

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease where muscle weakness and neuromuscular junction (NMJ) denervation precede motor neuron cell death. Although acetylcholine is the canonical neurotransmitter at the mammalian NMJ synapse, glutamate has recently been identified as a critical neurotransmitter for NMJ development and maintenance. One source of glutamate is through the catabolism of N-acetyl-aspartyl-glutamate (NAAG), which is found in mM concentrations in mammalian motoneurons, where it is released upon stimulation and hydrolyzed to glutamate by the glial enzyme glutamate carboxypeptidase II (GCPII). Using the SOD1G93A model of ALS, we found an almost fourfold elevation of GCPII enzymatic activity in SOD1G93A versus WT muscle and a robust increase in GCPII expression which was specifically associated with activated macrophages infiltrating the muscle. 2-(Phosphonomethyl)pentanedioic acid (2PMPA) is a potent GCPII inhibitor which robustly blocks glutamate release from NAAG but is highly polar with limited tissue penetration. To improve this, we covalently attached 2PMPA to a hydroxyl polyamidoamine (PAMAM-G4-OH) dendrimer delivery system (D-2PMPA) which is known to target activated macrophages in affected tissues. Systemic D-2PMPA therapy (20 mg/kg 2PMPA equivalent; IP 2 × /week) was found to localize in muscle macrophages in SOD1G93A mice and completely normalize the enhanced GCPII activity. Although no changes in body weight or survival were observed, D-2PMPA significantly improved grip strength and inhibited the loss of NMJ innervation in the gastrocnemius muscles. Our finding that inhibiting elevated GCPII activity in SOD1G93A muscle can prolong muscle function and delay NMJ denervation may have early therapeutic implications for ALS patients.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-021-01159-7.
Keywords: Amyotrophic lateral sclerosis, Glutamate carboxypeptidase II (GCPII), N-acetyl-aspartyl-glutamate (NAAG), mGluR3, Glutamate, NMDA, Neuroinflammation, Dendrimer, Neuromuscular junction (NJM)
Introduction
Amyotrophic lateral sclerosis (ALS) is a debilitating, fatal neurodegenerative disease characterized by progressive muscle weakness, eventually leading to paralysis and ultimately death [1]. Only two drugs are approved for use in ALS patients, riluzole and edaravone [2], both providing modest benefit. ALS involves both peripheral nervous system (PNS) and central nervous system (CNS) degenerations, where the earliest detectable signs of degeneration, in both humans and mouse models, occur in the periphery with motor axons degenerating before neuronal cell death [3–6]; however, whether this is a result of muscle-driven pathology or from the neurons themselves is an area of active debate.
The canonical neurotransmitter at the mammalian neuromuscular junction (NMJ) is acetylcholine (ACh). More recently, glutamate has been described as being critically involved in developmental NMJ synaptic pruning [7]. One source of glutamate is through the catabolism of the abundant neuropeptide N-acetyl-aspartyl-glutamate (NAAG) by glutamate carboxypeptidase II (GCPII) to N-acetyl-aspartate (NAA) and glutamate [8, 9]. Intact NAAG has been described as an endogenous agonist at mGluR3 [10]. The liberated glutamate can activate multiple excitatory receptors including N-methyl-d-aspartate (NMDA) receptors [11]. Glutamate’s role during developmental plasticity in the CNS is well documented [12]; however, its role in the mammalian PNS is less well characterized. Under basal conditions, GCPII is expressed peripherally on perisynaptic Schwann cells [7, 13]. During muscle development, multiply-innervated NMJs are pruned to be singly innervated. This process was attenuated with either GCPII inhibition or NMDA receptor antagonism in mice [7], suggesting NMJ glutamate influences development. However, the role of GCPII and glutamate at the diseased adult NMJ has not been explored.
2-(Phosphonomethyl)pentanedioic acid (2PMPA) is a potent GCPII inhibitor (Ki = 0.3 nM) [14] that has shown efficacy in over thirty in vivo preclinical models of neurological diseases wherein excess glutamate is presumed pathogenic [15, 16]. Despite preclinical successes, 2PMPA has not been developed for clinical use due to poor pharmacokinetics including short half-life (< 1 h), low oral bioavailability (F < 2%), and poor brain penetration (AUCbrain/AUCplasma < 0.02) [17]. These properties necessitate high peripheral doses (≥ 100 mg/kg) or direct brain injection to observe efficacy [16]. The only GCPII inhibitor identified which progressed to clinical trials was 2-(3-mercaptopropyl)pentanedioic acid (2-MPPA; also termed GPI5693). Although well tolerated in human volunteers [18], its chemical instability as well as concerns related to thiol-based immune toxicities terminated its development [19, 20]. Therefore, identifying a GCPII inhibitor or inhibitor conjugate which would enable clinical translation is of paramount importance.
Systemically administered generation 4 hydroxyl PAMAM dendrimers (PAMAM-G4-OH) can penetrate tissues and be preferentially engulfed by activated immune cells. This intrinsic targeting ability has shown tremendous promise for targeted drug delivery in multiple preclinical large and small animal models (reviewed in [21, 22]). When small molecule drugs are conjugated to PAMAM-G4-OH dendrimers, they exhibit enhanced efficacy at much lower doses with less toxicity compared to their respective free drug. These promising results have led to the initiation of two recent PAMAM-G4-OH dendrimer-drug clinical studies (NCT03500627; NCT04458298). Employing this strategy to circumvent the pharmacokinetic liabilities of 2PMPA, we covalently attached 2PMPA to PAMAM-G4-OH dendrimers (D-2PMPA) using a versatile and efficient click chemistry approach. We hypothesized that dendrimer-enabled targeted delivery of 2PMPA to activated immune cells within sites of injury would enhance its efficacy and decrease the required dose.
In this report, we describe, for the first time, robust upregulation of GCPII activity and expression in activated infiltrating macrophages in adult muscle of SOD1G93A mice. Using a dendrimer-conjugated potent and selective GCPII inhibitor, we demonstrate that targeted inhibition of this upregulated activity could significantly delay neuromuscular junction denervation and loss of muscle function.
Methods
Synthesis of D-2PMPA Conjugate
The detailed synthetic protocol for D-2PMPA is published elsewhere [23]. In brief, the terminal hydroxyl groups of PAMAM-G4-OH dendrimers were partially reacted with hexynoic acid using 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDC) and 4(dimethylamino)pyridine (DMAP) coupling reaction at room temperature for 24 h to create ~ 11 alkyne groups. The hexyne-terminating dendrimer was subsequently purified using dialysis against dimethylformamide (DMF) followed by water. The aqueous solution was then lyophilized to afford hexyne-terminating dendrimers. With regard to 2PMPA, the inhibitor was reacted with azido-PEG-11-alcohol using EDC/DMAP reaction at room temperature for 12 h. PMPA-PEG-azide was then purified using preparative HPLC to obtain the PMPA-PEG-azide fraction with modification at γ-carboxylate. Hexyne-terminating dendrimer was reacted with PMPA-PEG-azide using copper (I) catalyzed alkyne-azide (CuAAC) click reaction in the presence of a catalytic amount of copper sulfate pentahydrate and sodium ascorbate in a microwave reactor for 8 h at 50 °C. Upon completion, the trace amount of copper was removed by performing dialysis against EDTA followed by water dialysis to obtain D-2PMPA as final product as white solid.
Synthesis of Cyanine 5-D-2PMPA
The detailed synthetic protocol for cyanine 5 (Cy5)-D-2PMPA is published elsewhere [23]. In brief, the synthesis of Cy5-D-2PMPA was achieved by first synthesizing a trifunctional dendrimer using our previously published protocol [24]. The alkyne groups in the trifunctional dendrimers were reacted with PMPA-PEG-azide using CuAAC click reaction as described above. The resulting product was then reacted with Cy5-monoNHS ester at pH 7.5 using a previously published protocol [25] to obtain Cy5-D-2PMPA. All the intermediates and final conjugates were characterized using 1H NMR, mass spectroscopy, HPLC, and dynamic light scattering using methods we have described previously [25–27].
Animals
All experiments and animal care carried out in these studies were completed in accordance with the Johns Hopkins University Animal Care and Use Committee guidelines. Male mice had a more severe disease burden, and for this reason, male and female data were analyzed independently. SOD1G93A transgenic mice (B6.Cg-Tg(SOD1*G93A)1Gur/J) [28] and C57BL/6 J WT littermates were obtained from Jackson Labs (Strain #004,435; Bar Harbor, Maine, USA). This particular model was selected as the C57BL6/6 J background has a milder disease progression with prolonged survival (157.1 ± 9.3 days) compared with the B6SJL mixed background (128.9 ± 9.1 days; Jackson Labs strain #002,726) [29]. Animals were allowed to age to 12 weeks, a time when the earliest clinical phenotypes begin to manifest, before initiating treatment. Mice were randomly sorted into a vehicle (empty dendrimer; total n = 38; n = 18 female; n = 20 male) or treatment (20 mg/kg D-2PMPA; total n = 40; n = 19 female; n = 21 male) group and intraperitoneally dosed 2 times per week. This dosing scheme was selected based on previous studies done using dendrimer-conjugated molecules, including D-2PMPA [23, 30].
