. J. Am. Chem. Soc 2013, 135, 3387–3390. [DOI] [PubMed] [Google Scholar]; (b) Fu H; Shen P-X; He J; Zhang F; Li S; Wang P; Liu T; Yu J-Q Ligand-enabled alkynylation of C(sp3)–H bonds with Pd(II) catalysts. Angew. Chem. Int. Ed 2017, 56, 1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yoo EJ; Wasa M; Yu J-Q Pd(II)-catalyzed carbonylation of sp3 C–H bonds: a new entry to 1,4-dicarbonyl compounds. J. Am. Chem. Soc 2010, 132, 17378–17380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) He J; Jiang H; Takise R; Zhu R-Y; Chen G; Dai H-X; Murali Dhar TG; Shi J; Zhang H; Cheng PTW; Yu J-Q Ligand-promoted borylation of C(sp3)–H bonds with Pd(II) catalysts. Angew. Chem. Int. Ed 2016, 55, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Shao Q; Wu Q; Yu J-Q Pd(II)-catalyzed enantioselective C(sp3)–H borylation. J. Am. Chem. Soc 2017, 139, 3344–3347. [DOI] [PubMed] [Google Scholar]
- (29).He J; Shigenari T; Yu J-Q Palladium(0)/PAr3-catalyzed intermolecular amination of C(sp3)–H bonds: synthesis of β-amino acids. Angew. Chem. Int. Ed 2015, 54, 6545–6549. [DOI] [PubMed] [Google Scholar]
- (30).(a) Zhu R-Y; Tanaka K; Li G-C; He J; Fu H-Y; Li S-H; Yu J-Q Ligand-enabled stereoselective β-C(sp3)–H fluorination: synthesis of unnatural enantiopure anti-β-fluoro-α-amino acids. J. Am. Chem. Soc 2015, 137, 7067–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu R-Y; Saint-Denis TG; Shao Y; He J; Sieber JD; Senanayake CH; Yu J-Q Ligand-enabled Pd(II)-catalyzed bromination and iodination of C(sp3)–H bonds. J. Am. Chem. Soc 2017, 139, 5724–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shao Q; Wu K; Zhuang Z; Qian S; Yu J-Q From Pd(OAc)2 to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C–H functionalization reactions. Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Shi BF; Maugel N; Zhang YH; Yu J-Q PdII-catalyzed enantioselective activation of C(sp2)–H and C(sp3)–H bonds using monoprotected amino acids as chiral ligands. Angew. Chem. Int. Ed 2008, 47, 4882–4886. For representative studies on the mode of enantioselectivity arising from MPAA-type ligands, see: [DOI] [PubMed] [Google Scholar]; (b) Plata RE; Hill DE; Haines BE; Musaev DG; Chu L; Hickey DP; Sigman MS; Yu J-Q; Blackmond DG A role for Pd(IV) in catalytic enantioselective C–H functionalization with monoprotected amino acid ligands under mild conditions. J. Am. Chem. Soc 2017, 139, 9238–9245. [DOI] [PubMed] [Google Scholar]; (c) Haines BE; Yu J-Q; Musaev DG Enantioselectivity model for Pd-catalyzed C–H functionalization mediated by the mono-N-protected amino acid (MPAA) family of ligands. ACS Catal 2017, 7, 4344–4354. [Google Scholar]
- (33).For a review on the progress of ligand development for C–H activation, see: Engle KM; Yu J-Q Developing ligands for palladium(II)-catalyzed C–H functionalization: intimate dialogue between ligand and substrate. J. Org. Chem 2013, 78, 8927–8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wasa M; Engle KM; Lin DW; Yoo EJ; Yu J-Q Pd(II)-catalyzed enantioselective C–H activation of cyclopropanes. J. Am. Chem. Soc 2011, 133, 19598–19601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Xiao K-J; Lin DW; Miura M; Zhu R-Y; Gong W; Wasa M; Yu J-Q Palladium(II)-catalyzed enantioselective C(sp3)–H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc 2014, 136, 8138–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Notably, the parent carboxylate-containing MPAA ligand afforded low yield and poor ee, indicating the importance of N-methoxyamide motif.