Grip Strength
Animals were subjected to grip strength testing before treatment to assess baseline measurements and every 2 weeks following treatment. Grip strength was determined as previously described [31]. Briefly, mice were allowed to grab a rod with their forelimbs for stability while their hind limbs were grabbing a bar attached to a force meter (DFIS-2 Series Digital Force Gauge; Columbus Instruments, OH, USA). Mice were then pulled away from the bar until it was released, and the measured force was recorded. Three separate trials where the mice properly gripped the bar, defined as both feet equally clasping the rod, were recorded, and the average of the three trials was recorded. Mice were allowed to rest for several minutes between each trial.
Survival Analysis
Following the completion of the grip strength analysis, mice were continually dosed until they were euthanized (female vehicle n = 15, D-2PMPA n = 16; male vehicle n = 17, D-2PMPA n = 18). At the end stage of life following the onset of paralysis, animals were provided with hydragel and easy access to food. Animals were euthanized via CO2 inhalation when they could no longer right themselves after 30 s following being placed on their sides. The age of each mouse was recorded at this time as the age of death. Logrank survival analysis was completed using GraphPad Prism software V 9.1 (GraphPad Software, San Diego, CA, USA).
Mouse Perfusion
Mice were deeply anesthetized with isoflurane (Piramal Enterprises Ltd., India) before being transcardially perfused with 1 × PBS (Life Technologies, Carlsbad, CA, USA) followed by 2% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA, USA). Gastrocnemius muscles and the lumbar region of the spinal cord (L1–L5) were carefully dissected, post-fixed in 2% PFA for up to 24 h at 4 °C, and transferred to 1 × PBS for long-term storage at 4 °C. PFA (2%) was used to reduce background fluorescence at the green wavelength for imaging studies.
GCPII Western Blot Analysis
Fourteen-week-old SOD1G93A and WT littermates were deeply anesthetized with isoflurane before being transcardially perfused with 1 × ice-cold PBS. Lumbar spinal cord and gastrocnemius muscle tissue were quickly dissected and snap frozen with dry ice before being stored at −80 °C. The tissue was mechanically homogenized using a frosted glass mortar and pestle in ice-cold 1 × RIPA buffer (Millipore-Sigma) with cOmplete protease inhibitor cocktail (Millipore-Sigma). The tissue lysate was centrifuged at 12,000 rpm for 15 min, and the supernatant was collected and aliquoted before being frozen on dry ice and stored at −80 °C. Total protein concentrations were determined using Detergent Compatible Protein Assay as per the manufacturer’s instructions (Bio-Rad, CA, USA). Twenty micrograms of protein was loaded into each well of a NuPAGE 4–12% Bis–Tris Gel (Invitrogen) and then transferred onto an iBlot 2 PVDF membrane using an iBlot 2 apparatus (Thermo Fisher Scientific, Waltham, MA, USA). Blots were blocked in 5% nonfat dry milk for 1 h at room temperature before being incubated at 4 °C overnight with a GCPII primary antibody (Novus Biologicals, CO, USA). The blots were incubated with an appropriate secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. The membranes were then incubated with Clarity Western ECL substrate (Bio-Rad) and imaged using a western blot imager (Bio-Rad). Loading controls were determined using an anti-GAPDH HRP-conjugated antibody. GCPII protein band levels were normalized to GAPDH protein band levels using ImageJ software.
GCPII Localization Studies
Symptomatic 14-week-old SOD1G93A mice were deeply anesthetized with isoflurane and transcardially perfused with 1 × ice-cold PBS followed by 2% PFA. Gastrocnemius muscles were dissected out and post-fixed for 1 h at room temperature in 2% PFA before being cryoprotected in 30% sucrose for 48 h at 4 °C. The muscles were embedded in Tissue-Tek O.C.T. medium (Sakura, CA, USA) and snap frozen in 2-methylbutane (Thermo Fisher Scientific) on dry ice. Tissue was then cryosectioned using a cryostat (Microm HM 505E, International Medical Equipment, MI, USA) at 50 µm onto gelatin-coated glass slides. The muscle sections were then blocked in 5% goat serum in 0.1% Triton X-100 + PBS for 1 h before being incubated at 4 °C overnight with primary antibodies against GCPII (GCP-04, Novus Biologicals) and CD68 (Cell Signaling Technology, MA, USA). To reduce nonspecific binding, an anti-mouse IgG1 secondary antibody (Life Technologies) was used for the GCPII staining and an appropriate anti-rat secondary was used for CD68 staining together with an α-bungarotoxin (α-BTX) Alexa Fluor 647 (Invitrogen) for 1 h at room temperature. Sections were cover-slipped using prolong glass antifade mountant (Life Technologies) before being imaged on a Zeiss LSM800 confocal microscope at 40 × . Images were process using Zen Blue software (Zeiss).
Cy5-D-2PMPA Imaging
Fourteen-week-old SOD1G93A mice were given 2 doses of 55 mg/kg Cy5-D-2PMPA, 4 days apart. Twenty-four hours following the final dose, mice were transcardially perfused using ice-cold 1 × PBS. Gastrocnemius muscles were dissected and post-fixed in 2% PFA for 48 h at 4 °C. The muscles were then cryoprotected in 30% sucrose (Thermo Fisher Scientific) for 48 h and stored at −80 °C for a long term. They were then sectioned at a thickness of 50 µm using a cryostat (International Medical Equipment). The sections were permeabilized and blocked for 1 h at room temperature in 0.1% Triton (Sigma-Aldrich)/5% goat serum (Jackson labs) in 1 × PBS. Sections were stained overnight at 4 °C with primary antibodies against CD68 (Cell Signaling Technology, MA, USA). Sections were then stained with the appropriate secondary antibodies for 1 h at room temperature, stained with Hoechst 33,342 (Thermo Fisher Scientific) for 10 min at room temperature, before being cover-slipped with prolong glass antifade mountant (Life Technologies). The sections were then imaged at 40 × using a LSM800 confocal microscope (Zeiss, Germany). Images were processed using Zen Blue software (Zeiss).
GCPII Enzymatic Activity Assay
GCPII activity measurements were carried out based on a modification of previously published protocols [8, 32]. Briefly, spinal cords and gastrocnemius muscles were homogenized in ice-cold Tris buffer (40 mM, pH 7.5) containing protease inhibitors (Roche, Complete Protease Inhibitor Cocktail) using Biomasher II (seven pulses of 10-s duration on ice, 10 s between pulses) and then sonicated using Kontes Micro Ultrasonic Cell Disrupter (three pulses of 15-s duration on ice, 30 s between pulses). The resulting homogenates were spun down (16,000 × g for 2 min at 4 °C), and the supernatants were collected for both GCPII activity and total protein analysis. GCPII reactions were carried out for 1-h minimum at 37 °C, in 50 µl reaction volumes, in 96-well microplates, and were initiated upon the addition of prewarmed (37 °C) cobalt chloride (1 mM) and [3H]-NAAG (0.02 µM, 48.6 mCi/µmol). At the end of the reaction period, the assay was terminated with ice-cold sodium phosphate buffer (100 mM, pH 7) containing EDTA (1 mM). Ninety-six-well spin columns packed with anion exchange resin (Bio-Rad, AG® 1-X8 Resin, 200–400 mesh, formate form) were used to separate the substrate and the reaction product. [3H]-Glutamate, the reaction product, was eluted with 1 M formic acid and analyzed for radioactivity using Perkin Elmer TopCount instrument in conjunction with their 96-well LumaPlates. Finally, total protein measurements were assessed as per the manufacturer’s instructions using Bio-Rad DC Protein Assay kit and data were presented as fmol/mg/h.
Gastrocnemius Innervation
Fixed gastrocnemius muscles were cryoprotected in 30% sucrose in 1 × PBS for 48 h. The gastrocnemius muscles were then frozen into Tissue-Tek O.C.T. (Electron Microscopy Sciences) before being sectioned at 50 µm of thickness on a cryostat (International Medical Equipment). Tissue sections were treated similarly to the D-Cy5 imaging; however, to visualize axons, primary antibodies against neurofilament (NF) H (1:200; Millipore) and synaptic vesicle 2 (SV2; 1:50; Developmental Studies Hybridoma Bank) were used. α-Bungarotoxin conjugated to Alexa Fluor 647 (α-BTX) was used to label NMJs. α-BTX and the corresponding Alexa Fluor 488–conjugated secondary antibodies for NF and SV2 were incubated for 1 h at room temperature before mounting and cover-slipping the slides with prolong diamond mounting reagent. A Zeiss LSM800 confocal microscope was used to image the sections. In order to avoid the same NMJ being scored, every 6th section was imaged. To maintain blinded innervation analysis, treatment status of the slides was unknown at the time of imaging and NMJs were selected randomly using α-BTX staining without looking at the axonal staining. Innervated NMJs were scored as having at least half of the NF and SV2 staining overlapping with the α-BTX staining.
Motor Neuron Quantification
The lumbar region of the spinal cords were cryoprotected in 30% sucrose for 48 h before being embedded in Tissue-Tek O.C.T. and then frozen and stored at −80 °C. The tissue was sectioned at 30 µm thickness using a cryostat (International Medical Equipment). The sections were similarly stained as the D-Cy5 imaging, and only the primary antibody used was against NeuN (1:200; Abcam; Cambridge, UK). Following secondary antibody incubations, slides were cover-slipped with prolong diamond mounting reagent plus DAPI (Life Technologies). Every 6th section was imaged using a Zeiss LSM800 confocal microscope, and both ventral horns were imaged. Motor neurons were counted using Zen Blue software (Zeiss) by manually tracing the outline of the cell bodies within the ventral horn. Only those with a cell body area > 250 µm2 and a large nucleus with a prominent nucleolus were scored [33, 34]. Motor neurons with large vacuoles were excluded. The average number of motor neurons present per ventral horn was quantified.
RNA-Seq Analysis
Samples were sent to Qiagen Genomic Services (Frederick, MD, USA) for RNA isolation, quality control (QC), library prep, and UPX 3′ transcriptome sequencing. RNA was isolated from 10 mg of gastrocnemius muscle using the RNeasy Plus Universal (Qiagen) according to the manufacturer’s instructions. The library preparation was done using the QIAseq UPX 3′ Transcriptome Kit (Qiagen). A total of 10 ng purified RNA was converted into complementary DNA (cDNA) NGS libraries. Library preparation was quality-controlled using capillary electrophoresis (Agilent DNA 7500 Chip). Based on quality of the inserts and the concentration measurements, the libraries were pooled in equimolar ratios and quantified using qPCR. The library pool was then sequenced on a NextSeq (Illumina, Inc.) sequencing instrument with 100 bp read length for read 1 and 27 bp for read2. Raw data was de-multiplexed, and FASTQ files for each sample were generated using the bcl2fastq software (Illumina, Inc.). The reads were then mapped to the Mouse genome GRCm38 v. 80 and annotated using the ENSEMBL GRCm38 v. 86 messenger RNA (mRNA) annotation. The “Empirical analysis of DGE” algorithm of the CLC Genomics Workbench 20.0.4 (https://digitalinsights.qiagen.com) was used for differential expression analysis with default settings. It is an implementation of the “Exact Test” for two-group comparisons developed by Robinson and Smyth [35] and incorporated in the edgeR Bioconductor package [36].
RNA Extraction and RT-qPCR Analysis
Total RNA was isolated from the gastrocnemius muscle of SOD1G93A mice using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s guidelines. RNA concentration was determined using a NanoDrop ND1000 spectrophotometer. Expression of Tnnc1, Tnni1, and Tpm3 on an mRNA level was confirmed by RT-qPCR analysis using our published protocols with some modifications [37, 38]. In brief, cDNA synthesis was performed using SuperScript® III CellsDirect™ cDNA Synthesis Kit (Life Technologies, Grand Island, NY, USA) from total RNA. Real-time PCR reaction contained diluted cDNA from the synthesis reaction and 200 nM specific forward and reverse TaqMan primers specific to targeted genes (Assay IDs for Tnnc1, Tnni1, and Tpm3 are Mm00437111_m1, Mm00502426_m1, and Mm04336671_g1, respectively) (Applied Biosystems, Foster City, CA, USA). GAPDH was used to normalize the expression data. The real-time PCR reaction and measurement were carried out with Applied Biosystems PRISM 7900 HT. PCR reaction conditions were as follows: 50 °C, 2 min; 95 °C, 2 min; and 50 cycles of 95 °C, 1 s and 60 °C, 20 s. Quantification was performed using the ΔCt method (2−ΔΔCt).
Statistical Analysis
Cohort sizes were determined using power calculations and previous experience with the assays. Blinded analysis was ensured by randomly assigning each mouse to a treatment group (vehicle or D-2PMPA), and the specific treatment condition of each mouse was unknown at the time of data collection for all experiments performed. All statistical analyses between two data sets were performed using a two-tailed Student’s t test. Data collected over time were subject to a mixed-effects ANOVA analysis. Survival outcomes were determined using logrank survival analysis. All statistical analysis was performed using GraphPad Prism software, and any value of p < 0.05 was scored as statistically significant.
Results
GCPII Expression Is Elevated in SOD1G93A Muscle
While GCPII expression has been well-described in the central and peripheral nervous systems of normal wild-type mice, its expression in the SOD1G93A mouse model of ALS has not been explored. Since ALS has both central and peripheral components to the disease, we determined the relative protein expression in both the spinal cord and muscle of 14-week-old symptomatic mice and age-matched controls (Fig. 1). We observed a robust increase in GCPII protein levels in the SOD1G93A gastrocnemius muscle compared with those in the WT gastrocnemius muscle (Fig. 1A, B; WT = 0.31 ± 0.041 relative intensity, SOD1G93A = 0.96 ± 0.17 relative intensity; p = 0.019; n = 3/group). In contrast, there were no differences in the relative protein expression levels of GCPII in the lumbar region of the spinal cord between WT and SOD1G93A mice (Fig. 1C, D; WT = 0.53 ± 0.031 GCPII/GAPDH intensity, SOD1G93A = 0.56 ± 0.034 GCPII/GAPDH intensity; p = 0.76; n = 3/group).
Fig. 1.

GCPII expression is elevated in SOD1G93A muscle. A Western blot from WT and SOD1G93A gastrocnemius muscles. GAPDH shown as a loading control. B Quantification of the blot shown in A. n = 3/group. C Western blot from the lumbar region of the spinal cord from WT and SOD1G93A mice. GAPDH is shown as a loading control. D Quantification of the blot from C. n = 3/group. Bars represent mean ± SEM. Black bars represent WT values. Blue bars represent SOD1G93A values. *p < 0.05
GCPII Expression Is Selectively Enhanced in Infiltrating Macrophages in SOD1G93A Muscle
We next explored which cell types may be responsible for the dramatic increase in GCPII protein expression in the SOD1G93A gastrocnemius muscle. Although GCPII expression has been previously reported at the neonatal NMJ [7], its expression has not been characterized in the adult mammalian muscle. In the gastrocnemius muscle of WT mice, we observed very few CD68 + macrophages (Fig. 2A); in contrast, there was a large influx of CD68 + macrophages in the SOD1G93A gastrocnemius muscle (Fig. 2B). Of these infiltrating macrophages, highly activated and foamy macrophages had the most robust GCPII staining (Fig. 2C–E). We also observed punctate GCPII staining at the NMJs (Fig. 2F–H); this staining was similar between WT and SOD1G93A mice (Fig. 2I–K). These data demonstrate that GCPII is expressed in adult murine NMJs and supports infiltrating macrophages as the likely source of elevated GCPII expression in the gastrocnemius muscles of SOD1G93A mice.
Fig. 2.
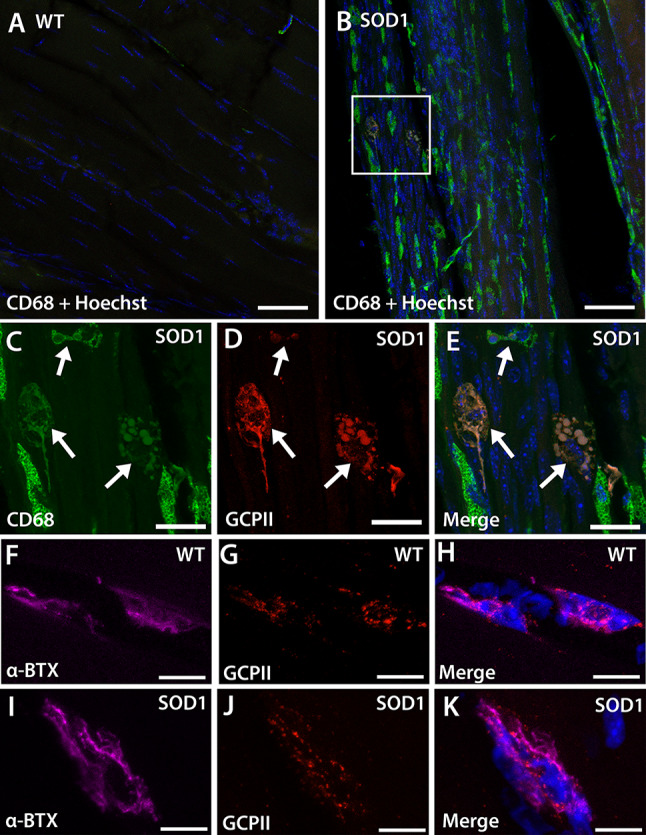
GCPII expression is localized to macrophages in SOD1G93A gastrocnemius muscle. A, B Representative images demonstrating that WT muscle (A) has limited CD68 + macrophage staining while SOD1G93A muscle (B) has a large influx of CD68 + macrophages. Scale bar, 50 µm. C–E High magnification of the boxed area from SOD1G93A muscle showing CD68 + macrophages (C, arrows) with GCPII expression (D, arrows). Merged image with Hoechst-stained nuclei (blue) shown in E. Scale bar, 20 µm. F, H WT NMJs stained with α-bungarotoxin (F and H, α-BTX, purple) have GCPII staining colocalization (G and H, red). I–K SOD1G93A NMJs stained with α-bungarotoxin (I and K, α-BTX, purple) also have GCPII staining colocalization (J and K, red). Nuclei stained blue. Scale bar, 10 µm
Synthesis and Characterization of a Dendrimer-2PMPA Conjugate
After determining that the increased GCPII muscle expression was localized to activated infiltrating macrophages, we rationalized the use of hydroxyl PAMAM dendrimers known to target small molecules drugs to activated immune cells [21, 22]. The detailed synthesis and characterization is described elsewhere [23]; however, we have briefly summarized it here. 2PMPA was conjugated to the surface of PAMAM-G4-OH through flexible and biocompatible polyethylene glycol (PEG) linkers via enzyme-sensitive ester linkages (Fig. 3A). We used a highly efficient copper(I)-catalyzed click chemistry (CuAAC) tool to carry out the drug conjugation on the periphery of the dendrimer [39]. Briefly, the hydroxyl groups on the surface of PAMAM-OH were partially modified to attach ~ 11 alkyne linkers. 2PMPA was modified at the γ-position to attach a short orthogonal PEG linker with azide-terminating group. The PEG was used as a linker to provide flexibility for the drug to interact with the GCPII enzyme. The alkyne-terminating dendrimer and azide-terminating drug were then reacted with CuAAC to achieve the D-2PMPA conjugate. In order to confirm that 2PMPA conjugation did not alter the dendrimer’s inherent potential of reactive macrophage targeting, we further attached Cy5 dye as an imaging agent on the dendrimer surface and constructed a fluorescently labeled Cy5-D-2PMPA conjugate using a similar synthetic approach (Fig. 3B). The structure and drug loading of the D-2PMPA conjugate were characterized using 1H NMR spectroscopy which revealed the attachment of ~ 11 molecules of 2PMPA per dendrimer with a 10% w/w drug loading. We have previously demonstrated that the drug loading of less than 20% w/w does not affect the overall targeting properties of PAMAM-OH [40, 41]. D-2PMPA demonstrated a high purity of > 99% as analyzed by HPLC (Fig. 3C). The molecular weight assessment of D-2PMPA by MALDI-ToF agreed with the theoretical molecular weight of 22.5 kDa. The conjugate displayed a size of ~4.7 nm, slightly more than the size of starting dendrimer (4 nm) as expected with a nearly neutral zeta potential (−2.07 mV) (Fig. 3C). The small size and neutral charge are the key factors contributing to the targeting ability of these dendrimers [42, 43].