- (37).Jerhaoui S; Chahdoura F; Rose C; Djukic J-P; Wencel-Delord J; Colobert F Enantiopure sulfinyl aniline as a removable and recyclable chiral auxiliary for asymmetric C(sp3)–H bond activation. Chem. Eur. J 2016, 22, 17397–17406. [DOI] [PubMed] [Google Scholar]
- (38).Jerhaoui S; Djukic J-P; Wencel-Delord J; Colobert F Asymmetric, nearly barrierless C(sp3)–H activation promoted by easily-accessible N-protected aminosulfoxides as new chiral ligands. ACS Catal 2019, 9, 2532–2542. [Google Scholar]
- (39).Enantioselective C-H arylation of benzylic C-H bonds has previously been reported using strong-coordinating bidentate directing groups. For representative examples, see:; (a) Yan S-B; Zhang S; Duan W-L Palladium-catalyzed asymmetric arylation of C(sp3)–H bonds of aliphatic amides: controlling enantioselectivity using chiral phosphoric amides/acids. Org. Lett 2015, 17, 2458–2461. [DOI] [PubMed] [Google Scholar]; (b) Tong H-R; Zheng S; Li X; Deng Z; Wang H; He G; Peng Q; Chen G Pd(0)-catalyzed bidentate auxiliary directed enantioselective benzylic C–H arylation of 3-arylpropanamides using the BINOL phosphoramidite ligand. ACS Catal 2018, 8, 11502–11512. [Google Scholar]
- (40).(a) Yang Y-F; Chen G; Hong X; Yu JQ; Houk KN The origins of dramatic differences in five-membered vs six-membered chelation of Pd(II) on efficiency of C(sp3)–H bond activation. J. Am. Chem. Soc 2017, 139, 8514–8521. [DOI] [PubMed] [Google Scholar]; (b) Romero EA; Chen G; Gembicky M; Jazzar R; Yu JQ; Bertrand G Understanding the Activity and Enantioselectivity of Acetyl-Protected Aminoethyl Quinoline Ligands in Palladium-Catalyzed β-C(sp3)-H Bond Arylation Reactions. J. Am. Chem. Soc 2019, 141, 16726–16733. [DOI] [PubMed] [Google Scholar]
- (41).Uttry A; van Gemmeren M Direct C(sp3)–H activation of carboxylic acids. Synthesis 2020, 52, 479–488. [Google Scholar]
- (42).Kao L-C; Sen A Platinum(II) catalysed selective remote oxidation of unactivated C–H bonds in aliphatic carboxylic acids. J. Chem. Soc., Chem. Commun 1991, 1242–1243. [Google Scholar]
- (43).Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunders LB; Yu J-Q Palladium-catalyzed methylation and arylation of sp2 and sp3 C–H bonds in simple carboxylic acids. J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]
- (44).Mei TS; Giri R; Maugel N; Yu J-Q PdII-catalyzed monoselective ortho halogenation of C–H bonds assisted by counter cations: a complementary method to directed ortho lithiation. Angew. Chem. Int. Ed 2008, 47, 5215–5219. [DOI] [PubMed] [Google Scholar]
- (45).A solid state structure verifying this hypothesis is presented in Giri R; Yu J-Q Synthesis of 1,2- and 1,3-dicarboxylic acids via Pd(II)-catalyzed carboxylation of aryl and vinyl C–H Bonds. J. Am. Chem. Soc 2008, 130, 14082–14083. [DOI] [PubMed] [Google Scholar]
- (46).Chen G; Zhuang Z; Li G-C; Saint-Denis TG; Hsiao Y; Joe CL; Yu J-Q Ligand-enabled β-C–H arylation of α-amino acids without installing exogenous directing groups. Angew. Chem. Int. Ed 2017, 56, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zhu Y; Chen X; Yuan C; Li G; Zhang J; Zhao Y Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids. Nat. Commun 2017, 8, 14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ghosh KK; van Gemmeren M Pd-catalyzed β-C(sp3)–H arylation of propionic acid and related aliphatic acids. Chem. Eur. J 2017, 23, 17697–17700. [DOI] [PubMed] [Google Scholar]
- (49).Zhuang Z; Herron AN; Yu J -Q. Syntheses of cyclic anhydrides via ligand-enabled C–H carbonylation of simple aliphatic acids. Angew. Chem. Int. Ed 2021, 60, 16382–16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ghosh KK; Uttry A; Koldemir A; Ong M; van Gemmeren M Direct β-C(sp3)–H acetoxylation of aliphatic carboxylic acids. Org. Lett 2019, 21, 7154–7157. [DOI] [PubMed] [Google Scholar]
- (51).Zhuang Z; Yu J-Q Lactonization as a general route to β-C(sp3)–H functionalization. Nature 2019, 577, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhuang Z; Herron AN; Fan Z; Yu J-Q Ligand-enabled monoselective β-C(sp3)–H acyloxylation of free carboxylic acids using a practical oxidant. J. Am. Chem. Soc 2020, 142, 6769–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zhuang Z; Herron AN; Liu S; Yu J-Q Rapid construction of tetralin, chromane, and indane motifs via cyclative C–H/C–H coupling: four-step total synthesis of (±)-russujaponol F. J. Am. Chem. Soc 2021, 143, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).For other recently developed free acid-directed aliphatic C-H functionalization reactions, see:; (a) Ghiringhelli F; Uttry A; Ghosh KK; van Gemmeren M Direct β- and γ-C(sp3)–H alkynylation of free carboxylic acids. Angew. Chem. Int. Ed 2020, 59, 23127–23131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fan Z; Zhao S; Liu T; Shen P-X; Cui Z-N; Zhuang Z; Shao Q; Chen JS; Ratnayake AS; Flanagan ME; Kölmel DK; Piotrowski DW; Richardson P; Yu J-Q Merging C(sp3)–H activation with DNA-encoding. Chem. Sci 2020, 11, 12282–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q Pd(II)-catalyzed enantioselective C(sp3)–H arylation of free carboxylic acids. J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hu L; Shen P-X; Shao Q; Hong K; Qiao JX; Yu J-Q PdII-catalyzed enantioselective C(sp3)–H activation/cross-coupling reactions of free carboxylic acids. Angew. Chem. Int. Ed 2019, 58, 2134–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Pd-catalyzed deuteration of methylene C–H bonds has been demonstrated using MPAAM ligands, see: Uttry A; Mal S; van Gemmeren M Late-stage β-C(sp3)–H deuteration of carboxylic acids. J. Am. Chem. Soc 2021, 143, 10895–10901. [DOI] [PubMed] [Google Scholar]
- (58).Li Z; Wang Z; Chekshin N; Qian S; Qiao JX; Cheng PT; Yeung KS; Ewing WR; Yu J-Q A tautomeric ligand enables directed C‒H hydroxylation with molecular oxygen. Science 2021, 372, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]