Fig. 3.
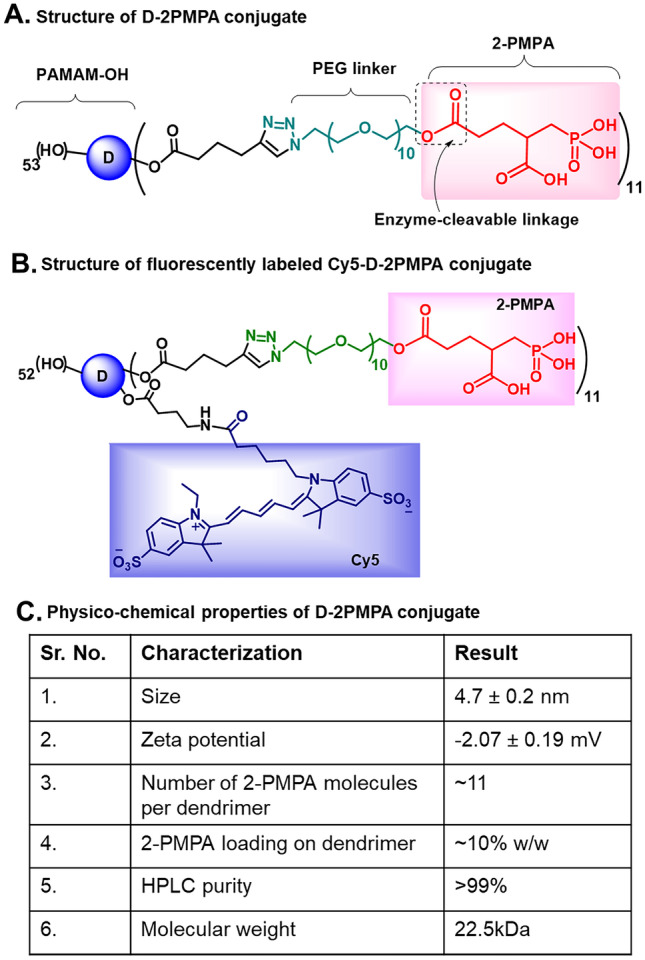
The structures and physicochemical characterization of dendrimer-2PMPA conjugates. A The structure of D-2PMPA conjugate having 2PMPA covalently attached to the dendrimer surface through PEG linkers via enzyme-sensitive ester linkages. B The structure of fluorescently labeled Cy5-D-2PMPA conjugate with the attachment of both the drug and the fluorophore for imaging purposes. C Table showing the physicochemical characteristics of D-2PMPA
Enzyme Inhibition and Fluorescent Imaging Confirmed D-2PMPA Targeting to Activated Macrophages in SOD1G93A Muscle
Before carrying out efficacy studies in SOD1G93A mice, we first ensured target engagement in the gastrocnemius muscle, employing both enzymatic activity and fluorescent imaging. GCPII activity in SOD1G93A muscle was increased 3.7-fold compared to WT muscle. D-2PMPA treatment of the SOD1G93A mice completely normalized this elevation (Fig. 4A; WT = 2090 ± 262 fmol/mg/h, vehicle = 7648 ± 1078 fmol/mg/h, D-2PMPA = 2723 ± 468 fmol/mg/h; WT vs. vehicle p = 0.0003, vehicle vs. D-2PMPA p = 0.0008, WT vs. D-2PMPA p = 0.80; n = 5/group). We also verified that the D-2PMPA conjugate was targeted to activated macrophages in the gastrocnemius muscle using fluorescent imaging. Twenty-four hours post dose, we observed punctate Cy5 signal specifically localizing with CD68 + -staining macrophages in the gastrocnemius muscles of SOD1G93A mice (Fig. 4E–G). There was no Cy5-D-2PMPA signal localizing with macrophages in WT gastrocnemius muscles (Fig. 4B–D). These data confirmed that D-2PMPA was being targeted to activated macrophages in SOD1G93A gastrocnemius muscles.
Fig. 4.

Cy5-labeled D-2PMPA is targeted to macrophages in SOD1G93A gastrocnemius muscle. A GCPII activity in gastrocnemius muscles excised from WT and SOD1G93A mice treated with vehicle or D-2PMPA for 2 weeks. Bars represent mean ± SEM. Black bars represent WT values. Blue bars represent SOD1G93A vehicle–treated mouse values. Red bars represent SOD1G93A D-2PMPA–treated mouse values. ***p < 0.001. n = 5/group. B, D Representative images from gastrocnemius muscles in WT mice showing no Cy5-D-2PMPA staining (B) localizing with CD68 + macrophages (C). Merged image shown in D with nuclei stained blue. E, G Representative images from gastrocnemius muscles in SOD1G93A mice showing Cy5-D-2PMPA staining (B) localizing with CD68 + macrophages (C). Merged image shown in D with nuclei stained blue. Scale bar, 10 µm
D-2PMPA Treatment Did Not Improve Weight Loss or Survival in SOD1G93A Mice
After confirming GCPII activity was inhibited with D-2PMPA treatment in SOD1G93A muscle, we next determined whether there were any treatment effects on disease outcome in the SOD1G93A mouse. Body weight was measured weekly with no treatment effect observed (Fig. 5A, B; male p = 0.501, female p = 0.76). Additionally, no survival benefit was observed with D-2PMPA treatment (Fig. 5C, D; male p = 0.42, female p = 0.55).
Fig. 5.

D-2PMPA treatment does not improve disease outcome in SOD1G93A mice. A, B Weekly body weight is not changed in male mice (A) or female mice (B) following treatment. Points represent mean ± SEM. C, D Overall survival is not altered in male mice (C) or female mice (D) following treatment. Blue points represent SOD1G93A vehicle–treated mouse values. Red points represent SOD1G93A D-2PMPA–treated mouse values. E, J Representative images of lumbar spinal cord ventral horns. Male WT mice (E) have more dense neuronal staining than vehicle (F) or D-2PMPA mice (G). Female WT mice (H) also have more dense neuronal staining than vehicle (I) or D-2PMPA mice (J). Neurons labeled red with NeuN and nuclei labeled blue with Hoechst. Arrows pointing to examples of α motor neurons. Scale bar, 100 µm. K, L Quantification of the number of α motor neurons (cell body area > 250 µm2) per ventral horn from males (K) and females (L). Bars represent mean ± SEM. Black bars represent WT values. Blue bars represent SOD1G93A vehicle–treated mouse values. Red bars represent SOD1G93A D-2PMPA–treated mouse values. A–D Male vehicle n = 20, D-2PMPA n = 21; female vehicle n = 18, D-2PMPA n = 19. I Male WT n = 4; female WT n = 4; male vehicle n = 5; male D-2PMPA n = 6; female vehicle n = 4; female D-2PMPA n = 4. *p < 0.05. **p < 0.01
We also evaluated motor neuron survival in the lumbar region of the spinal cord in SOD1G93A mice that were treated for either 4 weeks (males) or 6 weeks (females), to account for the 2-week delay in female symptom onset. There were no observable differences in the number or morphology of the large α motor neurons in either the males (Fig. 5F, G, K; vehicle = 22.13 ± 0.904, n = 4; D-2PMPA = 22.69 ± 1.123, n = 4; p = 0.921) or the females (Fig. 5I, J, L; vehicle = 22.71 ± 0.750, n = 5; D-2PMPA = 21.71 ± 1.210, n = 6; p = 0.733). There was a small, albeit significant, reduction in large α motor neurons in both vehicle-treated and D-2PMPA–treated SOD1G93A mice compared with WT mice for both males (Fig. 5K; WT = 28.51 ± 1.171, n = 3; compared to vehicle p = 0.0095; compared to D-2PMPA p = 0.0137) and females (Fig. 5L; WT = 27.58 ± 0.704, n = 3; compared to vehicle p = 0.0211; compared to D-2PMPA p = 0.0079). These results were unlikely to have been due to the drug not reaching the spinal cord as we observed Cy5-D-2PMPA signal in the spinal cord of SOD1G93A mice (Supp. Fig. 1). Further biodistribution was not studied as several studies in animal models, including mice, have demonstrated low levels of dendrimer accumulation up to 72 h post-dose in the liver and spleen, with the highest off-target accumulation observed in the kidney as that is the route of elimination [44–46]. Taken together, these data indicate that there was no motor neuron protection or overall survival benefit with D-2PMPA treatment in SOD1G93A mice.
D-2PMPA Selectively Slowed Progression of Limb Weakness and NMJ Denervation in SOD1G93A Mice
Given the selective increase in GCPII expression in SOD1G93A muscle, we also investigated whether D-2PMPA treatment affected muscle strength. D-2PMPA–treated male mice had significantly higher grip strength measurements over an 8-week test period (Fig. 6A; repeated measures ANOVA p = 0.0018; n = 20–21/group). Treated female mice also had significantly elevated grip strength measurements over a 10-week treatment period compared to vehicle-treated mice (Fig. 6E; repeated measures ANOVA p = 0.015; n = 18–19/group).
Fig. 6.
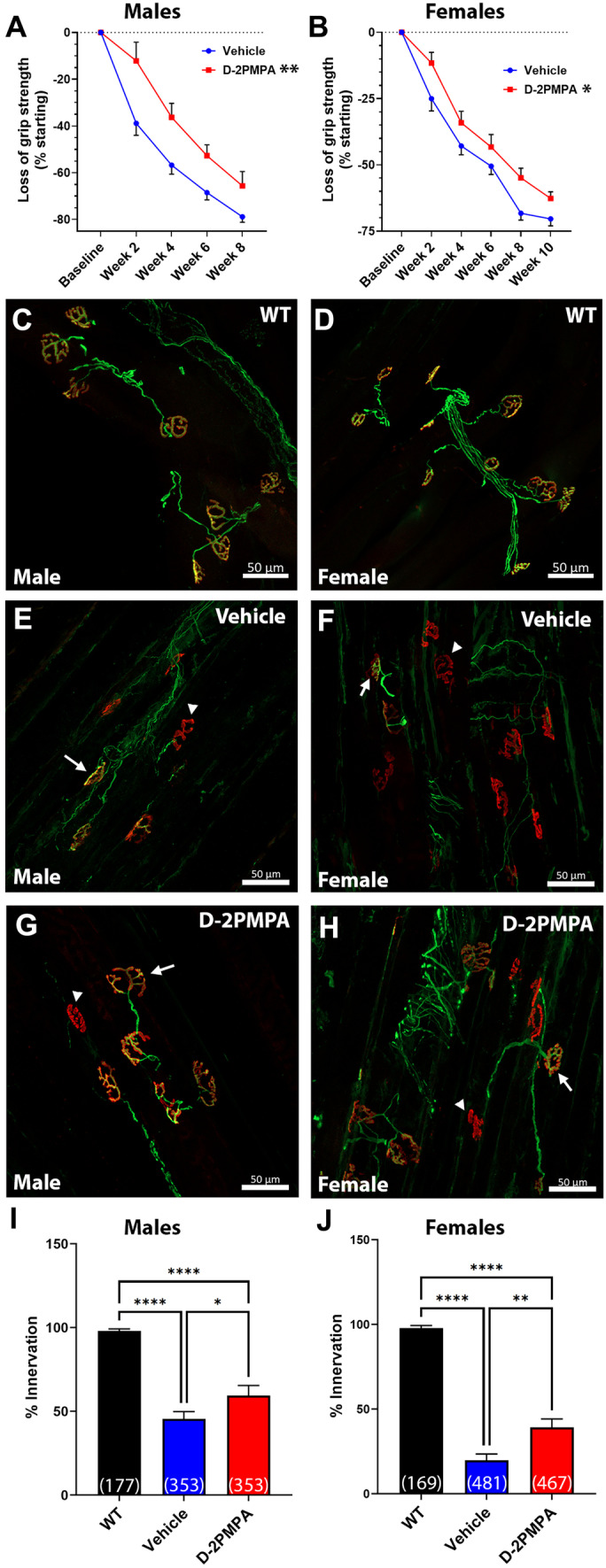
D-2PMPA treatment selectively improves muscle function and innervation in SOD1G93A mice. A, B Grip strength data from male (A) and female (B) mice. D-2PMPA–treated mice have significantly higher grip strength over 8 weeks or 10 weeks, respectively. Vehicle male n = 20, female n = 18; D-2PMPA male n = 21, female n = 19. Points represent mean ± SEM. Blue points represent SOD1G93A vehicle–treated mouse values. Red points represent SOD1G93A D-2PMPA–treated mouse values. C, H Representative images of WT (male C; female D), vehicle (male E; female F), and D-2PMPA (male G; female H)–treated SOD1G93A gastrocnemius muscle after 4 weeks (male) or 6 weeks (female). Axons stained green. NMJs stained red. Scale bar, 50 µm. Arrows indicate innervated NMJ. Arrowheads indicate denervated NMJs. I, J Quantification of the percentage of innervated NMJs from male (I) and female (J) mice. Bars represent mean ± SEM. Black bars represent WT values. Blue bars represent SOD1G93A vehicle–treated mouse values. Red bars represent SOD1G93A D-2PMPA–treated mouse values. Numbers in parenthesis indicate the total number of NMJs. N = 6/group. *p < 0.05. **p < 0.01. ****p < 0.0001
We then determined whether these improvements were reflected in gastrocnemius muscle innervation. Muscle innervation was determined after 4 weeks on drug for males and 6 weeks for females to correspond with the 2-week delay in symptom onset. In both male and female mice, there were observable differences in the innervation of the NMJs, with vehicle-treated SOD1G93A mice having more denervated NMJs than WT mice (Fig. 6B–C, F, G). Quantification of the percentage of total NMJs that were either fully or partially innervated revealed that D-2PMPA–treated male SOD1G93A mice had significantly more innervated NMJs versus vehicle-treated male SOD1G93A mice (Fig. 6D; vehicle = 45.44 ± 4.35%; D-2PMPA = 59.41 ± 2.98%; p = 0.041; n = 6/group). This effect was even more pronounced in the D-2PMPA-treated female SOD1G93A mice where the percentage of innervated NMJs was almost double that of the vehicle-treated mice (Fig. 6H; vehicle = 19.80 ± 3.68%; D-2PMPA = 39.23 ± 4.96%; p = 0.0085; n = 6/group). These data strongly suggest that D-2PMPA treatment has a selective effect on reducing muscle function loss and denervation in SOD1G93A mice.
We then conducted RNA-seq analysis on muscle from vehicle-treated and D-2PMPA–treated animals to determine if there were gene expression changes. We observed that several genes associated with slow twitch muscle fiber contractility were significantly downregulated in D-2PMPA–treated mice (Supp. Fig. 2A; e.g., ATPase sarcoplasmic/endoplasmic reticulum Ca2+-transporting 2 (Atp2a2), myosin heavy chain 7 (Myh7), myosin light chain 2 (Myl2), troponin C1, slow skeletal and cardiac type (Tnnc1), troponin I1, slow skeletal type (Tnni1), and tropomyosin 3 (TPM3)). We conducted RT-qPCR on muscle from a subset of animals and observed a significant reduction in Tnni1 expression (Supp. Fig. 2B; 3.8-fold reduction; p = 0.03). TPM3 had a 2.4-fold reduction in mRNA expression; however, it did not reach significance (Supp. Fig. 2C; p = 0.162). Protein expression for Tnni1 and TPM3 trended to be decreased (1.2-fold and 1.3-fold, respectively) however neither reached significance (Supp. Fig. 2D, E). These results indicate there may be reduced muscle fiber type restructuring occurring in the D-2PMPA–treated animals from type II fast twitch to type I slow twitch; however, further studies are needed to confirm this.
Discussion
We discovered a robust increase in GCPII activity and expression specifically associated with infiltrating macrophages in the denervated SOD1G93A mouse gastrocnemius muscle. To test if this upregulated activity had functional consequence, we treated SOD1G93A mice with a hydroxyl PAMAM dendrimer-GCPII inhibitor conjugate known to target activated macrophage [21, 22]. As stated in the “Introduction,” despite being a very potent compound, 2PMPA suffers from poor pharmacokinetics and therefore has never been clinically developed. Clinical development of 2-MPPA [47, 48], the only orally bioavailable GCPII inhibitor with reasonable pharmacokinetics [18, 49], was halted due to its racemic nature, poor chemical stability, and toxic immune complexes formed during chronic GLP primate studies [18]. Importantly, toxicity was not due to the GCPII mechanism, but rather 2-MPPA’s thiol moiety, which is known to induce hypersensitivity reactions [19, 20]. Our strategy of conjugating 2PMPA to a dendrimer molecule enabled the use of a fivefold lower drug dose and 3.5-fold reduced frequent dosing regimen than had been previously reported with free 2PMPA (daily 100 mg/kg vs. twice weekly 20 mg/kg) [16].
The use of dendrimer-conjugated 2PMPA was also advantageous as it was able to selectively target the compound to areas of active inflammation. It has been reported that dendrimer conjugates do not accumulate in uninjured tissue (e.g., liver, spleen) and are actively cleared by the kidneys within 24 h [44–46]. We observed dendrimer targeting to activated macrophages in the muscle and microglia in the spinal cord. We report that chronic D-2PMPA treatment, initiated at the onset of symptoms, completely attenuated the elevated GCPII activity in the muscle and resulted in a selective behavioral delay in muscle strength loss and a significant preservation of muscle innervation. That we observed dendrimer targeting to the spinal cord with the absence of elevated GCPII expression, prolonged survival, or neuronal protection further supports our observation that GCPII inhibition has a selective effect on the NMJ.
While our study demonstrated a selective delay on muscle strength and innervation, a previous study examining GCPII inhibition in an ALS mouse observed significant improvements in survival and a delay in clinical symptom onset [50]. It is important to note that our study differs from the previous study in several key ways. First, the previous study utilized 2-MPPA instead of 2PMPA due to its superior pharmacokinetic profile. Unfortunately, as stated above, 2-MPPA’s clinical development was halted due to issues with its thiol moiety. Second, the previous study utilized the low-expressing SOD1G93A mouse model of ALS, which has a milder disease progression [51], versus our study utilizing the high-expressing C57Bl/6 SOD1G93A mouse model which has a more aggressive disease progression with an earlier onset and shorter median survival [52]. Additionally, we initiated our study at a time point closer to the onset of clinical symptoms as this more accurately reflects when patients would begin treatment. The previous study began treatment at ~37 days, much earlier than clinical disease onset. It is also important to note that we, and others, have observed sex differences in the time of disease onset and survival in ALS mouse models [29, 53, 54]. The previous study may have been underpowered as they did not indicate the sex and number of animals used in the study, which could significantly impact both disease onset and survival outcomes. Lastly, the disease outcomes used looked at overt behavioral clinical symptoms; treatment effects on muscle innervation were not evaluated. Our study directly measured muscle health by quantifying muscle strength and innervation, as well as quantifying the number of large α-motor neurons. We believe that these key differences could account for the discrepancies between the two studies. Despite these methodological differences, however, both studies observed significant muscle function improvements, highlighting the validity of our findings. Given its clinical use albeit with modest efficacy, it is important to compare these results with those of riluzole in the same mouse model. Riluzole was initially reported to prolong survival in the SOD1 at around 8% [55, 56]; however, these early studies lacked rigorousness [57]. In recent, more rigorous studies, riluzole was shown to have no significant effects in SOD1G93A mice [57–59]. That our study was able to replicate significant results in a portion of this earlier study, while others have been unsuccessful in replicating findings of an approved therapeutic, further supports our findings as valid.
While our study did observe a selective improvement in grip strength and NMJ innervation, it was limited in the methodologies used. Future studies examining single muscle tension and electrophysiological endpoints, together with a more extensive study of axonal sprouting and muscle fiber size, are warranted and are an area of active investigation.
This study underscores an interesting new role for GCPII in the periphery. Typically, GCPII expression is observed on astrocytes (CNS) [60] and perisynaptic Schwann cells (PNS) [61], with skeletal muscle having relatively low expression [62]. Under normal conditions, microglia, the most dominant immune cells in the brain, do not express GCPII. However, once these immune cells are activated via inflammatory injury, GCPII activity increases dramatically (> 100-fold) [63]. It has been well described that macrophages infiltrate muscle following axonal injury (reviewed in [64, 65]); however, their role in disease progression is complex. Typically, the early influx of macrophages in response to axonal injury in the nerve and muscle is thought to be helpful to clear out cellular debris while their chronic presence is most likely harmful [66, 67]. Macrophage influx specifically in the muscle tissue of SOD1G93A rats was found to destabilize the perisynaptic Schwann cells, which are critical for NMJ health. In contrast, reducing macrophage activation was shown to prolong the NMJ integrity, indicating that activated macrophages can be detrimental to perisynaptic Schwann cells [68].
One possible mechanism for infiltrating macrophages to exacerbate the axonal withdrawal is through the upregulation of GCPII activity and increased glutamate production. While ACh is the canonical neurotransmitter at the neuromuscular synapse, recently, it was demonstrated that glutamate also plays a critical role during neuromuscular synapse development [7]. Blocking NMDA receptors or inhibiting GCPII was shown to delay developmental synaptic pruning [7]. In the present study performed on adult mice, administering D-2PMPA and normalizing GCPII activity may have delayed neuromuscular synapse destabilization, resulting in prolonged innervation and muscle strength. This theory is supported by data in ALS patients showing that circulating macrophage activation increases with disease severity [69], and that activated macrophages may, in part, elicit neurotoxic effects through enhanced release of glutamate [70]. Additionally, axons innervating fast muscle fibers are known to degenerate earlier in the disease [71], leading to compensatory sprouting from surviving axons innervating slow muscle fibers which causes fiber type grouping [72]. We observed trends of reduced gene and protein expression of slow muscle fiber–associated proteins in our 2PMPA-treated animals, suggesting that this compensatory reorganization may have been reduced due to delayed degeneration. Further studies are required to confirm this.
The role of upregulated GCPII activity on infiltrating macrophages is likely secondary to axonal degeneration and not involved in the underlying ALS pathophysiology which is probably why we did not observe improvements in motor neuron nor animal survival. However, this mechanism could be applicable to other neuromuscular degenerative disorders where activated macrophages have detrimental effects on Schwann cells and axonal integrity such as Charcot-Marie-Tooth disease [73] or even the general aging process of axonal withdrawal [74].
Interestingly, GCPII inhibition may be altering the immune response and affecting disease progression by reducing harmful immune cell activation. GCPII inhibition has been shown to be effective in both inflammatory bowel disease [75] and inflammatory pain models (reviewed in [76]). As mentioned previously, chronic immune cell activation in the muscle can be pathological. Additionally, immune cells are known to modulate the extracellular matrix [77], which is a key component of nerve regeneration and repair [78]. Our group has reported that GCPII cleaves specific peptides derived from the extracellular matrix protein laminin which contain carboxy-terminal glutamate moieties (LQE, IEE, LNE) and that these liberated peptides can alter the ECM architecture [79].
In summary, the present study describes, for the first time, upregulated GCPII activity and expression in SOD1G93A mice, specifically associated with activated macrophages infiltrating the muscle. Normalization of this enhanced activity was shown to delay both muscle function loss and NMJ denervation, suggesting that targeted delivery of the potent GCPII inhibitor 2PMPA to activated macrophage using hydroxy-dendrimers may represent a new therapeutic strategy for neuromuscular disease.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Mohamed H Farah and Nicholas J Maragakis for their advice and support.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This worked was funded by NIH NCI grant R01CA16105 (to BSS), NIH NIA grant R01 AG068130 (to BSS), NIH NINDS grants R01NS093416 (to SK, RMK, and BSS) and R01NS113140 (to SK), and the National MS Society (to BSS). The work done by TJ was funded by NIH R25GM109441 (Hopkins PREP).
Disclosures
Under license agreements involving Ashvattha Therapeutics, Inc., and the Johns Hopkins University, Drs. Slusher, Rangaramanujam, and Kannan, and the Johns Hopkins University are entitled to royalty distributions and share ownership related to technology involved in the study discussed in this publication. Drs. Slusher, Rangaramanujam (co-founder), and Kannan (co-founder) hold equity in Ashvattha Therapeutics and serve on its Board of Directors. Additionally, the study discussed in this publication was funded by and involved a drug manufactured by Ashvattha Therapeutics, Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cleveland DW, Rothstein JD. From charcot to lou gehrig: deciphering selective motor neuron death in als. Nature Reviews Neuroscience. 2001;2(11):806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 2.Oskarsson B, Gendron TF, Staff NP. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin Proc. 2018;93(11):1617–1628. doi: 10.1016/j.mayocp.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Azzouz M, Leclerc N, Gurney M, Warter J-M, Poindron P, Borg J. Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle & Nerve. 1997;20(1):45–51. doi: 10.1002/(SICI)1097-4598(199701)20:1<45::AID-MUS6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Experimental Neurology. 2004;185(2):232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiology of Disease. 2007;28(2):154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Tallon C, Russell KA, Sakhalkar S, Andrapallayal N, Farah MH. Length-dependent axo-terminal degeneration at the neuromuscular synapses of type II muscle in SOD1 mice. Neuroscience. 2016;312:179–189. doi: 10.1016/j.neuroscience.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Personius KE, Slusher BS, Udin SB. Neuromuscular NMDA Receptors Modulate Developmental Synapse Elimination. J Neurosci. 2016;36(34):8783–8789. doi: 10.1523/JNEUROSCI.1181-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson MB, Blakely RD, Couto R, Coyle JT. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. Journal of Biological Chemistry. 1987;262(30):14498–506. [PubMed]
- 9.Slusher BS, Robinson MB, Tsai G, Simmons ML, Richards SS, Coyle JT. Rat brain N-acetylated alpha-linked acidic dipeptidase activity. Purification and immunologic characterization. Journal of Biological Chemistry. 1990;265(34):21297–301. [PubMed]
- 10.Neale JH. N-acetylaspartylglutamate is an agonist at mGluR3 in vivo and in vitro. Journal of neurochemistry. 2011;119(5):891–895. doi: 10.1111/j.1471-4159.2011.07380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 12.Volianskis A, France G, Jensen MS, Bortolotto ZA, Jane DE, Collingridge GL. Long-term potentiation and the role of N-methyl-D-aspartate receptors. Brain research. 2015;1621:5–16. doi: 10.1016/j.brainres.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger UV, Carter RE, Coyle JT. The immunocytochemical localization of N-acetylaspartyl glutamate, its hydrolysing enzyme NAALADase, and the NMDAR-1 receptor at a vertebrate neuromuscular junction. Neuroscience. 1995;64(4):847–850. doi: 10.1016/0306-4522(95)92578-8. [DOI] [PubMed] [Google Scholar]
- 14.Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzanti BA, et al. Design, Synthesis, and Biological Activity of a Potent Inhibitor of the Neuropeptidase N-Acetylated α-Linked Acidic Dipeptidase. Journal of Medicinal Chemistry. 1996;39(2):619–622. doi: 10.1021/jm950801q. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nature Reviews Drug Discovery. 2005;4(12):1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
- 16.Vornov JJ, Hollinger KR, Jackson PF, Wozniak KM, Farah MH, Majer P, et al. Chapter Nine - Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. In: Schwarcz R, editor. Advances in Pharmacology. 76: Academic Press; 2016. p. 215–55. [DOI] [PubMed]
- 17.Rais R, Wozniak K, Wu Y, Niwa M, Stathis M, Alt J, et al. Selective CNS Uptake of the GCP-II Inhibitor 2-PMPA following Intranasal Administration. PLOS ONE. 2015;10(7):e0131861. [DOI] [PMC free article] [PubMed]
- 18.van der Post JP, de Visser SJ, de Kam ML, Woelfler M, Hilt DC, Vornov J, et al. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br J Clin Pharmacol. 2005;60(2):128–136. doi: 10.1111/j.1365-2125.2005.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffe IA. Adverse effects profile of sulfhydryl compounds in man. The American Journal of Medicine. 1986;80(3):471–476. doi: 10.1016/0002-9343(86)90722-9. [DOI] [PubMed] [Google Scholar]
- 20.Hari NP, Umashankar D, Rajendra KS, Jonathan RD. Cytotoxic Thiol Alkylators. Mini-Reviews in Medicinal Chemistry. 2007;7(2):131–139. doi: 10.2174/138955707779802642. [DOI] [PubMed] [Google Scholar]
- 21.Smith ES, Porterfield JE, Kannan RM. Leveraging the interplay of nanotechnology and neuroscience: Designing new avenues for treating central nervous system disorders. Advanced drug delivery reviews. 2019;148:181–203. doi: 10.1016/j.addr.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, Lin Y-A, Kannan S, Kannan RM. Targeting specific cells in the brain with nanomedicines for CNS therapies. J Control Release. 2016;240:212–226. doi: 10.1016/j.jconrel.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollinger KR, Sharma A, Tallon C, Lovell L, Thomas AG, Zhu X, et al. Dendrimer-2PMPA selectively blocks upregulated microglial GCPII activity and improves cognition in a mouse model of multiple sclerosis. Nanotheranostics. 2022;6(2):126–142. doi: 10.7150/ntno.63158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma A, Porterfield JE, Smith E, Sharma R, Kannan S, Kannan RM. Effect of mannose targeting of hydroxyl PAMAM dendrimers on cellular and organ biodistribution in a neonatal brain injury model. Journal of Controlled Release. 2018;283:175–189. doi: 10.1016/j.jconrel.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra MK, Beaty CA, Lesniak WG, Kambhampati SP, Zhang F, Wilson MA, et al. Dendrimer Brain Uptake and Targeted Therapy for Brain Injury in a Large Animal Model of Hypothermic Circulatory Arrest. ACS Nano. 2014;8(3):2134–2147. doi: 10.1021/nn404872e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A, Sharma R, Zhang Z, Liaw K, Kambhampati SP, Porterfield JE, et al. Dense hydroxyl polyethylene glycol dendrimer targets activated glia in multiple CNS disorders. Science Advances. 2020;6(4):eaay8514. [DOI] [PMC free article] [PubMed]
- 27.Sharma R, Kim S-Y, Sharma A, Zhang Z, Kambhampati SP, Kannan S, et al. Activated Microglia Targeting Dendrimer-Minocycline Conjugate as Therapeutics for Neuroinflammation. Bioconjugate Chemistry. 2017;28(11):2874–2886. doi: 10.1021/acs.bioconjchem.7b00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu. Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 29.Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, et al. Background and gender effects on survival in the TgN(SOD1-G93A)1Gur mouse model of ALS. Journal of the Neurological Sciences. 2005;236(1):1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Nance E, Kambhampati SP, Smith ES, Zhang Z, Zhang F, Singh S, et al. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. Journal of neuroinflammation. 2017;14(1):252-. [DOI] [PMC free article] [PubMed]
- 31.Lepore AC, Haenggeli C, Gasmi M, Bishop KM, Bartus RT, Maragakis NJ, et al. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Research. 2007;1185:256–265. doi: 10.1016/j.brainres.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rojas C, Frazier ST, Flanary J, Slusher BS. Kinetics and inhibition of glutamate carboxypeptidase II using a microplate assay. Anal Biochem. 2002;310(1):50–54. doi: 10.1016/S0003-2697(02)00286-5. [DOI] [PubMed] [Google Scholar]
- 33.Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, et al. Motor Neuron Disease Occurring in a Mutant Dynactin Mouse Model Is Characterized by Defects in Vesicular Trafficking. The Journal of Neuroscience. 2008;28(9):1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, et al. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1G93A mice. Experimental Neurology. 2006;201(1):120–130. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2007;9(2):321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 36.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu X, Nedelcovych MT, Thomas AG, Hasegawa Y, Moreno-Megui A, Coomer W, et al. JHU-083 selectively blocks glutaminase activity in brain CD11b(+) cells and prevents depression-associated behaviors induced by chronic social defeat stress. Neuropsychopharmacology. 2019;44(4):683–694. doi: 10.1038/s41386-018-0177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakamoto S, Mallah D, Medeiros DJ, Dohi E, Imai T, Rose IVL, et al. Alterations in circulating extracellular vesicles underlie social stress-induced behaviors in mice. FEBS Open Bio. 2021;11(10):2678–2692. doi: 10.1002/2211-5463.13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angewandte Chemie International Edition. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Sharma R, Sharma A, Kambhampati SP, Reddy RR, Zhang Z, Cleland JL, et al. Scalable synthesis and validation of PAMAM dendrimer-N-acetyl cysteine conjugate for potential translation. Bioeng Transl Med. 2018;3(2):87–101. doi: 10.1002/btm2.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kambhampati SP, Mishra MK, Mastorakos P, Oh Y, Lutty GA, Kannan RM. Intracellular delivery of dendrimer triamcinolone acetonide conjugates into microglial and human retinal pigment epithelial cells. Eur J Pharm Biopharm. 2015;95(Pt B):239–249. doi: 10.1016/j.ejpb.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, Nance E, Zhang Z, Jasty V, Kambhampati SP, Mishra MK, et al. Surface functionality affects the biodistribution and microglia-targeting of intra-amniotically delivered dendrimers. J Control Release. 2016;237:61–70. doi: 10.1016/j.jconrel.2016.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nance E, Zhang F, Mishra MK, Zhang Z, Kambhampati SP, Kannan RM, et al. Nanoscale effects in dendrimer-mediated targeting of neuroinflammation. Biomaterials. 2016;101:96–107. doi: 10.1016/j.biomaterials.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma R, Liaw K, Sharma A, Jimenez A, Chang M, Salazar S, et al. Glycosylation of PAMAM dendrimers significantly improves tumor macrophage targeting and specificity in glioblastoma. Journal of Controlled Release. 2021;337:179–192. doi: 10.1016/j.jconrel.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang F, Trent Magruder J, Lin Y-A, Crawford TC, Grimm JC, Sciortino CM, et al. Generation-6 hydroxyl PAMAM dendrimers improve CNS penetration from intravenous administration in a large animal brain injury model. J Control Release. 2017;249:173–182. doi: 10.1016/j.jconrel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lesniak WG, Mishra MK, Jyoti A, Balakrishnan B, Zhang F, Nance E, et al. Biodistribution of fluorescently labeled PAMAM dendrimers in neonatal rabbits: effect of neuroinflammation. Mol Pharm. 2013;10(12):4560–4571. doi: 10.1021/mp400371r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferraris DV, Majer P, Ni C, Slusher CE, Rais R, Wu Y, et al. delta-Thiolactones as prodrugs of thiol-based glutamate carboxypeptidase II (GCPII) inhibitors. J Med Chem. 2014;57(1):243–247. doi: 10.1021/jm401703a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majer P, Jackson PF, Delahanty G, Grella BS, Ko Y-S, Li W, et al. Synthesis and Biological Evaluation of Thiol-Based Inhibitors of Glutamate Carboxypeptidase II: Discovery of an Orally Active GCP II Inhibitor. Journal of Medicinal Chemistry. 2003;46(10):1989–1996. doi: 10.1021/jm020515w. [DOI] [PubMed] [Google Scholar]
- 49.Vornov JJ, Wozniak KM, Wu Y, Rojas C, Rais R, Slusher BS. Pharmacokinetics and pharmacodynamics of the glutamate carboxypeptidase II inhibitor 2-MPPA show prolonged alleviation of neuropathic pain through an indirect mechanism. J Pharmacol Exp Ther. 2013;346(3):406–413. doi: 10.1124/jpet.113.205039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghadge GD, Slusher BS, Bodner A, Canto MD, Wozniak K, Thomas AG, et al. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9554–9559. doi: 10.1073/pnas.1530168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science (New York, NY). 2009;326(5959):1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wooley CM, Sher RB, Kale A, Frankel WN, Cox GA, Seburn KL. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle & nerve. 2005;32(1):43–50. doi: 10.1002/mus.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acevedo-Arozena A, Kalmar B, Essa S, Ricketts T, Joyce P, Kent R, et al. A comprehensive assessment of the <em>SOD1<sup>G93A</sup></em> low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Disease Models & Mechanisms. 2011;4(5):686–700. doi: 10.1242/dmm.007237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cacabelos D, Ramírez-Núñez O, Granado-Serrano AB, Torres P, Ayala V, Moiseeva V, et al. Early and gender-specific differences in spinal cord mitochondrial function and oxidative stress markers in a mouse model of ALS. Acta Neuropathologica Communications. 2016;4(1):3. doi: 10.1186/s40478-015-0271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, et al. Benefit of vitamin E, riluzole, and gababapentin in a transgenic model of familial amyotrophic lateral sclerosis. Annals of Neurology. 1996;39(2):147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- 56.Snow RJ, Turnbull J, da Silva S, Jiang F, Tarnopolsky MA. Creatine supplementation and riluzole treatment provide similar beneficial effects in copper, zinc superoxide dismutase (G93A) transgenic mice. Neuroscience. 2003;119(3):661–667. doi: 10.1016/S0306-4522(03)00212-4. [DOI] [PubMed] [Google Scholar]
- 57.Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotrophic Lateral Sclerosis. 2008;9(1):4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 58.Hogg MC, Halang L, Woods I, Coughlan KS, Prehn JHM. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2018;19(5–6):438–445. doi: 10.1080/21678421.2017.1407796. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Sung M, Rutkove SB. Electrophysiologic Biomarkers for Assessing Disease Progression and the Effect of Riluzole in SOD1 G93A ALS Mice. PLOS ONE. 2013;8(6):e65976. [DOI] [PMC free article] [PubMed]
- 60.Šácha P, Zámečník J, Bařinka C, Hlouchová K, Vícha A, Mlčochová P, et al. Expression of glutamate carboxypeptidase II in human brain. Neuroscience. 2007;144(4):1361–1372. doi: 10.1016/j.neuroscience.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 61.Berger UV, Carter RE, McKee M, Coyle JT. N-acetylated alpha-linked acidic dipeptidase is expressed by non-myelinating Schwann cells in the peripheral nervous system. Journal of Neurocytology. 1995;24(2):99–109. doi: 10.1007/BF01181553. [DOI] [PubMed] [Google Scholar]
- 62.Knedlík T, Vorlová B, Navrátil V, Tykvart J, Sedlák F, Vaculín Š, et al. Mouse glutamate carboxypeptidase II (GCPII) has a similar enzyme activity and inhibition profile but a different tissue distribution to human GCPII. FEBS Open Bio. 2017;7(9):1362–1378. doi: 10.1002/2211-5463.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Z, Bassam B, Thomas AG, Williams M, Liu J, Nance E, et al. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiology of Disease. 2016;94:116–128. doi: 10.1016/j.nbd.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scheib J, Höke A. Advances in peripheral nerve regeneration. Nature Reviews Neurology. 2013;9(12):668–676. doi: 10.1038/nrneurol.2013.227. [DOI] [PubMed] [Google Scholar]
- 65.Liu P, Peng J, Han G-H, Ding X, Wei S, Gao G, et al. Role of macrophages in peripheral nerve injury and repair. Neural Regen Res. 2019;14(8):1335–1342. doi: 10.4103/1673-5374.253510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murdock BJ, Bender DE, Segal BM, Feldman EL. The dual roles of immunity in ALS: Injury overrides protection. Neurobiology of Disease. 2015;77:1–12. doi: 10.1016/j.nbd.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 67.Sakaguchi S, Shono J-i, Suzuki T, Sawano S, Anderson JE, Do M-KQ, et al. Implication of anti-inflammatory macrophages in regenerative moto-neuritogenesis: Promotion of myoblast migration and neural chemorepellent semaphorin 3A expression in injured muscle. The International Journal of Biochemistry & Cell Biology. 2014;54:272–85. [DOI] [PubMed]
- 68.Van Dyke JM, Smit-Oistad IM, Macrander C, Krakora D, Meyer MG, Suzuki M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS) Experimental neurology. 2016;277:275–282. doi: 10.1016/j.expneurol.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, Hadlock K, et al. Evidence for systemic immune system alterations in sporadic amyotrophic lateral sclerosis (sALS) Journal of Neuroimmunology. 2005;159(1):215–224. doi: 10.1016/j.jneuroim.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A. Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sciences. 2008;82(21):1111–6. [DOI] [PubMed]
- 71.Saxena S, Caroni P. Mechanisms of axon degeneration: From development to disease. Progress in Neurobiology. 2007;83(3):174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Jensen L, Jørgensen LH, Bech RD, Frandsen U, Schrøder HD. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. Biomed Res Int. 2016;2016:5930621-. [DOI] [PMC free article] [PubMed]
- 73.Groh J, Klein I, Hollmann C, Wettmarshausen J, Klein D, Martini R. CSF-1-activated macrophages are target-directed and essential mediators of schwann cell dedifferentiation and dysfunction in Cx32-deficient mice. Glia. 2015;63(6):977–986. doi: 10.1002/glia.22796. [DOI] [PubMed] [Google Scholar]
- 74.Yuan X, Klein D, Kerscher S, West BL, Weis J, Katona I, et al. Macrophage depletion ameliorates peripheral neuropathy in aging mice. The Journal of Neuroscience. 2018:3030–17. [DOI] [PMC free article] [PubMed]
- 75.Rais R, Jiang W, Zhai H, Wozniak KM, Stathis M, Hollinger KR, et al. FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight. 2016;1(12). [DOI] [PMC free article] [PubMed]
- 76.Neale JH, Yamamoto T. N-acetylaspartylglutamate (NAAG) and glutamate carboxypeptidase II: An abundant peptide neurotransmitter-enzyme system with multiple clinical applications. Progress in Neurobiology. 2020;184:101722. [DOI] [PubMed]
- 77.Tomlin H, Piccinini AM. A complex interplay between the extracellular matrix and the innate immune response to microbial pathogens. Immunology. 2018;155(2):186–201. doi: 10.1111/imm.12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Luca AC, Lacour SP, Raffoul W, di Summa PG. Extracellular matrix components in peripheral nerve repair: how to affect neural cellular response and nerve regeneration? Neural Regen Res. 2014;9(22):1943–1948. doi: 10.4103/1673-5374.145366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conway RE, Rojas C, Alt J, Nováková Z, Richardson SM, Rodrick TC, et al. Prostate-specific membrane antigen (PSMA)-mediated laminin proteolysis generates a pro-angiogenic peptide. Angiogenesis. 2016;19(4):487–500. doi: 10.1007/s10456-016-9521-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Under license agreements involving Ashvattha Therapeutics, Inc., and the Johns Hopkins University, Drs. Slusher, Rangaramanujam, and Kannan, and the Johns Hopkins University are entitled to royalty distributions and share ownership related to technology involved in the study discussed in this publication. Drs. Slusher, Rangaramanujam (co-founder), and Kannan (co-founder) hold equity in Ashvattha Therapeutics and serve on its Board of Directors. Additionally, the study discussed in this publication was funded by and involved a drug manufactured by Ashvattha Therapeutics, Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.